TABLE 32-4
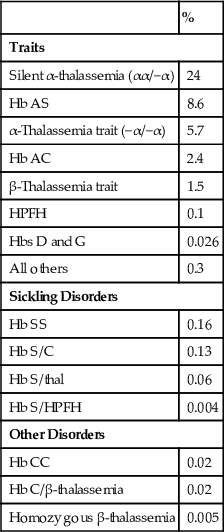
Prevalence of Common Hemoglobin Disorders Among African Americans
| % | |
| Traits | |
| Silent α-thalassemia (αα/−α) | 24 |
| Hb AS | 8.6 |
| α-Thalassemia trait (−α/−α) | 5.7 |
| Hb AC | 2.4 |
| β-Thalassemia trait | 1.5 |
| HPFH | 0.1 |
| Hbs D and G | 0.026 |
| All others | 0.3 |
| Sickling Disorders | |
| Hb SS | 0.16 |
| Hb S/C | 0.13 |
| Hb S/thal | 0.06 |
| Hb S/HPFH | 0.004 |
| Other Disorders | |
| Hb CC | 0.02 |
| Hb C/β-thalassemia | 0.02 |
| Homozygous β-thalassemia | 0.005 |
Abnormal Hemoglobin Syndromes
In homozygous β-chain structural variants, both allelic genes for the abnormal β-chains are present, so that no normal β-chains (hence, no Hb A) are produced. Because α-, γ-, and δ-genes are normal, Hb F and Hb A2 are structurally normal, although they may be increased in amount.
In heterozygous β-chain structural variants, the abnormal Hb is present, in addition to Hb A, F, and A2. Its level depends on its rate of synthesis and stability, but also on its readiness to form dimers with α-chains. This latter process is driven by electrostatic forces between the positively charged α-globin and the negative β-globin. Because most β-globin variants acquire positive charge, their assembly with the positive α-chain is slower. As a result, the percentage of abnormal Hb is usually less than that of Hb A. In the presence of coexistent α-thalassemia, when β-chains compete for an insufficient number of α-globins, these effects are exaggerated and the level of structural variant decreases further. Similar forces influence the levels of Hb A2. δ-chain subunit of Hb A2 has a significant positive charge, not favorable for dimer formation. Positively charged β-chain variants also have little affinity for α-chains, leaving excess α-chains for assembly with δ-globin and resulting in a slight increase in Hb A2. This has been described in Hbs S and C trait, but the increase is so minimal that its existence was always questioned.
Compound heterozygotes for a β-chain structural variant and β+-thalassemia are well known. Here, the quantity of abnormal Hb exceeds that of Hb A, in contrast to the heterozygous β-structural hemoglobinopathies, in which the reverse is true.
In heterozygous α-chain structural variants, the abnormality in the α-chain will affect all three Hb types. Therefore, six different hemoglobins are found—the three normal hemoglobins and the three abnormal forms.
Compound heterozygotes for two β-chain abnormalities produce two different abnormal β-chains; therefore, there are two abnormal Hbs and no Hb A. An example of this is Hb SC disease. Compound heterozygotes for β- and δ-chain abnormalities and for α- and β-chain abnormalities are rare but have provided important information. The latter will have four major Hb types on electrophoresis.
Sickling Disorders
Sickle Cell Trait (Hb AS)
The heterozygous state for Hb S is the most common hemoglobinopathy in the United States. It is present in about 8% of African Americans (Schneider et al, 1976) and in as many as 45% of the population in certain areas of West Africa. The gene is found throughout sub-Saharan Africa and, at a much lower frequency, in the Mediterranean, Middle East, and India. Hb S trait is a benign condition without clinical symptoms or hematologic abnormalities. Added risk associated with sickle cell trait is minimal at the most, and is seen only in very rare circumstances. Extreme low O2 tensions can trigger sickling, and a few people have sustained splenic infarction when flying at very high altitude in unpressurized airplanes. Slightly increased incidences of hematuria, impaired ability to concentrate urine, and bacteriuria in women have been reported. Sickle cell trait confers protection to children from the lethal effects of falciparum malaria, which accounts for the major distribution of Hb S in central Africa.
The stained blood film appears normal, except perhaps for a few target cells. Blood cell counts are normal. The sickle cell slide preparation is positive, and almost all red cells eventually sickle. The solubility test is positive.
Hemoglobin separation typically shows 60% Hb A, 40% Hb S, normal Hb F, and normal to slightly increased Hb A2, up to 4.0%.
The proportion of Hb S is decreased in the presence of α-thalassemia; Hb S is less than 35% when one α-gene is deleted and less than 29% when two genes are lost (Head et al, 2004). In the latter, cells are hypochromic and microcytic. Because 27% of African Americans carry the α+-thalassemia gene, it is not surprising that the combination is quite common. Hb S level may also be diminished in iron and folate deficiency. Clinically, the combination of sickle cell trait and α-thalassemia trait is benign, with possible mild microcytic anemia due to the thalassemia trait.
Sickle Cell Disease (Hb SS)
Homozygous Hb S disease is a serious chronic hemolytic anemia, first manifested in early childhood and often fatal before 30 years of age. With modern medical care, many patients live longer, but the median age of death in the United States is still only in the 40s. Hb S is found mostly in the black population; 1 of every 600 black persons in the United States has sickle cell anemia (Steinberg, 1999).
In Hb S (β6 glu→val), the glutamic acid in the sixth position on the β-chain is replaced by valine. Hb S is freely soluble when fully oxygenated; when O2 is removed, Hb S polymerizes, with formation of tactoids (fluid crystals) that are rigid and deform the cell into the shape that gave the disease its name. A strong interaction between the side chain of β6 valine and the hydrophobic pocket of β85 phenylalanine and 88 leucine of another Hb S molecule is probably the basis of polymer formation. In homozygous Hb S disease, sickling occurs at O2 tensions that are physiological in the peripheral microcirculation. The rigidity of the red cells is responsible for the hemolysis, as well as for most of the complications. These irreversibly sickled cells result from membrane reorganization during repeated episodes of sickling and unsickling, in addition to cell dehydration that markedly reduces cellular deformability. Sickle cells contain high calcium levels, which stimulate potassium and water loss (Gardos effect) and exaggerate cell dehydration (Weatherall & Clegg, 1999). The hemolytic component is mostly extracellular and is caused by clustering of band 3 of the cell membrane, with the consequence of increased IgG binding and recognition by macrophages. Integrins (α4β1) on the sickle cell surface attach to fibronectin and adhesion molecules expressed on endothelial cells; this is enhanced by inflammatory cytokines, von Willebrand factor, and platelet activation, and together these interactions cause vaso-occlusion.
Complications.
In early childhood, bilateral painful swelling of the dorsa of the hands or feet occurs as a result of sickling and capillary stasis; this is known as hand-foot syndrome, or sickle cell dactylitis. It lasts about 2 weeks, is accompanied by changes of periostitis as observed by X-ray, and does not occur after 4 years of age.
The spleen is central to three complications: A sequestration crisis refers to sudden pooling of blood and rapid enlargement of the spleen, resulting in hypovolemic shock. This may occur in early childhood when splenomegaly is present and is often preceded by infection. Functional asplenia consists of inadequate antibody responses under some conditions and an impaired ability of the reticuloendothelial system to clear bacteria and particulate material from the blood, probably owing to reticuloendothelial blockade. This may explain in part the increased risk for infection in children with the disease. Salmonella and pneumococcal infections are unusually prevalent in children with sickle cell anemia. Vaso-occlusive episodes result in progressive infarction, fibrosis, and contraction of the spleen—so-called autosplenectomy. Although splenomegaly is present in childhood, a small fibrotic remnant is the rule in the adult.
From early childhood, patients cannot produce concentrated urine, apparently as a result of anoxic damage in the medullae of the kidneys. Hematuria as a result of papillary necrosis is common. Renal insufficiency occurs in 5% to 20% of adults (Steinberg, 1999).
Vaso-occlusive crises are debilitating episodes of abdominal and bone or joint pain, accompanied by fever, which are probably due to plugging of small blood vessels by masses of sickled cells. Bone necrosis occurs in 10% to 50% of patients and may be a focus for salmonella osteomyelitis. Acute chest syndrome represents episodes of acute chest pain often associated with a new infiltrate in the chest film. Approximately 40% of patients experience at least one episode of acute chest syndrome. Although an infectious origin had been suspected in earlier studies, fat embolism is now considered to play a major role in this syndrome. The various complications that result from recurring vaso-occlusive crises involve many body organs (Steinberg, 1999).
Aplastic crises occasionally can afflict any patient with chronic hemolytic anemia. Temporary failure of red cell production that would not be noticed in a person with a normal red cell life span will cause a serious fall in Hb concentration in chronic hemolytic anemia. This may be the result of infection, particularly parvovirus B19, exposure to toxic drugs, or folic acid deficiency; sometimes no cause can be found. Hemolytic crises due to a further increase in hemolysis are rare.
Diagnosis.
The anemia is normochromic and normocytic; polychromasia is increased, and nucleated red blood cells are present. Sickle cells are almost always found in the stained smear (Fig. 32-22). Target cells are numerous, and Howell-Jolly and Pappenheimer bodies are regularly seen in older children and adults as a result of asplenia. The microhematocrit as an estimate of degree of anemia is unreliable because of excessive plasma trapping. Neutrophilia and thrombocytosis are usual. The marrow shows erythroid hyperplasia.
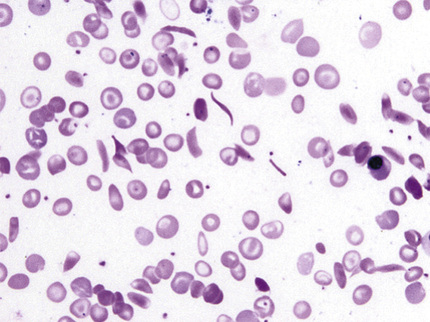
No Hb A is found if the patient has not been transfused recently; more than 80% of the hemoglobin is Hb S, 1% to 20% Hb F, and 2% to 4.5% Hb A2 (Wrightstone & Huisman, 1974). The fetal hemoglobin is distributed unevenly among the red cells. Hb S and several D and G hemoglobins have the same electrophoretic mobility at alkaline pH, but of these, only Hb S gives a positive sickling test.
Hemoglobin SC Disease
The frequency of Hb SC disease is almost the same as that of Hb SS disease in African Americans. It causes a mild hemolytic anemia. Crises are less frequent and less painful than in sickle cell anemia. Onset is usually in childhood, but the disease might be undetected until later in life. The life expectancy is only modestly shortened. The body habitus is normal or stocky in contrast to the asthenic features in sickle cell anemia. Splenomegaly might be the only finding on physical examination. Fatigue, dyspnea on effort, frequent upper respiratory infections, attacks of mild jaundice, and arthralgias are seen. Constant hip and low back pain may be present with aseptic necrosis of the head of the femur. Hematuria from renal medullary infarction and splenic infarcts have been described. In pregnancy, crises are more frequent and there is an increase in thrombotic tendency, which can cause massive thromboembolism and sudden death following childbirth. A higher incidence of retinopathy is seen in Hb SC disease than in SS disease.
Anemia varies from moderate to very mild and is normochromic-normocytic. Anisocytosis and poikilocytosis are mild to severe, and target cells are numerous—up to 85% of the erythrocytes. Plump and angulated sickled cells are often present on the film. The sickling test is positive. Hb S is 50% and Hb C is slightly less. Hb F is usually under 2%. The proportions of Hbs are the same in patients with coexistent α-thalassemia (Steinberg et al, 1983).
Hb S/β-Thalassemia
This is the third most common sickling disorder after Hb SS and Hb SC in African Americans, and the most common one in people from the Mediterranean. It usually runs a milder course in black people (usually S/β+-thalassemia) but causes a severe sickling disorder with manifestations similar to those of sickle cell anemia in people of Italian, Turkish, or Greek descent.
In Hb S/β0-thalassemia, Hb A is absent; Hb S is 75% to 90%, Hb F is 5% to 20%, and Hb A2 is 4% to 6%. This disorder clinically and hematologically resembles sickle cell disease, except for the spleen, which remains enlarged after childhood and into adult life. The main difference is that in Hb S/β0-thalassemia, the MCV and MCH are decreased, and the Hb A2 might be significantly increased. Family study is often necessary for a clear distinction (Wrightstone & Huisman, 1974). On the blood smear, pronounced microcytosis, variable hypochromia, and many target cells are present. As a consequence of reduced cellular hemoglobin, Hb S polymers are formed more slowly, and fewer sickled cells are present on the smear.
In Hb S/β+-thalassemia, Hb A is 15% to 30%; Hb S is over 50%, Hb F is 1% to 20%, and Hb A2 is 4% to 6%. Although these individuals clinically may resemble those with sickle trait (Hb AS), in S/β+-thalassemia, the amount of Hb S always exceeds Hb A, while in sickle cell trait Hb A always exceeds Hb S.
Hb SS/α-Thalassemia
Thirty to forty percent of patients with sickle cell anemia are heterozygous for α+-thalassemia, and 2% to 3% are homozygous (Higgs et al, 1982). The MCV (83 fL in heterozygotes and 72 fL in homozygotes) and MCH are decreased. The Hb level is higher and the reticulocyte count is significantly lower as compared with sickle cell anemia. On the smear, sickled cells are uncommon, similarly to Hb S/β0-thalassemia. Although the anemia is less severe, the vaso-occlusive disease is not, and some studies even show increased morbidity and mortality. As described earlier, δ-chains successfully compete with positively charged S-chains for a limited number of α-chains, and Hb A2 levels increase. Hematologically, this cannot be separated from Hb S/β0-thalassemia (Table 32-5). Whenever microcytosis and increased Hb A2 are present in sickle cell anemia, family or molecular studies are done to differentiate these entities.
TABLE 32-5
Differential Diagnosis of Sickle Cell/Thalassemia Syndromes
| Genotype | Clinical Expression | Hb | MCV | Hb F | Hb A2 | Hb A |
| SS | Severe | 7-8 | 85-95 | 2-20 | 2-4 | |
| S/β0 thal | Moderate to severe | 8-10 | 65-75 | 5-20 | 4-6 | |
| SS/α thal | Moderate to severe | 8-10 | 70-85 | 2-20 | 3-5 | |
| S/HPFH | Asymptomatic to mild | 13-14 | 75-85 | 20-30 | 1-3 | |
| S/β+ thal | Mild to moderate | 11-12 | 70-80 | 1-13 | 3-6 | 10-30 |
Hemoglobin SD Disease (Hb S/D–Los Angeles).
SD disease simulates but is less severe than sickle cell anemia, and thus may also resemble SC disease. It is occasionally seen in the African American population. On routine (alkaline) electrophoresis, the pattern is indistinguishable from sickle cell anemia because Hb S and Hb D cannot be separated. Agar gel electrophoresis at pH 6.2 will separate Hb S and Hb D.
Hb S/O Arab.
Compound heterozygotes of Hb S and Hb O Arab (β121 glu→lys) have a severe sickling disorder. Hb O Arab is found in black and Arabic people.
Other Compound Heterozygosities with Hb S.
Hb E and a few other, less frequent β-variants also cause sickling disorders with Hb S. Coexistence of an α-chain variant and a sickle cell trait does not cause a sickling disorder.
Other Common β-Chain Variants
Hb C Trait (β6 glu→lys).
Hemoglobin C is prevalent in West Africans and in about 2% to 3% of black people. The heterozygous state (Hb AC) is asymptomatic, without anemia, with normal or minimally reduced MCV and normal red cell life span. The MCHC might be slightly elevated. Target cells are present on blood film. Hb C makes up about 40% of the total hemoglobin. When significant microcytosis is present, it is usually caused by coexistent α-thalassemia, and Hb C is reduced to 38%, 32%, and 24% in patients with three, two, and one α-gene, respectively (Huisman, 1977).
Hb C Disease.
Homozygous Hb C disease is a mild hemolytic anemia with splenomegaly that is often asymptomatic but occasionally results in jaundice and abdominal discomfort. Life expectancy is normal. In the United States, 0.02% of black people have Hb C disease (Schneider et al, 1976). Reticulocytosis is low for the degree of anemia. The anemia is largely a consequence of low O2 affinity of Hb C (Bunn & Forget, 1986) and should not be treated. The MCV is usually decreased, and the MCHC is normal or increased. Numerous target cells with an admixture of microspherocytes and minimal polychromasia are seen in the blood. Osmotic fragility is biphasic, with both increased and decreased fragility, but is not used in the diagnosis today. Hexagonal or rod-shaped crystals may be seen in erythrocytes in the stained smear, especially after splenectomy or after slow drying of the smear (Fig. 32-23). As opposed to Hb S, crystals of Hb C tend to melt at low partial pressure of oxygen (pO2) and do not cause vaso-occlusive disease. The red blood cells are dehydrated (causing the increased MCHC), owing to loss of cations and water as a result of interaction of Hb C with the red cell membrane. As a consequence, the cells are more rigid and less deformable than normal, increasing their likelihood of being trapped and destroyed in the spleen. Hb F is elevated at 2% to 4%, and Hb A2, when measured by HPLC, might be slightly increased; the remainder of hemoglobin is Hb C.
Hb C/β+-Thalassemia.
This occurs mainly in black people, in whom it tends to result in little disability, except for anemia in pregnancy. Red cell indices are typical of β-thalassemia trait, but there is anisocytosis and 20% to 50% target cells. Usual values are 65% to 80% for Hb C, 16% to 30% for Hb A, and 2% to 5% for Hb F.
Hb C/β0-Thalassemia.
People of Mediterranean extraction usually have a moderately severe hemolytic anemia with β0 or a severe β+ genotype. This combination is extremely rare in the African population. Hb C/β0-thalassemia may be difficult to distinguish from Hb C disease in that Hb A is absent in both, and there is an overlap in Hbs A2 and F levels. Hb A2 is elevated to about twice normal, and Hb F is elevated at 3% to 10%.
Hb E (β 26 glu→lys).
This is probably the most prevalent hemoglobin variant worldwide and the third most common in the United States, behind S and C. It is found primarily in Southeast Asia, especially in people of Thai and Burmese extraction, but is also found in blacks and Caucasians. Hb E is associated with a β-thalassemia phenotype, as well as with a structurally abnormal globin chain. The mutation that causes amino acid substitution also activates a cryptic splice site that competes with the normal RNA splice donor site, and normally processed RNA is decreased. In the laboratory, Hb E can be demonstrated to be unstable. It precipitates abnormally in the heat denaturation test and with isopropanol, yet this has no in vivo significance; the red cell survival is normal (Fairbanks et al, 1980).
Hb E Trait (Hb AE).
Hb AE is asymptomatic, with borderline microcytosis (MCV = 84 ± 5) and no anemia. Hb A is 65% to 70% of the total hemoglobin, Hb E is 30%, and Hb F is normal. Coexistence of one α-thalassemia gene does not change any of the above parameters and can be proven only by DNA analysis. If two or three α-thalassemia genes are also present, the proportion of Hb E decreases to 21% and 14%, respectively. The proportion of Hb E is also significantly lower in iron deficiency.
Hemoglobin E Disease.
