Spinal Cord Plasticity and Pain
Some Useful Definitions
Nociception versus Pain
Nociception includes all forms of information processing triggered by noxious stimuli (i.e., stimuli that are damaging to normal tissues). In awake animals or human subjects, nociception may lead to withdrawal or vegetative responses and/or to the sensation of pain. Pain as defined by the International Association for the Study of Pain (IASP) is “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.” Although pain is always a subjective sensation, nociception can be measured in terms of objective parameters.
Nociceptive Neurons
Defined by their excitatory input, nociceptive neurons include all neurons that are excited by noxious stimuli. This classification is irrespective of the function or functions that the neurons might serve. Nociceptive-specific neurons are excited by nociceptive afferents only. Some spinal nociceptive neurons, however, receive convergent input from high- and low-threshold sensory fibers (wide–dynamic range neurons), some have projections to the brain, whereas other nociceptive neurons are inhibitory. Still others are excitatory interneurons or motoneurons that trigger withdrawal responses. Thus, excitation of a given type of nociceptive neuron may be irrelevant for the sensation of pain (e.g., nociceptive flexor motoneurons) or may dampen pain intensity (e.g., some inhibitory interneurons), whereas activity in other nociceptive neurons (e.g., spinal dorsal horn projection neurons) may lead to the sensation of pain (see also the next definitions).
Antinociceptive Neurons
Antinociceptive neurons are defined by their output and inhibit nociceptive neurons or nociceptive responses. Some antinociceptive neurons are excited by noxious stimuli and thus constitute a subgroup of nociceptive neurons.
Principal Pain Neurons
Defined by their function, principal pain neurons trigger the sensation of pain when activated. At present, no principal pain neurons have been identified with certainty. It appears, however, that in the peripheral nervous system, some (but not all) nociceptive nerve fibers are principal pain neurons. Excitation of a subgroup of primary afferent nociceptive nerve fibers is sufficient to trigger pain sensation in human subjects (Van Hees and Gybels 1981). This supports the specificity theory of pain (von Frey 1894), which states that pain is a “specific sensation, with its own sensory apparatus independent of touch and other senses.” It is possible that principal pain neurons also exist in the central nervous system (CNS). Good candidates are nociceptive-specific spinal dorsal horn neurons (Christensen and Perl 1970) with a projection to the thalamus, the midbrain periaqueductal gray, and/or the parabrachial area. Some of these ascending nociceptive pathways could constitute “labeled lines” for pain (Craig 2003). An alternative hypothesis states that the activity pattern of an ensemble of CNS neurons determines the type of sensation that is experienced by an individual. This is proposed by the pattern theory of pain (Goldscheider 1894). See also discussions by McMahon and Koltzenburg (1992), Craig (2003), and Liu and colleagues (2011).
Pro-nociceptive Helper Cells
Activation of pro-nociceptive helper cells does not directly mediate pain sensation but rather facilitates nociception. Pro-nociceptive helper cells may be excitatory interneurons, descending tract neurons, or non-neuronal cells such as glial cells or blood cells that facilitate discharges in nociceptive neurons.
Hyperalgesia
Hyperalgesia was originally defined as “a state of increased intensity of pain sensation induced by either a noxious or ordinarily non-noxious stimulation of peripheral tissue” (Hardy et al 1950). Later, the term allodynia was coined for pain in response to normally non-painful stimuli. A very common cause of pain, sensitization of nociceptive nerve endings (“peripheral sensitization”) will, however, always induce both hyperalgesia and allodynia by shifting the stimulus–response curve to lower intensities. Thus, a single neuronal mechanism would be described in terms of two different phenomena—“hyperalgesia” and “allodynia.” Use of these terms is further confused in the literature, where lowering of mechanical withdrawal thresholds (e.g., assessed with von Frey hairs) is typically described as mechanical “allodynia” whereas lowering thermal withdrawal thresholds (e.g., in the hot plate or Hargreaves’ tests) is labeled heat “hyperalgesia” (see also discussion in Sandkühler 2009). In 2008 the IASP task force for nomenclature suggested that “hyperalgesia” be used for all forms of increased pain sensitivity (similar to the original definition) and that the term “allodynia” be restricted to pain that is not elicited by nociceptive nerve fibers, that is, pain induced by low-threshold Aβ or C fibers (Koltzenburg et al 1992, Seal et al 2009).
When caused by a peripheral insult, hyperalgesia or allodynia are labeled “primary” at the site of the injury, “secondary” in the immediately surrounding area, and “spreading hyperalgesia” at remote sites. Referred hyperalgesia is localized to the corresponding dermatome of the affected inner organ. Most often no distinction is made between hyperalgesia in humans and “hyper-nociception” in surrogate pain models in animals. In this chapter the new definitions of hyperalgesia and allodynia are used, but not the rather unusual term hyper-nociception.
Neuronal Plasticity
Neuronal plasticity is defined as changes in the properties or functions of neurons or neuronal nets that outlast the stimulus that caused these changes. Note: In the context of pain, neuronal plasticity most often refers to the neural mechanisms of hyperalgesia or allodynia. Lasting forms of analgesia, such as after counter-stimulation, physical therapy, or psychotherapy, do, however, also involve some forms of neuronal plasticity.
Central Sensitization
Stemming from the original term central excitatory state (Hardy et al 1950), central sensitization has become a popular phrase in the literature. Unfortunately, it is now used in different and often incompatible definitions that also suffered from considerable metamorphosis over time. The newly proposed definition by the IASP describes “central sensitization” as the “increased responsiveness of nociceptive neurons in the central nervous system to their normal or subthreshold afferent input.” Nociceptive neurons may, however, serve very diverse functions (see earlier). Thus, “central sensitization” in the IASP definition may in some cases lead to enhanced pain sensitivity. In other cases, central sensitization may cause analgesia or may simply be unrelated to the experience of pain. Other definitions of central sensitization are equally problematic (see, e.g., discussion in Sandkühler 2010). The term “central sensitization” is therefore not used in this chapter. It is replaced by more specific technical terms that specify the location (spinal, brain stem, or cortical mechanisms), the underlying mechanism (e.g., synaptic long-term potentiation [LTP] or disinhibition), and the proposed functional meaning (e.g., pain amplification or generation).
Information Flow in Spinal Nociceptive Pathways
Noxious stimuli activate free nerve endings of thinly myelinated Aδ or unmyelinated C fibers. In contrast, most Aβ fibers have low thresholds and respond to innocuous stimuli (see Chapter 1). Sensory nerve fibers terminate in the spinal dorsal horn in a modality-specific fashion. Nociceptive Aδ fibers terminate largely in the most superficial lamina I and in the deep lamina V of the spinal dorsal horn, whereas most nociceptive C fibers terminate in laminae I and IIouter. Low-threshold Aβ fibers terminate in lamina IIinner and in laminae III and IV (Willis and Coggeshall 2004) (see Chapter 5). On excitation, apparently all sensory nerve fibers release glutamate. Glutamate is the major fast excitatory neurotransmitter in all nociceptive afferent nerve fibers. Glutamate binds to three types of ionotropic glutamate receptors and to G protein–coupled metabotropic glutamate receptors. Ionotropic glutamate receptors of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) subtype are highly permeable to Na+ ions. AMPA receptors lacking the GluR2 subunit are, in addition, also permeable to Ca2+. Activation of AMPA receptors mediates much of the post-synaptic excitatory currents at glutamatergic synapses. Spinal ionotropic kainate receptors are present pre- and post-synaptically and become activated with strong noxious stimuli. Glutamate receptors of the N-methyl-D-aspartate (NMDA) subtype are highly permeable to Ca2+ ions. NMDA receptors open when glutamate binds to the receptor and when in addition the membrane is sufficiently depolarized to remove the Mg2+ block from the channel pore. NMDA receptors composed of NR1/NR2D subunits have, however, a much-reduced Mg2+ block that renders them permeable to Ca2+ at resting membrane potential (Larsson 2009). These types of NMDA receptors are expressed in the superficial laminae of the spinal dorsal horn (Tong et al 2008). Activation of NMDA receptors and the resultant Ca2+ influx play pivotal roles in plasticity in the spinal dorsal horn (Sandkühler 2009). Some fibers in addition release various neurotransmitters and neuromodulators. The composition of substances released in the spinal dorsal horn depends on the fiber types activated and their discharge frequencies. For example, peptidergic fibers release the neuropeptides substance P or calcitonin gene–related peptide. On high-frequency discharge, some sensory nerve fibers also release brain-derived neurotrophic factor (BDNF). Primary afferents may also release adenosine triphosphate (ATP). After nerve injury some afferents synthesize and release the neuronal chemokine CCL2 and tumor necrosis factor-α (TNF-α). Once released, they bind to their respective synaptic and extrasynaptic receptors on spinal dorsal horn neurons and/or to receptors expressed by spinal glial cells (Basbaum et al 2009).
Projection neurons within spinal laminae I and V account for most of the nociceptive input to the brain. Spinal projections to thalamic nuclei are considered relevant for the somatosensory–discriminative aspects of pain, whereas projections to the parabrachial area are believed to contribute to the emotional–aversive components of pain. Subconscious and homeostatic reactions to pain, including autonomic responses, are triggered by nociceptive spinal neurons with projections to the nucleus tractus solitarius, the parabrachial area, and the midbrain periaqueductal gray (Craig 2003).
A large proportion of nociceptive spinal dorsal horn neurons are interneurons that do not project to the brain. This includes nociceptive spinal dorsal horn neurons that are components of reflex arcs mediating nocifensive withdrawal responses. About one-third of spinal nociceptive neurons use γ-aminobutyric acid (GABA) and/or glycine as neurotransmitters and inhibit other spinal neurons, including projection neurons (Todd 2010).
Like neurons, glial cells also respond to nociceptive input. Depending on the type of activation, glial cells release a distinct mélange of neuroactive substances and thereby modify the flow of information in spinal nociceptive pathways. See Chapter 5 of this textbook for more details on the neuroanatomical organization in the spinal dorsal horn.
Spinal processing of nociceptive information is under powerful inhibitory and facilitatory control from brain stem sites with long descending projections to the spinal dorsal horn. Descending neurons use mainly glutamate or the monoamines serotonin or noradrenaline as neurotransmitters in the spinal cord. The inhibition by noradrenaline is either direct or indirect via activation of spinal inhibitory interneurons. Descending modulations of spinal nociception are tonically active, display a circadian rhythm, and can be boosted on demand (Heinricher et al 2009) (see Chapter 8 for details).
Plasticity in the Spinal Dorsal Horn Contributes to Hyperalgesia or Allodynia
In 1950 the first systematic experimental evidence for central mechanisms of postinjury pain hypersensitivity was provided by Hardy and colleagues. These authors studied the properties of secondary hyperalgesia in human volunteers and came to the conclusion that “secondary hyperalgesia … is the result of a central excitatory state” where subliminal excitations of interneurons in the spinal dorsal horn become suprathreshold and thereby open pre-existing pathways linking the area of primary and secondary hyperalgesia in skin or deep tissues (see Fig. 6-1, a reproduction of the original figure by Hardy et al from 1950). These authors stated that the “central excitatory state causing secondary hyperalgesia” also leads to longer-lasting pain sensations and can be prevented or reversed by local anesthetic block of impulses in primary afferents from the injured area. In 1977 Lynn concluded that “referred hyperalgesia … must be occurring in the central nervous system … at the first synaptic relay area in the dorsal horn of the spinal cord.”
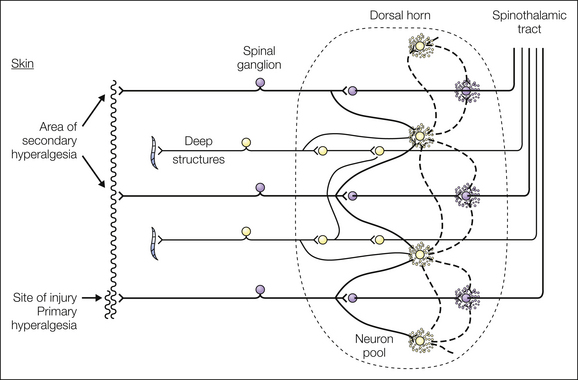
Figure 6-1 Schematic diagram of pain fiber connections within the neuron pool showing foci of excitation (stippled area) that result from the continuous barrage of noxious impulses from the site of injury. (From Hardy JD, Wolff HG, Goodell H 1950 Experimental evidence on the nature of cutaneous hyperalgesia. Journal of Clinical Investigation 29:115–140.)
The concept of Hardy (1950) and Lynn (1977) of a “central excitatory state” was later experimentally adopted by Woolf (1983). In unanesthetized decerebrate rats he found that unilateral thermal foot injury induces uni- or bilateral amplification and prolongation of nociceptive flexor reflexes. Furthermore, the discharges of motoneurons and the size of their receptive fields increase. Thereafter, a large number of laboratories have extended the early findings from the spinal ventral horn by exploring potential molecular, cellular, and network mechanisms of hyperalgesia and allodynia first in spinal dorsal horn lamina I projection neurons (McMahon and Wall 1984) and later also in the brain.
