Leukocytosis and Leukopenia
Nancy Berliner
The normal peripheral white blood cell count (WBC) ranges between 4500/µL and 10,000/µL, with a mean of 7500/µL, and is composed of neutrophils, lymphocytes, monocytes, basophils, and eosinophils. Because neutrophils usually represent about 60% of the peripheral WBC, derangement in the WBC usually reflects elevation or reduction in the absolute neutrophil count. Leukocytosis, an elevated WBC, and leukopenia, a depressed WBC, may be secondary to an underlying disease or exposure, or they may be manifestations of a primary hematologic disorder. This chapter outlines both the primary and secondary causes of leukocytosis and leukopenia, focusing particularly on neutrophilia and neutropenia.
 Normal Neutrophil Dynamics
Normal Neutrophil Dynamics
Neutrophils arise from multipotent progenitor cells in the bone marrow that also give rise to erythrocytes, megakaryocytes, eosinophils, basophils, and monocytes. Neutrophil precursors in the marrow mature over 6 to 10 days to form a storage pool of mature neutrophils (Fig. 167-1). Together the marrow populations make up about 95% of the body's total granulocyte mass (20% neutrophil precursors, 75% mature bands and neutrophils). The circulating neutrophil pool thus represents only the remaining approximately 5% of the body's total neutrophils, just over half of which at any given time are adherent to the vascular endothelium and the spleen, a phenomenon termed margination. These marginated neutrophils are poised for immediate release into the circulation during times of stress. The remaining neutrophils circulate freely in the blood. The lifespan of neutrophils in the peripheral blood was thought to be very short, only 6 to 12 hours; however, newer in vivo studies suggest that they circulate for up to 3 to 4 days.1 They subsequently migrate into tissues, where they can survive for 1 to 4 days. Changes in neutrophil number reflect these dynamics. Neutrophilia can occur as the result of increased marrow production, increased release of neutrophils from the storage pool, or mobilization of neutrophils from the marginated pool. Neutropenia, on the other hand, may be due to decreased marrow production, increased margination with or without sequestration by the spleen, or increased destruction of peripheral cells.
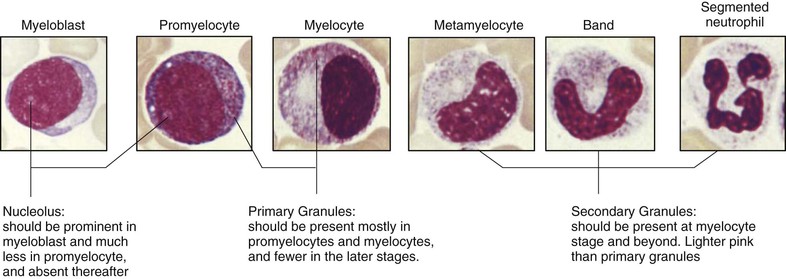
Neutrophilia
Most cases of neutrophilia are reactive or secondary to an underlying inflammatory process. This includes neutrophilia due to infection, chronic inflammation, stress, drugs, nonhematologic malignancy, marrow stimulation (as in hemolysis or idiopathic thrombocytopenic purpura), or splenectomy. Primary causes of neutrophilia may be congenital, including hereditary neutrophilia, Down syndrome, and leukocyte adhesion deficiency (LAD), or acquired as in the case of chronic myelogenous leukemia and other myeloproliferative neoplasms (Table 167-1).
TABLE 167-1
DIFFERENTIAL DIAGNOSIS OF NEUTROPHILIA

 Etiology
Etiology
Secondary Causes of Neutrophilia
Many acute bacterial infections can present with a modest leukocytosis with a “left shift,” referring to the circulation of more immature myeloid cells. This left shift most commonly is restricted to release of an increased number of band forms; however, in severe stress, one may see circulating metamyelocytes and even earlier cells (see Fig. 167-1) in the peripheral blood. Leukocytosis occurs within minutes to hours of infection owing to release of neutrophils from both the marrow and marginated pools. Examination of these neutrophils on peripheral smear may reveal evidence of toxic granulation (see Fig. 157-18), Döhle bodies (see Fig. 157-19), and cytoplasmic vacuoles. Certain infections (e.g., Clostridium difficile or tuberculosis in particular) are known to cause elevations in the WBC to greater than 30,000/µL in about one fourth of infected patients and may result in a leukemoid reaction, defined as a WBC of greater than 50,000/µL with a pronounced left shift (Fig. 167-2).
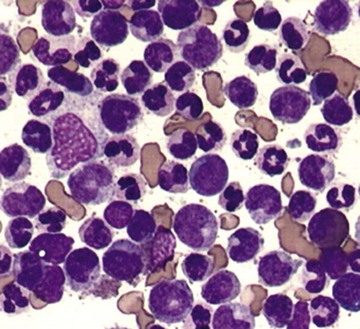
Chronic Inflammation
Leukocytosis due to chronic inflammation results from increased leukocyte (specifically neutrophil and monocyte) production as opposed to altered neutrophil distribution. Mature neutrophil pools become depleted with ongoing inflammation, and the myeloid compartment of the marrow expands to increase neutrophil production. Myriad cytokines, including tumor necrosis factor-α (TNF-α), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage inflammatory protein-1 (MIP-1), interleukin-1 (IL-1), IL-6, and IL-8, have been implicated in this marrow stimulation (Chapter 156). Chronic inflammatory conditions that are particularly associated with leukocytosis and neutrophilia include juvenile rheumatoid arthritis, rheumatoid arthritis, Still disease, Crohn disease, ulcerative colitis, granulomatous infection, and chronic hepatitis. The WBC and neutrophil elevation in these cases is typically more modest than that seen in acute infection or inflammation.
Cigarette Smoking
Cigarette smoking can cause a leukocytosis and neutrophilia in about 25 to 50% of chronic smokers that can persist for even up to 5 years after quitting smoking. The mechanism by which this occurs is unknown, although there is recent evidence that cigarette smoke slows neutrophil apoptosis.2
Stress
Within minutes of exercise, surgery, or stress, one can see an elevation in circulating neutrophils. This is presumed to be due to the effects of catecholamines on marginated neutrophils, with release of neutrophils into the circulation. Some cases of stress-induced neutrophilia can be prevented by pretreatment with β-adrenergic antagonists (e.g., propranolol), supporting the role of catecholamines in the process. Exercise-induced neutrophilia, however, is not blocked by propranolol, suggesting that it may instead be due to flow and mechanical perturbation of neutrophils in the lungs. An elevated WBC has also been noted in the setting of acute myocardial infarction, but whether this is a risk factor for cardiac ischemia or a result of inflammation is unclear.
Drug Induced
Probably the most well-known and widely used drugs associated with leukocytosis are corticosteroids. Other drugs that are associated with elevations in the neutrophil count include β-agonists and lithium. Lithium causes neutrophilia by increasing the production of endogenous colony-stimulating factors (CSFs). G-CSF or GM-CSF treatment likewise may result in neutrophilia, and although this is the desired effect, the neutrophilia can be quite pronounced if not appropriately monitored.
Nonhematologic Malignancy
Leukocytosis can be seen in a number of nonhematologic malignancies. Some tumors (lung, tongue, kidney, bladder) are thought to secrete G-CSF as an ectopic hematopoietic growth factor. Other tumors (lung, stomach, breast), when metastasized to the bone and bone marrow, can cause a leukoerythroblastic reaction, characterized by left-shifted leukocytosis, thrombocytosis, and red cell abnormalities including nucleated and teardrop-shaped red blood cells (Fig. 167-3). The presence of nonhematopoietic entities invading the bone marrow (metastatic cancer, fibrosis, granulomatous disease) is termed myelophthisis.
