Neuropathic Pain
Pathophysiological Response of Nerves to Injury
Introduction
Injury or disease affecting peripheral nerves frequently results in the development of chronic, often intractable pain. The clinical importance of neuropathic pain syndromes (Breivik et al 2006, Bouhassira et al 2008) and the intellectual challenge that they represent provide strong incentive for revealing the underlying mechanisms. There is general consensus today that both peripheral and central nervous system (PNS, CNS) processes play a role. The CNS changes, however, are driven largely by changes in the PNS. Thus, controlling pathophysiological change in the periphery is likely to have greater therapeutic impact than approaches that target CNS processes. PNS processes also tend to be more accessible to therapeutic intervention. The aim of this chapter is to summarize the state of knowledge on PNS pathophysiology associated with neuropathic pain, with special emphasis on processes relevant to clinical symptoms.
The Paradox of Neuropathic Pain
Chronic pain resulting from nerve injury and disease is paradoxical. Just as cutting a telephone wire leaves the line dead, cutting axons should deaden sensation. Sure enough, denervation of a body part does result in hypoesthesia or complete numbness, the hallmark “negative” symptoms of neuropathy. However, nerve pathology is also associated with “positive” symptoms and signs, including the following:
Neuropathic pain is frequently described in terms of natural stimuli—burning, stabbing, or cramping, for example. However, these stimuli may be accompanied by peculiar sensations that are more or less unique to neuropathy, such as pins and needles, electric shock–like paroxysms, “aftersensation” (persistence of the sensation after the stimulus has ended), and “hyperpathic” phenomena such as the spread of sensation beyond the site of stimulation or pain that starts dull but with repeated stimulation “winds up” to an unbearable crescendo (Kugelberg and Lindblom 1959, Noordenbos 1959, Gottrup et al 2003). These peculiar sensations are sufficiently distinctive that their presence may be sufficient to diagnose a chronic pain as being neuropathic in origin (Bouhassira et al 2005). Progress in animal and clinical research has advanced enough that it can now provide a reasonable framework for understanding the pain in neuropathy, including its bizarre peculiarities.
Chronic Pain Depends on the Properties of Neurons
Sensation, including pain, is the domain of the nervous system. Although this may seem trivially obvious, it is sometimes forgotten that stimuli delivered to skin, muscle, bone, and viscera give rise to pain only by virtue of the nerve fibers that innervate them. Completely denervated tissue is numb. On the other hand, sensations that feel as though they originated in a particular body part can be due to impulses generated in nerves, sensory ganglia, or the CNS, even if the tissue itself is numb or completely absent. Examples are anesthesia dolorosa and phantom limb pain. In these cases the sensations are “referred to” the numb or absent body part. Understanding neuropathic pain means understanding how neuropathy affects the generation of sensory signals and their transmission to the CNS for processing. Correspondingly, when planning a treatment strategy the first question that needs to be asked is “Where are the pain-provoking impulses coming from?”
”Normal,” “Inflammatory,” and “Neuropathic” Pain
Normally, pain is felt when signals originating in thinly myelinated (Aδ) and/or unmyelinated (C) nociceptive afferents reach a conscious brain. Examples are pinprick or a stubbed toe. The pain is always evoked by stimuli; spontaneous pain is not normal. The sensation felt (pain) corresponds in location, time, and quality to the stimulus (noxious) in the expected manner. This is “normal” (or nociceptive) pain.
In addition to evoking acute nociceptive pain, burns, abrasions, chemical irritations, and infections often cause more prolonged pain, both spontaneous and evoked by stimuli. Pain in response to weak, normally innocuous stimuli is “allodynia”; exaggerated pain in response to stimuli expected to be (moderately) painful is “hyperalgesia” (Merskey and Bogduk 1994). In the case of allodynia, at least, tenderness in the “sensitized” tissue (pain) no longer corresponds to the stimulus (non-noxious). This type of pain is usually called inflammatory pain since it is typically accompanied by an immune response and mediated by pro-inflammatory molecules. It should be noted, however, that pungent substances such as capsaicin and (non-injuring) electrical shocks can provoke pain with these characteristics even though they do not damage tissue or elicit an immune response.
Neuropathic pain, like inflammatory pain, involves pathology; it is not “normal,” and it often features sensitization and allodynia and hence mismatch between stimulus (non-noxious) and response (pain). It differs, however, in that it is caused by a lesion or disease of the somatosensory nervous system itself. This distinction is not without problems. Pain from inflammation in a major nerve trunk (“neuritis”) is generally considered neuropathic. On the other hand, even minor trauma to skin, muscle, or joint always injures the terminal part of some nerve fibers. Despite the neuropathy present, however, such pain is rarely neuropathic. The reason is that except in rare cases, the minor neural injury is not the cause of the pain. Neuropathic pain is also distinguished from inflammatory pain by the frequent presence of unique sensory features such as electric shock–like sensations and hyperpathia.
Normal (nociceptive) pain and inflammatory pain are adaptive design features of the intact pain system—an alarm bell. Neuropathic pain, in contrast, reflects faulty, maladaptive functioning of a pain system that has been damaged. Consider a defective alarm system constantly producing false alarms. All three types of pain can and often do co-exist. For example, a space-occupying tumor may simultaneously apply noxious force to otherwise healthy tissue evoking nociceptive pain, trigger an inflammatory response, and directly injure nerves. The relationships among these different pain terms are illustrated in Figure 61-1.
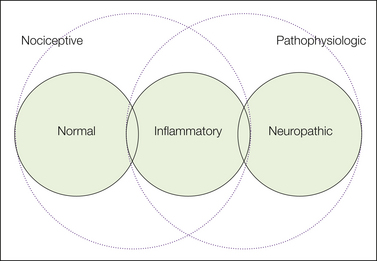
Figure 61-1 Pain terms.
“Nociceptive pain” refers to the normal, acute pain sensation evoked by noxious stimuli in intact tissue in the absence of peripheral or central sensitization. Some authors include in this category the subacute spontaneous pain and hypersensitivity that arise in inflamed tissue when nociceptor endings have undergone peripheral sensitization (central sensitization may also be present). Others group inflammatory and neuropathic pain under the heading “pathophysiological” because both involve sensitization of the pain system as a result of tissue or nerve pathology, respectively.
Sensitization
Sensitization by chemical substances, inflammation, and neuropathy may result from PNS and/or CNS processes. The classic explanation of tissue hypersensibility is the “sensitized nociceptor” hypothesis (Lewis 1942). According to this hypothesis, hypersensibility is due to a reduction in the threshold of nociceptive sensory endings, such as in the skin (“peripheral sensitization”). This is probably the right explanation in the case of heat allodynia. Bradykinin and many other inflammatory mediators are known to cause thermosensitive nociceptors to respond to modest warming at temperatures normally too low to evoke pain. The result is “heat allodynia.” If the threshold falls below the ambient temperature, impulse firing and burning pain will appear to be spontaneous. Likewise, sensitized nociceptors show an exaggerated response to suprathreshold heat and mechanical stimuli, including de novo responses of previously insensitive C fibers (Schmidt et al 1994). This yields “heat and mechanical hyperalgesia.” However, the sensitized nociceptor hypothesis does not explain “tactile allodynia” i.e., pain evoked by light touch. The mechanical response thresholds of Aδ and C nociceptors rarely drop into the range that evokes tactile allodynia no matter what inflammatory mediator is applied (Kocher et al 1987; Reeh et al 1987; Koltzenburg et al 1994a; Schmidt et al 1995; Andrew and Greenspan 1999; Banik and Brennan 2004, 2008; Tsuboi et al 2004, Shim et al 2005). Rather, a considerable body of evidence indicates that tenderness to touch is signaled by low-threshold Aβ touch afferents, not sensitized nociceptors.
The radical idea that pain can be signaled by non-nociceptive low-threshold afferents comes from many observations. First and foremost is response time. If sensitized C-fiber nociceptors were to blame, there ought to be a long delay between the tactile stimulus and the pain, about a second for an inflamed finger (≈1-m conduction distance at ≈1 m/sec) and longer for an inflamed toe. One could argue that the immediate response actually experienced is due to sensitized Aδ nociceptors. However, one would then expect that each touch would evoke two volleys of pain, a rapid Aδ-fiber response and then a later C-fiber response (first and second pain). Tactile allodynia is a common, almost an everyday event, and a 1-second delay between stimulus and response could not be missed. Such delays do not occur (Campbell et al 1988). A second argument is that sensitized nociceptors show only a small reduction in the tactile response threshold. As noted above, few if any come to respond to the light brush, touch, and air puff stimuli that evoke allodynic pain. A variety of additional observations involving afferent-selective nerve block, intraneural electrical stimulation, absence of flare, and others support the conclusion that the signal that evokes tactile allodynia is carried centrally by rapidly conducting, thickly myelinated, Aβ, low-threshold mechanoreceptive touch afferents (Campbell et al 1988, Koltzenburg et al 1994b, Torebjork et al 1992).
The existence of “Aβ pain” constitutes a revolution in our understanding of both inflammatory and neuropathic pain. Indeed, since tactile allodynia is an important cause of suffering and disability in patients with neuropathic pain, pain signaled by Aβ touch afferents may be as important as pain signaled by nociceptors. However, how can Aβ fibers, which normally evoke touch, come to evoke pain? This is due largely to altered central processing of the peripheral signal, or central sensitization (Hardy et al 1952, Woolf 1983, Devor et al 1991, Woolf 2011). Central sensitization, in contrast to peripheral sensitization, is not simply a threshold-lowering process. Indeed, in tactile allodynia there is no noticeable change in the response threshold of Aβ touch-sensitive fibers to touch stimuli. Moreover, the sensation that they evoke is not strong touch. It is pain. Rather than simply amplifying, central sensitization changes the modality of the response from touch to pain. This is accompanied by a corresponding change in the cortical areas activated (Maihofner et al 2006). A large variety of electrophysiological mechanisms have been proposed to explain this transformation. Examples include activation of previously blocked N-methyl-D-aspartate (NMDA)-type glutamate receptors, imbalance in chloride ion equilibrium, loss of inhibitory interneurons, activation of glia, and altered descending control. Mechanisms of central sensitization will not be discussed here, although the concept will be referred to frequently.
How Does Nerve Injury and Disease Trigger Neuropathic Pain?
Neuropathic pain can be induced by trauma, vascular and metabolic disorders, bacterial and viral infection, inflammation, autoimmune attack, genetic abnormalities, chemotherapeutic agents and other neurotoxins, burns, and a wide variety of other pathological processes that affect peripheral nerves, sensory ganglia, spinal roots, and CNS structures. The specific precipitating event appears to be less important than its common pathological effects: (1) axonopathy, ranging from deficits in axoplasmic transport to frank transection of the axon (axotomy), and (2) segmental dysmyelination or demyelination.
It is obvious how nerve damage–induced failure of signal conduction can cause hypoesthesia and numbness (negative symptoms), but why does it cause positive symptoms such as dysesthesia and pain? Not long ago the positive symptoms defied explanation. Now, we count too many potential explanations. Neuropathy triggers a large number of distinct cellular and molecular changes in the PNS and CNS. The need is to rank the importance of the numerous contenders. This challenge is illustrated by results from a relatively new technology, the expression microarray (“gene chip”).
Microarrays are devices that permit one to quantify the level of expression of large numbers of genes or even all genes simultaneously. Until their advent, molecular changes in axotomized dorsal root ganglion (DRG) cells were generally identified one at a time by using immunohistochemistry or molecular separation methods. These approaches revealed that the levels of dozens of molecules relevant to pain are affected by axotomy. That is, axotomized neurons undergo “phenotypic switching.” The peptide neurotransmitter substance P, for example, is depleted from many DRG neurons and their intraspinal terminals after nerve injury (expression of the corresponding gene is “down-regulated”). In parallel, expression of neuropeptide Y and galanin is increased (“up-regulation”; Hokfelt et al 1997). Interestingly, different neuron types may respond differently. For example, although the substance P level is reduced in small (nociceptive) neurons in the DRG, it is increased in large (touch-sensitive) neurons (Noguchi et al 1995, Weissner et al 2006). The expression and synaptic release of substance P from the central terminals of Aβ fibers secondary to phenotypic switching could itself explain how touch-sensitive afferents could come to signal pain after nerve injury (Devor 2009, Nitzan-Luques et al 2011).
Using gene chips we now know that upward of 10% of all genes expressed in the DRG are significantly up- or down-regulated in some neuropathic pain models. The fraction is even higher if one considers only genes directly related to neuronal excitability. Levels of at least 2000 genes expressed by sensory neurons, and perhaps as many as 4000 or more, are changed by neuropathy (Persson et al 2009a, Hammer et al 2010, Michaelevski et al 2010). However, this is not the end. Many genes contribute to the synthesis of more than one protein product, either through alternative splicing, by generating transcription factors, or by serving as enzymes in biosynthetic pathways. This multiplies the true molecular effects of neuropathy. Finally, beyond DRGs, massive changes also occur in the skin, nerve, spinal cord, and brain. The potential complexity is enormous.
Even though each one of the thousands of changes that constitute phenotypic switching can, in principle, be translated into a theory of neuropathic pain, it is a safe bet that not all are in fact related to pain. In addition to pain, nerve injury also triggers cell survival programs, responses to metabolic stress, regeneration, and other cellular processes. It is therefore essential to evaluate neuropathy-induced changes and identify those with functional importance for pain. Strategies for doing this will require creative thinking.
In this chapter I confront the problem of too many pain theories with a simple overriding principle. Pain perception occurs in the brain, but the precipitating injury occurs in the periphery. Since impulse discharge is the only way that pain signals can be conveyed rapidly from peripheral generators to the brain, changes that lead to abnormal electrogenesis deserve special attention. The slow signaling processes associated with axoplasmic flow also need to be considered. I place emphasis on the PNS because this is the location of the primary lesion and the primary pathophysiology. As stated, the CNS also contributes. However, as we shall see, the central changes involved in neuropathic pain are mostly triggered and maintained by abnormal input from the periphery. Thus, controlling the peripheral process can also reverse the central ones. The exception is pain caused by direct injury to the brain or spinal cord, or “central neuropathic pain.” Here, quite different processes come into play. Mechanisms of central pain will not be discussed in this chapter.
Ectopic Impulse Discharge Is A Fundamental Cause of Spontaneous and Evoked Neuropathic Pain
The structural changes caused by nerve injury and disease form an important backdrop for understanding neuropathic pain, but they do not in themselves explain the pain. A priori, axotomy and loss of myelin are expected to block conduction and yield hypoalgesia. Classic studies that focused on histopathology failed to reveal a consistent link between structural change and pain. They could not account for the reason why some neuropathies are painful whereas others are not (Dyck et al 1976). The reasons are now emerging. The link is altered electrogenesis. It is not enough that neural injury has occurred. One must determine whether the injured afferent neuron has become electrically hyperexcitable.
Structural Changes following Axotomy: Sprouting, Dying Back, and Neuroma Formation
When an axon is severed traumatically or as a consequence of disease, the proximal stump, the part still connected to the cell body, seals off and forms a terminal swelling, or “end-bulb” (Fig. 61-2). It may also die back for a few millimeters. The myelin sheath near the cut end is invariably disrupted (Fawcett and Keynes 1990, Fried et al 1991). Within hours or a day or two, numerous fine processes (“sprouts”) may start to grow out from the end of the axon. Under optimal conditions, blunt nerve compression or freezing, for example, many or all of these regenerating sprouts elongate within their original endoneurial tube and re-form connections with their original peripheral targets. Excess sprouts are culled and function is restored.