In principle, all forms of neuronal plasticity that have been identified in the CNS can also be relevant for nociceptive pathways in the spinal cord and brain. This includes various forms of synaptic plasticity, intrinsic (i.e., membrane) plasticity, and network plasticity in excitatory and inhibitory pathways.
How Plasticity is Induced in the Spinal Dorsal Horn
Plasticity in spinal nociceptive pathways may be induced in various ways. Most often strong and/or sustained activation of nociceptive C fibers, especially from deep tissues (Woolf and Wall 1986), is involved. Ablation of a subgroup of nociceptive afferent fibers that express the transient receptor potential vanilloid-1 (TRPV1) receptor channel (Shir and Seltzer 1990) or ablation of virtually all nociceptive afferents identified by Nav1.8 expression (Abrahamsen et al 2008) does not, however, prevent induction of mechanical hyperalgesia after peripheral nerve injury. This indicates that activation of nociceptive afferents is not always necessary for induction of plasticity in the spinal dorsal horn. Indeed, a recent study claimed that activity in low-threshold mechanosensitive C fibers that express the vesicular glutamate transporter-3 (VGLUT3) triggers mechanical hyperalgesia after injury (Seal et al 2009). Hyperalgesia and allodynia can be further induced in humans in the absence of any sensory stimuli, such as during prolonged application of opioids (Chen et al 2009a) or on abrupt withdrawal of opioids (Angst et al 2003). Nonetheless, expression of opioid-induced hyperalgesia requires the presence of TRPV1 receptor-expressing nociceptive afferents (Vardanyan et al 2009).
Substances that Induce Hyperalgesia and/or Allodynia on Spinal Application
The literature provides a large and steadily growing list of molecules that are released into the spinal cord by neuronal or non-neuronal pro-nociceptive helper cells. When applied experimentally onto the spinal cord, some substances are sufficient for the induction of hyperalgesia and/or allodynia. Other substances do not induce but rather facilitate the induction of hyperalgesia. Up to now the effects of individual modulators have almost exclusively been studied in isolation, but their clinically relevant action takes place jointly. It is likely that during the pathogenesis of pain the composition of relevant spinal mediators continuously changes and will be distinct for different types of pain. The presently known signaling pathways probably just reflect the tip of an enormous iceberg of spinal signaling molecules contributing to the pathogenesis of hyperalgesia and allodynia. The matter is further complicated by the fact that the pro-nociceptive effects of a given mediator may be absent or revert to antinociception, depending on the experimental context. Examples are galanin, nociceptin/orphanin FQ, and nerve growth factor (NGF), which may have either pro- or antinociceptive effects in different animal models of hyperalgesia and allodynia (Mogil and Pasternak 2001, Lang et al 2007, Virk and Williams 2008). Pro-nociceptive spinal mediators include but are not limited to the following substances.
Endogenous Substances
Drugs
These substances trigger different, only partially overlapping spinal signaling pathways, thereby leading to distinct forms of hyperalgesia or allodynia. See Sandkühler (2009) for review and references. The complexity of interacting signaling pathways in the neuronal and non-neuronal cellular networks is only beginning to unfold. Unraveling their mutual interactions will constitute a major challenge for the future.
Spinal Cord Cells Indispensible in the Induction of Hyperalgesia and/or Allodynia
A number of cellular elements in the spinal cord have been identified that appear to be necessary for full expression of hyperalgesia or allodynia. C fibers that express either the TRPV1 receptor (Meller et al 1992), the marker isolectin B4 (Tarpley et al 2004), or VGLUT3 (Seal et al 2009) are, in different contexts, claimed to be essential for the induction of hyperalgesia in some animal models. Selective destruction of dorsal horn neurons that express the neurokinin 1 (NK1) receptor also prevents full expression of hyperalgesia after inflammation or nerve injury (Mantyh et al 1997). Many of these neurons are located in lamina I and project to the brain. Substances that block the metabolism of microglia and astrocytes (e.g., fluorocitrate) (Meller et al 1994) or microglia only (minocycline) (Raghavendra et al 2003) also block the development of hyperalgesia and allodynia. Spinal fiber tracts that are essential for full expression of hyperalgesia or allodynia include the anterior lateral quadrant (Foerster 1913), the lateral funiculus (Palecek et al 2002), and the dorsal columns (Houghton et al 1999).
Playing Schedules on the Spinal Stage of the Pain Theater
The specificity theory of pain assigns the leading part to principal pain neurons. Although the leading actor or actors on the spinal stage have not yet been identified, a number of good candidates have been proposed—namely, nociceptive spinal dorsal horn neurons with a projection to the thalamus, the midbrain periaqueductal gray, and/or the parabrachial area. Excitatory and inhibitory interneurons in the spinal dorsal horn play important roles in modulating the perception of pain. Supporting actors include microglia and astrocytes, blood cells such as T lymphocytes, and perhaps also dural mast cells in their roles as pro-nociceptive helper cells. The roles played by individual actors may change during the pathogenesis of pain. For example, antinociceptive neurons may become excitatory in the course of neuropathy (Coull et al 2003, 2005); that is, they may then play the role of pro-nociceptive helper cells. Likewise, glial cells that otherwise fulfill housekeeping functions may become pro-nociceptive helper cells when releasing pro-nociceptive substances after activation. The actors on the spinal stage of nociception communicate with each other (see Fig. 6-2). The modes of their interactions will ultimately determine the outcome of the performance (i.e., whether pain is felt by an individual).
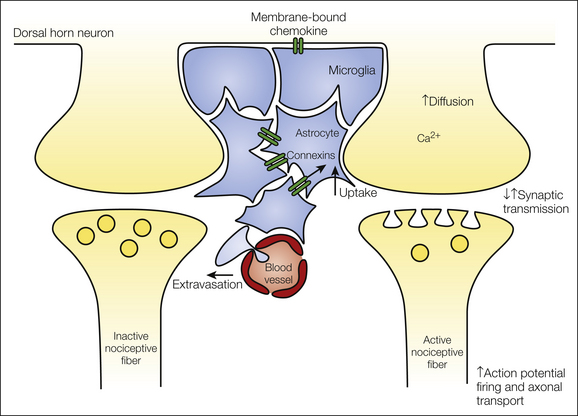
Figure 6-2 During nociception, neuronal and non-neuronal cells communicate with each other. The type of nociceptive afferent barrage determines which substances are synthesized and/or released from pre-synaptic nerve terminals, post-synaptic neurons, and glial cells. Under some pathological conditions, the integrity of the blood–spinal cord barrier may be interrupted and thereby allow extravasation of large molecules and blood cells into the spinal parenchyma. Together, the unique mixture and sequence of events determine the type of plasticity that is induced in the spinal dorsal horn.
The repertoire of plasticity in spinal nociceptive circuits ranges from subtle changes in ion channel conductance to drastic changes in the morphology of cells and the connectivity and functions of neuronal networks. The various forms of plasticity include but are not limited to molecular and cellular changes in the number and functional state of synaptic, somal, and axonic ion channels, receptors, enzymes, transporter molecules, and transcription factors. This may lead to alterations in the synthesis, release, and uptake of neurotransmitters and neuromodulators; to synaptic or intrinsic plasticity; and to changes in the cytoskeleton of cells and modifications in cell morphology. Plasticity at the cellular level feeds into functional changes at the network and systemic levels, some of which ultimately lead to altered pain experiences.
In this chapter presently known forms of plasticity in spinal nociceptive pathways are described. It should be kept in mind that the very same type of plasticity may have a different impact on pain, depending on the functions of the affected neurons. For example, disinhibition of principal pain neurons causes hyperalgesia, whereas disinhibition of antinociceptive neurons may have the opposite effect.
Plasticity in Excitatory Nociceptive Pathways of the Spinal Dorsal Horn
Synaptic strength describes the magnitude of post-synaptic currents or potentials in response to a pre-synaptic action potential. Synaptic strength may be modified in an activity-dependent manner. For example, high-frequency discharges in pre-synaptic fibers and concomitant strong synaptic activity may lead to a long-lasting increase in synaptic strength. This LTP is a form of synaptic plasticity that can be induced at many, if not all, synapses in the CNS, including spinal synapses of nociceptive primary afferents (Randic et al 1993, Ikeda et al 2003, Ikeda et al 2006, Drdla et al 2009) and excitatory synapses between spinal lamina II interneurons (Santos et al 2009).
Synaptic Long-Term Potentiation
In nociceptive pathways, LTP may be triggered by enhanced synaptic activity (i.e., in a use-dependent manner) or in the absence of any pre-synaptic activity; for example, by abrupt withdrawal from opioids).
Activity-Dependent Forms of Long-Term Potentiation in Nociceptive Pathways
LTP has been induced at synapses between primary afferent C fibers, many of which are nociceptive, and spinal dorsal horn neurons in vitro and in vivo (see Sandkühler 2009 for a review). Conditioning stimuli that induce LTP include injection of capsaicin or formalin into a hindpaw, peripheral inflammation, acute nerve injury, and direct electrical nerve stimulation at high (100 Hz) (Randic et al 1993, Ikeda et al 2003) or low (around 2 Hz) frequencies (Ikeda et al 2006). All these stimuli also induce hyperalgesia in awake animals or human subjects. Some forms of LTP could be induced selectively in spinal lamina I projection neurons (Ikeda et al 2003, 2006) that are essential for full expression of hyperalgesia in awake animals (Mantyh et al 1997, Nichols et al 1999) and are thus prime candidates for principal pain neurons.
Importantly, perceptual correlates of use-dependent LTP in nociceptive pathways were identified in human volunteers. When cutaneous, peptidergic afferents were stimulated at either high or low frequencies or with capsaicin, primary or secondary hyperalgesia was induced that lasted for several days (Klein et al 2004). Like LTP at spinal lamina I projection neurons, this form of hyperalgesia requires activation of NMDA receptors for its induction (Klein et al 2007).
It is presently unknown whether LTP in nociceptive pathways is homosynaptic in nature (i.e., affects synapses that were activated during the conditioning stimulus). Homosynaptic LTP at principal pain neurons would lead to primary hyperalgesia. If LTP is also expressed at other synapses that impinge on the same principal pain neuron, secondary hyperalgesia would then result. A recent study suggested that activation of C fibers activates silent synapses of Aδ fibers via heterosynaptic facilitation (Torsney 2011). (See the section entitled Mechanisms of Secondary or Widespread Hyperalgesia, later.)
Drug-Induced Long-Term Potentiation in Nociceptive Pathways
In addition to postinjury forms of hyperalgesia, drug-induced forms are also of considerable clinical relevance. For example, hyperalgesia may develop in human subjects and in experimental animals during the continuous use of opioids (Chen et al 2009a) or after abrupt withdrawal of opioids (Angst et al 2003). Opioids acutely depress synaptic strength at C fibers, mainly by pre-synaptic inhibition via interference with N- and P/Q-type voltage-gated calcium channels (VGCCs) (Heinke et al 2011). Acute depression of release of neurotransmitters from nociceptive afferents is a major mechanism underlying opioid analgesia. On abrupt withdrawal from opioids (remifentanil, fentanyl, or morphine), synaptic strength not only returns to normal but also may become potentiated for prolonged periods (Drdla et al 2009, Heinl et al 2011). Induction of this opioid withdrawal LTP is post-synaptic in nature because it requires activation of post-synaptic G proteins and post-synaptic NMDA receptors and a rise in post-synaptic Ca2+ concentration. It is presently unknown whether LTP also develops during the prolonged application of opioids. The signaling pathways for activity-dependent forms of LTP and opioid withdrawal LTP largely overlap both each other and opioid-induced hyperalgesia. Whereas induction of activity-dependent and opioid withdrawal LTP requires post-synaptic signaling (Ikeda et al 2003, 2006; Drdla et al 2009), consolidation and maintenance of LTP may involve pre-synaptic mechanisms (Heinl et al 2011, Luo et al 2012).
Additional mechanisms, including descending facilitation from brain stem sites, clearly also play a role in opioid-induced hyperalgesia (Chang et al 2007, Vera-Portocarrero et al 2007, Heinl et al 2011). (See also the section entitled Plasticity in Descending Pathways, later.)
Drugs other than opioids can likewise induce LTP in nociceptive pathways when applied directly onto the spinal cord in vivo. Such substances include ATP, BDNF, the dopamine receptor D1/D5 agonist SKF 38393, and the protein kinase A (PKA) activator 8-Br-cyclic adenosine monophosphate (cAMP). In spinalized animals (i.e., when the descending inhibitory pathways are blocked), spinal application of NMDA, substance P, or neurokinin A also induces LTP at C-fiber synapses, whereas TNF-α is effective in neuropathic animals only. For review and references, see the review by Ruscheweyh and associates (2011).
LTP at the synaptic relay between nociceptive nerve fibers and potential principal pain neurons in the superficial spinal dorsal horn shares induction protocols, pharmacological profiles, and signaling pathways with various forms of hyperalgesia and allodynia. LTP is thus considered a synaptic mechanism of pain amplification (Sandkühler 2009, Ruscheweyh et al 2011). It is quite likely that similar forms of plasticity exist at synapses upstream in nociceptive pathways, including synapses in the cerebral cortex (Li et al 2010, Zhuo 2011).
