Microglia
Critical Mediators of Pain Hypersensitivity after Peripheral Nerve Injury
Introduction
The world of pain research and therapy owes a great deal to the little-known German anesthetist and surgeon Carl Ludwig Schleich (1859–1922) for two specific contributions. First, Schleich was a pioneer of regional anesthesia and refined the technique considerably by introducing a new, safer method of infiltration anesthesia, as detailed in his book Schmerzlose Operationen (Painless Operations) (Schleich 1899). However, within that book are contained his theories of brain function, which make for remarkable reading. In what would be an extremely prescient proposal, Schleich rejected the accepted neural network concept of the time being championed by Sigmund Exner (1894) and suggested an active role for glial cells. It was while listening to a piano recital that he was struck with inspiration and announced “glia as a damper pedal, an apparatus for switching registers … an inhibition regulator” (Schleich 1921). Schleich postulated that glial cells control neuronal excitation in the brain, a theory now widely held and of intense research interest throughout neuroscience. Schleich lived in less enlightened times; both he and his theory were ignored, and he suffered the final ignominy of being described as though his own brain were “turning into glue” (Schleich 1921)—a reference to the Greek word for glue being the etymological root of glia.
Study of the nervous system has been a story of controversy since the first revelations of the inner structure of this “black box” were revealed, pioneered by discovery of the reazione nera or “black reaction,” the revolutionary tissue-staining technique of Camillo Golgi in 1873. Golgi’s silver staining revealed a new world of cellular structures, and it was immediately clear that other structures, distinct from neurons, were present in the tissue samples. These non-neuronal structures had no place in the prevalent theory of the day and were put to rest by one of the foremost physiologists of the time, Rudolf Virchow, who had previously dismissed these cells as Nervenkitt, or “nerve glue” (Virchow 1862). The glue cells were deemed to not contribute to the “physiological explanation of mental phenomena” (Exner 1894) and were subsequently ignored.
Though recognized as structural elements in their own right, this was the time of the advent of the “neuron doctrine.” This was established by the great Spanish histologist and founder of modern neuroanatomy Santiago Ramon y Cajal, who posited the nervous system as being made up of discrete individual cells. It was in opposition to Golgi himself who had developed his reticular theory proposing that every neuron in the entire nervous system is physically linked with its neighbors (ironically, a system that has more resonance with the structure of astrocytes than neurons). Cajal’s improvement of Golgi’s pioneering staining techniques showed clear differentiation of neurons from neuroglia (now known as astrocytes and the source of Schleich’s fascination), but it also revealed a further population of cells that he termed the “third element” (Andres-Barquin 2002). It was to be the work of Cajal’s student Pio del Rio Hortega to unravel the mystery of this third element (Penfield 1965, Rezaie and Male 2002). By further refining the metallic impregnation techniques of Cajal, he was able to successfully stain this cell population and in 1919 identify and define it as two distinct populations that he named microglia and oligodendroglia (Penfield 1965). This was an extremely controversial claim at the time, and debate raged between Rio Hortega and Cajal about the nature of the third element and stimulated a surge of research on the function of these enigmatic cells. However, this interest soon dwindled, and by the mid-20th century, microglia were once again the neglected cell population in the central nervous system (CNS) (Rezaie and Male 2002). Interest was sparked anew toward the end of the century with the realization that microglia are the resident macrophage population and therefore the immune effectors of the CNS, and in more recent years the role of microglia in particular in CNS function and malfunction has been revisited (Hanisch and Kettenmann 2007, Ransohoff and Perry 2009, Graeber 2010, Kettenmann et al 2011). Given their immune role, microglia represent the first line of defense against damage to the CNS, and with the understanding that peripheral neuropathy is manifested as a pathological state of the CNS (Costigan et al 2009b, Woolf 2010), there is intense interest in their role in pain pathophysiology.
Microglia in the Modern Era of Neuroscience
Microglia are now known to derive from a distinct macrophage population that comes from embryonic myeloid progenitors in the yolk sac (Ginhoux et al 2010), and they invade and populate the CNS through the pial membranes (Cuadros and Navascués 1998, Ginhoux et al 2010). Microglia have been likened to the electricians of the CNS; they exist outside the neuronal circuit and are able to delve in and modulate the electrical activity within (Graeber 2010). Unlike the reticular-like syncytial system of astrocytes throughout the CNS, microglia are not physically connected but reside within their own adjacent, non-overlapping microdomains within the brain and spinal cord (Fig. 4-1). Such a spatially restricted system allows the microglial response to react in an anatomically precise fashion after pathology or damage. Under physiological conditions, quiescent microglia are not “resting” but are in a state of motility and surveillance, with cellular processes continuously scanning their microenvironment (Exner 1894, Wake et al 2009).
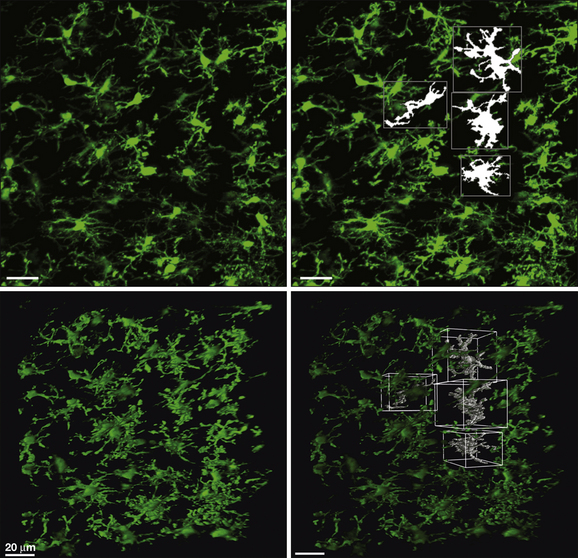
Figure 4-1 Microglia and their microdomains in the intact spinal cord.
Two photon photomicrographs of dorsal horn microglia taken from the intact in situ spinal cord of CX3CR1GFP mice. Top left: Extended focus image showing the grid-like distribution of microglia in the parenchyma of the normal spinal cord dorsal horn. Top right: Four microglia surface-rendered with Volocity (Perkin Elmer) software to show the adjacent, non-overlapping microdomains of individual cells. The bottom panels are three-dimensional projections of the top panels.
How this grid-like network of microglia that extends throughout the CNS interacts and modulates the underlying cellular circuitry of the CNS is of considerable interest as a fundamental cellular mediator underlying the pathophysiology of neuropathic pain. The extensive toolbox that microglia possess in terms of cytokines, chemokines, neurotrophins, and neurotransmitters has made this population of glial cells a rich seam of research, and a wealth of knowledge now exists and also remains to be discovered.