This is also asymptomatic; it resembles a thalassemia trait, with microcytosis (average MCV = 70 fL), erythrocytosis, normal MCHC, and slight anemia. Thus, Hb E behaves as an extremely mild thalassemia. The reticulocyte count is normal, but 20% to 80% target cells are evident on the blood film. Hb E accounts for more than 90% of the hemoglobin, with Hb F from 1% to 10% and no Hb A.
Hb E/β-Thalassemia.
In sharp contrast with Hb E disease, this is a severe condition. It is one of the most important thalassemia syndromes, comprising close to half of the severe β-thalassemia cases (Weatherall, 2010). It is most common in Southeast Asia. The clinical variability is marked; rarely, there are very few symptoms, but the usual picture is that of thalassemia intermedia or thalassemia major. Most of the thalassemia alleles are of the β0 or severe β+ variety. Ineffective erythropoiesis, a consequence of excess α-globins, red cell indices, red cell morphology, and clinical manifestations are similar to those in homozygous β-thalassemia. Hbs E, F, and A2 are present. Hb F shows extreme variation, from 5% to 85%; the mean Hb F is 42%, and the mean Hb E is 58% (Steinberg et al, 2009).
Hb D Los Angeles (Punjab) (β121 glu→gln).
This constitutes the most common D variant in African Americans (<0.02%). In the Punjab region of India, heterozygosity reaches 3%. Homozygotes have normal red cell indices, no evidence of hemolysis, and 95% Hb D with normal Hbs F and A2 (Bunn & Forget, 1986). Double heterozygotes for Hb D Punjab/β0-thalassemia have a mild hemolytic anemia with thalassemic red cell indices and increased Hbs F and A2.
The significance of Hb D Punjab is that compound heterozygosity with Hb S produces a moderately severe sickling disorder (see the Hemoglobin SD Disease section earlier in the chapter). Other, less frequent D hemoglobins (D Iran, D Ibadan) and G hemoglobins do not cause sickling—a major difference for genetic counseling. D and G hemoglobins migrate with Hb S on alkaline electrophoresis but are separated from S at acidic pH, migrating with Hb A. It is more difficult to separate them from each other. Careful analysis of isoelectric focusing and HPLC are helpful in most cases.
Common α-Chain Variants
Hb G Philadelphia (α 68asn→lys).
This is the most common α-chain variant in black people (Schneider et al, 1976). Almost invariably, the linked α-gene is deleted (−αG). Simple heterozygotes (−αG/αα) have 30% Hb G and normal red cell indices, but double heterozygotes with α-gene deletion on the other chromosome as well (−αG/−α) have 45% Hb G and a thalassemia trait phenotype. As with other α-chain variants, a minor hemoglobin representing a combination of αG with δ is present close to Hb A2 and helps to differentiate Hb G Philadelphia from β-chain variant G and D hemoglobins (sometimes called G2 hemoglobin).
Disorders of Hemoglobin Function and Stability
A number of amino acid substitutions occur in the heme pocket, where they may cause oxidation of ferrous iron to ferric form producing methemoglobin (Hb M) or alter the affinity of the heme for O2; the latter usually alters the stability of the molecule as well. Other substitutions affect the αβ contact sites; these also can change stability and O2 affinity of the molecule (Bunn, 1994).
These functionally significant hemoglobinopathies are heterozygous; usually, the concentration of the abnormal Hb is less than 50%. Generally, Hbs with abnormal α-chains form a smaller proportion of the total (10% to 25%) than those with abnormal β-chains (35% to 50%).
Hemoglobins Associated with High Oxygen Affinity and Polycythemia
Eighty-six abnormal Hbs with high O2 affinity that are associated with familial erythrocytosis are known today (Globin Gene Server, 2015). Some are listed in Table 32-3. The O2 dissociation curve is shifted to the left. The p50, the pO2 at which Hb is 50% saturated, is decreased. Under physiologic conditions, the normal p50 of whole blood is 26 mm Hg; in this disorder, it has ranged from 5 to 23 mm Hg. Because the Hb has a high affinity for O2, it cannot unload the oxygen and the tissues are relatively hypoxic, resulting in increased EPO production and polycythemia. These disorders are autosomal dominant; only heterozygotes have been described. The Hb concentration has ranged from 15 to 23.8 g/dL. Measurement of O2 affinity is required to establish the diagnosis (Bunn & Forget, 1986). Because the amino acid substitution is inside the molecule, often the abnormal hemoglobin is indistinguishable from Hb A on electrophoresis or HPLC.
Hemoglobins Associated with Low Oxygen Affinity
There are far fewer variants with decreased O2 affinity (see Table 32-3; Bunn & Forget, 1986). The O2 dissociation curve is shifted to the right (increased p50). These individuals have mild “anemia,” as they can unload more O2 to the tissues and they simply do not need that much Hb. A handful of variants with markedly decreased affinity are associated with cyanosis. In these, O2 uptake is impaired in the lung and the level of deoxyhemoglobin is more than 5 g/dL, causing cyanosis. The skin and mucous membranes have a slate gray color. There is no anemia. Many of the unstable Hbs also have decreased O2 affinity; however, the hemolytic state dominates the clinical picture.
M Hemoglobins: Pseudocyanosis
Nine abnormal Hbs are associated with clinical methemoglobinemia and cyanosis that do not respond to methylthioninium chloride (methylene blue) (Globin Gene Server, 2015; Bunn, 1994). The color is similar to cyanosis—a brownish color, caused by methemoglobin. The common feature is that all have an amino acid substitution at or near the heme group, so that methemoglobin is unusually stable, and reduction to ferrous heme and hence reversible binding of O2 are prevented. Methemoglobin constitutes no more than 3% of the total Hb in normal humans.
Cyanosis from birth is seen in Hb M disease with α-chain abnormalities, or in fetal Hb M (Hb FM Osaka). In the latter, cyanosis will disappear after the γ-chains have been replaced by β-chains by 6 months of age. Cyanosis does not appear until nearly 6 months of age in Hb M variants with β-chain abnormalities, for the same reason (Bunn, 1994). Of course, the cyanosis is not associated with enzyme abnormalities in the red cell, toxic drugs, or cyanotic heart disease—conditions that must be considered in the differential diagnosis. Patients usually have no other symptoms.
All Hb M disorders thus far discovered have been reported in heterozygotes, probably because homozygosity is lethal. Some types of Hb M do not separate from Hb A on alkaline electrophoresis. If the hemolysate is first converted to methemoglobin, the Hb M will migrate differently from normal methemoglobin at pH 7.1. The absorption spectra of the eluted Hb M, which may be distinctive, can be compared with those of normal methemoglobin (Bunn, 1994).
Unstable Hemoglobins
More than 100 Hbs, mostly β-chain variants have been described in which the Hb stable tetrameric structure is not maintained and Hgb precipitates within the red cell as Heinz bodies (Bunn, 1998). Some are listed in Table 32-3. Rare unstable hemoglobins such as Hb F Poole are γ-chain variants. The precipitated Hb attaches to the cell membrane making the cells inflexible. Heinz bodies are removed by the spleen that further damages the cells and causes shortened survival. The O2 affinity is usually abnormal but may be increased or decreased. Some of these unstable Hbs cause “congenital Heinz body hemolytic anemias.”
All patients have been heterozygous. Clinical features have shown considerable variation, from severe hemolytic anemia in the first year of life (e.g., Hb Hammersmith, Hb Bristol) to a very mild chronic hemolytic anemia (e.g., Hb Louisville, Hb Hasharon) that may be exacerbated by drugs (e.g., Hb Zurich). A few unstable Hbs have been discovered incidentally in clinically normal individuals (e.g., Hb Tacoma, Hb Sogn).
Jaundice and splenomegaly are common, as in other hemolytic anemias. More distinctive in some cases is the excretion of darkly pigmented urine (only during hemolytic crises in mild variants). Cyanosis is present in some patients and is due to methemoglobinuria and sulfhemoglobinemia or to low O2 affinity.
The anemia is normocytic and normochromic to hypochromic, the latter because of the removal of precipitated Hb from aging red cells by macrophages of the spleen and other reticuloendothelial organs. Prominent basophilic stippling, probably related to excessive clumping of ribosomes, is a common feature. Occasional “bite cells” may be seen. Patients with relatively high Hb concentrations in the steady state usually have Hb variants with a high O2 affinity and an unexpectedly high reticulocyte count (e.g., Hb Köln, Hb Gun Hill). On the other hand, patients with rather low Hb concentrations may be relatively asymptomatic if their Hb has a low O2 affinity; their reticulocyte counts are unexpectedly low for the Hb concentration (e.g., Hb Hammersmith). Heinz bodies are rarely seen in circulating red cells before splenectomy, although sometimes they may be generated by incubating the red cells with brilliant cresyl blue or new methylene blue. After splenectomy, Heinz bodies are readily demonstrable in a large proportion of cells; the blood film shows irregularly contracted cells and basophilic stippling that may be pronounced.
In splenectomized patients, the Heinz bodies may interfere with Hb determinations and with electronic platelet and white blood cell counts. Before the absorbance of the hemolysate is measured, it should be centrifuged to remove the Heinz bodies. Platelet and leukocyte counts should be performed by visual methods. Hb electrophoresis is normal in about one fourth of patients. Hb A2 may be elevated in β-chain variants because of the loss of abnormal Hb from the cells; this phenotype may resemble thalassemia intermedia. Hb F may be increased to a level of 10% to 15%. Key laboratory determinations include heat instability and isopropanol precipitation tests.
Heat Instability Test.
Most unstable Hbs precipitate more rapidly than normal Hbs when incubated at 50° C (Dacie & Lewis, 1991). In a hemolysate in Tris buffer, an easily visible precipitate forms within 1 hour if unstable Hb is present; the control sample is clear or slightly cloudy. Slight precipitation is equivocal; the test should be repeated and the isopropanol precipitation test performed as well. Precipitates accounting for 10% to 40% of the total Hb are found in unstable Hb disorders.
Isopropanol Precipitation Test.
A relatively nonpolar solvent weakens the internal bonds of Hb and decreases its stability (Carrell & Kay, 1972). An unstable Hb precipitates within 20 minutes in the nonpolar solvent isopropanol, whereas a normal hemolysate remains clear for 30 to 40 minutes. False-positive results occur with high levels of Hb F.
Thalassemias
In thalassemias, globin chains, usually of normal structure, are produced at a decreased rate. β-thalassemia refers to decreased production of β-chains; α-thalassemia, δβ-, δ-, and γδβ-thalassemias refer to reduced synthesis of the respective polypeptide chains. As a result, there is an overall deficit of Hb tetramers in the red cells, and MCV and MCH are reduced. However, it is not the lack of the affected globin chain, but the accumulation of the unaffected one, that causes hemolysis and, primarily in β-thalassemia, ineffective hematopoiesis in severe forms of the disease.
Thalassemia occurs predominantly in persons of Mediterranean, African, and Asian ancestry as, similarly to Hb variants or glucose-6-phosphate dehydrogenase (G6PD) deficiency, thalassemia genes are under selective pressure by malaria. In Greece and Southern Italy, the prevalence of β-thalassemia is around 10%, and that of α-thalassemia is 5%. Twenty-five percent to 30% of black people and 20% of Thai people carry an α-thalassemia gene. Because structural variants and thalassemias occur in the same population, a wide variety of diseases emerge from their interactions.
Several classifications are used. The clinical classification defines thalassemia major, a severe and transfusion-dependent form; thalassemia intermedia, with less severe symptoms and generally transfusion independent; and thalassemia minor (carrier state or trait), without clinical symptoms but with hematologic abnormalities. The genetic classification is based on gene(s) affected by the mutation, heterozygous/homozygous state, absent/reduced rate of globin synthesis, and so forth. Finally, specific mutations cause well-defined syndromes and can be used for classification.
Molecular Defects
In β-thalassemia, considerable heterogeneity in molecular defects is noted. One hundred ninety-seven different mutations have been identified as the cause of β-thalassemia. Most are associated with single base substitutions that produce defects in promoter activity, RNA processing/splicing, or translation, resulting in decreased or unstable mRNA. Large deletions are uncommon. In rare structural variants, the production of highly unstable β-chains results in the phenotype of β-thalassemia (Bunn, 1998). Despite this diversity, 20 common mutations account for 80% of β-thalassemia alleles in the world population (Weatherall & Clegg, 1999). In β0-thalassemia, β-chain synthesis is absent on the affected chromosome. Messenger RNA (mRNA) is absent or may be present but nonfunctional. In β+-thalassemia, β-globin chains are present but reduced in quantity because molecular defects have resulted in the production of unstable or decreased amounts of mRNA.
In δβ0-thalassemia, large deletions involve the δβ– or Aγδβ–gene complex. As a result, no Hb A or Hb A2 synthesis is supported from the affected chromosome, but the γ-gene is upregulated and Hb F production is increased. Lepore Hbs have δβ-fusion globins that are the result of unequal crossover between δ- and β-globin genes during meiosis.
The α-thalassemias are generally due to gene deletion of various lengths. Two α-globin genes are present on each chromosome 16, surrounded by two highly homologous duplication units, each containing three homologous segments (Z, X, and Y). Unequal crossing over between the Z segments produces a chromosome with one α-gene (−α3.7) and another with three (ααα). Similar nonreciprocal crossing over between X boxes causes another common deletion (−α4.2) (Fig. 32-24). This recombination has a high probability, and chromosomes with missing or extra α-genes are found in every civilization; nonetheless, only the thalassemic alleles became frequent in certain populations, under pressure from malaria. These defects, affecting only one of the genes, are called α+-thalassemia. The heterozygous genotype can be written (−α/αα). Nondeletion defects are less common (αTα/αα). α0-Thalassemia results from deletion of both α-globin genes on the chromosome (Fig. 32-25), which leads to no α-chain synthesis (− −/αα).
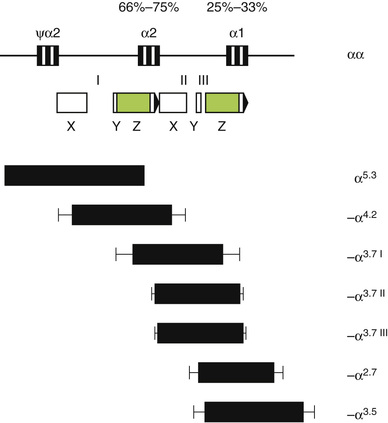
Hb Constant Spring is due to an abnormal termination codon in an α-globin gene that results in an elongated α-chain with 31 extra amino acids. Because of marked reduction in mRNA stability, the clinical phenotype of α-thalassemia is seen.
β-Thalassemias
Clinical and Hb findings in the β-thalassemias are summarized in Tables 32-6 and 32-7. The disorders are very heterogeneous phenotypically, as well as at the level of the molecular defects. The terms thalassemia major, thalassemia intermedia, and thalassemia minor refer to clinical severity and are not genetic designations.
TABLE 32-6
β-Thalassemias and Their Associated Biochemical and Molecular Defects
| Typical DNA Defect | β-Chain | δ-Chain | γ-Chain | Hb F Distribution | α: Non–α-Globin Imbalance | |
| β+ thalassemia | Mutation | ↓ | + | + | Heterocellular | +++ |
| β0 thalassemia | Mutation | 0 | + | + | Heterocellular | ++++ |
| δβ thalassemia | Deletion | 0 | 0 | +++ | Heterocellular | ++ |
| HPFH | Deletion | 0 or ↓ | 0 | ++++ | Pancellular | + |
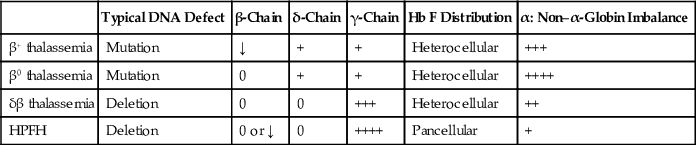
TABLE 32-7
Major Categories of β-Thalassemia Syndromes
| Syndrome | Genotype | Clinical Features | Hemoglobin Pattern |
| Homozygous States | |||
| β+-thalassemia | β+/β+ | Thalassemia major or intermedia | ↓↓Hb A, ↑↑Hb F, variable Hb A2 |
| β0-thalassemia | β0/β0 | Thalassemia major | >95% Hb F, rest Hb A2 |
| δβ0-thalassemia | δβ0/δβ0 | Thalassemia intermedia | 100% Hb F |
| Hb Lepore | Lepore/Lepore | Thalassemia major | 85% Hb F, 15% Hb Lepore |
| Heterozygous States | |||
| β+-Thalassemia | β+/β | Thalassemia minor | Hb A, ↑Hb A2, ±↑ Hb F |
| β0-Thalassemia | β0/β | Thalassemia minor | Hb A, ↑Hb A2, ±↑ Hb F |
| δβ0-Thalassemia | δβ0/δβ | Thalassemia minor | Hb A, 5%-20% Hb F, ±↓Hb A2 |
| Hb Lepore | Lepore/β | Thalassemia minor | Hb A, ↑Hb F, ↓Hb A2, 10% Hb Lepore |

Homozygous β-Thalassemia (Thalassemia Major; Cooley's Anemia)
In the absence (β0) or a marked decrease (β+) of β-chain production, there is an excess of α-chains. Aggregates of α-chains are unstable and precipitate in the normoblast or erythrocyte and damage the cells. Excess α-chains and their degradation products—heme, hemin, and iron, which serve as foci for the generation of reactive O2 species—result in the partial oxidation of band 4.1 and a reduced spectrin/band 3 ratio in red blood cell precursors. Precipitates and cells are removed, causing ineffective erythropoiesis and severe hemolytic anemia. Furthermore, clustering of band 3 in the membrane may be followed by opsonization with autologous IgG and complement and removal by macrophages (Weatherall & Clegg, 1999).
Clinical findings include jaundice and splenomegaly, which become evident early in childhood. Prominent frontal bones, cheekbones, and jaws impart a mongoloid appearance. These changes and X-ray findings of a thinned cortex of the long and flat bones and thickening of the skull with osteoporosis (“hair-on-end” appearance) reflect extreme bone marrow hyperplasia. Growth is stunted, and puberty is delayed. Most patients require regular transfusions and develop problems caused by iron loading. Iron overload commonly develops, and the major cause of death is cardiac failure due to myocardial siderosis by the end of the third decade.
Unlike most hemolytic diseases, the anemia is hypochromic and microcytic. Extreme poikilocytosis with bizarre shapes, target cells, ovalocytosis, Cabot rings, Howell-Jolly bodies, nuclear fragments, siderocytes, anisochromia, anisocytosis, and often extreme normoblastosis are present (Fig. 32-26). Poikilocytosis is more striking in patients with intact spleen, while nucleated red blood cells are more numerous after splenectomy. Nucleated red blood cells have hypochromic cytoplasm and, especially after splenectomy, aggregates of densely staining Hb, which probably represent precipitated α-chains. Incubation of the blood with methyl violet stains these precipitates in both red cells and erythroblasts. The reticulocyte count is less elevated than expected for the degree of anemia because of destruction of erythroid precursors in the marrow. Osmotic resistance of the red cells, serum iron, and indirect-reacting bilirubin are increased.
