Disorders of Peripheral Nerves
Bashar Katirji
Clinical Approach to Disorders of Peripheral Nerves
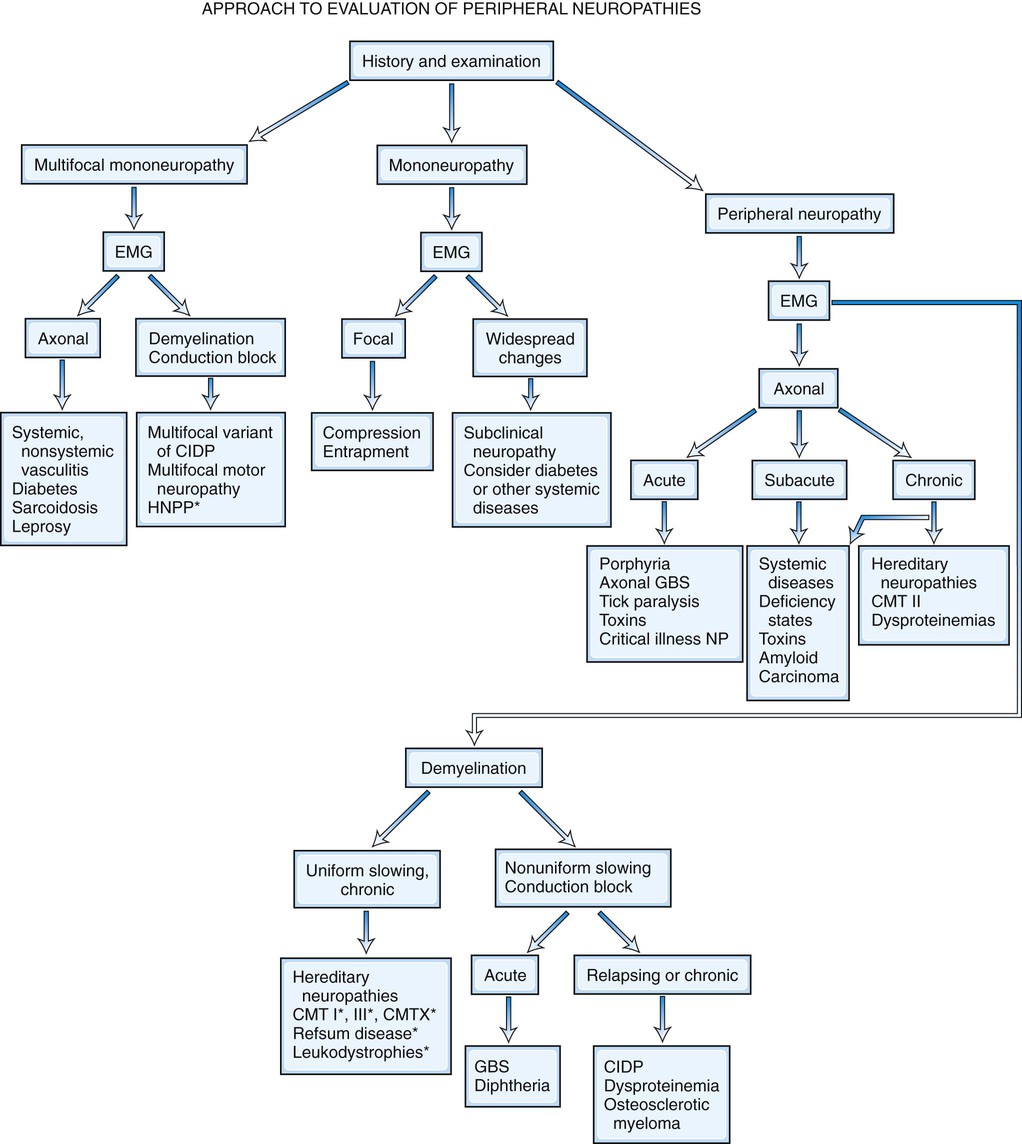
Peripheral nerve disorders are common neurological problems caused by dysfunction of peripheral motor, sensory, or autonomic nerves. The causes of neuropathies are disparate and their clinical presentations highly variable. The main causes of neuropathy are entrapment, systemic diseases, inflammatory and autoimmune disorders, inherited disorders, ischemic settings, paraneoplastic conditions, deficiency states, infections, and toxins. The “shotgun” approach of ordering several panels of diagnostic tests without an adequate understanding of their significance and usefulness should be avoided. A logical systematic diagnostic approach to peripheral neuropathies consists of a detailed history, comprehensive physical and neurological examinations, detailed electrodiagnostic (EDX) studies, and possibly additional ancillary testing (such as autonomic testing, skin biopsy and nerve biopsy). This approach confirms the presence of a peripheral nerve disorder; characterizes the fiber type, pattern, time course, and type of deficit of the peripheral nerve disease; and shortens the list of diagnostic and etiological possibilities. Further laboratory or pathological studies to determine a specific diagnosis are sometimes performed based on the outcome of the initial evaluation.
Structure of Peripheral Nerves
The peripheral nerve is a cable-like structure containing bundles of both unmyelinated and myelinated fibers and their supporting elements. The unmyelinated axons are surrounded only by the plasma membrane of a Schwann cell. The myelinated axons are engulfed by a Schwann cell that wraps around the axons multiple times, thereby insulating the axon with multiple layers of lipid-rich cell membrane. The myelinated axon is surrounded completely by myelin and Schwann cells except at regular gaps called the nodes of Ranvier, which measure approximately 1 µm in adults (see Fig. 64.2 in Chapter 64, Peripheral Nerve Trauma). The propagation of action potentials from one node of Ranvier to the next (saltatory conduction) is maintained by a thick myelin sheath with low capacitance and high resistance to electric current and by a high concentration of voltage-gated sodium channels at the nodes of Ranvier.
Pathological Processes Involving Peripheral Nerves
Despite the large number of causes for neuropathy, the pathological reactions of peripheral nerves to various insults remain limited. In general, these pathological processes are divided into four main categories: (1) wallerian degeneration, which is the response to axonal interruption, (2) axonal degeneration or axonopathy, (3) primary neuronal (perikaryal) degeneration or neuronopathy, and (4) segmental demyelination or myelinopathy. The patient's symptoms, the type and pattern of distribution of signs, and the characteristics of nerve conduction study abnormalities provide information about the underlying pathological changes.
Compression, traction, laceration, thermal, chemical or ischemic nerve injury that causes interruption of axons leads to wallerian degeneration—that is, distal degeneration of axons and their myelin sheaths. Immediately following injury, motor weakness and sensory loss occur in the distribution of the damaged nerve. On needle electromyography (EMG), there is complete loss of voluntary activity (with a complete lesion) or a decrease in motor unit action potential (MUAP) recruitment (with a partial lesion). However, the axons remain excitable distally, since distal conduction failure is not completed until 10 to 11 days later as the distal nerve trunk becomes progressively unexcitable. On nerve conduction studies, the amplitude of the compound muscle action potential (CMAP), evoked by stimulation distal to the lesion site, begins to decline by the second day after injury and reaches its nadir by the fifth to sixth. For sensory axons, the loss of sensory nerve action potential (SNAP) is delayed by another 2 to 3 days; distal SNAP remains normal for 5 to 6 days and then decreases rapidly to reach its nadir by 10 to 11 days after injury (see Fig. 35.9 in Chapter 35). The temporal sequence of wallerian degeneration is length dependent, occurring earlier in shorter than in longer distal nerve stumps. Denervation potentials (fibrillation potentials) are typically seen on needle EMG in some affected muscles (mostly proximal ones) 10 to 14 days after injury and become full after 3 weeks from acute nerve injury. Axonal interruption initiates secondary morphological changes of the nerve cell body, termed chromatolysis, and the proximal axonal caliber becomes smaller. Regeneration from the proximal stump begins as early as 24 hours following transection but proceeds slowly at a maximal rate of 2 to 3 mm/day and is often incomplete. Sprouting of intact axons starts also locally in partial lesions, becoming noticeable on needle EMG after 1 month of axonal injury. The quality of recovery depends on the degree of preservation of the Schwann cell/basal lamina tube and the nerve sheath and surrounding tissue, the distance of the site of injury from the cell body, and the patient's age.
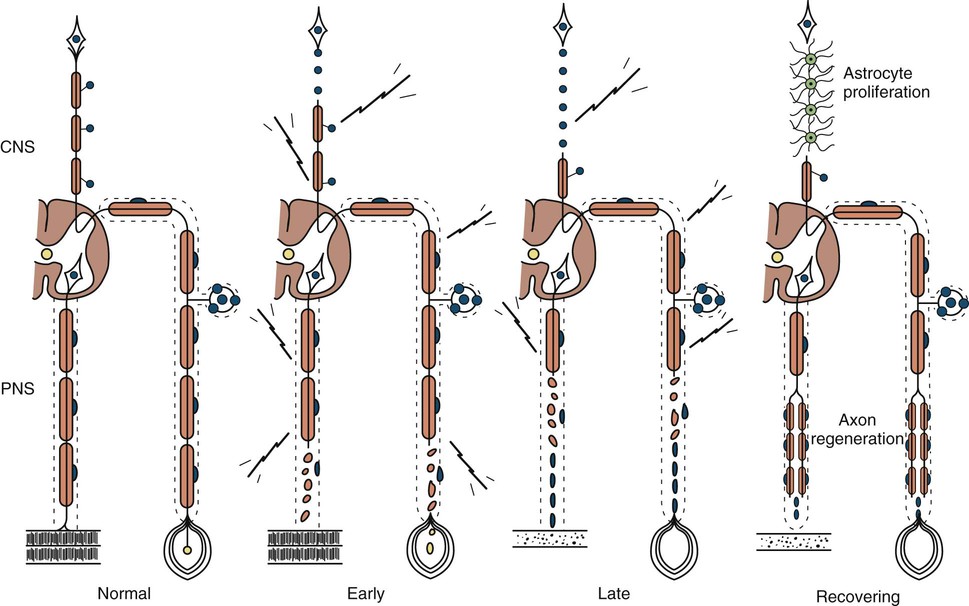
Axonal degeneration (or axonopathy), the most common pathological reaction of peripheral nerve, signifies distal axonal breakdown and is presumably caused by metabolic derangement within neurons or vascular compromise leading to ischemia. Systemic metabolic disorders, toxin exposure, vasculitis, and some inherited neuropathies are the usual causes of axonal degeneration. The myelin sheath breaks down concomitantly with the axon in a process that starts at the most distal part of the nerve fiber and progresses toward the nerve cell body, hence the term dying-back or length-dependent polyneuropathy (Fig. 107.1). A similar sequence of events may occur simultaneously in centrally directed sensory axons, resulting in distal degeneration of rostral dorsal column fibers. The selective length-dependent vulnerability of distal axons could result from failure of the perikaryon to synthesize enzymes or structural proteins, from alterations in axonal transport, or from regional disturbances of energy metabolism. In some axonopathies, alterations in axon caliber, either axonal atrophy or axonal swelling, may precede distal axonal degeneration. Clinically, dying-back polyneuropathy presents with symmetrical distal loss of sensory and motor function in the lower extremities that extends proximally in a graded manner. The result is sensory loss in a stocking-like pattern, distal muscle weakness and atrophy, and loss of distal limb myotatic reflexes. As the polyneuropathy ascends, it affects the hands and distal upper extremities giving a glove-like sensory loss (hence the term stocking and glove sensory loss), and hand weakness and atrophy. Axonopathies result in low-amplitude SNAPs and CMAPs, but they affect distal latencies and conduction velocities only slightly. Needle EMG of distal muscles shows acute and/or chronic of denervation changes (see Chapter 35). Because axonal regeneration proceeds at a maximal rate of 2 to 3 mm/day, recovery may be delayed and is often incomplete.
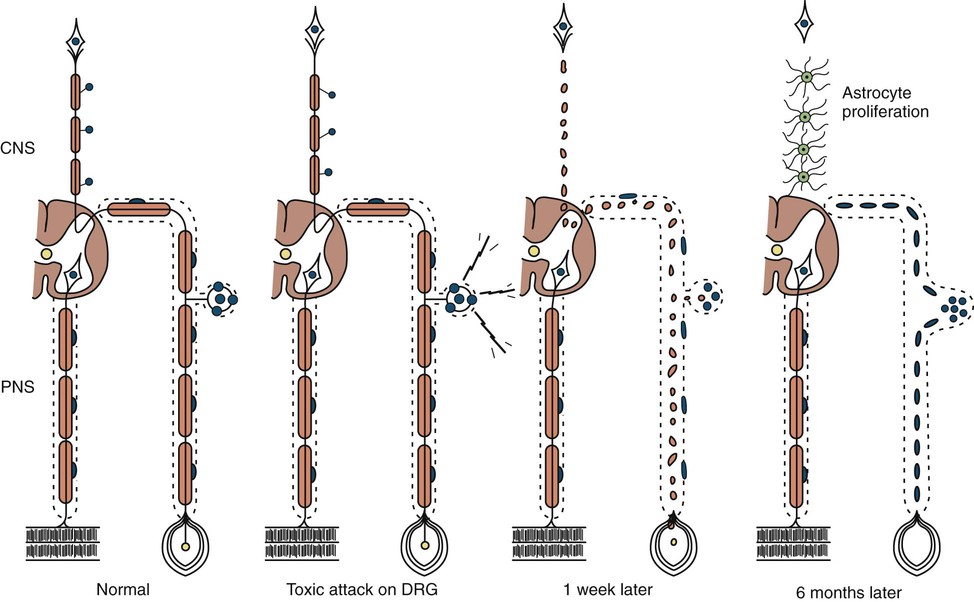
Neuronopathy designates loss of nerve cell bodies with resultant degeneration of their entire peripheral and central axons. Either anterior horn or dorsal root ganglion cells may be affected. Focal weakness without sensory loss occurs when anterior horn cells are affected, as in anterior poliomyelitis or motor neuron disease. Sensory neuronopathy, or dorsal polyganglionopathy, means damage to dorsal root ganglion neurons that results in sensory ataxia, sensory loss, and diffuse areflexia (Fig. 107.2). A number of toxins, such as organic mercury compounds, doxorubicin, and high-dose pyridoxine, or deficiency states, such vitamin E deficiency, produce primary sensory neuronal degeneration. Immune-mediated inflammatory damage of dorsal root ganglion neurons occurs in paraneoplastic sensory neuronopathy (anti-HU syndrome) and Sjögren syndrome (Hlubocky and Smith, 2014). It is often difficult to distinguish between neuronopathies and axonopathies on clinical grounds alone. Once the pathological processes are no longer active, sensory deficits become fixed, and little or no recovery takes place.
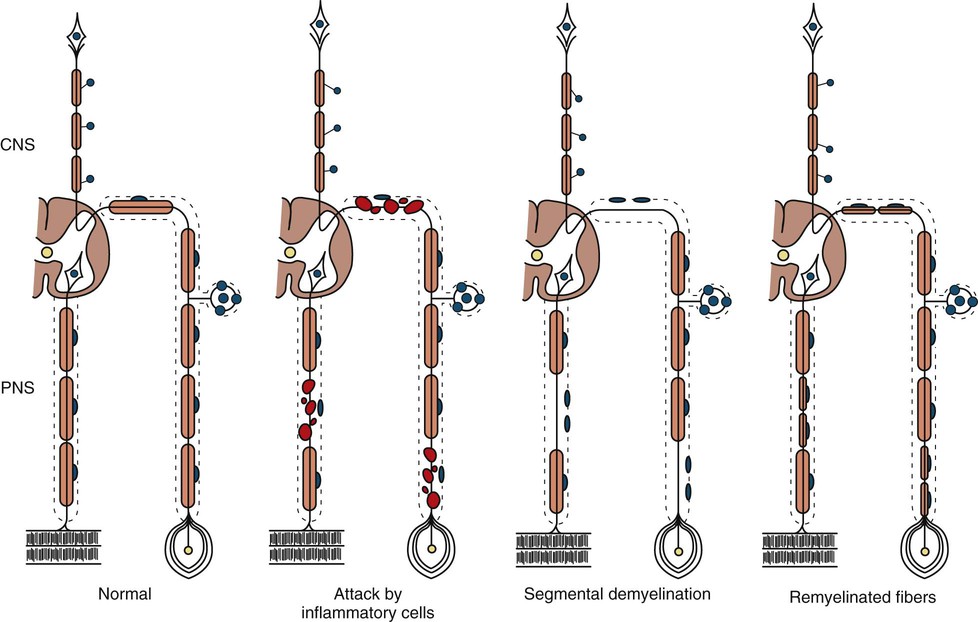
The term segmental demyelination (or myelinopathy) implies injury of either myelin sheaths or Schwann cells, resulting in breakdown of myelin with sparing of axons (Fig. 107.3). This occurs mechanically by acute nerve compression or chronic nerve entrapment and in immune-mediated demyelinating neuropathies and hereditary disorders of Schwann cell/myelin metabolism. Primary myelin damage may be produced experimentally by myelinotoxic agents such as diphtheria toxin or by acute nerve compression. Remyelination of demyelinated segments usually occurs within weeks. The newly formed remyelinated segments have thinner-than-normal myelin sheaths and internodes of shortened length. Repeated episodes of demyelination and remyelination produce proliferation of multiple layers of Schwann cells around the axon, termed an onion bulb. The physiological consequence of acquired demyelination, such as in inflammatory or compressive demyelination but not hereditary myelinopathies, is conduction block, which results in loss of the ability of the nerve action potential to reach the muscle, thereby producing weakness. Because the axon remains intact, there is little muscle atrophy. Relative sparing of temperature and pinprick sensation in many demyelinating polyneuropathies reflects preserved function of unmyelinated and small-diameter myelinated fibers. Early generalized loss of reflexes, disproportionately mild muscle atrophy in the presence of proximal and distal weakness, neuropathic tremor, and palpably enlarged nerves are all clinical clues that suggest demyelinating polyneuropathy. Nerve conduction studies or analysis of single teased nerve fiber preparations stained with osmium can confirm demyelination. Demyelination is present if motor and sensory nerve conduction velocities (NCVs) are reduced to less than 70% of the lower limits of normal, with relative preservation of CMAP and SNAP amplitudes. The presence of partial motor conduction block, temporal dispersion of CMAPs, and marked prolongation of distal motor and F-wave latencies are all features consistent with acquired demyelination (see Chapter 35). Recovery depends on the extent of remyelination, and therefore clinical improvement may occur within weeks. In many demyelinating neuropathies, axonal degeneration may also coexist, as evidenced by some distal limb atrophy and active deneravtion and reinnervation changes on needle EMG.