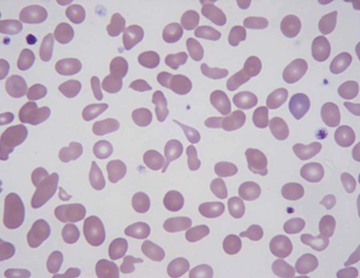
Marrow Stimulation
Peripheral destruction of red cells and platelets, as seen with hemolytic anemia and idiopathic thrombocytopenic purpura, can result in stimulation of the bone marrow and a “spillover” leukocytosis. Recovery of cell counts following marrow suppression, as in the case of chemotherapy, can result in a rebound leukocytosis that may last several weeks.
Primary Causes of Neutrophilia
Hereditary neutrophilia is an autosomal dominant genetic disease that is characterized by an elevated WBC in the 20,000 to 100,000/µL range with splenomegaly and widened diploe of the skull. The neutrophils in this disorder appear to function normally, and patients have no increased risk for bacterial infection or other sequelae. Hereditary neutrophilia caused by an autosomal-dominant GCSF3 gene mutation has been reported, causing constitutive activation of the G-CSF receptor.
Chronic Idiopathic Neutrophilia
Chronic idiopathic neutrophilia is a condition marked by leukocytosis in the 11,000 to 40,000/µL range with a normal bone marrow biopsy. In one series with a 20-year follow-up, patients with this condition had no medical sequelae from this elevated WBC.
Pelger-Huët Anomaly
Patients with the Pelger-Huët anomaly (PHA) are often misdiagnosed as having a left-shifted WBC because many of their mature neutrophils are misinterpreted as band forms. Although these patients do not actually have leukocytosis, the anomaly often raises suspicion for an acute infection or inflammatory process because of this apparent left shift. PHA is due to a mutation in the lamin B receptor gene and manifests with mature neutrophils and condensed, clumped chromatin within a bilobed nucleus (see Fig. 157-20). These neutrophils, however, function normally. A number of drugs can reversibly induce pseudo-PHA, including colchicine, sulfonamides, ibuprofen, taxoids, and valproate. Pseudo-PHA is also seen in some patients with myelodysplasia (Chapter 182). Vitamin B12 or folate deficiency can cause increased nuclear lobation of neutrophils in patients with PHA, perhaps leading to a missed diagnosis. With correction of the vitamin deficiency, however, the aberrant neutrophil nuclear morphology returns.
Down Syndrome
Up to 10% of patients with Down syndrome develop transient myeloproliferative disorder (TMD) related to peripheral blood leukocytosis with blasts in association with an accumulation of megakaryoblasts in the blood, liver, and marrow. Similar reactions have also been reported in patients with trisomy 21 mosaicism who are phenotypically normal. TMD resolves spontaneously in most patients but can progress to acute megakaryoblastic leukemia (AMKL) in 23 to 30% of affected patients. This disorder is attributable to acquisition of mutations in the GATA1 gene, which encodes a key transcription factor for hematopoietic regulation, leading to loss of normal GATA-1 expression and expression of a truncated GATA-1 protein. Evidence supports that these mutations are acquired during fetal life, and patients present in early infancy with TMD. The pathogenesis of progression to AMKL presumably requires additional genetic events and sequential epigenetic changes, and is the focus of intense study.3,4
Leukocyte Adhesion Deficiency
Patients with leukocyte adhesion deficiency (LAD) (see also Chapter 169) have persistent leukocytosis, defects in stimulus-dependent activation of neutrophils, recurrent infections, and delayed separation of the umbilical cord. LAD is an abnormality of leukocyte adhesion reflecting the loss of surface adhesion molecules. LAD-1 is due to absence or marked reduction in the common β chain of β2 integrins, resulting in loss of expression of leukocyte function−associated antigen 1 (LFA-1), the C3bi receptor, and GP150;95. This results in a failure to ingest and kill microbes opsonized by C3bi. In LAD-2, neutrophils lack sialyl Lewis X, the ligand for L-selectin expressed on endothelial cells. Neutrophils appear morphologically normal but are defective in chemotaxis, adherence, and phagocytosis.5
Familial Cold Urticaria
Familial cold urticaria is marked by episodic fevers, leukocytosis, urticaria, rash, conjunctivitis, and muscle and skin tenderness with cold exposure. The rash is composed of infiltrating neutrophils. The syndrome appears to be related to decreased levels of C1-esterase inhibitor and is associated with mutations in the CIAS1 gene on chromosome 1q.
Chronic Myelogenous Leukemia and Other Myeloproliferative Disorders
Chronic myelogenous leukemia (CML), chronic neutrophilic leukemia (CNL), and the other myeloproliferative neoplasms (namely polycythemia vera [PV] and essential thrombocythemia [ET]) are discussed in detail in Chapters 184 and 166. They are the principal acquired primary hematologic disorders associated with neutrophilia. They are marked by clonal expansion of myeloid precursors and increased release of both immature and mature myeloid cells into the peripheral blood. CML on presentation often has to be distinguished from a leukemoid reaction. In contrast to a leukemoid reaction, CML is characterized by the presence of abnormalities of other blood cell lines (“panmyelosis”) and by the presence of specific abnormalities. Therefore, the peripheral blood smear in CML (but not leukemoid reaction) demonstrates increased numbers in all cells of the neutrophilic series, classically with a greater proportion of myelocytes to metamyelocytes, and may display concomitant basophilia, eosinophilia, anemia, and thrombocytosis. CNL, a rare myeloproliferative neoplasm, is characterized by hepato/splenomegaly and leukocytosis of at least 25,000/µL, with more than 80% of leukocytes being segmented neutrophil/band forms and less than 10% being immature granulocytes, in contrast to CML.6 CML is characterized by the diagnostic presence of the Philadelphia chromosome [t(9;22)], which can be identified in the peripheral blood by the detection of the BCR-ABL translocation by fluorescence in situ hybridization (FISH) or reverse transcription–polymerase chain reaction (RT-PCR). At least 50% of patients with CNL, in contrast, harbor mutations in the receptor for CSF-3 (CSF3R; GCSFR).6 PV and ET, on the other hand, are notable for also having a marked increase in red cell mass and a marked thrombocytosis, respectively, which is often accompanied by leukocytosis.
The leukocyte alkaline phosphatase (LAP) score was historically used in the laboratory evaluation of granulocytosis as a diagnostic marker for myeloproliferative neoplasm. The LAP score is very low (usually 0) in the setting of CML and elevated in PV. The LAP score has a very wide normal range and in practical terms was only definitively helpful in the setting of CML because “high” LAP scores can also be seen in infectious and inflammatory settings. With the availability of direct molecular genetic diagnosis of CML by assay for the BCR-ABL fusion gene, it can no longer be recommended as a diagnostic test.
Post-splenectomy
Patients develop leukocytosis following splenectomy, and this may be long-standing, reflecting the loss of a major site of neutrophil margination. This is of no clinical importance, except insofar as it leads to unnecessary evaluation for other pathology.
