Central Pain
Introduction
Injury to the nervous system causes loss of sensation in the territory innervated by the damaged structure (nerve root, fascicle, peripheral nerve, spinal segment, cortical structure, etc.). In a limited number of patients, such damage is followed by long-lasting and occasionally persistent pain in the affected area—called neuropathic pain. Though primarily described for diseases and lesions in the peripheral nervous system, neuropathic pain may also be a feature of certain central pain disorders. According to the International Association for the Study of Pain (IASP), central pain was defined as pain initiated or caused by a primary lesion or dysfunction in the central nervous system (CNS) (Merskey and Bogduk 1994). Although not specified, it is generally assumed that the lesion causing central neuropathic pain affects parts of the CNS that either conduct or process spontaneous or evoked somatosensory information. Also, the word dysfunction creates confusion because prolonged activation of the nociceptive system in itself causes functional changes (Jensen and Baron 2003, Woolf 2004, Costigan et al 2009). For this reason, a new definition of central neuropathic pain has now been adopted by the IASP: pain caused by a lesion or disease of the central somatosensory nervous system (www.iasp-pain.org). There are multiple causes and mechanisms that induce central pain (Bowsher 1996; Jensen et al 2001; Koltzenburg and Scadding 2001; Boivie 2006a, 2006b; Baron et al 2010), but the consequence of the injury to the CNS giving rise to the pain seems to include at least two main changes: (1) partial or complete loss of sensation to one or several sensory modalities and (2) development of hypersensitivity in body parts that have been deprived of their normal patterned somatosensory information (Jensen and Lenz 1995). This chapter describes the lesions and diseases that affect the somatosensory system and thereby cause central pain. Pain following spinal cord injury (SCI) is dealt with only briefly; a more detailed description is provided in Chapter 68.
Overview of Central Pain Conditions
The first descriptions of central pain were probably provided by Greiff (1883) and Edinger (1891). However, the first clear account of central pain came in 1906 when the French neurologist Déjerine and his student Roussy reported eight patients with lesions in the thalamus associated with “persistent, paroxysmal often intolerable pain on the hemiplegic side.” Excellent detailed clinical descriptions were given in the first half of the 20th century (Garcin 1937; Riddoch 1938a, 1938b, 1938c), and it became clear that central pain may be seen following a number of CNS disorders (Table 69-1), the most common of which are stroke, multiple sclerosis (MS), SCI, syringobulbia and syringomyelia, spinal cord infarction and hemorrhage, and certain inflammatory conditions (Boivie 2006b, Klit et al 2009). More controversial and less documented conditions include brain tumors, Parkinson’s disease (PD), epilepsy, and traumatic brain injuries (Bowsher 1996, Boivie 2006b). Pain following central lesions is often neglected initially because the main causes, such as stroke, spinal diseases, and MS, cause severe motor and cognitive handicaps, thereby rendering pain a less important symptom that is either forgotten or reduced to a minor problem by patients and their physicians (Jensen and Lenz 1995). The main clinical feature of central pain is pain or dysesthesia (i.e., unpleasant sensations) in a body area with partial or complete sensory loss with respect to one or several sensory modalities. This is because the body area with abnormal sensory function has a representation in a CNS structure that has been damaged by a specific disease or lesion. Spontaneous pain can be accompanied by allodynia (i.e., evocation of pain by a non-painful mechanical or thermal stimulus) or hyperalgesia (i.e., increased pain response to a stimulus that normally produces pain) and after-sensations (i.e., pain continuing after the stimulation has ceased), all symptoms that are characteristic of these types of pain. The complexity of central pain conditions is underscored by the fact that not all types of pain in these patients can be ascribed to a specific disorder of the somatosensory system (Widar and Ahlström 2002, Siddall et al 2003, Attal and Bouhassira 2006, O’Connor et al 2008); associated musculoskeletal types of pain, headaches, spasticity, sleep disturbances, and mood disorders may either alone give rise to pain or aggravate an already existing condition.
Table 69-1
Etiology of Central Neuropathic Pain
Infarction or hemorrhage of the brain or spinal cord
Spinal cord injury
Multiple sclerosis
Syringomyelia or syringobulbia
Subacute combined degeneration
Surgical lesions of the brain or cord
Neoplasm of the brain or spinal tissue
Compression myelopathy
Parkinson’s disease?
Epilepsy?
Inflammation of brain or spinal cord tissue
Epidemiology of Central Pain
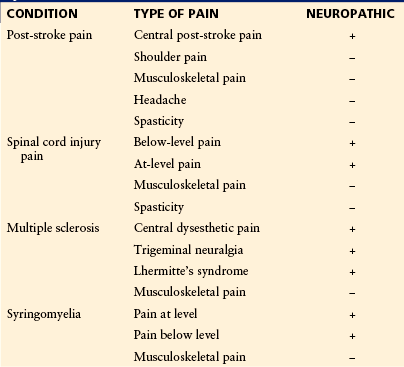
Information about the frequency of pain after stroke is limited. Studies have shown that pain occurs in anywhere between 1 and 50% of patients after a stroke, both ischemic and hemorrhagic, but not all pain is related to stroke (Boivie 2006a, 2006b; Klit et al 2009; Kumar and Soni 2009), and it is also clear that different criteria may account for some of the variations. The most common forms of chronic pain after stroke are musculoskeletal pain, shoulder pain, central post-stroke pain (CPSP), tension-type headache, and spasticity (Table 69-2). Estimates of the proportions of the different types of pain are 40% for musculoskeletal pain, 20% for shoulder pain, 10% for headache, 10% for CPSP, 7% for spasticity, and 13% for other types of pain (Klit et al 2009). No criteria for CPSP have been universally accepted thus far, but the following seem to be the minimal criteria, and they correspond to those used in recent publications (Klit et al 2011a, 2011b):
A prospective study found that CPSP developed in 8% of stroke patients within the first year after stroke (Andersen et al 1995). Bowsher (2001) found an 11% frequency of CPSP in a group of elderly patients. In a recent population-based study of 964 stroke patients (Klit et al 2011a, 2011b) that used a grading system and in which patients were classified as having either definite or probable neuropathic pain (Treede et al 2008), 7.3% were found to have either probable or definite CPSP. CPSP can occur after lesions at different levels of the somatosensory pathways, but interestingly, the prevalence of CPSP is dependent on the anatomical location of the lesion (MacGowan et al 1997, Kim and Choi-Kwon 1999, Kumar and Soni 2009). A higher frequency of pain has been observed with brain stem infarctions. Thus, MacGowan and colleagues (1997) found that CPSP developed in 16 (25%) of 63 patients with lateral medullary infarctions. Fitzek and colleagues (2001) found two-thirds of patients with lateral medullary infarctions to have CPSP. Bogousslavsky and co-workers (1988), looking at thalamic lesions, found the prevalence of CPSP to be higher in persons with lesions affecting the inferolateral part of the thalamus (16.7%) than in those with thalamic infarcts in general (7.5%). One study found that CPSP developed in 4 of 13 patients after intracerebral hemorrhage (≈30%). This high prevalence may be the result of a predilection for hemorrhage in the basal ganglia and thalamic structures.
Multiple Sclerosis
The reported prevalence of pain in patients with MS varies, with figures ranging from 29–86% (O’Connor et al 2008). The large span in reported frequencies probably reflects the use of different methodologies and only few of the studies being population based. In addition, the difficulty distinguishing spasticity and nociceptive types of pain from central pain may contribute to the variation in reported frequency of pain. In a postal survey of 627 patients with definite MS who were compared with a sex- and age-matched control group, 79% of the MS patients and 75% of the control group had some form of pain in the preceding month, but patients with MS had higher pain intensity and higher consumption of analgesics than did their respective controls (Svendsen et al 2003). Pain may be an initial symptom of MS in about 10–25% of patients, but it is otherwise seen as part of a chronic condition during the course of MS in approximately 50% of patients. Entities considered to represent central pain conditions include central dysesthetic pain, trigeminal neuralgia (TN), Lhermitte’s sign, and tonic spasms (Nurmikko et al 2010). In an Italian study involving 1627 patients, the following prevalence of central pain was found: central dysesthetic pain, 18%; TN, 2%; Lhermitte’s sign, 9%; and tonic spasms, 11% (Solaro et al 2004). In the aforementioned postal survey of 627 patients with definite MS and a similarly sized age- and gender-matched group, Svendsen and colleagues (2003) found that 38% in the MS group and 12% in the control group had touch-evoked pain. In another study by Svendsen and associates (2005) in which all types of pain in the past month were considered, 79% in the MS group and 75% in the control group had pain complaints. In another study of 364 MS patients, 27.5% were considered to have central neuropathic pain (defined as a pain distribution consistent with a CNS lesion and with simultaneous exclusion of a peripheral neuropathic or nociceptive cause) (Osterberg et al 2005).