Spinal Microglia as Intermediaries in the Pathobiology of Neuropathic Pain
Considerable progress has been made since microglia were heralded as “sensors of pathology” (Kreutzberg 1996), but it is important to reject the inflexible notion of microglial “activation” as a causative factor underlying peripheral nerve injury (PNI)-induced pain behavior. The classic morphological and proliferative responses of microglia within the spinal cord following PNI (Fig. 4-2) are a signifier of microglia reactivity but do not necessarily constitute a “pro-pain” phenotype. A common misconception is that resolving these microglial signatures following nerve damage will resolve the behavioral changes. That being said, microglia in the spinal dorsal horn do respond to and are critical mediators of the pathobiology of PNI; all existing neuropathic pain models now envisage some requisite degree of spinal microglial response (Beggs and Salter 2006, 2007) (Fig. 4-3). However, such microglial changes are less evident in inflammatory and chemotoxic models of pain (Honore et al 2000, Clark et al 2007, Lin et al 2007), and the role of microglia in the pain states that result from these insults remains to be elucidated (Li et al 2010). As stated above, microglia exist throughout the neuraxis, including at the level of primary afferents, spinal nociceptive circuitry, and projections to the brain. For a definitive causal role of microglia in pain to be stated, tests of both sufficiency and necessity must be proven.
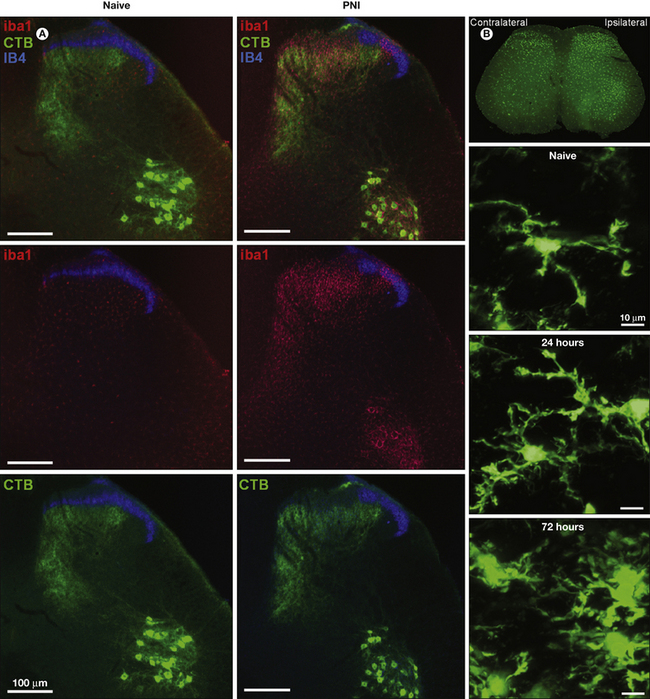
Figure 4-2 Proliferation of spinal microglia after peripheral nerve injury.
Spinal microglia proliferate around the central terminals of peripherally axotomized primary afferents. A, Iba1-immunostained microglia (red) are densely packed around the central terminals of primary afferents and retrogradely labeled with fluorescent cholera toxin B subunit (CTB; green). B, Two photon microscopic images of microglia in the intact in situ spinal cord of CX3CR1GFP mice. Proliferative and morphological changes can be seen by 24 hours after peripheral nerve injury.
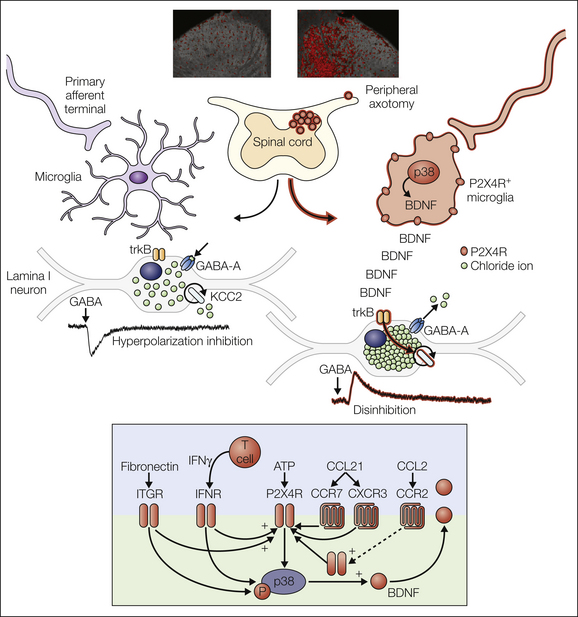
Figure 4-3 The P2X4R+ microglial phenotype mediates a core pain hypersensitivity cascade following peripheral nerve injury.
Chronic neuropathic pain is generated in part by pathological amplification of input to the nociceptive network of the central nervous system. A growing literature has established that neuron–glial interactions within the spinal cord are responsible, at least in part, for the enhanced output of this network. Some of the key molecular components of these interactions are summarized here. A specific microglial phenotypic state characterized by up-regulated P2X4R expression (P2X4R+) is induced by peripheral nerve injury and has been shown to play a critical role in the pathological changes in nociceptive processing that underlie neuropathic pain. The lower panel shows the complex modulation of P2X4R expression by various elements of the parenchymal environment: extracellular matrix (fibronectin; Tsuda et al 2008a, 2009c), infiltrating T cells (Costigan et al 2009a, Tsuda al 2009b), and cytokines and chemokines (Zhang et al 2007, Abbadie et al 2009, Clark et al 2009, Biber et al 2011, Toyomitsu et al 2012).
What are the Upstream Regulators of the Spinal Microglia Response to Peripheral Nerve Injury?
The initial critical event for changes in the spinal cord microglial phenotype following PNI is elicited in the injured primary afferents themselves. It is known that the activity of nociceptors is sufficient to elicit a microglial response inasmuch as electrical stimulation of a peripheral nerve at C-fiber intensity will induce microglial proliferation in otherwise naïve animals (Hathway et al 2009). This microgliosis is contiguous with the onset of mechanical hypersensitivity in these animals. Electrical stimulation at such intensity will stimulate all fibers, and Suter and colleagues further refined this effect to show that blockade of transient receptor potential vanilloid 1 (TRPV1)-expressing afferents (the majority of C fibers) did not diminish the microglial proliferation whereas complete blockade of all discharge activity in the nerve with bupivacaine did (Wen et al 2007, Suter et al 2009). It is possible that the residual non–TRPV1-expressing C fibers are responsible for the microglial response. However, a more parsimonious explanation would suggest a role for A-fiber activity (Suter et al 2009). Corroborating evidence for afferent discharge activity driving microglial changes comes from the observation that the discrete boundaries of microglial proliferation map the anatomical boundaries of the central terminal fields of the injured nerve (Beggs and Salter 2007). Spinal microglia express receptors for many neurotransmitters (Pocock and Kettenmann 2007) and are therefore ideally placed to respond to sustained release of pro-inflammatory neurogenic factors following PNI. However, these features are not evidence of a causative role for spinal microglial proliferation in the development or maintenance of neuropathic pain, and it is perhaps more pertinent to describe the proliferative microgliosis as a signifier of peripheral nerve damage than in terms of being explicitly algogenic. Indeed, early studies of microglial proliferation following PNI suggested that microglia were involved in the transganglionic degenerative response (Graeber et al 1988, Eriksson et al 1993, Svensson et al 1994).