Classification of Peripheral Nerve Disorders
There are several patterns of peripheral nerve disease (Box 107.1). Brachial, lumbar, and sacral plexopathy are discussed in Chapter 106, and radiculopathies are discussed in Chapter 98.
A mononeuropathy means focal involvement of a single nerve and implies a local process. Direct trauma, compression, entrapment, vascular lesions, and neoplastic compression or infiltration are the most common causes. Electrophysiological studies provide a more precise localization of the lesion than may be possible by clinical examination, distinguish axonal loss from focal segmental demyelination, and sometimes may reveal a more widespread change indicating an underlying generalized polyneuropathy that has made the nerve susceptible to entrapment, as occurs in diabetes mellitus, hypothyroidism, acromegaly, alcoholism, and hereditary neuropathy with liability to pressure palsy (HNPP).
Multiple mononeuropathies, or mononeuropathy multiplex, signify simultaneous or sequential damage to multiple noncontiguous nerves. Confluent multiple mononeuropathies may give rise to motor weakness with sensory loss that can simulate a length-dependent peripheral polyneuropathy.
Polyneuropathy is most commonly characterized by symmetrical distal motor and/or sensory deficits that typically have a graded increase in severity distally and distal attenuation of reflexes. The sensory and motor deficits generally follow a length-dependent stocking-glove pattern. Most polyneuropathies are fairly symmetrical, but some are asymmetrical and sometimes the result of a confluent mononeuropathy multiplex. A small number of polyneuropathies (e.g., that associated with acute intermittent porphyria [AIP]) may be predominantly proximal. It is helpful to determine the relative extent of sensory, motor, and autonomic neuron involvement, although most polyneuropathies produce mixed sensorimotor deficits and some degree of autonomic dysfunction.
Diagnostic Clues from the History
The symptoms of peripheral nerve disorders are due to motor, sensory, or autonomic disturbances. The inquiry should seek both negative and positive symptoms. Negative motor symptoms are weakness, atrophy, and walking difficulties. Muscle cramps, fasciculations, myokymia, or tremor are positive motor manifestations. In polyneuropathies, negative motor symptoms include early distal toe and ankle extensor weakness, resulting in tripping on rugs or uneven ground. However, a complaint of difficulty walking in itself does not distinguish muscle weakness from sensory, pyramidal, extrapyramidal, or cerebellar disturbance. If the fingers are weak, patients may complain of difficulty opening jars or turning a key in a lock.
Positive sensory symptoms include prickling, searing, burning, and tight band-like sensations. Paresthesias are unpleasant sensations arising spontaneously without apparent stimulus. The presence of spontaneously reported paresthesias is helpful in distinguishing acquired (>60% of patients) from inherited (<20% of patients) polyneuropathies. Allodynia refers to the perception of nonpainful stimuli as painful, and hyperalgesia is painful hypersensitivity to noxious stimuli. Neuropathic pain, the extreme example of a positive symptom, is a cardinal feature of many neuropathies. Neuropathic pain often has a deep, burning, or drawing character that may be associated with jabbing or shooting pains that typically increase at night or during periods of rest. Negative sensory manifestations include loss or reduction of pain, temperature or touch sensation. Imbalance and gait disturbance are common negative sensory symptoms of polyneuropathy, implying loss of proprioception. However, the negative sensory symptoms may be caused by a central disorder including dorsal column dysfunction such as with vitamin B12 deficiency.
Symptoms of autonomic dysfunction are helpful in directing attention toward specific neuropathies that have prominent autonomic symptoms. It is important to ask about orthostatic intolerance (lightheadedness, presyncopal symptoms or syncope), reduced or excessive sweating, heat intolerance, and bladder, bowel, and sexual dysfunctions. Anorexia, early satiety, nausea, and vomiting are symptoms suggestive of gastroparesis. The degree of autonomic involvement can be documented by noninvasive autonomic function studies (see Chapter 108).
Historical information regarding onset, duration, and evolution of symptoms provides important clues to diagnosis. Knowledge about the time course of disease (acute, subacute, or chronic) and the course (monophasic, progressive, or relapsing) narrows diagnostic possibilities. Guillain–Barré syndrome (GBS), acute porphyria, vasculitis, neuralgic amyotrophy and some forms of toxic neuropathies have acute presentations. A relapsing course is found in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), acute porphyria, Refsum disease, hereditary neuropathy with liability to pressure palsies (HNPP), familial brachial plexus neuropathy, and repeated episodes of toxin exposure.
In patients with a chronic indolent course over many years, inquiries about similar symptoms and bony deformities (such as pes cavus) in immediate relatives often point to a familial polyneuropathy. Inherited polyneuropathies are a major cause of undiagnosed polyneuropathies, accounting for about 30% of patients referred to tertiary centers for diagnosis. Molecular genetic testing or the clinical and electrophysiological evaluation of relatives of patients with undiagnosed neuropathy may corroborate that the disorder is familial. The presence of constitutional symptoms such as weight loss, malaise, and anorexia suggests an underlying systemic disorder as a cause of the polyneuropathy. Inquiry should be made about preceding or concurrent associated medical conditions (diabetes mellitus, hypothyroidism, chronic renal failure, liver disease, intestinal malabsorption, malignancy, connective tissue diseases, human immunodeficiency virus [HIV] seropositivity); drug use, including over-the-counter vitamin preparations (vitamin B6); alcohol and dietary habits; and exposure to solvents, pesticides, or heavy metals.
Diagnostic Clues from the Examination
The first step in the examination of patients with neuropathy is to determine the anatomical pattern and localization of the disease process and whether motor, sensory, or autonomic nerves are involved.
In mononeuropathy, the neurological deficit follows the distribution of a single nerve. For example, in a patient with foot drop due to a common fibular (peroneal) nerve lesion, the neurological examination reveals weakness of ankle and toe dorsiflexion and ankle eversion, but ankle inversion, toe flexion, and plantar flexion are normal, since muscles controlling these functions are innervated by the tibial nerve. Similarly, sensory loss is restricted to the lower two-thirds of the lateral leg and dorsum of the foot, but sensation on the sole of the foot is normal.
In multiple mononeuropathies (mononeuropathy multiplex), the neurological findings should point to simultaneous or sequential damage to two or more noncontiguous peripheral nerves. Confluent multiple mononeuropathies, such as with involvement of the fibular and tibial nerves or median and ulnar nerves, may give rise to motor weakness with sensory loss that can simulate a length-dependent peripheral polyneuropathy. EDX studies ascertain whether the primary pathological process is axonal degeneration or segmental demyelination (Box 107.2). Approximately two-thirds of patients with multiple mononeuropathies display a picture of axonal damage. Ischemia caused by systemic or nonsystemic vasculitis or microangiopathy in diabetes mellitus should be considered. Other less common causes are disorders affecting interstitial structures of nerve, namely infectious, granulomatous, leukemic, or neoplastic infiltration, including Hansen disease (leprosy) and sarcoidosis. In the event focal demyelination or motor conduction block leads to multiple mononeuropathies, multifocal acquired demyelinating sensory and motor neuropathy (Lewis-Sumner syndrome), multifocal motor neuropathy, or HNPP should be considered.
In polyneuropathy, the sensory deficits generally follow a length-dependent stocking-glove pattern. By the time sensory disturbances of the longest nerves in the body (lower limbs) have reached the level of the knees, paresthesias are usually noted in the distribution of the second-longest nerves (i.e., those in the upper limbs) at the tips of the fingers. When sensory impairment reaches the midthigh, involvement of the third-longest nerves, the anterior intercostal and lumbar segmental nerves, gives rise to a tent-shaped area of hypoesthesia on the anterior chest and abdomen. Involvement of the recurrent laryngeal nerves may occur at this stage, with hoarseness. Motor weakness follows a dying-back pattern and usually is greater in extensor foot muscles than in corresponding flexors. For example, heel walking is affected earlier than toe walking in most polyneuropathies. It is helpful to determine the relative extent of sensory, motor, and autonomic fiber involvement, although most polyneuropathies produce mixed sensorimotor deficits and some degree of autonomic dysfunction.
Motor deficits tend to dominate the clinical picture in acute and chronic inflammatory demyelinating polyneuropathies, hereditary motor and sensory neuropathies, and in neuropathies associated with osteosclerotic myeloma, porphyria, lead toxicity, organophosphate intoxication, and hypoglycemia (Box 107.3). The distribution of weakness provides important information. Asymmetrical weakness without sensory loss suggests a motor neuronopathy such as motor neuron disease or multifocal motor neuropathy. The facial nerve can be affected in several peripheral nerve disorders (Box 107.4). In most polyneuropathies, the legs are more severely affected than the arms, with several notable exceptions (Box 107.5). Polyradiculoneuropathies cause both proximal and distal muscle weakness. For example, proximal and distal weakness is encountered in acute and chronic inflammatory demyelinating polyradiculoneuropathies, osteosclerotic myeloma, porphyria, and diabetic lumbar radiculoplexopathy. Nerve root involvement is confirmed by denervation in paraspinal muscles on needle EMG.
Autonomic dysfunction of clinical importance is seen in association with specific acute (e.g., GBS) or chronic (e.g., amyloid and diabetic) sensorimotor polyneuropathies. Rarely, an autonomic neuropathy may be the exclusive manifestation of a peripheral nerve disorder, without somatic nerve involvement (Box 107.6).
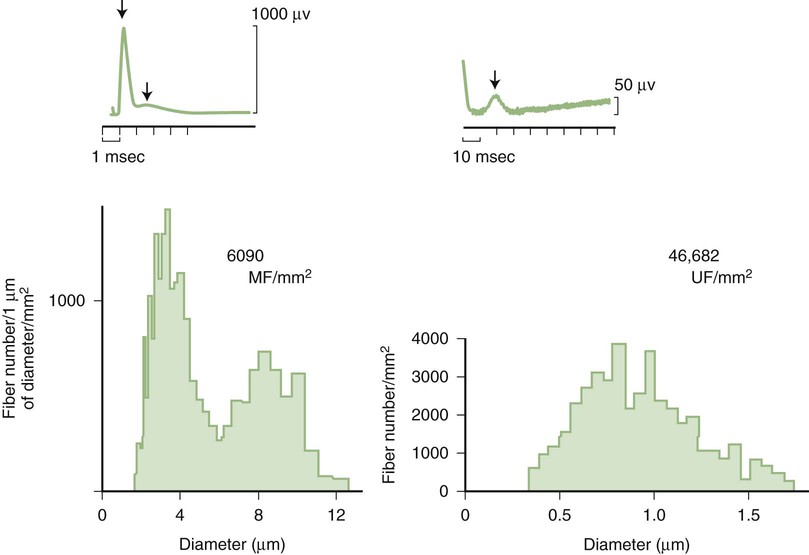
Predominant sensory involvement may be a feature of polyneuropathies caused by diabetes, carcinoma, Sjögren syndrome, dysproteinemia, acquired immunodeficiency syndrome (AIDS), vitamin B12 deficiency, celiac disease, inherited and idiopathic sensory neuropathies, and intoxications with cisplatin, thalidomide, or pyridoxine. Loss of sensation in peripheral neuropathies often involves all sensory modalities. However, the impairment may be restricted to selective sensory modalities in many situations, which makes it possible to correlate the type of sensory loss with the diameter size of affected afferent fibers (Fig. 107.4). Pain and temperature sensation are mediated by unmyelinated and small myelinated Aδ fibers, whereas vibratory sense, proprioception, and the afferent limb of the tendon reflex are subserved by large myelinated Aα and Aβ fibers. Light touch is mediated by both large and small myelinated fibers. In polyneuropathies preferentially affecting small fibers, diminished pain and temperature sensation predominate, along with spontaneous burning pain, painful dysesthesias, and autonomic dysfunction. There is preservation of tendon reflexes, balance, and motor strength, and hence few abnormal objective neurological signs are found on examination. A pattern of sensory loss that is very characteristic is distal loss of pinprick sensation, above which is a band of hyperalgesia (exaggerated pain from noxious stimuli), with normal sensation above this level. Relatively few disorders cause selective small-fiber neuropathies (Mendell and Sahenk, 2003) (Box 107.7). Selective large-fiber sensory loss is characterized by areflexia, sensory ataxia, and loss of joint position and vibration sense. Loss of joint position may also manifest as pseudoathetosis (involuntary sinuous movements of fingers and hands when the arms are outstretched and the eyes are closed) and/or a Romberg sign (disproportionate loss of balance with eyes closed compared with eyes open). Striking sensory ataxia, together with pseudoathetosis or asymmetrical truncal or facial sensory loss, directs attention to a primary disorder of sensory neurons or polyganglionopathies. The differential diagnosis of ataxic sensory neuropathies is limited (Box 107.8).
Palpation of peripheral nerves is an important though unreliable part of the examination. Hypertrophy of a single nerve trunk suggests either a neoplastic process (e.g., neurofibroma, schwannoma, malignant nerve sheath tumor) or localized perineurial hypertrophic neuropathy. Generalized or multifocal nerve hypertrophy is found in a limited number of peripheral nerve disorders including leprosy, neurofibromatosis, Charcot–Marie–Tooth (CMT) disease types 1 and 3, acromegaly, Refsum disease, and rarely CIDP.
Certain telltale signs of the skin and its appendages may direct the experienced examiner to a specific diagnosis (Table 107.1): alopecia is seen in thallium poisoning; tightly curled hair in giant axonal neuropathy; white transverse nail bands termed Mees lines in arsenic or thallium intoxications; purpuric skin eruptions of the legs in cryoglobulinemia and some vasculitides; skin hyperpigmentation or hypertrichosis in POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes); telangiectasias over the abdomen and buttocks in Fabry disease; enlarged yellow-orange tonsils in Tangier disease; pes cavus and hammer toes in CMT disease; and overriding toes and ichthyosis in Refsum disease.
TABLE 107.1
Neuropathies with Skin, Nail, or Hair Manifestations
| Disease | Skin, nail, or hair manifestations |
| Vasculitis | Purpura, livedo reticularis |
| Cryoglobulinemia | Purpura |
| Fabry disease | Angiokeratomas |
| Leprosy | Skin hypopigmentation |
| Osteosclerotic myeloma (POEMS syndrome) | Skin hyperpigmentation |
| Variegate porphyria | Bullous lesions |
| Refsum disease | Ichthyosis |
| Arsenic or thallium intoxication | Mees lines |
| Thallium poisoning | Alopecia |
| Giant axonal neuropathy | Curled hair |
Electrodiagnostic Studies
It is helpful to follow a decision-making pathway based initially on the overall pattern of distribution of deficits, followed by the electrophysiological findings, and finally the clinical course (Fig. 107.5). EDX studies, carefully performed and directed to the particular clinical situation, play a key role in the evaluation by (1) confirming the presence of neuropathy, (2) precisely locating focal nerve lesions, and (3) giving information as to the nature of the underlying nerve pathology (Gooch and Weimer, 2007; Wilbourn, 2002; Shapiro et al., 2014) (see Chapter 35).
Because routine sensory nerve conduction studies assess only large myelinated fibers, such studies may be entirely normal in selective small fiber neuropathies. Quantitative sensory testing assessing cold and heat-pain thresholds, tests of sudomotor function, and skin biopsy with analysis of intraepidermal nerve fiber density may be helpful in confirming the unmyelinated nerve fiber abnormalities (Devigili et al., 2008). Since sweating mediated by unmyelinated sympathetic cholinergic fibers is often impaired, the quantitative sudomotor axon reflex (QSART) that evaluates sweating is a highly specific and sensitive method (sensitivity of 80%) to confirm small nerve fiber damage. Quantitative sensory testing assessing both vibratory and thermal detection thresholds has become a useful addition to the bedside sensory examination in controlled clinical trials. Its use in routine clinical practice remains limited because the test is still subjective in that it requires patient cooperation and is time consuming.