Syringobulbia and Syringomyelia
The prevalence of syringomyelia and syringobulbia was previously noted to be between 3 and 8/100,000 (Kurland 1958, Gudmundsson 1968), but mainly because of improved neuroimaging methods, it is now reported to be higher. Brickell and colleagues (2006), in a study from New Zealand, reported a prevalence of 8.2/100,000 and higher in certain ethnic groups. Pain is considered the most important symptom in syringomyelia (Attal and Bouhassira 2006), but no population-based studies have provided reliable data on this. In a consecutive study of 46 patients with syringomyelia with and without pain, 67% had spontaneous pain and 64% had pain evoked by one or several stimulus modalities (Ducreux et al 2006).
General Symptoms and Signs of Central Pain
The symptoms and signs in patients with central neuropathic pain can, as in other cases of neuropathic pain, be divided into negative and positive phenomena (Jensen et al 2001, Jensen and Baron 2003, Finnerup and Jensen 2006, Baron et al 2010). The negative symptoms and signs reflect damage to the CNS resulting in partial or complete sensory loss and numbness in the body part corresponding to the nervous structure that has been damaged (Jensen and Baron 2003). The positive phenomena, such as allodynia, hyperalgesia, and hyperpathia, are all manifestations of central sensitization with hyperexcitability in parts of the nervous system (Woolf 2004, Baron et al 2010). The complexity of central pain is emphasized by the fact that many patients have concomitant pain conditions such as headache, musculoskeletal types of pain, spasticity, and co-morbid conditions, including anxiety, mood disturbances, and other emotionally charged states, which makes it difficult to distinguish central pain from other types of pain. Clinically, central neuropathic pain is characterized by the presence of spontaneous ongoing pain and various types of evoked pain. These symptoms often occur in different combinations.
Ongoing Pain
Ongoing pain is spontaneous and may be continuous or paroxysmal. The character differs, but it can be shooting, shock-like, aching, cramping, crushing, smarting, and burning, among other descriptions. Episodic, paroxysmal types of pain are short-lasting shooting, electric, shock-like, or stabbing in character.
Evoked Pain
Stimulus-evoked pain is classified according to the type of stimulus that provokes it, such as mechanical, thermal, or chemical stimuli. In some patients all these symptoms may be present; in others only one type of hypersensitivity is present. So a series of stimuli need to be applied to document or exclude abnormality. Evoked pain is usually brief and lasts only for the duration of stimulation, but it may sometimes persist even after cessation of stimulation because of after-sensations, which can last for minutes, hours, or even days. In such cases, distinction between evoked and spontaneous types of pain can be difficult. Allodynia and hyperalgesia may be seen with central pain disorders; however, it is clinical experience that in CPSP, dysesthetic MS, and perhaps also other central pain conditions, the sensory quality of the abnormal evoked sensation more often has the character of dysesthesia rather than true pain (i.e., allodynia). Hyperpathia (i.e., explosive pain evoked in areas with an increased sensory threshold when the stimulus exceeds the threshold) is also a feature of central pain, as in peripheral neuropathic pain. It has been claimed that a specific set of descriptors is associated with the various types of central pain, just as it has been suggested for peripheral neuropathic types of pain (Bouhassira and Attal 2011).
Specific Features
The lesion responsible for pain in CPSP was initially claimed to be located in the contralateral thalamus, and the syndrome was for years called “thalamic syndrome” (Déjerine and Roussy 1906; Garcin 1937; Riddoch 1938a, 1938b, 1938c). It is now well established that lesions causing central pain after stroke can be located anywhere along the somatosensory projection system, such as in the lateral medulla, in other parts of the brain stem, in the thalamus, or beyond the thalamus, including the cortex and the operculum (Boivie et al 1989; Leijon et al 1989; Andersen et al 1995; Bowsher 1996; Peyron et al 1998, 2000; Klit et al 2009; Garcia-Larrea et al 2010). For this reason the term “thalamic pain” has been abandoned and substituted by the broader and more appropriate term CPSP (Boivie 2006a). The pain in CPSP can be both superficial and deep, and in the latter case it may be difficult to distinguish from musculoskeletal types of pain, which often accompany CPSP. No set of specific pain descriptors is associated with CPSP, but burning, pricking, lancinating, icy, tearing, cutting, and squeezing types of pain are often present in CPSP. Using the McGill Pain Questionnaire, the median number of words chosen was eight with a median pain rating index of 21.5% and a median pain intensity of 2 on a scale of 0–10 (Vestergaard et al 1995). The onset of pain in CPSP varies and can occur anytime from immediately after the stroke to several months or, in some cases, years after the stroke (Boivie 2006a, Klit et al 2009, Kumar and Soni 2009). In a prospective study it was found that two-thirds of patients had an onset of CPSP during the first month, 19% within the first 6 months, and the remaining after 6 months (Andersen et al 1995). Lampl and co-workers (2002) found that pain developed about 10 months after the stroke in their population. The pain of CPSP can be spontaneous or evoked; in particular, spontaneous dysesthesia seems to be common and is reported by more than 75% of CPSP patients. Some studies have found higher pain intensity with lesions located in the brain stem and thalamus (Leijon et al 1989, Fitzek et al 2001) rather than outside these areas, but this does not seem to be a consistent finding (Klit et al 2009). The intensity of spontaneous pain often fluctuates and can be increased by emotional as well as physical distress and reduced by rest and distraction (Leijon et al 1989, Bowsher 1996, Boivie 2006a), similar to other types of neuropathic pain in which internal and external factors can modulate the pain experience. Pain often represents a great burden to the patient, even when the intensity is low. There has been interest in the distribution of pain, which can involve anything from small areas to the entire half of the body. In patients with medullary infarctions, crossed pain distributions are characteristic and have specific significance. In patients with pain located unilaterally in the orofacial area and in the ipsilateral finger digits, the pain is likely to originate in the contralateral thalamus. The sensory loss may involve all sensory abnormalities, but loss of spinothalamic functions (cold, warmth, pinprick) appears to be essential and is found in more than 90% of patients with CPSP, whereas sensory loss with respect to other modalities is less common (Boivie et al 1989, Andersen et al 1995, Vestergaard et al 1995). The area of pain is within the territory of the sensory abnormalities and typically occupies only a fraction of the sensory deficit. Another characteristic of CPSP is the paradoxical presence of hyper-phenomena in the painful area. Hyper-phenomena may be manifested either as hypersensitivity with allodynia or merely as dysesthesia to one or several sensory modalities. In a study by Andersen and associates (1995), cold allodynia or dysesthesia was present in 94% of patients with CPSP but in only 3% of stroke patients with sensory abnormalities but without pain. Similarly, 75% had touch-evoked allodynia or dysesthesia, but none in the group without pain had these abnormalities. Abnormal temperature and pain sensibility are the most consistent abnormalities in CPSP. In two studies it was found that 81% had reduced sensibility to temperature (Boivie et al 1989, Andersen et al 1995). The presence of hypersensitivity within the same territory of the sensory deficit can, for obvious reasons, occasionally obscure the detection of sensory loss or the extent of such.
Multiple Sclerosis
MS is a chronic progressive disease characterized by multiple sites of focal demyelination in the CNS as a result of inflammatory processes of assumed autoimmune origin in individuals with a susceptible genetic and environmental background. The focal inflammation gives rise to plaques scattered throughout the CNS, but with a preference for the periventricular white matter, the brain stem, and the spinal cord. The consequences of these plaques are a wide spectrum of neurological symptoms and signs, including motor, coordinative, sensory, autonomic, and cognitive abnormalities. One symptom—and an often neglected one—is pain. Central dysesthetic pain occurs, according to a study by Osterberg and colleagues (2005), in 28% of cases, but it is rarely an initial symptom. The quality is variable; burning and aching types of pain are often reported, but they are not specific for the central pain in MS. There are only a few studies on the sensory abnormalities in MS patients with pain. In a random sample of 50 MS patients with pain, 50 MS patients without pain, and 50 controls, more than 50% of the patients had central pain. The presence of cold allodynia and abnormal temporal summation was higher in the MS group than in the non-painful MS group (Svendsen et al 2005). In this study no difference was found between the two groups in terms of abnormalities in dorsal column and spinothalamic function. Osterberg and Boivie (2010), on the other hand, in a group of 62 MS patients with central pain and 16 MS patients without pain found sensory abnormalities in 97 and 81%, respectively. Abnormalities in spinothalamic function were more common in the pain group. Pain usually affects the legs and trunks uni- or bilaterally, and according to its origin it has a distribution that is compatible with a brain or spinal segmental localization. TN is more frequent and more often bilaterally localized when seen in MS patients than in the general population. Painful tonic seizures or tonic spasms, not to be confused with spasticity, are paroxysms of painful attacks lasting seconds and usually less than 2 minutes with pain in the face, arm, or leg associated with abnormal, often dystonic postures. They may start in one body part and spread either unilaterally in a segmental fashion or occasionally bilaterally. These attacks may last days or a few weeks and then disappear. The underlying mechanisms are unclear, but they have been related to an acute inflammatory lesion in the internal capsule or the cerebral peduncles (Nurmikko et al 2010). Lhermitte’s sign refers to a sudden and short-lasting electrical sensation spreading down the spine from the cervical region, usually in response to flexion of the neck. These attacks are assumed to be due to acute demyelination or inflammation in plaques in the cervical cord. They generally occur for a period of days or weeks and then disappear. For TN in patients with MS, it has been suggested that it could be related both to intrapontine pathology of the trigeminal axons and to concomitant peripheral vascular compression (Cruccu et al 2009). As in other cases of CNS lesions associated with pain that is spasm related, pain is also seen with MS. Spasm-related pain represents a difficult problem in terms of classification. Spasms can be spontaneous or provoked by different stimuli, including tactile stimulation, urinary tract infection, a full bladder, or emotional factors. The frequency is variable, but in MS it has been estimated that about 10% have such spasms (Moulin et al 1988). Flexor spasms are generally explained by disinhibition of the normal flexor withdrawal response (Sherrington 1948), and from that point of view it may be argued that spasm-related pain is a central pain phenomenon (Osterberg et al 2005). Others would argue that the pain with flexor spasms is related to repeated muscular contractions, movements, and postures and is therefore to be considered a musculoskeletal type of pain.