Interpreting Findings with so-Called Glial Inhibitors
An important consideration in studying the potential role of microglia—or other types of glial cells—in pain is the use of compounds that are widely touted as “glial inhibitors.” The compounds most commonly used for their glia-inhibiting activity are minocycline and propentofylline. It is important to note that these are broad-spectrum agents with anti-inflammatory properties. Although these compounds are often described as being glial-specific inhibitors, it is perhaps too narrow a description of the activity of these drugs. It has been shown that administration of minocycline and propentofylline is more effective in preventing than in reversing nerve injury–induced chronic pain behavior (Raghavendra et al 2003a, 2003b; Ledeboer et al 2005). One interpretation could be that microglia have only a transient role in neuropathic pain. An alternative is that these compounds are active elsewhere, for example, attenuating the discharge activity of primary afferents following injury (Gong et al 2010). In either case, the clear conclusion is to exercise caution in attributing erroneous cellular specificity to these compounds. These most commonly used “glial inhibitors” also function by probably non-specific anti-inflammatory activity.
The Critical Role of Microglial P2X4 Receptors in Peripheral Nerve Injury–Induced Pain Hypersensitivity
Chronic neuropathic pain is characterized by altered afferent input to the spinal cord and amplification of that input within the nociceptive network in the spinal cord at the segmental level (Woolf and Salter 2000). Sensory processing within the dorsal horn involves a complexly organized network of local and descending inhibitory and excitatory modulation (Costigan et al 2009b). Generation of pathological pain arises from a distorted output from the spinal cord to higher areas of the CNS involved in sensory and affective processing. The “distortion” is achieved through the suppression of inhibition and enhancement of excitatory transmission (De Koninck 2007; see Chapter 6). Conventional neurocentric bias has concentrated on neuron–neuron signaling underlying these effects, but there is now intense interest in a neuroimmune contribution, and the cell population that represents the immune side is microglia (Beggs and Salter 2010). How do neuron–glial interactions contribute to the enhancement of nociceptive output? There is now a considerable canon of literature detailing a plethora of potential molecular links between neurons and microglia and their involvement in the pathogenesis of neuropathic pain (Andres-Barquin 2002, Inoue and Tsuda 2009, Beggs and Salter 2010, Gosselin et al 2010, Calvo and Bennett 2012). However, converging lines of evidence currently point to enhanced expression of the purinergic receptor P2X4 (P2X4R) as playing a key role in neuropathic pain pathophysiology (see Fig. 4-3). Although many molecules have been implicated in mediating the microglial changes underlying the initiation and/or maintenance of chronic pain states, it is the P2X4R-expressing microglial phenotype, its intra- and intercellular signaling pathways, and consequent transformation of spinal output that have been systematically verified and describe a causal role of spinal microglia in the development of PNI-induced chronic pain (Tsuda et al 2003, 2008a; Coull et al 2005; Ulmann et al 2008; Trang et al 2009; Beggs et al 2012).
The initial series of observations that identified P2X4Rs as a critical molecular element of the neuroglial signaling underlying neuropathic pain came from Tsuda and colleagues (2003). The first indicator was a progressive increase in P2X4R protein in the ipsilateral spinal cord of rats with a PNI. Moreover, this increase correlated with the emergence of tactile allodynia (Tsuda et al 2003). Of considerable surprise at the time, it was revealed immunohistochemically that increased expression of P2X4Rs was confined to microglia. An ongoing problem with parsing the actions of P2X subtypes has been the lack of functional pharmacological tools (Jarvis and Khakh 2009). However, exploitation of the differences in pharmacological profiles of the P2X antagonists TNP-ATP and PPADs, the former reversing tactile allodynia in neuropathic rats and the latter having no effect, indicated that P2X4R was the active receptor mediating the pain behavior. Furthermore, because the behavioral allodynia could be transiently reversed by intrathecal administration of the antagonist, it could be surmised that ongoing P2X4R activation is required to maintain nerve injury–induced allodynia. The demonstration that P2X4R antisense oligonucleotide treatment had a similar action (Tsuda et al 2003) provided further confirmation. The pharmacological and behavioral evidence was subsequently corroborated by a genetic approach: mice deficient in P2X4R have dramatically reduced pain behavior following PNI (Ulmann et al 2008, Tsuda et al 2009a). This latter study also contained the surprising revelation that the proliferative response of microglia to PNI was undiminished in P2X4R−/− mice yet their behavioral responses were absent. In other words, although tonic P2X4R activation is required for maintenance of PNI-induced allodynia, proliferation and up-regulation of microglial markers in the spinal dorsal horn are independent. These experiments demonstrated the necessity of P2X4Rs as an active component in neuropathic pain but do not preclude the possibility of intermediary factors being required (i.e., sufficiency had not been demonstrated). This was definitively shown in experiments in which P2X4R-stimulated microglia were injected intrathecally into naïve rats and induced tactile allodynia similar to that seen in neuropathic rats (Tsuda et al 2003, Coull et al 2005). The pharmacological, genetic, and behavioral battery of experiments provided the requisite evidence to show sufficiency and necessity of P2X4Rs in mediating neuropathic pain behavior in rats and therefore logically a causative role. The question then turned to the effectors and effects of P2X4R up-regulation in the development and maintenance of neuropathic pain.