Nerve and Skin Biopsy
Skin punch or blister biopsies that demonstrate loss of intraepidermal nerve fibers are alternative methods for documenting small fiber neuropathy (Panoutsopoulou et al., 2009). Only unmyelinated intraepidermal networks of nerve fibers can be demonstrated by immunostaining with the panaxonal marker protein gene product 9.5, studied best with the use of confocal microscopy. Age, gender, and site of skin biopsy have a profound effect on epidermal nerve fiber density. The density of intraepidermal nerve fibers is reduced in skin biopsies obtained from patients with idiopathic, HIV-associated, diabetic, and other sensory neuropathies (Kennedy, 2004). Skin punch biopsy is most useful in patients with suspected small fiber neuropathy, when nerve conduction studies, are normal. The diagnosis of small-fiber neuropathy is best accomplished when at least two abnormal results are met, including positive clinical findings, quantitative sensory testing, QSART, and skin biopsy examinations (Devigili et al., 2008). Skin punch biopsy only detects the presence of skin nerve abnormalities and rarely leads to a specific etiological diagnosis. The skin biopsy also does not permit the study of myelinated fibers unless a thicker biopsy including dermis is obtained. Finally, unlike sural nerve biopsy, the interstitial pathological processes of the nerves cannot be studied.
Nerve biopsy (other than for the diagnosis of vasculitis and neoplasia) should be performed only in centers with established experience with the surgical procedure, handling of nerve specimens, and pathological technique; otherwise little useful information is likely to be obtained. The sural nerve is selected most commonly for biopsy, because the resultant sensory deficit is restricted to a small area over the heel and dorsolateral aspect of the foot, and because its morphology has been well characterized in health and disease. The superficial fibular nerve is an alternative lower-extremity cutaneous nerve suitable for biopsy and has the advantage of allowing simultaneous biopsy of the peroneus brevis muscle through the same incision. This combined distal nerve and muscle biopsy procedure increases the yield of identifying suspected vasculitis (Collins et al., 2000; Vital et al., 2006). In contrast, adding a proximal muscle (e.g., quadriceps) to a cutaneous nerve biopsy (e.g., sural) does not significantly increase the diagnostic yield compared to nerve biopsy alone (Bennett et al., 2008). In patients with proximal involvement of the lower limbs, the intermediate cutaneous nerve of the thigh combined with a muscle biopsy can be performed. When the risk of complication is increased from a biopsy of the lower limbs (e.g., in significant distal leg ischemia, edema) or the neuropathy is preferentially more pronounced in the upper limbs, a cutaneous nerve biopsy of superficial radial or antebrachial nerves may be performed. When the imaging studies indicate a plexus or nerve root pathological process (e.g., inflammatory, infiltrative), a fascicular biopsy of the affected nerve by an expert surgeon may provide invaluable information. Nerve biopsy has proved to be particularly informative when techniques such as single teased fiber preparations, semi-thin sections, ultrastructural studies, and morphometry are applied to quantitate the nerve fiber pathology. Nowadays, relatively few disorders remain in which a nerve biopsy is essential for diagnosis (Pleasure, 2007; Said, 2002) (Box 107.9). In general, nerve biopsy is most frequently diagnostic in suspected vasculitis, amyloid neuropathy, and leprosy. It is helpful in the recognition of CIDP, inherited disorders of myelin, and some rare axonopathies in which distinctive axonal changes occur, such as in giant axonal neuropathy and polyglucosan body disease. The availability of molecular genetic tests for several CMT neuropathies, HNPP, and familial transthyretin amyloidosis has decreased the necessity for nerve biopsy in these conditions.
Nerve biopsy is an invasive procedure and is associated with as high as 15% complication rate—particularly minor wound infections, wound dehiscence, and stump neuromas. Approximately one-third of patients (particularly those without much sensory loss initially) report unpleasant sensory symptoms at the sural nerve biopsy site that are still present 1 year after the biopsy (Gabriel et al., 2000). The area of the original sensory deficit declines by 90% after 18 months because of collateral reinnervation (Theriault et al., 1998). The complications may be greater if substantial foot ischemia is present or if the patient smokes cigarettes.
Other Laboratory Tests
The clinical neuropathic patterns and the results of EDX studies guide the experienced clinician to select the most appropriate laboratory tests. A few laboratory tests should be obtained routinely in all patients with peripheral polyneuropathy. These include complete blood cell count (CBC), sedimentation rate (with or without C-reactive protein), renal functions, fasting blood sugar, thyroid studies, vitamin B12 level, and serum protein electrophoresis with immunofixation electrophoresis. It is important to screen for monoclonal proteins in all patients with chronic undiagnosed polyneuropathy, particularly those older than 60 years, because 10% of such patients have a monoclonal gammopathy. Cerebrospinal fluid (CSF) examination is helpful in the evaluation of suspected demyelinating neuropathies and polyradiculopathies related to meningeal carcinomatosis or lymphomatosis.
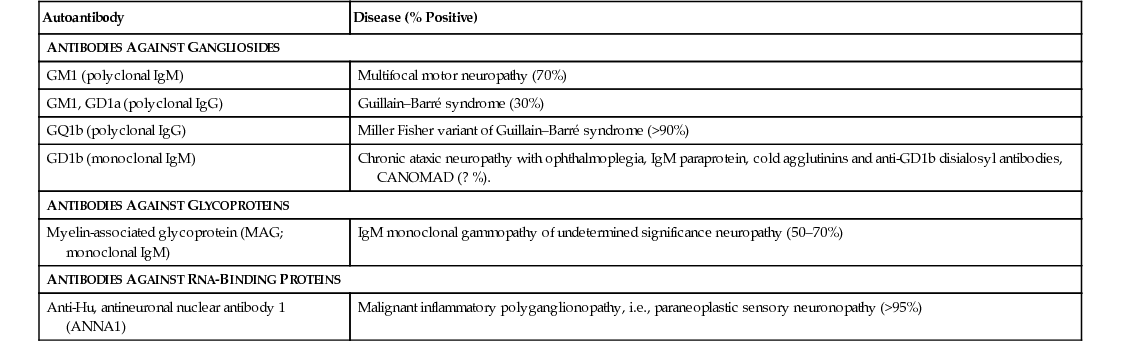
Several serum autoantibodies with reactivity to various components of peripheral nerve have been associated with peripheral neuropathy syndromes, and reference laboratories offer panels of nerve antibodies for sensory, sensorimotor, and motor neuropathies. It must be emphasized that the clinical relevance of most autoantibodies has not been established for patient treatment, and their use is not cost-effective (Vernino and Wolfe, 2007). Those of greatest clinical utility are listed in Table 107.2 (Kissel, 1998). An ever-increasing number of molecular genetic tests for inherited neuropathies is available at reference laboratories (see Hereditary Neuropathies, later).
TABLE 107.2
Neuropathies Associated with Serum Autoantibodies
| Autoantibody | Disease (% Positive) |
| ANTIBODIES AGAINST GANGLIOSIDES | |
| GM1 (polyclonal IgM) | Multifocal motor neuropathy (70%) |
| GM1, GD1a (polyclonal IgG) | Guillain–Barré syndrome (30%) |
| GQ1b (polyclonal IgG) | Miller Fisher variant of Guillain–Barré syndrome (>90%) |
| GD1b (monoclonal IgM) | Chronic ataxic neuropathy with ophthalmoplegia, IgM paraprotein, cold agglutinins and anti-GD1b disialosyl antibodies, CANOMAD (? %). |
| ANTIBODIES AGAINST GLYCOPROTEINS | |
| Myelin-associated glycoprotein (MAG; monoclonal IgM) | IgM monoclonal gammopathy of undetermined significance neuropathy (50–70%) |
| ANTIBODIES AGAINST RNA-BINDING PROTEINS | |
| Anti-Hu, antineuronal nuclear antibody 1 (ANNA1) | Malignant inflammatory polyganglionopathy, i.e., paraneoplastic sensory neuronopathy (>95%) |
In patients with initially undiagnosed peripheral neuropathy referred to specialized centers, a definite diagnosis can be made in more than 75% of cases. Inherited neuropathies, CIDP, and neuropathies associated with other systemic diseases accounted for most diagnoses. The improved diagnostic rate resulted in large measure from detailed clinical, EDX and laboratory evaluations and study of relatives of patients with undiagnosed neuropathy.
Mononeuropathies
Definition and Classification of Mononeuropathies
Mononeuropathy is defined as a disorder of a single peripheral nerve. This may result from compression, traction, laceration, thermal, or chemical injury. The damage may involve one or more structural components of the peripheral nerve, while the pathophysiological responses to peripheral nerve lesions include axon loss, demyelination, or a combination of both.
Peripheral nerve injuries are classified based on functional status of the nerve and histological findings. Seddon divided peripheral nerve injury into three classes: neurapraxia, axonotmesis, and neurotmesis. This classification remains popular, particularly among surgeons, because of its correlation to outcome (Seddon, 1975). Later, Sunderland (1991) revised the classification into five degrees that have better prognostic implications.
Neurapraxia (First-Degree Nerve Injury)
Neurapraxia, or first-degree nerve injury, usually results from brief or mild compression on the nerve that distorts the myelin, resulting in segmental demyelination but leaving the axons intact. The nerve conducts normally distal to but not across the lesion, resulting in conduction block, which is the electrophysiological correlate of neurapraxia. With this type of injury, recovery is usually complete following remyelination that occurs within 1 to 3 months if the offending cause (such as a compression) is removed.
Axonotmesis
Axonotmesis injury is characterized by axonal damage that results in wallerian degeneration; distal to the injury, the axons and their investing myelin sheath degenerate (wallerian degeneration) and the end-or;gans (muscle fibers and sensory receptors) become denervated. Sunderland divided this type of nerve lesion into three further subtypes based on the disruptions of the supporting structures (endoneurium, perineurium, and epineurium).
Second-Degree Nerve Injury.
The axonal loss is associated with intact endoneurial tubes as well as intact perineurium and epineurium. These lesions have fairly good prognosis, since nerve regeneration between the site of nerve injury and the target organs is well guided by the intact endoneurial tubes.
Third-Degree Nerve Injury.
The axons and endoneurium are damaged while leaving the perineurium and epineurium intact. These lesions have fair prognosis and may require surgical intervention, mostly because of axonal misdirection and formation of neuromas.
Fourth-Degree Nerve Injury.
The axons, endoneurium, and perineurium are disrupted, but the epineurium is intact. These lesions have poor prognosis and often require surgical repair, and requires surgical repair.
Neurotmesis (Fifth-Degree Nerve Injury)
Neurotmesis, or fifth-degree nerve injury, is the most severe type of nerve injury. It involves complete disruption of the nerve and all supporting structures. The nerve is transected, with loss of continuity between its proximal and distal stumps.
Entrapment neuropathy is defined as a mononeuropathy caused by focal compression or mechanical distortion of a nerve within a fibrous or fibro-osseous tunnel or less commonly by other structures such as bone, ligament, other connective tissues, blood vessels, or mass lesions. Compression, constriction, angulation, and stretching are important mechanisms that produce nerve injury at certain vulnerable anatomical sites (Tables 107.3 and 107.4). The term entrapment is a useful one in that it implies that compression occurs at particular sites where surgical intervention is often required to release the entrapped nerve, such as in the case of the median nerve at the wrist in moderate to severe carpal tunnel syndrome. Overuse has been implicated as the cause of entrapment neuropathies in certain occupations, including the playing of musical instruments by professional musicians.
TABLE 107.3
Entrapment/Compressive Neuropathies of Upper Limbs
| Nerve | Site of compression | Predisposing factors | Major clinical features |
| Median | Wrist (carpal tunnel syndrome) | Tenosynovitis, arthritis, repetitive wrist motions | Nocturnal Paresthesia, pain, thenar atrophy |
| Anterior interosseous | Strenuous exercise, trauma, neuralgic amyotrophy | Abnormal pinch sign, normal sensation | |
| Elbow (pronator teres syndrome) | Repetitive elbow motions | Tenderness of pronator teres, no weakness, sensory loss | |
| Ulnar | Elbow (cubital tunnel syndrome) | Elbow leaning, remote trauma (tardy ulnar), trauma | Clawing, Froment's sign, sensory loss of fourth and fifth fingers |
| Guyon canal | Mechanics, cyclists | Interosseous atrophy, normal sensation of dorsum of fourth and little fingers | |
| Radial | Axilla | Crutches | Wrist drop, triceps involved, sensory loss extending into forearm and sometimes arm |
| Spiral groove | Abnormal sleep postures | Wrist drop, Triceps spared, sensory loss of dorsum of hand only | |
| Posterior interosseous | Elbow synovitis, neuralgic amyotrophy | Paresis of finger extensors, radial wrist deviation | |
| Superficial sensory branch (cheiralgia paresthetica) | Wrist bands, hand cuffs | Paresthesias in dorsum of hand | |
| Suprascapular | Suprascapular notch | Blunt trauma, neuralgic amyotrophy | Atrophy of supraspinatus and infraspinatus muscles |
| Axillary | Axilla | Shoulder dislocation or surgery | Weakness of arm abduction |
| Lower trunk of the brachial plexus or T1 roots | Thoracic outlet | Cervical rib, cervical band with enlarged C7 transverse process | Atrophy of intrinsic hand muscles (Mostly thenar), paresthesias of medial hand and forearm |
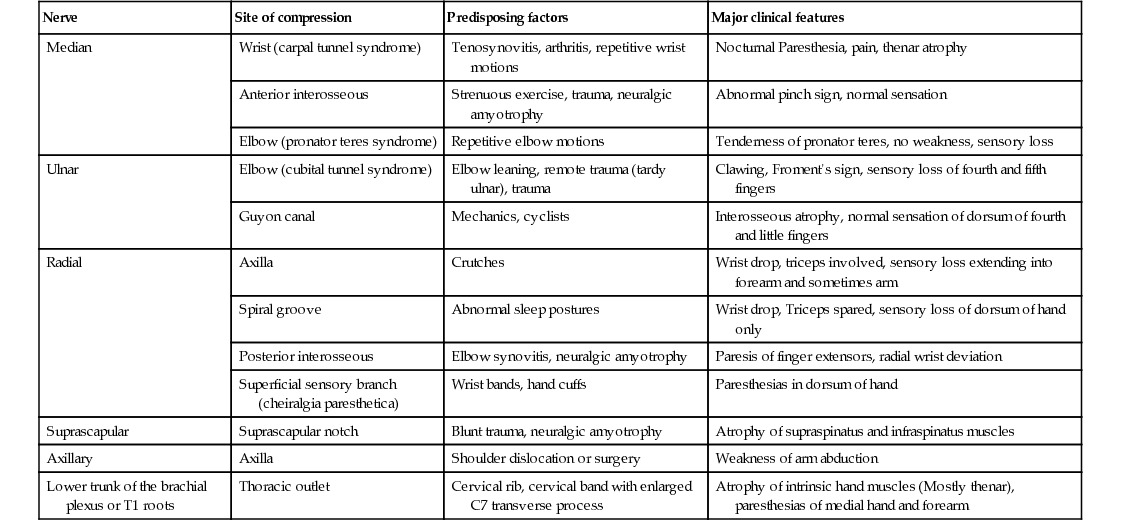
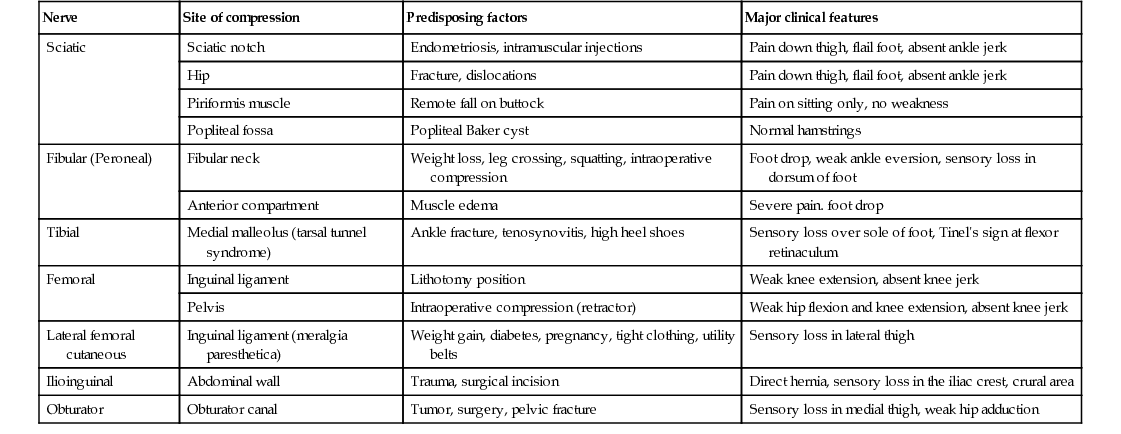
TABLE 107.4
Entrapment/Compressive Neuropathies of Lower Limbs
| Nerve | Site of compression | Predisposing factors | Major clinical features |
| Sciatic | Sciatic notch | Endometriosis, intramuscular injections | Pain down thigh, flail foot, absent ankle jerk |
| Hip | Fracture, dislocations | Pain down thigh, flail foot, absent ankle jerk | |
| Piriformis muscle | Remote fall on buttock | Pain on sitting only, no weakness | |
| Popliteal fossa | Popliteal Baker cyst | Normal hamstrings | |
| Fibular (Peroneal) | Fibular neck | Weight loss, leg crossing, squatting, intraoperative compression | Foot drop, weak ankle eversion, sensory loss in dorsum of foot |
| Anterior compartment | Muscle edema | Severe pain. foot drop | |
| Tibial | Medial malleolus (tarsal tunnel syndrome) | Ankle fracture, tenosynovitis, high heel shoes | Sensory loss over sole of foot, Tinel's sign at flexor retinaculum |
| Femoral | Inguinal ligament | Lithotomy position | Weak knee extension, absent knee jerk |
| Pelvis | Intraoperative compression (retractor) | Weak hip flexion and knee extension, absent knee jerk | |
| Lateral femoral cutaneous | Inguinal ligament (meralgia paresthetica) | Weight gain, diabetes, pregnancy, tight clothing, utility belts | Sensory loss in lateral thigh |
| Ilioinguinal | Abdominal wall | Trauma, surgical incision | Direct hernia, sensory loss in the iliac crest, crural area |
| Obturator | Obturator canal | Tumor, surgery, pelvic fracture | Sensory loss in medial thigh, weak hip adduction |
In chronic entrapment, mechanical distortion of the nerve fibers leads to focal demyelination or, in severe cases, to wallerian degeneration. Morphological studies show a combination of active demyelination, remyelination, wallerian degeneration, and axonal regeneration at the site of entrapment. Endoneurial swelling, collagen proliferation, and thickening of perineurial sheaths accompany the nerve fiber changes. Ischemia is not a significant contributing factor to nerve fiber damage in chronic compression. In contrast, ischemia plays a more significant role in nerve injury associated with acute compression secondary to space-occupying lesions such as hematoma or compartment syndromes.
