Molecular Diagnosis of Hematopoietic Neoplasms
Ali Gabali, Martin H. Bluth
• Overview of the role of molecular investigations in the diagnosis, classification, prognosis, and monitoring of hematolymphoid disorders.
• Detailed summary of the major genetic abnormalities and molecular diagnosis of the acute leukemias and chronic myeloproliferative neoplasms.
• Applications and detailed discussion of molecular minimal residual disease monitoring in chronic myeloid leukemia and acute promyelocytic leukemia in the era of targeted therapeutic options.
• Concept of risk-adapted therapy for optimized management of childhood acute lymphoblastic leukemias.
• Molecular pathologic origins of childhood leukemias.
• Description of antigen receptor gene rearrangements and application of molecular clonality assays to identify monoclonal lymphoid proliferations.
• Detailed summary of the major genetic abnormalities and molecular diagnosis of the non-Hodgkin lymphomas and chronic lymphoid leukemias.
• New research and technology developments that will impact molecular diagnosis and prognosis of hematolymphoid cancers.
Role of Clinical Molecular Diagnostics in Hematologic Cancers
The diagnosis of hematolymphoid neoplasms continues to undergo a dramatic transformation with the advent and application of new technologies. Whereas traditional morphologic (light microscopic) evaluation of glass slides still occupies the centerpiece of pathologic diagnosis, the judicious application of ancillary methods, such as special cytochemistry, immunohistochemistry, flow cytometry, cytogenetics (including fluorescence in situ hybridization [FISH]), and molecular genetics, has allowed for much-improved diagnostic reproducibility and refinement. The 2008 update of the World Health Organization (WHO) classification of hematopoietic and lymphoid neoplasms (Swerdlow et al, 2008) incorporates genetic abnormalities and clinical information in refining the diagnostic criteria of hematolymphoid neoplasms, including previously described and provisional diseases. The relative ease of sampling sites such as the lymph nodes and bone marrow, as well as the provision of easily disaggregated viable cell samples for analysis, has driven this paradigm of diagnostic evaluation. The nature of the hematolymphoid system is also conducive to relatively easy monitoring of disease status following therapy, which represents another important facet in the management of patients with these illnesses.
Given this plethora of diagnostic tools, what role does molecular genetic evaluation or molecular diagnostics play? There are perhaps four major scenarios in which molecular diagnostic analysis of a hematolymphoid proliferation is important. First, molecular investigation can establish a diagnosis in situations wherein morphologic details and results of other lab tests are inconclusive for malignancy. The presence of specific genetic abnormalities, or the demonstration of tumor genetic “homogeneity” of a cell population (e.g., by cell receptor gene rearrangement studies) can establish the presence of a clonal pathologic process. Second, molecular methods are used to subclassify disease entities. To this end, the current WHO classification of hematologic neoplasms includes molecular characterization as a defining feature of many leukemias and lymphomas (Swerdlow et al, 2008; Vardiman et al, 2009). Next, specific genetic anomalies in hematolymphoid malignancies have important prognostic value, which in turn can influence the initial treatment of certain tumors to produce an optimal clinical outcome. Finally, being potentially of high sensitivity, molecular techniques can be used to assess patients after the onset of therapy by monitoring for the presence and extent of minimal residual disease (MRD). Table 76-1 summarizes some of the key instances in which molecular diagnostic evaluation is sought in hematopathology practice. As the synergy between expanding basic biologic information and rapidly progressing technology continues unabated, the future holds promise for many additional applications of molecular diagnostics in the pathology laboratory, including the prediction of disease susceptibility, severity, or treatment response based on polygenic factors; pharmacogenomic profiling of patients to determine drug sensitivity and toxicity profiles; and the assessment of individual responses to increasingly “targeted” therapies.
TABLE 76-1
Applications of Molecular Diagnostics in Hematologic and Lymphoid Neoplasia
| Indication | Examples |
| Diagnosis and subclassification | JAK2 mutations in myeloproliferative neoplasms |
| CCND1-IGH@ abnormality in mantle cell lymphoma | |
| PML-RARA abnormality in APL | |
| FLT3, NPM1, and CEPBA mutations in normal karyotype AML | |
| Determination of tumor clonality | Assessment of abnormal B and T cell lymphoid proliferations |
| Prognosis and/or therapeutic monitoring | Quantitative PCR monitoring of BCR-ABL1 mRNA in CML patients on tyrosine kinase inhibitor therapy (e.g., imatinib) |
| Quantitative PCR monitoring of PML-RARα mRNA in APL | |
| Multiparameter cytogenetic and molecular assessment of chronic lymphocytic leukemia | |
| Relapse risk prediction in childhood acute lymphoblastic leukemias | |
| Outcome prediction of diffuse large B cell lymphoma subtypes by gene expression profiling |

This chapter focuses on the background, clinical rationale, and fundamental technical considerations underlying the molecular diagnostic evaluation and monitoring of hematopoietic and lymphoid tumors. Because the large majority of these laboratory assays are polymerase chain reaction (PCR) based, the emphasis is accordingly placed on methods concerning either DNA or RNA (i.e., reverse-transcription) PCR techniques. Of note, it is apparent that the detection of tumor-specific genetic abnormalities can be achieved by different analytic means—for example, cytogenetics, FISH, or PCR. Although at first glance these various methods may seem redundant, these laboratory techniques should be considered complementary and the choice of methodology is determined by a number of factors, including knowledge of the detection rates, analytic sensitivity, and limitations of various techniques; understanding of the disease pathobiology; the level of interpretive experience and expertise with these procedures; the type of sample; and the phase of the disease under investigation. Table 76-2 indicates comparative analytic sensitivities of the principal modalities used to detect hematolymphoid tumor-related genetic abnormalities.
TABLE 76-2
Relative Sensitivities of Major Techniques Used to Detect Leukemia or Lymphoma-Associated Abnormalities
| Method | Analytic Sensitivity (%)* | Notes |
| Cytogenetics | 5 | Global genomic assessment |
| Requires fresh, sterile cells | ||
| Detects numerical and structural chromosome aberrations | ||
| Difficult or impossible to detect alterations in DNA below band resolution | ||
| FISH | 1-5 | Targeted assessment of relatively large genomic abnormalities |
| Technically straightforward and rapid; can frequently be performed on interphase nuclei | ||
| Applicable to a variety of tissue sources | ||
| Requires rigorous quality standards to avoid false-positive results at low levels of the abnormality in question | ||
| SBH | 5-10 | Targeted assessment for structural abnormalities in genomic DNA |
| Technically laborious and time-intensive | ||
| Requires samples with well-preserved high molecular weight DNA (e.g., fresh or frozen cells) | ||
| PCR | 10–2-10–4 | Targeted assessment of small genomic or mRNA abnormalities |
| Detection of unique chimeric fusion gene mRNA species is highly sensitive | ||
| Technically straightforward and rapid but may require post-PCR detection procedures | ||
| Applicable to a broad range of sample types, including fixed paraffin-embedded tissues | ||
| Can be optimized for high sensitivity, minimal residual disease monitoring (real-time quantitative PCR), or other specialized applications such as allele-specific discrimination | ||
| DNA sequencing (Sanger method) | 20-30 | Targeted assessment of single nucleotide or small base alterations in template (DNA or RNA) sequences |
| Typically performed on PCR-amplified products | ||
| Modifications or special methods (e.g., pyrosequencing) can improve sensitivity to 5% |
The increasingly universal need for molecular genetic evaluation of hematolymphoid cancers has initiated earnest attempts to address and achieve better interlaboratory standardization, and such efforts can be expected to continue to improve the benchmarks of sensitivity and specificity in this field. Nonetheless, in the application and interpretation of molecular diagnostic tests, one caveat that cannot be emphasized enough is that the results of these investigations must never be considered in isolation but rather in conjunction with the clinical presentation, morphologic, and additional laboratory data concerning the individual patient. Regardless of the remarkable specificity and sensitivity of molecular markers, the evident complexity of hematolymphoid neoplasia mandates such a fully integrated approach in order to diminish the possibility of diagnostic error.
Molecular Diagnosis of Leukemias
Gene Fusion Concept in Leukemia and the Basis for Reverse-Transcription Polymerase Chain Reaction Analysis
The leukemias are hematologic cancers with a diverse pathogenesis and pathobiology. However, a significant proportion of these tumors are characterized by nonrandom, balanced (usually) chromosomal translocations, resulting in the formation of fusion or chimeric genes. In general, although two such fusions are created in this abnormal event (i.e., one on each reciprocal translocated chromosome), typically only one of these derivative loci gives rise to a leukemogenic “hybrid” gene. At the molecular level, the intronic breakpoint and fusion sites in the involved genes are highly variable among patients with the same type of leukemia, although there may be evidence of clustered breakpoint “hotspots” in certain types of leukemia-related rearranging genes. Despite the variability in breakpoint sites in both of the involved genes, a consistent feature of the leukemic gene fusions is the production of a chimeric messenger ribonucleic acid (mRNA) molecule. This abnormal fusion transcript is further translated to a fusion protein, presumed to be operative in disrupting many cellular pathways involved in regulating normal proliferative capacity, differentiation, and survival in immature (i.e., precursor) marrow stem or progenitor cells.
Although the formation of a translocation fusion gene, such as the well-known BCR-ABL1 abnormality, is known to be necessary for disease production, results from animal models have suggested that, at least for some types of leukemia, this may not be a sufficient event, requiring secondary incompletely characterized genetic insults as well. This concept has been formalized into a general scheme in which leukemogenic abnormalities are considered to be class I or class II, depending, respectively, on whether they provide a proliferative advantage or disturb the complex processes of hematopoietic differentiation and maturation. In this conceptualization, the development of leukemia becomes more likely when both class I and class II derangements cooperate to alter the fate of the cell (Ishikawa et al, 2009). In the molecular hematopathology laboratory, most leukemia-related assays are directed toward chimeric gene fusions identification that generally fall into the class II category. However, testing for class I mutations (e.g., FLT3 internal tandem duplication) has more recently become integral to more comprehensive evaluations. The challenge for the molecular hematopathology laboratory is thus to incorporate an adequate spectrum of assays directed to the detection of both class I and class II category genetic abnormalities, according to the (ever-changing) understanding of disease molecular pathogenesis.