Modulators of P2X4R Expression and Function
For microglia resident within the protected confines of the spinal dorsal horn parenchyma to contribute to altered spinal output to the brain following peripheral nerve damage, there must be a signaling event or events between the injured primary afferent and the spinal environment. If a key central component of the neuroglial signaling pathway is P2X4R up-regulation in spinal microglia, what signals that up-regulation? There have been a number of advances recently that address this question, and several signaling molecules have been implicated, including members of the chemokine, cytokine, extracellular matrix molecule, and protease families. These include CCL21, a neuronally released chemokine (de Jong et al 2005, 2008; Biber et al 2011). Importantly, the authors showed this signaling event to be dependent on neuronal injury, and crucially, the details of transport of CCL21 from the neuronal to the microglial environment and signals via the chemokine receptor CXCR3 (de Jong et al 2005) that precede P2X4R up-regulation have been elucidated. Of further interest to future translational studies, it has also been demonstrated that the same signaling event occurs with the human chemokine homologues (Dijkstra et al 2004). The cytokine interferon-γ has been shown to transform quiescent spinal microglia into a P2X4R-expressing phenotype (Tsuda et al 2009b). P2X4R expression levels seem to be critically dependent on the extracellular matrix molecule fibronectin (Nasu-Tada et al 2006; Tsuda et al 2008a, 2009c). Further studies revealed the Lyn kinase signaling pathway as mediating this event, in turn modulating the transcriptional and post-transcriptional levels of microglial P2X4R expression (Tsuda et al 2008b). Mast cells have also been shown to modulate P2X4R expression through release of the protease tryptase, which activates proteinase-activated receptor 2 (PAR-2) in microglia (Yuan et al 2010). Several other signaling pathways have been described that involve a primary afferent–microglial signaling component. These pathways include CCL2 (also known as monocyte chemoattractant protein 1 [MCP-1]), which is expressed on primary sensory neurons and is present in their central terminals and whose cognate receptor is present on microglia (Zhang et al 2007, Abbadie et al 2009), activation of which promotes membrane expression of P2X4Rs (Toyomitsu et al 2012). Whether these examples represent converging pathways that are mechanistically intertwined or else exist alone as independent signaling pathways remains to be resolved.
Cell Biology of Microglial P2X4R Expression and Function
Although mechanistically relevant changes in receptor expression are often couched in the simplistic language of up- and/or down-regulation, the functionally meaningful activity of receptors such as P2X4R occurs on the cell surface, regulated by constitutive internalization and reinsertion of the receptors into the cell membrane (Bobanovic et al 2002, Toulmé et al 2006, Fujii et al 2011). Much is known of P2X4R dynamics within the cell (which is beyond the scope of this review), but of considerable importance has been elucidation of the crystal structure of zebra fish P2X4R and subsequent details of the extracellular domain, putative adenosine triphosphate (ATP) binding site, transmembrane regions, and ion permeation pathway (Kawate et al 2009). Of potentially greatest importance with respect to P2X4R signaling is discovery of the ability of the receptor to adopt two distinct structural conformations. Using fast-scanning atomic force microscopy, Shinozaki and colleagues (2009) showed that ATP stimulation in the presence of extracellular Ca2+ causes the P2X4R to open a non-selective cation-permeable channel but that in the absence of extracellular Ca2+, the receptor undergoes pore dilatation and forms a macropore, which renders the receptor permeable to larger molecules. Importantly, microglial P2X4Rs have been shown to possess this ability to function in both conformations (Bernier et al 2008). Clearly, the implications for enhanced signaling capability, especially given the phenotypic change to high P2X4R expression following PNI, are enormous. However, the question of whether this function is physiologically relevant in the etiology of neuropathic pain remains unanswered.
p38 Mitogen-Activated Protein Kinase Mediates Microglial Signaling
A critical question for microglial signaling mechanisms is what are the intracellular pathways that mediate the myriad molecular systems? Does convergence account for the parallel signaling events occurring through activation of different populations of receptors? Considerable evidence has identified the mitogen-activated protein kinases (MAPKs) as a candidate family of intracellular mediators (Ji and Suter 2007, Ji et al 2009, Wen et al 2009). Of the MAPKs, p38 appears to be heavily implicated in microglia-mediated, post–nerve injury pain states (Jin et al 2003, Tsuda et al 2004) and has been identified as the intracellular mediator of P2X4R–brain-derived neurotrophic factor (BDNF) signaling in microglia (Trang et al 2009).
P2X4R-Mediated Release of Brain-Derived Neurotrophic Factor
Parallel or converging pathways from a variety of cellular and molecular substrates regulate and influence microglial P2X4R expression. Considerable evidence points to p38 as a common intracellular mediator (see Fig. 4-3). The step from altered primary afferent input to spinal microglia modulation has been bridged. However, it is pertinent to reiterate that given the considerable interplay between neuronal and glial components at the spinal level, ultimately it is neuronal signaling, via spinal projection neurons to centers in the brain stem and brain, that completes the necessary “geography” of the pathway that ultimately leads from nerve injury in the periphery to “pain” in the brain. Again, it should be reiterated that a common misconception is to imbue altered microglial activity in the spinal dorsal horn with a causative role. This is too broad an interpretation and skews the logic of correlation and causation. A signaling component from microglia to second-order dorsal horn neuron is required to complete the circuit. In spinal microglia, influx of Ca2+ through the P2X4R is a critical step that fulfils this requirement by linking stimulation of these receptors to the synthesis and release of BDNF (Trang et al 2009). In vivo, P2X4R-deficient mice exhibit impaired microglial BDNF release and altered BDNF signaling in the spinal cord (Ulmann et al 2008), and given the disruption of the P2X4R–BDNF pathway, mechanical allodynia does not develop in P2X4R null mice following PNI. Where the microglial transfer experiments earlier satisfied the conditions of sufficiency of microglial-derived BDNF to drive neuropathic pain behavior, studies by Ulmann and associates demonstrated that release of BDNF, driven by activation of P2X4R , is necessary for the development of neuropathic pain. Microglia as a source of BDNF within the spinal cord following PNI was countercurrent to contemporary thinking. Even though BDNF was considered a probable mediator of central cellular processes involved in pain states, primary afferents were the default source. Although a number of studies showed promising changes in expression of BDNF in spinal ganglia following PNI (Obata et al 2003a, 2003b; Pezet and McMahon 2006), it was subsequently demonstrated that primary afferent-evoked release of BDNF in the spinal cord was unaltered after nerve injury (Lever et al 2003). Convincing support came from a conditional knockout mouse study in which selective deletion of BDNF from primary afferent neurons resulted in no effect on nerve injury–induced mechanical allodynia (but strong suppression of pain behavior in a number of inflammatory pain models) (Zhao et al 2006). This presented something of a conundrum until a series of experiments provided the first evidence that in neuropathic pain states, it is indeed BDNF that affects nociceptive processing in the spinal cord but that the source of the BDNF was from microglia (Coull et al 2005).