Syringomyelia and Syringobulbia
Syringomyelia is characterized by a cystic cavity in the central canal within the spinal cord. If extending into or manifested at the brain stem level, it is termed “syringobulbia.” Approximately 90% of cases of syringomyelia and syringobulbia are congenital and often associated with Arnold-Chiari malformations (herniation of the cerebellar tonsils into the spinal canal). Non-congenital cases of syringomyelia are almost exclusively of traumatic origin and secondary to SCI. The central pain in syringomyelia and syringobulbia is similar in nature to that seen in other central pain conditions (i.e., a combination of ongoing/spontaneous and evoked types of pain). The pain can be bilateral or hemiform and is frequently located in the hands, shoulder, and thoracic areas. The pain may precede other symptoms and signs of syringomyelia by many years (Garcin 1937; Riddoch 1938a, 1938b, 1938c). In addition to central pain, these patients also suffer from musculoskeletal types of pain, visceral pain, and headache. The latter is particularly common in patients with Arnold-Chiari malformation type I. Pain may persist even when spinothalamic functions (pinprick and thermal sensation) have been completely abolished. In an interesting study looking at syringomyelia patients with and without pain, it was found that the extent of thermal sensory loss was not different between patients with and without pain, thus suggesting that a lesion of the spinothalamic system, although it may be necessary, is not sufficient to drive the pain (Ducreux et al 2006). In the same study a direct relationship was found between the degree of thermosensory deficit and the intensity of burning pain in syringomyelia patients, which suggests that deafferentation or loss of input into a central projection territory may be a driving mechanism for the spontaneous pain. It is of interest to note that a similar pattern is not seen in CPSP (TS Jensen, unpublished observations).
Other Causes
Central pain was formerly seen after thalamic destruction, cordotomy, mesencephalic and medullary tractotomy, myelotomy, lesions of the Lissauer tract, and other such surgeries (for review see Tasker et al 1991). Surgical lesions of the CNS are rarely done today, and pain as a consequence of such operations is accordingly reported infrequently. Destruction of the posterolateral and medial ventral nuclei of the thalamus has been carried out for the relief of pain (Pagni 1989); it sometimes causes transient anesthesia and analgesia on the contralateral body half, which after a few months is replaced by a burning pain with some recurrence of sensibility. Commisural myelotomy can be followed by bilateral girdle pain. Hemisection of the spinal cord, as in Brown-Sequard syndrome, may be associated with short-lasting pain immediately after injury on the paralytic but not on the analgesic limb side. This can be followed by late-developing pain in the non-paralytic but analgesic body part, below the lesion. These latter types of deafferentation pain are probably similar to those seen after anterolateral cordotomy, in which pain or dysesthesia often develops months after the cordotomy (White and Sweet 1969, Nathan and Smith 1979). A bizarre condition is occasionally seen following uni- or bilateral cordotomy: referred pain to normal sensory territories if thermal or painful stimuli are applied to analgesic body parts (Nathan 1956). Neoplasms of the spinal cord, either extra- or intramedullary, may be accompanied by pain, and some of these types of pain may have a central pain component, but they are usually difficult to distinguish from other neuropathic or nociceptive types of pain.
Parkinson’s Disease
It estimated that about 40–75% of patients with PD have sensory symptoms with pain as the main complaint (Wasner and Deuschl 2006). The pain can be related or unrelated to the motor symptoms and signs of PD. Pain related to the motor phenomena has been divided into early and late PD. In early PD, the musculoskeletal type of pain is seen as a manifestation of muscle rigidity, and in the late type of PD, the pain can be of musculoskeletal, nociceptive, or neuropathic character. In some cases, pain may also accompany the fluctuations in PD patients with on–off phenomena. In most cases of pain related to the motor symptoms, the pain improves following regulation with antiparkinson medication (Wasner and Deuschl 2006). To what extent some of the pain in PD patients can be linked to deafferentation similar to other central pain conditions is still speculative, but interesting observations suggesting that central pain can be a manifestation of PD have appeared. Recent studies have shown changes in heat pain thresholds that support a central mechanism for the pain. In a neurophysiologic study, patients with PD and pain but no other obvious reason for their pain had normal warm sensory thresholds but lower heat pain and laser pinprick thresholds and higher amplitudes of laser-evoked potentials in the “off” condition than did patients with no pain. These abnormalities were improved by administration of levodopa during the “on” condition (Schestatsky et al 2007). These changes suggest enhanced responsiveness to painful stimuli and a relationship between the hypersensitivity to painful stimuli and dopaminergic activity. The mechanisms underlying this type of pain in PD are unknown, but recent experimental and clinical neurophysiological, pharmacological, and brain-imaging studies indicate that the basal ganglia and dopaminergic system are involved in the gating of nociceptive information to higher areas for central pain processing and modulation and in the development of neuropathic pain (Ertas et al 1998, Pertovaara and Wei 2008). Involvement of striatal dopamine D2 receptors in the noradrenergic pain inhibitory circuitry is one of the mechanisms by which the basal ganglia may be involved in nociceptive integration and thus offers a possible explanation for the pain associated with PD (Pertovaara and Wei 2008), as well as perhaps explaining the efficacy of levodopa on pain thresholds in this pain condition (Djaldetti et al 2007, Gerdelat-Mas et al 2007).
Traumatic Brain Injury
Central pain may also be a feature in patients with brain trauma. In a systematic study of patients with brain trauma, it was shown that the chronic pain in patients after traumatic brain injury resembles that in other patients with central pain (Ofek and Defrin 2007). The time of onset was 0.5–30 months after injury, and the pain persisted. The pain was often unilateral, corresponding to the side of the body with more severe motor and sensory dysfunction. Pain descriptors included pricking, cold, freezing, numb and wretched, pressing, and burning, and all described allodynia to cold, touch, physical effort, or movement. A decrease in thermal sensitivity was demonstrated, thus supporting a lesion of the somatosensory pathways and the presence of central pain.
Epilepsy
Pain has been reported in association with epilepsy, but it is undoubtedly rare. Young and Blume (1983) reported painful seizures in 24 of 858 patients, but not all the cases were linked to the epileptic seizure activity. The mechanisms for such pain are unknown, but according to Gates and colleagues (1984), they can in some cases be considered focal or generalized seizure phenomena.
Diagnosis of Central Pain
Diagnosis of central pain is based on the history, clinical examination with emphasis on sensory findings, and imaging of relevant lesions with computed tomography (CT) or magnetic resonance imaging (MRI), with occasional supplementation by other measures (Table 69-3) (Cruccu et al 2010). The pain history should include information about the onset of pain, quality of the pain, presence of dysesthesia or allodynia, and a pain drawing. The clinical examination should include sensory testing in which both negative and positive sensory findings are recorded and mapped on a sensory phantom chart (see Table 69-3 and Figs. 69-1 and 69-2). Quantitative sensory testing (QST) is not necessary in all cases of central pain, but it can be helpful for documenting sensory loss and the presence of hypersensitivity. QST permits detailed testing of controlled and graded physiological stimuli, such as thermal, mechanical, and vibratory stimuli, which are used to document common or dissociated sensory findings in patients with pain from CPSP, MS, SCI, and syringomyelia. The usefulness of such examinations in clarifying mechanisms is illustrated by a study of Greenspan and colleagues (2004). In a group of 13 patients with CPSP, they demonstrated that tactile and cold allodynia was present both in those with normal and in those with abnormal touch sensation, but that allodynia was more common in patients with normal tactile sensibility. Most of their patients (85%) had reduced sensibility to cold stimuli, but only few of them exhibited cold allodynia. These findings suggest that cold hypoesthesia is neither necessary nor sufficient for cold allodynia in CPSP and that tactile allodynia may occur in patients with disturbed thermal pathways when the tactile pathways are preserved.