Clinical Manifestations and Diagnosis
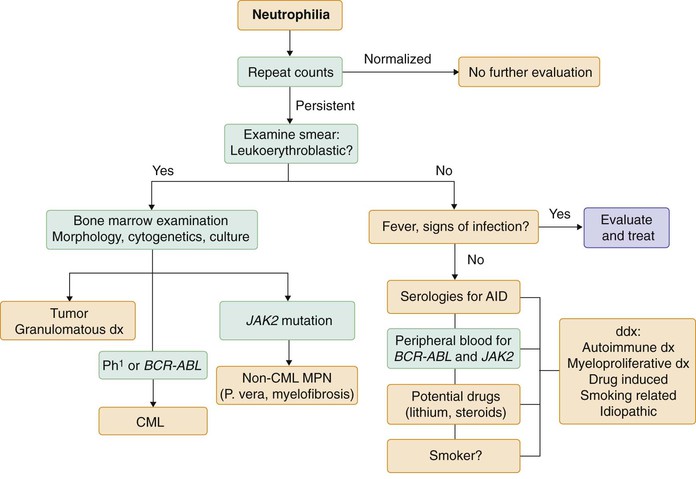
As outlined previously, acquired leukocytosis is most commonly the result of acute or chronic infection or inflammation. When it occurs in the absence of fever, aberrations in acute phase reactants, effusions and edema, or other signs and symptoms of inflammation, it still may be secondary to drugs or an underlying nonhematologic malignancy. As such, it should be seen as the sign of a healthy hematopoietic system responding to an outside stress. Bone marrow evaluation is therefore rarely indicated. However, persistence of leukocytosis in the absence of signs and symptoms of inflammation or infection, nonhematologic malignancy, and offending drugs should prompt an evaluation for a primary myeloproliferative disease or clonal hematologic neoplasm, particularly when there is evidence of a leukoerythroblastic reaction. CML and other myeloproliferative neoplasms can be ruled out by molecular diagnosis on the peripheral blood, as described previously and in Chapter 166. In this setting, bone marrow examination is indicated to evaluate for marrow infiltration by infection, tumor, or fibrosis and should include cultures for tuberculosis and fungal infection as well as cytogenetics and flow cytometry (Fig. 167-4).
Leukocytosis Due to Expansion of Other Cell Lines
Monocytosis and lymphocytosis can also lead to elevations of the WBC. Monocytosis is defined by an absolute monocyte count of greater than 500/µL and usually occurs in the setting of chronic inflammation resulting from infections like tuberculosis, syphilis, or subacute bacterial endocarditis, autoimmune or granulomatous disease, and sarcoidosis. It can also be seen in malignancies, such as preleukemic states, nonlymphocytic leukemia including acute myelomonocytic and monocytic leukemia, histiocytosis, Hodgkin disease, non-Hodgkin lymphoma, and various carcinomas. Finally, it can be seen in the setting of chronic neutropenia, after splenectomy, and in the setting of recovery from marrow suppression (Table 167-2).
TABLE 167-2
DIFFERENTIAL DIAGNOSIS OF MONOCYTOSIS
Lymphocytosis is defined by an absolute lymphocyte count of more than 5000/µL. The most common causes of an elevated lymphocyte count are viral infections such as Epstein-Barr virus and the hepatitis viruses. Although most bacterial infections cause neutrophilia, pertussis and cat-scratch disease due to Bartonella henselae can cause an impressive lymphocytosis. Other infections that may cause a secondary lymphocytosis include toxoplasmosis and babesiosis. Hypersensitivity reactions due to drugs or serum sickness may also be associated with lymphocytosis. Primary disorders that cause a lymphocytosis include chronic lymphocytic leukemia (CLL) and monoclonal B-cell lymphocytosis (Table 167-3; see also Table 184-2 and Chapter 184).

Eosinophilia is defined by an absolute eosinophil count of more than 400/µL. Eosinophils proliferate under the influence of IL-5 and play a role in phagocytosis and modulating toxicity due to mast cell degranulation in hypersensitivity reactions. Eosinophilia is therefore most often seen in the setting of drug reactions, allergy, atopy, and asthma. A variety of infections, particularly parasitic infections and, to a lesser degree, fungal infections, can be associated with an increased number of circulating eosinophils. Eosinophilia can also be the result of autoimmune and inflammatory conditions, as in Churg-Strauss vasculitis. Atheroembolic disease and adrenal insufficiency may also cause eosinophilia. A number of cancers have been associated with polytypic expansion of eosinophils, including lymphomas and solid tumors. There are also a number of clonal disorders of eosinophils that occur in the setting of some leukemias. Finally, there is a heterogeneous group of disorders termed hypereosinophilic syndromes. A FIP1L1-PDGFRA fusion gene has confirmed that some of these are primary clonal disorders of eosinophils; the clonality of other hypereosinophilic syndromes can be difficult to establish (see Table 170-1).
Neutropenia
The risk for infection in the setting of neutropenia is highly dependent on the size of the neutrophil storage pool. Although neutropenia is defined by an absolute neutrophil count of less than 1500/µL, patients with neutrophil counts below this number may have different risks and rates of infection depending on the cause of neutropenia. For instance, patients who are neutropenic owing to chemotherapy, marrow failure, or marrow exhaustion experience infection at much higher rates than those with chronic neutropenic syndromes and immune-mediated neutropenia. Neutropenia may be congenital or acquired. The following sections first discuss congenital neutropenic disorders, the study of which has provided critical insights into normal myelopoiesis, and then the secondary causes of neutropenia (Table 167-4).
TABLE 167-4
DIFFERENTIAL DIAGNOSIS OF NEUTROPENIA

Etiology
Ethnic and Benign Familial (Constitutional) Neutropenia
The normal range of the neutrophil count is genetically determined and can be variable. A number of racial and ethnic groups have been observed to have a relatively large proportion of members who are neutropenic by comparison to the published normal range, usually based on young, largely white individuals. This is termed constitutional neutropenia and is seen among a variety of ethnic groups, including African Americans, Yemenite Jews, Falasha Jews, and African Bedouins. Single-nucleotide polymorphisms in the gene for the Duffy antigen receptor for chemokine (DARC) have been shown to associate with race and are a postulated candidate to explain racial and ethnic differences in neutrophil counts.7 There is also an autosomal dominantly inherited condition called benign familial neutropenia that is characterized by neutrophil counts in the 800 to 1400/µL range. Neither ethnic nor benign familial neutropenia has been shown to be associated with any increased risk for infection.
Severe Congenital Neutropenia
First described by Rolf Kostmann in 1956, severe congenital neutropenia (SCN) is a disorder of severe neutropenia with neutrophil counts of less than 500/µL, presenting in the neonatal period with recurrent bacterial infections. These infections can occur as early as the first months of life and often include omphalitis and perirectal abscesses. There is often an increase in other myeloid cell lines, including monocytes and eosinophils. Bone marrow biopsy in SCN patients reveals a “maturation arrest” with an absence of mature neutrophil elements. SCN can follow autosomal dominant and recessive and X-linked patterns of inheritance and has been shown to be associated with mutations in a variety of genes, as summarized in Table 167-5.