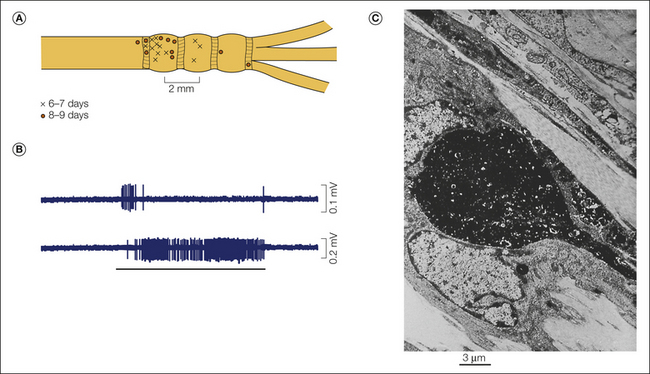
Figure 61-2 Punctate mechanosensitive “hot spots” in an injured nerve.
A, Hot spots recorded from the sciatic nerve 6–9 days after chronic constriction injury (distal tributaries of the nerve are to the right). B, Traces show rapidly adapting (above) and slowly adapting (below) responses. C, A neuroma end-bulb 7 days after injury visualized by anterograde filling with WGA-HRP/DAB reaction product. This is the probable appearance of a mechanosensitive hot spot. (B Reproduced from Chen Y, Devor M 1998 Ectopic mechanosensitivity in injured sensory axons arises from the site of spontaneous electrogenesis. European Journal of Pain 2:165–178; C Reproduced from Fried K, Govrin-Lippmann R, Rosenthal F, et al 1991 Ultra-structure of afferent axon endings in a neuroma. Journal of Neurocytology 20:682–701.)
In contrast, when forward growth is blocked, such as after limb amputation or when the severed nerve ends are separated by a gap, regeneration fails and end-bulbs and aborted sprouts form a tangled knot at the proximal nerve end with spread into nearby tissues. This is a “nerve-end neuroma.” Tight ligation of the end of the nerve suppresses sprouting and leaves a neuroma with end-bulbs but few sprouts (Fried et al 1991). Intermediate states also occur, such as when the nerve sheath (perineurium) is breached but the cut ends do not separate or when they are surgically reapproximated. Here, a fraction of fibers successfully elongate into the distal end of the nerve, although many reach inappropriate end structures. For example, skin grafts become reinnervated, with some return of sensation, but the epidermis is left with a reduced complement of sensory endings resulting in hyposensibility. Fibers that fail to regenerate become trapped in a “neuroma-in-continuity” at the injury line (Nedelec et al 2005). Pain is associated with less successful regeneration (Taylor et al 2010).
Individual elongating sprouts may get caught up on their way to the peripheral target and form disseminated “microneuromas” scattered along the distal nerve trunk, its tributaries, and distal target tissues. Disseminated microneuromas can also form when the cell body is unable to support a long sensory axon because of metabolic disease or DRG infection (e.g., in diabetic sensory polyneuropathy or post-herpetic neuralgia). The soma may survive, but the distal axon “dies back” and leaves the epidermis partially or completely denervated and subdermal nerve branchlets awash in retracted axon end-bulbs. Hypersensitive sprouts and intradermal end-bulbs can account for the seeming paradox of spontaneous pain and allodynia in the skin coupled with axonal loss in the epidermis (Oaklander 2001, Vlckova-Moravcova et al 2008).
In neonates, axotomy usually leads to rapid death of the cell soma. This is due to disruption in the retrograde transport of sustaining neurotrophic molecules (e.g., nerve growth factor [NGF] and glial-derived neurotrophic factor [GDNF]) normally supplied by innervated target tissues. A consequence of loss of the cell body is the loss of axons, absence of neuroma formation, and minimal neuropathic pain (e.g., phantom limb pain; Simmel 1962). In adults, sensory neurons are less dependent on trophic support. Few succumb to axotomy except after long intervals, but as noted, gene expression is reprogrammed and function altered.
Injured Nerve Fibers May Generate Ectopic Impulse Discharge
Neuromas, sprouts, and patches of dysmyelination are structural entities. Whether they contribute to pain depends on whether their formation is accompanied by the development of electrical hyperexcitability. In classic studies, electrophysiological recordings were made from sensory axons that terminate in an experimental nerve-end neuroma, and massive spontaneous discharge of impulses was observed. The firing was generated in the neuroma and was eliminated (transiently) by resection of the neuroma and by local anesthetic block of the nerve end. Likewise, it was enhanced by mechanically probing the neuroma (Fig. 61-3; also see Fig. 61-2; Wall and Gutnick 1974, Govrin-Lippmann and Devor 1978). Similar ectopic electrogenesis (ectopia), both spontaneous and evoked by stimuli, also occurs at mid-nerve locations such as neuromas-in-continuity, disseminated microneuromas, and sites of demyelination such as experimental entrapment neuropathies (Burchiel 1980, Smith and McDonald 1980, Calvin et al 1982, Baker and Bostock 1992, Kajander and Bennett 1992, Wallace et al 2003). Subsequent research revealed ectopic spontaneous and evoked electrogenesis at additional pacemaker locations in injured nerves: in regenerating sprouts (Scadding 1981, Pinault 1995, Chen and Devor 1998, Han et al 2000, Gorodetskaya et al 2003); at sites of nerve inflammation (neuritis) (Eliav et al 2001, Bove et al 2003); in experimental diabetic polyneuropathy (Burchiel et al 1985, Dobretsov et al 2001, Khan et al 2002); after viral infections (Mayer et al 1986, Kress and Fickenscher 2001); after vincristine, taxol, and methylmercury intoxication (Delio et al 1992, Tanner et al 1998, Xiao and Bennett 2008b); and in hereditary demyelinating polyneuropathies (Martini 1997, Gillespie et al 2000). The specific agent that causes neural injury may have some effect, but not a critical one. Pathophysiological discharge can emerge no matter how axons are injured.
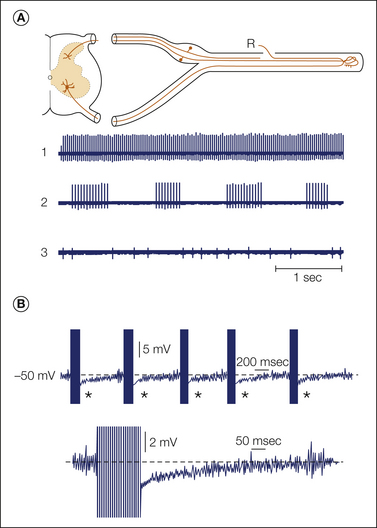
Figure 61-3 Patterns of spontaneous ectopic discharge recorded from sensory neurons ending in a neuroma.
A, Fine axon bundles were microdissected from an injured nerve and placed on a recording electrode (R). Spontaneously active fibers fire tonically (1), in bursts (2), or irregularly (3). B, Intracellular recording from a dorsal root ganglion neuron with ectopic burst discharge (asterisks; spike height is truncated). One burst is shown in detail below. Bursts are triggered when ongoing membrane potential oscillations reach threshold; they are maintained by post-spike depolarizing afterpotentials. The burst initiates a hyperpolarizing shift, which stops firing and resets the oscillations. (Reproduced from Amir R, Devor M 1997 Spike-evoked suppression and burst patterning in dorsal root ganglion neurons. Journal of Physiology 501:183–196.)
In all these cases the discharge is “ectopic” because it originates away from the normal location of sensory impulse generation, the peripheral sensory ending. As expected, activity generated ectopically drives spinal and higher-order neurons in the CNS in the normal way. This has been confirmed by electrical recording, imaging, and the use of activity markers (Mao et al 1992, Chi et al 1993, Sotgiu et al 1994b, Pitcher and Henry 2004). Note that the terms “ectopic” and “ectopia” do not imply that the abnormal discharge necessarily arises spontaneously. Discharge evoked by the application of stimuli at locations where impulse initiation does not normally occur, at the wrist in carpal tunel syndrome, for example, is also ectopic. With one known exception, heat (Hoffmann et al 2008, 2009; Teliban et al 2010), natural non-traumatic stimuli applied at a mid-nerve location do not evoke impulse discharge or refered sensation. In the presence of neuropathy, however, they frequently do. Nerve injury triggers a fundamental change in the midportion of axons that renders them locally hyperexcitable and capable of ectopic electrogenesis.
Firing Rates and Patterns
Spontaneous ectopic discharge occurs in both myelinated (A) and unmyelinated (C) fibers, although with certain differences. Activity in A fibers tends to commence earlier, is found in a higher proportion of neurons at its peak, and is characterized by higher firing rates and more bursting than is activity in C fibers (Fig. 61-4). In A fibers the ectopia is usually rhythmic; that is, there is a fixed interval between subsequent impulses within a train (usually 65–35 msec, which translates to an instantaneous discharge rate of 15–30 Hz). In many fibers the impulse train is interrupted by silent pauses, which results in a bursting, on–off pattern (“interrupted autorhythmicity”; see Fig. 61-3). The remaining A fibers, as well as most C fibers, fire in a slow, irregular pattern (0.1–10 Hz; Govrin-Lippmann and Devor 1978). Spike patterning, particularly bursting, can have an effect on post-synaptic neurons over and above the average firing rate by virtue of temporal summation of excitatory post-synaptic potentials during bursts (Burke et al 1970, Lever et al 2001).
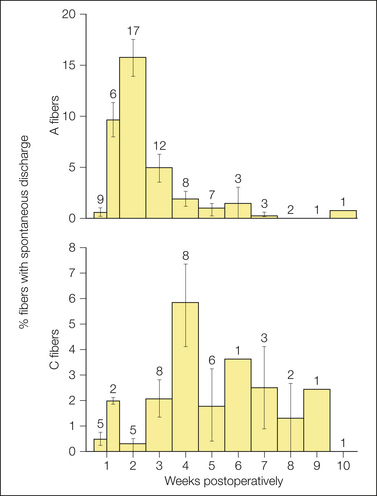
Figure 61-4 Incidence (means ± standard deviation) of spontaneous ectopic firing in myelinated (A) and unmyelinated (C) fibers in experimental sciatic nerve neuromas as a function of time after injury.
The value atop each column is the number of animals studied within the time window indicated. (Data from Devor M, Govrin-Lippmann R 1985 Spontaneous neural discharge in neuroma C-fibers in rat sciatic nerve. Neuroscience Letters. Supplement 22:532; Govrin-Lippmann R, Devor M 1978 Ongoing activity in severed nerves: source and variation with time. Brain Research 159:406–410.)
Different afferent types differ in their tendency to develop spontaneous firing or to become ectopically mechanosensitive. However, it is difficult to identify in which functional types ectopia preferentially develops. The problem is that axotomy disconnects the receptor ending from the rest of the axon. Heroic efforts are required to preserve information about the original receptor type over the many hours or days that elapse before ectopic firing begins. Partial information has been obtained from conduction velocity, from the ability to follow tetanic stimuli (the “marking method”), by comparing recordings from dorsal versus ventral roots, and from examination of cutaneous nerves versus nerves serving muscle, viscera, and other tissues. Injured sensory axons are much more likely than injured motor axons to generate spontaneous ectopic activity. Aβ and Aδ afferents are represented roughly in proportion to their numbers in the nerve. Interestingly, although injured cutaneous afferents frequently show ectopic mechanosensitivity without spontaneous firing, muscle afferents are more likely to fire spontaneously, at least after distal axotomy (Johnson and Munson 1991, Tal et al 1999, Michaelis et al 2000). Perhaps this is because muscle nerves normally contain many tonic and slowly adapting afferent fiber types (muscle spindle afferents, proprioceptors). Neuroma end-bulbs and sprouts tend to develop the same sensitivities that they had before injury, presumably because of fiber type–specific gene expression at the level of the cell soma (Devor et al 1990, Koschorke et al 1991, Teliban et al 2010).
Neurolysis As a Therapeutic Strategy
Since neuromas are an important source of painful ectopia, it seems logical that surgical resection or neurolysis should bring relief. Unfortunately, shortly after neuroma resection the same pathophysiological processes that caused pacemaker activity in the first place are re-engaged at the freshly cut nerve end, perhaps even in intensified form as a result of priming. Surgical mobilization of a neuroma to a site with a reduced likelihood of mechanical compression, however, may provide long-term relief in cases in which mechanosensitivity rather than spontaneous firing is the main problem (Campbell 2007). Different nerves behave differently. An incision for thoracotomy, for example, which frequently damages intercostal nerves, is much more likely than a comparable incision in the abdomen to be followed by neuropathic scar pain. For reasons that are not entirely clear, destruction of the tooth pulp in root canal treatment or tooth extraction does not commonly induce a painful neuroma. The same seems to be true of the intrinsic innervation of long bones. In hip replacement surgery, the femur is cut across and a prosthetic joint and bone cement are introduced into the bone marrow chamber. Yet despite the massive destruction of intrinsic bone afferent axons, the development of chronic neuropathic pain is infrequent. This suggests that pain relief might be achieved in patients with osteoarthritis by intrinsic denervation of the epiphyseal end of the bone, just as in dental root canal treatment (Niv et al 2003).
Dorsal Root Ganglia Are Also a Source of Ectopic Spontaneous Firing and Mechanosensitivity
Cutting the spinal nerve just peripheral to the DRG evokes ectopia in the dorsal root, altered dermatomal borders, and cutaneous hypersensibility. This does not occur when the same axons are cut just central to the DRG, in the dorsal root (Kirk and Denny-Brown 1970, Kirk 1974). The authors concluded that the DRG is the source of the spontaneous firing, although they did not adequately rule out the most obvious alternative source, the spinal nerve neuroma. It later became clear that the DRG is indeed a major source, along with the neuroma (Wall and Devor 1983, Amir et al 2005, Ma and LaMotte 2007). In fact, head-to-head comparisons in both the sciatic neuroma model and the spinal nerve ligation model in rats have shown that about 75% of the overall spontaneous discharge generated in the injured nerve originates in the DRG and 25% in the neuroma (Babbedge et al 1996, Liu et al 2000a).
In addition to the presence of spontaneous firing, activity in DRG neurons is initiated or exacerbated by the same physical and chemical stimuli that drive ectopia in neuromas. Despite the fact that the DRG is protected from direct stimulation by the rigid walls of its bony foramen and that DRG neurons have (almost) no synaptic input, there are nonetheless many factors capable of producing depolarization. For example, DRG neurons can be excited by mechanical stimulation during movement or straight-leg raising (which pulls on the sciatic nerve), by sympathetic efferent activity, by excitatory substances released within the ganglion as a result of activity in neighboring neurons (chemical “cross-excitation”), by agents in the systemic circulation, and by changes in temperature. Each of these factors will be considered below in more detail.