The characteristic electrophysiological feature of entrapment neuropathy is either short-segment conduction delay (i.e., focal slowing) or conduction block across the site of entrapment (see Chapter 35). In severe cases, wallerian degeneration gives rise to denervation and reinnervation in affected muscles. Nerve conduction studies together with needle EMG are essential for diagnosis and reliable documentation of the site and severity of nerve entrapment. Although plain radiography, computed tomography (CT), ultrasound, and magnetic resonance imaging (MRI) may be of occasional value in identifying rare structural abnormalities, these imaging procedures are not necessary for routine diagnosis.
Mononeuropathies of the Upper Extremities
Entrapment neuropathies of the upper extremities are shown in Table 107.3.
Median Nerve
Applied Anatomy.
The median nerve, formed from contributions of the lateral cord (C6 and C7 fibers) and medial cord (C8 and T1 fibers) of the brachial plexus, runs into the forearm between the two heads of the pronator teres. It then gives off branches to the pronator teres, flexor carpi radialis, flexor digitorum sublimis, and the palmaris longus muscles and the anterior interosseous nerve. The anterior interosseous nerve is the largest branch of the median nerve and is a pure motor nerve. It arises from the median nerve distal to these motor branches in the upper forearm and innervates the flexor pollicis longus, pronator quadratus, and median part of the flexor digitorum profundus muscles of the index and middle fingers. The median nerve then enters the wrist through the carpal tunnel, formed by the carpal bones and the transverse carpal ligament, the latter forming its roof. Before reaching the wrist, the median nerve gives off the palmar cutaneous sensory branch which runs subcutaneously (not through the carpal tunnel) to innervate the skin over the thenar eminence. Distal to the carpal tunnel, the median nerve divides into motor and sensory divisions. The motor division innervates the first and second lumbricals and most muscles of the thenar eminence including the opponens pollicis, abductor pollicis brevis, and superficial head of the flexor pollicis brevis. The sensory fibers of the median nerve innervate the skin of the thumb, index, middle, and lateral half of the ring fingers.
Median Nerve Entrapment at the Wrist (Carpal Tunnel Syndrome).
Carpal tunnel syndrome (CTS) is by far the most common entrapment neuropathy. This entrapment occurs in the tunnel through which the median nerve and long finger flexor tendons pass. Because the transverse carpal ligament is an unyielding fibrous structure forming the roof of the tunnel, tenosynovitis or arthritis in this area often produces pressure on the median nerve. The syndrome is frequently bilateral and usually of greater intensity in the dominant hand.
Symptoms consist of nocturnal pain and paresthesias, most often confined to the thumb, index, and middle fingers, but may be reported to involve the entire hand. Patients complain of tingling numbness and burning sensations, often awakening them from sleep. Referred pain may radiate to the forearm and even as high as the shoulder (Stevens et al., 1999). Symptoms are often provoked after excessive use of the hand or wrist or during ordinary activities such as driving or holding a phone, book, or newspaper, in which the wrist is assumed in either a flexed or extended posture. Objective sensory changes may be found in the distribution of the median nerve, most often impaired two-point discrimination, pinprick and light touch sensation, or occasionally hyperesthesia in the thumb, index, and middle fingers, with sparing of the skin over the thenar eminence. Thenar (abductor pollicis brevis muscle) weakness and atrophy may be present in advanced cases of CTS (Fig. 107.6). Flexing the patient's hand at the wrist for 1 minute (Phalen maneuver) or hyperextension of the wrist (reversed Phalen maneuver) often reproduces the symptoms, is present in about 80% of patients, and is rarely false positive. A positive Tinel sign, in which percussion of the nerve at the carpal tunnel causes paresthesias in the distribution of the distal distribution of the median nerve, is present in approximately 60% of affected patients but is not specific for CTS and may be false positive.

Work-related wrist and hand symptoms (repetitive motion injury) from cumulative trauma in the workplace have received increasing attention by the general public in recent years (Thomsen et al., 2002). Although a proportion of these cases have bona fide CTS, longitudinal natural history data suggest that the majority of industrial workers do not develop symptoms of CTS (Nathan et al., 1998). Symptoms consistent with hand and wrist arthritis in a variety of occupational settings are now recognized as being much more common than CTS (Dillon et al., 2002). CTS appears to occur in work settings that include repetitive forceful grasping or pinching, awkward positions of the hand and wrist, direct pressure over the carpal tunnel, and the use of handheld vibrating tools. Increased risk for the syndrome has been found in meat packers, garment workers, butchers, grocery checkers, electronic assembly workers, musicians, dental hygienists, and housekeepers. The highest reported incidence of work-related CTS, based on the number of carpal tunnel surgeries performed, was 15% among a group of meat packers. Although computer keyboard use has long been thought to be related to developing carpal tunnel symptoms, recent data provide no convincing correlation between intensive keyboard use and the subsequent development of CTS (Papanicolaou et al., 2001; Stevens et al., 2001).
Diseases and conditions that have been found to predispose to the development of CTS include pregnancy, diabetes, obesity, age, rheumatoid arthritis, hypothyroidism, amyloidosis, gout, acromegaly, certain mucopolysaccharidoses, arteriovenous shunts for hemodialysis, old fractures at the wrist, and inflammatory diseases involving tendons or connective tissues at the wrist level (Becker et al., 2002). On rare occasions, CTS may be familial, and some patients with CTS have carpal canals that are significantly narrower than average.
The most commonly performed EDX tests for CTS are the median nerve sensory and motor conduction studies, which exhibit delayed sensory or motor latencies across the wrist in about 70% of patients. However, these studies are not sensitive enough in the diagnosis of CTS and fail to detect up to a third of patients with CTS, particularly those with mild and early symptoms. Recording the median latency at short distances over the course of the median nerve from palm to wrist and/or comparing this latency with the latency for the ulnar or radial nerve at the same distance (internal comparison nerve conduction studies) increase the sensitivity of these nerve conduction studies (Stevens, 1997) (Table 107.5).
TABLE 107.5
Internal Comparison Nerve Conduction Studies in the Evaluation of Carpal Tunnel Syndrome
| Study | Median-ulnar palmar mixed study | Median-ulnar sensory study to ring finger | Median-ulnar motor study to second lumbrical interossei | Median-radial sensory study to thumb |
| Technique | Palm stimulation of median and ulnar nerves, recording at the wrist | Median and ulnar nerves stimulation at the wrist, recording ring fingers | Median and ulnar nerves stimulation at the wrist, recording second lumbrical and second interossei, respectively (second interosseous space) | Median and radial nerves stimulation at the wrist, recording thumb |
| Abnormal values | Median-ulnar peak latency difference ≥0.4 msec | Median-ulnar peak latency difference ≥0.4 msec | Median-ulnar onset latency difference ≥0.5 msec | Median-radial peak latency difference ≥0.4 msec |

Mild CTS must be distinguished from proximal median neuropathies, upper brachial plexopathy, C6 or C7 radiculopathies, and polyneuropathy involving the hands. Occasionally, a transient ischemic attack may mimic the symptoms of CTS.
Ultrasound is increasingly used in the diagnosis of CTS. Thickening of the median nerve, best expressed as an increase in the cross-sectional area of the median nerve at the carpal tunnel inlet (more than 13 mm; normal <10–13 mm), or flattening of the nerve at the level of the hamate are the best diagnostic criteria (Tai et al., 2012). Comparing the sensitivity of ultrasound versus electrodiagnostic studies in the diagnosis of CTS has been difficult since the electrodiagnostic studies have long been considered the gold standard. However, the majority of studies have found that the diagnostic utility of these two modalities are equal (Mondelli et al., 2008). MRI is most useful when a space-occupying lesion, such as ganglion, hemangioma or bony deformity, is suspected in patients with CTS.
In cases with only mild sensory symptoms, treatment with splints in neutral position, nonsteroidal anti-inflammatory drugs (NSAIDs), and local corticosteroid injection often suffice. Withdrawal of provoking factors is also important. Although nonoperative treatments have been advocated (Osterman et al., 2002), a comparison of splinting versus surgery suggested that the latter may have a better long-term outcome than the former (Gerritsen et al., 2002). Use of a range of devices and appliances to protect the hand against CTS, including gel-padded gloves, has shown little if any improvement in objective measures of nerve function. There is conflicting evidence that nonsteroidal anti-inflammatory agents, diuretics, laser and ultrasound are effective. Exercise therapy is not useful (Piazzini et al., 2007). Methylprednisolone injections for CTS significantly relieve symptoms for a few months and reduce the need for surgery, but a significant number of patients will ultimately need surgical treatment (Atroshi et al., 2013). Oral steroids are also effective but are associated with side effects. Severe sensory loss, thenar atrophy and active denervation on needle EMG of thenar muscles suggest the need for surgical carpal tunnel release. Open surgical sectioning of the volar carpal ligament or fiberoptic techniques are often successful, with more than 90% of patients having prompt resolution of pain and paresthesias (Mirza and King, 1996). Improvement in distal latencies may lag behind the relief of symptoms. Comparing with preoperative values, nerve conduction studies demonstrate improvement in those with moderate abnormalities preoperatively, whereas patients with severe or no abnormalities on baseline nerve conduction studies have poorer results (Bland, 2001). A correlation between patients seeking workers' compensation who hire attorneys and poorer operative outcomes has also been reported (Katz et al., 2001a). Older individuals may not improve as much as younger patients (Porter et al., 2002), and factors such as poor mental health, significant alcohol consumption, longer disease duration, and male gender also portend a poorer outcome. Rarely, symptoms persist after operation. Poor surgical results usually are associated with incomplete sectioning of the transverse ligament, surgical damage of the palmar cutaneous branch of the median nerve by an improperly placed skin incision, scarring within the carpal tunnel, or an incorrect preoperative diagnosis. Surgical re-exploration may be required in diagnostically certain cases with poor response to the initial operation (Steyers, 2002).
Median Nerve Compressions at the Elbow
Anterior Interosseous Nerve Syndrome.
Isolated acute involvement of the anterior interosseous nerve is often a partial neuralgic amyotrophy (idiopathic brachial plexus neuropathy, Parsonage-Turner syndrome) (England and Sumner, 1987; Katirji,1986). The majority of these lesions are fascicular lesions of the median nerve in the arm involving the anterior interosseous nerve fascicle selectively (Pham et al., 2014). Fascicular torsion of the anterior interosseous fascicle in the lower arm has also been advocated to be due to the high mobility of the anterior interosseous nerve fascicles during elbow flexion leading to torsion injury or inflammation/edema followed by intraneural adhesions. These cases showed good recovery after interfascicular neurolysis. In chronic lesions, a restricted form of multifocal motor neuropathy with conduction block should be considered and a careful and detailed electrophysiological study may reveal involvement of other nerves. The anterior interosseous nerve may be externally compressed following anterior elbow dislocations or complex elbow fractures, or rarely by fibrous bands attached to the flexor digitorum superficialis muscle, an anomalous muscle such as accessory head of the flexor pollicis longus (Gantzer muscle).
Patients often complain of an acute or subacute onset of pain in the forearm or elbow. The patient is unable to flex the distal phalanges of the thumb and index finger, making it impossible to form a circle with those fingers (pinch or O sign). Sensory and motor nerve conduction studies of the median nerve are usually normal. Needle EMG reveals denervation in muscles innervated by the anterior interosseous nerve, including the flexor pollicis longus, pronator quadratus, and flexor digitorum profundus muscles of the index and middle fingers. Spontaneous recovery usually occurs within 3 to 12 months, and therefore surgery may not be necessary unless penetrating injury, fracture, or progressive deterioration and weakness are detected.
Pronator Teres Syndrome.
In the pronator teres syndrome, the median nerve is compressed in the proximal forearm between the two heads of the pronator teres muscle, a fibrous arcade of the flexor digitorum superficialis muscle, or the lacertus fibrosus (a thick fascial band extending from the biceps tendon to the forearm fascia). This extremely rare and controversial entrapment may develop in individuals engaged in repetitive pronating movements of the forearm. Patients usually experience a vague aching pain in the volar aspect of the elbow and forearm, beginning or worsening during activities involving grasping or pronation or both. There is also an insidious onset of paresthesias and numbness of the palm of the hand, mimicking CTS but without the nocturnal symptoms. Resistance to pronation produces pain in the proximal forearm. The pronator teres may be firm and tender on palpation, and the Tinel sign may be elicited over the median nerve in the region of the elbow. Weakness of median-innervated muscles such as the flexor pollicis longus, pronator quadratus, and abductor pollicis brevis (but not of pronator teres) is rarely demonstrated, in contrast to traumatic cases such as following elbow dislocation, forearm fracture, or intracompartmental hemorrhage. Nerve conduction studies in the median nerve are usually normal and do not show the distal median motor and sensory latencies at the wrist that accompany CTS. Needle EMG is also usually normal, with no definite signs of denervation. Injection of corticosteroids into the pronator teres muscle, NSAIDs, and immobilization of the arm with the elbow flexed to 90 degrees and in mild pronation often provide relief of symptoms. On occasion, surgery is controversial but may be necessary, though patients may gain only partial relief.
Median Nerve Entrapment at the Ligament of Struthers.
An often bilateral supracondylar spur of the humerus is present in approximately 1% of normal individuals. This beadlike bony or cartilaginous projection arises from the anteromedial surface of the humerus, located about 5 cm above the medial epicondyle. A fibrous band, the ligament of Struthers, extends from this spur to the medial epicondyle and may rarely compromise the median nerve and the brachial artery above the elbow. Clinical symptoms resemble the pronator teres syndrome, but sometimes the radial pulse diminishes when the forearm is fully extended in supination because of the concomitant entrapment of the brachial artery. Elbow extension causes aggravation of the pain. On needle EMG, there is usually denervation in the abductor pollicis brevis, flexor pollicis longus, pronator quadratus, and pronator teres. Involvement of the pronator teres muscle theoretically allows differentiation of the ligament of Struthers syndrome from the pronator teres syndrome. Treatment consists of surgical excision of the spur and ligament.
Ulnar Nerve
Applied Anatomy.
The ulnar nerve derives its fibers from the C8 and T1 roots via the lower trunk and medial cord of the brachial plexus. The medial brachial and antebrachial cutaneous sensory nerves originate from the medial cord as well. The ulnar nerve gives no branches in the arm. At the elbow, the nerve becomes superficial and enters the ulnar groove formed between the medial epicondyle and the olecranon process. Normally, the ulnar nerve remains in the groove, but in some individuals or when there is an unusual degree of physiological cubitus valgus, the nerve may be unduly mobile, tending to slip forward (sublux) over the medial epicondyle when the elbow is flexed. In a small number of individuals, a dense fibrotendinous band and/or an accessory epitrochleoanconeus muscle may be present between the medial epicondyle and the olecranon process. Slightly distal to the groove in the proximal forearm, the ulnar nerve travels under the tendinous arch of the two heads of the flexor carpi ulnaris muscle, known as the humeral-ulnar aponeurosis, which forms the entrance of the cubital tunnel. Muscular branches originate to the flexor carpi ulnaris and the flexor digitorum profundus (ulnar part to the little and ring fingers). The ulnar nerve continues under the flexor carpi ulnaris and then exits in the distal forearm between the deep fascia separating the flexor carpi ulnaris and flexor digitorum profundus. Some 5 to 8 cm proximal to the wrist, the dorsal ulnar cutaneous sensory branch exits to innervate skin on the dorsal medial hand and the dorsal little and ring fingers. The palmar cutaneous sensory branch originates at the level of the ulnar styloid to supply sensation to the proximal medial palm. The ulnar nerve then enters the wrist through the Guyon canal, which is formed between the pisiform bone and the hook of the hamate and is covered by the volar carpal ligament and the palmaris brevis muscle. Within the Guyon canal, the ulnar nerve divides into its terminal deep palmar and superficial ulnar branches. Because the palmar cutaneous sensory and dorsal cutaneous sensory branches do not pass through the Guyon canal, the deep palmar branch is purely motor and supplies muscular innervation to the hypothenar muscles, the palmar and dorsal interossei, the third and fourth lumbricals, and two muscles in the thenar eminence, the adductor pollicis and the deep head of the flexor pollicis brevis.