Previous studies had proposed that a central mechanism underlying pain behavior was disruption of anion homeostasis manifested as a depolarizing shift in the equilibrium potential of γ-aminobutyric acid (GABA)-mediated chloride currents (Coull et al. 2003). This disruption, which affects the intrinsic circuitry of the spinal dorsal horn, results in a weakening of inhibitory tone within the nociceptive circuitry of the spinal cord. The underlying molecular mechanism was shown to be a rapid nerve injury–induced down-regulation of the neuronal chloride transporter KCC2 (Coull et al 2003, Prescott et al 2006). It was known that BDNF can down-regulate KCC2 levels under pathophysiological conditions in the hippocampus (Rivera et al 2002, 2004), therefore raising the possibility of a mechanistic link between P2X4R-dependent BDNF release and spinal KCC2 down-regulation. It was subsequently shown that intrathecal administration of P2X4R-stimulated microglia into naïve, uninjured rats causes a depolarizing shift in Eanion in spinal lamina I neurons that was sufficient to reduce inhibition and, furthermore, that in approximately one-third of neurons, GABAergic responses became excitatory. The consequence of this altered inhibitory response, as will be described later, is to produce a phenotypic switch in spinal lamina I neurons such that they relay innocuous mechanical input, increase discharge when presented with a noxious stimulus, and display spontaneous activity (Coull et al 2005, Keller et al 2007). The signaling role of BDNF in this observation was shown by a series of findings. First, intrathecal BDNF mimicked both the mechanical allodynia and depolarized Eanion caused by PNI or administration of P2X4R-stimulated microglia. Second, blocking BDNF-TrkB (tyrosine kinase receptor B) signaling either with a function-blocking antibody or by sequestration of free BDNF with TrkB–Fc fusion proteins prevented the mechanical allodynia evoked by P2X4R-stimulated microglia. Third and crucially, knockdown of BDNF expression in microglia with small interfering RNA (siRNA) abolished the effects of intrathecally administered ATP-stimulated microglia and resulted in an absence of both depolarized Eanion and pain behavior. Taken together, these findings provide compelling evidence that BDNF from microglia is a critical signaling molecule mediating the central pathophysiological effects of PNI.
Transformation of Lamina I Output May Underlie Symptoms of Neuropathic Pain
The final stage in nociceptive processing within the dorsal horn involves the transmission of sensory information from the spinal segmental level rostrally to higher centers in the CNS. There are broadly two populations of dorsal horn neurons, located in lamina I and lamina V of the dorsal horn, that project to the brain stem and thalamus and provide a nociceptive output pathway from the spinal cord. Lamina I neurons differ from their deeper counterparts in that they receive limited direct input from low-threshold A fibers under normal conditions. This nociceptive bias and specificity (in control animals, less than 25% of lamina I projection neurons respond to low-threshold innocuous stimuli; Keller et al 2007) therefore ensures that in the normal situation, lamina I neurons preferentially encode noxious and thermal input only (Bester et al 2000). However, tactile allodynia essentially requires that innocuous input elicit a nociceptive response. Because lamina I projection neurons are effectively nociceptive-specific output neurons, a mechanism is required that allows these neurons to become responsive to innocuous peripheral stimulation and elicit a noxious sensation supraspinally. Keller and co-workers (2007) showed that PNI causes a functional switch in lamina I projection neuron specificity such that the majority of the cells recorded, rather than being nociceptive specific, now responded to low-threshold tactile stimulation. The mechanism for this change has been the cause of some debate. It may represent an unmasking of polysynaptic connectivity within the dorsal horn such that low-threshold input can functionally activate lamina I projection neurons (Baba et al 2003, Kohno et al 2003) by either strengthening them, reducing their inhibition, inducing excitatory interneuron input, lowering the threshold of excitation of projection neurons via pre-existing subthreshold input, or switching the action of a subpopulation of inhibitory interneurons from inhibitory to excitatory (Coull et al 2003). Compelling evidence for the latter comes from the observation that acute disruption of chloride homeostasis in naïve animals in vivo switches the phenotype of identified lamina I projection neurons from nociceptive specific to wide dynamic range, in essence unmasking innocuous afferent input to the nociceptive spinal circuitry (Keller et al 2007). More pertinently, the same effect could be mimicked by acute spinal application of ATP-stimulated microglia (Keller et al 2007). This finding is the principal evidence that microglia functionally affect the output characteristics of the spinal cord nociceptive network to higher centers in the CNS, a requirement for a causal role of microglia in the ontogeny of nerve injury–induced pain states (Fig. 4-4).
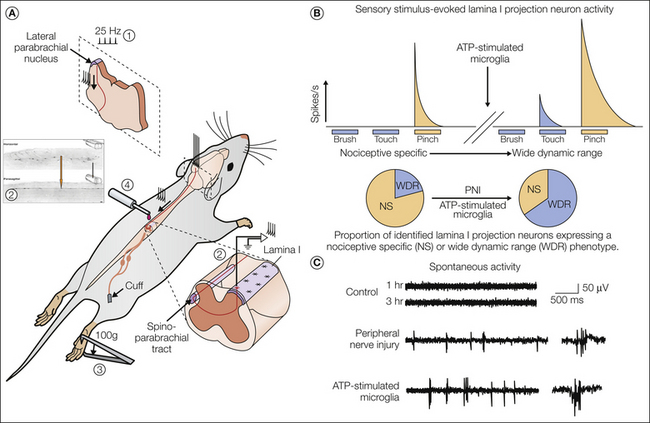
Figure 4-4 Transformation of lamina I output neurons reveals the symptoms of neuropathic pain.
A, Recording of single antidromically identified (1) lamina I projection neurons (2) in vivo. Neuronal responses to natural stimuli (brush, touch, pinch) (3) were made before and after adenosine triphosphate (ATP)-stimulated microglia were acutely administered into the spinal cord (4) (Keller et al 2007). B, The top panel shows the transformation of a single lamina I projection neuron from nociceptive specific (NS; no responses to innocuous tactile stimuli) to wide dynamic range (WDR; emergence of responsivity to tactile stimuli) after acute exposure of the spinal cord of an anesthetized rat to ATP-stimulated microglia. These changes mimic those seen following peripheral nerve injury (PNI). The lower panel shows a reversal in the relative proportions of NS and WDR phenotypes of a population of identified lamina I projection neurons after either PNI or acute microglial exposure. C, Lamina I projections in naïve animals are generally quiescent and show little or no activity. However, following PNI or microglial treatment, these neurons start to fire spontaneously. The responsivity of previously NS neurons to innocuous stimuli and the development of spontaneous activity may represent physiological correlates to the allodynia and spontaneous pain experienced by patients with neuropathic pain.
Preclinical models of pain typically use evoked responses as behavioral readouts of mechanical and thermal sensitivity. However, clinically, neuropathic pain is characterized by ongoing spontaneous pain, as well as exaggerated evoked pain responses (Baron et al 2010). Such a sensation requires ongoing, or episodic, activity at some point in the nociceptive pathway in the absence of an aberrant stimulus. Lamina I projection neurons are quiescent in the absence of nociceptive input (Craig and Kniffki 1985, Keller et al 2007) and display no spontaneous activity. However, following PNI, epileptiform-like burst activity can be seen in these neurons. This same activity can be reproduced in naïve animals by direct disruption of chloride homeostasis or by the acute spinal application of ATP-stimulated microglia (Keller et al 2007). These observations of changes in the response properties, selectivity, and discharge activity of spinal cord lamina I neurons may provide a biologically plausible mechanism for the cardinal symptoms of neuropathic pain: hyperalgesia, allodynia, and spontaneous pain.