There is specific evidence that ectopia originating in DRG neurons contributes to neuropathic pain. In animal models, increasing or decreasing spontaneous ectopia by delivery of pharmacological agents direct to the ganglion has corresponding effects on pain behavior (Sukhotinsky and Devor 1997, Zhang et al 2000, Xie et al 2006, Naik et al 2008, Thacker et al 2009). The mechanosensitivity of the DRG appears to be particularly significant because of its major role in movement-evoked pain in disorders of the vertebral column. Kuslich and colleagues (1991) used a local anesthetic technique to expose the spinal nerves and DRGs in patients with sciatica that permitted them to talk to the patients during the procedure. Mechanical stimulation of the spinal nerve and DRG capsule consistently provoked the patients’ characteristic shooting sciatica pain, whereas probing the local fascia, annulus fibrosus, periosteum, and other tissues produced only local sensations. The nerve root and DRG are subjected to tensile stress during everyday movement and during maneuvers such as straight-leg raising (Nowycky 1992). Normally, this does not evoke any sensation. However, if ectopic mechanosensitivity has developed as a result of neuropathy, radiculopathy, or ganglionopathy, these forces are translated into ectopic impulse discharge and pain (Nordin et al 1984).
Interestingly, a small number of DRG neurons fire spontaneously even in the absence of nerve injury (Wall and Devor 1983). This activity might contribute to the background sense of body schema. For example, when all nerves to a limb are blocked in healthy subjects, most feel a non-painful “normal phantom” rather than absence of the limb (Melzack and Bromage 1973). Likewise, dental anesthesia is followed by the sensation of a swollen lip, not a hole in the face. These phantom sensations may be due to background DRG discharge. Given that ectopic electrogenesis in the DRG is a significant factor in the etiology of neuropathic pain, it may also be an effective target for therapeutic intervention. This includes cases of nerve trauma, as well as conditions such as post-herpetic neuralgia and a herniated intervertebral disc, in which the DRG itself is directly affected by the disease process. The implications of the DRG as a therapeutic target for pain control in patients with neuropathic pain have not yet been widely realized.
Ectopia in Residual “Uninjured” Afferents and in Collateral Sprouts
Within nerves and in skin and other tissues, sensory fibers tend to intermingle such that any given tissue volume is innervated by afferents from more than one nerve branch and more than one DRG. For this reason, when a fraction of the axons in a nerve are injured and undergoing anterograde (wallerian) degeneration, the residual “uninjured” axons in the nerve and its target tissues are exposed to degeneration products. In addition, wallerian degeneration evokes an inflammatory response and the appearance of immune cells and diffusible pro-inflammatory mediators in the nerve and target tissue. These mediators, which include interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and NGF, are released by fibroblasts, mast cells, endothelial cells, Schwann cells, locally activated or invasive immune cells, and in the skin, keratinocytes (Rotshenker 1997, Li et al 2010, Radtke et al 2010). It has been reported that such “uninjured” afferents begin to fire spontaneously, albeit at extremely low discharge rates, often less than 1 spike/min (Ali et al 1999, Wu et al 2001a). The location of the electrogenesis has not been determined; if it comes from sensitized sensory endings, it would not be ectopic. However, wherever these impulses originate, they add to the overall ectopic barrage that drives spontaneous pain. This spontaneous activity may make a special contribution to the spontaneous burning pain that is so common in neuropathy. It might also play a role in tactile allodynia by contributing to the maintenance of central sensitization.
Beyond their potential for contributing to spontaneous ectopia, “uninjured” afferents also emit “collateral sprouts.” This is probably a result of elevated tissue levels of NGF (Devor et al 1979, Diamond et al 1987, Rajan et al 2003). Collateral sprouting, which also occurs in humans (Inbal et al 1987), is easiest to detect at innervation boundaries. Here, C fibers sprout and invade neighboring denervated territory, thereby restoring nociceptive sensation. But, sprouting also occurs at the center of partially denervated tissue (Fig. 61-5). Residual sensory endings and collateral sprouts, all bathed in inflammatory mediators, have long been suspected as being contributors to allodynia and hyperalgesia. This is uncertain, however, because collateral sprouts do not respond readily to weak tactile stimuli (Devor et al 1979, Koltzenburg et al 1994a; but see Shim et al 2005). Sprouts do show enhanced sensitivity to circulating and applied adrenaline and to sympathetic efferent activity (Sato and Perl 1991; Ali et al 1999, 2000; Jorum et al 2007). This may drive activity, and consequent pain, that appears to be spontaneous. Topically applied lidocaine and adrenergic blockers probably provide pain relief by suppressing the spontaneous drive originating in the skin and the central sensitization that it maintains.
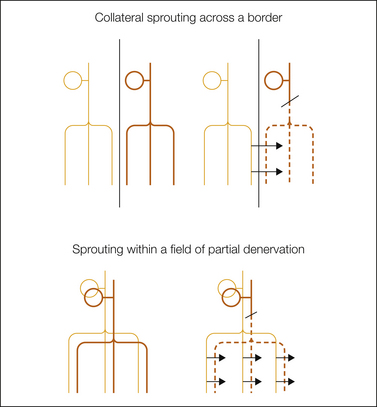
Figure 61-5 Anterograde (“wallerian”) degeneration of afferent axons distal to a nerve injury (broken lines) promotes sprouting of residual intact neighboring afferents. Such sprouting is well documented in the case of nerves sharing a common border (collateral sprouting) and very likely also occurs within the field of innervation of a single nerve following incomplete nerve damage. In each sketch, the sensory cell somata in the dorsal root ganglion are indicated at the top, and afferent innervation fields in the skin are indicated at the bottom. Arrows indicate the main direction of sprouting.
Ectopic Firing Is a Key Factor In Painful Neuropathies
The method of percutaneous microneurography has extended observations on ectopia to awake humans, including those with neuropathic pain. It remains a research rather than a diagnostic tool, however, because of its technical difficulty and intrinsic risk. Practitioners have been justifiably reluctant to insert microelectrodes into already problematic nerves. Nonetheless, enough studies have appeared to make it clear that ectopic hyperexcitability occurs in patients as in nerve-injured animals and that it is a fundamental contributor to many clinical neuropathic pain conditions.
Not long after the first observations in animals, Nystrom and Hagbarth (1981) carried out a pioneering study in which they documented ongoing firing in the peroneal nerve in a lower extremity amputee who had ongoing phantom foot pain. Percussion of the neuroma evoked stabbing pain (the Tinel sign) and an intense burst of spike activity. The evoked bursts and the evoked pain were eliminated by local anesthetic block of the neuroma. Interestingly, however, much of the ongoing discharge persisted. The DRG is the most likely source of this persistent activity, but direct evidence of this in humans is still lacking. Subsequent microneurographic studies documented a direct relationship between ectopia and pain in a variety of other neuropathic conditions (e.g., Nordin et al 1984, Campero et al 1998, Burchiel and Baumann 2004, Ochoa et al 2005, Jorum and Schmelz 2006). For example, painful dysesthesia triggered by straight-leg lifting in a patient with radiculopathy (Lasegue’s sign) was accompanied by evoked ectopic bursts recorded in the sural nerve. The ectopic source was the injured spinal root or DRG (Nordin et al 1984).
More recent studies have used the “marking method” to resolve activity in individual C fibers. The results provide evidence that the ongoing, often burning pain characteristic of many peripheral neuropathies is due to spontaneous discharge in C-fiber nociceptors (Serra 2010, Kleggetveit et al 2012). Multiplet and burst firing, afterdischarge, and other interesting peculiarities of ectopia in animal models (below) have also been seen in patients, thus further strengthening the clinical relevance of the experimental models (Weidner et al 2002, Bostock et al 2005). Other lines of evidence support these electrophysiological data. For example, pain is evoked by the application of substances known from animal preparations to excite ectopic pacemaker sites, including K+ channel blockers and adrenergic agonists (Chabal et al 1989b, 1992). Likewise, blockers of ectopia such as local, regional, and systemic anesthetics suppress neuropathic pain (Wallace et al 1996). Even the most severe pain, such as occurs in complex regional pain syndrome (CRPS), is reliably stopped by peripheral nerve or brachial plexus block, thereby permitting physiotherapy, albeit only for the duration of the block.
Animal Models of Spontaneous Pain
The foregoing observations leave little doubt that spontaneous ectopic discharge is a primary driver of spontaneous pain in humans. Spontaneous ectopia also occurs in neuropathy models in animals. Although this implies that the animals also experience spontaneous pain, proving it is not trivial. This issue is important because animal models are essential for screening novel analgesic drugs. This topic is discussed in more detail in Chapter 62.
Spontaneously emitted behaviors such as vocalization, abnormal posture and gait, and unprovoked paw lifting occurs in neuropathy models and have been put forward as potential markers of spontaneous pain. However, follow-up study has failed to provide convincing support for this conjecture (Mogil et al 2010, Urban et al 2011). Instinctive behavioral biomarkers of spontaneous pain may not even exist in prey species such as mice and rats since they would signal vulnerability to predators and natural selection is likely to have excluded them. Convincing evidence for spontaneous pain is available from a conditioning paradigm in which animals were trained to show place preference for a chamber in which they were provided with analgesia (King et al 2009).
Autotomy behavior in the neuroma model provides another option (Wall et al 1979). This behavior has been validated as a biomarker of spontaneous neuropathic pain (anesthesia dolorosa) via a number of approaches (Devor 2007). For example, it is reduced by appropriate drugs, it is associated with prominent ectopia, augmenting the ectopia augments the autotomy, and suppressing it suppresses autotomy (Devor et al 1982, Levitt 1985, Coderre et al 1986, Blumenkopf and Lipman 1991, Seltzer 1995, Kauppilla 1998, Liu et al 2001a, Abdulla and Smith 2002, Devor 2007, Nissenbaum et al 2008, Radtke et al 2010). Recently, it has been shown that a gene variant that predisposes to autotomy in rodents is associated with neuropathic pain in humans (Nissenbaum et al 2010). Two other end points proposed recently also offer some hope: the coding of facial expressions in mice (Langford et al 2010) and a new procedure for tracking ultrasonic calls (Kurejova et al 2010).
Mechanosensitivity: Hot Spots, Trigger Points, and the Tinel Sign
Exploring the surface of an injured nerve with a fine probe reveals clusters of tiny mechanosensitive “hot spots.” They do not occur in normal nerves. Such probing excites silent axons and also accelerates discharge in already active ones (Fig. 61-2A and B). Spontaneous and evoked discharges almost certainly arise from the same cellular locus and pacemaker process (Burchiel 1984, Chen and Devor 1998). Mechanosensitivity underlies the Tinel sign, the often stabbing or electric shock–like dysesthesia evoked by percussion of neuromas, along nerves in painful diabetic neuropathy, and in carpal tunnel syndrome, and it is responsible for the shooting leg pain in sciatica (Nystrom and Hagbarth 1981, Kuslich et al 1991, Dorsi et al 2008). Like normal mechanosensitive endings, hot spots typically respond either with a short spike burst at the onset and/or release of a stimulus or with sustained firing for the duration of application of the force (rapid and slow adaptation). Interestingly, however, in some hot spots firing persists beyond the end of the stimulus (“mechanical afterdischarge,” Fig. 61-6A). In effect, the brief stimulus triggers a period of spontaneous firing. This clearly abnormal pattern accounts for the frequent persistence of evoked sensation in neuropathy (Noordenbos 1959, Gottrup et al 2003). Locations where nerves run adjacent to tendons and bone (e.g., the carpal tunnel) or where small nerve branches cross tough fascial planes are particularly at risk for sustaining focal trauma and developing ectopic mechanosensitivity. Pain evoked at such trigger points by local palpation, weight bearing, and untoward movements may represent a neuropathic contribution to chronic musculoskeletal pain, perhaps including fibromyalgia.
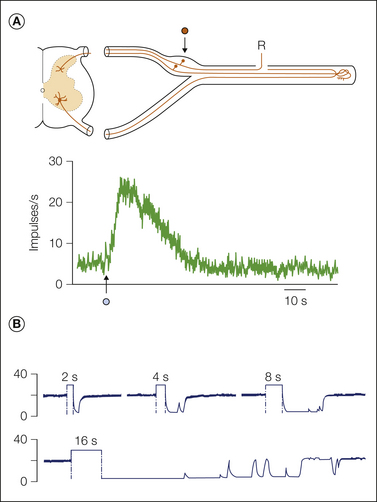
Figure 61-6 A, A dorsal root ganglion neuron responds to a momentary mechanical stimulus (arrow, 0.5 second, 150 mg), with afterdischarge lasting more than 30 seconds and including about 500 extra spikes. Much more prolonged responses may also occur. The sciatic nerve had been cut 11 days previously (Lisney and Devor 1987). B, This neuroma ending fired spontaneously at about 20 impulses/sec (experimental setup as sketched in Fig. 61-3). Electrical stimulation of the nerve central to the injury site (100 Hz) for 2, 4, 8, and 16 seconds silenced the firing for periods that increased with increasing duration of stimulation. An additional example of activity-provoked suppression is shown in Figure 61-10A (Amir and Devor 1997).
Ectopia and Tactile Allodynia
Tactile allodynia, unlike hyperalgesia and tender points, is probably not a result of excessive mechanosensitivity of nociceptive sensory endings for reasons noted above (under Sensitization). Even though tender collateral sprouts and intradermal disseminated microneuromas may well contribute, the major cause of tactile allodynia is thought to be spike activity in Aβ touch afferents processed by sensitized spinal cord circuitry. Here too, however, spontaneous ectopia in the periphery plays a crucial role: it induces and maintains the central sensitization. Thus, tactile allodynia results from pathophysiology in the PNS and in the CNS. Note that the sensory effect of spontaneous ectopia in Aβ fibers is also exacerbated by central sensitization, thereby adding to the spontaneous pain. Finally, central sensitization may amplify the sensory consequences of spontaneous and evoked activity in at least some types of nociceptors (Klein et al 2004).
In Figure 61-7 the sequence of events believed to underlie the tactile allodynia in neuropathy is illustrated by using the spinal nerve ligation model of neuropathic pain (Kim and Chung 1992). In this model the L5 (or L5–6) spinal nerve is cut; about half the afferents in the sciatic nerve are severed and the hindpaw is partially denervated. Cutting the nerve creates two permanent sources of spontaneous ectopic activity, the L5 spinal nerve-end neuroma and the L5 DRG. “Uninjured” nociceptors in the L4 segment may also contribute. This activity drives central pain pathways and also triggers central sensitization, hence augmenting spontaneous pain and inducing allodynia.
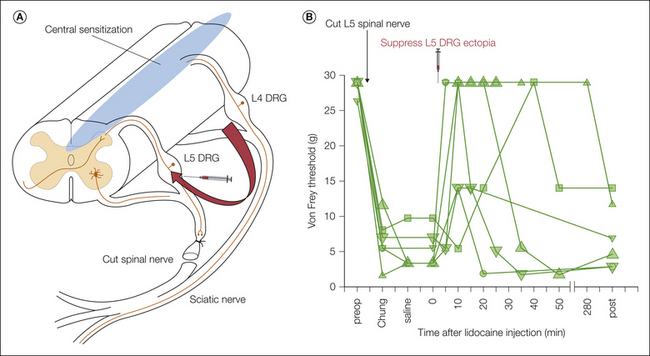
Figure 61-7 Proposed mechanism of tactile allodynia in neuropathy, in this case based on the spinal nerve ligation (Chung) model (see explanation in text).