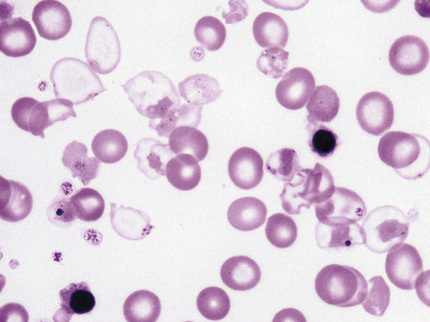
In the marrow, marked erythroid hyperplasia is present. Many late normoblasts show inclusion bodies, as in the blood. Gaucher-like cells are present. Storage iron and sideroblasts are increased.
In β0-thalassemia, Hb A is absent, Hb F is as high as 98%, and Hb A2 is about 2%. In β+-thalassemias (Mediterranean), Hb F is 60% to 95%, with Hb A present. Although Hb A2 may or may not be increased, the ratio of A2 to A is always increased. In black people with β+-thalassemia, the clinical features are less severe (thalassemia intermedia), and transfusion is usually unnecessary; Hb F is 20% to 40%, Hb A2 is 2% to 5%, and the rest is Hb A (see Table 32-7).
Heterozygous β-Thalassemia (β-Thalassemia Trait; Thalassemia Minor; Cooley's Trait)
This is caused by the β0-thalassemia gene with absent, or the β+-thalassemia gene with reduced, β-globin chain synthesis. There are usually no symptoms or abnormal physical signs. The only clinical presentation might be a refractory anemia of pregnancy (Weatherall & Clegg, 2001).
Most β-thalassemia heterozygotes have a mild anemia but, occasionally, Hct and Hb might be normal. Those of African origin have higher Hb levels than people from the Mediterranean region, reflecting their milder genotype. Characteristically, the RBC is elevated (5 to 7 M/µL), the MCH is low (usually less than 22 pg), and the MCV is low (between 55 and 70 fL). The MCHC is sometimes low but often normal. The reticulocyte count is twice the normal value. On stained films, the cells have a moderate degree of microcytosis, hypochromia, and poikilocytosis; target cells and basophilic stippling are often, but not always, present (Fig. 32-27). In the marrow, there is mild erythroid hyperplasia with ragged cytoplasmic borders—a sign of defective hemoglobinization.
Hb A2 is elevated in the 3.5% to 7% range, and Hb F is slightly elevated (1% to 3%) in about half of cases. In the few cases where Hb F exceeds 4%, it is likely that a gene for HPFH is also present. The relatively rare deletional forms tend to have higher levels of Hb F (up to 9%), and in a few families, in which the deletion included the promoter region, Hb F was found to be unusually high (up to 14%) (Weatherall & Clegg, 2001). In infants, a slower than normal decline in Hb F level is observed, and the adult steady-state level is not reached until adolescence. This is a particularly important consideration in double heterozygosity for β-thalassemia and Hb S, when Hb F level is used to predict prognosis.
β-Thalassemia Trait with Normal Hb A2
In a few cases, both Hb A2 and F are normal. These are difficult to distinguish from α-thalassemia trait, and only molecular studies might be definitive. The bulk of these cases results from the coinheritance of δ-thalassemia (Weatherall, 1994), either in trans or in cis, to the β-thalassemia gene. In other patients with normal Hb pattern, only minimal hematologic changes are noted (the “silent” β-thalassemia gene), and only a more severe β-thalassemia syndrome in a family member suggests the presence of a very mild β-thalassemia mutation.
Iron studies are little different from normal (Weatherall & Clegg, 2001), although iron deficiency often complicates thalassemia trait in childhood and during pregnancy. In severe iron deficiency, the level of Hb A2 may fall into the normal range, thus obscuring the diagnosis of β-thalassemia trait, but this is unusual. Most often, although the level of A2 falls, it remains elevated above the normal range (Weatherall & Clegg, 2001). Nevertheless, when iron deficiency is present, repeat measurement of Hb A2 is recommended after replenishment of iron.
The differential diagnosis between iron deficiency and β- or α-thalassemia trait can be difficult. Increased RBCs in the presence of decreased MCV is the hallmark of thalassemia trait. An MCV/RBC ratio less than 13 suggests thalassemia trait; a ratio greater than 13 is more consistent with iron deficiency, but this and other formulas are not conclusive enough for diagnosis.
δβ0-Thalassemia
This is sometimes called F-thalassemia, a helpful name, as you have to think of it when there are thalassemic indices and significantly increased levels of Hb F. β- and δ-chains are not produced; this is nearly, although not completely, compensated for by increased output of γ-chains. The heterozygous state is similar to a mild β-thalassemia trait, except that Hb A2 is not increased or even slightly reduced (mean level is 2.4%), and Hb F is significantly increased (5.4% to 20%). In the homozygous state, hemoglobin consists of only Hb F.
Clinically, (δβ)0-thalassemia behaves as a mild form of β-thalassemia. In the heterozygous state, the Hb is normal or slightly reduced, the MCH is between 21 and 26 pg, and the MCV is 65 to 79 fL. There are no clinical symptoms. Homozygotes have a mild form of thalassemia intermedia with a Hb level of 10 to 13 g/dL, mildly thalassemic red cell indices, and only minimal hepatosplenomegaly. It is most common in the Mediterranean population. The mild phenotype is the result of increased production of γ-chains, which compensate to some degree for the lack of β-chains.
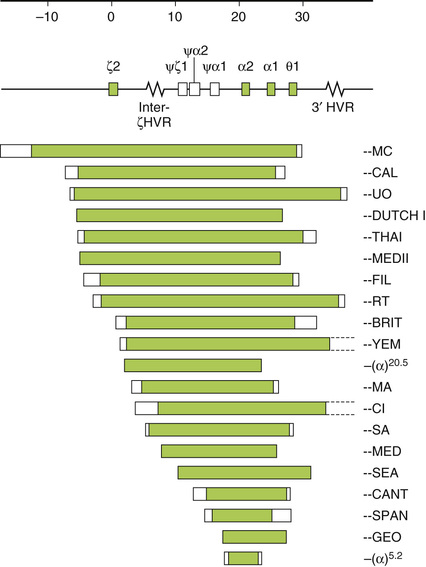
The molecular defect is a long deletion involving the β- and δ- and often also the Aγ-gene. Twenty-one different deletions have been described (Globin Gene Server, 2015), but the hematologic findings are essentially the same. When the Aγ-gene is also deleted, the accurate nomenclature is (Aγδβ)0-thalassemia; homozygotes have a somewhat more severe phenotype.
δβ+-Thalassemia: Lepore Hemoglobins
In the Lepore hemoglobins, an abnormal δβ-fusion chain is produced, a result of chromosome crossing-over and fusion of genetic material at the δβ-genes. No normal δ- or β-chain synthesis is directed from the affected chromosome. Because Hb F production is only slightly increased and the composite δβ-chain is synthesized at a very slow rate, a severe thalassemic phenotype is produced. Hematologic abnormalities are similar to those seen in β0-thalassemia. Hb Lepore migrates slightly faster than Hb S on alkaline electrophoresis and usually constitutes about 10% of total Hb in the heterozygotes; Hb A2 averages 2%, and Hb F is 2% to 3% (Efremov et al, 1978). In homozygotes, Hb Lepore is 10% to 15%, and the rest is Hb F. Different Hb Lepores have been described, depending on the point of fusion, but they behave similarly. This is a much more severe thalassemia gene than the (δβ)0 form and causes transfusion-dependent thalassemia major in the homozygous state.
In a similar mechanism, anti-Lepore hemoglobins have βδ-fusion gene. Several of these hemoglobins have been described, including P-Nilotic.
Hereditary Persistence of Fetal Hemoglobin
A group of conditions with persistence of fetal hemoglobin production beyond infancy, but without significant hematologic abnormalities, is known as HPFH. There are two major types: pancellular and heterocellular (or Swiss) HPFH.
The pancellular form of HPFH is closely related to β- and δβ-thalassemias, with which it forms a continuous spectrum. At one end of the spectrum is minimal γ-chain production with no compensation for deficiency of β-chains in β-thalassemia; at the other end, Hb F production is up and almost entirely compensates for the deficit in HPFH (Weatherall & Clegg, 2001). As a result, at least in heterozygotes, there is no clear evidence of thalassemia (except maybe borderline microcytosis in a few cases), and this serves as a criterion in diagnosis. In clinical practice, a patient with significantly elevated Hb F and reduced Hb A2 is suspected to have HPFH if the red cell indices are normal, but is diagnosed with δβ0-thalassemia if the indices are thalassemic. Nevertheless, there is a slight α/non–α-chain imbalance, and HPFH could be considered an extremely mild thalassemia. Hb F is homogeneously (evenly) distributed among the red cells (pancellular). This is in contrast to β- or δβ-thalassemia, in which the distribution is heterocellular. Pancellular HPFH is relatively rare. It is found in about 0.1% of African Americans and even less frequently in other ethnic groups.
Deletional Pancellular HPFH
In the six deletional forms, the δβ-gene complex is deleted. The black and Ghanaian forms are the most common, but Indian, Italian, and Southeast Asian forms are also well documented. Homozygotes have slightly microcytic, hypochromic red cells, but no anemia. Hb F is 100%; no Hb A or Hb A2 is present. The Hct can even be high—a result of high O2 affinity of Hb F. In the heterozygote, no hematologic abnormalities are found. Hb F is 15% to 30%, and Hb A2 is decreased at 1% to 2.1%.
Hb Kenya
This is a Hb analogous to the Lepore Hbs, which is associated with an HPFH phenotype. It contains an Aγβ-fusion gene. In the heterozygote, Hb Kenya is around 10%, F is 7%, and A2 is reduced.
Nondeletional Pancellular HPFH
A mutation in the promoter region of one of the γ-genes results in increased synthesis of Hb F. Output from the δ- and β-genes in cis is reduced. Levels of Hb F range from 3% to 31% in the different forms, Hb A2 is invariably low, and the red cell indices are close to normal. There are two particulars to keep in mind. First, β-chain synthesis is decreased, but not absent from the affected chromosome, and compound heterozygotes of this form of HPFH and Hb S on the other chromosome do have Hb A (≈30%). The hemoglobin composition is similar to that seen in Hb S/β+-thalassemia, except that Hb A2 is reduced. Second, it is not rare to find it together with an α-thalassemia gene, which is highly prevalent in African Americans.
Heterocellular or Swiss-type HPFH
Heterocellular or Swiss-type HPFH has less Hb F, ranging from 2% to 5%. The distribution of Hb F is uneven (heterocellular): Both F cells and erythrocytes completely lacking Hb F are present. It was first described among healthy Swiss army recruits but is present in other populations and is quite common. It is caused by polymorphic variations at three well-known and other lesser known loci that regulate adult Hb F production (Thein & Menzel, 2009). In normals or in carriers of β-thalassemia or Hb S, there is only a minimal increase in Hb F. When inherited with Hb SS or homozygous β-thalassemia, however, the result could be an unusually high level of Hb F and milder disease.
α-Thalassemias
α-thalassemia is probably the most common single-gene disorder in humans. Its distribution is largely limited to tropical and subtropical regions of Asia and Africa and the Mediterranean (Higgs et al, 1989), where it reaches extremely high frequencies.
Two α-globin genes are present on each chromosome 16. α-thalassemias are classified according to the total output of these two linked α-globin genes. In α0-thalassemia, both genes are inactive (− −/), but in α+-thalassemia, only one gene is defective as the result of deletion (−α/) or, less frequently, of mutation (αTα/). Nondeletional forms usually result in less globin output from the linked α-gene and more severe phenotypes. Rarely, the chromosome has a deletion and a separate mutation (αT−/). Previously, α0 and α+ were called α-thalassemia 1 and 2. This was rather confusing, as the milder defect with one affected gene was called α-thalassemia 2, and the more severe genotype with two defective genes, α-thalassemia 1. Alternatively, these terms were also used to describe clinical phenotypes.
Unlike the extremely unstable α-chains in β-thalassemia, excess β- and γ-chains can form stable tetramers, hemoglobin H (β4), and Bart's (γ4). These precipitate in aging red cells and, through interaction with the cell membrane, cause hemolysis. This is mostly a hemolytic anemia, whereas in β-thalassemia, ineffective erythropoiesis predominates.
α-Thalassemia Syndromes
Four α-thalassemia syndromes result from the combination of these genotypes (Bunn & Forget, 1986), which roughly correspond to the loss of 4, 3, 2, or 1 genes from the normal complement (αα/αα) (Table 32-8). In the following discussion, nondeletion forms are not always depicted separately, for the sake of simplicity.
TABLE 32-8
α-Thalassemia Syndromes
| Syndrome | Defective Genes | Genotype | Clinical Features | Newborn | After First Year |
| Hydrops fetalis | 4 | −−/−− | Fetal or neonatal death with severe anemia | Hb Bart's >80% Hb H, Hb Portland | |
| Hb H disease | 3 | −−/−α (−−/ααCS) | Chronic hemolytic anemia | Hb Bart's 20%-40% (Hb CS) | Hb H 5%-30% (Hb CS 2%-3%) |
| Thalassemia minor | 2 | −−/αα −α/−α αTα/−α | Asymptomatic, mild anemia, thalassemic indices | Hb Bart's 5%-10% | None |
| Silent carrier | 1 | −α/αα (αα/ααCS) | No clinical or hematologic abnormality | Hb Bart's ±1%-2% (Hb CS) | None (Hb CS 1%) |
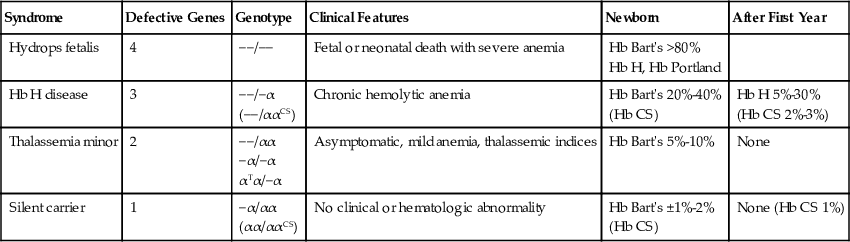
Hemoglobin Bart's Hydrops Fetalis (− −/− −).
Complete absence of α-chains is incompatible with life. Infants are stillborn with severe edema, marked anemia, and marked hepatosplenomegaly. The blood shows marked anisocytosis, poikilocytosis, microcytosis, and erythroblastosis. ABO or Rh incompatibility is absent. Because of the absence of α-chains, no Hb A or Hb F is present. Large quantities of Hb Bart's (γ4), a variable amount of Hb Portland, and traces of Hb H (β4) are present; all of these migrate faster than Hb A on alkaline electrophoresis. Hb Bart's is functionally useless for O2 transfer, causing extreme intrauterine hypoxia.
Hemoglobin H Disease (−α/− −).
Three of the four α-genes are absent. A chronic hemolytic anemia occurs with the clinical picture of thalassemia intermedia in a minority of cases, although the severity varies, and most patients do well. Hb H disease is very common in Southeast Asia but is also seen in the Mediterranean and the Middle East; it is very rare, however, in black people, as α0-thalassemia is uncommon in this group. Splenomegaly and sometimes hepatomegaly are present. Hb values average 3 g/dL less than in age- and sex-matched controls. Transfusion is rarely needed. The anemia may become more severe during pregnancy, but the Hb rarely falls below 7 g/dL. The MCV (60 to 70 fL) and MCH (17 to 21 pg) are decreased (Higgs et al, 1989), and RBC is increased (6 to 6.2 M/µL). The blood film shows hypochromia, basophilic stippling, and anisopoikilocytosis with target cells (Fig. 32-28). Reticulocytes range from 4% to 5%.

Hemoglobin electrophoresis shows a rapidly migrating band of Hb H (β4), accounting for 1% to 40% (average 9%) of the hemoglobin, and the slightly less rapidly migrating Hb Bart's in half of the cases. Hb H can precipitate and be lost from the hemolysate in vitro by careless handling or prolonged storage. In old hemolysates, Hb H appears as a series of bands on electrophoresis. The percentage of Hb Bart's is 2% to 40% at birth; it gradually falls thereafter, averaging 4.8%, but the level in adults is variable. As in other α-thalassemia syndromes, more Hb Bart's is present at birth than Hb H in adult life. Hb A2 is diminished.
Hemoglobin H preparation.
Vital staining of the blood with an oxidizing dye such as brilliant cresyl blue induces inclusion bodies (Hb H precipitates) in many of the red blood cells. During incubation of two parts of blood in one part of 1% brilliant cresyl blue stain, the unstable Hb H (β4) gradually precipitates as multiple small pale blue inclusions uniformly distributed on the red cell membrane (Fig. 32-29) (Jones et al, 1981). Hb H inclusions must be distinguished from (1) the granules and reticular networks of reticulocytes, which are darker blue in color and (2) preformed Heinz bodies, which are larger, also darker blue, and are often attached to the membrane. After 20 minutes of incubation at room temperature, Hb H inclusions are present in at least half of the red cells in Hb H disease, and in rare red cells in α-thalassemia trait. The larger, single Heinz bodies may be found after splenectomy in Hb H disease.

α-Thalassemia Trait: Heterozygous α0-Thalassemia (− −/αα) or Homozygous α+-Thalassemia (−α/−α).
Absence of two α-genes results in clinical features similar to β-thalassemia trait with very mild anemia and thalassemic indices, with MCV ranging from 65 to 75 fL (Higgs et al, 1989). The α-chain/β-chain synthesis ratio is decreased (≈0.6). Diagnosis is best made at birth, by finding 5% to 10% Hb Bart's in cord blood; in normals only trace amounts (<0.5%) are present. In adults, Hb Bart's is undetectable, and Hb studies are perfectly normal, except that Hb A2 might be slightly reduced. Hb H inclusions are found in α0-thalassemia, but rarely in heterozygous or homozygous α+-thalassemia, and only in a very small percentage of red cells, if exhaustively sought after (Wasi et al, 1974), and if the sample is enriched for Hb H-containing red cells (Jones et al, 1981). Otherwise, no evidence of Hb imbalance is detectable by standard techniques, and the diagnosis is one of excluding iron deficiency, anemia of chronic disease, and β-thalassemia trait. In contrast to β-thalassemia, Hb F is normal, and Hb A2 is normal or decreased. This condition is absolutely benign, and most patients are diagnosed on routine screening. Hematologic findings are identical in the two distinct genotypes found in different populations: (− −/αα), common in Southeast Asia and the Mediterranean and exceedingly rare in black people, and (−α−α), most common in those of African descent.
Silent Carrier α-Thalassemia (Heterozygous α+-Thalassemia) (αα/−α).
In this condition, one of four α-globin genes is absent. Hematologic findings are normal, except that the MCV might be slightly reduced, with a mean value of 81 fL and a range of 75 to 85 fL, and the MCH might be minimally decreased; many times, however, the red cell indices are perfectly normal. During the neonatal period, heterozygous α+-thalassemia can be diagnosed by a raised level of Hb Bart's (1% to 2%) in the cord blood. Hb Bart's disappears by 6 months of age, and the diagnosis can be made only by molecular or globin chain synthesis studies. Because only 40% of newborns with the heterozygous α+-thalassemia genotype have detectable Hb Bart's in their cord blood (Higgs et al, 1982), failure to detect Hb Bart's in the newborn does not rule out silent carrier α-thalassemia, and newborn screening should not be used to rule out this entity.
From large studies comparing hematologic findings of different genotypes, it became apparent that there is a continuum between normal, silent carrier, and α-thalassemia trait. Because there is no clear separation between one- and two-gene defects, some authors group these together as milder α-thalassemia phenotypes. None of the α-thalassemia genes or their combinations can be identified with certainty without molecular studies.