One practical aspect of the chimeric gene concept is that regardless of the variable location of the genomic (DNA-level) breakpoints in a given translocation gene fusion, the presence of this abnormality can be confirmed by detecting the corresponding invariant chimeric mRNA species. In the molecular diagnostic laboratory, we can thus utilize the reverse-transcription polymerase chain reaction (RT-PCR) method to first convert the fusion mRNA transcript to its complementary deoxyribonucleic acid (cDNA) and then amplify this specific molecule. The ability to perform RT-PCR analysis obviates the difficulties imposed by the often very large flanking regions of intronic DNA surrounding the genomic breakpoint-fusion loci, which would normally not be amenable to amplification by standard DNA-based PCR approaches. Furthermore, the presence of such an aberrant mRNA species is highly specific for the disease, in that it should not theoretically be present in normal cells. Table 76-3 lists the major leukemia-associated translocations along with their resultant fusion gene and mRNA products. Two related concepts are important in the further understanding of chimeric genes and transcripts. Breakpoint heterogeneity refers to the presence of two (or more) common breakpoint loci in a particular gene, which can be targeted by the translocation event. In some examples, different breakpoint sites may be correlated with a presenting disease phenotype or particular clinical features (e.g., the BCR-ABL1 fusion in chronic myeloid leukemia [CML] versus acute lymphoblastic leukemia [ALL]), but this is not always the case. Second, alternative exon splicing of a single chimeric transcript may occur, such that additional, related mRNA products may be observed during PCR amplification of the principal target (e.g., the PML-RARA fusion in acute promyelocytic leukemia). RT-PCR–based assays require fresh bone marrow aspirate or blood cells, although cryopreserved tissue or cells can also be used effectively for RNA isolation (Gabert et al, 2003). RNA extraction and RT-PCR from paraffin tissue block samples have also been successfully described; however, fixed cellular material is often subjected to degradation or the presence of PCR inhibitors, and consequently a higher PCR detection failure rate. Finally, given the highly sensitive nature of PCR amplification, these laboratory assays must be carefully controlled to avoid false-positive contamination artifacts. The following sections describe the molecular genetic and diagnostic features of the most common, prognostically important leukemic diseases.
TABLE 76-3
Common Leukemia-Associated Translocations and Gene Mutations in Myeloid Neoplasms
| Genetic Abnormality | Disease Associations | Basic Molecular Pathogenesis | Diagnostic Detection* |
| t(9;22)/BCR-ABL1 | Chronic myeloid leukemia (100%) Ph+ adult acute lymphoblastic leukemia (20% to 25%) Ph+ pediatric acute lymphoblastic leukemia (3%) | Chimeric fusion protein with deregulation of Abl tyrosine kinase; effects on cell proliferation, apoptosis, adhesion | RT-PCR; FISH |
| JAK2 V617F mutations | Polycythemia vera (>95%); primary myelofibrosis (75%); essential thrombocytosis (50%) | Aberrant activation of cell signaling tyrosine kinase; activation of JAK-STAT pathway | DNA PCR |
| t(15;17)/PML-RARA | Acute promyelocytic leukemia (100%) | Chimeric fusion protein; interference with myeloid maturation and differentiation | RT-PCR; FISH |
| t(8;21)/RUNX1-RUNX1T1 | AML (10%) | Chimeric fusion protein affecting the CBF transcriptional pathway | RT-PCR; FISH |
| inv(16) or t(16;16)/CBFB-MYH11 | Acute myelomonocytic leukemia with abnormal eosinophils (AMML-Eo) | ||
| FLT3 mutations | AML with normal karyotype (20% to 25%) | ITD or point mutation resulting in constitutive activity of FLT3 tyrosine kinase and cell proliferation | DNA PCR |
| NPM1 mutations | AML with normal karyotype (50%) | Complex effects of mutated NPM1 nuclear/cytoplasmic transport protein | DNA PCR |
| KIT mutations | Systemic mastocytosis | Aberrant activation of receptor for hematopoietic stem cell factor | DNA PCR |
| PDGFRA, PDFRB, and FGFR1 translocations | Myeloid and lymphoid neoplasms with eosinophilia | Deregulation of growth factor receptor tyrosine kinases | FISH > RT-PCR |
| t(12;21)/ETV6-RUNX1 | Childhood B-precursor acute lymphoblastic leukemia (20%) | Chimeric fusion protein; disruption of CBF transcriptional pathway | RT-PCR; FISH |
| 11q23/MLL translocations | Childhood (mainly infant) B-lineage ALL (5%) Adult de novo AML, usually monocytic (subset of cases) Subset of secondary AML (after DNA topoisomerase II agent exposure) | Chimeric MLL fusion proteins; disruption of MLL-mediated regulation of normal hematopoiesis | FISH; RT-PCR |
Acute Myeloid Leukemias
The importance of tumor genetic markers is reflected in the present WHO classification (Swerdlow et al, 2008), in which acute myeloid leukemias (AMLs) are increasingly defined by their characteristic genetic abnormalities in relation to immunophenotypic features, prognosis, and clinical outcome. The identification of specific genetic markers can thus establish a definitive AML subclassification, provide important prognostic information, guide therapeutic intervention, and reveal insights into tumor biology (Patel et al, 2012).
AML can be broadly classified as being of relatively “favorable,” “intermediate,” or “poor” prognosis type, based on the type of balanced translocation, degree of tumor cytogenetic complexity, or the nature of specific tumor genetic markers. For example, extensive numerical and structural karyotypic abnormalities, or the presence of “myelodysplasia-background” genetic findings (e.g., abnormalities of chromosomes 5 or 7), are indicative of very aggressive subtypes of AML. Several nonrandom recurring chromosomal translocations characterize another significant subset of AML and can be detected by PCR-based molecular diagnostic techniques. These translocations usually occur without additional cytogenetic findings and often show distinct genotype-phenotype correlations, and many are associated with relatively favorable outcome. In particular, the t(15;17), t(8;21), and inv(16) or related t(16;16) translocations together account for approximately 25% to 30% of de novo AML in both adults and children. In addition to these three, the current WHO classification recognizes four additional rare, recurrent cytogenetically defined subtypes of AML: t(9;11) MLLT3-MLL, t(6;9) DEK-NUP214, inv(3) RPN1-EVI1, and t(1;22) RBM15-MKL1 (Swerdlow et al, 2008; Lugthart et al, 2010; Ramchandren et al, 2013).
Historically, consideration of numerical and structural karyotypic abnormalities tended to dominate the discussion of AML subcategorization, because relatively little was known about “cytogenetically normal” cases. In addition, characteristic gene mutations have been recognized that appear to carry important etiologic and prognostic information in the latter group of AML. These alterations include mutations of CEBPA, NPM1, and FLT3. Whereas the relevance of these mutations is most pronounced in cases of AML with normal karyotype, some of these genetic changes (e.g., FLT3 mutation) may also accompany cases with characteristic translocations, such as the t(15;17), or other cytogenetic abnormalities. Thus, whereas AML with NPM1 and CEBPA mutations have been designated as provisional entities in the 2008 WHO classification, FLT3 alterations, by virtue of their relatively ubiquitous distribution in AML generally, remain an aid to prognostication but not to specific disease subclassification (Marcucci et al, 2005; Swerdlow et al, 2008; Green et al, 2010; Balusu et al, 2011; Parmar et al, 2011; Patel et al, 2012; Man et al, 2012).
This section focuses on the common AML-related molecular genetic abnormalities that are currently considered to be the most clinically relevant due to frequency of occurrence and impact on therapy or prognosis. The major abnormalities in AML are summarized in Table 76-3.
Acute Promyelocytic Leukemia: t(15;17)(q22;q21)/PML-RARA Abnormality
Acute promyelocytic leukemia (APL), or AML-M3 (according to the previous French-American-British [FAB] classification of AML) accounts for 5% to 10% of de novo AML. Patients typically present with symptoms from peripheral blood cytopenias (e.g., anemia, absolute neutropenia, and thrombocytopenia) and have a high propensity for life-threatening coagulopathy. Morphologically, APL consists of a proliferation of hypergranular promyelocytes and myeloblasts; however, a microgranular variant form is also frequently encountered (Mantadakis et al, 2008). Immunophenotypically, the cells are characterized by a lack of CD34 and HLA-DR. APL is genetically defined by the presence of the t(15;17)(q22;q12-21) abnormality, resulting in the fusion of the retinoic acid receptor-α gene, RARα (also called RARA) [17(q21)] with the PML gene [15(q22)] to form a PML-RARA chimeric gene on the derivative chromosome 15q (Grignani et al, 1994; Jurcic et al, 2007).
The RARA gene product is a subunit of a heterodimeric nuclear receptor for the naturally occurring ligand retinoic acid, or vitamin A. The retinoid nuclear receptor pathway is functional in many aspects of normal cell proliferation and differentiation (Chambon, 1996; Collins, 2008; Mark et al, 2009). PML encodes a DNA-binding zinc finger protein and associates with several other proteins in a macromolecular complex, forming discrete nuclear bodies (Dyck et al, 1994; Weis et al, 1994; Reineke & Kao, 2009). This interesting protein appears to have multiple possible cellular functions, including transcriptional regulation, apoptosis, and possibly immune surveillance (Quignon et al, 1998; Wang et al, 1998; Grimwade, 1999; Zhong et al, 2000; Borden & Culjkovic, 2009; Reinecke & Kao, 2009; Gabali, 2013). The hybrid PML-RARα protein is clearly involved in disrupting numerous intracellular processes, primarily resulting in a lack of terminal differentiation of neoplastic myeloid precursors beyond the promyelocytic stage. Nonetheless, the significance of APL as a “therapeutic paradigm” for leukemia resides in the ability of pharmacologic doses of all trans retinoic acid (atRA) to induce leukemic cell differentiation in vivo. When used in combination with cytotoxic chemotherapy, the synergistic effects of atRA induce complete remission in 90% to 98% of patients and prolonged remission in more than 80% of patients (Tallman et al, 1997; 2002; Clavio et al, 2009; Licht, 2009; Sanz et al, 2009; Patel et al, 2012). This remarkable finding has intensified efforts to identify other avenues for “cytodifferentiative” therapy in AML. Even though the effect of atRA would seem superficially related to the interaction with the RARα moiety in the abnormal PML-RARα protein, further investigations have unraveled the basic molecular mechanisms underlying the success of this agent. The normal retinoic acid receptor, when not bound to its retinoic acid ligand, associates with histone deacetylase (HDAC) in a nuclear protein corepressor complex (Guidez et al, 1998; Melnick & Licht, 1999; Lefebvre, 2001; Wei, 2004). This interaction reduces the accessibility of chromatin to transcription factors (and thus locus-specific transcriptional activity), yet is reversible in normal marrow cells upon exposure to physiologic concentrations of retinoic acid. The PML-RARα oncoprotein stabilizes and enhances the state of transcriptional repression by this multiprotein complex; however, therapeutic doses of atRA appear to overcome this abnormal condition and relieve the cellular differentiation block (Melnick & Licht, 1999; Hormaeche & Licht, 2007; Collins, 2008; Licht, 2009). Whereas some evidence suggests that the PML-RARα oncoprotein may also disrupt the normal proapoptotic functions of PML, thus potentiating transformation via pathways involving both partners in the translocation (PML-RARA and RARA-PML), the direct contribution of altered PML to leukemogenesis remains controversial (Strudwick & Borden, 2002; Collins, 2008; Brown et al, 2009). Functional haploinsufficiency of one remaining normal PML gene may also be important in the pathobiology of APL.