A, Ectopic firing generated in injured L5 afferent axons and dorsal root ganglion (DRG) somata, also with a probable contribution from non-axotomized L4 afferents, triggers and maintains a broad range of central amplification processes collectively known as “central sensitization.” The tactile input carried by residual neighboring hindlimb afferents, notably in the L4 segment, is amplified by central sensitization to yield a sensation of pain (tactile allodynia). The L5 ectopia can be suppressed by applying local anesthetics such as lidocaine (lignocaine) to the injured L5 afferents through a previously implanted indwelling catheter (curved arrow). B, When this is done, the tactile allodynia of the hindpaw is transiently suppressed. However, the allodynia returns within a few hours when the lidocaine block fades and ectopic firing is restored. Each line and symbol type represents an individual rat. (Data from Sukhotinsky I, Ben-Dor E, Raber P, et al 2004 Key role of the dorsal root ganglion in neuropathic tactile hypersensibility. European Journal of Pain 8:135–143.)
Corresponding observations have been made in patients. For example, Gracely and colleagues (1992) observed that when a focal source of ectopia from an old scar was identified and blocked with local anesthetic, not only was the focal pain temporarily eliminated but also the widespread allodynia that the patient suffered. The same principle is also relevant to conditions such as migraine and visceral and musculoskeletal pain (Burstein et al 2000, Giamberardino 2008, Graven-Nielsen and Arendt-Nielsen 2010, Woolf 2011). The focal impulse source dynamically maintains central sensitization. Remove it and hypersensibility fades, usually within minutes or at most a few hours.
Exacerbation of Ectopia by Temperature and by Chemical Mediators
Cold intolerance and cold allodynia are common symptoms in neuropathy; people who live in cold climates suffer in particular during the winter (Lindblom 1994). On the other hand, in some conditions, notably CRPS, patients may obtain relief by wrapping the painful limb in a cold, wet towel. This difference might reflect which primary afferents are involved (Matzner and Devor 1987, Hoffmann et al 2008). Ectopic activity in C fibers tends to be excited by cooling and suppressed by warming. Collateral sprouts (which derive mostly from C fibers) and many intradermal disseminated microneuroma endings are therefore expected to be hypersensitive to cold and to provoke pain when the skin is cooled. In contrast, ectopia in A fibers is enhanced by warming, whereas cooling suppresses it. If the pain in CRPS were primarily due to Aβ fibers, this could explain why cooling provides some relief in this condition. As discussed below, the abnormal sensitivity of injured axons reflects altered regulation and trafficking of ion channels and receptors, including thermoresponsive transient receptor potential (TRP) channels. This varies with fiber type (Schmidt et al 2009).
In addition to temperature, metabolic stress and a wide array of chemical factors can also depolarize sensory neurons and directly excite discharge at ectopic pacemaker sites in neuropathy. All these factors can also sensitize pacemaker sites and make them hyper-responsive to mechanical and other stimuli. Examples include tissue ischemia and hypoxia, changes in blood gases, elevated blood glucose, altered ion concentrations, and numerous endogenous neuroactive substances, including catecholamines, adenosine triphosphate (ATP), nitric oxide, peptides, cytokines, small lipids, neurotrophins, histamine, bradykinin, prostaglandins, TNF-α, and many other pro-inflammatory mediators (Wall and Gutnick 1974, Zimmermann et al 1987, Devor et al 1992b, Noda et al 1997, Levy et al 2000, Rivera et al 2000, Zhou et al 2000, Chen et al 2001, White et al 2005, Binshtok et al 2008, Patwardhan et al 2010, Xu et al 2010). New mediators continue to appear regularly, which suggests that the current inventory is far from complete. The chemical milieu of afferent neurons is a major factor in ectopia and neuropathic pain, but only if ectopic pacemaker capability has developed first.
Systemic and Focal Inflammation (“Neuritis”)
In Guillain-Barré syndrome (GBS) and related inflammatory polyneuropathies, nerves suffer widespread myelin destruction and axonopathy as a result of autoimmune attack. This causes paralysis and sensory loss. However, as in other forms of neuropathy, these negative symptoms are often accompanied by dysesthesias and ongoing pain (Moalem-Taylor et al 2007, Luongo et al 2008, Koike and Sobue 2010). Membrane remodeling and pain behavior have been documented in the animal model of GBS (experimental allergic neuritis), and ectopic hyperexcitability is probably present in this model (Novakovic et al 1998, Sorkin 2007). There is direct electrophysiological evidence that ectopic hyperexcitability occurs at sites of focal nerve inflammation. In the chronic constriction injury model of neuropathic pain, for example, chromic gut ligatures are applied loosely to the sciatic nerve. This causes local inflammation and nerve swelling, which partially strangles the nerve (Bennett and Xie 1988). Variants of this model include focal constriction of trigeminal nerve branches and compression of dorsal root and trigeminal ganglia (Vos et al 1994, Hu and Xing 1998, Zhang et al 2000, Ma and LaMotte 2007, Ahn et al 2009). Such injury causes frank destruction of many axons, focal demyelination of others, and the release of inflammatory mediators.
Peripheral nerve injury also triggers an inflammatory reaction in associated ganglia, including activation of intrinsic glial cells and attraction of invasive immune cells. An inflammatory milieu contributes to the development of ectopic firing at both the nerve injury site and the ganglia, with consequent spontaneous pain and hypersensibility (Kajander et al 1992, Tal and Eliav 1996, Homma et al 2002). The animal constriction and compression models emulate the many clinical conditions that feature focal neuropathy exacerbated by inflammation. Among these are carpal tunnel syndrome, disc herniation with sciatica, and solid tumors that press on nerves. Indeed, a contribution of inflammation in conditions involving neural trauma may be the rule rather than the exception.
There is also tentative evidence that inflammatory mediators might be able to act on mid-nerve fibers directly without concurrent (structural) neuropathy. Specifically, it has been reported that when TNF-α or complete Freund’s adjuvant is applied to the surface of healthy nerves or DRGs, a focus of spontaneous firing and mechanosensitivity may emerge rapidly (Sorkin et al 1997, Eliav et al 2001, Homma et al 2002, Wang et al 2007, Amaya et al 2009). By inference, in systemic and focal inflammatory and toxic neuropathies in which there is minimal concurrent nerve trauma, widespread pain might result from the direct action of inflammatory mediators on midaxon and intraganglionic receptors.
Sympathetic–Sensory Coupling and Sympathetically Maintained Pain
Sympathetic–sensory coupling is a special case of ectopic chemosensitivity. In some patients neuropathic pain appears to be exacerbated by sympathetic efferent activity and relieved by sympathetic block or sympatholysis. This is “sympathetically maintained pain” (SMP). Pain may be evoked by direct electrical stimulation of sympathetic efferents or by maneuvers that increase sympathetic drive, such as the Valsalva maneuver and whole-body cooling (Harden et al 2001, Schattschneider et al 2008). Pain also occurs in animals and in humans when adrenergic agonists are injected into nerve-end neuromas or into the skin in patients with SMP, and it may be rekindled in individuals whose hyperalgesia was previously relieved by treatments that reduce the endogenous sympathetic drive (Chabal et al 1992, Torebjork et al 1995, Ali et al 2000). These clinical observations correspond to the abnormal adrenosensitivity of ectopic pacemaker sites (injured axons and DRG cell somata). As noted, residual “uninjured” afferent endings in the skin also become adrenosensitive, perhaps in association with collateral sprouting (Wall and Gutnick 1974; Devor and Janig 1981; Sato and Perl 1991; Ali et al 1999, 2000).
SMP does not result from excess sympathetic drive as was once assumed. If anything, sympathetic drive is decreased in nerve-injured patients (Harden et al 2001). Rather, sympathetic–sensory coupling reflects the increased adrenergic sensitivity of sensory neurons, presumably caused by either an excess of adrenoreceptors, elevated baseline excitability, or both. Studies using receptor-selective agents indicate the predominance of α2 pharmacology, but α1-adrenoreceptors also contribute (Janig 1990, Devor et al 1994a, Chen et al 1996). Two recent studies based on microneurography investigated adrenosensitivity in cutaneous C nociceptors innervating painful skin in patients with CRPS. Although CRPS has long been considered to be the prototypical SMP, this is uncertain. Unfortunately, the results of the two studies were discordant (Campero et al 2010, Orstavik and Jorum 2010).
A unique and potentially important form of sympathetic–sensory coupling occurs within the DRG. In nerve-injured animals and in humans, sympathetic efferents that normally serve local blood vessels proliferate within the ganglion and form unique basket-like skeins around sensory somata. Since the sympathetic axons have not been cut, this is an instance of collateral or “reactive” (not regenerative) sprouting. The sprouting (and associated proliferation of satellite glial cells) may be induced by NGF released by axotomized DRG neurons. Axotomy triggers both elevated NGF synthesis and ectopia, which is expected to enhance NGF release (Devor 1983b, McLachlan et al 1993, Shinder et al 1999).
Role of Ectopia in Tissue Edema and Trophic Changes
Impulses that originate in nerve-end neuromas can travel only centrally, toward the CNS. However, impulses that originate in the mid-nerve region can also propagate distally (antidromically). Mid-nerve pacemaker sites include patches of demyelination and the DRG. Antidromic impulses also arise near sensory endings when one of several preterminal axon branches is abnormally active (“axon reflex”). In each of these cases the pacemaker site drives a sustained barrage of impulses centrally, which causes pain, and an identical barrage antidromically to invade peripheral tissue. The antidromic discharge may play a significant role in neuropathy by releasing vasoactive peptides from the axonal shaft (Sauer et al 2001) and from sensory endings (Ninian 1913, Lembeck and Holzer 1979, Lotz et al 1988, Hsieh et al 1996, Harden et al 2001). Some, notably substance P and calcitonin gene–related peptide (CGRP), cause “neurogenic inflammation,” that is, local vasodilation and hence warming and reddening of the skin, combined with plasma extravasation from postcapillary venules and hence edema. If such activity is sustained over time, it might induce the trophic changes in skin, nails, and bone that are so characteristic of CRPS and some other neuropathies. It might even sensitize nociceptors (but see Reeh et al 1986).
The Blood–Brain Barrier and Neurogenic Inflammation in the Spinal Cord
It has recently been noted that peripheral nerve injury triggers a breakdown of the capillary endothelial barrier of the intrinsic vasculature of the spinal cord. This permits plasma proteins and even circulating immune cells (T lymphocytes) to enter the cord (Gordh et al 2006, Costigan et al 2009, Beggs et al 2010). The process parallels and might even cause “glial activation” in the cord. Glial activation with associated intraspinal release of inflammatory cytokines is thought to be a significant contributor to central sensitization (Watkins and Maier 2002). The mechanism whereby nerve injury brings about breakdown of the spinal blood–brain barrier (BBB) has not been established yet. A likely possibility is the sustained central release of neuropeptides as a result of ectopic pacemaker discharge. This is the central equivalent of cutaneous neurogenic inflammation in the skin.
How Does Ectopia Induce Central Sensitization?
Induction by Nociceptive C Fibers
Most of the evidence available indicates that the signal that induces and maintains central sensitization in neuropathy is electrical impulse activity originating in the periphery. A less likely possibility is a molecule or molecules delivered centrally by axoplasmic transport independent of spiking. Acute tissue injury and electrical nerve stimulation at C-fiber strength reliably trigger central sensitization within seconds or minutes (Woolf 1983). This is far too fast for axoplasmic transport. Likewise, neuropathic conditions in which there is sustained firing in afferent C fibers often feature tactile allodynia. C-fiber activity originating in inflamed skin also evokes central sensitization, as reflected by tactile allodynia in the region in and surrounding the injury (“primary and secondary hyperalgesia”). In each of these cases silencing the ongoing nociceptor activity (e.g., by cooling or local anesthesia) rapidly reverses central sensitization and the consequent allodynia (Gracely et al 1992, Sheen and Chung 1993, Koltzenburg et al 1994b, Yoon et al 1996, Zhang et al 2000, Sukhotinsky et al 2004, Wen et al 2007, Pitcher and Henry 2008, Thacker et al 2009, Xie et al 2009).
The rapidity of the change, often within minutes, indicates that maintenance of central sensitization by ongoing impulse activity is dynamic. There is little solid evidence that spinal cord sensitization can become “centralized” and independent of peripheral drive. Consider childbirth, the passage of a kidney stone, or hip replacement surgery. Even after weeks or years of sustained nociceptor input, the pain and allodynia resolve quickly when the definitive peripheral driver is identified and removed. There is no “transition to chronicity” except perhaps in the sense of psychosocial deterioration. Likewise, prior afferent block does not prevent the later emergence of pain (“pre-emptive analgesia”) in cases in which peripheral drive continues to be present after the block has worn off (Niv et al 1999, Dahl and Kehlet 2011). Unfortunately, definitive removal of peripheral pain drivers is often beyond our reach.
Gently stroking healthy skin, or nerve stimulation at intensities that activate Aβ fibers only, does not induce central sensitization or pain. For this reason, conventional thought is dominated by the idea that nociceptor input is both sufficient and necessary for triggering and maintaining central sensitization. The presumed mechanism is that neurotransmitters released from nociceptors, but not from Aβ touch afferents, are required to initiate the sensitizing process. However, in animal models of traumatic neuropathy, ectopic spontaneous activity occurs overwhelmingly in Aβ afferents, at least in the early stages of pain when tactile allodynia first appears (see Fig. 61-4; Boucher et al 2000; Han et al 2000; Liu et al 2000a, 2000b; Xie et al 2005; Tal et al 2006). What, then, triggers and maintains central sensitization and tactile allodynia in these models?
A possible answer is C-fiber ectopia in residual “uninjured” neurons or collateral sprouts, but this is uncertain. The exceedingly low firing rates involved may be insufficient to drive central sensitization. Likewise, nerve injury–induced central sensitization and allodynia persist in animals in which most C nociceptors are eliminated with capsaicin and related neurotoxins (Shir and Seltzer 1990, Ossipov et al 1999). In contrast, a variety of procedures that suppress ectopia in axotomized Aβ fibers, without affecting neighboring “uninjured” C fibers, do relieve tactile allodynia (Sheen and Chung 1993, Yoon et al 1996, Na et al 2000, Sukhotinsky et al 2004, Naik et al 2008). Ectopic activity does occur in some Aδ afferents soon after axotomy, presumably including Aδ nociceptors, but the bulk of the activity takes place in Aβ afferents. Can we be certain that afferent nociceptors are the only afferents capable of triggering and maintaining central sensitization?
A Role for Ectopic Activity in Aβ Afferents in the Induction of Central Sensitization
Normally, Aβ touch fibers clearly play no role in the induction of central sensitization and allodynia. However, there is tantalizing evidence that cellular changes triggered by inflammation and axotomy might render Aβ afferents capable of doing so (Devor 2009). For example, following axotomy, Aβ afferents rapidly begin to express substance P and CGRP, the peptides present in nociceptors that are thought to mediate C fiber–induced pain and central sensitization (Noguchi et al 1995, Weissner et al 2006). This is one aspect of the cellular reprogramming (“phenotypic switching”) noted above. Once switching has occurred, impulse activity in injured Aβ fibers releases these peptides in the spinal cord. This, in turn, triggers central sensitization by a direct action on spinal neurons, via glial intermediates, or by opening the BBB (Pitcher and Henry 2004). Interestingly, phenotypic switching in touch afferents appears to account for the genotype-selective neuropathic pain behavior in rats selected for high versus low predisposition to neuropathic pain in the neuroma model (Nitzan-Luques et al 2011). CGRP in Aβ afferents is up-regulated in the pain-prone line but not in the pain-protected line. Phenotypic switching also occurs in animal inflammation models. In these models, activation of cutaneous Aβ fibers by gently brushing the inflamed skin triggers central sensitization (Ma and Woolf 1996).