TABLE 167-5
CONGENITAL NEUTROPENIA SYNDROMES
| SYNDROME | INHERITANCE PATTERN | GENE |
| SCN | Autosomal dominant | ELANE (~60%) |
| Autosomal recessive | HAX1 (~5%) | |
| X-linked | WASP (~5%) | |
| X-linked | TAZ (rare) | |
| Autosomal recessive | G6PC3 (~2%) | |
| Autosomal dominant | Gfi1 (rare) | |
| Autosomal dominant | G-CSFR (rare) | |
| CYCLIC NEUTROPENIA | Autosomal dominant | ELANE |
| OTHER CONGENITAL SYNDROMES | ||
| Shwachman-Diamond syndrome | X-Linked | SBDS |
| Fanconi anemia | Autosomal recessive | FANCA-FANCO |
| Dyskeratosis congenita | Variable | Telomerase and related genes |
| Glycogen storage disease type 1b | Autosomal recessive | G-6-PT |
| Myelokathexis | Autosomal dominant | CXCR4 |
| Chédiak-Higashi syndrome | Autosomal recessive | LYST |
| Griscelli syndrome type 2 | Autosomal recessive | RAB27A |
| Hermansky-Pudlak syndrome type 2 | Autosomal recessive | AP3B1 |
Neutrophil elastase (ELA2, now ELANE) is a serine protease that is synthesized at high levels in the promyelocyte stage of neutrophil maturation and is packaged in primary granules. It was originally hypothesized that mutations in ELANE led to its defective cellular trafficking and cytoplasmic accumulation, subsequently triggering neutrophil apoptosis. Newer evidence, however, supports a mechanism by which accumulation of the mutant neutrophil elastase in the endoplasmic reticulum activates the unfolded protein response, leading to apoptosis. SCN due to these mutations is inherited in an autosomal dominant fashion. Hax-1 is a mitochondrial protein with weak homology to bcl-2, and its absence results in mitochondrial-dependent apoptosis. It is the mutated protein implicated in the original autosomal recessive cases of SCN described by Kostmann. Although all SCN cases were originally referred to as Kostmann syndrome, that term is now reserved for this subgroup of autosomal recessive SCN. Wiskott-Aldrich syndrome protein (WASP) regulates actin polymerization in hematopoietic cells, and deficiency in this protein results in the Wiskott-Aldrich syndrome, characterized by small platelets in low number, sinopulmonary infections, and eczema. Another phenotype of mutated WASP, however, is X-linked thrombocytopenia and neutropenia. Mutations in the glucose-6-phosphatase catalytic subunit 3 (G6PC3) are the most recently discovered cause of a subset of SCN patients; homozygous loss of this metabolic enzyme also appears to lead to activation of the unfolded protein response and increased apoptosis of neutrophil precursors.8
SCN was formerly a disease of infancy and early childhood because few, if any, patients survived to adulthood. However, the advent of the availability of recombinant G-CSF and the observation that G-CSF is able to raise neutrophil counts and prevent infection in most patients have allowed these children to survive. Some patients require very high doses of G-CSF, but responses are seen in 80 to 90% of individuals. Increased survival led to the emerging realization that SCN predisposes to the development of myelodysplasia and acute leukemia (MDS/AML), with the development of MDS/AML at a rate of approximately 2% per year, and a cumulative risk of about 30% over 10 years.9 Patients refractory to, or requiring very high doses of, G-CSF appear to have a higher risk for leukemic transformation. As discussed later, development of MDS/AML is often associated with the acquisition of a mutation in the gene encoding for the G-CSF receptor. Considerable controversy exists concerning the potential contribution of G-CSF administration to the risk for malignant transformation in children with SCN who receive lifelong treatment with G-CSF (see later under Treatment).
Two classes of G-CSF receptor mutations have been associated with SCN and either hyper-responsiveness or hyporesponsiveness of the receptor. Initial studies aimed at demonstrating that SCN was caused by mutation of the G-CSF or G-CSF receptor gene did not implicate such mutations in the pathogenesis of a significant number of patients with SCN. However, a small number of patients have been shown to have mutations in the G-CSF receptor gene that block ligand binding and produce a G-CSF-resistant form of SCN. More important, however, the studies identified an acquired missense mutation that introduces a stop codon and leads to the deletion of the distal intracellular domain of the receptor known to be responsible for differentiation signaling. This has been hypothesized to cause proliferation of hematopoietic progenitors at the expense of maturation and to be associated with hypersensitivity to G-CSF, suggesting that this mutation may play an important role in the development of MDS/AML in the setting of SCN. However, whether this mutation is in fact of pathogenetic importance to the development of MDS/AML and whether G-CSF influences the risk for developing the mutation and influencing leukemic transformation remain subjects of significant controversy.
Cyclic Neutropenia
Cyclic neutropenia is defined as periods of neutropenia (≤200/µL) lasting 3 to 5 days and occurring at approximately 21-day intervals. These periods of neutropenia may be marked by recurrent fevers, mouth sores, and infections of the skin, upper respiratory tract, and ears. The disorder can be dominantly inherited or sporadic. Congenital cyclic neutropenia has also been shown to be associated with mutations in the gene for neutrophil elastase in virtually all cases tested to date.10 The diagnosis formerly required demonstration of transient neutropenia through frequent blood counts over a course of 6 weeks but now can be established by sequencing of the neutrophil elastase gene. It is successfully treated with G-CSF and is not associated with an increased risk for leukemic transformation. Rare cases of cyclic neutropenia acquired in adulthood have been associated with systemic diseases such as large granular lymphocytosis or T-cell lymphoma.
Other Congenital Syndromes with Associated Neutropenia
A number of other congenital syndromes are associated with neutropenia as one of the constellation of disease-associated abnormalities. These include Shwachman-Diamond syndrome, Fanconi anemia, dyskeratosis congenita, glycogen storage disease Ib, myelokathexis, Chédiak-Higashi syndrome, Griscelli syndrome II, and Hermansky-Pudlak syndrome II.11
Shwachman-Diamond syndrome usually begins as an isolated neutropenia but progresses to marrow failure and is also associated with pancreatic dysfunction and skeletal abnormalities. The responsible gene, the Shwachman-Bodian-Diamond syndrome gene (SBDS), is involved in the regulation of ribosomal RNA. These patients carry an increased risk for leukemic transformation.
Fanconi anemia is due to mutations in genes involved in DNA repair, and as such, it takes more time for marrow failure to develop in these patients (median age, 7 years). Patients with Fanconi anemia often have, in addition to marrow failure, short stature with upper limb anomalies and hyperpigmented cafe au lait spots, although about one third have no physical abnormalities. Screening is done by chromosomal fragility testing following exposure to diepoxybutane or mitomycin C as well as direct assessment for known Fanconi gene mutations. Stem cell transplantation is curative but carries a high risk for morbidity and mortality owing to the toxicity of preparative regimens.12
Dyskeratosis congenita (DKC) is a syndrome of nail dystrophy, leukoplakia, and skin pigmentation abnormalities with associated neutropenia or aplastic anemia, or both. It can be inherited in an autosomal dominant or recessive or X-linked fashion and has been shown to be associated with mutations in several genes that are implicated in telomere maintenance. It typically does not present until the second decade of life. Recent studies have demonstrated that DKC is one of several diseases associated with telomere abnormalities, with wide-ranging manifestations including pulmonary fibrosis and hepatic cirrhosis as well as bone marrow failure.
Glycogen storage disease Ib is inherited in an autosomal recessive fashion and characterized by intermittent neutropenia due to defects in the neutrophil respiratory burst with subsequent apoptosis of circulating neutrophils. Hepatomegaly and metabolic crises are also the hallmarks of this disease and are due to mutations in the gene for the glucose-6-phosphatase translocase enzyme.
The neutropenia of myelokathexis is due to retention of mature neutrophils in the bone marrow despite a low peripheral neutrophil count. During infection, however, patients with myelokathexis typically have a sudden rise in their neutrophil count, which makes their clinical course relatively more benign. There is an association between this condition and hypogammaglobulinemia and warts, the WHIM syndrome (warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis). It has been shown to be caused by heterozygous mutations in the gene encoding chemokine receptor CXCR4.