Synaptic Long-Term Depression and Depotentiation
In nociceptive pathways, synaptic strength may also decrease for prolonged periods after conditioning stimulation (Sandkühler et al 1997, Liu et al 1998). This is called long-term depression (LTD) when synaptic strength was normal before conditioning and depotentiation when synaptic strength was potentiated before conditioning. Both LTD and depotentiation may underlie analgesia after some forms of counter-stimulation in human subjects (Ellrich and Schorr 2004).
Homosynaptic Long-Term Depression at Synapses of Aδ Fibers
At synapses between Aδ fibers and neurons in spinal cord lamina II, conditioning low-frequency stimulation (at 1 Hz) in vitro leads to a homosynaptic, NMDA receptor–dependent LTD that requires a rise in post-synaptic Ca2+ (Sandkühler et al 1997). Long-lasting depressions of pain-related cerebral activation and perceptual correlates of LTD in nociceptive pathways have been identified in human volunteers (Ellrich and Schorr 2004).
Intrinsic Plasticity of Nociceptive Spinal Dorsal Horn Neurons
Three parameters essentially determine the discharges of a neuron: (1) excitatory synaptic input, (2) inhibitory synaptic input, and (3) membrane excitability. The latter determines how a given membrane depolarization translates into firing of action potentials. Membrane excitability is thus key to the input–output function of any neuron. Long-lasting changes in membrane excitability are called “intrinsic plasticity.” Intrinsic plasticity includes changes in thresholds for action potential firing, changes in discharge patterns, and accommodation of firing (Morisset and Nagy 1996, Beck and Yaari 2008). In contrast to the comprehensive literature on modulation of the membrane properties of primary afferent nerve fibers, little is presently known about the intrinsic plasticity of nociceptive neurons in the CNS and their potential role in hyperalgesia and allodynia.
At least some nociceptive spinal dorsal horn neurons have the capacity to express intrinsic plasticity when challenged with specific experimental stimuli such as activation of PKA, PKC, and group I metabotropic glutamate receptors or by removing tonic inhibition via GABAA receptors (Derjean et al 2003). A subset of deep spinal dorsal horn neurons display plateau potentials on depolarization that lead to accelerated firing of action potentials, afterdischarges, and “wind-up” (see later), which amplifies nociceptive responses (Reali et al 2011). Conditioning stimulation of dorsal root afferents facilitates or induces plateau potentials in some dorsal horn neurons by activating group I metabotropic glutamate receptors and NK1 receptors (Russo et al 1997). Induction of intrinsic plasticity during nociceptive or neuropathic pain models has been found in some studies (Dougherty and Hochman 2008) but not in others (Müller et al 2003, Schoffnegger et al 2006, Chen et al 2009b).
Plasticity in Inhibitory Nociceptive Pathways of the Spinal Dorsal Horn
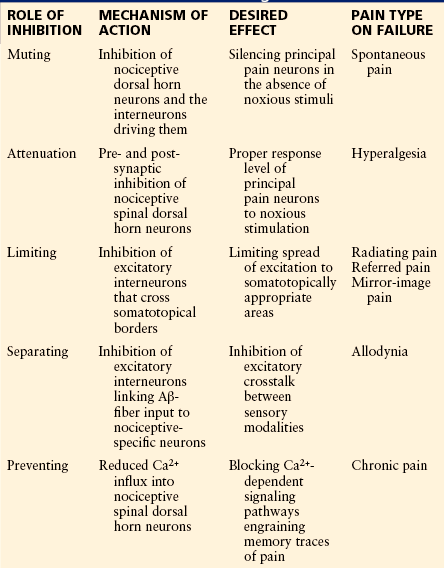
About one-third of all neurons in the spinal dorsal horn are inhibitory interneurons that use GABA and/or glycine as transmitters (Todd 2010). Long descending projections from brain stem sites also exert inhibitory control over spinal nociception, either directly or through intercalated inhibitory interneurons. Inhibition in the spinal dorsal horn serves five important functions, as summarized in Table 6-1.
To ensure normal spinal nociception, all elements of the inhibitory chain need to work properly, as illustrated in Figure 6-3. This includes a sufficient number of inhibitory interneurons; an appropriate level of excitatory input to drive inhibitory neurons; normal membrane excitability to convert excitatory synaptic input into action potential discharges; synthesis, reuptake, and release of inhibitory neurotransmitters such as GABA and glycine; activation of inhibitory pre- or post-synaptic receptors; and, in the case of binding to ionotropic GABAA or glycine receptors, a normal anion gradient across the post-synaptic cell membrane to allow influx of Cl− ions into the neuron.
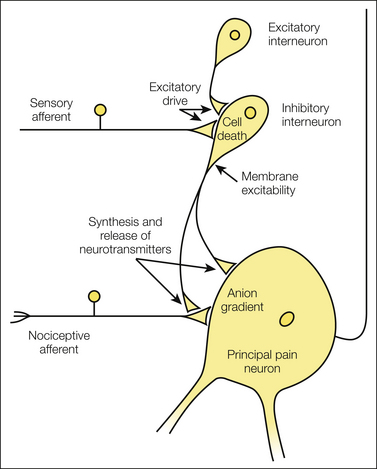
Figure 6-3 Proper inhibition of spinal principal pain neurons is indispensable for normal nociception but may be impaired by various modulations of inhibitory interneurons, including cell death, reduced excitatory drive, reduced membrane excitability, reduced synthesis and/or release of inhibitory neurotransmitters for pre- or post-synaptic inhibition, and changes in the anion gradient of post-synaptic neurons.
Cell Death of Inhibitory Interneurons?
It had been proposed that spared nerve injury led to programmed cell death of GABAergic neurons (Moore et al 2002). This hypothesis has, however, been challenged; quantitative stereological analysis has revealed that in neuropathic animals, neither the number of neurons nor the number of GABAergic profiles is different from controls (Polgár et al 2003).
Excitatory Drive to Inhibitory Interneurons
Inhibitory interneurons in the spinal dorsal horn apparently do not display any pacemaker activity. They thus require an excitatory drive from other neurons to become active. Primary afferents, excitatory spinal interneurons, and long descending excitatory pathways converge onto spinal GABAergic neurons and provide an excitatory drive to activate them. GABAergic neurons have the capacity to finely tune their own excitatory input, apparently by releasing a retrograde messenger. The nature of this messenger is still unknown, but it pre-synaptically controls the release of glutamate, which drives the GABAergic neuron. In animals with a chronic constriction injury (CCI) of the sciatic nerve, both the global excitatory input to GABAergic neurons and excitation by primary afferents are diminished, thereby leading to reduced activation of inhibitory neurons (Leitner et al 2012, unpublished observations).
Stability of Membrane and Discharge Properties of GABAergic Neurons
The principal parameters of membrane excitability are the resting membrane potential, input resistance, and threshold for firing of action potentials. These parameters do not change in spinal dorsal horn GABAergic neurons of animals with CCI of the sciatic nerve. The distribution of typical discharge patterns also does not differ between neuropathic and sham-treated animals (Schoffnegger et al 2006). The apparent stability of membrane excitability suggests that the reduced excitatory input to these neurons as described earlier faithfully translates into diminished inhibitory output.
Synthesis, Reuptake, and Release of Inhibitory Neurotransmitter
GABA immunoreactivity is temporarily reduced in the spinal dorsal horn after injury to peripheral nerves. The reversibility of these changes suggests that not the number of GABAergic neurons but rather their GABA content is reduced. In line with this, glutamic acid decarboxylase 65 (GAD65; one of the two GABA-synthesizing enzymes) levels drop in the spinal dorsal horn ipsilateral to a CCI (Eaton et al 1998). Furthermore, reuptake of GABA after its release may also be impaired in neuropathic animals since GABA transporter subtype 1 (GAT-1) is down-regulated after CCI of the sciatic nerve. This possibly leads to depletion of GABA from the terminals (Miletic et al 2003). Consistently, release of GABA is significantly reduced in spinal cord slices of rats after spinal nerve ligation (Lever et al 2003). Another study, however, found no changes in levels of GABA or the vesicular GABA transporter in animals with a spared nerve injury (Polgár and Todd 2008). Some authors found that the spinal content of GABA is even enhanced bilaterally 1 to 30 days after unilateral CCI of the sciatic nerve in the rat (Satoh and Omote 1996). The reasons for these discrepant results are presently unknown.
Activation of Inhibitory Neurotransmitter Receptors
The number, the subunit composition, the sensitivity of post-synaptic receptors, and the driving force for Cl− all determine the effects of GABA or glycine on post-synaptic neurons or pre-synaptic nerve terminals. There is evidence that all these parameters may change in the course of neuropathy or inflammation.
Neurectomy of the sciatic nerve causes complex changes in the spinal GABAergic systems. GABA content and GABAB receptor binding in lamina II of the spinal cord are down-regulated. In contrast, GABAA receptor binding is enhanced (Castro-Lopes et al 1995). Not only the cell surface expression of inhibitory neurotransmitter receptors but also their functions may be modulated; for example, by prostaglandins released in the spinal dorsal horn during inflammation. Prostaglandin E2 leads to activation of the prostaglandin E2 receptor, cholera toxin–sensitive G proteins, and cAMP-dependent protein kinase. This signaling cascade then depresses glycine receptor subtype α2 function (Harvey et al 2004) and reduces inhibitory glycinergic currents (Ahmadi et al 2002). This pathway seems to be relevant for inflammatory but not for neuropathic pain (Hösl et al 2006).
Altered Driving Force for Chloride Ions
Even if all the elements of the inhibitory chain described earlier and illustrated in Figure 6-3 were to remain the same, inhibition through ionotropic GABAA or glycine receptors could still be reduced, eliminated, or converted into paradoxical excitation by changes in the driving force for chloride ions.
Diminished Post-synaptic Inhibition
GABAA and glycine receptors are Cl−-permeable ion channels that open on binding of their respective ligands. The direction of Cl− flux is generally determined by the level of the Cl− equilibrium potential (ECl−) with respect to the resting membrane potential (VRest) of the cell. In most neurons of mature animals, 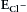 is more negative than VRest, partly because of continuous removal of Cl− from the cells (e.g., via potassium–chloride co-transporter-2 [KCC2]). Because of the low Cl− concentration in neurons, Cl− will normally move into the cell when the Cl− channels open. The influx of negatively charged ions then causes membrane hyperpolarization and thus inhibition of neuronal excitability. may change in some spinal dorsal horn neurons of animals with a neuropathy. This change involves the release of BDNF from spinal microglia. BDNF depresses the function of KCC2 and thereby leads to enhanced Cl− concentration within the neurons. This in turn results in a reduced (or even inverted) driving force for Cl− and thus reduces post-synaptic inhibition (Coull et al 2005).
is more negative than VRest, partly because of continuous removal of Cl− from the cells (e.g., via potassium–chloride co-transporter-2 [KCC2]). Because of the low Cl− concentration in neurons, Cl− will normally move into the cell when the Cl− channels open. The influx of negatively charged ions then causes membrane hyperpolarization and thus inhibition of neuronal excitability. may change in some spinal dorsal horn neurons of animals with a neuropathy. This change involves the release of BDNF from spinal microglia. BDNF depresses the function of KCC2 and thereby leads to enhanced Cl− concentration within the neurons. This in turn results in a reduced (or even inverted) driving force for Cl− and thus reduces post-synaptic inhibition (Coull et al 2005).
Paradoxical Post-synaptic Excitation
Activation of post-synaptic GABAA or glycine receptors may even lead to depolarization and eventually excitation if is less negative than VRest. This situation may occur when the function of KCC2 is severely impaired. Cl− then moves out of the neuron and the membrane potential depolarizes rather than hyperpolarizes. In other words, it is the level of the Cl− concentration gradient across the cell membrane that actually determines whether GABA and glycine are inhibitory or excitatory.
Paradoxical Pre-synaptic Excitation
The anion gradient across the membrane of primary afferent nerve terminals is different from that of other neural compartments in mature animals. In nerve terminals, the activity of the sodium–potassium–chloride co-transporter-1 actually enhances the Cl− concentration. ECl− is therefore normally less negative than VRest. This situation results in efflux of Cl− and membrane depolarization on opening of the Cl− channel. Thus, under normal conditions, activation of GABAA receptors will slightly depolarize the terminals of primary afferent nerve fibers. Moderate primary afferent depolarization inactivates VGCCs, which are required for release of neurotransmitters from the terminals, and it shunts the currents of the incoming action potentials (Rudomin and Schmidt 1999). In this manner, moderate depolarization of nerve terminals inhibits the release of neurotransmitters. It has been proposed that under conditions of inflammation or neuropathy, depolarization at primary afferent C fibers by GABA may become steeper and stronger and eventually reach the thresholds for activating voltage-gated sodium channels and for triggering firing of action potentials. This event would cause paradoxical pre-synaptic excitation by GABA (Cervero et al 2003, Pitcher and Cervero 2010). Direct proof of this mechanism is, however, still lacking.
It would truly constitute the worst-case scenario in nociception if normal pre- or post-synaptic inhibition of principal pain neurons would eventually turn into paradoxical excitation.