Conclusion
Microglia are able to respond in different ways to different stimuli and adopt an appropriate phenotype for a given stimulus. This phenotypic diversity includes proliferative, migrational, and phagocytic responses associated with a canon of expression of pro- and anti-inflammatory molecules. These responses are in addition to the immune role of microglia in antigen presentation and T-cell recruitment. The degree to which these phenotypes represent extant responses with discrete molecular signaling components is not clear, but a detailed picture of the molecular complexity involved is emerging (Hanisch and Kettenmann 2007). The most important message is that microglial responses are complex and tuned to the nature of the stimulus. A common conceptual error in the role of glial responses in mechanistic studies of neuropathic pain is too broad an interpretation being drawn from these responses. Given the multimodal capability of microglia to respond to peripheral nerve damage, correlative and causative conclusions are often muddled. Advances in understanding the microglial molecular machinery have only highlighted the dearth of precise tools required to identify the specific processes and specific cell populations involved in the etiology of neuropathic pain. Furthermore, the possibility of over-interpretation through experimental design imprecision remains ever present. It is still the case that quantification and analysis of microglial changes in the literature are generally limited to a very small number of cellular proteins (e.g., iba1, ox42). Even though antibodies against these proteins generally have a high degree of sensitivity and specificity, thereby providing useful immunohistochemical tools for visualizing changes in morphology and cell density, it is unknown whether these proteins are surrogates for any specific functional change in microglial activity relevant to pain or any other function. On the contrary, any phenotypic diversity within a proliferating microglial population in pathological states would be masked by the homogenizing labeling with these markers. Sophisticated optical and genetic techniques allow imaging of microglia in vivo, thus providing insight into the morphological and motile reactivity of microglia in the living animal (Exner 1894, Davalos et al 2005). Yet it is ironic that although the great controversy between the discoverer of microglia Rio Hortega and his mentor Cajal was based on histological clarity, both literally and figuratively, it is the histological evaluation of microglial morphology that remains one of the foremost signifiers of pathological processes in the CNS.
The incredible complexity of microglia activity and responsivity may represent the adoption of stimulus-specific phenotypes. Any potential therapeutic intervention without defined specificity, such as a general glial inhibitor, is not necessarily desirable and, given their role in the immune system, potentially catastrophic. A case in point may be a recent report on propentofylline, purported to be a non-specific glial inhibitor, which was found to not affect pain intensity in patients with long-standing post-herpetic neuralgia (Landry et al 2012, Watkins et al 2012). Although the reason for this lack of efficacy is unclear, it may be that this drug did not suppress the requisite specific glial pathway. Whether glia play a role in clinical pain remains an open question. However, we anticipate that any advance in tackling chronic pain therapeutically by targeting microglia will need to involve the development of molecules or compounds capable of targeting a specific microglial phenotype with the ability to reverse, as well as prevent, the adoption of that specific phenotype. It is important to also consider the important roles that microglia have within the immune system of the non-pathological CNS and the potential deleterious effects of inhibiting their function. However, the remarkable plasticity and phenotypic diversity of microglia offer a compelling opportunity to identify the molecular mediators of stimulus-dependent microglial changes and to tailor therapies targeted at them.
It has been more than a century since Carl Gustav Schleich proposed a non-neuronal “switching mechanism which organized and regulated the ebb and flow of nervous excitation” (Schleich 1921, Anonymous 2006). The full impact of that statement is only now beginning to be fully realized.
The references for this chapter can be found at www.expertconsult.com.
References
Abbadie C., Bhangoo S., De Koninck Y., et al. Chemokines and pain mechanisms. Brain Research Reviews. 2009;60:125–134.
Andres-Barquin P.J. Santiago Ramón y Cajal and the Spanish School of Neurology. Lancet Neurology. 2002;1:445–452.
Baba H., Ji R.-R., Kohno T., et al. Removal of GABAergic inhibition facilitates polysynaptic A fiber–mediated excitatory transmission to the superficial spinal dorsal horn. Molecular and Cellular Neurosciences. 2003;24:818–830.
Baron R., Binder A., Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurology. 2010;9:807–819.
Beggs S., Salter M.W. Neuropathic pain: symptoms, models, and mechanisms. Drug Development Research. 2006;67:289–301.
Beggs S., Salter M.W. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain, Behavior, and Immunity. 2007;21:624–633.
Beggs S., Salter M.W. Microglia-neuronal signalling in neuropathic pain hypersensitivity 2.0. Current Opinion in Neurobiology. 2010;20:474–480.
Beggs S., Trang T., Salter M.W. The P2X4R+ microglial state: an essential role in neuropathic pain. Nature Neuroscience. 2012. In press
Bernier L.-P., Ase A.R., Chevallier S., et al. Phosphoinositides regulate P2X4 ATP-gated channels through direct interactions. Journal of Neuroscience. 2008;28:12938–12945.
Bester H., Chapman V., Besson J.M., et al. Physiological properties of the lamina I spinoparabrachial neurons in the rat. Journal of Neurophysiology. 2000;83:2239–2259.
Biber K., Tsuda M., Tozaki-Saitoh H., et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO Journal. 2011;30:1864–1873.
Bobanovic L.K., Royle S.J., Murrell-Lagnado R.D. P2X receptor trafficking in neurons is subunit specific. Journal of Neuroscience. 2002;22:4814–4824.
Calvo M., Bennett D.L.H. The mechanisms of microgliosis and pain following peripheral nerve injury. Experimental Neurology. 2012;234:271–282.
Clark A.K., Gentry C., Bradbury E.J., et al. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. European Journal of Pain. 2007;11:223–230.
Clark A.K., Yip P.K., Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. Journal of Neuroscience. 2009;29:6945–6954.
Costigan M., Moss A., Latremoliere A., et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain–like hypersensitivity. Journal of Neuroscience. 2009;29:14415–14422.
Costigan M., Scholz J., Woolf C.J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual Review of Neuroscience. 2009;32:1–32.
Coull J.A.M., Beggs S., Boudreau D., et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021.
Coull J.A.M., Boudreau D., Bachand K., et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942.
Craig A.D., Kniffki K.D. Spinothalamic lumbosacral lamina I cells responsive to skin and muscle stimulation in the cat. Journal of Physiology. 1985;365:197–221.
Cuadros M.A., Navascués J. The origin and differentiation of microglial cells during development. Progress in Neurobiology. 1998;56:173–189.