Chédiak-Higashi syndrome, Griscelli syndrome II, and Hermansky-Pudlak syndrome II are all syndromes of albinism and neutropenia due to defects in vesicular trafficking. Chédiak-Higashi syndrome is due to mutations in a lysosomal trafficking regulatory gene (LYST) and is characterized by oculocutaneous albinism, bleeding, progressive neurologic disease, and increased susceptibility to hemophagocytic syndrome. Patients with Griscelli syndrome II also have an increased susceptibility to hemophagocytic syndrome as well as albinism and periodic neutropenia; Griscelli syndrome II is caused by mutations in the gene encoding the small guanosine triphosphatase RAB27A, which is involved in the release of myeloperoxidase from the primary granules of neutrophils. Hermansky-Pudlak syndrome II is due to mutations in the AP3B1 gene, which encodes a part of a protein transport complex that is involved in vesicular trafficking in many cell types and appears to be involved in the trafficking of neutrophil elastase. It is also marked by albinism, platelet abnormalities, and pulmonary fibrosis.
Barth syndrome is an X-linked autosomal recessive disorder characterized by neutropenia, cardiomyopathy, and growth retardation, with a high mortality rate through early childhood because of the heart disease. The causative mutation is in the TAZ gene, which encodes tafazzin protein that is critical to remodeling cardiolipin in the mitochondrial membrane.
Secondary Causes of Neutropenia
Several viral infections have been shown to cause a transient neutropenia that typically resolves as the viremia abates. These include varicella, measles, rubella, hepatitis A and B, Epstein-Barr virus, influenza, parvovirus, and cytomegalovirus. The mechanisms are diverse and can involve redistribution, decreased production, and immune destruction of neutrophils. Human immunodeficiency virus and acquired immunodeficiency syndrome can likewise cause mulitfactorial leukopenia and neutropenia (Chapter 393). Patients often have splenomegaly with increased sequestration, but more commonly the neutropenia reflects immune-mediated destruction. A myriad of atypical infections like Mycobacterium tuberculosis, ehrlichiosis, rickettsia, tularemia, brucellosis, and some staphylococcal infections can cause a moderate neutropenia. Any infection leading to overwhelming sepsis can cause neutropenia, but this is usually through consumption of the marrow neutrophil reserve and is typically seen in newborns and elderly patients and not in individuals with an otherwise healthy and mature marrow. There is also increased margination of neutrophils during sepsis due to systemic activation of complement, exacerbating the neutropenia.
Drug-Induced Neutropenia and Neutropenia Due to Marrow Injury
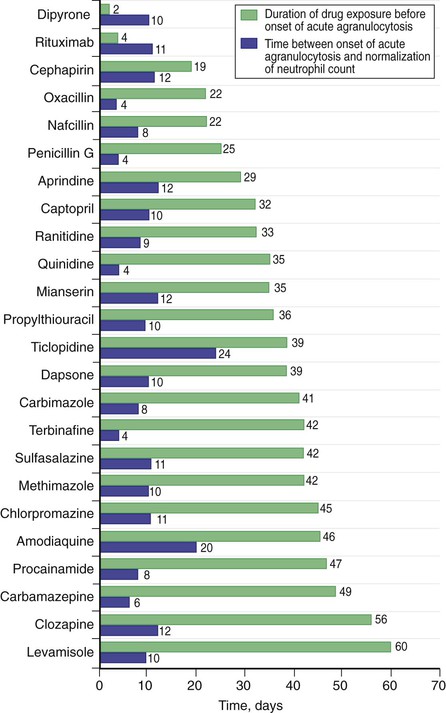
Drug-induced neutropenia is the most common cause of neutropenia. Multiple drugs have been implicated in neutropenia and agranulocytosis, in both predictable and idiosyncratic patterns. Drug-induced neutropenias may reflect either suppression of marrow granulopoiesis or increased destruction or clearance of peripheral neutrophils. Many drugs cause direct dose-dependent marrow suppression, which is predictable and often mild. Others incite an idiosyncratic immune-mediated destruction that can present with profound agranulocytosis. The typical pattern of drug-induced neutropenia is a marked decline in the neutrophil count that occurs after about 1 to 2 weeks of exposure to the drug with a recovery that begins within days of stopping the drug. However, atypical cases can present long after drug initiation, and others may be associated with a longer interval before recovery of the neutrophil count (Fig. 167-5). Patients with drug-induced agranulocytosis may present with acute sepsis, and it is associated with a significant risk for acute mortality. Recovery is often preceded by the appearance of monocytes and immature neutrophil forms. The more hypercellular the marrow is at diagnosis, the earlier marrow recovery may occur. Some common drugs known to cause neutropenia, in addition to antineoplastic, antiviral, and immunosuppressive agents, include clozapine, the antithryoidal thioamides including carbimazole, methimazole and propylthiouracil, quinidine, procainamide, sulfasalazine, and Levamisole, which is widely used as a cocaine adulterant, causing cocaine-associated neutropenia. Neutrophil recovery is speeded by G-CSF, although there are no definitive data that this improves survival in this setting.
Radiation can also result in marrow injury leading to an acute or chronic marrow failure state; in high doses, it is also a risk factor for the development of myelodysplasia and leukemia. These malignant hematopoietic diseases can themselves cause marrow failure because the malignant cells proliferate within the marrow-occupying space and can cause marrow fibrosis, both of which lead to cytopenias. These diseases are discussed in greater detail in Chapters 182 and 183, respectively. Likewise, metastatic carcinoma to the bone can also cause marrow failure because the marrow becomes increasingly replaced by the metastatic cells.
Immune Neutropenia
Infection and drugs cause immune-mediated neutrophil destruction. However, immune neutropenia can also occur as an isolated phenomenon (primary immune neutropenia) or as a manifestation of an underlying systemic autoimmune disease (secondary immune neutropenia). Primary autoimmune neutropenia is primarily a disease of children younger than 4 years; median age of onset is 6 to 12 months. Although infectious risk is increased, treatment is restricted to prophylactic antibiotics, with G-CSF reserved for acute infectious episodes. Ninety-five percent of patients undergo spontaneous remissions within 2 years. Nearly all of these patients have antineutrophil antibodies directed against antigens derived from the FcγIIIb receptor; these antibodies mediate neutrophil destruction by either sequestration in the spleen or complement-mediated neutrophil lysis.