Hemoglobin Constant Spring (αCSα/).
Hb Constant Spring is due to an abnormal termination codon in an α-gene that results in an elongated α-chain with 31 extra amino acids. It is by far the most common of the elongated α-chain variants. Because of marked reduction in mRNA stability, the clinical phenotype of α-thalassemia is seen. Similar to other nondeletional α+-thalassemia genes, it causes a more severe phenotype. The homozygous state appears as an asymptomatic, mild hemolytic anemia, with a Hb level of 9 to 11 g/dL. The red cell indices are unusual for thalassemia: The MCV is normal (88 fL), and the RBC is low (3.9 M/µL). Hemoglobin consists of 5% to 8% Hb CS, normal Hb A2, trace amounts of Hb Bart's, and the rest Hb A (Weatherall, 1994). Heterozygotes have a silent carrier phenotype with no hematologic abnormality and about 1% Hb CS. Hb CS undergoes proteolysis and appears as 1 to 4 faint bands migrating either slightly slower or slightly faster than Hb A2 at alkaline pH and is easily missed. Hb CS is common in Southeast Asia, where it is found in about 50% of cases of Hb H disease (αCSα/− −).
Screening and Prenatal Diagnosis of Hemoglobin Disorders
In populations in which there is a significant incidence of severe forms of thalassemia or sickle cell anemia, women should be screened early in pregnancy for thalassemia and the sickle cell trait (Weatherall, 1985, 2010; Alter, 1988). If both parents are carriers, prevention of severe disease is possible through genetic counseling and offering prenatal diagnosis with the option of therapeutic abortion. In many high-frequency regions, successful screening of schoolchildren or premarital counseling has been implemented. Initial tests include MCV (<80 fL), MCH (<27 pg), and HPLC to estimate Hb A2 (>3.5%) (cutoff values in parentheses) and to detect common Hb variants.
Hemolysis—Metabolic Disorders
Deficient enzyme activity in the erythrocyte may result in abnormalities that lead to premature destruction and hemolytic anemia; these disorders are usually inherited. However, interference with, or oxidative stress on, erythrocyte metabolism can sometimes result in hemolysis in individuals who have normal erythrocytes.
Erythrocyte Metabolism
The mature red blood cell lacks mitochondria and, therefore, lacks oxidative phosphorylation and Krebs cycle activity. Energy production is mainly glycolytic, 90% of which occurs through the Embden-Meyerhof pathway, as glucose goes to lactic acid with the net production of 2 mol of adenosine triphosphate (ATP) (Fig. 32-30). ATP is needed for energy-requiring reactions in the cell: for active cation transport across the membrane, for maintaining membrane deformability, and for preserving the cell's biconcave shape. Glucose uptake by red cells is independent of insulin. Approximately 90% of glucose is consumed in the glycolytic pathway, while 10% is utilized in the pentose phosphate pathway (hexose monophosphate [HMP] shunt). One step of the glycolytic pathway replenishes NADH (reducing agent derived from nicotinamide adenine dinucleotide [NAD]), which plays a major role in protecting Hb from oxidative stress. Most of the hemiglobin (methemoglobin) produced in the normal cell (about 3% of the total per day) is reduced by NAD-linked methemoglobin (Met Hb) reductase. The HMP shunt generates nicotinamide adenine dinucleotide phosphate (NADPH) in the first two steps through the enzymes G6PD and 6-phosphogluconate. NADPH production is linked to glutathione reduction and, through this mechanism, to preservation of vital enzymes and Hb from oxidation. Small amounts of oxidized hemoglobin (methemoglobin) are reduced by glutathione (GSH). The activity of the HMP shunt increases when the cell is exposed to an oxidant drug, probably as a result of increased NADP production. If an enzyme in this pathway lacks activity, GSH cannot be produced and it will remain in its oxidized form as GSSG, and thus Hb will not be protected from the oxidant stress. Oxidation in the red blood cells is mediated by high-energy derivatives of O2, referred to collectively as activated O2 (van Solinge & van Wijk, 2010). Oxidized globin chains denature and precipitate as Heinz bodies, which adhere to the membrane, inducing rigidity and a tendency to lyse. Moderate enzyme deficiencies in this pathway (e.g., in G6PD) may not be associated with anemia under normal conditions; however, an acute hemolytic episode occurs if the cells are challenged by oxidant stress (e.g., drugs, infection).
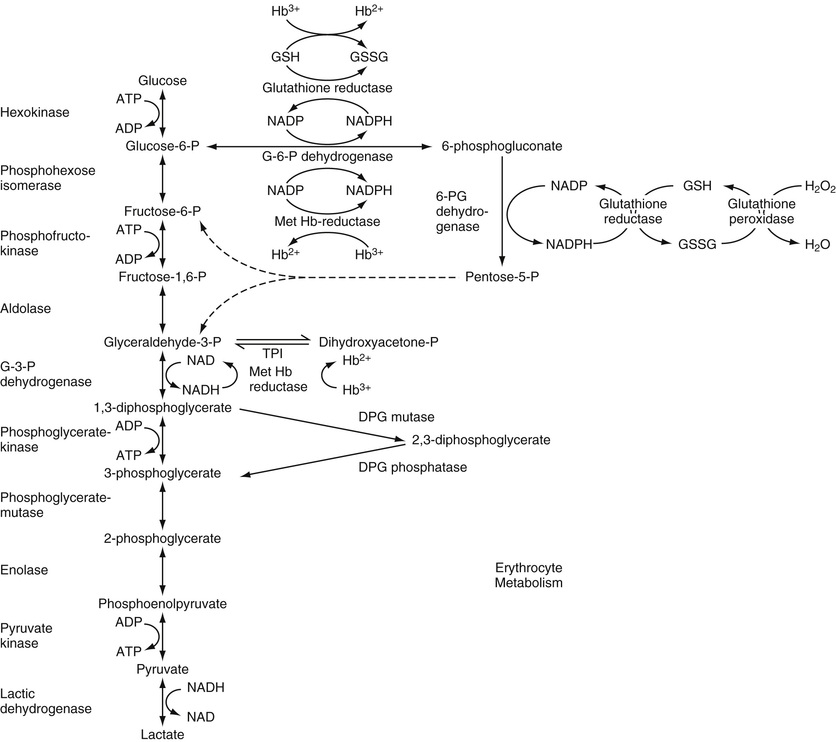
Deficiencies in the Embden-Meyerhof pathway result in impaired ATP generation and a chronic hemolytic anemia. The mechanism of red cell destruction here is less clear. Heinz bodies are not formed. Lack of cell deformability and impaired cation pumping are important in the hemolytic process. However, ATP deficiency is difficult to demonstrate in many patients, and other disorders associated with more severe ATP deficiency are not associated with significant hemolysis (van Solinge & van Wijk, 2010).
The Rapoport-Luebering shunt provides for the conversion of 1,3-diphosphoglycerate (1,3-DPG) to 2,3-DPG instead of directly to 3-phosphoglycerate (3-PG) (see Fig. 32-30). If this shunt is operating, generation of 2 mol of ATP (per mole of glucose) is bypassed; the result is no net energy production in glycolysis. However, 2,3-DPG combines with the β-chain of Hb and decreases the affinity of Hb for O2. At a given pO2, therefore, increased 2,3-DPG allows more O2 to leave Hb and go to the tissues; the O2 dissociation curve is shifted to the right. Increased activity of this shunt is apparently stimulated by hypoxia.
Glucose-6-Phosphate Dehydrogenase Deficiency
About 10% of male American blacks who were given the antimalarial drug primaquine during the Korean War developed a self-limited, acute hemolytic anemia (Beutler, 1994). The relationship between antimalarial drugs and hemolysis had been observed earlier in the 1920s (Beutler & Luzzatto, 1999). Only the older red cells were destroyed, and it was eventually determined that the deficiency in susceptible red cells was in G6PD. Reticulocytes have five times higher enzyme activity than the oldest erythrocyte population (van Solinge & van Wijk, 2010). It has since been found that G6PD deficiency is widespread throughout the world. Among whites, the highest incidence is in Kurdish Jews; the deficiency is common in the Middle East, in Mediterranean countries, and in Asia. G6PD deficiency is the most common human enzyme defect and is present in more than 400 million people worldwide (Cappellini & Fiorelli, 2008).
G6PD is present in all cells; however, its concentration varies in different tissues. In healthy red cells, the enzyme operates at only 1% to 2% of its maximal potential, allowing for a large reserve of reductive potential (Cappellini & Fiorelli, 2008). Because G6PD is determined by a gene on the X chromosome, full expression of the deficiency is found in the male hemizygote. Partial expression may be found in the heterozygous female, who has two populations of red cells—one normal and one deficient. The deficiency of G6PD limits the regeneration of NADPH, which renders the cell vulnerable to oxidative denaturation of Hb. Because, normally, G6PD is highest in young cells and decreases as the cell ages, in persons with G6PD deficiency, the older cells are preferentially destroyed.
Hemolytic susceptibility in affected persons can increase greatly during intercurrent illness or upon exposure to various drugs that have oxidant properties (Box 32-1).
The genetic heterogeneity is great, and approximately 400 biochemical variants have been defined. This genetic heterogeneity is expressed as variations in stability and the electrophoretic and catalytic properties of the enzymes, in the degree of deficiency, in the types of cells in the body affected, in the types of drugs that will produce hemolysis, and in susceptibility to chronic hemolysis or to neonatal jaundice. The most common (“normal”) G6PD isoenzyme in all population groups is designated as B. In blacks, an electrophoretically more rapid variant, A, is prevalent and has almost the same activity; 20% of black males have this variant. Eleven percent of black males have the A type of G6PD, which involves only 5% to 15% of normal enzyme activity; it is these individuals who are susceptible to hemolysis after ingesting oxidant drugs or during infection. The most common variant in whites is G6PD-Mediterranean, found in Mediterranean populations; the level of enzyme activity in affected males is low—often less than 1%. These individuals usually are not anemic but may have somewhat more severe and non–self-limited hemolytic anemia with infection, and with a wider variety of drugs than the black variant. In a subgroup of G6PD-deficient subjects, severe hemolysis may occur within hours after eating fava beans (“favism”). Although the vast majority of G6PD-deficient subjects worldwide are not anemic, a small proportion of persons with G6PD-Mediterranean (and persons with some rarer variants) have a chronic nonspherocytic hemolytic anemia. G6PD deficiency is a common cause of neonatal jaundice, occurring in approximately one third of all male neonates with jaundice (Cappellini & Fiorelli, 2008). Jaundice is usually evident by 1 to 4 days of age and is more severe in premature babies than in full-term infants.
Laboratory findings during active hemolysis are those of hemolytic anemia in general. In the blood film, one finds poikilocytes, some spherocytes, bite cells, and irregularly contracted cells that stain densely and have contraction of Hb from a part of the cell membrane. These probably are cells from which Heinz bodies have been removed by the spleen. After supravital staining with methyl violet, Heinz bodies may be present early in an acute hemolytic episode. G6PD deficiency may be detected by one of the screening tests: the dye reduction test, the ascorbate cyanide test, or a fluorescent spot test. Confirmation is made with a quantitative assay.
Heinz Bodies
When a globin chain denatures, it forms precipitates that are known as Heinz bodies (Dacie & Lewis, 1991). These precipitates cannot be detected in Romanowsky's-stained, air-dried blood films, but after vital staining with methyl violet or crystal violet, Heinz bodies stain deep purple. They vary from 1 to 4 µm in diameter and often attach to the red cell membrane. They also stain, but less intensely, as pale blue inclusions in reticulocyte stains (e.g., new methylene blue) (Fig. 32-31).
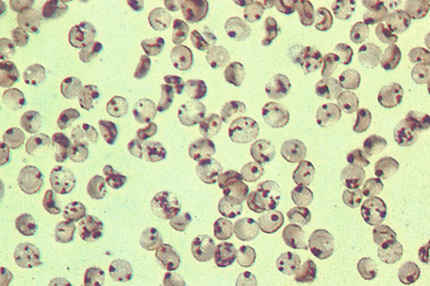
The presence of Heinz bodies in freshly drawn blood indicates that (1) an oxidizing drug or chemical (e.g., phenylhydrazine, chlorate, naphthalene, dapsone) has been ingested in sufficient amount to overwhelm the normal protective mechanisms of the red cell and denature Hb; (2) a drug such as primaquine has been ingested by an individual with G6PD deficiency (or another defect resulting in a deficiency of reduced glutathione), so that Hb is not protected from oxidative denaturation; or (3) the subject has an unstable Hb.
Ascorbate Cyanide Test
When blood is incubated with a solution of sodium cyanide and sodium ascorbate, hydrogen peroxide is generated from the coupled oxidation of ascorbate and oxyhemoglobin (Dacie & Lewis, 1991). Cyanide inhibits catalase, hydrogen peroxide is available to oxidize Hb, and the brown color of methemoglobin is discernible. This occurs more rapidly in G6PD-deficient cells than in normal cells.
The ascorbate cyanide test is not specific, in that abnormalities in the glutathione synthesis or maintenance pathways can produce positive results.
Fluorescent Spot Test
Whole blood is added to a mixture of glucose-6-phosphate (G6P), NADP, saponin, and buffer, and a spot of this mixture is placed on filter paper and is observed for fluorescence with ultraviolet light. If G6PD is present, NADP is converted to NADPH. Because phosphogluconate dehydrogenase is present in most hemolysates, further NADP is converted to NADPH (see Fig. 32-30). NADPH fluoresces, but NADP does not. The normal control sample fluoresces brightly, and lack of fluorescence indicates G6PD deficiency. By reoxidizing any small amounts of NADPH formed, oxidized glutathione (GSSG) enhances the ability of the test to detect mild G6PD deficiency. This is the recommended screening test for G6PD deficiency (Beutler et al, 1979).
Quantitative Assay of G6PD.
For G6PD, most assays are based on the rate of reduction of NADP to NADPH, measured spectrometrically at 340 nm, when a hemolysate is incubated with G6P (Beutler, 1984). In heterozygotes or in acute hemolysis in black subjects with G6PD deficiency, the diagnosis may be obscured even with the assay, because of increased levels of G6PD in reticulocytes and younger erythrocytes. Usually, however, the ascorbate cyanide screening test will be positive in these instances.
Pyruvate Kinase (PK) Deficiency
The most common red cell enzyme deficiency involving the Embden-Meyerhof glycolytic pathway, PK deficiency results in mild to moderately severe nonspherocytic chronic hemolytic anemia with splenomegaly. The prevalence of PK deficiency is estimated to be 1 : 20,000 in the general white population (Zanella et al, 2005). The anemia may be detected in infancy, or not until adult life in milder cases. Neonatal jaundice occurs in more than half of cases, which may require exchange transfusion. Patients tend to tolerate the anemia rather well because of high levels of 2,3-DPG, which occur as a result of the block in glycolysis. The blood film may show no notable red cell abnormalities until after splenectomy, when echinocytes, irregularly contracted cells, and crenated red cells may be prominent. Reticulocyte counts are elevated and increase further after splenectomy.
PK isoenzymes are produced by two separate genes: one on chromosome 15, encoding the M (muscle) isoforms, and the other on chromosome 1, encoding the L (liver) and R (red cell) isoforms. The complexity of PK isoforms is further complicated by the fact that functional enzyme is a tetramer. Inheritance is autosomal recessive, but this is probably true only in consanguineous families. PK mutants are numerous and are not detected in phenotypically normal heterozygotes that have one half the normal PK activity. Most individuals with PK-deficient hemolytic anemias are therefore probably double heterozygotes for two mutant genes (Valentine, 1979). Acquired PK deficiency occurs occasionally in myelodysplastic disorders and leukemias (Valentine, 1979; Miwa, 1981).
The autohemolysis test gives variable results. Some patients show only a mild increase in autohemolysis that is partially prevented by glucose (type I), and others have a greater increase that is not prevented by glucose (type II). Heinz bodies are not found. The diagnosis is made by a specific screening test or enzyme assay.
The reticulocyte count in unsplenectomized patients is usually increased, although not proportional to the degree of anemia, because younger PK-deficient erythrocytes are selectively sequestered in the spleen (Zanella et al, 2005). Splenectomy is indicated in cases requiring transfusions. After splenectomy, the Hb concentration usually increases by 1 to 2 g/dL and the reticulocytes increase sharply, although hemolysis persists (Miwa, 1981).
Fluorescent Spot Test.
Pyruvate kinase catalyzes the phosphorylation of adenosine diphosphate (ADP) to ATP by phosphoenolpyruvate (PEP) with the formation of pyruvate (Beutler, 1984). Pyruvate then reduces any NADH present to NAD with the formation of lactate (see Fig. 32-30). Loss of fluorescence of NADH under ultraviolet light is observed as evidence of the presence of PK.
Leukocytes must be removed from the sample because they normally contain about 300 times as much PK as red cells do, and in PK deficiency, the red cells but not the leukocytes are deficient.
Quantitative Assay of PK.
The same principle is employed as in the screening test, but the rate of decrease of absorbance at 340 nm is measured. A negative screening test or a normal PK assay (using the standard high substrate [PEP] concentrations) does not rule out PK-deficient hemolytic anemia. Because mutant PK enzymes may have normal activity at high PEP concentrations and decreased activity at low PEP concentrations, it is necessary to perform the assay both ways (Beutler, 1984).
Other Glycolytic Enzyme Deficiencies
Other enzyme deficiencies in the Embden-Meyerhof pathway are rarer (Beutler, 1994; van Solinge & van Wijk, 2010). When severe, they produce hemolytic anemia, with two exceptions: (1) LDH deficiency has no clinical manifestations; and (2) deficiencies of 2,3-DPG mutase and 2,3-DPG phosphatase activities occur together and result in erythrocytosis as a result of lack of 2,3-DPG, shifting the O2 dissociation curve of Hb to the left, and relative tissue hypoxia.
Other enzyme deficiencies in the hexose monophosphate shunt are rare. They include the two enzymes involved in glutathione synthesis: γ-glutamyl cysteine synthase and glutathione synthetase. As in G6PD deficiency, hemolysis increases with oxidant drug exposure or infection.
No good evidence is available, however, for the causation of chronic hemolytic anemia by 6-phosphogluconate deficiency, catalase, enolase, glutathione reductase (GR) deficiency, or glutathione peroxidase (GPx) deficiency (Beutler, 1994; van Solinge & van Wijk, 2010). GR contains flavine-adenine dinucleotide and is often partially deficient because of dietary riboflavin deficiency. GPx is one-half normal in about 30% of the Jewish population as a result of homozygosity for a gene with low GPx activity; in addition, GPx activity is dependent on selenium intake in the diet. In neither case is there an association with a hematologic disorder.
Pyrimidine-5′-Nucleotidase Deficiency
When RNA is degraded in the reticulocyte, pyrimidine nucleotides must be dephosphorylated by pyrimidine-5′-nucleotidase (PN) to cross the red cell membrane (Paglia & Valentine, 1981). Autosomal recessive PN deficiency results in accumulation of pyrimidines, and the impaired degradation of RNA results in pronounced basophilic stippling in red cells on the blood film. This is probably one of the more common enzyme deficiencies responsible for nonspherocytic hereditary hemolytic anemia, once G6PD and PK deficiencies are excluded (Rees et al, 2003).