Table 69-3
Assessment of Positive and Negative Sensory Symptoms or Signs with Central Pain
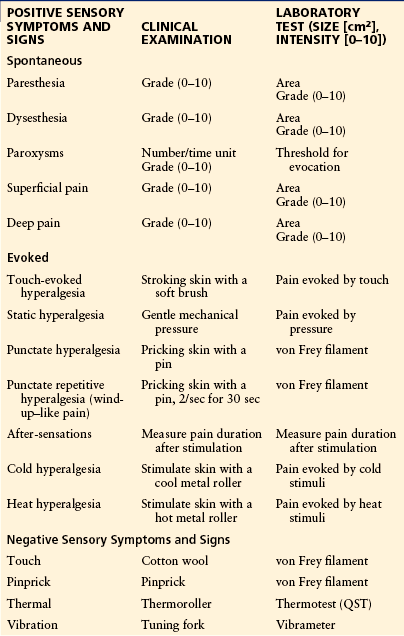
CPSP, central post-stroke pain; QST, quantitative sensory testing.
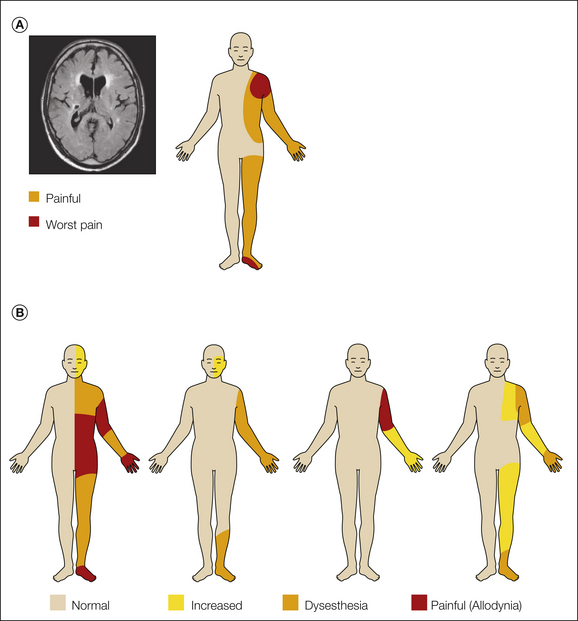
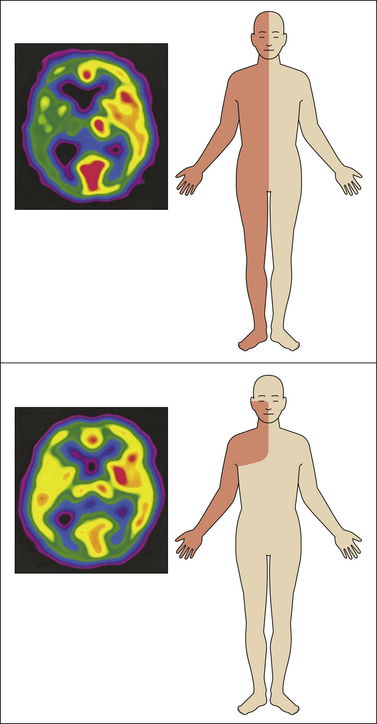
Figure 69-1 A, A 54-year-old man with a sudden onset of left-sided paralysis and sensory deficits. Magnetic resonance imaging showed an infarct in the crus posterior of the internal capsule extending into the dorsal part of the thalamus and several other minor ischemic lesions (left). Ten months after the stroke he complained of severe pain in the left part of his body (right). He described the pain as pressing and tight with a pins-and-needles sensation and a superficial burning sensation. The pain was constant with an average pain intensity of 7–8 (numerical rating scale of 0–10). B, Sensory examination and sensations elicited by touch (cotton ball), dynamic brushing (Somedic brush), pinprick (von Frey filament 5.88), and cold (thermoroller 20°C).
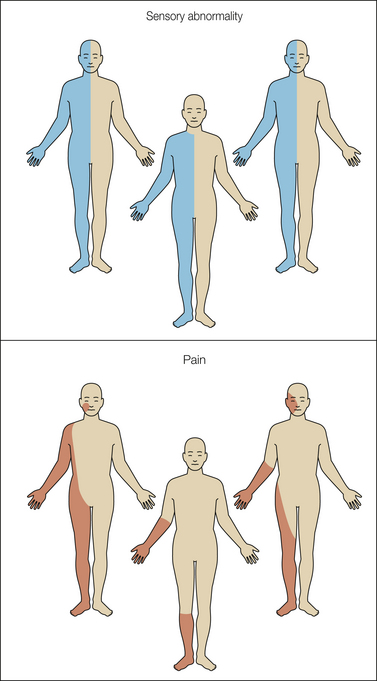
Figure 69-2 Distribution of sensory abnormality and pain in three cases of central post-stroke pain.
Abnormalities in somatosensory and laser evoked potentials are often seen in patients with central pain but thus far have been of limited diagnostic value (Casey et al 1996, Garcia-Larrea et al 2002). In cases in which CT or MRI has failed to demonstrate a clear lesion or in which multiple lesions are present in the CNS, it is possible that evoked potential measurements in combination with QST may aid in demonstrating both hypo- and hypersensitivity in the same body part. If further combined with mapping of the painful territory and the extent of sensory abnormalities, a central pain condition can usually be either confirmed or excluded. Table 69-3 presents a list of positive and negative sensory symptoms and signs and how they can be assessed or measured quantitatively. Several screening tools for neuropathic pain have been published within the past decade (Bennett et al 2007), but their diagnostic value for central pain conditions has not been determined in detail. It is clear that to postulate a central pain condition it is necessary to document the presence of a CNS disorder, and because of the frequent combination of non-neuropathic types of pain, it may often be difficult to determine a possible neuropathic component in these disorders. Pain scales, such as the visual analog scale and the numerical rating scale, are useful for the evaluation of pain intensity. No scales have been developed specifically for central pain.
Is Pain Associated with Central Nervous System Disease always Neuropathic?
According to the current IASP definition, central pain is a neuropathic pain disorder. In some CNS diseases such as stroke, SCI, MS, and syringomyelia, certain types of pain can be ascribed to damage to the classic central somatosensory pathways, whereas other types of pain such as headache, painful spasticity, and musculoskeletal pain in CNS disease are not necessarily linked to direct damage to somatosensory pathways. These latter types of pain should in our opinion not be considered central pain (Klit et al 2011a, 2011b). In other cases of pain associated with CNS disease such as PD, dementia, epilepsy, CNS neoplasms, and other conditions in which the lesion is not affecting the classic somatosensory pathways but the damage may affect systems shared by what is sometimes referred to as the “CNS pain matrix,” central neuropathic pain is indeed a possibility. Nevertheless, for the time being and in the interest of better classification and delineation of different pain types in patients with CNS disease, we propose to keep these types of pain apart from central pain.