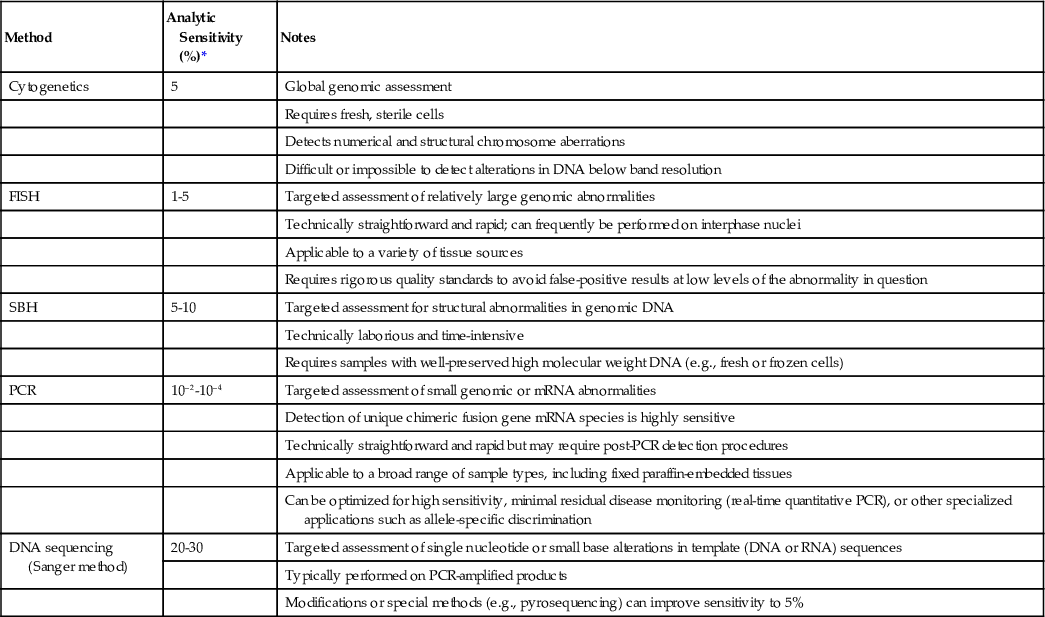
The molecular anatomy of the PML-RARA fusion gene and mRNA products in APL is summarized diagrammatically in Figure 76-1, A. The PML gene exhibits breakpoint heterogeneity in that one of three possible break sites can be encountered in any given patient, involving either intron 3 or intron 6, or occurring within exon 6 (Grignani et al, 1994; Gallagher et al, 1995; Reiter et al, 2003). In contrast, the RARA breakpoints are uniformly distributed in intron 2 of the gene. Thus one of three possible PML-RARα chimeric mRNAs can result from this genetic fusion: PML exon 6/RARα exon 3 (long (L)-form, or BCR 1), PML exon 3/RARα exon 3 (short (S)-form, or BCR 3) and PML exon 6Δ/RARα exon 3 (variable [V]-form, or BCR 2). The V-form transcript is unique in that the PML break occurs within exon 6 and a variable proportion of this exon is retained, although additional nucleotides may be added or deleted (Gallagher et al, 1995; Reiter et al, 2003). Notably, an in-frame fusion mRNA is produced in each case of APL, underscoring the critical requirement for the PML-RARα protein in leukemogenesis. The L-form (BCR 1) and S-form (BCR 3) PML-RARα fusions are most commonly found in APL (~45% to 50% of cases each), whereas the V-form (BCR 2) is only rarely encountered (~5% of APL). The strategy for RT-PCR amplification of these PML-RARα transcripts is depicted in Figure 76-1, B, as is a representative diagnostic PCR assay (Fig. 76-1, C). Of note, the L-form (BCR 1) and V-form (BCR 2) type transcripts show alternative splicing out of exons 5 and 6, producing three major amplified products when a PML exon 3 primer is employed.
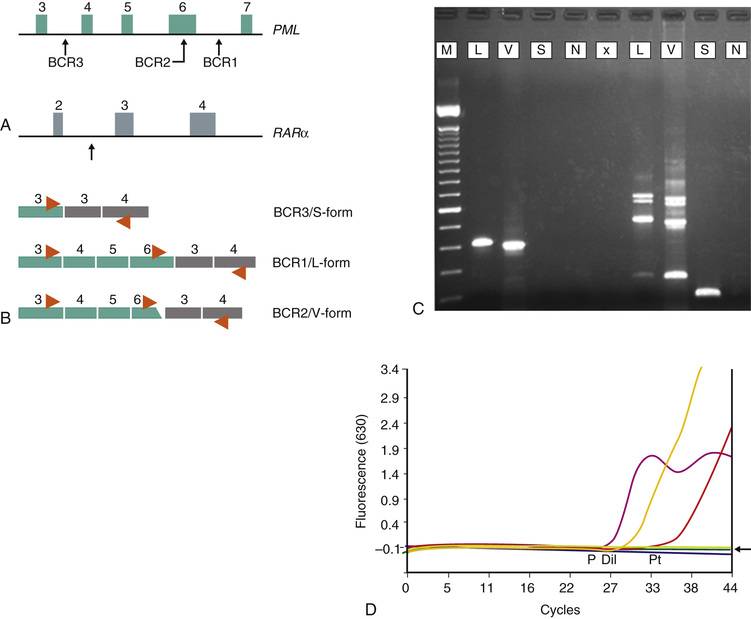
The clinical relevance in detecting the PML-RARα fusion abnormality in cases of suspected APL is evident from the efficacy of administering specific therapy (atRA plus anthracycline-based chemotherapy for induction and consolidation) with a high possibility of long-term remission and survival. To this end, despite the tight correlation between the t(15;17)/PML-RARA and APL morphology, it is recognized that other rare APL-like myeloid leukemias do occur. These morphologic mimics harbor alternative translocations involving the RARA gene with fusion partner genes other than PML. Examples of these variants include the t(11;17)/ZBTB16(PLZF)-RARA, t(11;17)/NUMA1-RARA, t(5;17)/NPM1-RARA, and the t(17;17) STAT5B-RARA acute myeloid leukemias (Melnick & Licht, 1999; Grimwade et al, 2000; Sainty et al, 2000; Zelent et al, 2001; Redner, 2002, Balusu et al, 2011). Notably, ZBTB16(PLZF)-RARA and STAT5B-RARA positive leukemias in particular are not responsive to the differentiating effects of atRA and are associated with less favorable outcome (Jansen & Löwenberg, 2001; Redner, 2002). Molecular assays specific for the common PML-RARα fusion will not detect these variant transcripts. Other morphologic AML subtypes characterized by increased promyelocytes are similarly atRA unresponsive and also require distinction from true PML-RARA positive APL. Several studies have assessed the possible clinical significance of the PML-RARα transcript type in APL patients. Both the S-form (BCR 3) and rare V-form (BCR 2) PML-RARα fusions have been associated with adverse features such as higher presentation white blood cell count, poor atRA response, and possibly shorter remission duration (Vahdat et al, 1994; Gallagher et al, 1995; 1997; Jurcic et al, 2001; Gupta et al, 2004; Patel et al, 2012); however, the independent prognostic value of molecular subclassification in APL has not been definitively established.
Finally, the PML-RARα transcript serves as a valuable molecular disease marker to follow therapeutic response (Fig. 76-1, D). Patients with APL treated with combination chemotherapy and atRA have a relatively favorable prognosis, yet disease relapses occur frequently. The use of qualitative RT-PCR methods to detect the PML-RARα fusion mRNA (with a typical sensitivity of 10–3 to 10–4) was initially found to be a very powerful tool for predicting relapse risk in individual patients (Jurcic et al, 2001; Grimwade & Lo Coco, 2002; Lo Coco et al, 2002). The timing, or phase of disease therapy, is an important consideration in this leukemia. Patients evaluated for PML-RARα at the end of induction are often found to be positive, and the predictive value at this time point is not significant. By the end of consolidation therapy, however, PCR positivity is strongly predictive of relapse in patients who have apparently achieved clinical complete remission, in that nearly all such patients will progress to hematologic relapse within months. In contrast, a single PCR-negative measurement at this time point is not necessarily predictive of favorable outcome, in that a significant number of such patients may also suffer relapse. This has led to recommendations for more frequent posttherapy monitoring using sensitive and precise real-time quantitative PCR methods (RQ-PCR). To this end, several APL study groups have reported results of serial PML-RARα monitoring using RQ-PCR techniques, and these efforts have shown a benefit for detecting low-level molecular disease and its value in predicting relapse risk (Grimwade & Lo Coco, 2002; Gallagher et al, 2003; Grimwade et al, 2009; 2010; Borthakur et al, 2011). Furthermore, several studies have validated the concept of “salvaging” or retreating patients with molecular residual disease, with good clinical outcomes. Given an array of additional treatment options for APL, including highly effective second-line intervention with arsenic trioxide (As2O3) and autologous or allogeneic stem cell transplantation, molecular residual disease assessment thus forms an integral aspect of the management of all APL patients (Santamaria et al, 2007; Kohno et al, 2008; Lo Coco et al, 2008; Grimwade et al, 2009; Paschka et al, 2012; Patel et al, 2012). Although no formal guidelines have been firmly established for RQ-PCR monitoring of PML-RARα in APL, proposed common sampling time points for assessment include the end of induction, the end of consolidation, and then every 2 to 3 months for the first year after therapy, because this is the window during which most relapses occur. Bone marrow aspirate samples are preferred for MRD assessment, in that the peripheral blood is less sensitive for transcript detection following treatment onset.