The disorder is characterized by mild to moderate chronic hemolysis, jaundice, reticulocytosis (≈10%), marked basophilic stippling (about 5% of the red cells), and splenomegaly without notable improvement after splenectomy. However, no cases of transfusion dependency associated with PN deficiency have been reported (Rees et al, 2003). A screening test compares the ultraviolet absorption of deproteinized extracts of red cells at 260 nm with that at 280 nm. In PN deficiency, the major absorption peak is shifted from the normal 260 nm to 280 nm, which is the maximum for the pyrimidines uridine diphosphate and cytidine diphosphate. Confirmation requires an assay showing decreased nucleotidase activity (Beutler, 1984).
Acquired PN deficiency occurs in lead poisoning and is probably responsible for the basophilic stippling in that condition. It has been also reported in β-thalassemia trait, acute leukemias, chronic myeloid leukemia, chronic lymphocytic leukemia, and lymphomas (Rees et al, 2003).
Hemolysis—Acquired; Extrinsic
Chemical Agents
Agents Hemolytic to Normal Cells
The action of chemical agents depends on the dose and on other factors, many of which are known only vaguely. They range from simple substances, such as water, to some that are highly complex. When used as irrigating fluid, distilled water may be found responsible for acute hemolytic anemia as a result of entry into venous channels during transurethral resection. Also, drowning in fresh water may be associated with red cell hemolysis, particularly in the lungs, although drowning in salt water could cause profound red cell hydration (Schrier & Price, 2009).
In addition to anemia, some chemicals produce methemoglobinemia, which can manifest as cyanosis (toluene, trinitrotoluene, nitrobenzene, acetanilide, and phenacetin). Some may lead to AA (toluene and trinitrotoluene). Promin, a sulfone derivative, makes blood turn chocolate brown because of the formation of sulfhemoglobins. As mentioned earlier, ribavirin, a drug used in the treatment of hepatitis C virus infection, may produce a dose-dependent hemolytic anemia in about one third of patients.
Lead toxicity may produce progressive anemia, with basophilic stippling, reticulocytosis, normoblastemia, Cabot rings, Howell-Jolly bodies, and leukocytosis. Lead not only causes damage to the red cell and hemolysis, but also produces defects in the heme synthetic pathway. In cases of chronic exposure to lead, basophilic stippling, more in the marrow than in the peripheral blood, ring sideroblasts, and coproporphyrinuria are the characteristic findings. These changes produce defective erythrocytes, which are removed by the spleen.
Agents Hemolytic to Abnormal Cells
Certain drugs and chemicals that have oxidizing activity may produce hemolytic anemia in individuals with G6PD deficiency or other defects resulting in glutathione deficiency. In addition, unstable hemoglobins, such as Hb Zürich, have a propensity for drug-induced hemolytic anemia. Premature infants, although they have high levels of G6PD, have glutathione instability and low levels of glutathione and may develop hemolytic anemia when given large doses of synthetic water-soluble analogs of vitamin K.
It must be remembered that, if exposure to these oxidant substances is great enough, acute hemolytic anemia may be produced in normal individuals.
During the acute hemolytic episode, Heinz bodies can be demonstrated frequently by direct staining of blood with methyl violet. Red cells with Heinz bodies are removed from the circulation by the spleen, or the Heinz bodies are extracted from the red cells by splenic action. Therefore, Heinz bodies may not be found in the blood if the spleen is effectively removing them, or after the acute hemolytic process has abated.
Tests for G6PD deficiency, the most common underlying cause of drug-sensitive hemolytic anemia, are described earlier in this chapter.
Physical Agents
Heat
Extensive burns produce hemolytic anemia, probably because of direct damage to red cells. Gross hemoglobinuria may occur in patients with severe burns. The blood film may show remarkable morphologic abnormalities of the red cells, including budding fragmentation of the membrane and microspherocytosis. The most severe abnormalities are often found immediately after extensive burns before a reticulocyte response has had time to develop. Badly damaged cells are rapidly removed from the circulation.
Traumatic Hemolysis
Hemolytic anemia characterized by striking morphologic abnormalities of the red cells, which include fragments (schistocytes) and irregularly contracted cells (triangular cells, helmet cells), has been attributed to physical trauma to the red cells. The International Council for Standardization in Haematology (ICSH) recommends the classification of schistocytes into four categories (crescentic/triangular, keratocytes, helmet cells, and microspherocytes), and it also recommends the use of 1% threshold to designate increased schistocytes (Zini et al, 2012). The basis of the hemolytic process is probably damage to the red cells in their contact with loose fibrin meshworks (intravascular coagulation) or with pathologic vascular lesions. Fragmentation of the cells results with or without intravascular lysis. Two general categories are recognized in this group of disorders, aptly termed red cell fragmentation syndrome: macroangiopathic and microangiopathic.
Macroangiopathic Hemolytic Anemia (Cardiac Valvular Disease and Prostheses).
Chronic intravascular hemolysis associated with low serum haptoglobin, hemosiderinuria, reticulocytosis, and red cell abnormalities (e.g., schistocytes, irregularly contracted cells) may occur after surgical replacement of a diseased heart valve with a prosthesis or after surgical repair of a septal defect with a plastic patch (Erslev, 2001b). This has been attributed to mechanical damage of red cells in the turbulent environment of a leaky valve or of a roughened surface uncovered by endothelial cells. Repair of the valve or coverage of the patch by endothelium has improved the hemolytic process. Other studies have shown that some patients with cardiac valvular disease have a hemolytic process that may be altered by surgery.
External mechanical trauma has been reported in march hemoglobinuria, a condition that is seen in soldiers after a long march and in joggers and other athletes following long practices, particularly on hard surfaces. In this condition, anemia and reticulocytosis are uncommon. The chronic intravascular hemolysis may lead to iron deficiency as a result of loss of Hb in the urine.
Thrombotic Microangiopathy (Microangiopathic Hemolytic Anemia).
Thrombotic microangiopathies (TMAs) are microvascular occlusive disorders characterized by systemic or intrarenal aggregation of platelets, thrombocytopenia, and mechanical injury to erythrocytes. Fragmented red cells (e.g., schistocytes, irregularly contracted cells) are probably produced when blood flows through turbulent areas in the microcirculation that are partially occluded by platelet aggregates (Moake, 2002). TMA may be idiopathic or precipitated by drugs such as mitomycin C, transplantation, malignancy, pregnancy, collagen vascular disease, or HIV (Rosove, 2014).
The classification of TMA has been controversial for many years. In routine clinical practice, the term thrombotic thrombocytopenic purpura (TTP) is often used for adult cases with predominant neurologic symptoms, whereas the term hemolytic-uremic syndrome (HUS) has been used to describe childhood cases with thrombotic microangiopathy and predominant renal involvement (Moake, 2002). However, the distinction is not always possible, and overlapping symptoms may be observed. Authors suggested the inclusion of both entities under TTP/HUS, presuming a similar pathogenetic mechanism with variable organ tropism. However, recent studies indicate a difference in pathogenetic mechanisms between HUS and TTP, as discussed later.
Hemolytic-Uremic Syndrome
HUS is classified into diarrhea-associated (D+) or typical form, and diarrhea-negative (D−) or atypical form. D+ HUS constitutes approximately 95% of cases in children and is less common in adults (Zheng & Sadler, 2008). D+ HUS occurs most commonly in infants younger than 2 years of age, but it affects all ages. It is caused by one of several serotypes of Escherichia coli that produce a verotoxin (named for its toxicity against African green monkey kidney [vero] cells). The most common is E. coli O157:H7, which produces Shiga toxins 1 and 2. This microorganism is one of the cattle gut commensals. E. coli O104:H4 has been associated with epidemic infection in Europe (Rosove, 2014). The annual incidence of infection with E. coli O157 is approximately 8 per 100,000 people in North America. The percentage of cases with bloody diarrhea that progress to HUS is 3% to 7% for sporadic cases and 20% to 30% for some outbreaks (Zheng & Sadler, 2008). HUS usually occurs 4 to 6 days after the onset of diarrhea. Shiga toxins cause bloody diarrhea, then enter the circulation and travel in the plasma on the surface of platelets or monocytes (Moake, 2002). Recent studies suggest that these toxins become attached to glomerular capillary endothelium and other renal epithelial cells. Shiga toxins, together with locally secreted cytokines, cause the release of unusually large multimers of von Willebrand factor (vWF), which promote platelet aggregation in renal vasculature (Moake, 2002). Endothelial cell damage is the hallmark of D+ HUS in children, and endothelial swelling may be severe enough to occlude the capillary lumen. The thrombi of D+ HUS are composed predominantly of fibrin with few platelets and little vWF (Zheng & Sadler, 2008). There is growing evidence that shiga toxin also activates complement component in the renal circulation, which may contribute to cell damage and thrombogenesis (Rosove, 2014). Hemolytic anemia with schistocytes (due to interaction of red cells with microvascular thrombotic lesions), variable thrombocytopenia, and uremia are the cardinal features. Death formerly occurred in almost half of cases; the renal pathology has included acute glomerulonephritis and thrombotic and necrotic vascular lesions associated with patchy, bilateral renal cortical necrosis. However, with advanced supportive therapy, including transfusions and dialysis, some investigators have reported mortality reduced to 5% to 15%. Nonetheless, minor to major renal impairment has been reported in approximately 50% of patients. Five to ten percent of patients with HUS have D− HUS, with no antecedent diarrhea of Shiga-toxin–producing organisms, and some of these patients have familial history of the disease (Zheng & Sadler, 2008). First presentations are more common in children, including neonates, although some cases present in adulthood (Rosove, 2014). Infections and pregnancy may trigger hemolytic crises. Approximately 50% of patients with D− HUS have mutations in one of the complement regulatory proteins: factor H, factor I, membrane cofactor protein (MCP), or factor B. These patients have a much higher mortality rate (Moake, 2002). Approximately 5% of D− HUS patients have antibodies against factor H, raising the possibility of using plasma exchange or immunosuppressants for therapeutic strategy (Zheng & Sadler, 2008).
Thrombotic Thrombocytopenic Purpura (TTP)
TTP may occur at any age but has a peak incidence in the third decade; it occurs more often in females than in males. The annual incidence is 3.7 cases per 100,000 with the acquired form being more common, with an incidence of 0.2 to 1 per 100,000 compared to the much less common familial form, which has an incidence of 0.05 to 0.4 per 100,000 (Rosove, 2014). The triad of clinical manifestations present in most patients includes hemolytic anemia, thrombocytopenia, and neurologic symptoms; in addition, fever and renal diseases are often present. This constitutes the classic pentad. Pathologically, microvascular occlusive lesions with hyaline thrombi and endothelial proliferation are widespread throughout the body. Until 1997, the origin was unknown. Different theories included endothelial injury by oxidant stress, antiendothelial antibodies, anti-CD36 (glycoprotein IV located on microvasculature) antibodies, platelet aggregating factors, and release of large vWF multimers into the plasma. In 1996, a protease that cleaves vWF subunits was identified in normal plasma. In 1997 and the following years, several reports indicated deficient vWF-cleaving protease in patients with TTP due to low enzyme levels (in familial cases) or to the presence of IgG enzyme inhibitor (in nonfamilial cases). These observations were confirmed by additional studies, which showed normal levels of vWF-cleaving protease in HUS (Moake, 2004). The enzyme has been further characterized as number 13 in a family of 19 distinct ADAMTS-type enzymes identified to date (a disintegrin and metalloprotease with eight thrmobospondin-1-like domains). Therefore, the vWF-cleaving metalloprotease is now referred to as ADAMTS13 (Moake, 2004). In the absence of vWF-cleaving protease, ultra-large vWF multimers secreted by endothelial cells would not be processed normally. These highly adhesive multimers may induce platelet clumping in the microcirculation under shear stress, resulting in the clinical manifestation of TTP. The disease is acute and until the 1980s was fatal in well over half of cases. With the therapy of plasma exchange by plasmapheresis and plasma transfusions (in addition to platelet inhibitors), the remission rate has improved, and long-term survival now approaches 90% (Moake, 2004). TTP can now be classified into three main categories: familial, idiopathic, and secondary. The familial form (Upshaw-Schulman syndrome) results from a mutation in the ADAMTS13 gene and is associated with a very low activity of the enzyme. More than 50 mutations have been described to date. Approximately 50% of patients with familial TTP experience their first episode during childhood, and exacerbations may occur in association with minor infections (Zheng & Sadler, 2008). The idiopathic form is caused by antibodies to ADAMTS13. The secondary form may occur along the course of other conditions, such as malignancies, organ transplantation, chemotherapy, infection, and certain medications, such as cyclosporine, quinine, ticlopidine, and clopidogrel. The mechanism of TTP under these circumstances is unknown, although direct endothelial cytotoxicity and drug-induced autoimmunity are possible mechanisms (Zheng & Sadler, 2008).
TMA after organ tranplantation is a well-recognized entity occurring as a complication of renal, lung, and allogeneic hematopoietic stem cell transplant. In this setting, medications, such as sirolimus and cyclosporin, infections, and graft-versus-host disease are important factors (Rosove, 2014).
Preeclampsia/Eclampsia
Preeclampsia and eclampsia are microangiopathic disorders occurring with pregnancy and sharing some features of HUS or TTP. Approximately 4% to 39% of patients with severe preeclampsia develop HELLP syndrome: hemolysis, elevated liver enzymes and low platelet count. The syndrome becomes manifest usually during the third trimester, although some patients may develop it during the postpartum period. Most patients recover within a few days after delivery. In a small subset of patients, severe persistent multisystem disease requires plasma exchange (McMinn & George, 2001). ADAMSTS13 has been reported to be low in 10% to 20% of cases (Rosove, 2014)
Infectious Agents
Destruction of erythrocytes by plasmodia is responsible for the anemia in malaria. This is supported by the observation that the osmotic and mechanical fragility of parasitized erythrocytes is increased. Inhibition of marrow activity may be an additional factor. Fulminant hemoglobinuria (blackwater fever) is a complication of P. falciparum malaria.
Oroya fever, a frequently fatal disease that occurs in Peru, is characterized by a hemolytic anemia and leukocytosis. Bartonella bacilliformis is the responsible agent.
Babesiosis, a protozoan infection transmitted by ticks from rodents or cattle, is associated with hemolysis; parasites may be seen in red cells in Romanowsky's-stained blood films.
Hemolytic anemia with cold agglutinins may complicate mycoplasmal pneumonia and infectious mononucleosis. This is due to the effect of antibody on the red cells.
Hemolytic anemia of varying severity is frequent in some bacterial infections. A notable example is Clostridium perfringens septicemia following septic abortion or biliary tract surgery, which may be accompanied by a dramatic and life-threatening hemolytic crisis.
Immune Hemolytic Anemias
Immune hemolytic anemias are disorders in which erythrocyte survival is reduced because of the deposition of immune globulin and/or complement on the red cell membrane. The immune hemolytic anemias can be grouped according to the presence of autoantibodies, alloantibodies, or drug-related antibodies (Table 32-9)
TABLE 32-9
Autoimmune Hemolytic Anemia (AIHA)*
| Condition | Warm AIHA | COLD AIHA | Mixed AIHA | |
| CHAD | PCH | |||
| Idiopathic | 282 (23) | 194 (16) | 5 (<1) | 47 (4) |
| Drug-induced disorders | 184 (15) | 0 | 0 | 2 (<1) |
| Neoplasia | 165 (14) | 81 (7) | 0 (0) | 26 (2) |
| Non-Hodgkin's lymphoma | 27 | 25 | 0 | 8 |
| Chronic lymphocytic leukemia | 65 | 4 | 0 | 8 |
| Hodgkin's lymphoma | 11 | 7 | 0 | 5 |
| Carcinomas | 37 | 30 | 0 | 4 |
| Miscellaneous | 25 | 15 | 0 | 1 |
| Infections | 9 (<1) | 76 (6) | 14 (1) | 2 (<1) |
| Pneumonia—Mycoplasma | 0 | 21 | 0 | 0 |
| Viral pneumonia | 2 | 19 | 0 | 0 |
| Infectious mononucleosis | 0 | 11 | 1 | 0 |
| Miscellaneous | 7 | 25 | 13 | 2 |
| Collagen diseases | 30 (2) | 15 (1) | 0 (0) | 20 (2) |
| Systemic lupus erythematosus | 7 | 4 | 0 | 16 |
| Rheumatoid arthritis | 21 | 6 | 0 | 4 |
| Others | 2 | 5 | 0 | 0 |
| Miscellaneous disorders | 45 (4) | 20 (2) | 0 | 6 (<1) |
| Totals | 715 (58) | 386 (32) | 19 (2) | 103 (8) |
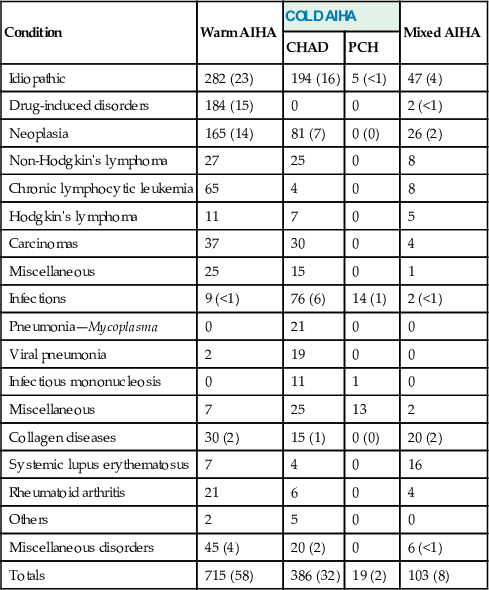
Autoimmune Hemolytic Anemia
The autoimmune hemolytic anemias (AIHAs) are due to an altered immune response, resulting in the production of antibody against the host's own erythrocytes, with subsequent hemolysis. The incidence of AIHA is estimated at 10 to 30 cases per 1 million population (Gehrs & Friedberg, 2002). The AIHAs can be classified according to serologic or clinical characteristics (Table 32-10). Some AIHAs are mediated by antibodies with maximum binding affinity at 37° C, and other AIHAs are mediated by antibodies with maximum binding affinity at 4° C. In addition, AIHAs could be viewed according to their association with other disorders. In a study of 1834 patients, approximately 40% of cases of AIHA have been associated with an underlying disease, while the remainder were idiopathic (Sokol & Booker, 1992). Lymphoproliferative disorders account for approximately half of cases of both warm and cold AIHA (Gehrs & Friedberg, 2002).
TABLE 32-10
Examples of Drugs Implicated in Causing Immune Hemolytic Anemia and Their Mechanism of Action
| Hapten or Drug Adsorption Mechanism | |
| Penicillins | Cephalosporins |
| Tetracyclines | Tolbutamide |
| Ternary Complex Mechanism | |
| Stibophen | Quinidine |
| Quinine | Cephalosporins |
| Rifampicin | Diethylstilbestrol |
| Chlorpropamide | Amphotericin B |
| Possible Autoantibody Mechanism | |
| α-Methyldopa | L-Dopa |
| Procainamide | Cephalosporins |
| Mefenamic acid | Fludarabine |

Etiology and Pathophysiology.