Additional evidence that activity in injured (but not normal) Aβ fibers may both drive pain and trigger central sensitization comes from studies using c-fos expression as an indicator of central sensitization. In healthy animals, stimulation of C fibers triggers c-fos expression in post-synaptic neurons in the superficial and deep laminae of the dorsal horn, but stimulation of intact Aβ fibers does not. However, if there has been previous nerve injury, Aβ-fiber stimulation does induce c-fos expression (Molander et al 1994, Shortland and Molander 1998, Day et al 2001). Again, the likely explanation is phenotypic switching. Overall, these data suggest that contrary to prevailing beliefs, ectopia in Aβ afferents might well participate in central sensitization in chronic pain conditions, both inflammatory and neuropathic (Devor 2009, Suter et al 2009).
Beyond Ectopia: Other Peripheral Nervous System Mechanisms That Distort Sensory Signals In Neuropathy
Sensory Peculiarities in Neuropathic Pain
Patients often describe neuropathic pain in terms of natural stimuli: burning, cramping, stabbing, crushing, intense cold, and other types. Painful CRPS type 2 (formerly causalgia) was named for its constant burning quality (caustos = burned). “Burning mouth” and “burning feet” syndromes are other examples. A logical inference is that these sensations derive from discharge in the corresponding modality-specific afferent types (i.e., thermonociceptors).
Other painful sensations are familiar but do not correspond to any particular afferent type. Examples are “crawling insect,” lancination, “pins-and-needles,” tingling and electrical or frankly electric shock–like sensations. These sensations presumably reflect ectopic activity in the ensemble of afferent types normally activated by the corresponding stimuli. Such sensations tend to be particular to neuropathy. Indeed, they form the basis for a remarkably simple pain-type classification scheme (Bouhassira et al 2005). Finally, the sensory experience in neuropathy is sometimes so bizarre that it defies description, with patients being left frustrated by an inability to find words to convey exactly what they are feeling. Any theory of neuropathic pain needs to account for these uniquely neuropathic sensations.
Somatic sensation is a reflection of the particular chorus of afferents that are active, as well as their central processing. With regard to neuropathic pain, one ultimately wants to map out the detailed correlation between sensation and ectopia in conscious humans. Indeed, some relevant information is already available from microneurographic recordings and suggestive hints have also been obtained from animal preparations.
Afterdischarge, “Extra Spikes,” and Aftersuppression
After responding to a stimulus, sensory endings normally return rapidly to rest. Brief electrical pulses trigger single-spike responses. Ectopic pacemaker sites, by contrast, have intrinsic rhythmicity. Some fire spontaneously, whereas others are “turned on” by brief stimuli and then continue to discharge for a time afterward (see Fig. 61-6A). This “afterdischarge” yields aftersensation, a common symptom in neuropathy (Gottrup et al 2003). Basically, the stimulus triggers a period of spontaneous firing. The result is amplification; a given stimulus now produces more spikes than normal. Why this happens is discussed below.
Another manifestation of afterdischarge is “extra spike” formation. Here, geometrical irregularities along axons, such as branch points and patches of dysmyelination, cause the duration of a propagating action potential to increase. If the duration comes to outlast the absolute refractory period of the axon, it might re-excite the membrane that it has just passed over and generate an extra spike (Amir and Devor 2003b). In principle, if there were many such geometrical anomalies along a diseased axon, a brief stimulus might trigger reverberating cascades of extra spikes. Calvin and co-workers (1982) and Amir and Devor (2003b) proposed this as a potential mechanism for the paroxysms of pain in trigeminal neuralgia (TN). Unidirectional conduction block in branched sensory endings might also generate extra spikes (Bostock et al 2005).
Episodes of high-frequency firing, whether spontaneous or evoked by stimulation, are often followed by suppression (Amir and Devor 1997). In the fiber illustrated in Figure 61-6B, for example, a few seconds of excitation above the baseline ectopic firing rate (20 impulses/sec) was followed by a period of silence before baseline firing resumed. The duration of this “aftersuppression” increased with increasing duration of the prior excitation. A clinical manifestation of activity-evoked aftersuppression is the “refractory period” that follows individual pain paroxysms in TN (Kugelberg and Lindblom 1959, Rappaport and Devor 1994). During the refractory period it is difficult or impossible to trigger another burst of pain. Pain relief by transcutaneous electrical nerve stimulation (TENS) might also work this way. Specifically, the aftersuppression caused by TENS activation is expected to silence ectopic pacemaker sites for a prolonged period. This might be the mechanism of pain relief by TENS rather than gate control as is widely presumed. The same could be true for the pain relief provided by high-frequency dorsal column/spinal cord stimulation.
Two mechanisms have been proposed for this effect. First, aftersuppression induced by high-frequency firing may result from the activation of Ca2+-activated K+ channels. During firing Ca2+ enters the cell and opens these K+ channels. The resulting hyperpolarization silences the discharge (see Fig. 61-3B). This process is apparently responsible for the bursting firing pattern frequently generated at ectopic pacemaker sites (Amir and Devor 1997). Excess firing also activates the Na+ pump. In fine axon endings, where the volume of cytoplasm is small in proportion to the membrane area, this can hyperpolarize the fiber and silence it (Rang and Ritchie 1968).
Shock-like Pain Paroxysms and Hyperpathic Sensory Spread: Outcomes of Cross-excitation among Sensory Neurons
Individual primary sensory neurons normally constitute independent signal conduction channels. In the event of neural injury, however, excitatory interactions develop in neighboring neurons. Such cross-excitation can amplify sensory signals and prolong them, permit the effects of repeated stimuli to accumulate and cause sensation to spread beyond the immediate region of stimulation. These sensory consequences of cross-excitation probably contribute to such phenomena as “wind-up” and sensory spread in hyperpathia, neuropathic stabbing/lancinating sensations and electric shock–like paroxysms such as occur in TN. PNS cross-excitation occurs in the absence of synapses. It is different from the network interactions familiar in the CNS. On the other hand, comparable non-synaptic cross-excitation phenomena probably also occur in CNS white matter, where they might contribute to central pain. There are several mechanisms of cross-excitation.
Ephaptic Crosstalk
The most widely cited, though not the most important in terms of the number of neurons affected, is “ephaptic” (i.e., electrical) crosstalk. In the mid-1940s Granit and Skoglund (1945) discovered that acute nerve transection short-circuits the insulation between adjacent axons and thereby permits ionic current from one fiber to directly excite neighboring fibers. This coupling vanishes within minutes and is therefore unlikely to be of much functional significance. However, several weeks later ephaptic crosstalk re-emerges, now in an enduring form (Fig. 61-8). Ephaptic crosstalk occurs when there is a sufficient surface area of close membrane apposition between adjacent neurons in the absence of the normal glial insulation (Fig. 61-9; Fried et al 1993). It has been documented in neuromas, regenerating sprouts, and patches of demyelination (Seltzer and Devor 1979, Rasminsky 1980).
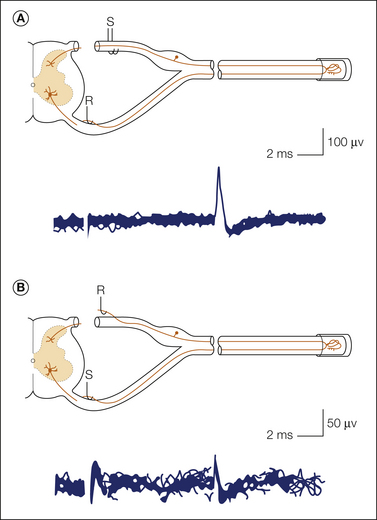
Figure 61-8 Bidirectional ephaptic crosstalk between a pair of fibers ending in an experimental nerve-end neuroma.
A, First, a fiber was found in the L5 ventral root that responded (R) at fixed latency to electrical stimulation (S) of ipsilateral dorsal roots (DRs). B, Stimuli (S) were then applied to the fiber, and fixed-latency responses were sought in the DRs. One responding fiber was found in the L4 DR. Note that response latency for conduction in the two directions is identical. (Modified from Seltzer Z, Devor M 1979 Ephaptic transmission in chronically damaged peripheral nerves. Neurology 29:1061–1064.)
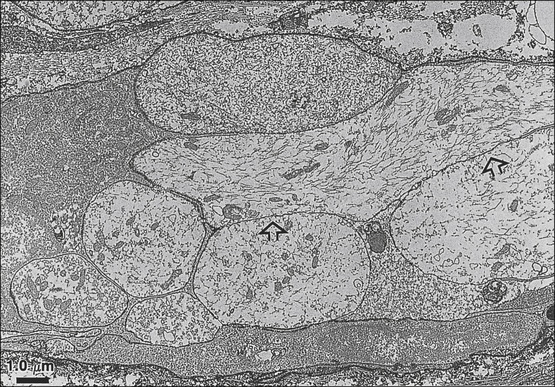
Figure 61-9 Close membrane apposition is the presumed structural basis of ephaptic crosstalk in injured nerves.
Arrows show regions of close apposition between neighboring demyelinated axons in the absence of an intervening glial (Schwann cell) process. Other axons are compartmentalized by (dark) Schwann cell cytoplasm as in normal nerves (lower left). A rat sciatic nerve neuroma 25 days after injury is shown (see Fried et al 1991). Scale bar = 1 μm.
Coupled fibers are frequently of different types. For example, nociceptors can be driven ephaptically by stimulation of low-threshold touch afferents (Amir and Devor 1992). Such coupling is an additional potential basis for tactile allodynia, independent of intracutaneous pacemaker sites and central sensitization. When multiple axons are coupled, discharge in one may activate many neighbors. The resulting signal amplification is instantaneous, and the sensation evoked is expected to be lancinating and paroxysmal. Interestingly, ephapsis does not occur in DRGs, intact or following traumatic nerve injury (Devor and Wall 1990), but it can occur after infection by certain strains of herpes simplex virus (Mayer et al 1986). This implicates ephaptic crosstalk as a factor in the pain of post-herpetic neuralgia. Ephaptic crosstalk does not appear to develop between sensory and sympathetic fibers (Devor and Janig 1981) thus undermining the old theory that such coupling accounts for SMP.
Chemically Mediated (Non-ephaptic) Cross-excitation, Crossed-depolarization, and Crossed-afterdischarge
Lisney and Devor (1987) discovered a very different type of cross-excitation among injured axons and termed it “crossed-afterdischarge.” Unlike electrical coupling, where spikes in the passive fibers follow spikes in the active fibers on a one-to-one basis (see Fig. 61-8), firing in the passive cross-excitated fiber outlasts firing in the active fiber (hence “afterdischarge”). Single-stimulus impulses have little effect on the passive fibers, but repetitive activity does have an effect (Fig. 61-10A). When more fibers are activated for a longer duration and at higher activation frequency, stronger cross-excitation is generated.
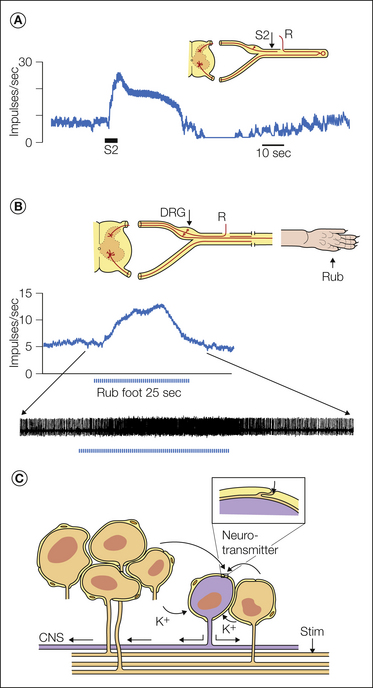
Figure 61-10 Non-ephaptic, chemically mediated cross-excitation in an experimental neuroma and dorsal root ganglion (DRG).
A, Electrophysiological recording (teased fiber method, R in the sketch) from a single spontaneously active axon that terminated in a nerve-ending neuroma. The baseline spontaneous firing rate was 8 impulses/sec. Brief tetanic stimulation of the sciatic nerve (S2 in the sketch and firing rate tracing) cross-excited the axon, which transiently increased its firing rate to 25 impulses/sec. Note that (1) the stimulation was delivered to neighboring axons in the neuroma but not to the recorded axon itself and (2) the enhanced firing long outlasted the stimulation itself. This is therefore an example of crossed-afterdischarge. Baseline spontaneous firing is not essential. Previously silent axons can be ignited in the same way (Lisney and Devor 1987). B, In this experiment the sciatic nerve was only partially injured and ectopic firing originating in the DRG was recorded (R) as shown in the sketch. Rubbing the rat’s foot increased the baseline spontaneous firing of this neuron as a result of DRG crossed-afterdischarge (Devor and Wall 1990). C, The presumed mechanism is that active neurons (orange) release K+ ions and a neurotransmitter or neurotransmitters into the extracellular space within the DRG. These substances access and excite passive (i.e., non-stimulated) neighbors. The inset on the right, which shows the junction point of two satellite glial cells, indicates that molecules in the extracellular space have direct access to the surface of the DRG neuron (Shinder and Devor 1994). K+ ions, at least, could also pass through the satellite cell sheath.
Cross-excitation also occurs in sensory and autonomic ganglia (Amir and Devor 1996, Matsuka et al 2001, Oh and Weinreich 2002). In both nerves and ganglia it depends on the passive neighbor having repetitive firing capability. In healthy DRGs, electrical stimulation of the axons of neighboring neurons induces depolarization in a large fraction of the cells sampled, but this “crossed-depolarization” is usually subthreshold for spike discharge (Fig. 61-10). However, in neuropathy models, crossed-depolarization reaches threshold for evoking repetitive discharge in many DRG somata (“cross-excitation”). A particularly salient situation is when pacemaker sites are present at mid-nerve locations (e.g., the DRG) but at least some of the axons in the nerve remain in continuity with the skin. In this situation, electrical nerve stimulation, or simply brushing the skin, can evoke crossed-afterdischarge amplification in the DRG (Fig. 61-10B). As in nerves, cross-excitation in the DRG can involve different fiber types and potentially render weak tactile stimulation of the skin painful (tactile allodynia).
Cross-excitation is thought to be due to the buildup of diffusible chemical mediators released into the extracellular space from the activated neurons. It is a unique form of paracrine communication. Repeated stimulation progressively winds up discharge in the passive neighboring fibers as the concentration of the paracrine mediator (or mediators) in the extracellular compartment increases (Fig. 61-10). The identity of the chemical mediator is not yet known with confidence. There is evidence implicating both a transient, stimuation-evoked rise in extracellular K+ concentrations (Utzschneider et al 1992) and released ATP acting through P2X7 receptors (Zhang et al 2007, Gu et al 2010). Non-synaptic chemically mediated neuron-to-neuron interaction of this sort is also known to occur in the CNS, where it is termed “volume transmission” (Agnati et al 2010). The phenomenon is difficult to study in the brain because of simultaneous synaptic interactions. The DRG is an advantageous preparation for studying volume transmission in isolation.