Heterosynaptic Long-Term Potentiation at Spinal GABAergic Synapses
The strength of GABAergic synapses in the superficial spinal dorsal horn not only may be diminished but can also be potentiated for prolonged periods. A recent study has demonstrated that conditioning stimuli of primary afferent C fibers that lead to LTP at C-fiber synapses also trigger heterosynaptic LTP at GABAergic synapses that impinge onto the same neurons in lamina I of the spinal dorsal horn (Fenselau et al 2011). This LTPGABA is expressed pre-synaptically and requires activation of spinal metabotropic but not ionotropic glutamate receptors and NO as a retrograde messenger. This novel form of synaptic plasticity in spinal nociceptive circuits may be an essential mechanism for maintaining the relative balance between excitation and inhibition and for improving the signal-to-noise ratio in nociceptive pathways (Fenselau et al 2011). It is presently unknown whether this mechanism is impaired in some chronic pain conditions.
Plasticity in Descending Pathways
Spinal nociception is under powerful control from a number of descending inhibitory and facilitatory pathways that originate in part from serotonergic neurons in the rostral ventromedial medulla or from adrenergic neurons in the nucleus locus coeruleus. Under normal conditions, descending inhibition appears to prevail over facilitation. The reverse may be true in neuropathic conditions.
Plasticity in Descending Inhibitory Pathways
Descending adrenergic and serotonergic pathways inhibit spinal dorsal horn neuronal responses to acute noxious stimuli and dampen the responses in the course of neuropathy or peripheral inflammation (Millan 2002, Benarroch 2008, Heinricher et al 2009). Furthermore, descending inhibitory systems impede induction of LTP at spinal synapses and other forms of spinal dorsal horn neuronal plasticity in animal models of inflammatory or neuropathic pain. On the other hand, descending inhibitory systems are themselves subject to neuroplastic changes. For example, peripheral inflammation leads to rapid and prolonged phosphorylation of AMPA receptors in the rostral ventromedial medulla and to a leftward shift of the dose–response curve for AMPA-induced descending inhibition (Guan et al 2002). Peripheral inflammation generally enhances the strength of tonic descending inhibition. See Vanegas and Schaible (2004) for a review.
Plasticity in Descending Facilitatory Pathways
Descending facilitatory pathways contribute to the development of primary and secondary hyperalgesia. In the rostral ventromedial medulla, “ON-cells” have been identified. Their discharges correlate with descending facilitation of spinal withdrawal reflexes and with enhanced activity of spinal nociceptive neurons in the course of peripheral inflammation. ON-cell activation involves opening of NMDA receptor channels and the production of NO. These circuits are apparently also subject to neuroplastic changes inasmuch as low doses of NMDA microinjected into the rostral ventromedial medulla trigger descending facilitation in the early phase of peripheral inflammation, but later, the same doses of NMDA trigger descending inhibition (Guan et al 2002). After unilateral ligation of the sciatic nerve, levels of noradrenaline and serotonin rise bilaterally in the spinal cord (Satoh and Omote 1996), thus suggesting that neuropathy leads to enhanced tonic descending facilitation and/or inhibition. In line with this conclusion, transection of fibers descending within the dorsolateral funiculus abolishes mechanical and thermal hyperalgesia in animals after spinal nerve ligation but does not affect response thresholds in sham-treated animals (Ossipov et al 2000). More specifically, depletion of functional phenotypes of serotonin in neurons of the rostral ventromedial medulla with regional short hairpin RNA (shRNA) interference of tryptophan hydroxylase-2, the rate-limiting enzyme in the synthesis of neuronal serotonin, reduces hyperalgesia in the formalin test but not the responses to acute noxious stimuli (Wei et al 2010).
Descending facilitation from the rostral ventrolateral medulla is also relevant to the hyperalgesia that develops during continuous application of opioids. Facilitation involves a loop consisting of spinal NK1 receptor–expressing (projection) neurons, activation of cholecystokinin-2 receptors in the rostral ventromedial medulla, spinal release of serotonin, and activation of spinal 5-HT3 receptors (Vera-Portocarrero et al 2007, Heinl et al 2011). See Porreca and colleagues (2002) for a review.
Other Proposed Mechanisms of Hyperalgesia and Allodynia
In some deep dorsal horn neurons with convergent nociceptive and low-threshold input, a “wind-up” phenomenon can be observed in response to ongoing discharges in C fibers. Wind-up is due to temporal summation of excitatory post-synaptic potentials. Action potential firing of some post-synaptic spinal dorsal horn neurons increases during the first seconds of ongoing C-fiber discharges at rates between 0.5 and 5 impulses per second (Mendell and Wall 1965). Thereafter, the discharges of spinal neurons remain constant or decrease again. This ceiling effect is due to adaptive changes. A biological function of wind-up in pro-nociceptive pathways could be that during the first seconds of an ongoing noxious stimulus, a progressive increase in the action potential discharges of some spinal dorsal horn neurons drives nocifensive responses. Wind-up is an aspect of the normal coding properties of some spinal dorsal horn neurons and per se neither a form of neuronal plasticity nor a mechanism of hyperalgesia or even chronic pain. All forms of synaptic, intrinsic, and network plasticity described in this chapter can, however, change the properties of wind-up. For example, LTP leads to higher amplitudes and longer durations of excitatory post-synaptic potentials and thus to stronger temporal summation and wind-up. Long-lasting changes in wind-up properties can be used as markers—rather than as mechanisms—of plasticity in spinal nociceptive pathways (Herrero et al 2000).
Sprouting of Low-Threshold Myelinated Afferents
In the adult spinal dorsal horn, nociceptive afferents terminate in laminae I and IIouter. In contrast, low-threshold mechanosensitive Aβ-fiber afferents terminate in laminae IIinner, III, and deeper. It has been proposed that after nerve injury, low-threshold mechanosensitive Aβ fibers would sprout into the superficial laminae I and IIouter and make synaptic contact with nociceptive-specific neurons. This would then lead to Aβ-fiber–mediated allodynia (Woolf et al 1992). The marker used in that study to label Aβ fibers (the B subunit of the cholera toxin) does, however, also label C fibers after axotomy. This phenotypic switch in C fibers rather than sprouting of Aβ fibers largely explains that the label was found massively in superficial layers after nerve injury (Bao et al 2002, Hughes et al 2003). Nonetheless, intracellular recording and labeling of individual Aβ-fiber afferents suggest that sprouting of Aβ fibers into superficial lamina might occur, but to a rather limited extent. It appears that disinhibition of pre-existing polysynaptic pathways from the deep to the superficial spinal dorsal horn is functionally more important for crosstalk between low-threshold Aβ-fiber afferents and the nociceptive system (Baba et al 2003, Schoffnegger et al 2008) than any sprouting of Aβ fibers.
Non-Neuronal Pro-Nociceptive Helper Cells
A number of neuronal (as described earlier) and non-neuronal mechanisms are in a prime position for modulating the response properties of potential principal pain neurons.
Some non-neuronal cells may temporarily take over the role of pro-nociceptive helper cells when they become activated: for example, glial cells (Meller et al 1994, McMahon and Malcangio 2009, Milligan and Watkins 2009) or white blood cells after infiltrating the spinal cord. Lymphocytes and dendritic cells may under certain conditions pass the blood–spinal cord barrier (Costigan et al 2009). Humoral mediators secreted by resident or infiltrating pro-nociceptive helper cells may diffuse through the spinal cord parenchyma and modify nociception.
Glial Cells
In the CNS three types of glial cells can be distinguished: microglia, astrocytes, and oligodendrocytes. The role, if any, of the latter for nociception is largely unknown and not considered further here. In this chapter the term glia therefore refers to astrocytes and microglia only. There is now convincing evidence that spinal glial cells not only fulfill housekeeping functions in normal and otherwise healthy subjects but also may contribute to hyperalgesia and allodynia under various experimental conditions (Milligan and Watkins 2009) and then function as pro-nociceptive helper cells. Glial cells may become activated by mediators such as glutamate, substance P, ATP, prostaglandins, bradykinin, and chemokines, which are released in the spinal dorsal horn in the course of peripheral nerve or spinal cord injury or by peripheral trauma or inflammation. Microglia may also become activated by the chemokine fractalkine, which is expressed on the surface of neurons and activates CX3C chemokinin receptor 1 receptors on microglia (Clark et al 2009). Activation of glial cells is not a unitary process. Depending on the activating stimulus, divergent signaling pathways are involved and lead to different forms of morphological and secretory reactions. It has been suggested that the term “activated glia” be dropped and be replaced with “pain-related enhanced response states” (McMahon and Malcangio 2009).
Microglia
Microglia are resident macrophages that constitute up to 10% of the glia in the CNS. Microglia express pattern recognition receptors (toll-like receptors) and receptors for complement components, and they perform immune surveillance. Microglia appear to respond quickly to injuries to the spinal cord or peripheral tissue or to nerve injuries with changes in their morphology, phagocytic activity, expression of receptors, and synthesis and release of cytokines, including TNF-α and IL-1β. Cytokines secreted by perivascular microglia weaken the integrity of the blood–brain barrier. Microglia possess a rich response repertoire, different aspects of which are activated by specific stimuli. On the other hand, very different types of stimuli may converge onto the same signaling pathways. For example, the glial toll-like receptor 4 may be activated by peripheral nerve injury, opioids, and lipopolysaccharides. See McMahon and Malcangio (2009) and Milligan and Watkins (2009) for reviews.
Astrocytes
Astrocytes are the most numerous cells in the CNS. They constitute about 50% of the glial cells and display considerable heterogeneity in their morphology and physiology (Matyash and Kettenmann 2010). Like microglia, astrocytes are also highly secretory cells. Individual astrocytes completely encapsulate thousands of synapses of different neurons (Haydon 2001), and they are also in intimate contact with blood vessels. Much of the extracellular glutamate is taken up by astrocytes; such uptake is essential for normal function of glutamatergic synapses (Tzingounis and Wadiche 2007). Astrocytes also control blood flow by releasing vasoconstrictors or vasodilators, depending on the context. Astrocytes may respond quickly to stimuli with elevated Ca2+ levels, and they are connected to each other via gap junctions to form extensive networks (Fig. 6-4; see also Fig. 6-2). Other responses of astrocytes (e.g., to peripheral or spinal cord injury) occur, however, with longer delays than those of microglia, and their responses are also more prolonged. Astrocytes express receptors for neurotransmitters, and they synthesize and release transmitters (such as glutamate) and pro-inflammatory substance (e.g., IL-1β, IL-6, prostaglandin E2, and NO) into the spinal cord following nerve or spinal cord injury or during peripheral inflammation.
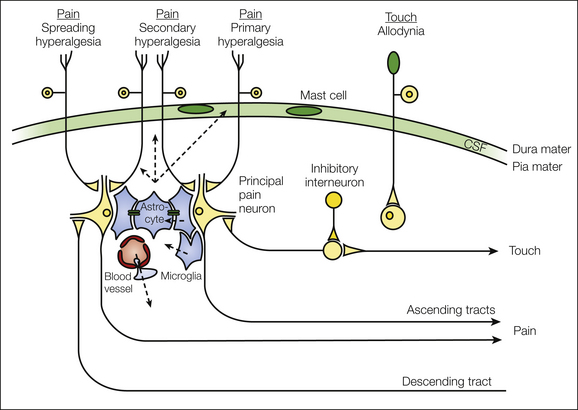
Figure 6-4 Under normal conditions, sensory stimulation leads to finely tuned excitation of an appropriate ensemble of spinal dorsal horn neurons. Somatotopic and modality borders are actively maintained by tonic inhibition and by anatomical constraints. In the course of neuropathy or inflammation, plasticity in spinal nociceptive pathways may affect remote neurons beyond the somatotopic and/or modality borders. Heterosynaptic long-term potentiation affects synapses that were inactive during the initial insult. Disinhibition opens pre-existing polysynaptic pathways beyond the somatotopic and/or modality borders. Astrocytes are connected via gap junctions, which allows spread of Ca2+ and small molecules throughout the astrocytic network. Glial and neuronal helper cells synthesize and release substances into the spinal cord parenchyma, some of which diffuse to remote sites through the interstitial space or cerebrospinal fluid (CSF). The integrity of the blood–spinal cord barrier is impaired under some pathological conditions, which can result in extravasation of molecules and blood cells. Descending fiber tracts originating in brain stem sites exert inhibitory and excitatory control of spinal nociception. Impaired balance of descending control contributes to the spread of excitation in the spinal dorsal horn. Dotted arrows indicate flow of information by diffusion or extravasation.
At least some of these glial cell responses are required for full expression of hyperalgesia and allodynia after nerve injury. Induction of hyperalgesia and allodynia can be prevented in animal models of neuropathy by blocking the metabolism of microglia with minocycline, by blocking both microglia and astrocytes with fluorocitrate, or by interrupting the Janus kinase signal transducer and activator of transcription-3 signal transduction pathway (Dominguez et al 2010). There are also reports suggesting that activation of glia contributes to inflammatory pain (Watkins et al 1997, Hua et al 2005). Furthermore, activation of glial cells (e.g., with intrathecal injections of lipopolysaccharides or ATP) induces signs of hyperalgesia and allodynia in experimental animals (Clark et al 2010). Astrocytes may release the NMDA receptor co-agonist D-serine, and this appears to be required for some forms of mechanical hyperalgesia (Miraucourt et al 2011). Thus, activation of spinal glial cells is necessary and sufficient for the induction of some forms of hyperalgesia and allodynia.