The cause of the production of autoantibody in patients with AIHA is unknown. However, several mechanisms have been suggested. Autoimmune antibodies, particularly cold-reacting antibodies, are sometimes produced following an infection. This is typically seen with the elaboration of anti-I in patients with Mycoplasma pneumoniae infections and anti-i in patients with infectious mononucleosis. The cold agglutinins of anti-I and anti-i specificity are strikingly similar to one another in the structure of antigen-binding sites. These antibodies react with antibodies that also identify the product of the VH4-34 gene segment in B cells. It has been hypothesized that infections can cause the production of antibodies of B cell, utilizing this gene segment. These antibodies will also have cold agglutinins activity against the I/i antigens (Rosse, 2004).
The amount of antibody, its avidity for the erythrocyte autoantigen, and its ability to fix complement are significant variables. Opsonization of red cells can destroy them in the circulation (intravascular hemolysis) or can cause their accelerated removal from the circulation by tissue macrophages (extravascular hemolysis). Major sites for these macrophages are the spleen and, to a lesser extent, the liver (Gehrs & Friedberg, 2002).
The development of AIHA in patients with lymphoproliferative disorders or with autoimmune disorders may relate to some abnormality with B cells, T cells, or macrophages, or the interaction among these cells. Perhaps loss of T cell suppressor function could result in unrestrained production of red cell antibody by B cells. This hypothesis is strengthened by the observation that methyldopa, a drug known to cause the development of anti–red cell antibodies, inhibits the activation of suppressor T lymphocytes (Gehrs & Friedberg, 2002).
In AIHA associated with warm-type antibody, there is IgG coating of erythrocytes with or without complement fixation. Clearance of red cells occurs mostly in the spleen. In the absence of complement fixation, it appears that the Fc portion of the red cell–bound IgG interacts with the Fc receptor present on the membrane of splenic macrophages located along the cords of Billroth. Thus, sensitized erythrocytes are retained, phagocytosed, or fragmented by splenic macrophages during their passage through the spleen.
In AIHA associated with the production of cold-type autoantibody, erythrocytes are usually coated with IgM. Under these circumstances, the fixation of complement frequently occurs. In paroxysmal cold hemoglobinuria, the offending antibody is an IgG that fixes complement. If the entire complement sequence is activated, there may be intravascular hemolysis. This phenomenon may occur in cases of cold hemagglutinin disease, as well as in paroxysmal cold hemoglobinuria. If complement activation fails to proceed to completion but is halted at an intermediate stage, intravascular lysis of the erythrocytes may not occur. However, extravascular hemolysis can still continue. In this situation, sensitized cells with C3b on the membrane are bound in the liver by the interaction of C3b and its receptors on Kupffer's cells. Erythrocytes may be phagocytosed entirely, or portions of the cells may be removed, resulting in fragmentation and spherocyte formation.
Approximately 7% of patients with AIHA satisfy diagnostic criteria for both warm and cold autoantibodies (Sokol & Booker, 1992). In these cases, IgG and C3d sensitize the erythrocytes. The serum contains IgM cold autohemagglutinins (optimally reactive at 4° C, but with a high thermal amplitude to 37° C) and IgG warm autoantibodies.
AIHA Associated with Warm Antibody.
The warm antibody type of AIHA is slightly more frequent in females than in males and is most likely to occur in individuals 40 years of age or older; peak incidence occurs around the seventh decade. This age distribution may be related to the higher frequency of lymphoproliferative disorders with age (Packman, 2010). Idiopathic warm-antibody AIHA accounts for approximately 50% of cases. Chronic lymphocytic leukemia and lymphomas account for about half of secondary warm-antibody AIHA, and autoimmune disorders, such as SLE, account for most of the remaining cases (Packman, 2010). Clinical signs and symptoms frequently are those of an underlying disorder. However, the patient with idiopathic AIHA may have noted the presence of a mild upper respiratory tract infection just before the onset of hemolysis. As the disorder progresses, weakness, dizziness, and fever may be noted. Jaundice can be a presenting complaint. Pregnancy carries a fivefold risk of developing autoantibodies compared with the control population (Gehrs & Friedberg, 2002).
Laboratory findings include the presence of moderate to severe anemia. The neutrophil count may be increased. In a small proportion of cases, thrombocytopenia can exist and if severe should raise the possibility of concomitant immune thrombocytopenia (Evans syndrome). The peripheral film frequently shows spherocytosis, red cell fragmentation, polychromasia, and a few normoblasts. Reticulocyte percentage is high in approximately 50% of patients and is often associated with increased MCV. However, as many as one third of patients have transient reticulocytopenia, despite the presence of normal or hyperplastic erythroid precursors in the marrow (Packman, 2010). The lack of reticulocytosis should not keep one from making a diagnosis of AIHA. The bone marrow exhibits erythroid hyperplasia, sometimes with mild megaloblastic changes. Reticulocytopenia may be related to immune destruction of reticulocytes or bone marrow suppression caused by marrow infiltration, infection, nutritional deficiencies, or chemicals (Packman, 2010).
There is usually a decrease in serum haptoglobin and an increase in unconjugated bilirubin and LDH. The osmotic fragility and autohemolysis test can be normal or increased.
Direct and indirect antiglobulin tests indicate the presence of erythrocyte antibodies. The specificity of the autoantibodies is usually directed against antigens of the Rh system, membrane protein band 3 and band 4.1, and glycophorin A. However, activity against U, LW, Kell, jka, and Fya antigens may also occur. The warm antibody is most likely an IgG with subclass IgG1 and less frequently with IgG3. When IgG2 or IgG4 is present on the red cells alone, no hemolytic reaction is associated (Packman, 2010). Occasionally, the antibody may be an IgA immune globulin, or rarely an IgM immune globulin. Complement may be detected on the erythrocyte membrane in slightly more than half of cases. Although many warm antibodies fix complement, intravascular complement–mediated hemolysis and hemoglobinuria are unusual, possibly because of the ability of complement regulatory proteins in plasma and on red cell surfaces to abort fixation of terminal complement components (Packman, 2010).
In some cases, sensitized red cells contain less immune globulin than can be detected using commercially prepared antiglobulins, which are normally sensitive to 250 to 500 molecules of IgG per red cell. Under these circumstances, the autoantibody at times can be detected with an antiglobulin consumption test or more automated enhanced agglutination techniques (Packman, 2010).
The clinical course of AIHA associated with warm antibody is characterized by periods of remission and relapse. In secondary AIHA, the course and prognosis are related to the nature of the underlying disorder. In idiopathic AIHA, complications of the hemolytic disorder may be severe, leading to the demise of the patient. Pulmonary emoboli, infection, and cardiovascular collapse are causes of death, and deep vein thrombosis and splenic infarcts are common complications during active hemolysis (Packman, 2010).
AIHA Associated with Cold Antibody.
AIHA associated with cold antibody can be mediated by an IgM and less frequently by an IgG. The IgM autoantibody is associated with a syndrome known as cold agglutinin disease, whereas the IgG autoantibody is seen with paroxysmal cold hemoglobinuria.
Cold Agglutinin Disease
Cold agglutinin disease occurs in individuals usually over 50 years of age and in females more often than in males. In some cases, cold agglutinin disease is associated with a lymphoproliferative disorder or infection (especially with M. pneumoniae or infectious mononucleosis). Cases unassociated with an underlying disorder are listed as idiopathic. Virtually all sera from healthy individuals contain low-titer cold agglutinins, regarded as benign or harmless, and are polyclonal. Postinfectious cold agglutinins are usually polyclonal. By contrast, monoclonal cold agglutinins are generally pathogenetic and may arise from B cell lymphoma (Gehrs & Friedberg, 2002). Cold agglutinin disease represents approximately 20% of AIHA.
Symptoms and signs vary widely. Some individuals may complain of acrocyanosis or Raynaud's phenomenon. Others will have episodes of hemolysis, including hemoglobinuria, following exposure to cold. Temperatures of 30° C and lower are normally attained in the superficial skin vessels of those body parts exposed to cold. The thermal range of the antibody is clinically more important than the agglutination titer (Petz, 2008).
Laboratory findings usually indicate an anemia. Spherocytes and polychromatophilic erythrocytes are present to a variable degree in the blood film. There may be marked red cell agglutination, which should be differentiated from rouleaux formation (see Figs. 30-25 and 30-26). A mild leukocytosis can exist. Red cell agglutination may interfere with automated hematology counts, particularly MCHC, which tends to be high.
The cold antibody is usually an IgM with anti-I or, less frequently anti-i, specificity. Rarely do other specificities exist. In the chronic idiopathic form of cold agglutinin disease, the antibody tends to be monoclonal IgM, k with anti-I specificity (Gehrs & Friedberg, 2002). The autoantibody is also capable of fixing complement. When the titer of cold antibody is very high, the thermal range of antibody activity may extend up to 37° C. The direct antiglobulin test is positive only if the reagents contain anticomplement activity. Thus, one usually observes a positive antiglobulin reaction with broad-spectrum and non–γ-reagents, but no agglutination with only the γ-reagent. On cold exposure and antibody binding to red cells, the complement may be activated to the stage of C3b, which adheres to the red cells after entering the systemic circulation. In the hepatic circulation, macrophages carry specific receptors for C3b. However, cells usually escape hepatic destruction. Most patients experience chronic mild to moderate anemia, which they tolerate well, and patient outcome is significantly better than with warm-antibody AIHA (Petz, 2008). Some authors remain with the opinion that chronic cold agglutinin disease is a variant of Waldenström's macroglobulinemia in which the IgM M-component has cold agglutinin activity.
Paroxysmal Cold Hemoglobinuria
Paroxysmal cold hemoglobinuria is a very rare disorder that can occur in an individual of any age. However, more cases are now being reported in young children (Petz, 2008). Females are as frequently involved as males. Patients have symptoms of acute hemolysis following exposure to the cold. Chills, fever, pain in the back and legs, and hemoglobinuria are reported. The acute form may follow an acute upper respiratory illness or immunization by 1 to 2 weeks, but the chronic form is associated with congenital syphilis. Hemoglobinuria following exposure to cold is rare in the acute form.
Laboratory features consist of anemia, elevated reticulocyte count, increased concentration of indirect bilirubin, and the presence of Hb in the urine. Anemia is frequently severe and rapidly progressive.
The serum contains a cold hemolysin with biphasic activity. This antibody, first described by Donath and Landsteiner (1904), is an IgG that fixes the first components of complement (C1–C4) in the cold (4° C). As the temperature rises to 25° to 37° C, the antibody dissociates, but the remainder of the complement proteins are activated, and erythrocyte lysis results; this biphasic hemolysis is the basis for the Donath-Landsteiner autohemolysis test for the diagnosis of this condition. The direct antiglobulin test is positive with anti-C3 but is generally negative with anti-IgG unless performed at colder temperature. The specificity of the antibody is directed against the P antigen. In general, the prognosis is good.
AIHA Associated with Warm and Cold Antibodies
AIHA associated with both warm and cold autoantibodies is mediated by IgG warm antibodies and complement, as well as IgM cold hemagglutinins. Females are more likely involved than males. There appears to be an association between the combined warm and cold antibody AIHA and SLE in that 15% to 42% of patients have SLE (Shulman et al, 1985; Sokol & Hewitt, 1985). In one study (Shulman et al, 1985), 6 of 12 patients (50%) had idiopathic AIHA, and 4 of these (33%) had concomitant thrombocytopenia (Evans syndrome); all 12 patients had severe hemolysis that responded dramatically to corticosteroid therapy.
Alloimmune Hemolytic Disease of the Newborn
Alloimmune hemolytic disease of the newborn (HDN) usually occurs following the transplacental passage of maternal antifetal red cell antibodies. HDN most frequently results from incompatibility in Rh and ABO erythrocyte antigens between mother and fetus. In rare cases, some other red cell antigen may be responsible for this disorder.
In HDN due to Rh incompatibility, prior sensitization is necessary to initiate the disease process. This sensitization usually occurs during pregnancy when Rh(D)-positive fetal red cells cross the placenta and enter the circulation of a mother with Rh-negative cells. Most women have less than 1 mL of fetal blood in their circulation following delivery. However, intrapartum fetomaternal hemorrhage of more than 30 mL occurs in approximately 1% of pregnancies (Ramasethu & Luban, 2010). Maternal sensitization can also occur by a previous incompatible transfusion. Under either of these circumstances, maternal IgG antibodies are produced against the fetal cells. If a subsequent pregnancy occurs in a sensitized mother, fetal erythrocytes again reach the maternal circulation and restimulate an antibody response, resulting in transfer of anti-Rh(D) antibody across the placenta and reduced fetal red cell survival.
In the ABO system, anti-A or anti-B antibodies of the IgG class may arise spontaneously in the mother; their presence does not require prior transfusion or pregnancy. As a result, first-born children may suffer from HDN when ABO incompatibility exists. Although ABO incompatibility occurs in 15% of group O pregnancies, ABO hemolytic disease is estimated to occur in only 3% of all births (Ramasethu & Luban, 2010).
The clinical features of HDN due to Rh incompatibility vary greatly. Some newborns experience only mild jaundice. Others initially appear markedly pale and then develop jaundice. They can have prominent hepatosplenomegaly. The disease may be complicated by a bleeding diathesis, marked acid-base abnormalities, and kernicterus. In very severe cases, patients can present with hydrops fetalis.
Early examination of the blood usually reveals an increase in nucleated erythrocytes, which may include forms as immature as pronormoblasts. Although this finding gave the disease its name, erythroblastosis fetalis, erythroblastosis is not always present, especially if the examination is not performed immediately after birth.
Up to 2.0 × 109 nucleated red cells per liter in term infants and up to 5.0 × 109/L in premature infants are commonly seen in this disorder. Normally, nucleated red cells average 0.5 × 109/L in term infants and 1.0 to 1.5 × 109/L in premature infants. Blood from the umbilical vein for early examination is more reliable than peripheral (capillary) blood because the erythrocyte count and the Hb may be significantly altered between birth and ligation of the cord.
Generally, there is macrocytic anemia of varying severity and an increase in reticulocytes. Occasionally, anemia may develop suddenly on the second or third day. The leukocyte count is frequently elevated, with immature leukocytes. Normoblastic hyperplasia of the marrow is pronounced.
In severely affected infants, thrombocytopenia, depression of prothrombin complex procoagulants, or diffuse intravascular coagulation may occur.
A direct antiglobulin test on fetal erythrocytes indicates the presence of an IgG antibody. When the maternal serum and an eluate from the fetal erythrocytes are incubated separately with a panel of O cells, one can usually demonstrate the presence of antibody with Rh(D) specificity. In Rh-negative pregnant women known to be sensitized to Rh(D), the titer of anti-Rh(D) antibody is measured periodically during pregnancy to serve as a guide for performing amniocentesis.
HDN associated with ABO incompatibility is less severe than that observed with Rh incompatibility. Occasionally, the diagnosis is suggested by the presence of unexplained hyperbilirubinemia in a group A or B newborn infant from a group O mother.
Laboratory findings usually show a mild anemia and modest reticulocytosis. In contrast to Rh isoimmune disease, spherocytosis in ABO isoimmune disease may be prominent. However, there may be no anemia. Fetal cells are usually weakly positive with the antiglobulin reagents. Serum from the newborn and eluates from the cells should contain anti-A or anti-B antibody. In addition, maternal serum should contain high titers of anti-A or anti-B antibodies of the IgG subclass.
Drug-Induced Immune Hemolytic Anemia
Immune hemolytic anemia may occur following the administration of drugs. Most cases in the 1970s were associated with methyldopa or high-dose intravenous penicillin therapy. Recently, this picture has changed, with second- and third-generation cephalosporins being the most common causative agent and accounting for more than 80% of drug-induced immune hemolytic anemia (Garratty & Arndt, 2007). Four mechanisms appear to mediate immune hemolysis (Packman, 2010).
Formation of Ternary Complexes (Neoantigen Formation).
Numerous drugs are known to weakly bind to red cell membrane, even when given in small doses (Table 32-11). Antibodies recognize the combined drug and red cell membrane components as antigen and bind to them, forming a ternary complex. The drug-induced antibody is usually IgM and tends to fix complement, resulting in lysis of cells. In addition, red cells may be sequestered in the liver and spleen because of their coating with C3b.
TABLE 32-11
Laboratory Features in Microcytic Hypochromic Anemias
| Serum Iron | Serum TIBC | % Saturation | MARROW | Serum Ferritin | ZPP | Hb A2 | Hb F | ||
| % Sideroblasts | Iron Stores | ||||||||
| Iron deficiency | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ↑ | N-↓ | N |
| β-Thalassemia trait | N (↑) | N | N | N | N-↑ | N-↑ | N | ↑ | N-↑ |
| ACD | ↓ | N-↓ | ↓ | ↓ | N-↑ | N-↑ | ↑ | N | N |
| Sideroblastic anemia | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ (↓) | N | N-↑ |
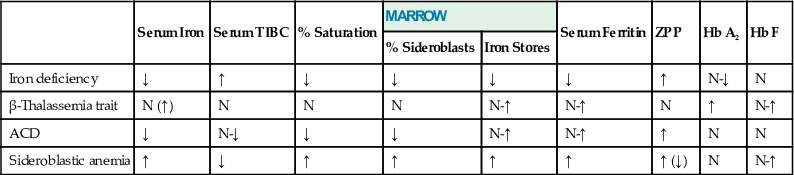
Patients present with acute intravascular hemolysis, hemoglobinemia, and hemoglobinuria. The direct antiglobulin reaction is positive if the reagents contain anticomplement activity. The reaction usually is negative with the γ-reagent because it contains little anti-IgM or complement specificity.
The diagnosis can be determined by incubating the patient's serum with the offending drug in the presence of target erythrocytes, while observing agglutination, lysis, or sensitization of the erythrocytes.
Adsorption of Drug to Red Cell Membrane.
Penicillin and cephalosporin, particularly in high doses, bind firmly to proteins, including red cell membrane. These drugs induce an antibody response to them, but the antibody specificity is not directed against a red cell membrane. Both IgM and IgG antibodies are made, but only the IgG antibodies are associated with immune hemolysis. Complement is not involved. The erythrocytes, coated with IgG antibody, are presumably removed via the Fc receptors on macrophages in the spleen.
The direct antiglobulin test is strongly positive. Antibody eluted from patients' erythrocytes will react only with red cells previously treated with penicillin or cephalosporins.
Induction of Autoantibody by Drugs.
In approximately 15% of patients using the antihypertensive drug methyldopa, a positive direct antiglobulin reaction is present. The antibody is of the IgG class and in some studies appears to have Rh specificity (Packman, 2010). The actual mechanism of autoantibody production is not known. However, it has been hypothesized that methyldopa causes activation of T cells and increased secretion of γ-IFN, which upregulates MHC class I and class II molecules on antigen-presenting cells and induces antibody production by B cells. The drug may also bind to hemoglobin-free proerythroblasts producing a neoantigen followed by the production of autoantibody (Packman, 2010).
Development of the positive antiglobulin reaction occurs after a lag period of 3 to 6 months and is dose dependent. Thirty-six percent of patients have a positive antiglobulin reaction when consuming 2 g or more of methyldopa per day, and 11% have a positive reaction when taking only 1 g daily. An immune hemolytic anemia occurs in less than 1% of patients and is not dose dependent. Methyldopa also decreases mononuclear phagocytic activity, which may explain the rarity of hemolytic anemia despite the common presence of autoantibodies (Sokol & Booker, 1992). Levodopa, procainamide, fludarabine, and mefenamic acid are additional drugs that have been reported to cause an AHA in a fashion similar to methyldopa.