It has recently been shown that nerve injury (and peripheral inflammation) causes the satellite glial cells that surround each neuron in the DRG to become coupled to one another. The coupling, which in this case is mediated by gap junctions, has the potential to affect the discharge of DRG neurons and hence to signal pain (Hanani 2005). In addition, in neuropathy models satellite glia are known to proliferate and become “activated,” much like spinal astrocytes and microglia. Cytokines, neurotrophins, and other mediators released by activated satellite glia can excite DRG (and trigeminal ganglion) neurons in a bidirectional neuron–glia interaction (Obata et al 2006; Takeda et al 2007, 2009; Ohara et al 2009; Xie et al 2009; Gu et al 2010).
Role of Cross-excitation in the Pain Paroxysms in Trigeminal Neuralgia
Crossed-afterdischarge provides a potential explanation for one of the most distinctive, peculiar and devastating of the chronic neuropathic pain states, TN (tic douloureux). The mechanism may also contribute to pain paroxysms in a wide variety of other neuropathies. Patients with TN suffer from dramatic, brief stabbing or electric shock–like “lightning” pains in the trigeminal distribution, either spontaneously or on gentle tactile stimulation of a trigger point on the face or in the oral cavity. The underlying pathology in most patients is thought to be demyelination in the trigeminal root secondary to microvascular root compression. The discovery 50 years ago that TN responds to the anticonvulsant drug carbamazepine (Tegretol) gave rise to the hypothesis that the pain paroxysms in TN reflect seizure-like activity in the trigeminal brain stem.
More recently, however, Rappaport and Devor (1994) proposed an alternative hypothesis more consistent with the known PNS pathology of TN. According to their “ignition hypothesis” pain paroxysms begin with discharge in a small cluster of trigeminal nerve afferents that are activated by cutaneous trigger point stimulation or spontaneously. Crossed-afterdischarge, which may occur at the trigeminal root compression site or in the trigeminal root ganglion, then “ignites” ectopic activity in previously passive neighboring neurons. This activity ignites additional passive neighbors, and they ignite still more neurons. The resulting positive-feedback chain reaction builds up rapidly to an intense, explosive peak. Since neurons of all types become active simultaneously, an event that otherwise occurs only with electric shocks, the sensation felt is electric shock–like. After a few seconds of massive high-frequency firing, activity-evoked aftersuppression dampens the paroxysm, thereby ending the pain attack and establishing a few minutes of refractoriness. The system is then reset, ready for the next pain attack. The ignition mechanism is expected to be sensitive to at least some of the membrane-stabilizing anticonvulsants that suppress CNS seizure activity, notably carbamazepine. However, the drug’s action is supposed to be on PNS ectopia rather than on CNS seizures and is achieved by membrane stabilization rather than by synaptic suppression.
DRG Electrogenesis: Spike Invasion and Repetitive Firing in Intact DRG Neurons
Axotomy enhances the excitability of DRG neuronal somata, but why are these neurons electrically excitable in the first place? Impulses that propagate along nerves on their way to the CNS bypass the DRG. There is no obvious need for these spikes to invade the DRG cell soma, and yet spike invasion is always observed in intracellular recordings from DRG somata. Moreover, the invasion is not passive; the intact DRG cell is electrically excitable (Amir and Devor 2003a). The DRG is protected from direct stimulation within the intervertebral foramen and it contains virtually no synapses. However, spikes that invade the ganglion are known to trigger (non-synaptic) vesicular fusion and the release a variety of neurotransmitters into the extracellular space (Huang and Neher 1996, Ouyang et al 2005). Even more remarkably, DRG somata are known to express a large variety of membrane receptor molecules sensitive to the neurotransmitters released. Finally, unlike the rest of the PNS, ganglia lack the blood–nerve, tissue–nerve, and adherent glial barriers that normally insulate neurons from molecules in the systemic circulation (Allen and Kiernan 1994, Shinder and Devor 1994, Byrod et al 2000). Thus, DRG neurons are also subject to excitation by receptor agonists present in the systemic circulation (e.g., glutamate, endorphins, ATP).
All these peculiarities suggest that the DRG is more than just a passive metabolic depot serving the sensory neuron. Chemosensory function, including chemically mediated paracrine communication, appears to be a part of the normal physiology of the DRG. However, except when neuropathy causes DRG neurons to become hyperexcitable, this communication remains subthreshold for spike initiation (Amir and Devor 1996). The role, if any, of intraganglionic and blood–ganglion communication remains unknown, although as discussed below, there are reasons to believe that it might be important in controlling the afferent excitability of intact DRG neurons (Devor 1999).
Cellular Processes In Nerve Pathophysiology
The importance of ectopic hyperexcitability to neuropathic pain, as well as the urgency of finding more effective treatments, motivates detailed consideration of its underlying cellular and molecular mechanisms. How do nerve injury and disease, in addition to causing axonopathy and demyelination, lead to abnormal impulse discharge? To understand the significant progress that has been made in recent years toward answering this question, it is important to consider the processes responsible for electrogenesis in normal neurons (Jack et al 1983). What follows is a brief and qualitative refresher.
Primary Sensory Neurons Are Regionally Specialized
Primary afferent axons run uninterrupted from their sensory transducer endings (e.g., in the skin) to their central synaptic terminals in the CNS. The cell soma lies along this path in the DRGs or cranial nerve ganglia (see Fig. 61-7A). The soma provides metabolic support for the axon, including synthesis of the various molecules required to carry out the neuron’s four key functions: to transduce, encode, conduct, and relay sensory messages. Consider, for example, a myelinated afferent innervating the skin of the fingertip (Fig. 61-11A). The distal few tens of microns are specialized for transducing mechanical, thermal, and chemical stimuli into a depolarizing generator potential. In close association, still at the end of the axon, is the “pacemaker” zone where the generator potential is encoded into a train of electrical impulses. Encoding is the most crucial of the four functions for neuropathic pain. The midaxon region, which may be a meter and more in length, is specialized for conducting the encoded sensory signal rapidly and accurately toward the CNS. Finally, the central terminal arbor is specialized for relaying the signal to synaptic partners in the CNS.
Figure 61-11 Principles of rhythmic firing.
A, Sketch of a sensory ending showing the generator, encoding, and propagating compartments. The generator and encoding functions might occupy the same membrane region. B, Frequency–stimulus intensity relationship (f-I or “encoding” function) in response to a prolonged depolarizing generator current (computer simulation). At the rhythmic firing threshold, the firing rate jumps from zero to the fiber’s minimum rhythmic firing frequency (mRFF). A small change in stimulus strength around threshold (region a) has a much larger effect on firing frequency than the same change in region b does. (From Matzner O, Devor M 1992 Na+ conductance and the threshold for repetitive neuronal firing. Brain Research 597:92–98.)
Each function imposes particular design demands that are met by a specific complement of molecules whose synthesis and delivery must be closely regulated. Uncontrolled changes in the sensitivity or specificity of sensory endings can deliver signals inconsistent with the original stimulus. Our current understanding of the regulatory process is rudimentary. It is clear, though, that injury and disease can disrupt the regulation.
Impulse Initiation (Electrogenesis)
Stimulus Transduction and Spike Encoding
Signal transduction depends on transducer molecules, large proteins that translate stretch, heat, cold, pH, and other stimuli into a “generator depolarization.” They do this by controlling the flow of ions across the membrane of the sensory ending in response to stimuli. A well-known example is the transient receptor potential vanilloid 1 (TRPV1) channel. In the case of TRPV1 the receptor apparatus, the molecular structure that actually senses the vanilloid (capsaicin) stimulus, is an integral part of the larger transducer channel. Alternatively, the receptor may be physically separate from the ion channel and coupled to it functionally. G protein–coupled receptors (GPCRs), for example, do not have an intrinsic ion channel.
The generator potential is not a nerve impulse. It fades to nearly nothing over the first millimeters of the axon and cannot deliver a sensory signal to the CNS, which is much farther away. Only if the generator potential is encoded into a train of impulses can the pain signal reach the brain. This requires a different set of molecules, voltage-sensitive ion channels, especially Na+ and K+ channels. With some possible exceptions, transduction and encoding molecules are not the same. Both types are required for electrogenesis.
Single-Spike Threshold
The ion channels responsible for encoding are proteins that respond to changing transmembrane voltage rather than to sensory stimuli. Na+ and K+ channels each come in different “types” (Jenkinson 2006, Dib-Hajj et al 2010). At rest, most Na+ channels are closed. The generator depolarization initiated by the sensory stimulus causes the most sensitive types to open. This results in an inward flow of Na+ ions, which augments the generator depolarization. If the inward Na+ current is small, it is quenched by an outward counter-flow of K+ ions. However, if it is a bit larger, the depolarization resulting from the inward Na+ current causes more Na+ channels to open, and this in turn causes still more to open. The resulting inward torrent of Na+ ions overwhelms the outward flow of K+ ions and results in an explosive net inward current, the “action potential” (nerve impulse, spike). The crossover point between quenching and spike generation is the “spike threshold.” Note that threshold is not a fixed value. It is the equilibrium point between inward (mostly Na+) and outward (K+) currents. Its value (in millivolts) is affected by the density and properties of the channels present, and importantly, it is also affected by the rate of depolarization. Gradual depolarization may not evoke a spike (“accommodation”), whereas rapid depolarization of the same magnitude does.
Having opened, Na+ channels rapidly close again and membrane polarity returns toward rest. The rate of repolarization is often accelerated by transient opening of voltage-sensitive K+-selective ion channels, although at some locations (e.g., the node of Ranvier in some species), only voltage-insensitive K+ “leak channels” are present. When voltage-sensitive K+ channels are present, the outward flow of K+ ions actually overshoots the resting potential and cause a brief post-spike hyperpolarization. In some cells the after-hyperpolarization is followed by a small rebound depolarization, the “depolarizing afterpotential” (DAP). DAPs bring the membrane potential of the cell back toward spike threshold. If large enough and rapid enough, they may trigger a second spike or even a third and fourth (i.e., a spike burst; Fig. 61-12A).
Figure 61-12 In dorsal root ganglion (DRG) neurons, subthreshold oscillations trigger repetitive bursting; depolarizing afterpotentials (DAPs) maintain firing within each burst.
A, Brief spike bursts are triggered at the beginning of prolonged depolarizing current steps in vitro. The trace shows three superimposed trials at fixed stimulus strength that yielded bursts of two, three, and four spikes. In each case the burst ended with a subthreshold DAP (asterisks). B, Subthreshold oscillations trigger repetitive spike bursts in vivo. The recording setup is shown above. Interspike intervals during each of the four bursts shown are fairly stable as demonstrated in the dot display (above the spikes). This rhythmic discharge is interrupted by pauses of variable duration. Each burst is initiated by an oscillation sinusoid, a pattern also visible in panel D. DR, dorsal root; R&Sic, record and stimulate intracellularly; SCN, sciatic nerve; VR, ventral root. C, Fourier and autocorrelation analysis of spontaneous subthreshold oscillations in a neuron such as the ones shown in B and D (during epochs without spiking). The oscillation frequency and amplitude (power) are voltage sensitive. D, This DRG neuron had subthreshold oscillations at −58 mV. When the membrane potential depolarized to −50 mV and −43 mV, simulating natural excitatory stimulation, the oscillation sinusoids increased in amplitude and the larger ones began to trigger single spikes and DAP sustained bursts. (Data from Amir R, Michaelis M, Devor M 1999 Membrane potential oscillations in dorsal root ganglion neurons: role in normal electrogenesis and in neuropathic pain. Journal of Neuroscience 19:8589–8596; and Amir R, Michaelis M, Devor M 2002b Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. Journal of Neuroscience 22:1187–1198.)
Pacemaker Capability: Threshold for Repetitive Firing
The individual action potential is the basic unit of neural signaling, but sensory experience and certainly chronic pain depend on sustained firing. Some types of sensory endings, including nociceptors, can generate sustained (pacemaker) activity. Others respond only transiently. However, away from the sensory ending, the axon can convey trains of spikes but cannot generate them (except if injury has occurred). Pacemaker sites (natural and ectopic) use two related electrogenic processes. The first is the classic Hodgkin–Huxley process whereby a persistent generator current repeatedly depolarizes the neuron following each spike, thereby drawing the membrane potential from rest back toward spike threshold (Jack et al 1983). K+ channels control the rate of depolarization and hence the probability and rate of repetitive firing. The second pacemaker process is an outcome of “resonance,” or the intrinsic tendency of the membrane potential in some cells to oscillate between minor depolarization and minor hyperpolarization. This property is reflected in damped oscillations at the beginning and end of depolarizing steps and in the DAP that follows single spikes in some cells (Fig. 61-12A). A unique “resurgent” current may contribute to resonance (Cummins et al 2005, Enomoto et al 2006, Kovalsky et al 2009).
Oscillations, in cells that have them, are usually subthreshold. However, when peaks are brought to threshold by gradual depolarization or moderate stimuli, they may initiate action potentials (Fig. 61-12D) (Amir et al 1999, Xing et al 2001, Ma and LaMotte 2007). Oscillations do not promote spiking merely because of the extra few millivolts of depolarization that they contribute. After all, depolarization in a slow ramp usually triggers no spikes at all, even if the depolarization is deep. The key property of oscillation sinusoids is their rapid rate of depolarization. This overcomes membrane accommodation. Thus, even though they are small in amplitude, oscillations riding on a slow depolarization create pacemaker capability (Kovalsky et al 2008).
The “Therapeutic Window”: Threshold for Impulse Initiation versus Threshold for Repetitive Firing
The emergence of pacemaker capability at ectopic locations is arguably the most important process underlying neuropathic hyperexcitability and pain. Figure 61-11B shows the typical relationship between firing frequency and (tonic) stimulus intensity (f-I curve, or “encoding function”) in a neuron that is able to fire repetitively. Below a certain stimulus intensity the neuron is silent, but once threshold is reached (8 μA in Fig. 61-11B), sustained firing begins with a sudden jump to a relatively high “minimum rhythmic firing frequency” (mRFF). If the pacemaker site rests well below its repetitive firing threshold, a small depolarizing stimulus will not evoke any spikes. If it is well above threshold (and already firing repetitively), the small stimulus will accelerate the firing a bit (see Fig. 61-11B, zone B). However, if the pacemaker at rest hovers just below threshold (Fig. 61-11B, zone A), the same small stimulus will cause a shift from silence to just above the mRFF, a major amplification (Matzner and Devor 1992, Kovalsky et al 2009). Correspondingly, if the pacemaker at rest is just above the rhythmic firing threshold (and firing spontaneously), slight suppression will cause silence and thereby “stabilize the membrane.” Straddling the threshold yields burst firing (see Fig. 61-3).
The threshold for initiating a single spike and the threshold for repetitive firing are very different. Both depend on the density of Na+ channels, but the rhythmic firing threshold is far more sensitive than the single-spike threshold to changes in channel density. The relationship between the two is plotted in Figure 61-13. When Na+ channels are abundant, the two thresholds are similar (see Fig. 61-13A right). As Na+ channel density is reduced, the repetitive firing threshold rises sharply until firing can no longer be evoked at all, no matter how strong the stimulus (Fig. 61-13; ≈80 mS/cm2). Single spikes, however, are still evoked normally. Complete conduction block occurs only when very few functioning Na+ channels remain and action potentials can no longer be sustained (Fig. 61-13A left). The gap between pacemaker and single-spike firing constitutes a “therapeutic window.” Within this window sustained ectopia (and neuropathic pain) can be suppressed without blocking normal sensory signaling. A variety of “membrane-stabilizing” drugs exploit this therapeutic window to selectively suppress ectopia. This is illustrated for lidocaine in Figure 61-14.