Secondary autoimmune neutropenia is a disease of adults and can be seen in association with hyperthyroidism, Wegener granulomatosis, rheumatoid arthritis, and systemic lupus erythematosus (SLE). The role of antineutrophil antibodies in these patients is less clear. More than 50% of patients with SLE, for example, have antineutrophil antibodies, but many have normal neutrophil counts, and there is a poor correlation between the presence of the antibodies and neutrophil number.13
Felty syndrome and large granular lymphocyte syndrome deserve separate mention. Felty syndrome occurs in association with long-standing rheumatoid arthritis (RA) (Chapter 264) and is characterized by splenomegaly and profound neutropenia. Large granular lymphocyte syndrome often occurs in the setting of RA but can also occur as an isolated phenomenon. Both Felty syndrome and large granular lymphocyte syndrome are associated with a proliferation of large granular lymphocytes, with a characteristic surface phenotype (CD3+, CD8+, CD16+, and CD57+). In the setting of RA, these two syndromes were originally thought to be separate diseases, with Felty syndrome being polyclonal and large granular lymphocyte syndrome representing a monoclonal proliferation of larger granular lymphocytes. However, with increasing sensitivity of detection of monoclonal populations of lymphocytes, this distinction has become blurred. It has been observed that more than 90% of RA patients with either syndrome are human leukocyte antigen (HLA)-DR4-positive, leading to the postulate that the two entities represent the extremes of a single spectrum of disease. This HLA restriction is not found among non-RA patients with large granular lymphocytes. Both syndromes cause immune-mediated neutrophil destruction by a wide array of mechanisms, including antineutrophil antibodies and cell-mediated destruction. Some patients may also have G-CSF resistance mediated by inhibitory G-CSF antibodies.14
There are other rare forms of immune neutropenia. Isoimmune neonatal neutropenia is a moderate to severe neutropenia of the newborn due to transplacental passage of maternal immunoglobulin G antibodies against alleles inherited from the father, resulting in neutropenia in a manner similar to the development of anemia in Rh hemolytic disease. Pure white cell aplasia is a rare disease associated with severe pyogenic infections, and also with thymoma in more than two thirds of cases. It has also occurred following ibuprofen therapy. There is a complete absence of myeloid precursors on bone marrow examination. It is immune mediated, but removal of the thymoma in thymoma-associated cases may not be sufficient for remission. Adjuvant therapy with cyclophosphamide, corticosteroids, cyclosporine, and intravenous immunoglobulin may be needed.
Neutropenia Due to Increased Margination and Hypersplenism
Complement activation can result in both acute and chronic neutropenia as a result of increased margination of the circulating neutrophil pool. This is attributed to the fact that C5a renders neutrophils more adherent and thereby prone to aggregation within the pulmonary vasculature. This has been seen in patients suffering from burns and transfusion reactions. Complement activation may also lead to neutrophil destruction, as in paroxysmal nocturnal hemoglobinuria. The circulating neutrophil pool can also be diminished in association with hypersplenism, although this is typically less common and less pronounced than the anemia and thrombocytopenia seen in patients with an enlarged spleen.
Neutropenia Due to Nutritional Deficiency
Several vitamin and mineral deficiencies, particularly B12, folate (Chapter 164), and copper, are associated with neutropenia. Copper deficiency is a particularly under-recognized cause of neutropenia (usually seen along with anemia), found especially in clinical situations like total parenteral nutrition without copper supplementation, protein-losing enteropathies, celiac disease, gastric bypass surgery and postgastrectomy, malabsorption syndromes, and zinc toxicity. In addition to low serum copper and ceruloplasmin levels, distinct morphologic findings include hypogranularity and hypolobation (PHA) on peripheral smear, and cytoplasmic vacuolization of myeloid as well as erythroid precursors and ringed sideroblasts.
Deficiencies in these vitamins and minerals result in ineffective myelopoiesis, maturation arrest, and megaloblastic changes with nuclear-cytoplasmic dyssynchrony. The characteristic finding in the setting of megaloblastic anemia is hypersegmentation of the neutrophil (Fig. 167-6).

Chronic Idiopathic Neutropenia
Chronic idiopathic neutropenia in adults (CINA) has been described as an acquired disorder of granulopoiesis characterized by prolonged neutropenia in the absence of any apparent underlying etiology.15 Patients present with chronic neutropenia that is often an incidental finding on routine blood tests, making it impossible to know how long the neutropenia has been present. They have no evidence of autoimmune disease, nutritional deficiency, or myelodysplasia. The syndrome is heterogeneous, with a wide range of neutrophil counts. A group of patients with CINA from Greece were originally found to have increased production of transforming growth factor-β (TGF-β) and consequent suppression of granulopoiesis by the bone marrow. These patients tend to have very mild neutropenia, with an absolute neutrophil count (ANC) that is rarely less than 800. They may have an ethnic predisposition to neutropenia; indeed, the neutropenia has been demonstrated to be linked to a genetic polymorphism in the TGF-β locus. It seems likely, however, that these patients should be distinguished from other CINA patients, many of whom have an ANC below 200. The etiology of neutropenia in these patients is completely unknown. However, the natural history of CINA, even in the face of very low neutrophil counts, is generally benign. Most patients require no therapy, although those with very low counts must be treated with G-CSF when they develop fever in the setting of infections. Some patients with recurrent infections or troublesome aphthous ulcers require chronic G-CSF treatment. These patients typically respond to fairly low doses of G-CSF, and there is no reported increase in the development of MDS/AML.
Clinical Manifestations and Diagnosis
Neutropenia by itself is not associated with many clinical signs and symptoms other than those of the condition that may be causing it. It becomes clinically evident, however, when it results in infection. Although patients with an ANC below 1000/µL do have a slightly increased risk for infection, the risk is substantially increased once the neutrophil count falls below 500/µL. Given the lack of neutrophils, the signs and symptoms of infection may be attenuated; as such, pneumonia may be present with minimal infiltrate on chest radiograph, or a urinary tract infection may yield only a very mild pyuria. Given this, fever in any neutropenic patient must be considered an emergency with prompt acquisition of cultures and administration of empirical antibiotic therapy.
When fever, infection or sepsis, and neutropenia present concomitantly for the first time, it can be difficult to determine whether the neutropenia predated the infection or, conversely, if it is the result of the infection. Examination of the peripheral blood smear can be helpful in this regard because an elevation of band forms and evidence of toxic granulation suggest the latter.
Because drug-induced neutropenia is the most common cause of acquired neutropenia, a careful inventory of all drug and toxin exposures is warranted.16 Likewise, there should be a careful evaluation for underlying malignant and inflammatory conditions as the precipitant of neutropenia. The time course of the neutropenia and infections can provide clues to the etiology of the neutropenia (acute versus chronic, persistent versus cyclic, neonatal versus childhood versus adult onset). Attention should be paid to the skin, bones, appendages, and nails because abnormalities in these may point toward one of the congenital neutropenia syndromes. Evaluation of the complete blood count, peripheral blood smear, and vitamin B12 and folate levels should also be performed.
When the neutropenia is not severe and is associated with anemia and thrombocytopenia, one should consider the possibility of hypersplenism. In many cases, the diagnosis can be made by the finding of palpable splenomegaly. However, especially in obese patients, abdominal imaging should be used to evaluate spleen size. Abdominal ultrasound allows the assessment of portal venous flow with Doppler studies. If splenic enlargement is confirmed, the etiology of the splenomegaly (Chapter 168) should be determined. It may reflect congestive splenomegaly secondary to portal hypertension (as a result of cirrhosis, fatty liver, or congestive heart failure, among others) or infiltrative splenomegaly due to a benign or malignant process. Felty syndrome should be considered in the setting of RA.
In patients with chronic neutropenia in the absence of a history of infection or drug or toxin exposure or an evident B12 or folate deficiency, bone marrow examination should be performed to rule out myelodysplasia, with assessment of morphology, flow cytometry for large granular lymphocyte syndrome, and cytogenetics. Once a normal marrow has been obtained, further bone marrow examination is not indicated in patients with CINA (Fig. 167-7).