Core Binding Factor–Related Acute Myeloid Leukemias: t(8;21)(q22;q22)/RUNX1-RUNX1T1 and Inv(16)(p13q22) or t(16;16)(p13;q22)/CBFB-MYH11 Abnormalities
Acute myeloid leukemias with the t(8;21)(q22;q22) and inv(16)(p13q22) or related t(16;16)(p13;q22) cytogenetic abnormalities together account for approximately 11% to 18% of de novo cases (Schnittger et al, 2007; Cheng et al, 2009). Core binding factor (CBF)–related AML cases are roughly evenly distributed between those with the t(8;21) and cases with inv(16)/t(16;16) (Dombret et al, 2009). Whereas the t(8;21) is generally associated with AML showing maturation (FAB type AML-M2), the inv(16) and t(16;16) are highly correlated with acute myelomonocytic leukemia with abnormal eosinophils (FAB type AML-M4Eo) (Le Beau et al, 1983; Cheng et al 2009). Clinically, these genetically defined AML subtypes are responsive to chemotherapy (especially high-dose cytarabine regimens) and are considered to be prognostically favorable compared with AML in general (Appelbaum et al, 2006; Dombret et al, 2009; Paschka et al, 2012; Patel et al, 2012).
Although different respective leukemia phenotypes derive from these translocations, a remarkable and common pathobiologic feature of both the t(8;21) and inv(16) or t(16;16) leukemias is disruption of the CBF transcriptional regulatory pathway (Speck et al, 1999; Paschka, 2008). CBF is a heterodimeric transcription factor that consists of a DNA binding α-subunit (CBFα) and a peptide-interacting β-subunit (CBFβ), which acts to stabilize CBFα at sites of DNA interaction (Fig. 76-2). In normal cells, CBF acts at “core” enhancer sequences in a number of genes required for proper myeloid and lymphoid cell differentiation and/or maturation. The binding of CBF facilitates the access of other transcription factor complexes to these chromatin sites, in part through increased acetylation of DNA-bound histone proteins by histone acetyltransferases (Lorsbach & Downing, 2001; Yamagata et al, 2005). In human acute myeloid leukemias with the t(8;21), the RUNX1 gene (formerly designated AML1, or CBFA2) encoding an isoform of CBFα is joined to a putative transcription factor, RUNX1T1 (formerly ETO, or MTG8) to form the RUNX1-RUNX1T1 fusion (Downing, 1999; Peterson et al, 2007; Patel et al, 2012). In the case of the inv(16) or t(16;16), the gene producing CBFβ (i.e., CBFB) is juxtaposed to a smooth muscle myosin heavy chain gene MYH11 to form the hybrid CBFB-MYH11 (Liu et al, 1995; Mrózek et al, 2008; Pratcorona et al, 2012). In either case, profound disruption of the normal cellular differentiation program is thought to be central to the causation of acute leukemia. The prominence of CBF pathway alterations in leukemogenesis is additionally emphasized by the finding of RUNX1 gene translocations in several other types of leukemia and myelodysplasia (see Fig. 76-2), including the t(12;21)/ETV6-RUNX1 abnormality, the most common single genetic abnormality in pediatric ALL.
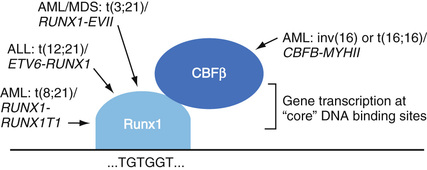
From a mechanistic viewpoint, the RUNX1-RUNX1T1 leukemic fusion protein is thought to act in a dominant negative manner to normal RUNX1 (i.e., CBFα) by recruiting or stabilizing elements of transcriptional repression, including histone deacetylase complexes, at critical DNA sites (Lorsbach & Downing, 2001; Yamagata et al, 2005). Normal gene expression patterns are also disrupted by the altered affinity of the abnormal fusion protein for specific core enhancer sequences, including an increased propensity to bind sites with such sequences in duplicate (Okumura et al, 2008). CBFβ-MYH11 chimeric protein sequesters normal CBFα protein in the cytosol, thereby abrogating heterodimeric CBF assembly in the nucleus, but may also create an abnormally repressive transcriptional complex in a manner analogous to that of RUNX1-RUNX1T1 (Shigesada et al, 2004; Pratcorona et al, 2012). In either case, the resultant inhibition of transcription at key genes alters the genetic program for normal proliferation and differentiation in hematolymphoid stem or progenitor cells.
In keeping with the concept outlined previously that an individual genetic aberrancy is of itself not likely sufficient for carcinogenesis, transgenic mice engineered to conditionally express a RUNX1-RUNX1T1 oncogene develop acute myeloid leukemia only on subsequent exposure to a promoting mutagen (Lorsbach & Downing, 2001; Peterson et al, 2007; Müller et al, 2008). In CBF leukemias, the pathognomonic translocations create a block in cellular differentiation (class II mutations), but affected cells often acquire secondary class I mutations that endow them with enhanced proliferative capacity. Indeed, there is a particularly strong association in CBF leukemias with secondary mutations to the gene encoding the c-KIT tyrosine kinase (stem cell factor receptor), a potent regulator of cell growth. Up to half of CBF leukemias carry KIT mutations; in contrast, approximately 5% of all AML cases show such mutations (Müller et al, 2008). Many of these KIT mutations involve the D816 “hot-spot” amino acid (that is also affected in systemic mastocytosis), but mutations have been described in other exons as well. The presence of KIT mutations in CBF leukemias has been associated with increased relapse risk and, in some studies, poorer overall survival, although the latter finding remains somewhat controversial at present (Cairoli et al, 2006; Paschka et al, 2006; Mrózek et al, 2008; Müller et al, 2008; Faderl et al, 2011). To date, the routine clinical utility of KIT mutation testing in CBF leukemias currently awaits validation by larger trials. Notably, deregulation of c-KIT expression as a consequence of KIT mutations in these leukemias may present a rational therapeutic target, given that the 5-year-survival for CBF leukemia patients, albeit better than for AML in general, does not substantially surpass 50%.
The molecular rearrangements underlying the RUNX1-RUNX1T1 fusion are such that the breakpoint-fusion sites invariably involve the same limited set of intron regions in both genes, resulting in a consistent in-frame RUNX1-RUNX1T1 mRNA molecule in each patient with this type of AML, although some degree of variant transcripts derived from alternative splicing and differential promoter usage can also be seen (Zhang et al, 2002; Lafiura et al, 2007; Pratcorona et al, 2012). In contrast, standard RT-PCR detection of the CBFβ-MYH11 chimeric transcript is complicated by the potential for marked breakpoint heterogeneity, mainly in the MYH11 gene, as well as by rare alternate breakage sites described in CBFB. In all, more than 10 CBFB-MYH11 fusion transcript forms have been identified to date (Liu et al, 1995; Viswanatha et al, 1998; Kadkol et al, 2004; Schnittger et al, 2007). Despite this complexity, the CBFB-MYH11 gene fusion can be readily detected by RT-PCR, because approximately 90% of these tumors harbor the so-called “type A” chimeric mRNA, characterized by fusion of CBFB nucleotide 495 to MYH11 nucleotide 1921 to form a transcript of relatively short length (Schnittger et al, 2007). More comprehensive PCR strategies have also emerged to detect the majority of the other rare CBFB-MYH11 fusion types, each of which account for the remainder of cases of inv(16) or t(16;16) AML (Kadkol et al, 2004). RT-PCR analysis for the CBFB-MYH11 abnormality is often advantageous, in that standard cytogenetic interpretation may be uninformative in some cases (Merchant et al, 2004; Monma et al, 2007). In this context, it is possible, although unlikely, that a case with a true non–type A CBFB-MYH11 fusion could be negative by both conventional cytogenetic and molecular analysis, if karyotyping fails to detect a cryptic translocation and RT-PCR technique targets only the type A transcript; this possibility underscores the general importance of maintaining awareness of the limitations of a specific molecular assay, as well as the central role that morphologic assessment must play even in the molecular era. Nevertheless, given the generally more favorable clinical outcome for both of these AML subtypes, molecular diagnosis can play a significant role both in initial identification and disease monitoring following treatment (Pratcorona et al, 2012).
Acute Myeloid Leukemia with Other Translocations
AML with 11q23/MLL
In AML the presence of rearranged MLL gene is considered an unfavorable prognostic indicator and is observed in AML, B-ALL (further discussed below), and myelodysplastic syndrome. The translocated MLL gene is reported in de novo AML, around 3% to 4%, as well as in therapy-related acute myeloid leukemia (t-AML). Numerous partners have been reported to partner with the MLL gene, but in around 80% of cases, the partner will include t(4;11)(q21;q23)/AF4-MLL, t(9;11)(q21;q23)/MLLT3-MLL, or t(11;19)(q23;p13)/MLLT1-MLL. The latter two MLL gene partners are also the most common balanced translocation involving the MLL gene in t-AML, particularly in patients with a previous, relatively short history of receiving topoisomerase II inhibitor therapy. The most common breakpoints reported in the translocated MLL gene are present between exon 5 and 11 of the gene. Some of the translocations/insertions can be too small and cryptic, thus limiting their detectability by conventional karyotyping. FISH analysis, using break-apart probes, is widely used to detect MLL abnormalities and can detect gene amplification or mutant MLL when the partner is unknown. RT-PCR is the most sensitive approach for detecting specific subtypes of MLL rearrangements when the partner gene is known (Ramchandren et al, 2013).
AML with t(6;9)(p23;q34)/DEK-NUP214
Acute myeloid leukemia with t(6;9)(p23;q34)/DEKNUP214 is a distinctive entity in the current WHO 2008 classification of hematopoietic malignancies and is identified in around 1% to 2% of acute myeloid leukemia. Characteristically, these AML cases are seen in children and younger adults and are associated with significant multilineage dysplasia, absolute basophilia in bone marrow, and/or peripheral blood, and the myeloblasts may express terminal deoxynucleotidyl transferase (TdT) protein. The presence of this translocation in AML is considered a poor prognostic indicator and tends to be accompanied by FLT3-ITD mutation. Detection of such translocations can be easily achieved by RT-PCR because the breakpoints in both genes are constant (Ramchandren et al, 2013).
AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2)/RPN1-EVI1
Recurring inv(3)(q21q26.2) or t(3;3)(q21;q26.2) is observed primarily in around 1% to 2% of acute myeloid leukemia and results in the formation of RPN1-EVI1 gene transcript that impairs cellular proliferation and differentiation. Around 10% to 20% of the patients with this translocation have peripheral thrombocytosis and dysplastic changes involving granulocytic and megakaryocytic series. This type of leukemia may be preceded by a myelodysplastic picture with or without chromosomal abnormalities, more commonly involving chromosome 7 than chromosome 5. Therefore, the presence of this translocation is critical for diagnosing this condition. However, t(1;3)(p36.3;q21.1), t(3;21)(q26.2;q22.1), t(2;3)(p15;q26.2), and t(3;12)(q26.2;p13) are excluded because they are more associated with myelodysplastic syndrome. The gene transcript can be detected by RT-PCR as well as by FISH analysis (Ramchandren et al, 2013).