Ulnar Nerve Entrapment at the Elbow.
Ulnar mononeuropathy is the second most common entrapment or compression mononeuropathy, although it is considerably less common than CTS. Compression of the ulnar nerve by a thickened, fibrotic flexor carpi ulnaris aponeurosis (humeral-ulnar aponeurosis) at the entrance of the elbow's cubital tunnel is a common cause of ulnar neuropathy (cubital tunnel syndrome). Patients with a subluxed ulnar nerve are at high risk for compression at the elbow. Also, prolonged and frequent resting of the flexed elbow on a hard surface such as a desk or armchair may result in external pressure to the nerve (ulnar groove syndrome). Occupations involving repeated flexion of the elbow may on occasion cause symptoms of ulnar neuropathy. A flexed elbow position increases both the intraneural and extraneural pressure on the nerve. The nerve at the site of repeated compression is associated with fibrous thickening, when a spindle-shaped swelling can often be felt. Other possible sources of injury of the ulnar nerve at the elbow include direct compression when the patient uses the arms to raise up in bed following surgical operations (Stewart and Shantz, 2003) or after periods of prolonged unconsciousness. The ulnar nerve at the elbow may be acutely injured as a result of fracture or dislocation involving the lower end of the humerus and the elbow joint. Occasionally, however, the nerve becomes chronically compressed years after such an injury, which often has led to cubitus valgus deformity (“tardy ulnar palsy”). The nerve may be damaged by osteophyte outgrowths resulting from arthritis of the elbow joint, by a ganglion or lipoma, by a Charcot elbow, and by the epitrochleoanconeus muscle and/or its dense fibrotendinous band. The ulnar nerve may also be involved in conditions that are known to increase the susceptibility of nerves to compression, such as diabetes mellitus or HNPP. Ulnar neuropathy at the elbow segment may also occur without any apparent cause.
Ulnar nerve lesions at the elbow result in numbness and tingling of the little and ring fingers, with variable degrees of hand weakness. Less commonly, patients present with weakness and wasting with no clear sensory symptoms. There is also variable weakness of the flexor carpi ulnaris and the flexor digitorum profundus of the ring and little fingers (ulnar part). Grip strength is reduced secondary to weakness of the adductor pollicis, flexor pollicis brevis, and palmar and dorsal interosseous muscles. To compensate for adductor pollicis weakness during an attempt to pinch a piece of paper between the thumb and index fingers, the flexor pollicis longus, a median nerve-innervated muscle, becomes involuntarily active and flexes the distal phalanx of the thumb (Froment sign). Weakness of the interossei muscles results in an inability to forcefully extend the interphalangeal joints, as is necessary in finger-flicking movements. Prominent atrophy of hand muscles ensues and is most noticeable at the first dorsal interosseous muscle. Lumbrical weakness leads to clawing of the fourth and fifth fingers and flexion of the proximal and distal interphalangeal joints, with secondary hyperextension of the metacarpophalangeal joints (benediction posture or ulnar clawing). Weakness of the third palmar interosseous muscle results in abduction of the little finger, which may get caught when the patient tries to put the hand in a pocket (Wartenberg sign). In chronic ulnar neuropathies, the weakness and atrophy of small muscles of the hand is always more severe than the weakness and atrophy of the forearm muscles. Sensory loss or hypoesthesia involves the fifth finger, part of the fourth finger, and the hypothenar eminence and includes the dorsum of the hand but does not extend above the wrist level. Pain around the elbow and tenderness of the ulnar nerve with deep palpation is common, but distal hand or finger pain is rare. A Tinel sign at the elbow may be elicited, but this sign as well as provocative tests (flexion compression test, palpating for local ulnar nerve tenderness and nerve thickening) have poor diagnostic values (Beekman et al., 2009).
Ulnar nerve lesions at the elbow should be distinguished from ulnar nerve lesions at the wrist, lower brachial plexus lesions (lower trunk or medial cord), and C8 radiculopathy. Confirmed sensory loss that extends more than 3 cm above the wrist into the medial forearm and arm, the territories of the medial brachial and antebrachial cutaneous nerves, is inconsistent with an ulnar neuropathy at the elbow and suggests a more proximal lesion of the lower plexus or C8 or T1 roots. Similarly, weakness of median and radial innervated C8 muscles such as the flexor pollicis longus or the long finger extensors points to a plexopathy or radiculopathy.
Compared to evaluating other entrapment neuropathies such as CTS, the EDX studies used to confirm and localize ulnar nerve entrapment at the elbow are more challenging. Localizing ulnar neuropathy at the elbow relies upon the demonstration of focal demyelination across the elbow, namely slowed motor conduction velocity (>10 meters per second) or conduction block (localized reduction in CMAP amplitude and area of >20%–30%) or both. Focal slowing or conduction block may be found in the elbow segment in more than 75% of cases (Azrieli et al., 2003). Performing an additional ulnar motor conduction study, recording the first dorsal interosseous muscle in addition to recording the abductor digiti minimi muscle, increases the yield of finding focal slowing or conduction block. In the remaining patients, localization becomes less precise because of predominant axonal loss. To provide the extra nerve length needed during elbow flexion, the ulnar nerve is anatomically redundant in the ulnar groove when the elbow is extended, and this can cause measurement errors. A flexed position of the elbow (70 to 90 degrees) is preferred to the extended position when doing ulnar motor conduction studies to localize an ulnar lesion at the elbow. Short-segment incremental studies (“inching”) by stimulating the ulnar nerve in successive 1-cm increments across the elbow, looking for either an abrupt drop in amplitude or increase in latency, is a useful technique that helps to precisely localize the ulnar nerve lesion (Visser et al.,2005). Electrophysiological tests are helpful in differentiating between an ulnar neuropathy and a C8 nerve root or brachial plexus lesion. Ulnar sparing in ulnar sensory studies points to C8 radiculopathy, and needle EMG of C8 muscles innervated by the median nerve (e.g., abductor pollicis brevis, flexor pollicis longus) and radial nerve (e.g., extensor indicis proprius) helps exclude a C8 root lesion or a lower brachial plexopathy. MRI of the elbow may reveal a space-occupying lesion or anomalous structures impinging on the nerve or demonstrate nerve enlargement and increased signal intensity, even in the absence of localizing electrophysiological abnormalities (Vucic et al., 2006). High-resolution sonography at the elbow is also useful by accurately detecting thickening of the ulnar nerve at the elbow (Beekman et al., 2004).
Conservative treatment should be attempted in patients with mild or intermittent sensory symptoms or in those with symptoms brought on by occupational causes. Avoidance of repetitive elbow flexion and extension or direct pressure on the elbow may alleviate the symptoms. Elbow protectors are helpful in patients with a history of excessive elbow leaning. Conservative treatment should be continued for at least 3 months before surgery is considered. Several surgical approaches to an ulnar nerve lesion at the elbow are possible, each with its proponents and critics. Techniques include simple release of the flexor carpi ulnaris aponeurosis, anterior transposition of the nerve trunk, and resection of the medial epicondyle. The choice of procedure should be tailored to the specific lesion found at surgery and may be assisted by short-segment incremental electrophysiological studies (“inching”). Transposition of the nerve trunk carries a higher rate of complications than ulnar neurolysis (Biggs and Curtis, 2006). Depending on the type of surgery and the severity and duration of neuropathy, response to these procedures will vary. Only about 60% of patients, especially those with symptoms of less than 1 year's duration, benefit from surgery; some experience worsening of symptoms. It appears that those with more thickening of the nerve at the time of diagnosis (as determined by sonography) have a more unfavorable outcome, and those with electrophysiological signs of demyelination across the elbow, specifically significant >50% conduction block, have a more favorable course (Beekman et al., 2004; Dunselman and Visser et al., 2008).
Ulnar Nerve Entrapment at the Wrist.
Distal entrapment of the ulnar nerve at the wrist (Guyon canal) or hand is a relatively uncommon condition. Ulnar nerve entrapment in the Guyon canal occurs much less frequently than at the elbow. Aside from direct trauma and laceration, the most common cause is a ganglion cyst. Other usual causes are chronic or repeated external pressure by hand tools, bicycle handlebars, the handles of canes, or excessive push-ups. Compression also may be caused by degenerative wrist joint changes, rheumatoid arthritis, or distal vascular anomalies.
Ulnar nerve entrapment at the wrist may present with a confusing array of sensory and motor symptoms or both, depending on which branches of the nerve are involved. The majority of cases of ulnar nerve entrapment at Guyon's canal, however, involve solely motor fibers and present with painless unilateral hypothenar and interossei weakness or atrophy. Because the palmar cutaneous and dorsal cutaneous branches leave the ulnar nerve in the distal forearm and do not enter the Guyon canal, sensation in the proximal hypothenar region and the dorsum of the little and ring fingers is not impaired in all cases of ulnar nerve lesions at the wrist or hand. The sensory loss, if present, is confined to the palmar surface of the ulnar-innervated fingers (the little finger and usually the ulnar half of the ring finger) and the distal hypothenar region. Compression at the distal portion of the Guyon canal (also referred to as the pisohamate hiatus) results in selective involvement of the deep motor branch, with interossei weakness and atrophy and complete or relative sparing of the hypothenar muscles as well as sensation (Katirji and Dokko,1996).
The diagnosis is confirmed by EDX studies, often by demonstrating low amplitude (with or without prolonged distal motor latencies) to the first dorsal interosseous or abductor digiti minimi muscles or both, along with denervation of the ulnar-innervated hand muscles that parallels the clinical manifestations. Ulnar SNAP may or may not be abnormal. These EDX studies are also important in excluding an ulnar neuropathy at the elbow. Several features on the EDX examination are inconsistent with an ulnar neuropathy at the wrist: low amplitude or absent dorsal ulnar SNAP, focal slowing or conduction block across the elbow, or denervation of the flexor carpi ulnaris or the flexor digitorum profundus (ulnar portion).
Plain radiograph of the wrist may reveal a fracture of the pisiform or hook of the hamate bone. MRI or ultrasound through the Guyon canal may demonstrate a structural lesion such as a ganglion cyst. Sources of occupational or recreational trauma should be eliminated. In patients with fractures, ganglia, or mass lesions, surgical intervention is necessary. The prognosis is usually good after surgical decompression with effective reinnervation.
Radial Nerve
Applied Anatomy.
The radial nerve is the largest nerve in the upper extremity. In the arm, lying medial to the humerus, the radial nerve innervates all three heads of the triceps muscle and the anconeus muscle. The nerve passes obliquely behind the humerus and then through the spiral groove, a shallow groove formed deep to the lateral head of the triceps muscle. Before entering the spiral groove in the midarm, it gives three sensory branches: the posterior cutaneous nerve of the arm (which innervates a strip of skin overlying the triceps muscle), the lower lateral cutaneous nerve of the arm (which innervates the lateral half of the arm), and the posterior cutaneous nerve of the forearm (which innervates the skin of the extensor surface of the forearm). In the anterior compartment of the arm, the radial nerve, lying lateral to the humerus, innervates the brachioradialis and the extensor carpi radialis longus. The nerve then passes anterior to the lateral epicondyle and innervates the extensor carpi radialis brevis and supinator. The “radial tunnel” is a space (not an anatomical tunnel) where the radial nerve travels in the upper forearm from the humeroradial joint past the supinator muscle. Within that space, the radial nerve divides into its terminal branches, the superficial radial and posterior interosseous nerves. The posterior interosseous nerve, a terminal pure motor branch, passes under the proximal edge of the supinator muscle (arcade of Frohse) and travels in the forearm and innervates all the remaining wrist and finger extensors. The superficial radial nerve is a terminal pure sensory nerve and innervates the skin of the proximal two-thirds of the extensor surfaces of the thumb, index, and middle fingers, and half of the ring finger, along with the corresponding dorsum of the hand.
Radial Nerve Compression in the Arm.
Radial nerve compression in the arm often occurs at the spiral groove of the humerus during drunken sleep wherein the arm is draped over a chair (Saturday-night palsy) (Spinner et al., 2002). The radial nerve may be also injured following fractures of the humerus. Radial nerve lesions at the axilla are much less common and may result from crutches or from the weight of a sleeping partner's head (honeymoon palsy). The radial nerve is also often involved in isolation or in combination with other single nerves in multifocal motor neuropathy with conduction block.
In radial nerve lesions in the spiral groove or midarm, there is weakness of the brachioradialis, wrist, and finger extensors, while the triceps is spared. Sensory abnormalities may occur over the dorsum of the hand, thumb, index finger, and middle finger. In lesions at the axilla, there is additional weakness of the triceps, and the sensory loss may extend into the extensor surface of the forearm and lateral half of the arm and over to the triceps owing to involvement of the posterior cutaneous nerve of the forearm and the lower lateral cutaneous and posterior cutaneous nerve of the arm.
The EDX studies are essential in confirming the site and extent of the lesion, excluding other causes of wrist drop, and estimating severity and prognosis. Low-amplitude or absent radial SNAP is common except when the pathology at the spiral groove is purely demyelinating. Conduction block across the spiral groove is seen in segmental demyelinating lesions, or the radial motor responses are low in amplitude in axon-loss lesions. Mixed lesions are also common. Needle EMG reveals denervation of all finger and wrist extensors, as well as the extensor carpi radialis and the brachioradialis. The triceps and anconeus are spared in midarm lesions and denervated in axillary lesions.
As with other peripheral nerve lesions, the prognosis depends on the primary pathology. Radial nerve lesions due to demyelinative conduction block, such as in most cases of Saturday-night palsy, usually improve in 6 to 8 weeks. Axon-loss lesions such as those often associated with humeral fracture, however, have a less favorable prognosis, with a protracted course and often incomplete recovery.
Posterior Interosseous Neuropathy.
Lesions of the posterior interosseous nerve (PIN) are uncommon and usually occur in association with trauma, fracture, soft-tissue masses (e.g., lipomas, gangliomas), or exuberant synovium motor neuropathy (i.e., rheumatoid arthritis). On rare occasions, a PIN lesion is a manifestation of neuralgic amyotrophy, with acute arm pain followed within a few days by weakness (Hashizume et al., 1996). The clinical manifestations of a PIN lesion are dropped fingers and inability to extend them at the metacarpophalangeal joints. Radial deviation of the wrist on wrist extension is often pathognomic and is due to weakness of the extensor carpi ulnaris muscle with sparing of the extensor carpi radialis muscle, the latter innervated by the main trunk of the radial nerve. EMG study confirms the diagnosis by demonstrating normal radial SNAP and denervation of the muscles supplied by the posterior interosseous nerve, with sparing of more proximal radial-innervated muscles including the brachioradialis, extensor carpi radialis, and triceps muscles.
In rheumatoid arthritis, local injection of corticosteroids may be helpful. If the syndrome is progressive, surgical exploration, including synovectomy or decompression of the posterior interosseous nerve, may become necessary (Shergill et al., 2001).
Radial Tunnel Syndrome.
Patients with persistent tennis elbow (lateral epicondylitis) are sometimes given a diagnosis of radial tunnel syndrome, an extremely rare and highly controversial entrapment of the radial nerve or its posterior interosseous branch within the radial tunnel (Rosenbaum, 1999). The nerve appears most vulnerable to entrapment at the level of the supinator muscle. These patients present with forearm pain and tenderness at the lateral epicondyle and slightly distally into the forearm, with no associated muscle weakness or sensory loss in the radial or PIN distribution. Pain is induced by extension of the middle finger or supination with the elbow extended. The EMG study, including radial nerve conduction studies recording the extensor digitorum communis and extensor indicis proprius, is almost always normal. Local steroid injection may temporarily relieve symptoms. In patients with persistent pain, surgical division of the supinator muscle has been advocated, with variable results.
Superficial Radial Sensory Neuropathy (Cheiralgia Paresthetica).