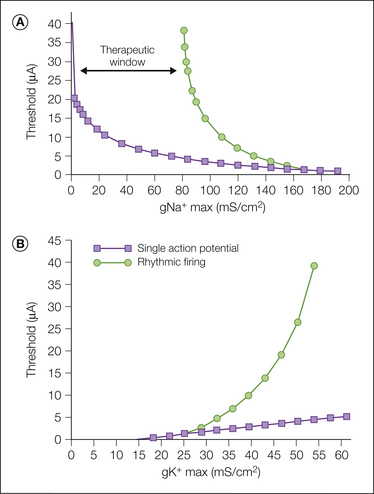
Figure 61-13 The threshold for repetitive firing is highly sensitive to the density of voltage-sensitive Na+ and K+ channels in the encoding compartment of the afferent axon end.
A, Computer simulation of the threshold for evoking a single spike (squares) and for evoking repetitive firing (circles) as the maximal Na+ current (gNa+ max) is varied. The gap between the threshold for repetitive firing and the threshold for evoking a single spike creates a “therapeutic window” (Matzner and Devor 1992). B, Same type of simulation as above but keeping gNa+ max constant and varying gK+ max.
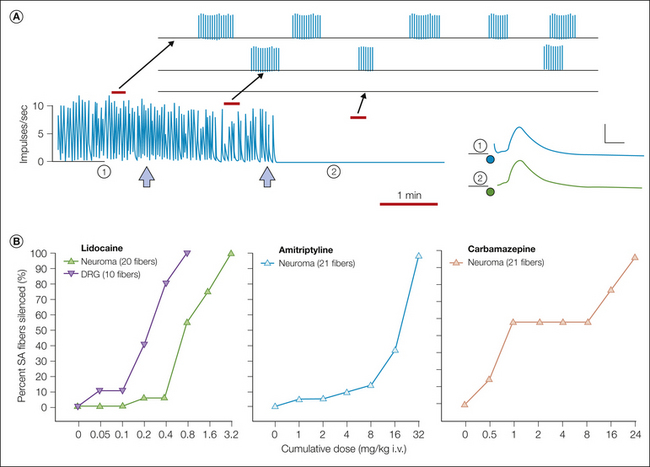
Figure 61-14 Membrane-stabilizing drugs administered systemically silence ectopia without blocking the ability of axons to conduct impulses.
A, The recording shows spontaneous burst discharge originating in an experimental nerve-end neuroma (8 days after surgery, experimental setup as in the sketch in Fig. 61-3A). The first injection of lidocaine (left arrow, 100 μL of 1% lidocaine) slowed the rate of bursting. The second injection stopped the firing altogether (right arrow). The continued response of the axon to electrical stimulation just central to the neuroma (lower right) proves that axonal conduction was not blocked. These action potentials were evoked at times 1 and 2. Calibration = 1.5 mV, 0.5 msec. B, Suppression of ectopic neuroma firing as the systemic dose of lidocaine, amitriptyline, and carbamazepine is gradually increased. Methods were as presented in A. For each drug, at the highest doses used all spontaneously active fibers (100%) were silenced. For lidocaine, recordings were made from the dorsal root ganglion (DRG), as well as from experimental neuromas. Ectopia originating in the DRG is considerably more sensitive than ectopia originating in the neuroma (Devor et al 1992a).
Subthreshold Oscillations and Ectopic Firing after Nerve Injury
Detecting stimuli is the specialized task of the sensory ending. At all other locations nerve fibers are essentially incapable of responding to natural stimuli (with the exception of direct heating and some chemical stimuli; Hoffmann et al 2009). Try applying pressure over the median nerve in your forearm. This does not evoke impulses in the midaxon portion of median nerve axons where the pressure is applied. If it did, you would feel tingling in the medial part of your hand. However, injury and disease change this. If there had been a neuroma or a patch of demyelination where you applied the pressure, it might well have produced a sensation referred to the hand, perhaps a painful sensation (e.g., Tinel’s sign at the carpal tunnel).
Research on the cellular mechanisms underlying this change is based mostly on recordings from DRG somata. Recording transmembrane potentials from axons remains technically challenging. However, the similarity of discharge patterns and responses to stimuli and drugs in DRGs and neuromas and the small number of quasi-intracellular recordings that have been made from axons (Kapoor et al 1997) suggest that the same basic electrogenic processes apply. Nearly all intact DRG neurons respond to prolonged step depolarization with a single spike or a brief burst lasting up to a few hundred milliseconds. They then fall silent even though the depolarization is still present (see Fig. 61-12A). Slow ramp depolarization, which better mimics natural generator potentials, rarely generates even a single spike. A few neurons, however, show sustained firing even with slow ramp depolarization. In these exceptional neurons subthreshold oscillations are present (Amir et al 1999, Wu et al 2001b).
In DRG neurons with subthreshold oscillations, each time an individual oscillation sinusoid crosses threshold, a single spike is generated. Such crossings occur at random intervals, which yields a slow, irregular ectopic firing pattern (Fig. 61-3A [3]). This changes if the oscillating neuron also generates DAPs. In this case the first evoked spike triggers a continuous DAP-sustained impulse train, or “tonic autorhythmicity” (see Fig. 61-3A [1]; Amir et al 2002b). If in addition to oscillations and DAPs factors are present that transiently stop the firing (e.g., an activity-induced hyperpolarizing K+ current), the impulse train will eventually stop, only to resume the next time that an oscillation sinusoid happens to reach threshold (see Figs. 61-3A [2] and B and 61-12B and D). The result is firing in bursts (“interrupted autorhythmicity”). Slow irregular firing is characteristic of ectopia in C fibers, whereas all three patterns, slow irregular firing, tonic firing, and burst firing at irregular interburst intervals, occur in A fibers (see Fig. 61-3A).
Axotomy and other forms of injury that induce neuropathic pain trigger a dramatic increase in the proportion of DRG neurons with subthreshold oscillations and hence an increased proportion with sustained discharge at rest and in the presence of tonic or slow ramp depolarization (Amir et al 1999, Xing et al 2001, Liu et al 2002, Ma and LaMotte 2007). Neuropathy also increases the likelihood of DAPs and hence the prevalence of high-frequency tonic and burst firing. Both changes are apparently due to remodeling of the intrinsic electrical properties of the neuronal membrane. The remodeling process appears to be a universal hallmark of ectopia in sensory neurons and is hence a key target for control of neuropathic pain.
Hyperexcitability and Membrane Remodeling in Injured Afferents
Electrogenesis: Excitation, Excitability, and Hyperexcitability
The terms “excitation” and “excitability” sound alike, but they are subtly different. Excitation refers primarily to stimulus transduction, the ability of a stimulus to depolarize the sensory neuron and create a generator potential. It depends on the transduction molecules present in the membrane of the sensory ending and both the properties of the stimulus and changes that occur in the process of its physical conveyance from the point of application to the transduction molecules (e.g., thermal conductivity and viscoelastic properties of the skin). Excitability, the capability of being excited, refers mainly to the process of encoding of the generator potential into a propagated impulse train. Encoding depends primarily on Na+ and K+ channels and the DAPs, subthreshold oscillations, and spikes that they generate. Spike electrogenesis is an outcome of both processes. Analgesic drugs can target either. However, stimuli that excite are numerous, and while eliminating any one of them might have a significant effect, all the others would remain in play. In contrast, if excitability is suppressed, the neuron loses its ability to signal pain in response to any stimulus. Excitation can be thought of as the wide mouth of a funnel whereas excitability is the narrow outlet. The latter is a uniquely powerful locus for controlling neuropathic pain signals. How is excitability modeled in healthy afferents and how do axotomy and demyelination lead to remodeling?
Retrograde Signaling and Control of Excitability in Normal Afferents
The membrane proteins responsible for transduction and encoding are synthesized on ribosomes in the DRG cell soma. They are then loaded on intracytoplasmic transport vesicles and vectorially transported down the axon. When the proteins arrive at the sensory ending, they are inserted into the membrane by vesicle exocytosis (Fig. 61-15). At first they are free to drift in the membrane plane, but after a short time, many become trapped and anchored to submembrane cytoskeleton-associated proteins such as ankyrin B and G and spectrin. The overall process, membrane “trafficking,” is highly complex and closely regulated in healthy neurons so that the correct molecules arrive at their correct addresses in appropriate numbers (Pfeffer 2007, Bennett and Healy 2009). In the event of nerve injury, regulation can fail, resulting in hyperexcitability and pain. This mechanism is discussed later.
Figure 61-15 Cellular mechanism underlying the development of ectopic pacemaker capability at the cut axon end of an injured afferent neuron.
Various ion channels, transducer molecules, and receptors (green symbols) are synthesized in the cell soma (left), axoplasmically transported distally within the axon on cytoplasmic vesicles, and incorporated in excess into the axonal membrane at end-bulbs and sprouts (see Fig. 61-16). The inset drawing illustrates the process whereby transported vesicles fuse with the axonal membrane, release their contents into the extracellular space, and deliver transmembrane proteins to the end of the axon.
Trafficking is dynamic. Vesicles carrying freshly synthesized transducer and ion channel molecules are delivered continuously to the sensory ending, and this delivery is balanced by endocytotic reuptake and degradation of molecules that were inserted previously (Leterrier et al 2010). The turnover half-life of Na+ channels, for example, is about 1–3 days. How trafficking is regulated and kept in balance is understood only sketchily. Control of receptor and channel biosynthesis and delivery is thought to involve neurotrophic molecules supplied, at least in part, by the innervated target tissues. Specific neurotrophins (e.g., NGF, GDNF) are taken up by axon endings that express the corresponding neurotrophin receptors and transported retrogradely to the cell soma. There they regulate gene expression and anterograde trafficking of the gene products to the axon’s sensory ending. This determines afferent sensitivity.
In embryonic development, neurotrophic signaling is critical for the very survival of sensory neurons. Interrupting retrograde flow (e.g., by axotomy) leads to rapid cell death. In adults, DRG neurons are less dependent on neurotrophins for survival, but they continue to play an important role in membrane modeling and neuronal phenotype. This role is illustrated in experiments in which adult DRG neurons were axotomized to block the retrograde flow of endogenous neurotrophins. Exogenous neurotrophins were then provided as a replacement. When the right neurotrophins were chosen, axotomy-induced hyperexcitability was prevented and even reversed (Boucher and McMahon 2001). A different, newly discovered mode of retrograde signaling may also play a role, at least in the event of axonopathy. Specifically, at the site where the axon is breached, importin proteins are locally translated and then transported back to the cell soma where they affect gene expression and neuronal phenotype (Hanz and Fainzilber 2006).
Regulation of Excitability
An important gap in our knowledge concerns how intact neurons of a given type maintain a stable level of excitability over long periods (a lifetime). How do nociceptors remain nociceptors and touch-sensitive fibers remain sensitive to touch? If there were an upward drift in the delivery of Na+ channels to nociceptor endings, for example, they might begin responding to weak stimuli and cause pain at every turn. A downward drift would make then unresponsive, thereby eliminating the protective role of pain. It is almost inconceivable that the control is “open loop”—that is, the cell soma delivers an unwavering quantity of channels and receptors to the end of the axon. There must be some sort of homeostatic feedback control.
One possible device is the dependence of gene regulation and trafficking on neuronal spike activity. For example, if afferent activity were too high over a particular integration window (hours, days?), excitability would be reduced; if too low, it would be increased (Devor 1999). In this way the sensitivity of the afferent ending would be stabilized around a functional set-point related to the message that it is intended to transmit. Evidence for homeostatic control of this sort exists for other systems (Dargent and Couraud 1990, Dargent et al 1994, Desai et al 1999, Fields et al 2001, Klein et al 2003, Schmidt et al 2009, Krol et al 2010, Kuba et al 2010, Savtchouk and Liu 2011, Turrigiano 2012). For example, in cardiac myocytes, excess spike activity has been shown to increase intracellular Ca2+ levels, which reduces Na+ conductance and hence excitability (Brodie et al 1989, Offord and Catterall 1989). Similarly, sound deprivation causes brain stem auditory neurons to increase their Na+ conductance and hence response sensitivity (Kuba et al 2010). In DRG neurons, cytoplasmic neurotrophin levels and release are both regulated by spike activity, as is TRPA1 trafficking from the cytoplasm to the surface of the membrane (Balkowiec and Katz 2000, Schmidt et al 2009).
Activity–excitability coupling, however, may not work for nociceptors because they rarely fire action potentials and therefore cannot provide activity-related feedback. Consider, for example, nociceptors in cardiac muscle. Even though such fibers may not fire at all for years, they are nonetheless reliably available to signal cardiac insufficiency by causing chest pain. Perhaps the activity-related feedback signal comes from firing in low-threshold afferents that share the same DRG as the nociceptors. The signal could be delivered by crossed-depolarization. That is, nociceptor phenotype could be regulated by the subthreshold depolarization driven by spike activity in the neighboring low-threshold afferents (Rusnak et al 2007, Sohya et al 2007). Such a feedback control system, if it exists, provides a possible rationale for why DRG somata are electrically excitable in the first place and why subthreshold cross-depolarization exists. There are indicators that control of excitability might also involve non-neuronal cells such as glia and Merkel cells, and extracellular matrix components (Kazarinova-Noyes et al 2001).
Membrane Remodeling after Nerve Injury
Regardless of whether excitability in healthy afferent neurons is regulated over the long run by activity-related signals, axonopathy and demyelination clearly disrupt the regulation. The resulting remodeling involves at least three interrelated cellular processes: membrane trafficking, up- and down-regulation of gene expression, and altered behavior of receptors and ion channels. Although the focus of research has been on receptors and channels, allied molecules such as lipids, anchoring proteins, and structural proteins must also be regulated.
Ion Channel Trafficking and Accumulation
Axotomy or functional block of axoplasmic transport (e.g., by cold or antimitotic drugs) leads to the local accumulation of channel-loaded transport vesicles. Some of these vesicles insert into the membrane and thereby increase local channel density and enhance electrogenicity (Fig. 61-16; also see Fig. 61-15). A variety of voltage-sensitive Na+ channel types have been shown to accumulate at neuroma endings and patches of demyelination in both animal neuropathy models and humans with neuropathic pain (Novakovic et al 1998, Devor et al 1993, Kretschmer et al 2002, Rasband et al 2003, Black et al 2008, Coggan et al 2010). This remodeling appears to be a causative factor in the creation of ectopic pacemaker sites following nerve injury.
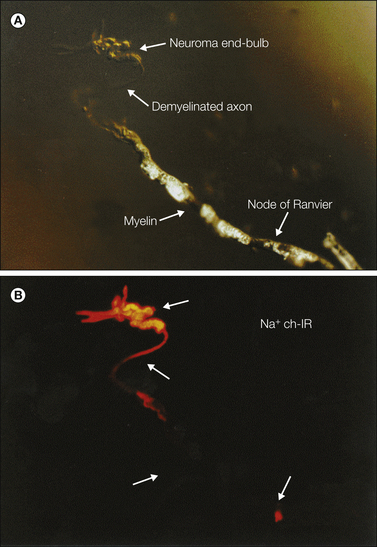
Figure 61-16 Na+ channel accumulation in neuroma end-bulbs.