AML (Megakaryoblastic) with t(1;22)(p13;q13)/RBM15-MKL1
This entity of AML is reported in around 1% of de novo AML. The classification of this AML is strict in that it occurs in non–Down syndrome and children younger than age 3. The translocation results in the formation of RBM15-MKL1 fusion transcript that causes defects in megakaryoblastic proliferation and differentiation. The bone marrow demonstrates increased megakaryoblast positive for CD41 and CD61, with increased bone marrow fibrosis. This AML is associated with poor outcome, and most patients present with hepatosplenomegaly. The t(1;22) can be detected by conventional cytogenetic or FISH analysis. However, some laboratories are using RT-PCR to identify the chimeric mRNA (Ramchandren et al, 2013).
Acute Myeloid Leukemias with FLT3, NPM1, and CEBPA Gene Mutations
The current WHO classification of AML has been broadened to include additional entities characterized by specific gene mutations (Swerdlow et al, 2008). Chief among these with regard to diagnostic and clinical significance are alterations of FLT3, NPM1, and CEBPA. These gene mutations involve relatively small-scale modifications to the specific DNA sequences, including short insertions, deletions, or single base pair changes, and therefore cannot be detected by conventional cytogenetic or FISH techniques. AML with gene mutations usually are characterized by the presence of a normal tumor karyotype and are discussed here in this context; however, similar mutations can also accompany AML with recurrent translocations or other cytogenetic abnormalities. Notably, the characteristic “genotype-phenotype” correlations among morphologic, clinical, and genetic findings observed in AML with recurrent translocations are not as obviously encountered in AML with FLT3, NPM1, or CEBPA gene mutations. Among the group of AML with gene mutations, FLT3 is an adverse prognostic factor, whereas NPM1 and CEBPA mutations indicate an outcome somewhat better than for AML in general (Dufour et al, 2012). Complicating this scenario is the fact that a significant subset of normal karyotype AML may harbor two or more gene mutations, producing variable effects on tumor prognosis (Grossman et al, 2011). Most significantly, FLT3 and NPM1 mutations may occur together, in which case the relative outcome benefit of the NPM1 mutation is abrogated by the coexisting FLT3 mutation. Furthermore, if these mutations are detected in the setting of cytogenetic abnormalities, or in other infrequent combinations, the prognostic significance is less clearly established. Whereas testing for FLT3, NPM1, and CEBPA gene mutations is currently indicated in cytogenetically normal AML cases, molecular genetic assays for these mutations should ideally not be considered in isolation. As noted earlier, FLT3 mutation was not designated as a specific subtype of AML in the 2008 WHO classification, and cases of AML with mutated NPM1 or CEBPA remain only provisional diagnostic entities at present (Green et al, 2010; Taskesen et al, 2011; Balusu et al, 2011).
FLT3 Mutations in Acute Myeloid Leukemia
Expression of the FLT3 gene (Fms-like tyrosine kinase 3; also known as FLK2 and STK1) produces a membrane-spanning signal transduction protein of the receptor tyrosine kinase (RTK) type III family, whose members also include the platelet-derived growth factor receptor (PDGFR) genes and the KIT gene (Agnes et al, 1994; Small, 2008; Fischer et al, 2010). FLT3 receptor-ligand interactions are important for the maintenance and propagation of early progenitor cells in normal myeloid and lymphoid hematopoiesis. Not surprisingly, the wild type receptor protein is also expressed in the majority of AML and B-lineage ALLs, emphasizing its role in the survival and proliferation of immature hematopoietic cells (Gilliland & Griffin, 2002; Stirewalt & Radich, 2003). FLT3 mutations in AML most frequently take the form of internal tandem duplications (FLT3-ITD) of part of the coding region for the juxtamembrane portion of this tyrosine kinase, producing an abnormal, constitutively active FLT3 protein. All ITDs preserve an intact reading frame, despite the introduction of additional nucleotides, consistent with abnormal activation of a largely functional protein. A second type of activating lesion involves point mutation of FLT3 at amino acid sites D835 or I836 in the “activation loop” domain of the protein, FLT3-TDK (Yamamoto et al, 2001; Bacher et al, 2008). The constitutively active FLT3 tyrosine kinase initiates increased activity within its downstream signaling cascade, which in turn provides a pro-proliferative stimulus to the myeloid cell (Kontzias et al, 2012). Thus FLT3 belongs to the “class I” group of AML-related genetic abnormalities. These FLT3 gene mutations are together estimated to occur in 20% to 40% of adult AML (as well as a smaller number of pediatric AML, therapy-related AML, and myelodysplasias), and appear to be distributed among all morphologic subtypes. FLT3 mutations are consequently one of the most common recurrent abnormal genetic findings encountered in AML (Pozdnyakova et al, 2008). FLT3-ITD mutations are most frequently observed in several cytogenetic settings, including t(15;17) PML-RARA, t(6;9) DEK-NUP214–associated AML, and in many cytogenetically normal cases; this suggests a key role for this genetic aberration in the pathophysiology of these subsets of AML (Schnittger et al, 2002; Thiede et al, 2002; Green et al, 2010).
In general, the presence of FLT3 gene mutations in AML has been associated with poor prognosis, and the prognostic impact of FLT3 mutations is particularly well established in cytogenetically normal cases (Kottaridis et al, 2001; Zwaan et al, 2003; Schlenk et al, 2008; also reviewed in Stirewalt & Radich, 2003). More refined prognostic information appears to be derived from an assessment of the “gene dosage,” or ratio of FLT3-ITD relative to wild type allele (FLT3-ITD : WT), with higher ratios correlated with poorer clinical outcome (Thiede et al, 2002; Baldus et al, 2006; Meshinchi et al, 2006; Green et al, 2010). Elevated FLT3-ITD : WT ratios could possibly arise from gene amplification of the ITD allele, biallelic mutations, or from the presence of a more prevalent subclone of leukemic cells with the FLT3-ITD (Stirewalt & Radich, 2003). However, some caveats remain notable regarding FLT3 mutations. Although the prognostic impact of FLT3-ITD mutations is well established, the influence of FLT3-TKD mutations remains controversial (Fröhling et al, 2002; Moreno et al, 2003; Yanada et al, 2005; Mead et al, 2007). Recent data suggest that FLT3-TKD mutations have no effect on prognosis in AML overall but may have differential effects on outcomes (i.e., better or worse) in particular subgroups of AML (Bacher et al, 2008). AML with FLT3-ITD and FLT3-TKD mutations also have distinct gene expression profiles, suggesting that these two mutations, although within the same gene, may act in biologically and perhaps prognostically dissimilar ways (Neben et al, 2005). The value of additional screening for FLT3-TKD mutations in the molecular hematopathology laboratory therefore awaits a clearer understanding of the clinical significance of this molecular target. Second, the role of cooperating FLT3 mutations in APL is well recognized and has been associated with proliferative features, such as leukocytosis (Callens et al, 2005); however, APL patients with FLT3 mutations do not have a significantly different outcome than those without, and mutation analysis is thus not routinely recommended in the setting of APL.
In contrast to the situation for translocation fusion genes with chimeric mRNA transcripts, FLT3-ITD can be detected by PCR amplification of genomic DNA. Amplification of exons 14 and 15 of FLT3 can identify the ITD, because these are variably larger in fragment size than expected for this region in the wild type gene. The presence of an ITD is typically established by PCR product sizing—for example, using capillary electrophoresis of fluorescent PCR amplicons. Different molecular approaches are employed to detect TKD mutations, including DNA PCR followed by amplicon digestion with informative restriction endonucleases, direct sequencing, or other sequence-specific methods.
NPM1 Mutations in Acute Myeloid Leukemia
NPM1, which encodes the protein nucleophosmin, is mutated in 50% to 60% of AML with a normal karyotype (Falini et al, 2005; Balusu et al, 2011; Falini et al, 2013). Many of these leukemias are characterized by a lack of CD34 antigen expression and features of monocytic differentiation. NPM1 is also the partner gene in the translocation characteristic of an unrelated hematologic malignancy—anaplastic large cell lymphoma (ALCL)—suggesting its broader importance in fundamental hematopoietic signaling pathways. Normal cellular nucleophosmin is largely present within the nucleolus, but the protein operates as a “shuttle,” escorting proteins, particularly ribosomal subunits, between the nucleus and the cytoplasm (Yun et al, 2003; Yu et al, 2006). Nucleophosmin also participates in cell cycle regulation by virtue of its ability to activate, via both direct and indirect mechanisms, p53 and other proteins involved in cell cycle control (Falini et al, 2007; Balusu et al, 2011; Falini et al, 2011; 2013). Yet another task of this multifaceted protein is the control of centrosome duplication before mitosis (Tsou & Stearns, 2006), and experimental depletion of NPM1 results in improper chromosome alignment, abnormal centrosomes, and disorganization of mitotic spindles (Amin et al, 2008). NPM1 mutations in AML involve small insertions of variable length and sequence in a specific region of the gene, at 5q35, producing a frameshift that alters the amino acid sequence at the C-terminus of the protein. This portion of nucleophosmin carries a nuclear localization signal (NLS) important for its nuclear retention. The altered protein, lacking the NLS and incorporating instead a nuclear export signal, is aberrantly retained in the cytoplasm (Chen et al, 2006; Balusu et al, 2011). The exact mechanisms by which cytoplasmic nucleophosmin contributes to leukemogenesis remain unclear, but the loss of the usual intranuclear interactions between nucleophosmin and cell cycle control proteins and tumor suppressor proteins (e.g., p19Arf) is hypothesized to play an important role in transformation (Falini et al, 2007). The presence of mutated NPM1 is significantly associated with a relatively good prognosis in normal karyotype AML, but only in the absence of FLT3 (Schlenk et al, 2008). However, nearly 40% of NPM1 mutated AML cases harbor a concomitant FLT3 mutation, and these patients have an inferior prognosis, essentially similar to AML with FLT3 mutations alone (Baldus et al, 2007).
Although the cytoplasmic localization of nucleophosmin in NPM1-mutated AML cases presents a potentially attractive target for simple detection via immunohistochemistry, such a technique may be insufficient for accurate prognostication based on the lack of quantitative standards and the problem of tumor heterogeneity (Konoplev et al, 2009). Therefore, molecular analysis is the preferred method for identifying NPM1 mutations. More than 25 mutations (all heterozygous) have been identified affecting exon 12 (Konoplev et al, 2009). The most common mutation, labeled type A, is seen in up to 80% of NPM1 positive cases and involves the insertion of the tetranucleotide sequence TCTG at positions 956 to 959 of the gene (Falini et al, 2007; Balusu et al, 2011). Because these NPM1 exon 12 mutations change the length of the DNA sequence relative to wild type and that NPM1 mutation is stable over the course of the disease, standard PCR amplification of the genomic region paired with fluorescent product size analysis by capillary electrophoresis suffices to detect the change. A distinct advantage of this strategy is that the same method can be applied to the detection of FLT3-ITD mutations, simplifying the technical approach to these genetic abnormalities; as indicated, NPM1 analysis should always be performed in conjunction with FLT3 testing to provide accurate prognostic information (Didier et al, 2008; Falini et al, 2011; Dolnik et al, 2012).