White Blood Cells
Under normal conditions, very few blood cells are present in the spinal cord parenchyma. T cells enter the spinal cord by leukocyte extravasation only at a low level. The situation changes if peripheral nerves are injured, and then chemoattraction by cytokines and chemokines released in the proximity of the spinal terminals of injured fibers recruits many more peripheral immune cells to the spinal cord (White et al 2007). Both CD2+ and CD4+ T cells but not B cells accumulate at the spinal termination zone of injured primary afferents. Th1 T cells synthesize and release interferon-γ, which activates microglia, enhances the excitability of spinal dorsal horn neurons, and induces hyperalgesia when injected intrathecally. This cascade apparently contributes to the hyperalgesia in neuropathy because CD4-knockout mice display less mechanical hyperalgesia after L5 nerve transection (Cao and DeLeo 2008). B and T lymphocyte–deficient mice, as well as mice deficient in interferon-γ receptors, likewise display less mechanical hyperalgesia after spared nerve injury (Costigan et al 2009).
Mechanisms of Secondary or Widespread Hyperalgesia
The spinal dorsal horn consists of a complex network of neuronal and non-neuronal cells that communicate with each other via chemical synapses, gap junctions, and interstitial fluid. Along these communication channels, messengers are exchanged that trigger neuronal plasticity at somatotopically remote sites. This may cause hyperalgesia and allodynia outside the primary area of insult (see Fig. 6-4).
Heterosynaptic Long-Term Potentiation
In principle, LTP may affect not only synapses that were activated by the conditioning noxious stimulus but also neighboring, inactive synapses impinging on the same post-synaptic neuron. Intracellular messenger molecules such as Ca2+ rise not just at activated but also at inactive synapses and may thereby trigger heterosynaptic forms of LTP. In addition, gaseous messengers such as NO synthesized either in neurons or in glial cells may freely diffuse tens of microns and thereby induce LTP at synapses that were not activated during the initial insult. If heterosynaptic LTP exists at synapses between nociceptive afferents and principal pain neurons, it will cause pain amplification outside but close to the area of injury or inflammation (i.e., secondary hyperalgesia).
Unblocking Neuronal Pathways
Within the neuronal network of the spinal dorsal horn, excitatory connections exist that are normally blocked by tonic inhibition. Impaired inhibition in the course of inflammation or neuropathy may unblock these pathways and then lead to spread of excitation across modality borders or to somatotopically inappropriate areas. When tonic inhibition in the spinal dorsal horn is impaired, activation of low-threshold mechanosensitive Aβ fibers leads not only to activation of low-threshold or wide–dynamic range neurons in the deep dorsal horn but also to excitation of normally nociceptive-specific neurons in the superficial spinal dorsal horn (Baba et al 1999, Schoffnegger et al 2008). Likewise, nociceptive input may spread to somatotopically inappropriate sites in the spinal cord when tonic inhibition is reduced (Schoffnegger et al 2008). Thus, unblocking excitatory pathways in the spinal dorsal horn probably leads to allodynia and to secondary and spreading hyperalgesia (see also Table 6-1).
Astrocytic Network
Astrocytes form a dense network throughout the CNS, including the spinal dorsal horn. The astrocytic network is connected via connexins (gap junctions), which are aqueous channels permeable by small molecules (up to 1.2 kDa). Spinal astrocytes react to noxious stimulation or spinal cord injury with an increase in [Ca2+]i. The increase in Ca2+ then triggers the synthesis and release of pro-inflammatory and pro-nociceptive substances. Ca2+ waves may spread through connexins across the astrocytic network and thus activate astrocytes at somatotopically remote sites (Kuga et al 2011). This may have profound effects on synaptic strength in remote nociceptive pathways because individual astrocytes wrap thousands of synapses. Indeed, blocking spinal p38 mitogen–activated protein kinase (p38 MAPK), spinal pro-inflammatory cytokines (IL-1, TNF-α, or Il-6), and spinal glial cell activation with fluorocitrate and uncoupling connexins (Spataro et al 2004) all prevent the development of mirror-image hyperalgesia induced by unilateral sciatic inflammatory neuropathy (Milligan et al 2003).
Volume Transmission
When messengers diffuse through the interstitial space or cerebrospinal fluid, it is called “volume transmission,” as opposed to “wired transmission,” which occurs at anatomically defined synapses (Syková and Nicholson 2008). Diffusible substances such as prostaglandin E2 and BDNF are sufficient for disinhibition and/or induction of LTP. They are released into and diffuse within the spinal parenchyma and perhaps gain access to cerebrospinal fluid. They may then trigger hyperalgesia and/or allodynia not only at the site of injury but also at neighboring sites (i.e., secondary hyperalgesia) or remote sites (spreading hyperalgesia or mirror-image hyperalgesia). Volume transmission could well be of clinical relevance since pro-inflammatory cytokines such as IL-1β and IL-6 are increased in the cerebrospinal fluid of some pain patients (Alexander et al 2005).
Impaired Blood–Spinal Cord Barrier
Under normal conditions the blood–spinal cord barrier securely prevents cells or large molecules such as peptides or proteins from leaving the intravascular space and gaining access to the spinal parenchyma. Peripheral neuropathy or spinal cord injury may, however, impair the integrity of this barrier (Whetstone et al 2003, Gordh et al 2006). This leads to exudation of plasma-derived proteins and to invasion of immune competent cells, including macrophages and T lymphocytes (Sweitzer et al 2002). Peripheral inflammation leads to the migration of polymorphonuclear monocytes into the spinal parenchyma (Mitchell et al 2008). Blood-derived inflammatory mediators and leukocytes then modify nociception within and outside somatotopic and modality borders.
Commissural Interneurons
Unilateral peripheral nerve lesions may also have symmetrical, albeit smaller effects on corresponding contralateral uninjured peripheral nerves. Koltzenburg and colleagues (1999) have argued that a system of commissural interneurons capable of both anterograde and retrograde transmedian trophic signaling in the spinal cord or brain stem may be involved.
Descending Facilitation
A number of studies have demonstrated that descending facilitatory pathways originating from the rostral ventromedial medulla contribute to the development of secondary or spreading hyperalgesia and to bilateral hyperalgesia in animal models of inflammatory and muscle pain (Wiertelak et al 1994, Urban et al 1996, Tillu et al 2008, You et al 2010). Enhanced and prolonged spinal release of excitatory amino acids (including glutamate), activation of cAMP-dependent signaling, activation of PKA, and bilateral phosphorylation of cAMP response element–binding protein (CREB) play a role (DeSantana and Sluka 2008).
Multiplicity of Signaling Pathways
A variety of cellular and network mechanisms in the spinal cord dorsal horn contribute to pain amplification, pain generation, and/or touch-evoked pain (Table 6-2). Not surprisingly, these different forms of plasticity also involve distinct, partially overlapping signal transduction pathways. In the following sections, some examples are provided to demonstrate the diversity of signaling pathways in spinal plasticity.
Table 6-2
Key Signaling Molecules in Spinal Nociceptive Networks

ATP, adenosine triphosphate; BDNF, brain-derived neurotrophic factor; iGluR, ionotropic glutamate receptor; IL-1β, interleukin-1β; mGluR, metabotropic glutamate receptor; NK1R, neurokinin 1 receptor; NMDAR, N-methyl-D-aspartate receptor; TNF-α, tumor necrosis factor-α; TNFR, tumor necrosis factor receptor.
Signaling Pathways for Induction or Maintenance of Long-Term Potentiation
Distinct forms of LTP exist at nociceptive synapses as outlined earlier. Their signaling pathways largely overlap, but they are not identical.
Induction of Long-Term Potentiation
A rise in post-synaptic Ca2+ concentration is an essential central step for virtually all forms of LTP. Post-synaptic Ca2+ rises by opening of post-synaptic NMDA receptors (Ikeda et al 2003), T-type VGCCs (Ikeda et al 2006), and Ca2+-permeable AMPA receptors (Hartmann et al 2004) as well as by release of Ca2+ from intracellular Ca2+ stores triggered by activation of metabotropic glutamate receptors or NK1 receptors (Drdla and Sandkühler 2008). Metabotropic receptors mobilize intracellular Ca2+ by activation of ryanodine and inositol 1,4,5-triphosphate (IP3) receptors via phospholipase C (Drdla and Sandkühler 2008) (Fig. 6-5). The post-synaptic Ca2+ apparently needs to be taken up by mitochondria to produce reactive oxygen species (Kim et al 2011) and then activates Ca2+-dependent signaling pathways involving PKC, calcium-calmodulin–dependent protein kinase II (CaMKII), nitric oxide synthase (NOS) (Ikeda et al 2006), and cyclooxygenase-2 (Cox-2). Other enzymes involved are extracellular signal–regulated kinase (ERK), followed by a lasting phosphorylation of CREB (Zhou et al 2008). A recent study suggests that in addition to the well-documented post-synaptic mechanisms, activation of pre-synaptic TRPV1 receptors is also required for induction of LTP at C-fiber synapses (Park et al 2011).
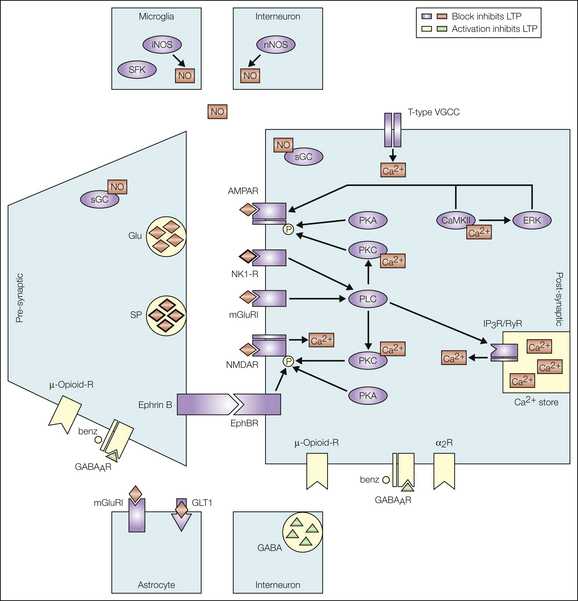
Figure 6-5 Signaling pathways for the induction of long-term potentiation (LTP) at the first synaptic relay of nociception. Pathways shown in red prevent induction of LTP when blocked. The signaling molecules in green inhibit induction of LTP. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptor; benz, benzodiazepine; CaMKII, calcium-calmodulin–dependent kinase II; ERK, extracellular signal–regulated kinase; GABA, γ-aminobutyric acid; GABAAR, GABAA receptor; Glu, glutamate; iNOS, inducible nitric oxide synthase; IP3R, inositol 1,4,5,-triphosphate receptor; mGluRI, metabotropic glutamate receptor I; NK1-R, neurokinin 1 receptor; NMDAR, N-methyl-D-aspartate receptor; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; RyR, ryanodine receptor; SFK, Src family kinases; sGC, soluble guanyly cyclase; SP, substance P; VGCC, voltage-gated calcium channel. (From Ruscheweyh R, Wilder-Smith O, Drdla R, et al 2011 Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Molecular Pain 7:20, Fig. 1.)
LTP induction may also require activation of spinal glial cells via release of ATP from primary afferents and activation of glial P2X4 receptors. This then leads to the activation of p38 MAPK. Activated glial cells release a myriad of pro-inflammatory cytokines, including but not limited to TNF-α and IL-6 (Milligan and Watkins 2009).
Not all forms of spinal LTP induction require the activation of all of these signaling elements. For example, LTP can be induced by spinal application of ATP or BDNF. These forms, but not high-frequency stimulation–induced LTP, depend on p38 MAPK. Likewise, high- and low-frequency stimulation–induced LTP, but not opioid withdrawal LTP, requires activation of CaMKII. Finally high- but not low-frequency stimulation–induced LTP involves activation of NOS and NO signaling.
Maintenance of Long-Term Potentiation
The signaling pathways required for maintaining LTP are different from those engaged in its induction. For example, neither activation of NK1 or NMDA receptors nor activation of NOS is required for maintaining LTP. The early phase of LTP (minutes to hours) apparently involves post-translational modifications of proteins, probably phosphorylation of synaptic AMPA receptors, which enhances their probability of being open, conductance, or trafficking. All this potentiates the strength at glutamatergic synapses (Wang et al 2010). Late-phase LTP develops slowly (over a period of hours) after LTP induction and persists for days, weeks, or even longer. Expression of late-phase LTP requires synapse-to-nucleus signaling via signaling molecules such as ERK1, ERK2, and cAMP, all of which may trigger the activation of CREB. The transcription factor CREB controls the expression of a myriad of proteins, many of which are relevant for nociception. Late-phase LTP can consequently be blocked by protein synthesis inhibitors (Zhou et al 2008) and may involve the incorporation of new AMPA receptors into the post-synaptic membrane (Galan et al 2004). Protein kinase Mζ is essential for late-phase LTP in the hippocampus and for long-lasting facilitation of spinal (Asiedu et al 2011) and cortical nociception (Li et al 2010). Its role in spinal LTP remains to be determined. See Figure 6-6 and Ruscheweyh and colleagues (2011) for a comprehensive review.