Mechanisms of Pain
The starting point in understanding the mechanisms of central pain should be based on clinical observations. The essential pathological feature in central pain is a lesion of the spinothalamic transmission system resulting in partial or complete loss of input to the CNS and the development of corresponding negative sensory phenomena such as partial or complete anesthesia in the damaged area. This dissociated sensory loss seems to be an almost consistent feature in central pain and in neuropathic pain associated with at least some types of CPSP, SCI, MS, and syringomyelia. In accordance with the neuropathic nature, there are both negative and positive sensory hypersensitivities reflecting loss of input to a cerebral structure and hypersensitivity in the same body part as a consequence of neuronal hyperexcitability in part of the CNS (Boivie et al 1989, Vestergaard et al 1995, Jensen and Lenz 1995). The distribution of the pain is typically within the territory with sensory abnormality and generally occupies only a fraction of the area with that sensory abnormality (Vestergaard et al 1995, Finnerup et al 2003, Finnerup and Jensen 2004) (Fig. 69-3). These observations have led to the proposal that central pain may not only be a consequence of loss of spinothalamic input into certain parts of the CNS but may also be a manifestation of neuronal hyperexcitability in neuronal populations that have lost their normal patterned input. Pain may under these circumstances be seen as a release phenomenon from cell populations that are normally under the control of other surrounding structures. It remains to be seen whether similar patterns also occur in other central neuropathic pain states. The role of the spinothalamic tract in the development of central pain has been a topic of key interest (Pagni 1989; Lenz et al 1989, 1994, 1998, 2004; Willis and Westlund 1997; Osterberg et al 2005; Dostrovsky 2006). The general observation that patients with central pain almost universally have partial loss of one or several spinothalamic tract functions, such as cold, warm, and pinprick sensitivity, suggests that a lesion of the spinothalamic tract is a necessary condition for the development of central pain. However, this is not a sufficient condition alone based on the simple observation that loss of these functions is not always accompanied by pain (Leijon et al 1989, Andersen et al 1995, Vestergaard et al 1995). Studies of patients with SCI and syringomyelia have likewise demonstrated that spinothalamic functions are also disturbed in patients without pain. In a recent study, Svendsen and colleagues (2011) used MRI to determine the location of plaques in 13 MS patients with pain and in 10 MS patients without pain. Allodynia and/or dysesthesia was more frequent in patients with pain. No difference was found in the number of patients with plaques in the spinothalamic tract, dorsal column–medial lemniscus, dorsolateral funiculus, gray substance, thalamus, or capsula interna. In parallel with loss of input, sensitization may occur. Clinical and experimental studies indicate the presence of sensitization of second- or third-order neurons in the CNS, and one candidate structure here is the thalamus, which is known to play an important role in processing somatosensory information, including stimuli in the noxious range. The spinothalamic and trigeminothalamic tracts project into the thalamus via a lateral and medial projection system (Ralston 2005). In the thalamus, the main terminations of the spinothalamic tracts consist of the ventrocaudal nucleus, the ventroposterior inferior nucleus, the ventral lateral nucleus, the central lateral nucleus, the parafascicular nucleus, and the medial dorsal nucleus (Dostrovsky and Craig 2006). The complexity of the subsequent processing is demonstrated by the fact that these nuclei project differently into the primary (SI) and secondary (SII) somatosensory cortices (associated with the sensory–discriminative aspects of pain), the anterior and posterior divisions of the insula subserving both the sensory–discriminative and affective components of pain, and the anterior cingulate cortex (associated with attention to the noxious activity).
Theories of Central Pain
Various theories have been advanced to explain central pain (Figs. 69-4 and 69-5). They can be summarized into three main categories: disinhibition, sensitization, and neuroplastic changes.
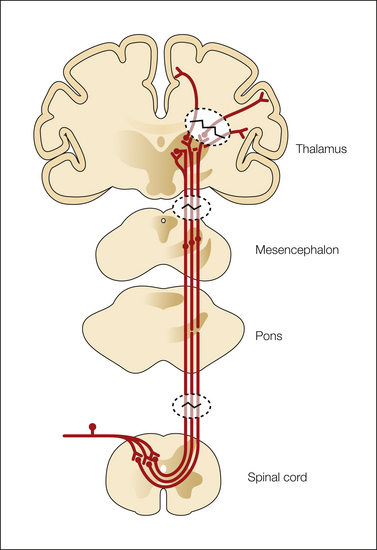
Figure 69-4 Loss of input to the thalamus or other brain structure because of lesions at lower levels may give rise to central pain.
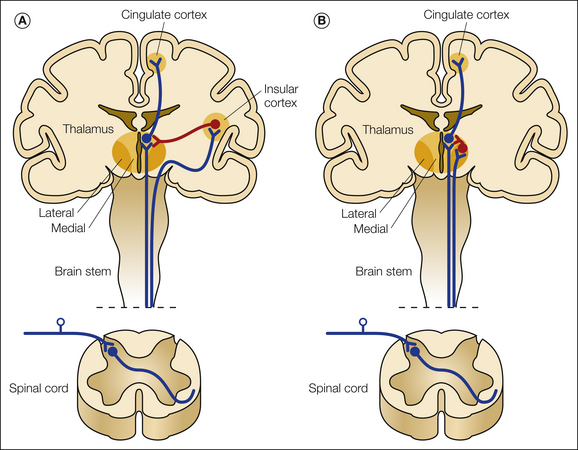
Figure 69-5 Potential mechanisms of central pain.
A, The thermosensory disinhibition theory by Craig suggests that a lesion in the lateral cool-signaling spinothalamocortical projections to a thermosensory area in the insula via the posterior part of the ventral medial nucleus causes disinhibition (red) of a medial limbic network involving the parabrachial nucleus and the periaqueductal gray of the brain stem, the medial thalamus, and the anterior cingulate cortex. B, Proposal by Head and Holmes in which loss of spinothalamic tract input to the posterior lateral part of the thalamus causes disinhibition (red) of the medial thalamus and leads to pain.
Disinhibition
According to the disinhibition hypothesis, which was originally formulated by Head and Holmes (1911), disruption of input into the lateral thalamus by a vascular lesion causes a disinhibition of the medial thalamus that results in pain (see Fig. 69-5). Head and Holmes did not explain exactly how the pain developed but based their hypothesis on the observation that in patients with central pain, the pain was located in the lateral parts of the thalamus whereas the medial thalamus was intact. Others have modified this hypothesis. A variant of the disinhibition hypothesis is the thermosensory disinhibition proposal (see Fig. 69-5). According to this hypothesis proposed by Craig (1998, 2002, 2003), central pain is a thermoregulatory disorder, and pain is the result of lost normal inhibitory input exerted by cool-signaling pathways from lamina I neurons projecting into the insula. Pathways from the dorsal posterior insula normally inhibit a limbic matrix involving the parabrachial nucleus, the periaqueductal gray, the medial thalamus, and the anterior cingulate cortex. A lesion of the lateral cool projection system is assumed to disinhibit the medial system of heat–pinch–cold neurons passing from lamina I to the medial part of the thalamus. This disinhibition will then result in the release of cold allodynia and burning and ongoing pain. Disruption of thermosensory integration leads to disinhibition of noxious-responding thalamocortical neurons and a sensation of burning pain. Another potential mechanism for the development of evoked pain that is also related to disinhibition is disturbance of the normal balance between a lateral spinothalamic system projecting into the insular region from the lateral thalamic nuclei and a medial system projecting into the anterior cingulate cortex. The medial and intralaminar thalamic nuclei as part of the “medial” pain system project to the anterior cingulate cortex, which is associated with attention to pain. Persistent activation of the anterior cingulate may then tend to evoke pain.
Sensitization
Lesions in the CNS imply a risk for the development of sensitization in certain parts of the brain. Loss of ascending input to the lateral thalamus has been shown to produce pain. Bogousslavsky and colleagues (1988) demonstrated in a series of thalamic infarctions that only those with lesions in the ventral posterolateral and medial nuclei were associated with pain, and in the study by Kim and co-workers (2007), lesions in the primary somatosensory nucleus were associated with pain. Consistent with a role of ascending input in the development of CPSP, positron emission tomography studies have demonstrated a reduction in regional cerebral blood flow in the thalamus contralateral to the side of pain in patients with spontaneous CPSP (Garcia-Larrea et al 2006) (see Fig. 69-3). Neuronal burst activity has been demonstrated in the ventral caudal part of the thalamus in patients with central pain, a phenomenon that may reflect sensitization of certain cell groups within the thalamus (Lenz et al 1989, 1994; Rinaldi et al 1991). Electrical stimulation of certain areas in both the lateral and medial thalamus by microelectrodes can elicit pain. There is an increased incidence of stimulus-evoked pain sites in the ventrocaudal parts of the thalamus, and microstimulation in these areas and other parts of the thalamus is more likely to cause a burning sensation in CPSP patients than in other chronic pain patients (Lenz et al 1998). Recent animal studies support some of these data by demonstrating that lesions of the spinothalamic tract produce hyperalgesia and mechanical allodynia in the paws of rats associated with increased burst activity in ventral posterior lateral nucleus brain slices. A T-type calcium channel blocker eliminates this bursting activity (Wang and Thompson 2008). Although these data suggest that partial or complete lesioning of the spinothalamic pathways may sensitize neurons in the thalamus, it is still unclear whether such neuronal activity has anything to do with pain because bursting is also found in patients without pain (Dostrovsky 2006). Sensitization phenomena may also be involved in the development and maintenance of pain in patients with SCI and syringomyelia (Ducreux et al 2006, Finnerup 2008). A series of mechanisms could trigger and maintain such hyperexcitability following SCI and syringomyelia, including increased release of excitatory amino acids, ectopic impulse generation, loss of tonic inhibition from γ-aminobutyric acid (GABA)-containing neurons, increased descending facilitation, and so on (Finnerup 2008). These mechanisms and possibly others may contribute to central sensitization with increased neuronal background activity and increased responses to external stimuli with changes in the receptive fields of dorsal horn neurons. Studies in patients have found a correlation between at-level evoked pain and spontaneous below-level pain and between larger gray matter lesions at the rostral end of the SCI and below-level pain when compared with pain-free patients (Finnerup et al 2003, 2004). There are also suggestions that neuroplastic changes may occur at supraspinal sites following lesions in the spinal cord. For example, it has been suggested that abnormal input from second-order neurons in the rostral end of the spinal cord lesion may activate thalamic neurons via multisynaptic systems and result in pain referred to areas below the injury level (Finnerup and Jensen 2004). In patients with incomplete injury, it is possible that the spinothalamic tract could be the source of abnormal impulse generation.