Cheiralgia paresthetica is a mononeuropathy of the superficial dorsal sensory branch of the radial nerve. It occurs as a result of trauma from tight wristbands or handcuffs, or may result from intravenous cannulation, fracture of the wrist, or wrist surgery (e.g., plating of forearm bones after fracture). The use of one-way (only tighten) ratcheting mechanism of handcuffs increases the risk of cheiralgia paresthetica (Grant and Cook, 2000). In up to 50% of nontraumatic cases, it is also associated with de Quervain tenosynovitis, an inflammatory condition of thumb extensor muscles, predominantly extensor pollicis brevis (Lanzetta and Foucher, 1995). In de Quervain tenosynovitis, there is tenderness of the anatomical snuffbox and thumb extensor tendons with forced ulnar deviation while holding the thumb wrapped in the palm. Paresthesias and pain in the distribution of the superficial sensory branch of the radial nerve characterize this benign self-limiting condition. A small area of hypoesthesia in the dorsoradial aspect of the hand is frequently identified. Nerve conduction study often shows a low-amplitude or absent dorsal radial SNAP with normal needle EMG including all radial innervated muscles.
Musculocutaneous Nerve
The musculocutaneous nerve arises from the lateral cord of the brachial plexus, with fibers originating from the C5 and C6 roots via the upper trunk. The nerve innervates and penetrates the coracobrachialis muscle and courses down the anterior aspect of the upper arm between the two muscles it innervates—the biceps brachii and brachialis. It then terminates as a sensory nerve, the lateral antebrachial cutaneous nerve, which innervates the lateral forearm to the base of thumb. This nerve may be damaged with shoulder dislocations, following general anesthesia, or with vigorous exercise such as weight lifting or repetitive movements such as occur in carpet carriers, where the nerve is repeatedly compressed by carrying carpets on the shoulder while held in place by the arm (Sander et al., 1997). The musculocutaneous nerve may also be involved in neuralgic amyotrophy (idiopathic brachial plexus neuropathy). The differential diagnosis includes C5 or C6 radiculopathy, upper trunk or lateral cord brachial plexopathy, and rupture of the biceps tendon.
Clinically, patients with musculocutaneous mononeuropathy present with weakness and atrophy of the biceps brachii and brachialis muscles, diminished biceps brachii reflex, and sensory loss over the lateral aspect of the forearm anteriorly. Nerve conduction studies show reduced musculocutaneous CMAP amplitude recording the biceps muscle and a low-amplitude or absent lateral antebrachial cutaneous sensory response. Needle EMG demonstrates denervation limited to the biceps brachii and brachialis muscles, often sparing the coracobrachialis muscle.
Spontaneous recovery is the rule. Local corticosteroid injection may provide some relief of pain. Surgical decompression is contemplated if no improvement occurs.
Suprascapular Nerve
The suprascapular nerve is a pure motor branch of the upper trunk of the brachial plexus with innervation from the C5 and C6 roots. It then passes through the suprascapular notch, covered by the transverse scapular ligament, to innervate the supraspinatus muscle. It wraps around the spinoglenoid notch of the scapular spine and innervates the infraspinatus muscle. Entrapment at the suprascapular notch occurs after repetitive forward traction of the shoulders—a condition seen in certain athletes, especially volleyball players. This nerve also may be involved in a restricted form of neuralgic amyotrophy (idiopathic brachial plexus neuropathy). Diffuse aching pain in the posterior aspect of the shoulder exacerbated by overhead activities is a cardinal symptom. The pain has an articular characteristic because the acromioclavicular joint and surrounding structures are innervated by the suprascapular nerve. Atrophy and weakness are confined to the infraspinatus and supraspinatus muscles. Slow and steady abduction of the arm starting from a vertical position alongside the chest is not possible with a severe lesion of the suprascapular nerve. Tendon ruptures of the rotator cuff have to be considered in the differential diagnosis. EMG shows denervation restricted to the supraspinatus and infraspinatus muscles. Local corticosteroid injection may give temporary relief of pain, although surgery is sometimes required (Antoniou et al., 2001). In entrapment at the spinoglenoid notch, pain is usually absent, and there is atrophy, weakness, and denervation of the infraspinatus muscle only.
Intercostobrachial Nerve
The intercostobrachial nerve is a cutaneous sensory nerve derived from the second and third thoracic nerve roots. It supplies the skin on the medial surface of the upper arm and axilla, as well as the adjacent chest wall. It may be injured in a modified radical mastectomy and other surgical procedures involving the axilla and lateral pectoral region (Wallace et al., 1996).
Double Crush Syndrome
When a sizable cohort of patients with EDX evidence of distal upper-limb entrapment neuropathies was found to have either electrophysiological or radiological and clinical evidence of cervical radiculopathy, Upton and McComas proposed that focal compression of single nerve fibers proximally might so alter axoplasmic transport as to render the distal nerve more susceptible to symptomatic entrapment neuropathy. They termed this the double crush syndrome. Although the concept of double crush syndrome has since been invoked in a wide variety of entrapment neuropathies, often as an explanation for failure of decompressive surgeries of the neck or limb or as a rationale to decompress a nerve in multiple proximal-to-distal sites along its course, this phenomenon is of uncertain validity (Wilbourn and Gilliatt, 1997).
Mononeuropathies of the Lower Extremities
Entrapment neuropathies of lower limbs are shown in Table 107.4.
Sciatic Nerve
Applied Anatomy.
The sciatic nerve is formed from nerve roots L4 through S3 and is composed of two distinct nerves, the common peroneal (renamed the fibular nerve by the Federative Committee on Anatomical Terminology [FCAT], owing to confusion between the terms peroneal and perineal) and tibial nerves, which share a common sheath from the pelvis to the popliteal fossa. Usually the sciatic nerve emerges from the pelvis by passing beneath the piriformis muscle, but sometimes the fibular division only passes through or above the piriformis muscle. The sciatic nerve innervates all the hamstring muscles via the tibial nerve except the short head of the biceps femoris, which is innervated by the common fibular nerve. The common fibular and tibial nerves separate completely in the upper popliteal fossa or slightly above.
Sciatic Neuropathy at the Sciatic Notch.
The sciatic nerve is occasionally vulnerable to entrapment as it crosses over the sciatic notch leaving the pelvis. Most sciatic nerve lesions result from trauma such as bullet and stab wounds, fractures, dislocations, hematomas in the posterior thigh compartment, misplaced intramuscular injections, and complications of hip replacement surgery (Plewnia et al., 1999). Recurrent sciatic mononeuropathy may be caused by endometriosis involving the nerve at the sciatic notch. Direct compression of the sciatic nerve is rare but occasionally occurs during coma, anesthesia, or prolonged sitting on a hard surface (toilet seat palsy). Either or both divisions of the nerve may be compressed by a Baker cyst in the popliteal fossa.
A complete sciatic nerve lesion results in weakness of knee flexors and all muscles below the knee, as well as sensory loss of the entire foot and leg below the knee except for a region supplied by the saphenous nerve over the medial leg. The fibular division is more commonly involved than the tibial in proximal lesions of the sciatic nerve. Partial sciatic nerve lesions often affect the fibular nerve more than the tibial nerve and may mimic a more distal common fibular neuropathy. This is explained by the fewer fascicles with limited supportive tissue within the fibular nerve, which is also taut and secured at the sciatic notch and fibular neck. In such patients, evidence of denervation in the short head of the biceps femoris and tibialis posterior muscles and abnormal sural or medial plantar SNAPs help localize partial proximal sciatic nerve lesions (Yuen and So, 1999). Occasionally, the common fibular nerve gets injured selectively (Katirji and Wilbourn, 1994).
Piriformis Syndrome.
On rare occasions, the piriformis muscle may entrap the sciatic nerve trunk as it passes through or over the piriformis muscle. Since the term was coined in 1947 by Robinson, the piriformis syndrome has been subject to controversy (Fishman and Schaeffer, 2003; Stewart, 2003). The syndrome fell out of fashion with the advancement of radiological techniques (myelography, CT, MRI) that often demonstrate that most patients with sciatica have nerve root compression and occasionally lesions of the sacral plexus or sciatic nerve at other locations.
The typical patient with piriformis syndrome has a history of buttock trauma and experiences maximal buttock pain during prolonged sitting (e.g., driving, biking), bending at the waist, or activity that requires hip adduction and internal rotation (e.g., cross-country skiing) (Kirschner et al., 2009). The neurological and EDX examinations are usually normal. A bedside test maneuver in which the hip is placed passively in adduction, internal rotation, and flexion of the hip (AIF maneuver) may reproduce the pain and is considered diagnostic. Imaging is usually normal but occasionally shows hypertrophy of the piriformis muscle or abnormal vessels or bands in the region of the piriformis muscle. MR neurography may show sciatic nerve hyperintensity at the sciatic notch, a more specific sign of nerve entrapment (Filler et al., 2005). Treatment consists of exercises that include prolonged stretching of the piriformis muscle by flexion, adduction, and internal rotation of the hip. CT- or MRI-guided corticosteroid injection into the piriformis muscle may alleviate the symptoms; a positive response is used as a confirmatory test. Surgical sectioning of the piriformis muscle is indicated in cases resistant to conservative therapy. Although good outcome is expected in carefully selected patients (Filler et al., 2005), severe postoperative sciatic nerve injury was recently reported (Justice et al., 2012).
Common Fibular (Peroneal) Nerve
Applied Anatomy.
As noted earlier, the confusing similarity between peroneal and perineal led FCAT to rename the peroneal nerve the fibular nerve. Soon after the sciatic nerve divides close to the popliteal fossa, the common fibular nerve gives off the lateral cutaneous nerve of the calf, which innervates the skin over the upper third of the lateral aspect of the leg, and the fibular communicating nerve which joins the sural nerve. The common fibular nerve then winds around the fibular neck and passes through the origin of the peroneus longus muscle (“fibular tunnel”). Near that point, the common fibular nerve divides into its terminal branches, the deep and superficial fibular nerves. The deep fibular nerve traverses the lateral and then anterior leg compartments and innervates the tibialis anterior, extensor hallucis longus, peroneus tertius, and extensor digitorum longus. It then divides close to the ankle joint to innervate the extensor digitorum brevis and the skin of the web space between the first and second toes. The superficial fibular nerve innervates the peroneus longus and brevis and the skin of the lower two-thirds of the lateral aspect of the leg and the dorsum of the foot (except for the first web space). An accessory deep fibular nerve, seen in up to 28% of individuals, arises from the superficial fibular nerve, passes behind the lateral malleolus, and innervates the lateral part of the extensor digitorum brevis.
Common Fibular (Peroneal) Neuropathy at the Fibular Neck.
Compression of the common fibular nerve is the most frequent compressive neuropathy in the lower extremity. This nerve is particularly vulnerable to direct pressure in the region of the fibular neck as it passes through the origin of the peroneus longus muscle. Intraoperative compression due to improper positioning or padding during anesthesia is the leading cause of acute common fibular neuropathy at the fibular neck (Katirji and Wilbourn, 1988). Weight loss, habitual leg crossing, or unrecognized pressure on the nerve in hospitalized critically ill, debilitated, or unconscious patients may also be responsible for this nerve injury (Aprile et al., 2005; Katirji, 1999). Devices that may compress the fibular nerve include casts, orthoses, pneumatic compression, antithrombotic stockings, bandages, and straps. Fibular nerve stretch injury may result from an acute forceful foot inversion or prolonged squatting (strawberry pickers palsy). Blunt trauma (e.g., post fibular fracture, knee dislocation) and open injury (e.g., lacerations) account for a significant number of cases. Postpartum peroneal neuropathy may be due to stirrups compression, prolonged squatting or direct hand compression. Fibular nerve injury is also a known complication of knee surgery, including arthroscopic surgery and lateral meniscus repair. Up to half of patients without a clear cause of fibular mononeuropathy across the fibular head have intraneural ganglia (Young et al., 2009). These are formed when disruption of the capsule of the superior tibiofibular joint results in dissection of synovial fluid along the articular branch of the fibular nerve into the tibialis anterior motor branch (Hebert-Blouin et al., 2010). Other mass lesions such as osteochondromas or schwannomas are much less common.
A common fibular nerve lesion leads to weakness of foot and toe extension and foot eversion, with a foot drop and steppage gait. Sensory impairment is found over the lateral aspect of the lower two-thirds of the leg and the dorsum of the foot. Foot eversion and sensory loss may be spared (except for the first web space of the foot) when the lesion is selective, involving the deep fibular nerve. Pain is rare except with intraneural ganglia.
Fibular mononeuropathy is most often confused with other causes of unilateral foot drop, including L5 radiculopathy, sciatic nerve lesions (especially when predominantly affecting the common fibular nerve), and lumbosacral plexopathy (particularly when involving the lumbosacral trunk). A fibular nerve lesion must be differentiated from anterior tibial compartment syndrome, in which the deep fibular nerve is compressed by muscle swelling within the anterior compartment secondary to injury, heavy exercise, trauma, or ischemia. This results in an acute syndrome of severe lower leg pain, swelling, and weakness of foot and toe extensors. The anterior tibial compartment must be decompressed rapidly by fasciotomy to prevent irreversible nerve and muscle damage.
EDX studies are useful for localizing lesions and may provide clues to the underlying cause and a guide to prognosis. Although it is often possible by nerve conduction studies to demonstrate focal conduction block across the fibular head, contrary to common belief, the most frequent pathophysiological process is axonal loss, regardless of the cause (Katirji and Wilbourn, 1988). Axon-loss lesions reveal diffusely low or absent fibular motor and sensory amplitudes. In contrast to ulnar lesions across the elbow and CTS, localized slowing in the region of the fibular head is not common. Needle EMG demonstrates denervation in common fibular-innervated muscles but not in the short head of the biceps femoris (innervated by the common fibular division of the sciatic nerve in the thigh), in the L5 nerve root-innervated muscles, such as the tibialis posterior, flexor digitorum longus, tensor fascia lata and gluteus medius, or the low lumbar paraspinal muscles. MRI and ultrasound are effective in visualizing intraneural ganglia and other soft-tissue masses or tumors.
The prognosis is uniformly good in cases of acute demyelinating lesions, whereas recovery is delayed in those with axonal lesions and stretch injuries. The distal fibular motor amplitude recording tibialis anterior serves as an accurate estimate of the extent of axonal loss and a good prognostic indicator of foot drop. Hence, fibular nerve studies should be performed bilaterally and compared. Bracing with a custom-made plastic ankle-foot orthosis is necessary to improve the gait in the presence of severe foot drop. The few patients who do not improve spontaneously after 3 months, or those who have pain or a slowly progressive fibular nerve lesion, may require MRI studies and surgical exploration (Kim and Kline, 1996).
Tibial Nerve
Applied Anatomy.
The tibial nerve innervates all the hamstring muscles except the short head of the biceps femoris. It then separates from the common fibular nerve, usually in the upper popliteal fossa, and gives off the sural sensory nerve, which is often joined by a branch from the common fibular nerve, the sural communicating nerve, to innervate the skin over the lateral aspect of the lower leg and foot, including the little toe. In the upper calf, the tibial nerve passes underneath the soleus muscle and innervates the gastrocnemius, soleus, tibialis posterior, flexor digitorum profundus, and flexor hallucis longus. At the ankle, the tibial nerve passes under the laciniate ligament, which covers the tarsal tunnel through which the nerve passes together with the tendons of the tibialis posterior, flexor digitorum longus, and flexor hallucis longus muscles and the tibial artery and veins.
Tarsal Tunnel Syndrome.
Entrapment of the tibial nerve occurs behind and immediately below the medial malleolus. Burning pain is experienced in the toes and the sole of the foot. If the calcaneal sensory branches are involved, pain and numbness also involves the heel. Examination usually reveals plantar sensory impairment and wasting of the intrinsic foot muscles. Percussion at the site of nerve compression or eversion of the foot often elicit pain and paresthesias. EDX study results should confirm entrapment of the tibial nerve at the tarsal tunnel by demonstrating slowing of motor fibers to the abductor hallucis and/or abductor digiti minimi muscles, as well as involvement of the medial and/or lateral plantar mixed potentials fibers, with sparing of the sural nerve sensory action potential. Unfortunately, medial and/or lateral plantar mixed (or sensory) potentials are technically very difficult and may be unelicitable in normal subjects with plantar calluses, foot edema, previous surgical procedures in the foot, or in adults over the age of 45. Needle EMG shows denervation of the abductor hallucis and/or abductor digiti minimi muscles and normal S1-innervated and proximal muscles such as the gastrocnemius, soleus, biceps femoris, and gluteus maximus muscles.
The majority of suspected cases of tarsal tunnel syndrome turn out to have generalized peripheral neuropathy, S1 radiculopathy, or non-neurological foot pain such as plantar fascitis, stress fracture, arthritis, or bursitis.
Local injection with corticosteroids underneath the laciniate ligament may temporarily relieve symptoms. Surgical decompression is needed for permanent results in those rare cases in which objective evidence of this syndrome exists.
Sural Nerve
Although the vast majority of sural nerve lesions are iatrogenic as the result of diagnostic sural nerve biopsy or sural nerve harvesting for nerve grafts, mononeuropathy of the sural nerve has been reported with a number of other conditions including lower-limb vein-stripping surgery, Baker cyst or ankle joint surgery, local trauma, tightly laced high-topped footwear such as ski boots or ice skates, and rarely as the initial presentation of vasculitic mononeuritis multiplex (Li and Lederman, 2014; Stickler et al., 2006).
Femoral Nerve
Applied Anatomy.