A, Nomarski differential interference contrast micrograph of a chronically injured afferent axon end-bulb with sprouts. Myelin is present until about 250 μm from the axon end. Arrows mark the location of a node of Ranvier, myelin, the demyelinated portion of the axons, and a neuroma end-bulb. B, The same axon immunofluorescently labeled with an antibody that recognizes voltage-sensitive Na+ channels. Label has accumulated on the axonal membrane in the region of demyelination just proximal to the end of the axon and on the membrane of the end-bulb and sprouts (Devor et al 1989, 1993). Scale bar = 100 μm.
Functional remodeling by channel accumulation does not require a de novo process, only a quantitative shift in the normal equilibrium between channel insertion and reuptake. Remodeling is a result of both permissive and promotional factors. In myelinated axons the axonal membrane under the myelin normally contains a very low density of Na+ channels, perhaps because myelin suppresses their insertion. Demyelination, sprouting, and end-bulb formation remove this suppression and permit excess channel insertion. Axotomy also promotes membrane remodeling. Normally, the large amount of Na+ channel protein transported down the axon supplies the rapid turnover of channels at downstream nodes of Ranvier, the internodal membrane, and sensory endings (Leterrier et al 2010, Persson 2010a). Following axotomy these targets no longer exist. As a result, in-transit channels are redistributed into whatever competent membrane sites remain, notably neuroma end-bulbs, sprouts, and plaques of demyelination (see Figs. 61-15 and 61-16). Sites far upstream of the injury might also receive excess channel protein (Gilly and Brismar 1989), a potential mechanism for hyperexcitability in axotomized DRG somata. The same principles hold for other membrane proteins, including K+ and Ca2+ channels and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (England et al 1998, Garry and Fleetwood-Walker 2004, Nissenbaum et al 2008, Mathie et al 2010, Nuwer et al 2010, Roza et al 2011). In principle, drugs that alter axoplasmic transport could be exploited for pain therapy (Devor and Govrin-Lippmann 1983).
A final mode of altered channel trafficking is in the sensory ending itself. Neurons contain stores of latent cytoplasmic ion channels that do not contribute to the transmembrane ionic current. Impulse traffic and diffusible mediators within innervated tissue can promote Ca2+-dependent vesicle exocytosis and local membrane insertion of these latent receptors and channels. The expected result is enhanced ionic currents and electrical hyperexcitability (Huang and Neher 1996, Bao et al 2003).
Regulation of Gene Expression
Axotomy also triggers changes in the expression of various transducer, receptor, and ion channel molecules in DRG neurons. Since more than 99% of the cytoplasmic mass of sensory neurons serving the extremities is in the axon, mid-nerve axotomy represents a major metabolic challenge for the neuron. Interestingly, neuronal gene expression may also change after demyelination (Craner et al 2003, Wallace et al 2003). As noted above, thousands of genes are up- or down-regulated in neuropathic pain models (Persson et al 2009a, Hammer et al 2010, Michaelevski et al 2010), and a much larger number of protein products are altered. For some genes the degree of regulation varies across mouse strains in a manner that correlates with strain-specific pain susceptibility (Persson et al 2009a, 2009b). Such genes are good candidates for being related to pain mechanisms. Likewise, as noted, regulation may differ among afferent types within a single strain (Persson et al 2010b).
It needs to be appreciated, however, that the levels of mRNA expressed and the size of cytoplasmic protein stores do not necessarily translate simply into the density of proteins in the membrane. For example, most Na+ channel types accumulate in neuroma endings despite the fact that their synthesis in the soma is down-regulated (Kretschmer et al 2002, Waxman et al 2002, Black et al 2008). Likewise, neurotransmitters may be depleted in the DRG without an obvious loss at the central terminals (Bailey and Ribeiro-da-Silva 2006). The phenotype of a sensory neuron reflects the entire ensemble of molecules present, including modifications as a result of the local environmental influences of nearby non-neural cells and extracellular matrix components. Given that the levels of thousands of interacting molecules may change following nerve injury and disease, it is difficult to intuit or to simulate how all aspects of a neuron’s behavior might be changed by such injury.
Understanding remodeling can be made more tractable by focusing on the facets of the neuronal phenotype that are most closely related to neuropathic pain. I have made the case above that membrane hyperexcitability and ectopic electrogenesis transcend issues of sensory transduction and abnormal synaptic processing in the CNS. If so, a focus on these channels is justified.
Excitability depends mostly on a relatively restricted set of ion channels. Mammalian DRG neurons express, at least in some measure, eight types of voltage-gated Na+ channel pore (α) subunits in combination with three modulatory (β) subunits. A similar variety of voltage-sensitive and voltage-insensitive (K2p leak) K+ channels are expressed. Some members of each type are selective for afferents and even for nociceptors. To these we need to add channels that are gated by ions and ligands rather than voltage. For example, activity-related entry of Ca2+ ions can activate Ca2+-gated K+ channels and cause hyperpolarization. Changes in the level and disposition of any of these molecules can alter excitability and pain.
Na+ Channels
Insight into the role of Na+ channels has been obtained by computer simulations, the use of channel-selective reagents, biophysical protocols, and genetic manipulations. As noted, simulation of increased Na+ conductance predicts a modest reduction in the single-spike threshold as well as a dramatic facilitation of resonance and repetitive spiking (see Fig. 61-13A). Particularly important for pacemaker capability is the slowly inactivating (2–20 msec) components of the neuronal Na+ current, although not necessarily the truly persistent, non-inactivating component (Wu et al 2001b, Waxman et al 2002, Dong et al 2008, Kovalsky et al 2009). This prediction is consistent with observations on neuromas and axotomized DRG neurons in neuropathic pain models. For example, electrophysiological studies have shown that the degree of increase in whole-cell Na+ conductance and excitability in axotomized DRG neurons correlates with the degree of pain behavior observed across different animal strains and even among individual animals of a single strain (Liu et al 2001a, Abdulla and Smith 2002).
Considerable research attention has been drawn to the possibility that one or another Na+ channel type might be of particular importance for pain control. The Nav1.3 channel stood out in this regard since its expression is up-regulated by axotomy whereas that of the other Na+ channels is down-regulated (Cummins and Waxman 1997, Abe et al 2002). However, hopes that Nav1.3 might be a uniquely effective therapeutic target were dashed with the discovery that mice in which expression of this channel is knocked down or knocked out by genetic manipulations have basically unchanged neuronal excitability and pain behavior in neuropathy models (Lindia et al 2005, Nassar et al 2006). The Nav 1.8 and 1.9 Na+ channels have also been highlighted.
An individual Na+ channel type that has emerged recently as potentially special for pain is Nav1.7. People with particular rare mutations in the gene that codes for this channel (SCN9A) suffer from one of two extreme pain conditions (familial erythromelalgia or paroxysmal extreme pain disorder), whereas individuals with other mutations are congenitally insensitive to pain (Cox et al 2006; Dib-Hajj et al 2008, 2010). When replicated in cellular expression systems in vitro, these mutations cause cells to be hyper- and hypo-excitable, respectively, thus suggesting a direct link between the mutation, afferent excitability, and pain. Interestingly, the gain-of-function (pain-causing) mutations slow the rate of inactivation of the channel and thus enhance slow components of the Na+ current. As noted previously, this specific change is implicated in enhanced pacemaker capability based on other evidence. It is very likely that subthreshold oscillations are also altered in SCN9A mutants.
In addition to the rare, disease-causing mutations just discussed, common polymorphisms in the Nav1.7 gene show genetic association with a variety of different chronic painful conditions including osteoarthritis, phantom limb pain, idiopathic small fiber neuropathy, and pancreatitis (Reimann et al 2010, Faber et al 2012). Together, these observations point to Nav1.7 as being uniquely important for pain processing. However, pregnant as the findings are, much remains to be learned. For example, deletion of the Nav1.7 gene in mice does not alter neuropathic pain sensation much (Nassar et al 2005). Likewise, in affected family members pain is affected selectively in particular parts of the body (e.g., the extremities in familial erythromelalgia) whereas the gene is altered everywhere. Finally, other aspects of nerve conduction (except for the sense of smell) remain normal. This includes the sense of touch, proprioception, and motor and autonomic functions. It is difficult to conceive of how global point mutations in Nav1.7 can affect pain sensation in such an idiosyncratic manner. It will be fascinating to see how this story plays out.
K+ Channels
K+ channels have been less studied, but theory also highlights their marshaling as being critically important to ectopic hyperexcitability and pain (see Fig. 61-13B). K+ conductances hyperpolarize and reduce excitability. Therefore, reduced K+ conductance is expected to enhance excitability and pain. Like Na+ channels, expression and trafficking of K+ channels are regulated by nerve injury. In addition, the resulting conductances are modulated by a variety of hormones and neuroactive peptides and cytokines. In neuropathic pain models, axotomy down-regulates the expression of certain voltage-sensitive K+ channel genes and reduces whole-cell K+ conductance (Devor 1983a, Kocsis and Devor 2000, Kim et al 2002). Likewise, K+ channel blockers injected into neuromas in animals and humans enhance ectopia and induce pain (Burchiel and Russell 1985, Chabal et al 1989a). Genetic polymorphisms in the K+ channel gene KCNS1 have been associated with pain susceptibility in humans (Costigan et al 2010).
A particularly interesting K+ channel implicated in neuropathic pain is one that is activated by ATP rather than by voltage. This conductance is suppressed in neuropathy models, a change that causes depolarization and enhances ectopia. Strikingly, as for voltage-sensitive Na+ conductance, this change occurs only in individual animals with prominent neuropathic pain behavior. It does not occur in nerve-injured animals in which pain does not develop and hence strongly implicates this change as being causally related to the pain (Kawano et al 2009). Interestingly, the change is specific to large Aβ neurons in the DRG, again pointing to a role of touch afferents in neuropathic pain. Reduced K+ conductance probably contributes as much to ectopic hyperexcitability and pain as elevated Na+ conductance does (Ocana et al 2004).
Among the distinct K+ channel types expressed in sensory neurons, some may offer unique advantages as therapeutic targets (Rasband et al 2001, Takeda et al 2011). Two quite different families have captured attention with reference to chronic pain, the voltage-sensitive K+ channels KCNQ2/3 (Kv7.2) and the two-pore voltage-insensitive K+ leak channels (K2p, KCNK). Openers of these channels are candidate pain relievers (Alloui et al 2006, Ma et al 2010, Mathie et al 2010, Peretz et al 2010, Rose et al 2011). Many K+ channels are expressed in DRG neurons, but one, the K2p channel TRESK (K2P18.1, KCNK18), appears to be selective to the DRG and spinal cord (Sano et al 2003, Kang and Kim 2006, Tulleuda et al 2011).
Other Ion Channels
Many other ion channels are regulated by nerve injury, but it is unclear which if any of these channels makes so fundamental a contribution to ectopic hyperexcitability and neuropathic pain as Na+ and K+ channels do. For example, members of the so-called pacemaker or HCN channel family (hyperpolarization cyclic nucleotide–gated ion channels) are regulated by axotomy (Chaplan et al 2003, Yao et al 2003). Ca2+-activated Cl− conductance is also regulated in DRG neurons following axotomy. This might affect firing by altering the Cl− equilibrium as occurs in the CNS (Price et al 2009). Also of potential interest are several varieties of Ca2+ channels (Cao 2006). T- and N-type Ca2+ conductance is reduced following axotomy (Abdulla and Smith 2001), a change that in principle could either reduce excitability or increase it (by reducing the activation of Ca2+-activated K+ channels). In practice, wide-spectrum pharmacological blockers of Ca2+ conductance have only minor effects on ectopia except at very high concentrations, so the significance of this change is uncertain (Matzner and Devor 1994, Liu et al 2001b, but see Dogrul 2003). Intrathecally administered neuronal Ca2+ channel blockers, notably ziconotide (SNX-111, Prialt), probably provide analgesia by suppressing Ca2+-dependent neurotransmitter release in the spinal cord rather than by suppressing ectopia.
Perhaps more important than overall levels of Ca2+ channel α-subunit expression and ion flow are changes in expression of the auxiliary Ca2+ channel subunits that participate in channel and receptor trafficking. The A2δ Ca2+ channel subunit, for example, is an auxiliary subunit up-regulated in neuropathic pain models. This is the binding site of the antineuropathic pain drugs gabapentin and pregabalin (Hendrich et al 2008, Taylor 2009).
Yet another hint that the trafficking of ion channels and receptors may be a key substrate for chronic pain comes from genomic screens. Pain, particularly neuropathic pain, is highly variable from individual to individual even when the precipitating nerve injury or disease is the same. Useful information can be derived from genetic comparison of cohorts (animals or patients) that have and do not have pain. At their best, genetic studies analyze the entire genome in an unbiased manner, free of investigator preconceptions about what genes might be important. Remarkably, a considerable proportion of such studies, covering painful conditions ranging from migraine headache to neuropathy, have converged on polymorphisms in genes coding for auxiliary Ca2+ channel subunits, including auxiliary subunits related to protein trafficking. These include the genes CACNA1A, CACNA1D3, CACNA2D1, CACNA2D2, CACNA2D3, CACNK1, and CACNG2. Polymorphisms in several of these genes are also implicated in epileptogenesis, a condition already allied with neuropathic pain on the grounds that anticonvulsant drugs often provide effective pain relief (Barclay et al 2001, de Vries et al 2009, Neely et al 2010, Nissenbaum et al 2010, Tselnicker et al 2010). These observations, which are unlikely to be just coincidental, focus attention on protein trafficking and distribution as a potentially productive avenue for future research.
Current-Carrying Capacity of Ion Channels Already in Place
Ion channels determine cell excitability by controlling the transmembrane flow of ionic currents. An increase in net flow through a fixed number of channels has much the same effect as an increase in the total number of channels present. Increased net flow can result from a higher probability of channels opening, extended channel open time, incomplete inactivation, or increased unitary channel conductance. Such changes in the intrinsic kinetics of ion channel behavior represent a third process that can contribute to neuronal hyperexcitability.
Evidence is accumulating that modulation of the current-carrying capacity of voltage-gated channels might contribute to neuropathic pain. For example, cyclic adenosine monophosphate–dependent phosphorylation of Na+ channels reduces Na+ current, wherease dephosphorylation returns it to normal (Li et al 1992, Gold et al 1998, Vijayaragavan et al 2004, Chahine et al 2005, Hudmon et al 2008). Certain hormones, trophic factors, neuromodulatory peptides (e.g., CGRP), and inflammatory mediators (notably prostaglandins) activate dephosphorylating enzymes, including protein kinase A and C. They are therefore positioned to affect afferent excitability. Some of these mediators are known to sensitize afferents by increasing Na+ current density, reducing K+ current density, and augmenting oscillations and neuronal firing even when the membrane potential remains unchanged (Nicol et al 1997, Gold et al 1998, Hu et al 2001, d’Alcantara et al 2002, Natura et al 2005, Binshtok et al 2008). They alter excitability without necessarily changing excitation. Transduction channels, of course, can also be sensitized.