Treatment and Management 
The management of neutropenia is dependent on the etiology of the depressed neutrophil count as well as its severity and the presence or absence of fever or infection.17 The approach to the patient with fever and neutropenia is discussed in more detail in Chapter 281. Neutropenia with fever is a clinical emergency because these patients are at risk for hemodynamic collapse and septic shock. Therefore, these patients should be evaluated thoroughly and cultured promptly, and antibiotics should be administered within 30 to 60 minutes of presentation before obtaining the results of the cultures. The timely empirical administration of a combination of antipseudomonal antibiotics in patients with neutropenia at the onset of fever results in significant clinical benefit, with respect to both response and survival. With the advent of newer generation cephalosporins, studies have shown that monotherapy with a third- or fourth-generation cephalosporin at the onset of fever may be sufficient. A systematic review to assess the evidence for combination therapy versus monotherapy in cancer patients with febrile neutropenia in clinical trials has been recently updated. A1 It concluded that randomized controlled trials have demonstrated the survival superiority of β-lactam monotherapy compared with β-lactam-aminoglycoside combination therapy.
The addition of a second antipseudomonal agent, vancomycin, or antifungal agent is warranted in patients considered at risk for resistant pseudomonal infection, resistant gram-positive infection, or fungal infections, respectively, or in the face of a failure to defervesce within 3 to 5 days of antibiotic administration. These considerations are discussed in more detail in Chapter 281.
How to manage the uninfected and afebrile neutropenic patient is more nuanced and dependent on the etiology of the neutropenia. Patients with immune-mediated neutropenia are typically treated with immunosuppressive therapy, including steroids, antithymocyte globulin, or cyclosporine, or a combination of these, aimed primarily at treatment of the underlying autoimmune disease. Patients usually respond to G-CSF, although in the setting of RA, this may induce a flare of joint symptoms. Patients with large granular lymphocyte syndrome often respond to therapy with low-dose methotrexate (10 mg/m2 orally once a week), cyclosporine (100 to 600 mg or 2 to 10 mg/kg orally daily), or low-dose cyclophosphamide (50 to 100 mg orally daily).18
Patients with congenital neutropenia, including idiopathic, severe congenital, or cyclic neutropenia, are usually successfully managed with G-CSF for years. Before the use of G-CSF, the mean age of death for patients with severe congenital neutropenia was 2 to 3 years. Since the advent of G-CSF, however, life expectancy has been extended by decades into adulthood. Therapy is daily and chronic, given by subcutaneous injection, with doses varying by the type of neutropenia and the individual responsiveness to therapy. It is usually well tolerated, although accelerated bone loss has been observed. Growth and development do not appear to be affected. Patients with SCN typically require the highest doses, whereas those with idiopathic neutropenia require the lowest, and patients with cyclic neutropenia fall somewhere in between. An increased rate of MDS/AML has been reported with the use of G-CSF in patients with SCN, but this has occurred coincidentally with improved survival. The increased incidence of MDS/AML may then be due to the fact that patients are living longer with a disease whose natural history includes a risk for developing MDS or AML. Certainly, the observation that acquired mutations in the G-CSF receptor are present in 65 to 80% of patients with SCN who develop MDS or AML suggests a potential mutagenic pathway toward leukemogenesis, but whether this is enhanced or accelerated by the administration of G-CSF has yet to be determined. Patients with idiopathic and cyclic neutropenia do not develop MDS or AML, despite therapy with G-CSF. However, recent evidence suggests that the risk for MDS/AML in SCN patients is much higher in those requiring high doses of G-CSF (>10 µg/kg/day), and patients with idiopathic and cyclic neutropenia are usually responsive to much lower G-CSF doses.
In patients with CINA, G-CSF should be reserved for acute febrile episodes unless the patient has recurrent infections. Patients with CINA treated with G-CSF may experience significant side effects, including fever, gastrointestinal symptoms, and splenomegaly. Consequently, in patients with CINA requiring chronic G-CSF administration, the cytokine should be administered at the lowest dose necessary to prevent infections; it is usually sufficient to treat to maintain the ANC in the range of 300 to 500.
For patients with an inflammatory, infectious, or drug-induced neutropenia, the recommendation is to treat the underlying condition or stop the offending agent. This, however, is not always immediately possible, as in the cases of HIV-infected patients with opportunistic infections or on antiretroviral therapy, solid organ and bone marrow transplant recipients on immunosuppression and antiviral prophylaxis and treatment, and cancer patients undergoing chemotherapy. Prophylactic use of G-CSF in these patients has been shown to be effective in improving the ANC and decreasing rates of infection and febrile neutropenia, but has not been associated with a proven or consistent survival advantage. A2 In light of these findings, many oncologic professional society guidelines recommend the use of prophylactic G-CSF in patients receiving chemotherapy who have a 20% or greater risk for developing febrile neutropenia based on age, comorbid illness, disease characteristics, and myelotoxicity of the chemotherapy regimen. In addition, it is recommended for use during hematopoietic stem cell transplantation, for patients receiving chemotherapy for non-Hodgkin lymphoma or dose-dense chemotherapy, and for patients with a history of febrile neutropenia receiving further chemotherapy.
The use of prophylactic antibiotics has also been investigated, predominantly in neutropenic patients receiving chemotherapy. Early studies in the 1980s and 1990s demonstrated an improvement in infection-related outcomes but not in infection-related or overall survival. Most recently, several randomized trials of prophylactic quinolones in patients receiving chemotherapy demonstrated an improvement in rates of infection and febrile neutropenia. A meta-analysis of studies investigating the use of prophylactic quinolones in patients receiving chemotherapy was the first to document an overall survival benefit as well, although most of these patients had hematologic as opposed to solid tumor malignancies. A3 The current Infectious Diseases Society of America guidelines do not recommend the use of prophylactic antibiotics in cancer patients undergoing myelosuppressive chemotherapy, with the exception of trimethoprim-sulfamethoxazole in patients at risk for Pneumocystis jirovecii pneumonia. Antibiotic prophylaxis is not generally used outside of the stem cell transplantation setting. The use of antibiotics to prevent infection in patients with neutropenia due to other causes has not been extensively studied but is typically not recommended and should be based on clinical context.
Stem cell transplantation (Chapter 178) can be curative for a number of the congenital neutropenia and bone marrow failure syndromes. It is, however, not without risk and should therefore be reserved for patients with severe neutropenia complicated by recurrent infection definitively shown to be due to marrow failure.

 Leukopenia Due to Deficiency of Other Cell Lines
Leukopenia Due to Deficiency of Other Cell Lines
Lymphocyte production takes place in a variety of anatomic sites, and lymphocyte trafficking from those sites is bidirectional, making it difficult to understand lymphocyte dynamics in the same way that we do for neutrophils. Despite this, the peripheral lymphocyte count seems to be maintained in a narrow range at 2000 to 4000/µL, 20% of which are B cells and 70% of which are T cells. Lymphocytopenia is a total lymphocyte count of less than 1500/µL. It can be the result of decreased production, defective trafficking, or increased loss or destruction. Decreased production can occur as a result of protein and calorie malnutrition; lymphocyte progenitor pool injury secondary to radiation, chemotherapy, or immunosuppressive agents; and congenital immunodeficiency states. Endogenous or exogenous glucocorticoid excess can cause lymphocytopenia by altering lymphocyte trafficking. This can also occur as the result of acute bacterial or fungal infections, certain viral infections, and granulomatous disease. Finally, many viruses can cause direct destruction of lymphocytes, as can antilymphocyte antibodies seen in patients with underlying autoimmune diseases. Lymphocytes can also be lost from intestinal lymphatics in cases of protein-losing enteropathy, primary disease of the gut or intestinal lymphatics, or gut edema secondary to severe heart failure. When lymphocytopenia is discovered, a comprehensive assessment of the immune system should be done, including lymphocyte subtyping, quantitative immunoglobulins, and skin testing to detect deficiencies of cell-mediated immunity. Treatment is typically aimed at the underlying disease, but intravenous immunoglobulin can be administered to patients who are hypogammaglobulinemic, and transplantation can be performed in patients with severe deficiencies of cell-mediated immunity due to impaired lymphocyte production and function.