Neuroplastic Changes
Altered connectivity may play a role in the development of central pain. In some patients the development of central pain is delayed for months or even years, thus suggesting that prolonged maladaptive changes might have occurred, possibly associated with an altered balance between input from noxious and non-noxious processing systems. In patients with SCI and syringomyelia, it has been suggested that activation of microglia (Carlton et al 2009) or abnormalities in sodium channel expression might play a role in the generation of pain (Hains et al 2003). These changes in Nav1.3 expression may also spread to rostral structures such as the thalamus (Hains et al 2005). The expansion of receptive fields or elicitation of pain by microstimulation in patients with lesions of ascending input into the thalamus is another finding that points to late neuroplastic changes with reorganization within the thalamus.
It is unlikely that any single mechanism can explain all aspects of central pain, and the mechanisms detailed previously may either alone or in combination play a role in pain generation in these central disorders. It is equally possible that other mechanisms yet to be determined may contribute to the different types of pain in patients with central pain.
Management
As with other chronic pain conditions, treatment of central pain is complex and involves different approaches. Whenever possible, the underlying disease should be treated; otherwise, symptomatic treatment of the pain and any related disability should be offered. Realistic expectations of the outcome of a given treatment should be discussed with the patient, and it should be explained that often only partial pain relief can be obtained. Another important aspect is the common association of nociceptive types of pain and co-morbid conditions such as depression, memory problems, motor disturbances, and others (see, for example, Klit et al 2011a, 2011b). Management can be divided into pharmacological and non-pharmacological types of treatment.
Pharmacological Treatment of Central Pain
Antidepressants are the classic compounds used for neuropathic pain conditions. Antidepressants inhibit monoamine transporter systems with a corresponding increase in noradrenaline and serotonin drive on post-synaptic receptors. Tricyclic antidepressants (TCAs) also exert a blocking effect on N-methyl-D-aspartate (NMDA) receptors and sodium channels (Sindrup et al 2005).
In central pain, TCAs were first studied in a three-way crossover study with amitriptyline, 75 mg/day, and carbamazepine, 800 mg/day, in 15 patients with CPSP (Leijon and Boivie 1989). Amitriptyline, but not carbamazepine, had a significant pain-relieving effect with a number needed to treat (NNT) for improvement of pain of 1.7 (1.2–3.1). This study demonstrated a correlation between efficacy and total plasma concentrations of amitriptyline and its active metabolite nortriptyline, with higher responses occurring if concentrations exceeded 300 nmol/L. A study of similar design also found effects of amitriptyline on non-paroxysmal central pain in patients with MS, but this study had a high dropout rate because of side effects (Österberg and Boivie 2005). In SCI, amitriptyline (up to 150 mg) was compared with gabapentin and the active placebo diphenhydramine (Rintala et al 2007). This study demonstrated a small but significant effect of amitriptyline. Amitriptyline did not relieve mixed types of pain (nociceptive and neuropathic) in patients with SCI (Cardenas et al 2002).
Duloxetine, a mixed serotonin and noradrenaline reuptake inhibitor (SNRI), was studied in patients with SCI pain and CPSP in doses of 60–120 mg/day (Vranken et al 2011). There was no statistically significant effect of duloxetine in comparison to placebo, but a trend toward a decrease in mean pain score at the end point (P = 0.056) and an effect on several secondary end points suggest that the study may have been underpowered and thus produced a false-negative result. Duloxetine relieved dynamic mechanical and cold allodynia. The selective serotonin reuptake inhibitor (SSRI) citalopram did not relieve CPSP in a small underpowered study of 13 patients (Vestergaard et al 1996), and trazodone did not relieve SCI pain (Davidoff et al 1987).
The preventive effect of early amitriptyline has also been studied in a small trial in which amitriptyline was given the first day after a stroke and titrated to 75 mg (Lampl et al 2002). No effect was found, but the study was also likely to be underpowered.
Side effects are related to anticholinergic actions (e.g., dry mouth, nausea, constipation, and orthostatic hypotension), cardiovascular effects (changes in conduction), lowering of the seizure threshold, and sedation. Many of these adverse effects may be of particular concern in patients with central pain since motor impairment, epilepsy, urinary retention, cardiovascular disease, cognitive disturbances, and other conditions are common in patients with spinal cord or brain lesions. SNRIs and SSRIs are generally better tolerated but may cause gastrointestinal side effects, sedation, and sexual dysfunction.
The pain-relieving effect of TCAs has been shown to be independent of their antidepressant effects (Sindrup et al 2005). Consistent with this finding, Leijon and Boivie (1989) found effects of amitriptyline on CPSP in patients with low depression scores and found similar depression scores in both the amitriptyline and placebo groups, whereas Rintala and associates (2007) found that amitriptyline was most effective in participants with many depressive symptoms at baseline. Because of this dual effect, antidepressants may be the first drug of choice in patients with co-existing depression.
α2δ Binding Agents
Gabapentin and pregabalin also have a well-established effect on various neuropathic pain conditions and are considered first-line drugs for neuropathic pain (Attal et al 2010, Dworkin et al 2010). These drugs are structurally related and act through presynaptic binding to the α2δ subunit of voltage-gated calcium channels.
In central pain, two small studies failed to demonstrate a statistically significant effect of gabapentin on SCI (Tai et al 2002, Rintala et al 2007), whereas another found reduced intensity and frequency of pain and improved quality-of-life measures with the administration of gabapentin, up to 3600 mg (Levendoglu et al 2004).
Pregabalin is approved by the European Medicines Evaluation Agency (EMEA) for the treatment of central neuropathic pain in adults (EMEA 2011). Pregabalin in doses of up to 600 mg has been shown to relieve SCI pain and CPSP (Siddall et al 2006, Vranken et al 2008). The improvement in pain score from baseline (pregabalin–placebo) in the two studies was 1.53 (0.92–2.15) and 2.18 (0.57–3.80), similar to values observed in studies in peripheral neuropathic pain. The NNT for 30% pain relief was 3.9 (2.5–9.1) and 4.0 (2.0–328). In addition to pain relief, amelioration of sleep problems and anxiety occurred. In a large trial involving CPSP, no effect of pregabalin was seen on the primary efficacy measure, but significant improvements were noted in sleep, anxiety, and clinician global impression of change (Kim et al 2011).
The most common side effects of gabapentin and pregabalin are nervous system related and consist of dizziness, somnolence, and ataxia. These side effects may be particularly problematic in patients with concomitant medications such as spasmolytics (Siddall et al 2006). Other side effects include peripheral edema, visual disturbances, and weight gain. The drugs are excreted mainly unchanged in urine, and dose reduction is needed in patients with impaired renal function (i.e., creatinine clearance <60 mL/min).
Pregabalin may be the first choice of drug in patients with co-existing anxiety because of its anxiolytic effects, but pregabalin also has analgesic effects in patients without clinically relevant levels of anxiety and depression (Siddall et al 2006). Gabapentin has some effect on spasticity (Mueller et al 1997) and may therefore be considered in patients with concomitant spasticity.
Opioids and Cannabinoids
Opioids, including tramadol, are considered second/third-line drugs for neuropathic pain (Attal et al 2010, Dworkin et al 2010). Cannabinoids have also been shown to have an effect on neuropathic pain.
Few studies have examined the efficacy of opioids for central pain. One study found that patients with CPSP are less likely than those with other neuropathic pain conditions to respond to opioids (Rowbotham et al 2003). Tramadol with its combined effect on μ-opioid receptors and monoamine transporters had a pain-relieving effect in patients with SCI and also a positive effect on anxiety, global life satisfaction, and sleep quality (Norrbrink and Lundeberg 2009). The study illustrated that tramadol was not well tolerated and the dropout rate was high because of adverse effects. A high initial dose and concomitant medication were thought to cause the high withdrawal rate (Norrbrink and Lundeberg 2009). The study does, however, support the use of tramadol (or other opioids) for central pain with the same indications as for peripheral neuropathic pain (i.e., for refractory pain, for intermittent pain, or as rescue medication) (Dworkin et al 2010).
Orally administered synthetic Δ9-tetrahydrocannabinol (dronabinol, up to 10 mg daily) and whole-plant cannabis-based oromucosal spray have been shown to relieve central pain in patients with MS (Svendsen et al 2004, Rog et al 2005), with relatively low NNT values of 3.4–3.7, but they failed to relieve SCI pain in a small study (Rintala et al 2010).
These drug classes have well-known side effects related to the CNS and gastrointestinal tract. Long-term side effects, as well as abuse and addiction liability, need to be addressed, and cannabinoids should probably not be administered to individuals with previous psychotic symptoms or a genetic predisposition to schizophrenia.