CEBPA Mutations in Acute Myeloid Leukemia
Acute myeloid leukemias with mutations of the CCAAT/enhancer binding protein α (also called CEBPA) gene, at 19q13.1, represent a third distinct group of tumors that are included in the 2008 WHO AML classification. CEBPA encodes a transcription factor essential to granulocytic differentiation, such that the production of mature granulocytes does not occur in its absence (Koschmieder et al, 2009). Corollary functions of normal CEPBA protein include transcriptional repression of genes involved in nonhematopoietic programs and the promotion of cell cycle arrest as a component of terminal differentiation. Although mutations of CEPBA are thought to promote leukemogenesis by blocking granulocytic differentiation and proliferation control, the exact pathogenetic mechanisms remain to be elucidated. Notably, some families with an inherited predisposition to the development of AML carry germline CEBPA mutations (Renneville et al, 2009a, 2009b; Dufour et al, 2012). Approximately 15% to 20% of cytogenetically normal cases of AML carry the CEBPA mutation (Baldus et al, 2007; Taskesen et al, 2011). In this setting, CEBPA mutations are associated with a relatively favorable prognosis (Fröhling et al, 2004; Bienz et al, 2005; Schlenk et al, 2008). Although the significance of CEBPA mutations in other types of AML is not completely clear, data suggest that the beneficial prognostic effect of CEBPA mutations applies only in cases with normal karyotype and without FLT3-ITD mutation (Renneville et al, 2009a, 2009b; Grossman et al, 2011). Paradoxically, though, coexisting FLT3-TKD mutations in particular may not affect the positive impact of mutated CEBPA (Bacher et al, 2008; Taskesen et al, 2011). Furthermore, studies suggest that the improved outcome in CEBPA mutation-positive AML appears to be limited only to those cases carrying biallelic mutations (Dufour et al, 2012). AML with single CEBPA allele mutation may have more risk for FLT3-ITD mutation (Wouters et al, 2009b; Green et al, 2010; Taskesen et al, 2011).
Prototypical CEBPA mutations are found widely separated within both N- and C-terminal regions of the gene, in contrast to the more clustered localization of mutations in FLT3 and NPM1. The N-terminal mutations prevent translation of the full-length p42 isoform, but an alternative start site downstream from the mutated N-terminal site permits continued production of a shorter p30 isoform (Wouters et al, 2009b; Dufour et al, 2012). In contrast, C-terminal mutations are in frame and are thought to impair protein function. Other mutations have been described throughout the intervening region. Consequently, molecular detection strategies must be capable of detecting many mutations over a potentially wide area. Direct sequencing of RT-PCR amplified CEBPA mRNA, both with and without initial screening via high-resolution melting curve analysis, has been successfully employed in this regard (Ahn et al, 2009; Rázga et al, 2009; Taskesen et al, 2011; Dufour et al, 2012).
Other Gene Mutations in Acute Myeloid Leukemias
Mutations involving many other genes, including WT1 (see below), MLL, TET2, JAK2, IDH1/IDH2, EZH2, PLK1, NRAS, and KRAS, have been associated with prognostic or biologic importance in AML. In addition, abnormal gene expression patterns of BAALC, ERG, and MN1 have also been tied to prognosis (Baldus et al, 2007; Ernst et al, 2010; Dang et al, 2010; Abbas et al, 2010; Hart et al, 2011; Chotirat et al, 2012; Ernst et al, 2012; Weissman et al, 2012; Benetatos et al, 2013). As this list continues to expand, a major challenge for the diagnostician and clinician will be in the rational interpretation of potentially many interacting genetic factors in order to determine appropriate risk stratification and therapy options for individual AML patients. As suggested from the inherent complexity of simultaneously assessing the three relatively common mutations considered previously (FLT3, NPM1, and CEBPA), the impact of multiple cooperating genetic events requires sophisticated bioinformatics analyses applied to well-designed and sufficiently powered clinical studies. Basically, these mutations are detected by PCR amplification and mutational analysis by sequencing and by comparison of these sequences with the published unmutated sequence (Ramchandren et al, 2013). The optimal strategy for evaluation of gene mutations in AML will thus continue to evolve as additional clinical data emerge (Hatzimichael et al, 2013).
WT1 Mutations in Acute Myeloid Leukemias
Wilms' Tumor 1 gene (WT1) is a zinc finger transcription factor, located at 11p13 allele, which is expressed normally by hematopoietic stem cells and embryonic kidney cells. Wild type WT1 is suggested to have tumor suppressing properties, and various gene mutations are identified in different malignancies, including those of hematopoietic origin. WT1 mutations are reported in about 10% to 14% of AML, and most mutations are located in exons 7 and 9 at the “hot spot” Cys-His zinc finger domains. Mutated WT1 in AML with normal cytogenetics is associated with poor prognosis and high rate of induction failure, particularly when accompanied by other mutations such as FLT-3 or cKIT. WT1 mutations may disappear after achieving complete remission; however, it has been shown that, in relapse, cases that acquired new or additional mutations may develop (Hou et al, 2010; Chou et al, 2010, Duncan et al, 2012). WT1 mutations are detected by using allele-specific PCR, followed by sequencing analysis for the presence of mutations.
Acute Lymphoblastic Leukemia/Lymphoma—B and T Cell Lineage
B Cell Lymphoblastic Leukemia/Lymphoma (Precursor B Cell Acute Lymphoblastic Leukemia, B-ALL)
Significant biologic and clinical differences exist between adult and childhood onset ALLs. Comparatively little is known about the pathogenesis of adult ALL, which overall is associated with a relatively unfavorable outcome. As such, cytogenetic and molecular genetic anomalies in adult ALL are incompletely characterized, with the exception of the very poor prognosis t(9;22)/BCR-ABL1 (i.e., Ph+ ALL). Beyond detection of the BCR-ABL1 gene fusion, molecular diagnosis is currently of limited utility in the management of adult ALL. In marked contrast, the molecular diagnostic evaluation of pediatric ALL is of substantial value in the stratification of patients to “risk-adapted” treatment strategies. Over the past 20 years, the improvement in therapeutic regimens for these children, coupled with a more profound understanding of ALL tumor biology, has resulted in cure rates of 80% or better overall (Pui et al, 2004; O'Leary et al, 2008; Pui et al, 2008; Vrooman & Silverman, 2009). In turn, this has led to the concept of tailoring therapy to defined subsets of patients based on a combination of presenting clinical and biologic features, including early treatment response and tumor genetics. The aim in childhood ALL has thus evolved to balance the highest probability of long-term remission with the lowest chance of therapy-related adverse events. Although clinical features (e.g., age, degree of leukocytosis, CNS involvement) are initially used to separate “standard-risk” from “high-risk” individuals, tumor genetics are also an integral component of the initial (i.e., pretreatment) evaluation of childhood ALL patients. Broadly, both numerical and structural chromosomal abnormalities are considered in this process (Table 76-4). In the former instance, “high hyperdiploidy,” or tumor aneuploidy with a chromosome number in excess of 52, has been associated with more favorable treatment response and outcome, particularly when certain chromosomal trisomies (e.g., +4, +10, +17) are present (Harris et al, 1992; Heerema et al, 2000; Moorman et al, 2003; Sutcliffe et al, 2005; Schultz et al, 2007). Conversely, tumor hypodiploidy, especially less than 44 chromosomes, is associated with a very high frequency of treatment failure (Heerema et al, 1999; Nachman et al, 2007; Schultz et al, 2007). Among the structural rearrangements, four chromosomal translocations account for approximately one-third of pediatric ALL cases, each with prognostic importance: the t(9;22)/BCR-ABL1, t(1;19)/TCF3-PBX1, t(12;21)/ETV6-RUNX1, and rearrangements involving the 11q23/MLL locus (Bartolo & Viswanatha, 2000; Harrison, 2001; Pui et al, 2004; Armstrong & Look, 2005). In general, numerical and structural alterations are mutually exclusive in any given case of childhood ALL; however, occasionally, these findings can coexist. Although not dealt with formally in this section, the detection of minimal residual disease at certain posttherapeutic time points (e.g., end-of-induction) has also emerged as a highly important component of outcome prediction in childhood ALL (Cave et al, 1998; Van Dongen et al, 1998; Nyvold et al, 2002; Campana, 2008; Flohr et al, 2008; Campana, 2009).