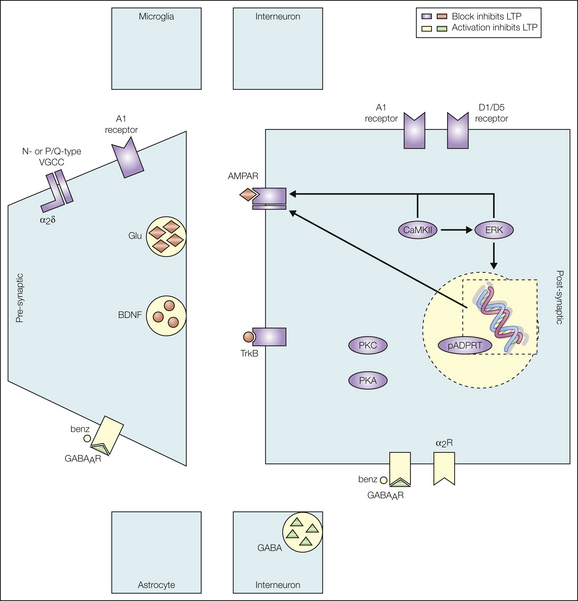
Figure 6-6 Maintenance of long-term potentiation (LTP) requires different signaling pathways from those for its induction. Pathways shown in red inhibit LTP maintenance when blocked. Signaling molecules shown in green inhibit LTP maintenance. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptor; BDNF, brain-derived neurotrophic factor; benz, benzodiazepine; CaMKII, calcium-calmodulin–dependent kinase II; ERK, extracellular signal–regulated kinase; GABA, γ-aminobutyric receptor; GABAAR, GABAA receptor; Glu, glutamate; pADPRT, poly (ADP-ribosyl) transferase; PKA, protein kinase A; PKC, protein kinase C; VGCC, voltage-gated calcium channel. (From Ruscheweyh R, Wilder-Smith O, Drdla R, et al 2011 Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Molecular Pain 7:20, Fig. 2.)
Signaling Pathways in Astrocytes
The rich repertoire of receptors expressed by astrocytes (or microglia) differs according to region, animal species, and activation status, and so do the signaling pathways recruited. Astrocytes express the four known ErbB tyrosine kinase receptors ErbB1–4. They may be activated directly by binding of their ligands, the epidermal growth factor (EGF)-related peptide growth factors, or they may indirectly be transactivated via metabotropic glutamate receptors. Transactivation can be induced by glutamatergic (mGluR5), adrenergic (α2 receptors), serotonergic (5-HT2B), or opioidergic μ or κ receptors. Depending on the type of activation, different ErbB-dependent signaling pathways are relevant. ErbB1 activation can stimulate expression of the glutamate transporter type 1 and the glial glutamate transporter and thus increase the uptake of glutamate and its conversion to glutamine in astrocytes (Sharif and Prevot 2010). Glutamine is then released by astrocytes, taken up by neurons, and converted into glutamate for synaptic release. Integrity of this pathway is essential for maintaining elevated synaptic activity at glutamatergic synapses.
Signaling Pathways Leading to Altered Chloride Homeostasis
Post-synaptic inhibition via GABAA or glycine receptors requires low intracellular Cl− concentrations. The chloride extrusion capacity of neurons largely relies on KCC2 transporter activity. Expression of KCC2 is bidirectionally controlled by BDNF. On activation of microglia by ATP, BDNF is released and binds to the TrkB receptor expressed on spinal dorsal horn neurons. This may lead to the activation of two different signaling pathways in neurons: the Shc/FRS-2 (src homology 2 domain–containing transforming protein/fibroblast growth factor receptor substrate 2 pathway) and the phospholipase Cγ–CREB pathway (Rivera et al 2004). When both pathways are activated simultaneously, KCC2 is down-regulated and chloride extrusion capacity is impaired, as is inhibition via GABAA or glycine receptors (Coull et al 2005). In contrast, KCC2 is up-regulated if only the Shc pathway is activated by BDNF. The former pathway is relevant for impaired inhibition in neuropathy, the latter during development.
Signaling Pathways of Impaired Glycinergic Inhibition during Peripheral Inflammation
In response to inflammation in peripheral tissues, Cox-2 and microsomal prostaglandin E synthase are induced in the spinal cord. These enzymes then produce prostaglandin E2, which binds to the EP2 receptor and increases cAMP levels. This activates PKA, a substrate of which is a specific type of glycine receptor that contains the α3 subunit and is expressed in superficial spinal dorsal horn neurons. Phosphorylation of these inhibitory (strychnine-sensitive) glycine receptors by PKA depresses their functions and thereby leads to disinhibition (Ahmadi et al 2002).
Sex Differences in Spinal Nociception and Antinociception
Sexual dimorphism in neuronal function has been demonstrated from fruit flies to humans (Jazin and Cahill 2010) and also has major effects on nociception. Females tend to have lower thresholds for heat-, mechanical-, inflammation-, and chemical-induced pain (Fillingim et al 2009). Some, but not all, studies have revealed that females are also more prone to the development of chronic pain in a number of nociceptive and neuropathic pain conditions or models (Carmichael et al 2009). Sex differences also exist for analgesia evoked by endogenous inhibitory systems (including descending monoaminergic pathways) and by pharmacological (e.g., opioids) and non-pharmacological (e.g., stress-induced analgesia) means (Fillingim et al 2009). This is described in more detail in Chapter 15 of this textbook. Here, some sex differences in spinal nociception are summarized.
In healthy young female volunteers, nociceptive reflex thresholds are lower and verbal pain ratings are higher than in males. When pain ratings are, however, corrected for differences in spinally mediated reflex thresholds, the sex differences disappear. This suggests that sex differences in spinal nociception substantially contribute to the sexual dimorphism in pain perception (Mylius et al 2005). In line with this, subcutaneous injections of formalin evoke stronger second-phase responses of spinal dorsal horn wide–dynamic range neurons in female than in male rats. This effect was observed in intact but not in spinalized animals, thus suggesting the contribution of a facilitatory supraspinal loop (You et al 2006).
Activation of G protein–coupled, inwardly rectifying potassium channels (GIRKs) mediates post-synaptic inhibition by a number of inhibitory neurotransmitters. This form of inhibition is apparently more effective in males than in females and is responsible for a substantial part of the elevated pain thresholds in male mice. In mice with GIRK2-null mutations, sex differences in spinal nociceptive withdrawal thresholds are no longer detectable (Mitrovic et al 2003).
Nerve injury leads to a myriad of changes in gene expression in spinal dorsal horn cells. Some of these changes depend on sex steroids, which regulate gene expression, particularly in astrocytes but also in other glial cells. Following spinal nerve ligation a number of genes are up-regulated specifically in female rats, including the growth factor neuregulin-1 and its high-affinity receptor ErbB4 (and also the mGluR6 and the tachykinin 1 receptor) (LaCroix-Fralish et al 2006). The up-regulation of neuregulin-1 is restricted to female rats with circulating progesterone. Spinal application of neuregulin-1 induces transient tactile allodynia, thus suggesting that it contributes to sex differences in neuropathic pain.
Female rats in proestrus exhibit stronger hyperalgesia in response to injections of complete Freund’s adjuvant into a hindpaw than do female rats in other estrous stages, ovariectomized rats, or male rats. This difference correlates with higher spinal preprodynorphin levels ipsilateral to the injection site (Bradshaw et al 2000).
Repetitive activation of spinal μ-opioid receptors leads to tolerance of systemic morphine application. This effect is stronger in female than in male rats (Hopkins et al 2004). Female rats express more μ-opioid/κ-opioid receptor heterodimers in spinal cord tissue than do males. Part of the difference is resistant to ovariectomy and thus probably of genetic and/or developmental origin. Another part, however, is affected by the stage of the estrous cycle and depends on the level of circulating ovarian sex steroids. The more extensive expression of μ-opioid/κ-opioid receptor heterodimers in females corresponds to the more robust antinociception by spinally applied κ-opioid receptor agonists in females (Chakrabarti et al 2010). Antinociception by spinal κ-opioid receptors and δ-opioid receptors also leads to elevated pain thresholds during gestation and following the simulation of pregnancy blood levels of estrogen and progesterone (so-called hormone-simulated pregnancy) (Dawson-Basoa and Gintzler 1998). On the other hand, antinociception by activation of spinal α2-adrenergic receptors requires testosterone in male rats and is attenuated in female rats by estrogen (Thompson et al 2008).
Reversal of Spinal Plasticity and Pain
LTP at synapses between nociceptive nerve fibers and principal pain neurons causes hyperalgesia. Reversal of LTP (i.e., “depotentiation”) thus constitutes a potential means of erasing a memory trace of pain. Indeed, brief (1-hour) systemic application of a high dose of the ultrashort-acting µ-opioid receptor agonist remifentanil reverses LTP induced either by low- or high-frequency conditioning stimulation of C-fiber afferents or by subcutaneous capsaicin (Drdla-Schutting et al 2012). The opioid-induced depotentiation involves activation of NMDA receptors, metabotropic glutamate receptors, release of Ca2+ from ryanodine-sensitive intracellular stores, and activation of protein phosphatase 1. AMPA receptor channels are phosphorylated at Ser831 by LTP-inducing stimuli. This leads to enhanced single-channel conductance and thus synaptic strength. AMPA receptors are dephosphorylated at Ser831 by protein phosphatase 1 after high-dose opioid administration. This probably constitutes a key mechanism for opioid-induced depotentiation (Drdla-Schutting et al 2012). Thus, in contrast to current beliefs, opioids not only may temporarily dampen pain but also might eliminate an important cause of hyperalgesia. Likewise, when benzodiazepines are applied directly onto the spinal cord during early-phase LTP, its consolidation is impaired (Hu et al 2006).
LTP at C-fiber synapses can be further reversed by repetitive electrical stimulation of Aδ fibers (Liu et al 1998). This depotentiation is reminiscent of the lasting analgesia following some types of counter-stimulation that are slightly painful, such as high-intensity transcutaneous electrical nerve stimulation or (electro-) acupuncture (Xing et al 2007).
Conclusion
Remarkable progress has been made in understanding the spinal mechanisms of pain generation and amplification. Chronic pain is not a nosological entity, and we must thus not assume that the underlying mechanisms in the spinal cord would be uniform. Rather, our current knowledge suggests that multiple players are involved, including powerful pro- and antinociceptive neuronal and non-neuronal elements. Together, they constitute a complex matrix in which spinal nociception is embedded. The various forms of neuropathy, trauma, and inflammation have differential and time-dependent effects on the nociceptive system and continuously modify its operational modes. Key elements of the spinal nociceptive system temporarily take over novel functions. For example, housekeeping glial cells may switch to pro-nociceptive helper cells and inhibitory synapses may turn into excitatory synapses. Changes within the spinal nociceptive matrix may initially be adaptive to allow better protection of injured or damaged tissue. Spinal plasticity may, however, also take a pathobiological path, overtax compensatory mechanisms, and become maladaptive. The list of key molecular and cellular elements involved is steadily and rapidly growing. It will be a major challenge for the future to unravel their mutual interactions and the operational sequences that define the pathogenesis of chronic pain states and its reversal.
The references for this chapter can be found at www.expertconsult.com.
References
Abrahamsen B., Zhao J., Asante C.O., et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705.
Ahmadi S., Lippross S., Neuhuber W.L., et al. PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nature Neuroscience. 2002;5:34–40.
Alexander G.M., van Rijn M.A., van Hilten J.J., et al. Changes in cerebrospinal fluid levels of pro-inflammatory cytokines in CRPS. Pain. 2005;116:213–219.
Angst M.S., Koppert W., Pahl I., et al. Short-term infusion of the μ-opioid agonist remifentanil in humans causes hyperalgesia during withdrawal. Pain. 2003;106:49–57.
Asiedu M.N., Tillu D.V., Melemedjian O.K., et al. Spinal protein kinase M ζ underlies the maintenance mechanism of persistent nociceptive sensitization. Journal of Neuroscience. 2011;31:6646–6653.
Baba H., Doubell T.P., Woolf C.J. Peripheral inflammation facilitates A β fiber–mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. Journal of Neuroscience. 1999;19:859–867.
Baba H., Ji R.-R., Kohno T., et al. Removal of GABAergic inhibition facilitates polysynaptic A fiber–mediated excitatory transmission to the superficial spinal dorsal horn. Molecular and Cellular Neurosciences. 2003;24:818–830.
Bao L., Wang H.F., Cai H.-J., et al. Peripheral axotomy induces only very limited sprouting of coarse myelinated afferents into inner lamina II of rat spinal cord. European Journal of Neuroscience. 2002;16:175–185.
Basbaum A.I., Bautista D.M., Scherrer G., et al. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284.
Beck H., Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nature Reviews. Neuroscience. 2008;9:357–369.
Benarroch E.E. Descending monoaminergic pain modulation: bidirectional control and clinical relevance. Neurology. 2008;71:217–221.