Other Drugs
The anticonvulsant drug lamotrigine and other sodium channel blockers have a questionable role in the treatment of neuropathic pain. Lamotrigine relieved pain and cold allodynia in stroke patients (Vestergaard et al 2001), but subsequent studies in patients with SCI and MS failed to show an effect (Finnerup et al 2002, Breuer et al 2007), although lamotrigine was suggested to relieve pain in a subgroup of patients with incomplete SCI injury and evoked pain (Finnerup et al 2002). Carbamazepine (Leijon and Boivie 1989, Osterberg and Boivie 2005), levetiracetam (Finnerup et al 2009), valproate (Drewes et al 1994), and mexiletine (Chiou-Tan et al 1996) did not relieve central pain in randomized controlled trials.
If oral drug treatment fails, spinal administration may be considered. Clonidine in combination with morphine has been shown to relieve SCI pain, particularly if the drugs are able to reach the rostral end of the lesion, but information on long-term side effects and tolerance is scarce (Siddall et al 2000). Ziconotide is a non-opioid intrathecal analgesic recommended for moderate to severe chronic pain that may be combined with morphine or baclofen, but ziconotide treatment may be associated with severe side effects, and experience in patients with central pain is limited (Wallace et al 2010). Intravenous treatment with lidocaine and ketamine also relieves central pain, but these treatments are not useful because of lack of long-term efficacy (Eide et al 1995, Attal et al 2000, Finnerup et al 2005, Amr 2010), and iontophoretic administration of S(+)-ketamine was not more effective than placebo in patients with central pain (Vranken et al 2005).
Non-pharmacological Treatment
As with other chronic pain conditions, treatment often needs a multidisciplinary approach. Concomitant anxiety, depression, and psychological distress should be evaluated and treated. Explanation of the underlying mechanisms of neuropathic pain is important. Multidimensional pain management programs consisting of educational, cognitive, and behavioral interventions (Norrbrink et al 2006) and hypnosis (Jensen et al 2008) may be useful. Physical therapy, though not directly effective in relieving central pain, may be useful to alleviate spasticity, overuse, or other effects of the neurological disease. Visual illusion, in which patients are placed in front of a screen aligned with an upper body mirror and a film of a lower body walking, has been shown to decrease pain in patients with SCI in double-blind trials (Moseley 2007, Soler et al 2010), but other studies have found increased pain following movement imagery in patients with complete SCI (Gustin et al 2008); more information is needed on the methodology, long-term efficacy, and predictors of response.
The effect of deep brain stimulation in patients with CPSP is still unclear. Motor cortex stimulation and transcranial magnetic stimulation are options for the treatment of neuropathic pain, but further information is needed on the safety, long-term effects, and predictors of response. According to published guidelines there is level C evidence (evidence obtained from retrospective cohort or case–control series) of the efficacy of motor cortex stimulation in patients with CPSP (Cruccu et al 2007). Transcranial magnetic stimulation is a technique in which analgesia is obtained by non-invasive cortical stimulation. There is level B evidence (evidence from case series only) that this technique provides slight pain relief in patients with CPSP (Cruccu et al 2007).
Summary and General Treatment Recommendations
Before choosing a neuropathic pain treatment, clinicians need to consider the benefits and hazards of a specific treatment. In general, the simpler and less harmful treatments should be chosen. The few and generally small randomized controlled trials of central pain support the finding that the non-topically applied drugs that are recommended as first- or second-line drugs for neuropathic pain are also useful for central pain. Based on results from randomized controlled clinical trials on efficacy (Fig. 69-6) and recent published guidelines, the recommendations for central pain are as follows: pregabalin (150–600 mg/day), gabapentin (1200–3600 mg/day), and TCAs (25–150 mg/day) are first-line treatments of central pain (Attal et al 2010). Tramadol (200–400 mg/day) is second-line and opioids third-line treatment. Lamotrigine may be used for CPSP or SCI with incomplete lesions and brush-evoked allodynia and cannabinoids for MS if all other treatments fail (Attal et al 2010). Clinicians should be aware of contraindications. This is particularly important in the elderly, in whom cardiac conduction abnormalities, congestive heart failure, and convulsive disorders are common and represent contraindications to treatment with TCAs. For the anticonvulsants, contraindications are fewer, but side effects such as sedation, dizziness, tremor, and rash are not uncommon, and again, for compounds with sodium-blocking properties, cardiac safety is important. Other types of side effects may also occur in patients with CNS lesions/diseases, such as dizziness and bowel and bladder problems with the use of TCAs, and TCAs may also increase spasticity. The sedative and dizziness-provoking effects of anticonvulsants such as gabapentin and pregabalin may be a clear limiting factor for weak and easily fatigued stroke patients already suffering from paresis or ataxia.
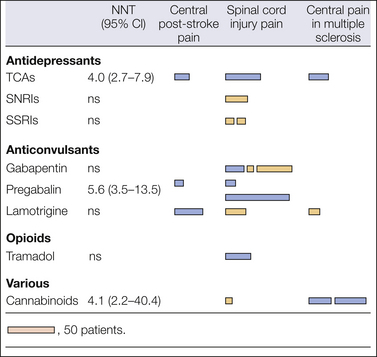
Figure 69-6 Randomized double-blind, placebo-controlled trials for central pain with combined numbers needed to treat (NNT) for 50% pain relief.
The figure indicates positive (dark bars) and negative (light bars) trials. The length of the bar illustrates the number of patients who had been allocated to active treatment in the trials. CI, confidence interval; ns, not significant; SNRIs, serotonin–noradrenaline reuptake inhibitors; SSRIs, mixed selective serotonin reuptake inhibitors; TCAs, tricyclic antidepressants.
Conclusion
Pain following SCI or stroke may significantly reduce the quality of life of patients who already suffer from multiple handicaps because of their CNS disease/lesion. Better understanding of the underlying mechanisms is needed to find better treatments for these groups of patients.
Acknowledgments
Dr. Henriette Klit is thanked for her contribution to this paper. This work was supported by a grant from the Velux Foundation. Ms. Helle O. Andersen is thanked for secretarial assistance.
The references for this chapter can be found at www.expertconsult.com.
References
Amr Y.M. Multi-day low dose ketamine infusion as adjuvant to oral gabapentin in spinal cord injury related chronic pain: a prospective, randomized, double blind trial. Pain Physician. 2010;13:245–249.
Andersen G., Vestergaard K., Ingeman-Nielsen M., et al. Incidence of central post-stroke pain. Pain. 1995;61:187–193.
Attal N., Bouhassira D. Pain in syringomyelia/bulbia. In: Cervero F., Jensen T.S., eds. Handbook of Clinical Neurology: PAIN, vol 81. Edinburgh: Elsevier; 2006:705–713.
Attal N., Cruccu G., Baron R., et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2009 revision. European Journal of Neurology. 17, 2010. 1113–e88
Attal N., Gaude V., Brasseur L., et al. Intravenous lidocaine in central pain: a double-blind, placebo-controlled, psychophysical study. Neurology. 2000;54:564–574.
Baron R., Binder A., Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurology. 2010;9:807–819.
Bennett M.I., Attal N., Backonja M.M., et al. Using screening tools to identify neuropathic pain. Pain. 2007;12:199–203.
Bogousslavsky J., Regli F., Uske A. Thalamic infarcts: clinical syndromes, etiology, and prognosis. Neurology. 1988;38:837–848.
Boivie J. Central post-stroke pain. In: Cervero F., Jensen T.S., eds. Handbook of Clinical Neurology: PAIN, vol 81. Edinburgh: Elsevier; 2006:715–730.
Boivie J. Central pain. In: McMahon S.B., Koltzenburg M., eds. Textbook of pain. London: Elsevier; 2006:1057–1074.
Boivie J., Leijon G., Johansson I. Central post-stroke pain—a study of the mechanisms through analysis of the sensory abnormalities. Pain. 1989;37:173–185.
Bouhassira D., Attal N. Diagnosis and assessment of neuropathic pain: the saga of clinical tools. Pain. 2011;152(Suppl 3):S74–S83.
Bowsher D. Central pain: clinical and physiological characteristics. Journal of Neurology, Neurosurgery, and Psychiatry. 1996;61:62–69.
Bowsher D. Stroke and central poststroke pain in an elderly population. Journal of Pain. 2001;2:258–261.
Breuer B., Pappagallo M., Knotkova H., et al. A randomized, double-blind, placebo-controlled, two-period, crossover, pilot trial of lamotrigine in patients with central pain due to multiple sclerosis. Clinical Therapeutics. 2007;29:2022–2030.
Brickell K.L., Anderson N.E., Charleston A.J., et al. Ethnic differences in syringomyelia in New Zealand. Journal of Neurology, Neurosurgery, and Psychiatry. 2006;77:989–991.