TABLE 76-4
Risk Stratification/Outcome Prediction in Childhood B-Lineage Acute Lymphoblastic Leukemia*
| Abnormality | Prognostic Significance | Notes |
| High hyperdiploidy (>52 chromosomes) | Favorable | Hyperdiploid tumors with particular trisomic chromosomes (e.g., +4, +10, +17) associated with very favorable outcome |
| t(12;21)/ETV6-RUNX1 | Favorable | Very favorable outcome, but appreciable incidence of late relapses |
| t(1;19)/TCF3-PBX1 | Intermediate | Intensive therapy results in good outcome |
| t(9;22)/BCR-ABL1 | Unfavorable | Very high risk for relapse/refractory disease |
| 11q23/MLL gene rearrangements | Unfavorable | Very high risk for infant ALL; also considered high risk factor for older children |
| Hypodiploidy (<44 chromosomes) | Unfavorable | Very high risk for relapse/refractory disease |
| High level molecular MRD at end-induction therapy | Unfavorable | Very high risk of relapse in patients with MRD above 10–2 to 10–3, detected by molecular or flow cytometric methods |
Major Translocation Fusion Gene Abnormalities in B Cell ALL
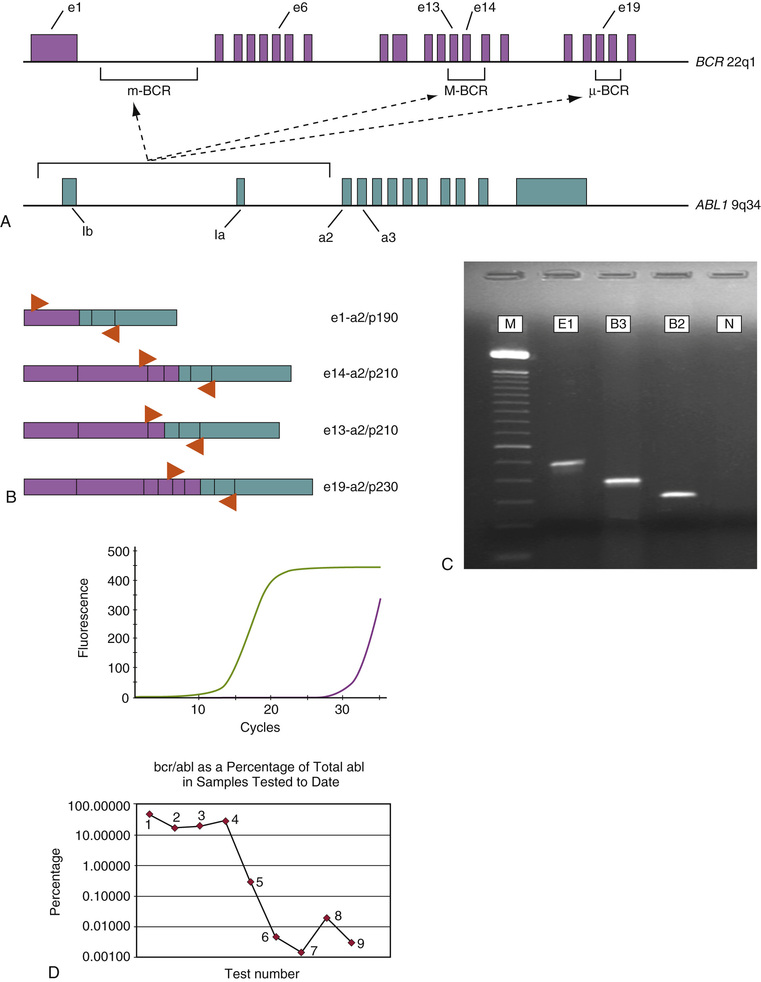
The t(9;22)(q34;q11.2)/1BCR-ABL1 is found in approximately 3% to 4% of childhood B-lineage ALL, but it occurs in 20% to 25% of adult B-ALL (Armstrong & Look, 2005). Typically, these patients present with markedly elevated lymphoblast counts and other adverse clinical features. Of the two common break-fusion events associated with the BCR-ABL1 fusion gene, the majority of cases of pediatric B-ALL (80% to 90%) have BCR breakpoints situated in the minor breakpoint cluster region (m-BCR), with production of an e1-a2 type chimeric BCR-ABL1 mRNA (p190). The e1-a2 mRNA type is also found in many adult BCR-ABL1 positive B-ALL, but approximately one-third of adult cases alternatively demonstrate e13-a2 or e14-a2 transcripts, characteristic of major breakpoint cluster region (M-BCR) disruption in the BCR gene (p210). The structure and molecular diagnostic aspects of the BCR-ABL1 gene fusion are presented in greater detail in the subsequent section on CML. The presence of the BCR-ABL1 in childhood ALL is an independently poor prognostic factor, placing these patients among the very highest at risk for primary treatment failure and relapse (Fletcher et al, 1991; Gaynon et al, 1997; Jones & Saha, 2005; Yanada et al, 2009). The majority of BCR-ABL1 positive ALL patients are candidates for more aggressive therapy, including early allogeneic hematopoietic stem cell transplantation. The introduction of the tyrosine kinase inhibitor imatinib mesylate may offer additional therapeutic benefit in the treatment of this disease; in adult Ph+ALL, imatinib in combination with conventional chemotherapy results in a very high (95%) rate of initial complete remission, although durable responses are often difficult to maintain (Jones & Saha, 2005; Gökbuget & Hoelzer, 2009; Ribera et al, 2009; Vrooman & Silverman, 2009; Yanada et al, 2009; reviewed in Gruber et al, 2009; Shabbir & Stuart, 2010). Studies of the efficacy of imatinib in childhood Ph+ ALL are promising (Jones & Saha, 2005; Vrooman & Silverman, 2009), so in 2013, the U.S. Food and Drug Administration (FDA) approved the use of imatinib to treat children newly diagnosed with Ph+ALL. Molecular analysis of the BCR-ABL1 transcript by the RT-PCR technique is critical for predicting risk in childhood ALL, as well as providing a marker for residual disease monitoring. Deletion of IKZF1, which codes for the transcription factor Ikaros, has been identified as a genetic change present in many cases of BCR-ABL1 ALL (Mullighan et al, 2008; 2009; Iacobucci et al, 2009; Martinelli et al, 2009), and Ikaros alterations appear to be an important cooperative lesion in this subclass of ALL, strongly influencing the aggressive behavior of BCR-ABL1 ALL (Martinelli et al, 2009; Mullighan et al, 2009).
B-ALL characterized by the t(1;19)(q23;p13.3) abnormality are uncommon, accounting for less than 5% of pediatric cases and only rare occurrences in adults (Armstrong & Look, 2005). This translocation is strongly associated with “pre-B” immunophenotypic features, typified by the presence of cytoplasmic IgM production in the tumor cells. Following its initial recognition in childhood ALL, the t(1;19) was associated with poor outcome. However, it has become apparent that more intensive treatment protocols largely negate the adverse effects of this genotype, resulting in stable remissions for most of these individuals. Recognition of this translocation is thus required for appropriate therapy (Schultz et al, 2007). The t(1;19) most frequently occurs as an unbalanced form of translocation (with resultant loss of some genetic material) and produces the chimeric TCF3 (formerly E2A)-PBX1 gene fusion, although balanced translocations with the same fusion product also occur (Hunger et al, 1991; Paulsson et al, 2007). The TCF3 gene encodes a series of basic helix-loop-helix transcription factors, which are involved in myocyte and B-lymphocyte development. PBX1 is a DNA-binding transcription factor that is not normally expressed in lymphoid cells. The fusion TCF3-PBX1 protein replaces the DNA binding and protein dimerization motifs of TCF3 with the DNA binding region of PBX1, predisposing the progenitor B cell to neoplastic proliferation. The location of genomic breakpoints within TCF3 and PBX1 introns is consistent in essentially all cases, resulting in the formation of a single TCF3-PBX1 transcript (Hunger et al, 1991; Wiemels et al, 2002a), with only few reported variants (Paulsson et al, 2007). This fusion mRNA is readily amenable to RT-PCR detection and can also serve as a disease-specific marker for sensitive detection in the bone marrow. Of note, rare cytogenetically detectable ALL cases with t(1;19) lack the TCF3-PBX1 fusion and appear to be associated with an inferior clinical outcome (Privitera et al, 1992; Prima & Hunger, 2007); these variant t(1;19)(6q23;p13.3) translocations instead create reciprocal oncogenic fusion proteins, DAZAP1/MEF2D and MEF2D/DAZAP1 (Prima & Hunger, 2007).
Overall, 20% to 25% of pediatric ALL patients are characterized by the presence of the t(12;21)(p13;q22) abnormality, establishing this lesion as the most common recurrent translocation in this patient group (Armstrong & Look, 2005). This genetic finding is essentially absent among adults with B-ALL. The t(12;21) is cytogenetically cryptic in almost all cases (Karrman et al, 2006), instead often being suggested by an apparent deletion of the short arm of one chromosome 12. Although patients with t(12;21) ALL are found distributed among different clinical risk groups, in general these individuals are between the ages of 2 and 7 and have diploid karyotype tumors (Rubnitz et al, 1997a; Forestier & Schmiegelow, 2006; Forestier et al, 2007). The t(12;21) results in the joining of the 12p locus ETV6 gene (also known as TEL) with the RUNX1 (or AML1, CBFA2) gene on 21q to form the chimeric gene fusion ETV6-RUNX1 on the derivative chromosome 12 (Romana et al, 1995; Zelent et al, 2004). Pediatric patients with ETV6-RUNX1 positive ALL have a favorable outcome, with excellent disease-free remissions approaching 80% (similar to the aforementioned high-hyperdiploid group with specific chromosomal trisomies) (Shurtleff et al, 1995; Borkhardt et al, 1997; Rubnitz et al, 1997a; 1997b; Forestier et al, 2008). However, reports of a substantial late relapse rate among some ETV6-RUNX1 ALL patients have refocused attention on more clearly defining the risk characteristics of this group, even though many such relapsed patients remain responsive to “salvage therapy” protocols (Harbott et al, 1997; Seeger et al, 1998; Ford et al, 2001; Forestier et al, 2008). The molecular pathology of ETV6-RUNX1 ALL is related to disruption of normal CBF activity, leading to enhanced transcriptional repression of many target genes, as similarly discussed for the RUNX1-RUNX1T1 abnormality in AML (Fig. 76-3) (Zelent et al, 2004). In almost 90% of ETV6-RUNX1 ALL cases, the remaining ETV6 allele is partially or completely deleted in a variable proportion of tumor cells, suggesting that this event may be an important secondary event for leukemogenic transformation, through complete loss of normal ETV6 function (Raynaud et al, 1996; Tsuzuki et al, 2007; Wiemels et al, 2008). At the DNA level, ETV6 and RUNX1 breaksites in ALL occur most often in the same genomic regions of ETV6 intron 5 and RUNX1 intron 1 (Romana et al, 1995; Von Goessel et al, 2009), with evidence of some propensity for breakpoint clustering. A second minor variant with breakpoints situated in RUNX1 intron 2 is also described. The in-frame transcribed ETV6-RUNX1 mRNA thus encompasses a novel region joining ETV6 exon 5 to RUNX1 exon 2 (most cases) or ETV6 exon 5 to RUNX1 exon 3 (fewer cases) (Nakao et al, 1996a; Von Goessel et al, 2009). Additional complexity is present in this gene fusion owing to alternative splicing out of RUNX1 exons 2 and/or 3, with the possibility of detecting up to four ETV6-RUNX1 amplified transcript forms by RT-PCR in any given case. Molecular (RT-PCR) or FISH detection of the ETV6-RUNX1 is frequently employed in pediatric ALL, given the inability to diagnose the t(12;21) by standard cytogenetics because of its cryptic nature. The chimeric mRNA further represents a highly specific molecular marker for residual disease assessment.