Davalos D., Grutzendler J., Yang G., et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8:752–758.
de Jong E.K., Dijkstra I.M., Hensens M., et al. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. Journal of Neuroscience. 2005;25:7548–7557.
de Jong E.K., Vinet J., Stanulovic V.S., et al. Expression, transport, and axonal sorting of neuronal CCL21 in large dense-core vesicles. FASEB Journal. 2008;22:4136–4145.
De Koninck Y. Altered chloride homeostasis in neurological disorders: a new target. Current Opinion in Pharmacology. 2007;7:93–99.
Dierig S. Beyond the temples of science: bohemian neuroscience in fin-de-siècle Berlin, 2006, The Virtual Laboratory (ISSN 1866-4784), http://vlp.mpiwg-berlin.mpg.de/references?id=art44.
Dijkstra I.M., Hulshof S., van der Valk P., et al. Cutting edge: activity of human adult microglia in response to CC chemokine ligand 21. Journal of Immunology. 2004;172:2744–2747.
Eriksson N.P., Persson J.K., Svensson M., et al. A quantitative analysis of the microglial cell reaction in central primary sensory projection territories following peripheral nerve injury in the adult rat. Experimental Brain Research. 1993;96:19–27.
Exner S. Entwurf zu einer physiologischen Erklärung der psychischen Erscheinungen. F. Deuticke. 1894.
Fujii K., Young M.T., Harris K.D.M. Exploiting powder x-ray diffraction for direct structure determination in structural biology: the P2X4 receptor trafficking motif YEQGL. Journal of Structural Biology. 2011;174:461–467.
Ginhoux F., Greter M., Leboeuf M., et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845.
Gong K., Yue Y., Zou X., et al. Minocycline inhibits the enhancement of antidromic primary afferent stimulation-evoked vasodilation following intradermal capsaicin injection. Neuroscience Letters. 2010;482:177–181.
Gosselin R.-D., Suter M.R., Ji R.-R., et al. Glial cells and chronic pain. Neuroscientist. 2010;16:519–531.
Graeber M.B. Changing face of microglia. Science. 2010;330:783–788.
Graeber M.B., Tetzlaff W., Streit W.J., et al. Microglial cells but not astrocytes undergo mitosis following rat facial nerve axotomy. Neuroscience Letters. 1988;85:317–321.
Hanisch U.-K., Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature Neuroscience. 2007;10:1387–1394.
Hathway G.J., Vega-Avelaira D., Moss A., et al. Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain. 2009;144:110–118.
Honore P., Rogers S.D., Schwei M.J., et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98:585–598.
Inoue K., Tsuda M. Microglia and neuropathic pain. Glia. 2009;57:1469–1479.
Jarvis M.F., Khakh B.S. ATP-gated P2X cation-channels. Neuropharmacology. 2009;56:208–215.
Ji R.-R., Gereau R.W., 4th., Malcangio M., et al. MAP kinase and pain. Brain Research Reviews. 2009;60:135–148.
Ji R.-R., Suter M.R. p38 MAPK, microglial signaling, and neuropathic pain. Molecular Pain. 2007;3:33.
Jin S.-X., Zhuang Z.-Y., Woolf C.J., et al. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. Journal of Neuroscience. 2003;23:4017–4022.
Kawate T., Michel J.C., Birdsong W.T., et al. Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature. 2009;460:592–598.
Keller A.F., Beggs S., Salter M.W., et al. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Molecular Pain. 2007;3:27.
Kettenmann H., Hanisch U.-K., Noda M., et al. Physiology of microglia. Physiological Reviews. 2011;91:461–553.
Kohno T., Moore K.A., Baba H., et al. Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. Journal of Physiology. 2003;548:131–138.
Kreutzberg G.W. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19:312–318.
Landry R.P., Jacobs V.L., Romero-Sandoval E.A., et al. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Experimental Neurology. 2012;234:340–350.
Ledeboer A., Sloane E.M., Milligan E.D., et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83.
Lever I., Cunningham J., Grist J., et al. Release of BDNF and GABA in the dorsal horn of neuropathic rats. European Journal of Neuroscience. 2003;18:1169–1174.
Li K., Lin T., Cao Y., et al. Peripheral formalin injury induces 2 stages of microglial activation in the spinal cord. Journal of Pain. 2010;11:1056–1065.
Lin T., Li K., Zhang F.-Y., et al. Dissociation of spinal microglia morphological activation and peripheral inflammation in inflammatory pain models. Journal of Neuroimmunology. 2007;192:40–48.
Nasu-Tada K., Koizumi S., Tsuda M., et al. Possible involvement of increase in spinal fibronectin following peripheral nerve injury in upregulation of microglial P2X4, a key molecule for mechanical allodynia. Glia. 2006;53:769–775.
Obata K., Yamanaka H., Dai Y., et al. Differential activation of extracellular signal–regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. Journal of Neuroscience. 2003;23:4117–4126.
Obata K., Yamanaka H., Fukuoka T., et al. Contribution of injured and uninjured dorsal root ganglion neurons to pain behavior and the changes in gene expression following chronic constriction injury of the sciatic nerve in rats. Pain. 2003;101:65–77.
Penfield W. Cytology & cellular pathology of the nervous system. New York: Hafner; 1965.
Pezet S., McMahon S.B. Neurotrophins: mediators and modulators of pain. Annual Review of Neuroscience. 2006;29:507–538.
Pocock J.M., Kettenmann H. Neurotransmitter receptors on microglia. Trends in Neurosciences. 2007;30:527–535.
Prescott S.A., Sejnowski T.J., De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Molecular Pain. 2006;2:32.
Raghavendra V., Tanga F., DeLeo J.A. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. Journal of Pharmacology and Experimental Therapeutics. 2003;306:624–630.
Raghavendra V., Tanga F., Rutkowski M.D., et al. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain. 2003;104:655–664.
Ransohoff R.M., Perry V.H. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145.
Rezaie P., Male D. Mesoglia & microglia—a historical review of the concept of mononuclear phagocytes within the central nervous system. Journal of the History of the Neurosciences. 2002;11:325–374.
Rivera C., Li H., Thomas-Crusells J., et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. Journal of Cell Biology. 2002;159:747–752.
Rivera C., Voipio J., Thomas-Crusells J., et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. Journal of Neuroscience. 2004;24:4683–4691.
Schleich C.L. Schmerzlose Operationen. Julius Springer; 1899.
Schleich C.L. Besonnte vergangenheit: Lebenserinnerungen (1859-1919). E Rowohlt; 1921.
Shinozaki Y., Sumitomo K., Tsuda M., et al. Direct observation of ATP-induced conformational changes in single P2X4 receptors. PLoS Biology. 2009;7:e103.