Nonimmunologic Adsorption of Immunoglobulins to Red Cell Membrane.
Drugs such as cephalosporins and cisplatin appear to alter the erythrocyte membrane, resulting in the nonspecific adsorption of plasma protein to its surface. As a result, IgG and IgM may be loosely bound to red cell membrane. This phenomenon can then cause a positive direct antiglobulin reaction. It is now believed that this mechanism can cause hemolytic anemia (Garratty & Arndt, 2007).
Laboratory Investigations of Anemia
The diagnosis and study of anemia require the proper use and interpretation of laboratory measurements. Prerequisites for the efficient use of the laboratory are a careful history and physical examination, both of which lead to initial laboratory measurements and provide important guidance in determining the nature of the anemia. Anemias that are of acute onset and rapidly progressive course are likely hemolytic in origin, while slowly progressive anemias are likely caused by bone marrow production defect.
Whether the patient is anemic can be ascertained by determining whether the Hb, Hct, or erythrocyte count lies below (1) the reference intervals for age, sex, and altitude or (2) the patient's previous values, even though these are within the reference intervals. The task then is to define the underlying cause or mechanism for the anemia.
Usually, complete blood count (WBC, RBC, Hb, Hct, MCV, MCH, MCHC, platelets) and examination of a Wright's-stained film are parts of the routine examination of blood. It is possible that all these values could be normal in the presence of a mild macrocytic anemia, in which the RBC count does not fall below the normal range and the macrocytes present (and detectable on the blood film) do not elevate the MCV above the normal range.
Once anemia is discovered, the basic examination of the blood should include the following: (1) Hb, Hct, RBC count, and erythrocyte indices; (2) blood film examination; (3) leukocyte count; (4) platelet count; and (5) reticulocyte count.
With current multichannel instruments, all red cell values and their indices have comparable precision. Indices are mean values, however, and will not detect different populations of cells that balance each other. For example, combined deficiencies of folate and iron may give rise to populations of macrocytic and hypochromic microcytic cells, which could yield normal indices. Careful examination of the blood film is essential, and red cell volume RDW is very useful in defining this type of abnormality (see Chapter 30).
The size of the erythrocytes, determined by the MCV and by examination of the blood film, will determine the morphologic type of anemia. In addition, certain findings on the blood film will suggest mechanisms that are involved.
Increased numbers of polychromatic macrocytes, with or without normoblasts, suggest increased erythropoiesis, and this is usually due to hemorrhage or hemolysis in the untreated patient. In this situation, the history (of blood loss) or physical examination (jaundice or splenomegaly) will help.
Findings suggestive of hemolysis are poikilocytes (abnormally shaped red cells), sickle cells, irregularly contracted forms (including red cell fragments or schistocytes), and spherocytes. Sometimes it is difficult to detect spherocytes in HS because of minimal anisocytosis. Morphologic findings in red cells that are helpful in this situation include the presence of a low MCD (mean cell diameter), between 6.0 and 6.5 µm (normal = 7.0 to 7.4 µm), red cells with small eccentric or no pallor, and red cells with slightly more intense staining due to Hb density.
Target cells may be found in hemoglobinopathies, especially in the presence of Hb C, Hb D, and Hb E, and in the thalassemias. They may be present in any hypochromic anemia, although usually in smaller numbers. Target cells without microcytosis are also found in liver disease and in the absence of the spleen or of splenic function.
Fine basophilic stippling (which is due to precipitation of RNA) may be found in polychromatic red cells associated with a significant increase in the generation of erythrocytes, as in response to hemorrhage or hemolysis. Coarse basophilic stippling suggests an abnormality in Hb synthesis. It is found in megaloblastic anemias, thalassemias, refractory anemias, some red cell enzyme deficiencies, unstable Hbs, and lead poisoning. In particular, hypochromasia or microcytosis with stippling is against the diagnosis of iron deficiency anemia and is more suggestive of thalassemia or lead poisoning. As mentioned earlier, some cases of iron deficiency may be associated with lead poisoning (see the Iron Metabolism section earlier in the chapter).
The combination of oval macrocytes (especially egg-shaped macrocytes) and hypersegmented neutrophils indicates the very likely existence of megaloblastic anemia.
Finally, examination of the blood film allows the evaluation of qualitative abnormalities in leukocytes and platelets, as well as an estimate of their numbers. Blood diseases that may be first suspected or detected in this manner are many and include compensated hemolytic anemia; early megaloblastic anemia; and anomalies of red cells, such as hereditary elliptocytosis, or of leukocytes, such as the Pelger-Hüet nuclear anomaly.
After the basic studies just mentioned, the choice of additional procedures depends on the morphologic type of the anemia, as determined by the indices and the blood film.
Macrocytic Anemia (Increased MCV)
The macrocytic anemias are normochromic, as determined by appearance on the film and by the MCHC. The first step is to ascertain whether the anemia is megaloblastic. Clues from the film have been mentioned. A bone marrow aspiration should be performed to confirm the presence of megaloblastosis.
Megaloblastic Marrow
If the marrow is megaloblastic, with characteristic changes in both red cell and white cell precursors, the anemia in all likelihood is due to folate or cobalamin deficiency.
See earlier sections on diagnosis of cobalamin deficiency and of folate deficiency. Once the type of deficiency is defined, the cause must be determined.
Nonmegaloblastic Marrow
If the marrow is not megaloblastic, conditions that can be associated with macrocytosis should be investigated. These include liver disease; hemolytic anemias; hypothyroidism; excessive alcohol intake; aplastic anemias; and myelodysplastic syndromes. Anemias associated with these disorders, although they may be macrocytic, are more usually normocytic, and thus are considered with the normocytic anemias as well.
Microcytic and Hypochromic Anemias (Decreased MCV and MCH)
If the counts are performed on a multichannel instrument, the MCHC is likely to be in the normal range, with slight to moderate degrees of hypochromia. Consequently, the MCV has assumed the leading role in the detection of microcytic hypochromic anemias. These anemias reflect a quantitative defect in Hb synthesis. Major causes of microcytic anemia include the following:
1. Iron deficiency anemias are due to increased requirement or blood loss not balanced by intake.
2. ACD is associated with infection, neoplasia, or collagen disease. This anemia may be normochromic and normocytic, but in long-standing disease is often hypochromic and microcytic.
3. Thalassemia is a genetically determined impairment in the rate of globin synthesis.
4. Sideroblastic anemia is that group of refractory anemias with erythroid hyperplasia of the marrow in which a defect in Hb synthesis creates a population of hypochromic microcytic cells. The blood film is dimorphic and macrocytes may prevail, making the MCV normal or high, particularly in acquired forms of sideroblastic anemia.
Because iron deficiency is the most common anemia, the first step is to determine whether the body lacks iron.
When blood loss cannot be documented, serum ferritin, serum iron and iron-binding capacity, or bone marrow study for iron should be performed. These will usually discriminate between the two most common anemias in this category: iron deficiency and simple chronic anemia associated with some other disease, frequently chronic infection or cancer. In both, the serum iron concentration is low, but in iron deficiency, the TIBC is elevated, whereas in simple ACD it is normal or decreased. Storage iron in the marrow is depleted in iron deficiency but is normal or elevated in anemia of chronic disease. Iron deficiency anemia in an adult male almost always means chronic blood loss; the source must be found and corrected, if necessary. If no obvious source for iron loss is identified, PNH should be considered, particularly when there is associated neutropenia or thrombocytopenia.
Hypochromic anemias (or hypochromic erythrocytoses) with basophilic stippling and normal or increased serum iron are most likely thalassemias, and the next examinations to perform are Hb electrophoresis and determination of Hb A2 and Hb F. Family studies are often necessary.
Sideroblastic anemias include refractory anemia with ring sideroblasts, which are part of myelodysplastic syndromes (see Chapter 33), as well as anemias that occur after therapy with certain drugs (e.g., isoniazid) or in chronic lead poisoning. Coarse basophilic stippling is common in this group of anemias.
Box 32-2 summarizes some laboratory distinctions within the microcytic anemias.
Normocytic and Normochromic Anemias (Normal MCV)
This large group of anemias has many causes. A useful approach is evaluation of the erythrokinetics in a given patient (see Chapter 31). Often, the reticulocyte production index (RPI) and examination of the bone marrow will suffice. The RPI is the simplest measure of effective erythropoiesis.
Optimal Marrow Response: Reticulocyte Production Index Greater Than Two
If the output of reticulocytes has exceeded two times normal, as determined by the absolute reticulocyte count, or RPI, it can be assumed that the marrow has reached an optimal response. The cause for the anemia is then acute blood loss or hemolysis. If blood loss cannot be proved, evidence that hemolysis is in fact present must be sought.
Erythroid hyperplasia of the marrow, serum bilirubin, and urine or fecal urobilinogen will indicate whether erythropoietic activity and destruction are increased. Red cell survival determination may be needed to prove hemolysis in some cases. Low serum haptoglobin and high LDH point to hemolysis, but a normal level does not exclude it. None of these measurements will specify whether hemolysis is intravascular or extravascular, but elevated plasma Hb, hemoglobinuria, and hemosiderinuria indicate intravascular hemolysis.
Once it is determined that excessive hemolysis is occurring, the type of hemolytic mechanism must be ascertained.
Direct Antiglobulin (Coombs') Test.
If the direct antiglobulin reaction is positive using broad-spectrum reagents, tests to determine the presence of IgG, IgM, or complement on the red cells should be undertaken. If immunoglobulin is present on the red cells, tests for antibody specificity, cold agglutinins, Donath-Landsteiner antibody, and serum protein electrophoresis may help to define the process.
If the direct antiglobulin reaction is negative, the examinations performed next will depend on the clinical findings and the results of measurements already made.
If HS is suspected, osmotic fragility before and after 24-hour incubation at 37° C and family studies will be necessary.
If a nonspherocytic congenital hemolytic anemia is suspected, screening for G6PD and PK deficiencies, and Hb electrophoresis will be helpful. If these are negative, the heat instability test, isopropanol solubility test, and autohemolysis test should be considered.
If thalassemia seems likely, determinations of Hb A2 and Hb F and perhaps looking for Hb H inclusions are appropriate. Thalassemias are usually microcytic and hypochromic anemias; β-thalassemia major, β-thalassemia intermedia, and Hb H disease are hemolytic disorders and may have an increased RPI.
If drug-induced hemolysis is suspected, a test for Heinz bodies, a screening test for G6PD, and, if possible, tests for a drug-dependent autoantibody are indicated.
If the nature of the hemolytic anemia is obscure, a flow cytometry test for PNH should be performed.
Inadequate Marrow Response: Reticulocyte Production Index Less Than Two
The mechanism of the anemia may be ineffective erythropoiesis. Conditions with the greatest degree of ineffective erythropoiesis appear in other categories (e.g., megaloblastic anemia, thalassemia), but most of the myelodysplastic syndromes have a hyperplastic bone marrow and impaired delivery of cells to the blood. In some of these, morphologic changes in erythroid precursors may overlap with megaloblastic anemia, but granulocytic and megakaryocytic changes usually seen in megaloblastic anemia are lacking.
A low reticulocyte count may indicate decreased production caused by inadequate stimulation of the marrow. Chronic renal disease may result in impaired production of EPO. Certain endocrinopathies, such as hypopituitarism or hypothyroidism, may result in regulation of Hb production at a lower level as a result of decreased tissue need for O2.
A large group of normochromic anemias associated with various chronic diseases form a heterogeneous group characterized by failure of the marrow to meet the need for slightly decreased red cell survival. Some of these are ACDs associated with infection, cancer, or rheumatoid arthritis; they have the defect in iron metabolism noted previously in the Microcytic and Hypochromic Anemias section earlier in the chapter.
Inability of the marrow to respond to EPO may be due to damage to the marrow by drugs or toxic chemicals, to unknown causes, or to infiltration of the marrow by neoplastic cells or fibrous tissue.
In those conditions with low reticulocyte counts in which the marrow is not effectively producing erythrocytes, it is usually helpful to examine the bone marrow. Other studies conducted to determine the underlying disease process can then proceed according to the marrow picture, the assessment of erythrokinetics, and the clinical findings.
Polycythemia
Polycythemia (erythrocytosis) is classically defined as an elevated Hct level above the normal range. In the clinical setting, polycythemia exists when Hb and red cell count are increased, reflecting an elevation of the total red cell volume (Maran & Prchal, 2004). Yet, the recent World Health Organization (WHO) classification of hematologic malignancies has defined polycythemia as Hgb >18.5 g/dL for men, >16.5 g/dL for women, Hct >99th percentile of the method-specific range, or lower values but with a documented unexplained Hgb increase of at least 2 g/dL (Swerdlow et al, 2008).
Absolute polycythemia refers to an increase in the total red cell mass in the body; in relative polycythemia, the total red cell mass is normal, but the Hct is elevated because the plasma volume is decreased. Polycythemia may be classified as in Box 32-3. Some classifications are based on EPO response (Cazzola, 2004), and others are based on the underlying mechanism (i.e., primary or secondary, and congenital or acquired) (Gordeuk et al, 2005).
Relative Polycythemia
Relative polycythemia refers to an increase in Hct or red cell count as a result of decreased plasma volume; total red cell mass is not increased. This occurs in acute dehydration (e.g., severe diarrhea, burns) and in patients on diuretic therapy.
In spurious polycythemia (apparent polycythemia, Gaisböck's syndrome), the red cell mass is often high normal and the plasma volume is low normal; these patients have been regarded as an extreme of the normal physiologic state. Almost all are men; they have a high incidence of tobacco smoking and tend to be obese and to have hypertension. Sleep apnea and diuretics may be contributory factors. Serum EPO level is normal (Cazzola, 2004).
Absolute Polycythemia
Appropriately Increased Erythropoietin Production Due to Hypoxia
Cellular response to hypoxia is controlled by a family of α-β heterodimeric transcription factors, known as hypoxia-inducible factors (HIFs), which regulate the transcription of EPO. There are three HIF isoforms—HIF1α, HIF2α, and HIF3α—with HIF2α being the major isoform regulating EPO expression. In the presence of normoxia, HIF1α is rapidly destroyed by the collaborative effect of O2, prolyl hydroxylase domain–containing enzymes, and the von Hippel–Lindau tumor suppressor protein (VHL). Under hypoxic conditions, degradation of HIFα is slowed, resulting in increased transcription of its target genes, which include EPO. Binding of EPO to its receptor results in autophosphorlyation of the receptor-associated Janus kinase 2 (JAK 2), which has tyrosine kinase activity (Patnaik & Tefferi, 2009).
Arterial Oxygen Unsaturation
Lack of O2 reaching the blood for whatever reason results in arterial unsaturation, impaired O2 delivery to the tissues, increased production of EPO, erythroid hyperplasia in the marrow, and resultant erythrocytosis. The red cell mass is increased. As a response to the hypoxia, the red cell 2,3-DPG and the p50 are increased. In contrast to polycythemia vera, there is usually no leukocytosis or thrombocytosis, and the neutrophil alkaline phosphatase is normal. Arterial O2 unsaturation may be the cause of polycythemia in persons living at high altitudes; in patients with chronic pulmonary disease and a block in diffusion of O2 into the blood; in cyanotic heart disease in which there is right-to-left shunt; and in caroboxyhemoglobinemia mostly related to cigarette smoking. In case of CO poisoning, hypoxia is caused by two mechanisms: direct reduction of O2 saturation and interference with O2 release from Hb (Landaw, 1990).
High Oxygen Affinity Hemoglobinopathy
Another cause of tissue hypoxia is the presence of structurally abnormal Hb that has a high affinity for O2 (Prchal, 2003) (see the Hemoglobins Associated with High Oxygen Affinity section earlier in the chapter). As in other functional hemoglobinopathies, the disorder occurs in the heterozygote. More than 100 Hb mutations associated with increased O2 affinity have been identified (Prchal, 2003). Abnormal Hb releases less O2 to the tissues than does normal Hb at the same pO2; the O2 dissociation curve is shifted to the left, and the p50 is decreased. The red cell 2,3-DPG is not increased. As in arterial O2 unsaturation, EPO production and erythrocytosis are increased. It must be emphasized that routine Hb electrophoresis often does not detect these Hb variants because the amino acid substitution occurs at one of the αβ-contact sites, or near the heme pocket. A low p50 therefore is presumptive evidence for a hemoglobinopathy. Some high-affinity Hbs associated with polycythemia are unstable; in these instances, the heat instability test is positive.
Other causes of altered O2 affinity include Hb M and deficiency of red cell enzyme 2,3-DPG mutase, which is involved in the generation of 2,2-DPG (Gordeuk et al, 2005).
Inappropriate Erythropoietin Production
Neoplasms
Neoplasms, benign or malignant, have been associated with polycythemia. Renal neoplasms account for the majority. In almost all cases, erythrocytosis has disappeared after resection of the tumor. These neoplasms have been shown to express high levels of EPO mRNA. These tumors include cerebellar hemangiomas, renal cell carcinomas, Wilms' tumors, some hepatomas, uterine leimyomas, pheochromocytoma, parathyroid adenomas, and meningiomas (Patnaik & Tefferi, 2009).
Renal Disorders
In other neoplasms or growths (e.g., renal cysts, hydronephrosis, ovarian carcinoma, some hepatomas), it appears that the mass impinging on the kidney induces increased renal production of EPO as a result of increased pressure or local hypoxia within the kidney. Renal artery stenosis is also associated with polycythemia. Posttransplant erythrocytosis occurs in 10% to 20% of renal transplant recipients. The prevelance is much higher in patients who receive a renal transplant without removing their original kidneys. The therapeutic effects of angiotensin-converting enzyme inhibitors in this condition suggest a role for angiotensin II in regulating erythropoiesis (Prchal, 2003).
Familial Polycythemia
The most common familial polycythemia is due to the presence of a high oxygen affinity hemoglobin, which is inherited as an autosomal dominant trait. Congenital polycythemia may be due to a defect in the hypoxia-sensing mechanism. Von Hippel–Lindau syndrome is an autosomal dominant disorder associated with mutations in the VHL gene. These mutations result in increased levels of HIF-1α and increased levels of EPO (Prchal, 2003; Gordeuk et al, 2005). Chuvash polycythemia is an autosomal recessive congenital polycythemia that is endemic in the Chuvash population of the Russian federation. It has been reported to be associated with a high mortality rate caused by thrombotic and hemorrhagic complications. VHL gene mutations have been recently described in Croatian population as well.
Congenital polycythemia may occur secondary to a defect in the EPO receptor in the presence of a normal hypoxia-sensing mechanism. Primary familial and congenital polycythemia (PFCP) is an autosomal dominant disorder that is present at birth. PFCP is associated with low serum EPO levels and in vitro hypersensitivity of erythroid progenitors to EPO. Sixteen germline mutations in the EPO receptor gene (EPOR) with resulting truncated receptors have been described in PFCP to date (Patnaik & Tefferi, 2009). As mentioned earlier, marked decrease in red cell 2,3-DPG associated with deficiency of 2,3-DPG mutase activity results in polycythemia and appears to be inherited as an autosomal recessive condition.