The femoral nerve is formed in the pelvis from the posterior divisions of the ventral rami of L2, L3, and L4 spinal roots, where it innervates the psoas muscle. It then passes within the iliacus compartment and innervates the iliacus muscle via a motor branch that originates 4 to 5 cm before the nerve crosses underneath the inguinal ligament. In the anterior thigh, the femoral nerve innervates the quadriceps and sartorius muscles and the skin of the anterior thigh and gives off the saphenous sensory nerve, which innervates the skin of the medial surface of the knee and medial leg.
Femoral Nerve Lesions.
The majority of femoral nerve lesions are iatrogenic (Al-Hakim and Katirji, 1993). Pelvic lesions follow a variety of gastrointestinal, vascular, urological, or gynecological operations such as abdominal hysterectomy, radical prostatectomy, renal transplantation, and abdominal aortic repair. During these procedures, the femoral nerve becomes compressed between the lateral blade of the retractor and the pelvic wall; the incidence of these lesions is significantly reduced when self-retractors are avoided. Acute retroperitoneal hematoma is often iatrogenic following anticoagulant therapy, pelvic operations, or femoral vessel catheterization such as for cardiac catheterization. At the inguinal ligament, the femoral nerve may become kinked during lithotomy positioning, particularly when the leg is held in extreme hip flexion and external rotation, used during vaginal delivery, vaginal hysterectomy, prostatectomy, and laparoscopy. Total hip replacement, particularly surgical revisions and complicated reconstructions, may result in femoral nerve injury.
Femoral nerve injury due to spontaneous retroperitoneal hematoma may occur in hemophiliacs, patients with blood dyscrasias, or following a ruptured abdominal aortic aneurysm. Pelvic lymphadenopathy, primary malignancy of the colon or rectum, and neurofibromas or schwannomas are rare causes of femoral neuropathies. Hip hyperextension, such as in dancers or during Yoga exercise, may also cause a femoral stretch injury.
Femoral nerve lesions manifest with acute thigh weakness and anterior thigh and medial leg numbness. Thigh weakness often leads to falls. Pain is usually absent except in cases due to retroperitoneal hematomas. On examination, there is weakness of knee extension, with absent or depressed knee jerk. Thigh adduction is normal. Hip flexion is usually weak when the lesion is within the pelvis, although it may be difficult to assess hip flexion in the setting of severe quadriceps weakness.
Needle EMG reveals denervation of the quadriceps muscle. The iliacus muscle is often normal in inguinal lesions but shows denervation in femoral nerve lesions in the pelvis. Needle EMG of the thigh adductor muscles, innervated by the L2, L3, L4 roots via the obturator nerve, helps distinguish femoral nerve lesions from upper lumbar radiculopathy or plexopathy. Nerve conduction studies have prognostic value, since the amplitude and area of the femoral CMAP is a very good quantitative measure of motor axonal loss (Kuntzer et al., 1997). CT or MRI of the pelvis are urgently indicated in patients with suspected retroperitoneal hematoma or pelvic mass lesion.
Apart from patients with confirmed retroperitoneal hematoma who may require emergent drainage, most other femoral nerve lesions are treated conservatively. A knee or knee-ankle-foot orthosis is helpful for patients with unilateral severe weakness of the quadriceps and will assist in walking and prevent falls.
Saphenous Nerve
Saphenous nerve lesions may follow stripping of a long saphenous varicose vein, harvesting the vein for a coronary artery bypass, or surgical and arthroscopic operations on the knee. Entrapment of the saphenous nerve is rare and may occur as it exits the subsartorial (adductor or Hunter) canal or by pes anserine bursitis. Patients with saphenous mononeuropathy have sensory loss or hyperesthesias of the medial leg that may extend into the medial arch of the foot.
Saphenous nerve lesions should be differentiated from L4 radiculopathy, lumbar plexopathy, and femoral mononeuropathy. In addition to the clinical examination, EDX studies can confirm that the quadriceps, iliacus, and thigh adductors are normal in patients with saphenous nerve lesions. Saphenous SNAP is often unilaterally absent or low in amplitude, but this response is difficult to elicit, particularly in elderly patients. Saphenous nerve lesions improve spontaneously with time; decompression underneath the subsartorial canal is occasionally performed.
Other Lower-Extremity Mononeuropathies
Lateral Femoral Cutaneous Nerve Entrapment (Meralgia Paresthetica).
The lateral femoral cutaneous nerve, which is a pure sensory nerve, passes medial to the anterior superior iliac spine under the inguinal ligament to enter the thigh under the fascia lata that it penetrates to supply the skin of the anterolateral part of the thigh. The site of entrapment is usually at the level of the inguinal ligament. Rarely, the nerve can be affected in its proximal segment by retroperitoneal tumors or be injured during appendectomy. The disorder occurs in about 4 per 10,000 individuals. It is most often seen in association with obesity, diabetes, and advancing age (Parisi et al., 2011). It is a common entrapment neuropathy during pregnancy, particularly the third trimester, and usually recovers after delivery. It may occur with ascites, or in other conditions that increase intra-abdominal pressure. Direct compression by a belt, corset, beeper, or cellular phone; fracture of the anterior portion of the ilium; or pelvic tilt causing undue stresses on the abdominal musculature are other causes.
Patients develop numbness, painful burning, and itching over the anterolateral thigh. Pressure at the inguinal ligament medial to the anterior superior iliac spine elicits referred pain and dysesthesias. Some patients report relief of pain when assuming a supine position.
Lateral femoral cutaneous nerve SNAP is technically difficult to measure and may be absent in healthy subjects, particularly women and obese individuals. Asymmetrical low-amplitude or absent potential on the symptomatic side is a confirmatory finding. Electrophysiological studies of the femoral nerve and quadriceps femoris and iliacus muscles are normal, which helps exclude lumbar radiculopathy and plexopathy. A local anesthetic nerve block may have diagnostic value (Haim et al., 2006). Treatment consists of symptomatic measures such as rest, analgesics, and weight loss. Postural abnormalities should be corrected. Neurolysis is rarely beneficial.
Ilioinguinal Neuropathy.
The ilioinguinal nerve is analogous to an intercostal nerve. Muscle branches innervate the lower portion of the transverse abdominal and internal oblique muscles. The cutaneous sensory nerve supplies the skin over the inguinal ligament and the base of the scrotum or labia. As the nerve takes a zigzag course, passing through the transverse abdominal and internal oblique muscles, it is subject to mechanical compression such as with a direct inguinal hernia. Trauma, surgical procedures, scar tissue, and increased abdominal muscle tone caused by abnormal posture are frequently responsible. Pain is referred to the groin, and weakness of the lower abdominal wall may result in the formation of an asymmetrical bulging of the lower abdominal wall.
Conservative treatment includes rest and NSAIDs. Neurolysis may be required in refractory cases when a mechanical lesion is suspected.
Obturator Neuropathy.
The obturator nerve is vulnerable to entrapment as it passes through the obturator canal (e.g., by an obturator hernia or osteitis pubis). An obturator neuropathy is most often associated with pelvic malignancies (prostate, cervical, or uterine cancers). It can also be seen with trauma and synovial cyst of the hip or as a surgical complication, especially with extensive retroperitoneal surgeries or laparoscopic pelvic lymphadenectomies and during total hip replacement.
Entrapment produces radiating pain from the groin down the inner aspect of the thigh, often difficult to distinguish from the pain of a recent procedure or trauma. There is weakness of hip adduction and sensory impairment in the upper medial thigh. Many patients appear to have hip-flexor weakness as a false localizing sign. Although this phenomenon may be explained by pain, it is more likely due to mechanical disadvantage of the hip flexors in the presence of weak thigh adductors. CT or MRI scanning of the pelvis is helpful in finding primary or metastatic pelvic tumors. EMG testing is essential for diagnosis by detecting selective denervation of the thigh adductor muscles, with normal quadriceps and iliacus muscles, thus excluding other causes of hip weakness including femoral nerve lesions, upper lumbar (L2, L3 or L4) radiculopathy or plexopathy, and diabetic amyotrophy (diabetic proximal neuropathy) (Sorenson et al., 2002).
This entrapment neuropathy is treated conservatively, which often provides good results, especially in those with acute onset of symptoms. If such treatment fails or if symptoms progress to involve other nerves in the region, a careful search for occult pelvic or retroperitoneal malignancy must be pursued.
Migrant Sensory Neuritis of Wartenberg
In this rarely reported but not uncommon condition, a pure and relapsing-remitting sensory mononeuritis multiplex is associated with loss of sensation and pain in the distribution of the affected nerves. The onset is usually sudden, and pain is precipitated by movements and (especially) stretching of the affected limbs. Many different cutaneous nerves can be involved. Motor nerve fibers are not affected. Laboratory tests fail to detect any underlying cause, but on occasion a sural nerve biopsy demonstrates inflammatory changes or a vasculitis, with patchy loss of nerve fibers and evidence of axonal degeneration suggestive of an ischemic process. Rarely, immunoglobulin (Ig)G deposits are also observed around blood vessels. The pain and areas of sensory loss often recover over weeks to months, but the improvement may be partial. Symptoms may recur at the same or other sites. The condition is often misdiagnosed as other diseases such as multiple sclerosis, but the discrete areas of sensory deficit and nerve irritation in several cutaneous nerves should indicate the proper diagnosis. The differential diagnosis should always include conditions like diabetes mellitus, leprosy, vasculitis, sarcoidosis, sensory perineuritis, and rarely HNPP (Nicolle et al., 2001; Zifko and Hahn, 1997).
Localized Perineurial Hypertrophic Mononeuropathy
A slowly progressive painless mononeuropathy that cannot be localized to entrapment sites and is caused by a focal fusiform enlargement of the affected nerve, termed localized hypertrophic neuropathy or perineurioma, is an uncommon condition affecting young adults (Simmons et al., 1999). Although any nerve may be involved, it often occurs in the radial, posterior interosseous, tibial, and sciatic nerves. The fusiform enlargement is mainly composed of “onion bulblike whorls” formed by layers of perineurial cells (Fig. 107.7, B). The lamellae of the whorls stain for epithelial membrane antigen. The cause of the perineurial cell proliferation is unknown. It typically results in painless, slowly progressive weakness and atrophy in the distribution of the affected nerve. Sensory symptoms are minor, although sensory nerve fibers are obviously involved. EDX study results show an axonal mononeuropathy and help in the precise localization of the focal nerve lesion. MRI shows a focal enlargement of the affected nerve, increased signal on T2-weighted images, and enhancement with gadolinium (see Fig. 107.7, A).
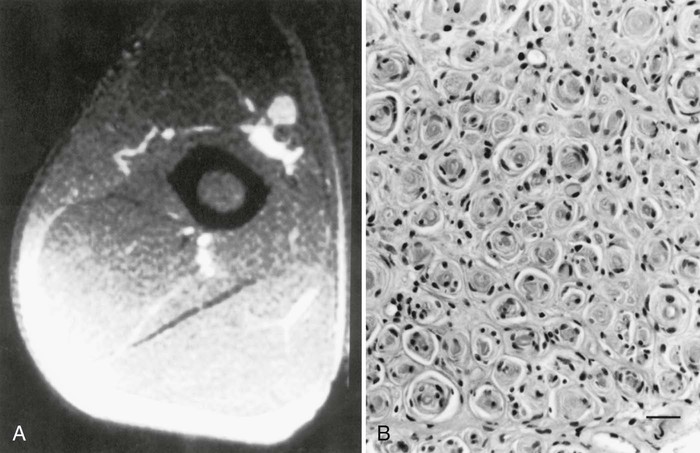
Surgical exploration and a fascicular biopsy by a surgeon experienced in peripheral nerve microsurgery may confirm the diagnosis and exclude malignant peripheral nerve sheath tumors, which are difficult to exclude without biopsy. Surgical resection of the involved nerve segment with graft repair has been proposed, but because of the benign nature of the “tumor” and its very slow progression, the involved nerve should be preserved if it has even partial function.
Hereditary Neuropathies
The hereditary neuropathies constitute a complex heterogeneous group of diseases that usually share the clinical features of insidious onset and indolent course over years to decades. The number of hereditary disorders for which a metabolic or molecular defect is known is rapidly increasing, allowing a more accurate classification. For those inherited neuropathies for which the underlying genetic abnormality has yet to be identified, the classification still depends on the clinical phenotype, mode of inheritance, and class of neurons predominantly affected. Major advances in understanding the molecular basis of inherited neuropathies have come from identifying chromosomal loci or causative genes for a given disease phenotype, leading to identification of an ever-increasing number of genes coding for a specific gene product essential to myelin or axonal function (Bassam, 2014; Berger et al., 2002; Kamholz et al., 2000; Lupski, 1998; Scherer, 2006).
Hereditary neuropathies are common disorders, accounting for nearly 40% of chronic polyneuropathies, and as many as 50% of previously unidentified peripheral polyneuropathies. Their inherited nature may go unrecognized in a surprisingly large percentage of patients (Klein, 2007). Eliciting historical evidence of long-standing neuromuscular symptoms; obtaining detailed family histories; looking for skeletal abnormalities such as hammer toes, high arches, or scoliosis; and more importantly, performing neurological evaluations in relatives of patients are essential in identifying a previously unsuspected inherited neuropathy. Because of the paucity of positive symptoms, patients may not volunteer information about their own or relatives' conditions. For example, paresthesias are spontaneously reported three times more commonly in acquired than in inherited neuropathies. Even in the face of a truly negative family history, the possibility of an inherited neuropathy cannot be dismissed. Such a situation may arise in cases of early death of one or both parents, few blood relatives, or autosomal recessive disease. Also, available diagnostic deoxyribonucleic acid (DNA) testing has shown that about a third of isolated cases of inherited neuropathies may arise from de novo gene mutations (Boerkoel et al., 2002). It is advisable to consider the possibility of an inherited neuropathy in any patient with a chronic “acquired” neuropathy that remains cryptogenic or refractory to treatment.
Charcot–Marie–Tooth Disease (Hereditary Motor and Sensory Neuropathy)
The syndrome of peroneal muscular atrophy, or CMT disease, was first described in 1886 by Charcot and Marie in Paris and Tooth in London (Charcot and Marie, 1886; Tooth, 1886). CMT disease is the most common inherited neuropathy, with an estimated prevalence of 10 to 41 per 100,000 (Martyn and Hughes, 1997). Clinical studies combined with electrophysiological and sural nerve biopsy findings of a large number of families with peroneal muscular atrophy have allowed a separation into two main groups: (1) the demyelinating form, or CMT1 (sometimes known as hereditary motor and sensory neuropathy [HMSN-I]), in which there are marked reductions in motor NCVs (<38 m/sec in forearm) and nerve biopsy findings of demyelination and onion bulb formation; and (2) the axonal form of CMT disease, or CMT2 (HMSN-II), in which motor NCVs are normal or near normal, and nerve biopsy reveals axonal loss without prominent demyelination (Harding, 1995). The peroneal muscular atrophy phenotype without sensory involvement on either clinical or electrophysiological examination has been classified as hereditary distal spinal muscular atrophy. A more severe form of demyelinating neuropathy with onset occurring in early childhood is referred to as Dejerine-Sottas disease (or syndrome, DSS).
CMT1 and the vast majority of subtypes of CMT2 display autosomal dominant inheritance. A minority of cases occur sporadically or in siblings only and have therefore been attributed to autosomal recessive inheritance or to de novo gene mutations. Because a great variability in clinical expression exists among affected kin in the dominant disorders, a recessive inheritance can only be accepted if the clinical and electrophysiological examinations of both parents have proved to be normal. Even when the cause is nonparental, most of these patients phenotypically resemble CMT1. For all of these groups, an ever-increasing number of genetic subtypes have been elucidated.
Major advances have been made in recent years in the molecular genetics of CMT disease (Bennett and Chance, 2001; Berger et al., 2002; Kamholz et al., 2000). CMT may be classified by mode of inheritance (autosomal dominant, X-linked, and rarely autosomal recessive), chromosomal locus, or causative genes (Table 107.6). The classification remains fluid and may change as experts alter and revise these designations based on new molecular findings. DSS, formerly CMT3, may no longer be a useful designation because it is genetically heterogeneous, caused by different structural myelin protein and transcription factor gene mutations. One proposal is to reserve CMT3 for rare axonal types of autosomal recessive neuropathy (Vance, 2000).