Monocytopenia, eosinopenia, and basophilopenia can be seen in the setting of bone marrow failure syndromes or as a result of acute infection, malignancy, or severe injury. This is thought to be due to elevations in glucocorticoids, prostaglandins, and epinephrine. A rise in these humoral factors has the greatest impact on eosinophils such that a lack of eosinopenia in any of these settings should prompt suspicion for adrenal insufficiency, a primary myeloproliferative syndrome, parasitic infection, or primary hypereosinophilic syndrome. Monocytopenia is less frequently seen, probably owing to the diverse roles monocytes play in normal human physiology; prolonged and extreme monocytopenia may not be compatible with life.
Review Questions
1. A 20-year-old African American college student receives a scholarship to spend her junior year abroad in France. She needs to have a health form completed before her departure. A complete blood count (CBC) reveals neutropenia, and her physician advises her to postpone the scholarship and to see you for evaluation. She is healthy with no history of excessive infections. Her physical examination is normal. CBC shows: white blood cell count 3400 w/ 30% neutrophils, hematocrit 41, platelets 200,000. What should you advise her?
A. She should have a bone marrow to rule out a bone marrow process causing neutropenia.
B. She should have serial blood counts to rule out cyclic neutropenia.
C. She should have chromosome analysis of her peripheral blood.
D. She should be evaluated for an elastase mutation.
E. She should go to France without any further evaluation.
Answer: E The normal neutrophil count is partially determined by ethnic background. African American males frequently have a neutrophil count of 1000 to 1500, and African American females may have counts that are even lower. Many may actually have counts below 1000. With a total neutrophil count of 1120, a normal hematocrit and platelet count, and an unremarkable history and physical examination, the patient should be diagnosed with constitutional neutropenia. This requires no further evaluation, although if DARC testing is available, confirming she is Duffy negative will confirm the diagnosis. The patient should be reassured that these counts are normal for her and present no undue risk. She should leave for France as planned. (See Ethnic and Benign Familial [Constitutional] Neutropenia.)
2. A 1-month-old baby boy presents with fever and cough and is found to have pneumonia associated with severe neutropenia. His parents have normal blood counts but had lost a baby girl at age 3 months under similar circumstances 2 years before. Which of the following statements is not true?
A. The patient likely has a mutation in neutrophil elastase.
B. The child should be treated with granulocyte colony-stimulating factor (G-CSF).
C. The child is at risk for developing myelodysplastic syndrome and acute myeloid leukemia (MDS/AML).
D. If the child survives, his offspring will probably have normal neutrophil counts.
E. The child's white blood cell count likely has a 21-day cycle in and out of the normal range.
Answer: A This child has a disease that is apparent in neither parent but has a sibling that died of the same disease. This is consistent with an autosomal recessive form of severe congenital neutropenia (SCN). SCN related to neutrophil elastase mutation is an autosomal dominant disorder. Cyclic neutropenia is a milder autosomal dominant disorder, also due to mutations in ELANE; it is clinical mild and rarely presents in infancy. Although it can appear sporadically, the occurrence in a sibling makes that virtually impossible. This case is most consistent with Kostmann syndrome, which is due to mutations in the HAX1 gene. Because it is a very rare recessive disorder, if the child lives to adulthood and has offspring, they will be obligate heterozygotes and, like the patient's parents, will have normal counts. Like patients with dominantly inherited SCN associated with neutrophil elastase mutations, these patients respond to G-CSF and have an increased risk for developing MDS/AML. (See Severe Congenital Neutropenia.)
3. A 45-year-old man presents to the emergency department after a motor vehicle crash. He is afebrile with a blood pressure of 160/95 mm Hg and a pulse of 110 beats per minute. He has several scrapes and bruises, but no major injuries. Admission complete blood count reveals: white blood cell count 16,000, hematocrit 45, and platelets 280,000. What is the most likely explanation for his leukocytosis?
B. Undiagnosed chronic myelogenous leukemia
C. Stress-induced demargination of neutrophils
D. Cytokine release stimulating increased marrow production and release of neutrophils
E. Unsuspected splenic rupture
Answer: C Acute stress is commonly associated with leukocytosis. Acute leukocytosis in almost any setting occurs by demargination, in this case in response to the release of catecholamines during the stress of the accident, also causing his tachycardia. Cytokine-induced marrow proliferation and release of neutrophils reflect more chronic inflammation and could not have occurred in less than about a week. Unsuspected splenic rupture would likely lead to hypotension and shock. There is no reason to consider a diagnosis of chronic myeloproliferative disease or sepsis in the absence of other evidence for a secondary condition. The patient should have his counts rechecked in several days or weeks to rule out any other pathology. (See Stress.)
4. A 3-year-old child presents with recurrent infections and a diagnosis of Chédiak-Higashi syndrome (CHS) is made. Which of the following is not true of his disease?
A. He is likely to have associated nystagmus.
B. His sibling has a 50% chance of having the disease.
C. He is likely to have abnormal neutrophil granules.
D. He is likely to develop hemophagocytic syndrome.
E. He is likely to have a defect in skin pigmentation.
Answer: A CHS is a generalized disorder of granule formation caused by abnormalities in the LYST gene, which is required for the correct trafficking of proteins into granules. Consequently, patients with CHS have other defects associated with abnormal granule formation, including oculocutaneous albinism, with decreased pigmentation and nystagmus, and abnormal-appearing neutrophil granules. As the disease is an autosomal recessive disorder, the heterozygous parents are usually totally unaffected, and siblings have a 25% chance of inheriting the disease. Patients with CHS often have a terminal phase of the disease associated with the development of hemophagocytic syndrome, manifesting as fever, splenomegaly, and pancytopenia, often as a result of Epstein-Barr virus infection. (See Other Congenital Syndromes with Associated Neutropenia.)
5. A 1-month-old infant presents with a temperature of 103° F and a white blood cell count (WBC) of 90,000. He has a history of delayed umbilical cord separation, poorly healing skin lesions, recurrent otitis media, and failure to thrive. He is admitted to the hospital, where he is documented to have methicillin-resistant Staphylococcus aureus (MRSA). He is placed on appropriate antibiotics, but he remains febrile, and his WBC is persistently elevated in the 80,000 to 100,000 range. This patient is likely to have a mutation in which of the following?
Answer: C The child has a congenital disorder of neutrophils that results in leukocytosis. The diagnosis is leukocyte adhesion deficiency. This disease arises from defects in leukocyte adhesion to endothelium. It can arise from defects in the integrin receptor common β chain (LAD-1), with loss of expression of leukocyte function−associated antigen 1 (LFA-1), C3bi receptor, and gp150;95, leading to a failure to ingest and kill microbes opsonized by C3bi. It can also arise from an abnormality of selectin glycosylation (LAD-2). ELANE and HAX1 mutations are causes of severe congenital neutropenia, and affected patients present with profound neutropenia. LYST mutations are the cause of Chédiak-Higashi syndrome, which is also associated with neutropenia. Congenital G-CSFR mutations are very rare, although two or three patients in the literature have been described to have such mutations as the basis for G-CSF−resistant severe congenital neutropenia. More commonly, G-CSFR mutations are somatic mutations that arise in patients with severe congenital neutropenia and may be a harbinger of the development of myelodysplastic syndrome or acute myeloid leukemia. (See Leukocyte Adhesion Deficiency.)