Polycythemia Vera
Polycythemia vera is a myeloproliferative neoplasm characterized by panmyelosis, that is, a condition in which excessive proliferation occurs in megakaryocytes and granulocytes, as well as in erythrocytes. It is manifested by erythrocytosis, leukocytosis, and thrombocytosis of varying degrees. Mutation in the JAK2 gene is now described as a constant finding. Polycythemia vera is discussed with the myeloproliferative neoplasms.
Measurement of Erythrocyte and Plasma Volume
The diagnosis of absolute polycythemia depends on reliable measurements of erythrocyte and plasma volumes. Erythrocyte and plasma volumes are measured with the use of radioactive isotopic tracers and the dilution principle. The most commonly employed tracers are 51Cr in the form of sodium chromate bound to erythrocytes for measurement of erythrocyte volume. 125I or 131iodine is bound to albumin and can be used to measure plasma volume.
For a detailed description of measurement of red cell and plasma volume, see the report of the International Committee for Standardization in Haematology (1980).
Erythrocyte Volume
In brief, blood is collected from the patient, and the erythrocytes are labeled with 51Cr. The chromated erythrocytes are washed in saline. An aliquot of the 51Cr erythrocytes diluted in saline is injected intravenously into the patient. After a period of equilibration, usually 10 to 20 minutes, a sample of blood is withdrawn from the opposite arm. In cases in which the equilibration time is likely to be prolonged (as in splenomegaly, heart failure, or shock), another sample should be withdrawn 60 minutes after injection.
Radioactivity of each sample is recorded by a scintillation counter. The erythrocyte volume (EV) is calculated using the following formula:
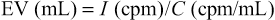
where I is the total injected radioactivity (counts/minute), and C is the radioactivity in erythrocytes after mixing is complete (counts/minute/mL of erythrocytes).
Plasma Volume
Approximately 20 mL of blood is withdrawn from a patient. After centrifugation, the plasma is removed, and radioiodine-labeled albumin is added. After mixing, the labeled plasma is injected intravenously into the patient. At 10, 20, and 30 minutes following injection, 5 mL of blood is removed and radioactivity is counted in a well-type scintillation counter. The radioactivity at zero time (P0) is determined by plotting the three points on semilogarithmic graph paper and extrapolating to zero time. A standard is prepared by diluting an aliquot of the radioiodine-labeled albumin with saline containing a small amount of detergent.
The plasma volume (PV) is calculated using the following formula:
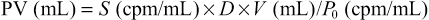
where
Interpretation
The normal erythrocyte volume for men is 20 to 36 mL/kg, and for women, it is 19 to 31 mL/kg. The plasma volume for men is 25 to 43 mL/kg; for women, 28 to 45 mL/kg. In newborns and premature infants, the red cell volume and plasma volume in milliliters per kilogram are higher than in adults.
Patients with polycythemia have red cell volumes exceeding 36 mL/kg for men and 32 mL/kg for women. Changes in erythrocyte volume and plasma volume in a variety of conditions are recorded in Table 32-12.
TABLE 32-12
Clinical Effect of Variable Relationship Between Red Blood Cell Volume and Plasma Volume
| Red Blood Cell Volume | Plasma Volume | Cause | Effect |
| Normal | High | Pregnancy Cirrhosis Nephritis Congestive cardiac failure | Pseudoanemia |
| Normal | Low | Stress Peripheral circulatory failure Dehydration Edema Prolonged bed rest | Pseudopolycythemia |
| Low | Normal | Anemia | Accurate reflection of degree of anemia |
| Low | High | Anemia | Anemia less severe than indicated by blood count |
| Low | Low | Hemorrhage Severe anemia (when hematocrit <0.2) | Anemia more severe than indicated by blood count |
| High | Normal to low | Polycythemia | Accurate reflection of polycythemia or polycythemia less severe than apparent |
| High | High | Polycythemia (when hematocrit >0.5) | Polycythemia more severe than apparent |
| Normal or even high | High | Marked splenomegaly | Pseudoanemia |

The Porphyrias
Physiology
Heme biosynthesis is an essential pathway that occurs in all metabolically active cells containing mitochondria and is most prominent in bone marrow and liver. The erythroid marrow is the major heme-forming tissue in the body, producing 85% of the daily heme requirement. Heme complexed with globin is preserved in circulating red blood cells for approximately 120 days, whereas heme produced in liver for cytochromes and enzymes is subject to much more rapid turnover, measurable in hours. A brief sequence of the heme biosynthesis pathway, with the disease states associated with specific enzyme deficiencies along the pathway, is shown in Figure 32-32.
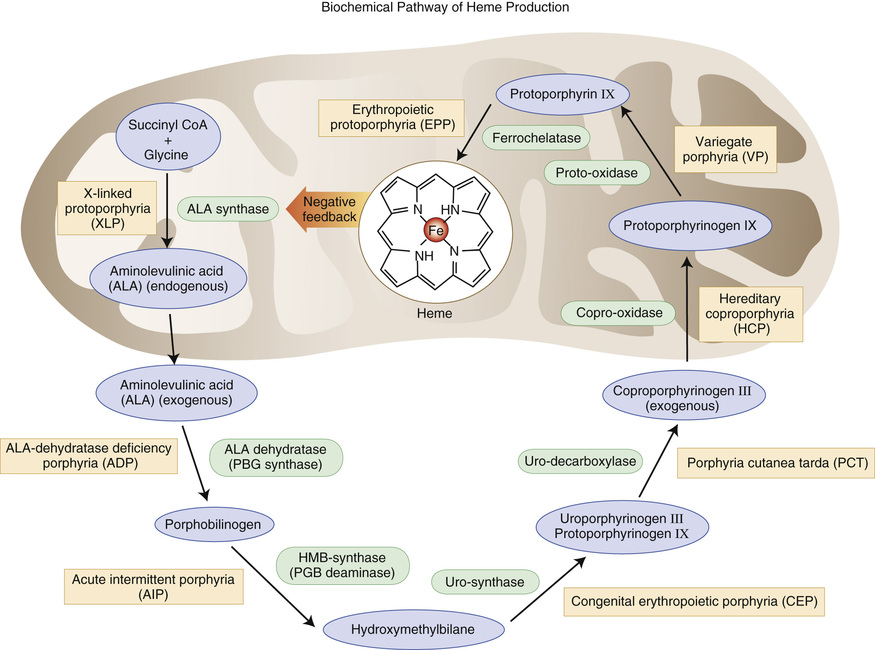
Porphyrins are compounds composed of four pyrrole rings connected by methene bridges, differentiated by substituents found in the eight peripheral positions. The arrangement of four nitrogen atoms in the center of the porphyrin ring enables porphyrins to chelate various metal ions, such as iron, or in disease states, zinc. The biosynthetic intermediates between porphobilinogen (PBG) and protoporphyrin are not porphyrins, but rather their reduced, precursor forms: the porphyrinogens. The porphyrias make up a group of inherited and acquired disorders of heme biosynthesis caused by a deficiency of a specific enzyme in the biosynthetic pathway, culminating in the excess production and increased excretion of precursors formed in the steps before the enzyme defect. Porphyrins that leak out of capillaries into the skin are the cause of photosensitivity, and the precursor substances have been linked to the neurovisceral manifestations. δ-Aminolevulinic acid seems to be the chief offender here (Sassa, 2006).
Approach to the Porphyrias
Consulting pathologists may take a three-step approach to this diverse group of uncommon diseases. First, review the clinical manifestations described by the referring provider. Classifying porphyrias by their clinical presentation is a helpful starting point, as clinicians will call with information about their patients' signs and symptoms, and then will ask for help with ordering laboratory studies (Tables 32-13 and 32-14). Second, suggest biochemical studies, as outlined in Table 32-13 and in the following section. A rapid diagnostic test for urine porphobilinogen is now available as a first step. Finally, coordinate genetic testing as guided by biochemical test results. Great strides have been made in identifying the genes and many mutations causing the porphyrias, and patients will likely wish to know this information as they discuss their diagnosis with family members. Websites helpful to both providers and patients include those of the Porphyrias Consortium of the National Institutes of Health Rare Diseases Clinical Research Network (http://rarediseasesnetwork.epi.usf.edu/porphyrias/index.htm), the American Porphyria Foundation (www.porphyriafoundation.com), and the Drug Database for Acute porphyrias (www.drugs-porphyria.com).
TABLE 32-13
Key Features of Major Porphyrias
| Porphyria | Clinical Features | Excess Metabolites | Rapid Test | Enzyme Deficiency | Genetic Mutations |
| Acute intermittent porphyria | Abdominal pain; psychiatric symptoms | PBG (U) ALA (U) Darkened urine on standing | ✔ | HMB synthase | HMBS (>375 mutations) |
| Congenital erythropoietic porphyria | Photosensitivity; red urine, teeth; hemolysis | UP (U) CP (U) UP (E) Red, fluorescent pigment in urine | UP I synthase, UPG III cosynthase | UROS (>35 mutations) | |
| Hereditary coproporphyria | Photosensitivity | CP III (F, U) PBG (U) ALA (U) Red, fluorescent pigment in urine | ✔ | Copro'ogen oxidase | CPOX (>60 mutations) |
| Variegate porphyria | Photosensitivity | PBG (U) ALA (U) UP (U, F) CP (U, F) Red, fluorescent pigment in urine | ✔ | Proto'ogen oxidase | PPOX (>165 mutations) |
| Porphyria cutanea tarda | Photosensitivity | UP I (U) UP III (U) Acid induces pink fluorescence | Uro'ogen decarboxylase | UROD (>105 mutations) |
TABLE 32-14
Test Results for the Porphyrias
| Porphyria | Urine | RBC | Feces |
| Acute intermittent porphyria | ↑ PBG, ALA | ↓ PBGD activity | Noncontributory |
| ALA dehydratase deficiency porphyria | ↑ porphyrins (ALA alone is increased) Note: Rule out organic acids and heavy metals in urine. | ↓ D-aminolevulinic acid dehydratase activity | Noncontributory |
| Variegate porphyria | ↑ porphyrins, PBG | Noncontributory | ↑ coproporphyrin III/I ratio (<10) |
| Hereditary coproporphyria | ↑ porphyrins, PBG | Noncontributory | ↑ coproporphyrin III/I ratio (>10) |
| Porphyria cutanea tarda | ↑ porphyrins | ↓ UROD in Type II, UROD WNL in Types I, III | Noncontributory |
| Congenital erythropoietic porphyria | ↑ porphyrins | ↓ UPG III cosynthase | Noncontributory |
| Erythropoietic protoporphyria | Noncontributory | ↑ free protoporphyrin | Noncontributory |

Clinical Porphyrias and Their Biochemical Basis
Neurovisceral Symptoms in Isolation
Acute Intermittent Porphyria (AIP)
AIP is the most common acute and probably the most common inherited porphyria (Chemmanur & Bonovsky, 2004). It is inherited in an autosomal dominant fashion with incomplete penetrance, making family studies more challenging, and over 375 mutations have been identified (Balwani & Desnick, 2012). PBG deaminase deficiency has been demonstrated in all examined tissues, at levels of about 50% during attacks and quiescent periods. Diagnosis may be made most easily with demonstration of elevated levels of ALA and PBG in urine during acute attacks (with PBG greater in amount than ALA), but the PBG enzyme assay can be performed at any time on RBCs. Frequently, patients have urine metabolite levels at or near the reference range during quiescent periods. Characteristically, the urine of AIP patients turns dark red upon exposure to air and light, as PBG is oxidized to a porphyrin spontaneously. The patient may be hyponatremic (20% of cases) or hypomagnesemic during acute attacks (Anyaegbu et al, 2012). Common symptoms include colicky abdominal pain (74%), nausea and vomiting (73%), mental confusion, tachycardia, and motor weakness (63%) (Bonkovsky et al, 2014). Symptoms typically begin at the time of puberty, reflecting the influence of steroid hormones on the disease, but the disease can present in children (Anyaegbu et al, 2012). Prevention is the mainstay of management, with avoidance of particular drugs (barbiturates and sulfonamides among many others), hormonal changes, and fasting. Specifically, a low-carbohydrate diet may trigger an attack. A recent study identified an increased risk for malnutrition in AIP patients compared to matched controls (Garcia-Diz et al, 2012). In the postoperative period, patients with AIP must be managed carefully to avoid hypoglycemia (Benassi et al, 2012).
Aminolevulinic Acid Dehydratase Deficiency Porphyria (ADP)
ADP is the rarest of the porphyrias, with just ten reported cases (Balwani & Desnick, 2012). Clinically indistinguishable from AIP, it may be differentiated in the laboratory by the presence of excess ALA alone in the urine and decreased d-aminolevulinic acid dehydratase in the red blood cells of affected patients. Of note, this type of porphyria must be distinguished from tyrosinemia and heavy metal intoxication.
Neurovisceral Symptoms Plus Photosensitivity
Variegate Porphyria (VP)
Similar to AIP and HCP, VP is inherited as an autosomal dominant disorder with low penetrance. It is sometimes referred to as South African porphyria because of its prevalence in the Caucasian population in this region. It is called variegate because of the spectrum of patient presentations. Some have only the cutaneous manifestations, others just the neurovisceral, and others both features. Thus, the clinician need not check both boxes to entertain this diagnosis, especially in patients of Afrikaner descent. Patients have approximately 50% activity of protoporphyrinogen oxidase. Thus, they excrete fecal protoporphyrins and coproporphyrins and urine ALA, PBG, and coproporphyrins. Induction of hepatic ALA synthase I by precipitating factors, such as barbiturates, oral contraceptives, or a low-carbohydrate diet, specifically results in increased ALA and PBG during acute attacks (Chemmanur & Bonovsky, 2004). Cutaneous manifestations in the form of erosions or bullae following trauma to sun-exposed skin are seen at a younger age than is observed with porphyria cutanea tarda. Diagnosis is made by conventional measurements of metabolites, family history, and DNA analysis. Avoidance of triggering factors remains the major approach to patient care.
Hereditary Coproporphyria (HCP)
Also inherited in an autosomal dominant fashion with incomplete penetrance, HCP is less common than AIP. It is due to a deficiency of coproporphyrinogen oxidase. Clinically, HCP resembles a milder form of AIP with its neurovisceral attacks, but cutaneous manifestations are seen in roughly one third of patients (Chemmanur & Bonovsky, 2004). Fecal coproporphyrinogen III is excreted both during and between attacks, and ALA and PBG are seen in urine during an acute crisis. ALA excretion usually exceeds that of PBG, and this, coupled with the cutaneous findings (if present) helps to differentiate HCP from AIP. Prevention, including sunscreen, is central to management.
Photosensitivity Plus Hemolytic Anemia
Porphyria Cutanea Tarda (PCT)
PCT is the most common of the porphyrias in the United States. Type 1 is sporadic; type 2 is inherited in autosomal dominant fashion. The deficient enzyme is uroporphyrinogen decarboxylase (UROD), and researchers have postulated the presence of an inhibitor of UROD, possibly secondary to iron and metabolites of uroporphyrinogen. Over 105 mutations have been inheritied in the UROD gene (Balwani & Desnick, 2012). It is interesting to note that patients with hemochromatosis are at increased risk for acquired PCT (Chemmanur & Bonovsky, 2004). Patients present with cutaneous findings alone, and these are due to mild trauma to sun-exposed areas, not simple photosensitivity. Diagnostic laboratory findings include increased uroporphyrins with smaller increases in urine coproporphyrins. Exacerbating factors, such as alcohol, an inducer of hepatic ALA synthase I, and estrogens, should be avoided. Additional management strategies involve phlebotomy to reduce the hepatic iron load, chloroquine to complex porphyrins for urinary excretion, and the use of sunscreen (Chemmanur & Bonovsky, 2004).
Congenital Erythropoietic Porphyria (CEP)
CEP is distinct from the other porphyrias discussed earlier in both its inheritance (it is a recessive disorder) and its severity. Patients present shortly after birth with red-pigmented urine, hemolytic anemia, and severe cutaneous photosensitivity. Erythrodontia is striking and may be a useful clue if the diagnosis has not already been made. The deficient enzyme is uroporphyrinogen III synthase with over 35 mutations identified, and ALA synthase activity is increased. Coproporphyrin I and uroporphyrin I are present in urine. The prognosis for CEP is significantly worse than for the other porphyrias, with death occurring at an early age in many cases.
Photosensitivity in Isolation
Erythropoietic Protoporphyria (EPP) and X-linked Porphyria (XLP)
EPP is due to a partial deficiency of the enzyme ferrochelatase, the final enzyme in the heme biosynthetic pathway. An autosomal recessive disorder, it is caused by one of over 135 mutations of the FECH gene (Balwani & Desnick, 2012). Clinical symptoms begin in childhood, earlier than in other porphyrias (which have their onset at puberty) and painful, itching erythema occurs within a short time (minutes) of sun exposure. Neuropsychiatric symptoms are absent, but a recent case series described a child who was sent for psychiatric care after screaming in pain when outside (Wahlin et al, 2006). This article makes a useful point regarding the emotional sequelae of painful skin lesions: Emotional distress follows many diseases and must be carefully distinguished from the specific, although myriad, neurovisceral symptoms that characterize other porphyrias. Uniquely among the porphyrias, EPP has a normal urine profile, and RBC testing must be performed to render a diagnosis. XLP is newly described and clinically indistinguishable from EPP. It is due to a gain-of-function of the ALAS2 gene (Balwani & Desnick, 2012).
Analytic Techniques
Diagnosis in the clinic or hospital laboratory may begin with the Trace PBG Kit (Trace America/Trace Diagnostics, Wheeling, Ill.) to identify urine porphobiloinogens. Table 32-13 shows which porphyrias will have elevated levels. After an initial test, analysis of urine, red blood cells, and feces for various porphyrins typically increased in the porphyrias, and for specific enzymes of the heme biosynthetic pathway, takes place in reference laboratories. The pathologist should select a laboratory that is well familiar with the porphyrias and that follows a logical algorithm, so that disease, if present, can be properly classified. Neither the ordering clinician nor the general pathologist needs to attempt classification of the porphyria by selecting one or two specific assays. The algorithmic approach is the rational one and begins with a quantitative evaluation of porphyrins: PBG and ALA in the urine, and porphyrins and PBGD (porphobilinogen deaminase) measurement in the erythrocytes. High-performance liquid chromatography (HPLC) and fluorometry are the mainstays of analytic technique. HPLC can separate individual porphyrins; this, combined with measurement of total fecal porphyrins, reliably distinguishes between AIP and HCP (Deacon & Elder, 2001). VP may be diagnosed by measurement of fecal and urine metabolites, and also by the fluorescence emission peak of 624 to 627 nm, which is unique among the porphyrias (Poh-Fitzpatrick, 1980). The proper samples to submit for testing are urine (a 24-hour sample is strongly preferred, with the appropriate preservative dictated by the reference laboratory) and blood. A stool sample should be provided if VP or HCP is being considered but is less critical for the other porphyrias. Genetic testing for specific mutations should be performed on patients diagnosed by clinical signs and urine/RBC/feces analysis. Recall that the porphyrias are characterized by incomplete penetrance. Family history may or may not suggest the diagnosis; an absence of family history in no way precludes the presence of a porphyria. One a specific mutation is identified, testing of kindreds can follow in a targeted fashion.