TABLE 107.6
Molecular Genetic Classification of Charcot–Marie–Tooth Disease and Related Disorders (2002)
| Disorder | Locus | Gene | Mechanism | Testing available |
| CMT1 | ||||
| CMT1A | 17p11.2 | PMP22 | Duplication > pm | Yes |
| CMT1B | 1q22-q23 | MPZ | Pm | Yes |
| CMT1C | 16p13.1 | LITAF | Pm | Yes |
| CMT1D | 10q21 | EGR2 | Pm | Yes |
| CMT1E | 17p11.2 | PMP22 | Pm | |
| CMT1F | 8p21 | NEFL | ||
| CMTX | ||||
| CMTX1 | Xq13.1 | GjB1 (Cx32) | Pm | Yes |
| CMTX2 | Xq24 | ? | ? | — |
| CMT2 | ||||
| CMT2A | 1p36.2 | MFN2 | Pm | Yes |
| CMT2A | 1p35 | KIFBß | Pm | — |
| CMT2B | 3q13-q22 | RAB7 | Pm | Yes |
| CMT2C | 12q24 | ? | ? | — |
| CMT2D | 7p15 | GARS | Pm | Yes |
| CMT2E | 8p21 | NEFL | Pm | Yes |
| CMT2F | 7q11-21 | HSPB1 | Pm | Yes |
| CMT2G | 12q12-q13.3 | ? | ? | — |
| CMT2H | 8q13/q21.3 | GDAP1? | ? | — |
| CMT2L | 12q24 | HSPB8 | Pm | — |
| HNPP | ||||
| 17p11.2 | PMP22 | Deletion > pm | Yes | |
| DSS | ||||
| DSS-A (CMT3) | 17p11.2 | PMP22 | Pm | Yes |
| DSS-B (CMT3) | 1q22-q23 | MPZ | Pm | Yes |
| DSS-C | 10q21-q22 | EGR2 | Pm | Yes |
| AR CMT (CMT4) | ||||
| CMT4A | 8q21 | GDAP1 | Pm | Yes |
| CMT4B | 11q22 | MTMR2 | Pm | — |
| CMT4C | 5q23-q33 | SH3TC2 | Pm | Yes |
| CMT4D | 8q24 | NDRG1 | Pm | Yes |
| CMT4E | 10q21-q22 | EGR2 | Pm | Yes |
| CMT4F | 19q13 | Periaxin | Pm | Yes |
| CMT4G | 10q23 | ? | ? | — |
| CMT4H | 12q11.1-q13.11 | FGD4 | Pm | — |
| CMT4J | 6q21 | FIGURE4 | Pm | Yes |
Charcot–Marie–Tooth Disease Type 1
In CMT1, symptoms often begin during the first or second decade of life. It is characterized by slowly progressive weakness, muscular wasting, and sensory impairment predominantly involving the distal legs. Foot deformities and difficulties in running or walking resulting from symmetrical weakness and wasting in the intrinsic foot, peroneal, and anterior tibial muscles are often present. In two-thirds of patients, the upper limbs are involved later in life. Inspection reveals pes cavus and hammer toes in nearly three-quarters of adult patients, mild kyphosis in approximately a tenth, and palpably enlarged hypertrophic peripheral nerves in a quarter (Fig. 107.8). The foot deformities occur because of long-term muscular weakness and imbalance between the intrinsic extensor and long extensor muscles of the feet and toes (a similar process causes clawing of the fingers in more advanced cases). Absent ankle reflexes are universal and frequently associated with absent or reduced knee and upper limb reflexes. Some degree of distal sensory impairment (diminished vibration sense and light touch in the feet and hands) is usually discovered by examination but rarely gives rise to positive sensory symptoms. Occasionally, patients have an essential or postural upper-limb tremor. Such cases have been referred to as Roussy-Lévy syndrome, but current evidence suggests that this is not a separate clinical or genetic entity.
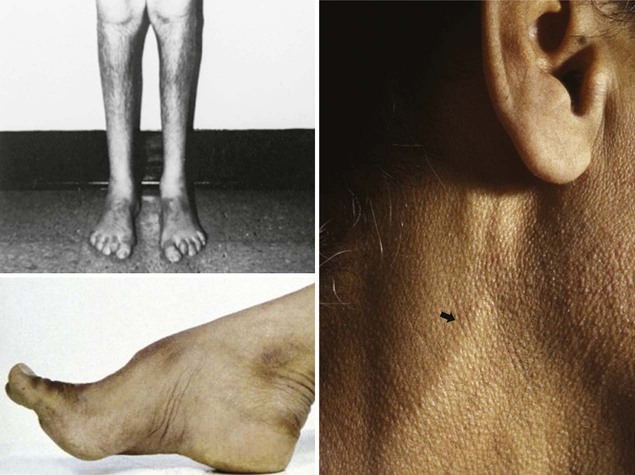
Severity of neuropathy in affected family members varies considerably. Approximately 10% of patients with slowed NCVs may remain asymptomatic. In women with CMT1, the disease may exacerbate during pregnancy. Such worsening is temporary in about a third of patients but becomes progressive in the remainder. Slow deterioration in strength and decline in axonal function continues throughout adulthood, although much of this deterioration likely represents the effects of aging superimposed on decreased reserves (Verhamme et al., 2009).
Motor nerve conduction studies show uniform slowing to less than 75% of the lower limits of normal in all nerves. Motor conduction of upper-limb nerves proves more useful than studies of lower-extremity nerves because distal denervation in the feet is often severe and virtually complete. A motor conduction velocity below 38 m/sec in the forearm segment of the median nerve is proposed as a cutoff value to distinguish between CMT1 and CMT2. Although this cutoff is useful, it can be misleading if applied too rigidly. SNAPs are usually absent with surface recordings. Uniform conduction slowing has been used to differentiate CMT1 from acquired demyelinating neuropathies. Uniform slowing along the entire length of nerves and among neighbouring nerves suggests an inherited myelinopathy affecting conduction in all nerves and nerve segments to the same degree. The conduction slowing evolves over the first 5 years of age and does not change appreciably afterward. Neurological deficits correlate with reductions in CMAP and SNAP amplitudes rather than conduction velocity, indicating that clinical weakness results from loss of axons. In contrast, acquired demyelinating neuropathies result in multifocal or nonuniform conduction slowing together with excessive temporal dispersion and conduction block. Uniform conduction slowing is found in CMT1A with PMP22 duplication or point mutations; CMT1B with MPZ point mutations; DSS, including PMP22, MPZ, and EGR2 gene mutations; as well as metachromatic leukodystrophy; Cockayne disease; and globoid cell leukodystrophy (Lewis et al., 2000).
Routine hematological and biochemical studies are normal. CSF is also normal, which helps differentiate the condition from chronic inflammatory demyelinating polyneuropathy, in which the CSF protein is usually elevated. Sural nerve biopsy typically shows the changes of a hypertrophic neuropathy, characterized by onion bulb formation, increased frequency of fibers with demyelinated and remyelinated segments, an increase in endoneurial area, and loss of large myelinated fibers (Fig. 107.9). Gene mutations, predominantly affecting genes for myelin and Schwann cell proteins, have been recognized that account for about three-quarters of families with CMT1 (Fig. 107.10). CMT1A is the most common CMT subtype, accounting for 70%–80% of CMT1 cases and more than 50% of all CMT cases. The disease is caused by duplication of a 1.5-Mb fragment in the short arm of chromosome 17p11.2-12 harboring peripheral myelin protein 22 (PMP22). Rarely, the disease is caused by PMP22 point mutation. PMP22 is a membrane glycoprotein found in the compact portion of the peripheral myelin sheath. The precise function of PMP22 in normal nerve remains unknown. Deletion of the same 1.5-megabase region on chromosome 17p11.2 results in a single copy of the normal PMP22 gene, a finding observed in 85% of patients with HNPP. The CMT1A duplication or HNPP deletion is caused by reciprocal recombination events that occur in male germ cell meiosis. The PMP22 duplication or deletion can be detected in blood samples using pulse-field electrophoresis followed by hybridization with specific CMT1A duplication junction fragments or cytogenetic testing with a PMP22 probe by fluorescence in situ hybridization.
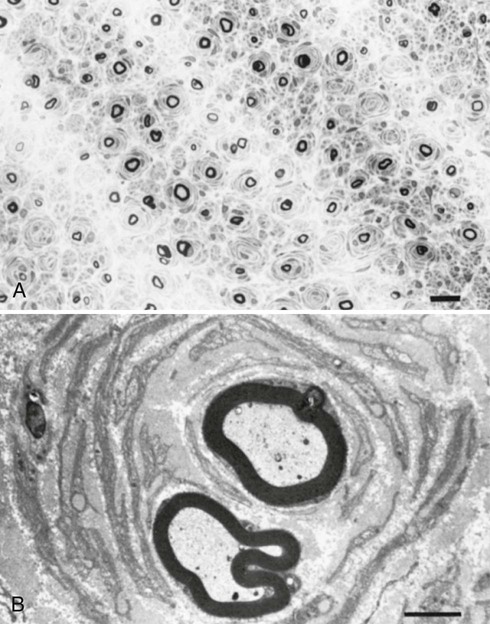
CMT1B is clinically indistinguishable from CMT1A but it only accounts for 10–20% of all CMT cases. It is caused by mutations in the myelin protein zero (P0; gene symbol, MPZ) gene, mapped to chromosome 1q22-23. MPZ is the major peripheral myelin glycoprotein and is thought to function as an adhesion molecule in the formation and compaction of peripheral myelin. It is a member of the immunoglobulin superfamily, with distinct extracellular transmembrane and intracellular domains. Mutations in the gene encoding for MPZ have been also associated with DSS, and congenital hypomyelination neuropathy. Different MPZ mutations result in divergent morphological effects on myelin sheaths, consisting of uncompacting of myelin or focal myelin foldings (Gabreëls-Festen et al., 1996). Motor conduction block was reported rarely in CMT1B patients with specific MPZ mutations (Street et al., 2002). Specific MPZ missense mutations have also been reported with a CMT2 phenotype, showing only mild slowing of NCVs (Marrosu et al., 1998). The Thr124 Met mutations in the MPZ gene have been detected in several families with a distinct CMT2 phenotype (CMT2J) characterized by late onset, marked sensory loss, and sometimes deafness, chronic cough, and pupillary abnormalities (De Jonghe et al., 1999).
CMT1C is caused by a mutation in lipopolysaccharide-induced tumor necrosis factor-alpha (LITAF/SIMPLE) gene, mapped to chromosome 16p13-12 expressed on Schwann cells. This gene encodes a lysosomal protein that may play a role in protein degradation pathways (Street et al., 2003). CMT1C used to be reserved for autosomal dominant CMT1 families not linked to either CMT1A or CMT1B. Affected individuals in these families manifest characteristic CMT1 symptoms.
CMT1D is mapped to chromosome 10q21-q22 and is due to mutation of the early growth response 2 gene (EGR2) which encodes a zinc-finger transcription factor expressed in myelinating Schwann cells that regulates the expression of myelin proteins including PMP22, P0, Cx32, and periaxin (Kamholz et al., 2000). EGR2 gene missense mutations have been also reported in patients with DSS, or congenital hypomyelination neuropathy (Timmerman et al., 1999; Warner et al., 1998). Respiratory compromise and cranial nerve dysfunction are commonly associated with EGR2 mutations (Szigeti et al., 2007). Other rare CMT1 subtypes include CMT1E and CMT1F.
Charcot–Marie–Tooth Disease Type 2
CMT2 constitutes one-third of all autosomal dominant CMT disease. It is associated with mutations in genes affecting intracellular processes such as axonal transport, membrane trafficking, and translation (see Chapter 50). Clinical symptoms begin later than in CMT1, most commonly in the second decade, but may be delayed until middle age or beyond. Foot and spinal deformities tend to be less prominent than in CMT1. The clinical features closely resemble those of CMT1 but differ in that peripheral nerves are not enlarged, and upper limb involvement, tremor, and general areflexia occur less frequently. However, in individual cases, it is often impossible to determine the type of CMT disease on the basis of clinical manifestation alone. Approximately 20% of affected individuals are asymptomatic.
CMT2A is the most common CMT2 subtype and accounts for 30% of CMT2 cases. CMT2A1, linked to chromosome 1p35, is caused by a mutation in kinesin protein involved in axonal transport of synaptic vesicles (Saito et al., 1997; Zhao et al., 2001). CMT2A2, which is responsible for most CMT2 families, shares clinical features of weakness and atrophy with CMT2A1 but has an earlier onset and is more severe. It may also be associated with optic atrophy. It is caused by mutations in the mitofusin 2 (MFN2) gene, with a locus on chromosome 1p36-p35. MFN2 protein is a mitochondrial fusion protein ubiquitously expressed in many tissues including peripheral nerves. In CMT2B, which is linked to chromosome 3q13-22, there is prominent sensory loss with foot ulcerations (De Jonghe et al., 1997). A mutation in the RAB7 gene, which encodes a small guanosine triphosphatase (GTPase) late endosomal protein, has been found to be causative (Verhoeven et al., 2003). This form of CMT is clinically very similar to hereditary sensory neuropathy type 1 (HSN1) but lacks spontaneous lancinating pain. Another distinct subgroup of severely affected patients, designated CMT2C (mapped to chromosome 12q24), develop vocal cord, intercostal, and diaphragmatic muscle weakness (Klein et al., 2003). Because of respiratory failure, the life expectancy of these patients is shortened. CMT2D, mapped to chromosome 7p14, is characterized by weakness and atrophy that is more severe in the hands than in the feet (Ionasescu et al., 1996b). In CMT2E, some patients within the same kindred and with an otherwise typical CMT2 phenotype may exhibit slowed motor nerve conduction that is much below the forearm cutoff value of 38 m/sec and a more severe clinical phenotype. This form of CMT is caused by mutations in genes that encode neurofilament light (NEFL) subunit, and patients may have axonal swelling (giant axons) and significant secondary demyelination on sural nerve biopsies (Fabrizi et al., 2006; Jordanova et al., 2003). CMT2F, caused by mutations in small heat shock protein 27 (Hsp27), is characterized by later onset (35–60 years), mild sensory impairment, and moderate to severely slowed NCVs of lower limbs but normal or mildly reduced velocities in the upper limbs. Mutation in Hsp27 may impair formation of the stable neurofilament network that is essential for the maintenance of peripheral nerves. CMT2G, reported in a Spanish family, has the same gene locus as CMT4H (see later discussion on type 4 disease) on chromosome 12q12-q13.3, with the age onset from 9 to 76 years. CMT2J, also designated as CMT2 with MPZ (myelin protein zero) gene mutation, is associated with pupillary abnormalities (Adie pupil) and hearing loss. CMT2L is caused by mutation in the HSPB8 gene and is associated with otherwise typical features of the CMT2 phenotype.
Motor NCV may be normal or mildly reduced. SNAPs are either absent or reduced in amplitude. Sural nerve biopsy specimens show preferential loss of large myelinated fibers, without significant demyelination; there may be clusters of regenerating myelinated fibers, a hallmark of axonal regeneration.
X-Linked Charcot–Marie–Tooth Disease
X-linked Charcot–Marie–Tooth disease (CMTX) is phenotypically similar to CMT1. Affected male subjects tend to be more severely affected, and females with the gene mutation may have a mild neuropathy or be asymptomatic. No male-to-male transmission occurs; hence, CMTX should be considered in any patient whose family history does not exhibit a male-to-male transmission. CMTX accounts for 7% to 16% of all forms of CMT, making it the second most common form of CMT (following CMT1A). It is caused by many mutations in GJB1, the gene that encodes connexin 32 (Cx32). The connexins are a family of highly related genes encoding a group of channel-forming proteins. Cx32 is a gap junction protein found in noncompacted paranodal loops and Schmidt-Lanterman incisures of Schwann cell cytoplasm, which is encoded by a four-exon gene located on chromosome Xq. As a gap junction protein, Cx32 forms small channels that facilitate transfer of ions and small molecules between Schwann cells and axons. More than 200 different mutations in Cx32 have been identified in CMTX families. Genotype-phenotype correlations among patients with Cx32 mutations suggest that most missense mutations result in a mild clinical phenotype, whereas nonsense and frameshift mutations produce more severe phenotypes (Ionasescu et al., 1996a).
Cx32 is expressed in Schwann cells and oligodendrocytes, regions of noncompact myelin (incisures and paranodes), as well as other non-neural cells. Some mutations of Cx32 have been reported to be associated with central nervous system (CNS) involvement with white-matter MRI and MR spectroscopy abnormalities, abnormal brainstem auditory evoked potentials, and deafness (Murru et al., 2006). An interesting phenomenon of transient and acute ataxia, dysarthria, and weakness occurring after visiting high altitudes and associated with CNS white-matter MRI abnormalities has been described in patients with two mutations: R142W and C168Y (Paulson et al., 2002). This suggests that CMTX patients should be cautioned about travel to high-altitude locations. It has been proposed that Cx32 mutations may cause these abnormalities by reducing the number of functional gap junctions between oligodendrocytes and astrocytes, making them more susceptible to changes in intercellular ions and small-molecule exchange that occur in situations of metabolic stress (e.g., high altitude).
Men with CMTX show significant slowing in NCV, whereas brainstem auditory evoked responses are often abnormal. A picture of both axonal loss and demyelination is revealed on nerve biopsy. Nerve conduction velocities in CMTX with Cx32 mutations range from near normal to intermediate slowing in the 30- to 40-m/sec range. There is debate as to whether CMTX should be classified as a primary axonal or demyelinating disorder (Birouk et al., 1998). However, careful studies of individual patients suggest nonuniform conduction slowing consistent with demyelination (Gutierrez et al., 2000; Lewis, 2000). Conduction slowing in heterozygous women, the slowing paralleling the loss of CMAP amplitude, may be subtle and frequently is in the range found in patients with CMT2. Before considering a diagnosis of CMT2 in such cases, it is important to review the family history. If there is no male-to-male transmission, the presence of intermediate conduction velocities (>42 m/sec) in female carriers and delayed brainstem auditory evoked response latencies in affected men is highly suggestive of Cx32 mutations (Nicholson et al., 1998).