Suter M.R., Berta T., Gao Y.-J., et al. Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Molecular Pain. 2009;5:53.
Svensson M., Eriksson P., Persson J., et al. Functional properties of microglia following peripheral nerve injury. Neuropathology and Applied Neurobiology. 1994;20:185–187.
Toulmé E., Soto F., Garret M., et al. Functional properties of internalization-deficient P2X4 receptors reveal a novel mechanism of ligand-gated channel facilitation by ivermectin. Molecular Pharmacology. 2006;69:576–587.
Toyomitsu E., Tsuda M., Yamashita T., et al. CCL2 promotes P2X4 receptor trafficking to the cell surface of microglia. Purinergic Signalling. June 8(2)301–310, 2012.
Trang T., Beggs S., Wan X., et al. P2X4-receptor–mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. Journal of. 2009;29:3518–3528.
Tsuda M., Kuboyama K., Inoue T., et al. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Molecular Pain. 2009;5:28.
Tsuda M., Masuda T., Kitano J., et al. IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8032–8037.
Tsuda M., Mizokoshi A., Shigemoto-Mogami Y., et al. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95.
Tsuda M., Shigemoto-Mogami Y., Koizumi S., et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783.
Tsuda M., Toyomitsu E., Komatsu T., et al. Fibronectin/integrin system is involved in P2X(4) receptor upregulation in the spinal cord and neuropathic pain after nerve injury. Glia. 2008;56:579–585.
Tsuda M., Toyomitsu E., Kometani M., et al. Mechanisms underlying fibronectin-induced up-regulation of P2X4R expression in microglia: distinct roles of PI3K-Akt and MEK-ERK signalling pathways. Journal of Cellular and Molecular Medicine. 2009;13:3251–3259.
Tsuda M., Tozaki-Saitoh H., Masuda T., et al. Lyn tyrosine kinase is required for P2X(4) receptor upregulation and neuropathic pain after peripheral nerve injury. Glia. 2008;56:50–58.
Ulmann L., Hatcher J.P., Hughes J.P., et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. Journal of Neuroscience. 2008;28:11263–11268.
Virchow R. Gesammelte Abhandlungen zur wissenschaftlichen Medicin, Berlin. G Grote; 1862.
Wake H., Moorhouse A.J., Jinno S., et al. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. Journal of Neuroscience. 2009;29:3974–3980.
Watkins L.R., Hutchinson M.R., Johnson K.W., Commentary on Landry, et al. Propentofylline, a CNS glial modulator, does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Experimental Neurology. 2012;234:351–353.
Wen Y.-R., Suter M.R., Ji R.-R., et al. Activation of p38 mitogen-activated protein kinase in spinal microglia contributes to incision-induced mechanical allodynia. Anesthesiology. 2009;110:155–165.
Wen Y.-R., Suter M.R., Kawasaki Y., et al. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321.
Woolf C.J. What is this thing called pain? Journal of Clinical Investigation. 2010;120:3742–3744.
Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769.
Yuan H., Zhu X., Zhou S., et al. Role of mast cell activation in inducing microglial cells to release neurotrophin. Journal of Neuroscience Research. 2010;88:1348–1354.
Zhang J., Shi X, Echeverry S., et al. Expression of CCR2 in both resident and bone marrow–derived microglia plays a critical role in neuropathic pain. Journal of Neuroscience. 2007;27:12396–12406.
Zhao J., Seereeram A., Nassar M.A., et al. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Molecular and Cellular Neurosciences. 2006;31:539–548.
Abbadie C., Bhangoo S., De Koninck Y., et al. Chemokines and pain mechanisms. Brain Research Reviews. 2009;60:125–134.
Beggs S., Trang T., Salter M.W. The P2X4R+ microglial state: an essential role in neuropathic pain. Nature Neuroscience. 2012. (In press)
Biber K., Tsuda M., Tozaki-Saitoh H., et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO Journal. 2011;30:1864–1873.
Calvo M., Bennett D.L.H. The mechanisms of microgliosis and pain following peripheral nerve injury. Experimental Neurology. 2012;234:271–282.
Clark A.K., Yip P.K., Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. Journal of Neuroscience. 2009;29:6945–6954.
Costigan M., Moss A., Latremoliere A., et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain–like hypersensitivity. Journal of Neuroscience. 2009;29:14415–14422.
Coull J.A.M., Beggs S., Boudreau D., et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021.
Coull J.A.M., Boudreau D., Bachand K., et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942.
Davalos D., Grutzendler J., Yang G., et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005;8:752–758.
Hanisch U.-K., Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nature Neuroscience. 2007;10:1387–1394.
Hathway G.J., Vega-Avelaira D., Moss A., et al. Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain. 2009;144:110–118.
Jarvis M.F., Khakh B.S. ATP-gated P2X cation-channels. Neuropharmacology. 2009;56:208–215.
Ji R.-R., Gereau R.W., 4th., Malcangio M., et al. MAP kinase and pain. Brain Research Reviews. 2009;60:135–148.
Jin S.-X., Zhuang Z.-Y., Woolf C.J., et al. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. Journal of Neuroscience. 2003;23:4017–4022.
Keller A.F., Beggs S., Salter M.W., et al. Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Molecular Pain. 2007;3:27.
Kettenmann H., Hanisch U.-K., Noda M., et al. Physiology of microglia. Physiological Reviews. 2011;91:461–553.
Lin T., Li K., Zhang F.-Y., et al. Dissociation of spinal microglia morphological activation and peripheral inflammation in inflammatory pain models. Journal of Neuroimmunology. 2007;192:40–48.
Pocock J.M., Kettenmann H. Neurotransmitter receptors on microglia. Trends in Neurosciences. 2007;30:527–535.
Prescott S.A., Sejnowski T.J., De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Molecular Pain. 2006;2:32.
Ransohoff R.M., Perry V.H. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145.
Rivera C., Li H., Thomas-Crusells J., et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. Journal of Cell Biology. 2002;159:747–752.
Suter M.R., Berta T., Gao Y.-J., et al. Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Molecular Pain. 2009;5:53.
Tsuda M., Kuboyama K., Inoue T., et al. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Molecular Pain. 2009;5:28.
Tsuda M., Masuda T., Kitano J., et al. IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8032–8037.
Ulmann L., Hatcher J.P., Hughes J.P., et al. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. Journal of Neuroscience. 2008;28:11263–11268.
Wen Y.-R., Suter M.R., Kawasaki Y., et al. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321.
Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769.
Zhang J., Shi X,., Echeverry S., et al. Expression of CCR2 in both resident and bone marrow–derived microglia plays a critical role in neuropathic pain. Journal of Neuroscience. 2007;27:12396–12406.