Bradshaw H., Miller J., Ling Q., et al. Sex differences and phases of the estrous cycle alter the response of spinal cord dynorphin neurons to peripheral inflammation and hyperalgesia. Pain. 2000;85:93–99.
Cao L., DeLeo J.A. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection–induced neuropathic pain. European Journal of Immunology. 2008;38:448–458.
Carmichael N.M., Charlton M.P., Dostrovsky J.O. Sex differences in inflammation evoked by noxious chemical, heat and electrical stimulation. Brain Research. 2009;1276:103–111.
Castro-Lopes J.M., Malcangio M., Pan B.H., et al. Complex changes of GABAA and GABAB receptor binding in the spinal cord dorsal horn following peripheral inflammation or neurectomy. Brain Research. 1995;679:289–297.
Cervero F., Laird J.M., García-Nicas E. Secondary hyperalgesia and pre-synaptic inhibition: an update. European Journal of Pain. 2003;7:345–351.
Chakrabarti S., Liu N.-J., Gintzler A.R. Formation of μ-/κ-opioid receptor heterodimer is sex-dependent and mediates female-specific opioid analgesia. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20115–20119.
Chang G., Chen L., Mao J. Opioid tolerance and hyperalgesia. Medical Clinics of North America. 2007;91:199–211.
Chen L., Malarick C., Seefeld L., et al. Altered quantitative sensory testing outcome in subjects with opioid therapy. Pain. 2009;143:65–70.
Chen Y., Balasubramanyan S., Lai A.Y., et al. Effects of sciatic nerve axotomy on excitatory synaptic transmission in rat substantia gelatinosa. Journal of Neurophysiology. 2009;102:3203–3215.
Christensen B.N., Perl E.R. Spinal neurons specifically excited by noxious or thermal stimuli: marginal zone of the dorsal horn. Journal of Neurophysiology. 1970;33:293–307.
Clark A.K., Staniland A.A., Marchand F., et al. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. Journal of Neuroscience. 2010;30:573–582.
Clark A.K., Yip P.K., Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. Journal of Neuroscience. 2009;29:6945–6954.
Costigan M., Moss A., Latremoliere A., et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain–like hypersensitivity. Journal of Neuroscience. 2009;29:14415–14422.
Coull J.A.M., Beggs S., Boudreau D., et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021.
Coull J.A.M., Boudreau D., Bachand K., et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942.
Craig A.D. Pain mechanisms: labeled lines versus convergence in central processing. Annual Review of Neuroscience. 2003;26:1–30.
Dawson-Basoa M., Gintzler A.R. Gestational and ovarian sex steroid antinociception: synergy between spinal κ and δ opioid systems. Brain Research. 1998;794:61–67.
Derjean D., Bertrand S., Le Masson G., et al. Dynamic balance of metabotropic inputs causes dorsal horn neurons to switch functional states. Nature Neuroscience. 2003;6:274–281.
DeSantana J.M., Sluka K.A. Central mechanisms in the maintenance of chronic widespread noninflammatory muscle pain. Current Pain and Headache Reports. 2008;12:338–343.
Dominguez E., Mauborgne A., Mallet J., et al. SOCS3-mediated blockade of JAK/STAT3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. Journal of Neuroscience. 2010;30:5754–5766.
Dougherty K.J., Hochman S. Spinal cord injury causes plasticity in a subpopulation of lamina I GABAergic interneurons. Journal of Neurophysiology. 2008;100:212–223.
Drdla R., Gassner M., Gingl E., et al. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210.
Drdla R., Sandkühler J. Long-term potentiation at C-fibre synapses by low-level pre-synaptic activity in vivo. Molecular Pain. 2008;4:18.
Drdla-Schutting R., Benrath J., Wunderbaldinger G., et al. Erasure of spinal memory traces of pain by a brief, high-dose opioid administration. Science. 2012;335:235–238.
Eaton M.J., Plunkett J.A., Karmally S., et al. Changes in GAD- and GABA-immunoreactivity in the spinal dorsal horn after peripheral nerve injury and promotion of recovery by lumbar transplant of immortalized serotonergic precursors. Journal of Chemical Neuroanatomy. 1998;16:57–72.
Ellrich J., Schorr A. Low-frequency stimulation of trigeminal afferents induces long-term depression of human sensory processing. Brain Research. 2004;996:255–258.
Fenselau H., Heinke B., Sandkühler J. Heterosynaptic long-term potentiation at GABAergic synapses of spinal lamina I neurons. Journal of Neuroscience. 2011;31:17383–17391.
Fillingim R.B., King C.D., Ribeiro-Dasilva M.C., et al. Sex, gender, and pain: a review of recent clinical and experimental findings. Journal of Pain. 2009;10:447–485.
Foerster O. Vorderseitenstrangdurchschneidung im Rückenmark zur Beseitigung von Schmerzen. Berliner Klinische Wochenschrift. 1913;50:1499–1500.
Galan A., Laird J.M., Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323.
Goldscheider A. Über den Schmerz in physiologischer und klinischer Hinsicht. Berlin: Hirschwald; 1894.
Gordh T., Chu H., Sharma H.S. Spinal nerve lesion alters blood–spinal cord barrier function and activates astrocytes in the rat. Pain. 2006;124:211–221.
Guan Y., Terayama R., Dubner R., et al. Plasticity in excitatory amino acid receptor–mediated descending pain modulation after inflammation. Journal of Pharmacology and Experimental Therapeutics. 2002;300:513–520.
Hardy J.D., Wolff H.G., Goodell H. Experimental evidence on the nature of cutaneous hyperalgesia. Journal of Clinical Investigation. 1950;29:115–140.
Hartmann B., Ahmadi S., Heppenstall P.A., et al. The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron. 2004;44:637–650.
Harvey R.J., Depner U.B., Wässle H., et al. GlyR α3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304:884–887.
Haydon P.G. GLIA: listening and talking to the synapse. Nature Reviews. Neuroscience. 2001;2:185–193.
Heinke B., Gingl E., Sandkühler J. Multiple targets of μ-opioid receptor mediated pre-synaptic inhibition at primary afferent Aδ- and C-fibers. Journal of Neuroscience. 2011;31:1313–1322.
Heinl C., Drdla-Schutting R., Xanthos D.N., et al. Distinct mechanisms underlying pronociceptive effects of opioids. Journal of Neuroscience. 2011;31:16748–16756.
Heinricher M.M., Tavares I., Leith J.L., et al. Descending control of nociception: specificity, recruitment and plasticity. Brain Research Reviews. 2009;60:214–225.
Herrero J.F., Laird J.M., Lopez-Garcia J.A. Wind-up of spinal cord neurones and pain sensation: much ado about something? Progress in Neurobiology. 2000;61:169–203.
Hopkins E., Rossi G., Kest B. Sex differences in systemic morphine analgesic tolerance following intrathecal morphine injections. Brain Research. 2004;1014:244–246.
Hösl K., Reinold H., Harvey R.J., et al. Spinal prostaglandin E receptors of the EP2 subtype and the glycine receptor α3 subunit, which mediate central inflammatory hyperalgesia, do not contribute to pain after peripheral nerve injury or formalin injection. Pain. 2006;126:46–53.
Houghton A.K., Hewitt E., Westlund K.N. Dorsal column lesion prevents mechanical hyperalgesia and allodynia in osteotomy model. Pain. 1999;82:73–80.
Hu X.-D., Ge Y.-X., Hu N.-W., et al. Diazepam inhibits the induction and maintenance of LTP of C-fiber evoked field potentials in spinal dorsal horn of rats. Neuropharmacology. 2006;50:238–244.
Hua X.-Y., Svensson C.I., Matsui T., et al. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. European Journal of Neuroscience. 2005;22:2431–2440.
Hughes D.I., Scott D.T., Todd A.J., et al. Lack of evidence for sprouting of Aβ afferents into the superficial laminas of the spinal cord dorsal horn after nerve section. Journal of Neuroscience. 2003;23:9491–9499.
Ikeda H., Heinke B., Ruscheweyh R., et al. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240.
Ikeda H., Stark J., Fischer H., et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662.
Jazin E., Cahill L. Sex differences in molecular neuroscience: from fruit flies to humans. Nature Reviews. Neuroscience. 2010;11:9–17.
Kim H.Y., Lee K.Y., Lu Y., et al. Mitochondrial Ca2+ uptake is essential for synaptic plasticity in pain. Journal of Neuroscience. 2011;31:12982–12991.
Klein T., Magerl W., Hopf H.-C., et al. Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. Journal of Neuroscience. 2004;24:964–971.
Klein T., Magerl W., Nickel U., et al. Effects of the NMDA-receptor antagonist ketamine on perceptual correlates of long-term potentiation within the nociceptive system. Neuropharmacology. 2007;52:655–661.
Koltzenburg M., Lundberg L.E., Torebjörk H.E. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51:207–219.
Koltzenburg M., Wall P.D., McMahon S.B. Does the right side know what the left is doing? Trends in Neurosciences. 1999;22:122–127.
Kuga N., Sasaki T., Takahara Y., et al. Large-scale calcium waves traveling through astrocytic networks in vivo. Journal of Neuroscience. 2011;31:22607–22614.
LaCroix-Fralish M.L., Tawfik V.L., Spratt K.F., et al. Sex differences in lumbar spinal cord gene expression following experimental lumbar radiculopathy. Journal of Molecular Neuroscience. 2006;30:283–295.
Lang R., Gundlach A.L., Kofler B. The galanin peptide family: receptor pharmacology, pleiotropic biological actions, and implications in health and disease. Pharmacology & Therapeutics. 2007;115:177–207.
Larsson M. Ionotropic glutamate receptors in spinal nociceptive processing. Molecular Neurobiology. 2009;40:260–288.
Lever I., Cunningham J., Grist J., et al. Release of BDNF and GABA in the dorsal horn of neuropathic rats. European Journal of Neuroscience. 2003;18:1169–1174.
Li X.-Y., Ko H.-G., Chen T., et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMζ in the anterior cingulate cortex. Science. 2010;330:1400–1404.
Liu M., Oh U., Wood J.N. From transduction to pain sensation: defining genes, cells, and circuits. Pain. 2011;152:S16–S19.
Liu X.-G., Morton C.R., Azkue J.J., et al. Long-term depression of C-fibre–evoked spinal field potentials by stimulation of primary afferent Aδ-fibres in the adult rat. European Journal of Neuroscience. 1998;10:3069–3075.
Luo C., Gangadharan V., Bali K.K. PloS Biology. 2012;10:e1001283.
Lynn B. Cutaneous hyperalgesia. British Medical Bulletin. 1977;33:103–108.
Mantyh P.W., Rogers S.D., Honoré P., et al. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279.
Matyash V., Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Research Reviews. 2010;63:2–10.
McMahon S.B., Koltzenburg M. Itching for an explanation. Trends in Neurosciences. 1992;15:497–501.
McMahon S.B., Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54.
McMahon S.B., Wall P.D. Receptive fields of rat lamina 1 projection cells move to incorporate a nearby region of injury. Pain. 1984;19:235–247.
Meller S.T., Dykstra C.L., Grzybicki D., et al. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478.
Meller S.T., Gebhart G.F., Maves T.J. Neonatal capsaicin treatment prevents the development of the thermal hyperalgesia produced in a model of neuropathic pain in the rat. Pain. 1992;51:317–321.
Mendell L.M., Wall P.D. Responses of single dorsal cord cells to peripheral cutaneous unmyelinated fibers. Nature. 1965;206:97–99.
Miletic G., Draganic P., Pankratz M.T., et al. Muscimol prevents long-lasting potentiation of dorsal horn field potentials in rats with chronic constriction injury exhibiting decreased levels of the GABA transporter GAT-1. Pain. 2003;105:347–353.
Millan M.J. Descending control of pain. Progress in Neurobiology. 2002;66:355–474.
Milligan E.D., Twining C., Chacur M., et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. Journal of Neuroscience. 2003;23:1026–1040.
Milligan E.D., Watkins L.R. Pathological and protective roles of glia in chronic pain. Nature Reviews. Neuroscience. 2009;10:23–36.
Miraucourt L.S., Peirs C., Dallel R., et al. Glycine inhibitory dysfunction turns touch into pain through astrocyte-derived D-serine. Pain. 2011;152:1340–1348.
Mitchell K., Yang H.-Y., Tessier P.A., et al. Localization of S100A8 and S100A9 expressing neutrophils to spinal cord during peripheral tissue inflammation. Pain. 2008;134:216–231.
Mitrovic I., Margeta-Mitrovic M., Bader S., et al. Contribution of GIRK2-mediated post-synaptic signaling to opiate and α2-adrenergic analgesia and analgesic sex differences. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:271–276.
Mogil J.S., Pasternak G.W. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacological Reviews. 2001;53:381–415.
Moore K.A., Kohno T., Karchewski L.A., et al. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. Journal of Neuroscience. 2002;22:6724–6731.
Morisset V., Nagy F. Modulation of regenerative membrane properties by stimulation of metabotropic glutamate receptors in rat deep dorsal horn neurons. Journal of Neurophysiology. 1996;76:2794–2798.