Translocations involving the “mixed lineage leukemia” (MLL) gene located on chromosome 11q23 are observed in 5% to 9% of pediatric ALL cases overall and are disproportionately represented in infants younger than 1 year of age (Kaneko et al, 1986; Bartolo & Viswanatha, 2000; Armstrong & Look, 2005; Silverman, 2007; Chowdhury & Brady, 2008; Ramchandren et al, 2013). The presence of MLL gene translocations is associated with a poor outcome for these very young children and is considered an adverse finding in older children as well, particularly in the setting of a poor prednisone response (Pui et al, 2003; Chowdhury & Brady, 2008). Infant MLL-positive leukemias are of B-lineage, lack CD10 positivity, and demonstrate aberrant expression of some myeloid-associated markers, particularly CD13 and CD33 (Chen et al, 1993; Burmeister et al, 2009). Clinically, these patients have high initial leukocyte counts, organomegaly, and a tendency for involvement of the central nervous system. Translocations involving 11q23/MLL also occur in de novo AML and secondary (therapy-related) AML, and in either case are characterized by monocytic morphologic features (i.e., FAB AML-M4 or M5 types). In de novo adult AML, MLL gene rearrangements, specifically the t(9;11) MLLT3-MLL abnormality, may not represent a high-risk tumor genotype. In contrast, secondary AML arises in subsets of both pediatric and adult patients who have had prior dose-intensive chemotherapy with DNA topoisomerase II inhibitors (e.g., epipodophyllotoxins and anthracyclines); these tumors occur abruptly within a few months to years after exposure and have a very aggressive treatment-resistant course (Godley & Larson, 2008).
The pathobiology underlying MLL gene translocations in these different types of de novo and secondary acute lymphoid and myeloid leukemias is becoming better understood. The MLL protein is a large (~430 kD) DNA binding protein, which is characterized by several functional domains, indicating its role as a multifunctional transcriptional factor with histone methyltransferase activity (Hess, 2004; Li et al, 2005; Krivtsov et al, 2007; Wang et al, 2009). MLL is known to regulate several HOX (homeobox) genes involved in the coordination of proper skeletal development during mammalian embryogenesis (Yu et al, 1995; Soshnikova & Duboule, 2008). Several HOX genes also appear to be central to the development and maintenance of myeloid and lymphoid progenitor cells, thus establishing a tight link for MLL in the genetic control of normal hematopoiesis as well (Hess, 2004; Ernst et al, 2004a; 2004b; McGonigle et al, 2008). MLL gene translocations produce an altered functional form of the protein; in each instance, the N-terminal “A-T hook” DNA binding motif of MLL is retained in the chimeric protein, typically with fusion to a portion of another transcription factor (Waring & Cleary, 1997; Chowdhury & Brady, 2008; Ramchandren et al, 2013). Accumulating data indicate that abnormal expression of MLL fusion proteins can constitutively deregulate certain HOX genes, leading to hematopoietic progenitor cell immortalization and predisposition to leukemogenesis (Armstrong et al, 2003; Hess, 2004; Ono et al, 2009). Indeed, studies using chromatin immunoprecipitation to identify portions of the genome bound by MLL have further detected its presence at numerous promoter regions, suggesting a more global role for MLL in transcriptional regulation.
Some additional key biologic insights have emerged from the structural analysis of MLL gene breakpoints in different subsets of leukemias. The majority of de novo AML with MLL translocations have breaksites situated 5′ in the MLL breakpoint cluster region, whereas the preponderance of secondary AML occur in a more distal (3′) segment of this locus (Stissel-Broeker et al, 1996; Zhang et al, 2006). Infant ALL cases strikingly also tend to associate with the 3′ genomic region, suggesting a commonality in pathogenesis with secondary AML. This 3′ “hotspot” area contains a strong consensus binding site for DNA topoisomerase II, as well as an adjacent “scaffold attachment region,” which is highly susceptible to cleavage by endonucleases. Although the presence of the topoisomerase II site presents an attractive explanation for the basis of developing secondary AML (e.g., via double strand break induction by topoisomerase II inhibitors and subsequent MLL translocation rearrangements), the hypersensitive 3′ scaffold site of the gene has received further attention, because it too appears to be particularly prone to double-strand breaks in experimental models (Stissel-Broeker et al, 1996). The latter MLL region is in fact selectively targeted for cleavage during cell death in response to diverse apoptogenic stimuli (e.g., following cellular stress or DNA damage) (Betti et al, 2001; 2003; 2005; Sim & Liu, 2001; Greaves & Wiemels, 2003a). These investigations imply that exposure to agents that can induce this site-specific MLL breakage potentially set the stage for MLL gene fusions, leading to the very aggressive forms of leukemia observed in infants and after distinct types of chemotherapy. In turn, because the causal etiology is already identified for secondary AML with MLL translocations, current studies in infant ALL seek to discover possible exogenous or naturally occurring transplacental factors that may predispose developing fetal hematopoietic stem cells to MLL gene breakage and illegitimate recombination (Spector et al, 2005; Pombo-de-Oliveira & Koifman, 2006).
The MLL gene is involved in translocation events with more than 60 known partner genes (Liu et al, 2009); however, the t(4;11)(q21;q23)/MLL-AF4 abnormality is the most frequently encountered in infant ALL (75% of cases). MLL gene locus breakpoints are distributed over an 8.3-kb region of DNA encompassing exons 5 to 11 (Chowdhury & Brady, 2008). This marked breakpoint heterogeneity may also occur in the translocated gene (e.g., AF4), resulting in the possibility of several MLL hybrid gene products, typically one of which is expressed in any given patient with this leukemia. RT-PCR method can be used to identify individual MLL fusion gene transcripts, such as the MLL-AF4 chimeric mRNA; however, the combination of multiple rearranging gene partners in these leukemias and the potential for substantial genomic breakpoint variability requires comprehensive and technically challenging laboratory approaches, such as multiplex PCR assays or, in the research setting, techniques such as long-distance inverse PCR (Pallisgaard et al, 1998; Andersson et al, 2001; Meyer & Marschalek, 2009; Meyer et al, 2009). MLL gene locus rearrangements have also been successfully detected by Southern blot analysis, although the FISH technique now provides a rapid and highly efficacious alternative for this purpose (Cuthbert et al, 2000; Cavazzini et al, 2006). FISH analysis can provide very rapid confirmation of MLL gene breakage, implying a translocation event, but it cannot give information regarding the particular translocation present. Thus RT-PCR, even with the limitations described, can serve a useful purpose in the diagnosis and monitoring of these leukemias, especially because the fusion gene type can be specifically and more sensitively identified in contrast to other analytic methods. Furthermore, mutation due to partial tandem duplication involving the MLL gene (MLL-PTD) is only detected by RT-PCR.
Prenatal Origins of Childhood Leukemias
Major advances continue to be made in understanding the molecular basis of the pediatric acute leukemias. These efforts now span the realms of basic science, translational and clinical research, and, increasingly, population-based methods. Studies of concordant ALL occurring in monozygotic twins first demonstrated that the leukemias arising in each paired affected individual were genetically identical based on molecular analysis of the genomic breakpoint sequences of involved MLL, ETV6-RUNX1, or clonotypic immunoglobulin or T cell receptor gene rearrangements (summarized in Greaves et al, 2003b; Taub & Ge, 2004). Notably, the propensity to develop concordant leukemias is variable for different leukemic subtypes in twin studies, approaching 100% for MLL translocation ALL and only 10% for ETV6-RUNX1 ALL. These findings suggest a variable transformation potential for these different gene fusions in vivo, raising the issues of both the nature and timing of secondary genetic insults. The results of these investigations have been corroborated using similar experimental techniques to “backtrack” leukemia-specific DNA abnormalities to archived neonatal blood samples of individual (nontwin) children of different ages, with genetically defined subtypes of ALL or AML (Gale et al, 1997; Wiemels et al, 1999; Fasching et al, 2000; Yagi et al, 2000; Wiemels et al, 2002a; Greaves & Wiemels, 2003a; Greaves et al, 2003b; Taub & Ge, 2004; Ross, 2008). Together, these data strongly suggest that many pediatric ALL (and some AML) have origins in utero; that is, prenatal acquisition of potentially leukemogenic translocation events, or other genetic abnormalities, can occur during the time of active fetal hematopoiesis, but in most cases, additional, later-occurring genetic alterations in susceptible individuals are required in order to produce overt leukemia. The rapid evolution of MLL-associated ALL versus the longer latency (i.e., older age at onset) of ETV6-RUNX1 ALL indicates that both the biologic effects of the primary (predisposing) genetic lesion, as well as the timing and type of promoting insult(s) are complex, although within defined subtypes of leukemia, it may now be possible to better elucidate the molecular pathways resulting in tumorigenesis. Not all types of pediatric ALL, however, can be traced back to a prenatal clonal cell origin. The retrospective neonatal blood data for the t(1;19)/TCF3-PBX1 ALL subgroup instead support the concept that the majority of these patients develop this genetic aberration in the postnatal period (Wiemels et al, 2002a).
Prospective RT-PCR and FISH screening of blood samples from healthy newborn children have subsequently revealed a detection frequency of 1% for the ETV6-RUNX1 abnormality and 0.2% for the RUNX1-RUNX1T1 fusion transcript, markedly higher than the incidence of either of these leukemia subtypes in childhood (Mori et al, 2002; Olsen et al, 2006). These data demonstrate that the occurrence of at least some chromosomal translocation fusion genes is a common event in utero; however, the presence of such an abnormality is clearly insufficient to cause overt leukemia in the vast majority of individuals. Although the translocation-type acute leukemias are karyotypically characterized in most instances by an apparent “single genetic anomaly,” it is becoming evident that these tumors also conform to the “two-hit” hypothesis of Knudson proposed for solid tissue cancers (Knudson, 1992; Schindler et al, 2009; Ramchandren et al, 2013), as discussed in further detail in Chapter 75. In this concept, the initiating translocation event must be stable enough in a long-lived progenitor hematopoietic cell to confer some advantage in survival or proliferation; the acquisition of one or more subsequent genotoxic events in a susceptible individual would then lead to complete malignant transformation. The fate of such silent “translocation-positive” cells is thus an important parameter, not only for understanding disease pathogenesis but also in a practical setting for the appropriate interpretation of molecular diagnostic assays in the context of detecting minimal residual disease.
Intriguingly, studies using genome-wide association have identified certain low-penetrance germline variants of IKZF1 (as well as other genes) that may increase susceptibility to the development of childhood ALL (Papaemmanuil et al, 2009). Even though much research remains directed at the basic molecular and cell biology of childhood acute leukemias, molecular epidemiologic methods will play an increasing role in identifying the interplay between environmental factors and host genetic polymorphisms, which underlie the predisposition to this disease in some individuals (Clavel et al, 2005; Spector et al, 2005; Lafiura et al, 2007; reviewed in Pui et al, 2008). Indeed, the relatively brief period of potential environmental exposure in utero may lend itself to the detection of specific risk factors in a manner impossible for adult-onset leukemias (Kim et al, 2006; Ross, 2008); mitigation strategies to decrease the risk of childhood leukemia is thus a goal of these efforts.