Müller F., Heinke B., Sandkühler J. Reduction of glycine receptor–mediated miniature inhibitory post-synaptic currents in rat spinal lamina I neurons after peripheral inflammation. Neuroscience. 2003;122:799–805.
Mylius V., Kunz M., Schepelmann K., et al. Sex differences in nociceptive withdrawal reflex and pain perception. Somatosensory & Motor Research. 2005;22:207–211.
Nichols M.L., Allen B.J., Rogers S.D., et al. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561.
Ossipov M.H., Hong Sun T., Malan P., Jr., et al. Mediation of spinal nerve injury induced tactile allodynia by descending facilitatory pathways in the dorsolateral funiculus in rats. Neuroscience Letters. 2000;290:129–132.
Palecek J., Paleckova V., Willis W.D. The roles of pathways in the spinal cord lateral and dorsal funiculi in signaling nociceptive somatic and visceral stimuli in rats. Pain. 2002;96:297–307.
Park C.-K., Lu N., Xu Z.-Z., et al. Resolving TRPV1- and TNF-α–mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. Journal of Neuroscience. 2011;31:15072–15085.
Pitcher M.H., Cervero F. Role of the NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain. 2010;151:756–762.
Polgár E., Hughes D.I., Riddell J.S., et al. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. 2003;104:229–239.
Polgár E., Todd A.J. Tactile allodynia can occur in the spared nerve injury model in the rat without selective loss of GABA or GABAA receptors from synapses in laminae I-II of the ipsilateral spinal dorsal horn. Neuroscience. 2008;156:193–202.
Porreca F., Ossipov M.H., Gebhart G.F. Chronic pain and medullary descending facilitation. Trends in Neurosciences. 2002;25:319–325.
Raghavendra V., Tanga F., DeLeo J.A. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. Journal of Pharmacology and Experimental Therapeutics. 2003;306:624–630.
Randic M., Jiang M.C., Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. Journal of Neuroscience. 1993;13:5228–5241.
Reali C., Fossat P., Landry M., et al. Intrinsic membrane properties of spinal dorsal horn neurones modulate nociceptive information processing in vivo. Journal of Physiology. 2011;589:2733–2743.
Rivera C., Voipio J., Thomas-Crusells J., et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. Journal of Neuroscience. 2004;24:4683–4691.
Rudomin P., Schmidt R.F. Pre-synaptic inhibition in the vertebrate spinal cord revisited. Experimental Brain Research. 1999;129:1–37.
Ruscheweyh R., Wilder-Smith O., Drdla R., et al. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Molecular Pain. 2011;7:20.
Russo R.E., Nagy F., Hounsgaard J. Modulation of plateau properties in dorsal horn neurones in a slice preparation of the turtle spinal cord. Journal of Physiology. 1997;499:459–474.
Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiological Reviews. 2009;89:707–758.
Sandkühler J. Central sensitization versus synaptic long-term potentiation (LTP): a critical comment. Journal of Pain. 2010;11:798–800.
Sandkühler J., Chen J.G., Cheng G., et al. Low-frequency stimulation of afferent Aδ-fibers induces long-term depression at primary afferent synapses with substantia gelatinosa neurons in the rat. Journal of Neuroscience. 1997;17:6483–6491.
Santos S.F., Luz L.L., Szucs P., et al. Transmission efficacy and plasticity in glutamatergic synapses formed by excitatory interneurons of the substantia gelatinosa in the rat spinal cord. PLoS ONE. 2009;4:e8047.
Satoh O., Omote K. Roles of monoaminergic, glycinergic and GABAergic inhibitory systems in the spinal cord in rats with peripheral mononeuropathy. Brain Research. 1996;728:27–36.
Schäfers M., Sorkin L. Effect of cytokines on neuronal excitability. Neuroscience Letters. 2008;437:188–193.
Schoffnegger D., Heinke B., Sommer C., et al. Physiological properties of spinal lamina II GABAergic neurons in mice following peripheral nerve injury. Journal of Physiology. 2006;577:869–878.
Schoffnegger D., Ruscheweyh R., Sandkühler J. Spread of excitation across modality borders in spinal dorsal horn of neuropathic rats. Pain. 2008;135:300–310.
Seal R.P., Wang X., Guan Y., et al. Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature. 2009;462:651–655.
Seybold V.S. The role of peptides in central sensitization. Handbook of Experimental Pharmacology. 2009;194:451–491.
Sharif A., Prevot V. ErbB receptor signaling in astrocytes: a mediator of neuron-glia communication in the mature central nervous system. Neurochemistry International. 2010;57:344–358.
Shir Y., Seltzer Z. A-fibers mediate mechanical hyperesthesia and allodynia and C-fibers mediate thermal hyperalgesia in a new model of causalgiform pain disorders in rats. Neuroscience Letters. 1990;115:62–67.
Spataro L.E., Sloane E.M., Milligan E.D., et al. Spinal gap junctions: potential involvement in pain facilitation. Journal of Pain. 2004;5:392–405.
Sweitzer S.M., Hickey W.F., Rutkowski M.D., et al. Focal peripheral nerve injury induces leukocyte trafficking into the central nervous system: potential relationship to neuropathic pain. Pain. 2002;100:163–170.
Syková E., Nicholson C. Diffusion in brain extracellular space. Physiological Reviews. 2008;88:1277–1340.
Tarpley J.W., Kohler M.G., Martin W.J. The behavioral and neuroanatomical effects of IB4-saporin treatment in rat models of nociceptive and neuropathic pain. Brain Research. 2004;1029:65–76.
Thompson A.D., Angelotti T., Nag S., et al. Sex-specific modulation of spinal nociception by α2-adrenoceptors: differential regulation by estrogen and testosterone. Neuroscience. 2008;153:1268–1277.
Tillu D.V., Gebhart G.F., Sluka K.A. Descending facilitatory pathways from the RVM initiate and maintain bilateral hyperalgesia after muscle insult. Pain. 2008;136:331–339.
Todd A.J. Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews. Neuroscience. 2010;11:823–836.
Tong C.-K., Kaftan E.J., MacDermott A.B. Functional identification of NR2 subunits contributing to NMDA receptors on substance P receptor–expressing dorsal horn neurons. Molecular Pain. 2008;4:44.
Torsney C. Inflammatory pain unmasks heterosynaptic facilitation in lamina I neurokinin 1 receptor–expressing neurons in rat spinal cord. Journal of Neuroscience. 2011;31:5158–5168.
Tzingounis A.V., Wadiche J.I. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nature Reviews. Neuroscience. 2007;8:935–947.
Urban M.O., Jiang M.C., Gebhart G.F. Participation of central descending nociceptive facilitatory systems in secondary hyperalgesia produced by mustard oil. Brain Research. 1996;737:83–91.
Vanegas H., Schaible H.-G. Prostaglandins and cyclooxygenases in the spinal cord. Progress in Neurobiology. 2001;64:327–363.
Vanegas H., Schaible H.-G. Descending control of persistent pain: inhibitory or facilitatory? Brain Research. Brain Research Reviews. 2004;46:295–309.
Van Hees J., Gybels J. C nociceptor activity in human nerve during painful and non painful skin stimulation. Journal of Neurology, Neurosurgery, and Psychiatry. 1981;44:600–607.
Vardanyan A., Wang R., Vanderah T.W., et al. TRPV1 receptor in expression of opioid-induced hyperalgesia. Journal of Pain. 2009;10:243–252.
Vera-Portocarrero L.P., Zhang E.-T., King T., et al. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007;129:35–45.
Verri W.A., Jr., Cunha T.M., Parada C.A., et al. Hypernociceptive role of cytokines and chemokines: targets for analgesic drug development? Pharmacology & Therapeutics. 2006;112:116–138.
Virk M.S., Williams J.T. Agonist-specific regulation of μ-opioid receptor desensitization and recovery from desensitization. Molecular Pharmacology. 2008;73:1301–1308.
von Frey M. Beiträge zur Physiologie des Schmerzsinns. Berichte über die Verhandlungen der Königlich Sächsischen Gesellschaft der Wissenschaften zu Leipzig. Mathematisch-Physische Classe. 1894;46:185–196.
Wang Y., Wu J., Wu Z., et al. Regulation of AMPA receptors in spinal nociception. Molecular Pain. 2010;6:5–12.
Watkins L.R., Martin D., Ulrich P., et al. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235.
Wei F., Dubner R., Zou S., et al. Molecular depletion of descending serotonin unmasks its novel facilitatory role in the development of persistent pain. Journal of Neuroscience. 2010;30:8624–8636.
Whetstone W.D., Hsu J.-Y., Eisenberg M., et al. Blood–spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. Journal of Neuroscience Research. 2003;74:227–239.
White F.A., Jung H., Miller R.J. Chemokines and the pathophysiology of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20151–20158.
Wiertelak E.P., Furness L.E., Horan R., et al. Subcutaneous formalin produces centrifugal hyperalgesia at a non-injected site via the NMDA–nitric oxide cascade. Brain Research. 1994;649:19–26.
Willis W.D., Jr., Coggeshall R.E. Sensory mechanisms of the spinal cord 1. New York: Kluwer Academic/Plenum; 2004.
Wood J.N. Nerve growth factor and pain. New England Journal of Medicine. 2010;363:1572–1573.
Woolf C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688.
Woolf C.J., Shortland P., Coggeshall R.E. Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature. 1992;355:75–78.
Woolf C.J., Wall P.D. Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. Journal of Neuroscience. 1986;6:1433–1442.
Xing G.-G., Liu F.-Y., Qu X.-X., et al. Long-term synaptic plasticity in the spinal dorsal horn and its modulation by electroacupuncture in rats with neuropathic pain. Experimental Neurology. 2007;208:323–332.
You H.-J., Cao D.-Y., Yuan B., et al. Sex differences in the responses of spinal wide–dynamic range neurons to subcutaneous formalin and in the effects of different frequencies of conditioning electrical stimulation. Neuroscience. 2006;138:1299–1307.
You H.-J., Lei J., Sui M.-Y., et al. Endogenous descending modulation: spatiotemporal effect of dynamic imbalance between descending facilitation and inhibition of nociception. Journal of Physiology. 2010;588:4177–4188.
Zhou L.-J., Zhong Y., Ren W.-J., et al. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Experimental Neurology. 2008;212:507–514.
Zhuo M. Cortical plasticity as a new endpoint measurement for chronic pain. Molecular Pain. 2011;7:54.
Christensen B.N., Perl E.R. Spinal neurons specifically excited by noxious or thermal stimuli: marginal zone of the dorsal horn. Journal of Neurophysiology. 1970;33:293–307.
Clark A.K., Staniland A.A., Marchand F., et al. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. Journal of Neuroscience. 2010;30:573–582.
Coull J.A.M., Beggs S., Boudreau D., et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021.
Craig A.D. Pain mechanisms: labeled lines versus convergence in central processing. Annual Review of Neuroscience. 2003;26:1–30.
Drdla R., Gassner M., Gingl E., et al. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210.
Fillingim R.B., King C.D., Ribeiro-Dasilva M.C., et al. Sex, gender, and pain: a review of recent clinical and experimental findings. Journal of Pain. 2009;10:447–485.
Hardy J.D., Wolff H.G., Goodell H. Experimental evidence on the nature of cutaneous hyperalgesia. Journal of Clinical Investigation. 1950;29:115–140.
Harvey R.J., Depner U.B., Wässle H., et al. GlyR α3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304:884–887.
Hughes D.I., Scott D.T., Todd A.J., et al. Lack of evidence for sprouting of Aβ afferents into the superficial laminas of the spinal cord dorsal horn after nerve section. Journal of Neuroscience. 2003;23:9491–9499.
Ikeda H., Stark J., Fischer H., et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662.
Klein T., Magerl W., Hopf H.-C., et al. Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. Journal of Neuroscience. 2004;24:964–971.
Mantyh P.W., Rogers S.D., Honoré P., et al. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279.
McMahon S.B., Malcangio M. Current challenges in glia-pain biology. Neuron. 2009;64:46–54.
McMahon S.B., Wall P.D. Receptive fields of rat lamina 1 projection cells move to incorporate a nearby region of injury. Pain. 1984;19:235–247.
Millan M.J. Descending control of pain. Progress in Neurobiology. 2002;66:355–474.
Milligan E.D., Twining C., Chacur M., et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. Journal of Neuroscience. 2003;23:1026–1040.
Pitcher M.H., Cervero F. Role of the NKCC1 co-transporter in sensitization of spinal nociceptive neurons. Pain. 2010;151:756–762.
Porreca F., Ossipov M.H., Gebhart G.F. Chronic pain and medullary descending facilitation. Trends in Neurosciences. 2002;25:319–325.
Randic M., Jiang M.C., Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. Journal of Neuroscience. 1993;13:5228–5241.
Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiological Reviews. 2009;89:707–758.
Seal R.P., Wang X., Guan Y., et al. Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature. 2009;462:651–655.
Todd A.J. Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews. Neuroscience. 2010;11:823–836.
Wood J.N. Nerve growth factor and pain. New England Journal of Medicine. 2010;363:1572–1573.
Woolf C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688.
Zhou L.-J., Zhong Y., Ren W.-J., et al. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. Experimental Neurology. 2008;212:507–514.