Cardenas D.D., Warms C.A., Turner J.A., et al. Efficacy of amitriptyline for relief of pain in spinal cord injury: results of a randomized controlled trial. Pain. 2002;96:365–373.
Carlton S.M., Du J., Tan H.Y., et al. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. 2009;147:265–276.
Casey K.L., Beydoun A., Boivie J., et al. Laser-evoked cerebral potentials and sensory function in patients with central pain. Pain. 1996;64:485–491.
Chiou-Tan F.Y., Tuel S.M., Johnson J.C., et al. Effect of mexiletine on spinal cord injury dysesthetic pain. American Journal of Physical Medicine & Rehabilitation. 1996;75:84–87.
Costigan M., Scholz J., Woolf C.J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual Review of Neuroscience. 2009;32:1–32.
Craig A.D. A new version of the thalamic disinhibition hypothesis of central pain. Pain Forum. 1998;7:1–14.
Craig A.D. How do you feel? Interoception: the sense of the physiological condition of the body. Nature Reviews. Neuroscience. 2002;3:655–666.
Craig A.D. A new view of pain as a homeostatic emotion. Trends in Neurosciences. 2003;26:303–307.
Cruccu G., Aziz T.Z., Garcia-Larrea L., et al. EFNS guidelines on neurostimulation therapy for neuropathic pain. European Journal of Neurology. 2007;14:952–970.
Cruccu G., Biasiotta A., Di Rezze S., et al. Trigeminal neuralgia and pain related to multiple sclerosis. Pain. 2009;143:186–191.
Cruccu G., Sommer C., Anand P., et al. EFNS guidelines on neuropathic pain assessment: revised 2009. European Journal of Neurology. 2010;17:1010–1018.
Davidoff G., Guarracini M., Roth E., et al. Trazodone hydrochloride in the treatment of dysesthetic pain in traumatic myelopathy: a randomized, double-blind, placebo-controlled study. Pain. 1987;29:151–161.
Déjerine J., Roussy G. La syndrome thalamique. Revue Neurologique. 1906;14:521–532.
Djaldetti R., Yust-Katz S., Kolianov V., et al. The effect of duloxetine on primary pain symptoms in Parkinson disease. Clinical Neuropharmacology. 2007;30:201–205.
Dostrovsky J.O. Brainstem and thalamic relays. In: Cervero F., Jensen T.S., eds. Handbook of Clinical Neurology: PAIN, vol 81. Edinburgh: Elsevier; 2006:127–139.
Dostrovsky J.O., Craig A.D. Ascending projection systems. In: McMahon S.B., Koltzenburg M., eds. Textbook of pain. London: Elsevier; 2006:187–203.
Drewes A.M., Andreasen A., Poulsen L.H. Valproate for treatment of chronic central pain after spinal cord injury. A double-blind cross-over study. Paraplegia. 1994;32:565–569.
Ducreux D., Attal N., Parker F., et al. Mechanisms of central neuropathic pain: a combined psychophysical and fMRI study in syringomyelia. Brain. 2006;129:963–976.
Dworkin R.H., O’Connor A.B., Audette J., et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clinic Proceedings. 2010;85:S3–S14.
Edinger L. Gibt es zentral entstehende Schmerzen? Deutsche Zeittschrift für Nervenheilkunde. 1891;1:262–282.
Eide P.K., Stubhaug A., Stenehjem A.E. Central dysesthesia pain after traumatic spinal cord injury is dependent on N-methyl-D-aspartate receptor activation. Neurosurgery. 1995;37:1080–1087.
EMEA 2011 Lyrica—scientific discussion.
Ertas M., Sagduyu A., Arac N., et al. Use of levodopa to relieve pain from painful symmetrical diabetic polyneuropathy. Pain. 1998;75:257–259.
Finnerup N.B. A review of central neuropathic pain states. Current Opinion in Anaesthesiology. 2008;21:586–589.
Finnerup N.B., Biering-Sorensen F., Johannesen I.L., et al. Intravenous lidocaine relieves spinal cord injury pain: a randomized controlled trial. Anesthesiology. 2005;102:1023–1030.
Finnerup N.B., Grydehoj J., Bing J., et al. Levetiracetam in spinal cord injury pain: a randomized controlled trial. Spinal Cord. 2009;47:861–867.
Finnerup N.B., Jensen T.S. Spinal cord injury pain—mechanisms and treatment. European Journal of Neurology. 2004;11:73–82.
Finnerup N.B., Jensen T.S. Mechanism-based classification of neuropathic pain: a critical analysis. Nature Clinical Practice. Neurology. 2006;2:107–115.
Finnerup N.B., Johannesen I.L., Fuglsang-Frederiksen A., et al. Sensory function in spinal cord injury patients with and without central pain. Brain. 2003;126:57–70.
Finnerup N.B., Sindrup S.H., Bach F.W., et al. Lamotrigine in spinal cord injury pain: a randomized controlled trial. Pain. 2002;96:375–383.
Fitzek S., Baumgartner U., Fitzek C., et al. Mechanisms and predictors of chronic facial pain in lateral medullary infarction. Annals of Neurology. 2001;49:493–500.
Garcia-Larrea L., Convers P., Magnin M., et al. Laser-evoked potential abnormalities in central pain patients: the influence of spontaneous and provoked pain. Brain. 2002;125:2766–2781.
Garcia-Larrea L., Maarrawi J., Peyron R., et al. On the relation between sensory deafferentation, pain and thalamic activity in Wallenberg’s syndrome: a PET-scan study before and after motor cortex stimulation. European Journal of Pain. 2006;10:677–688.
Garcia-Larrea L., Perchet C., Creac’h C., et al. Operculo-insular pain (parasylvian pain): a distinct central pain syndrome. Brain. 2010;133:2528–2539.
Garcin R. La douleur dans les affections organiques du systeme nerveux central. Revue Neurologique. 1937;68:105–153.
Gates P., Nayernouri T., Sengupta R.P. Epileptic pain: a temporal lobe focus. Journal of Neurology, Neurosurgery, and Psychiatry. 1984;47:319–320.
Gerdelat-Mas A., Simonetta-Moreau M., Thalamas C., et al. Levodopa raises objective pain threshold in Parkinson’s disease: a RIII reflex study. Journal of Neurology, Neurosurgery, and Psychiatry. 2007;78:1140–1142.
Greenspan J.D., Ohara S., Sarlani E., et al. Allodynia in patients with post-stroke central pain (CPSP) studied by statistical quantitative sensory testing within individuals. Pain. 2004;109:357–366.
Greiff F. Zur Localization der Hemichorea. Archiv für Psychologie und Nervekrankenheiten. 1883;14:598–624.
Gudmundsson K.R. The prevalence of some neurological diseases in Iceland. Acta Neurologica Scandinavica. 1968;44:57–69.
Gustin S.M., Wrigley P.J., Gandevia S.C., et al. Movement imagery increases pain in people with neuropathic pain following complete thoracic spinal cord injury. Pain. 2008;137:237–244.
Hains B.C., Klein J.P., Saab C.Y., et al. Upregulation of sodium channel NaV 1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. Journal of Neuroscience. 2003;2:8881–8892.
Hains B.C., Saab C.Y., Waxman S.G. Changes in electrophysiological properties and sodium channel NaV 1.3 expression in thalamic neurons after spinal cord injury. Brain. 2005;128:2359–2371.
Head H., Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–254.
Jensen M.P., Barber J., Hanley M.A., et al. Long-term outcome of hypnotic-analgesia treatment for chronic pain in persons with disabilities. International Journal of Clinical and Experimental Hypnosis. 2008;56:156–169.
Jensen T.S., Baron R. Translation of symptoms and signs into mechanisms in neuropathic pain. Pain. 2003;102:1–8.
Jensen T.S., Gottrup H., Bach F.W., et al. The clinical picture of neuropathic pain. European Journal of Pharmacology. 2001;429:1–11.
Jensen T.S., Lenz F.A. Central post-stroke pain: a challenge for the scientist and the clinician. Pain. 1995;61:161–164.
Kim J.H., Greenspan J.D., Coghill R.C., et al. Lesions limited to the human thalamic principal somatosensory nucleus (ventral caudal) are associated with loss of cold sensations and central pain. Journal of Neuroscience. 2007;27:4995–5004.
Kim J.S., Bashford G., Murphy T.K., et al. Safety and efficacy of pregabalin in patients with central post-stroke pain. Pain. 2011;152:1018–1023.
Kim J.S., Choi-Kwon S. Sensory sequelae of medullary infarction: differences between lateral and medial medullary syndrome. Stroke. 1999;32:2697–2703.
Klit H., Andersen G., Jensen T.S., et al. Central poststroke pain: a population-based study. Pain. 152, 2011. 828–824
Klit H., Finnerup N.B., Jensen T.S. Central post stroke pain: clinical characteristics, pathophysiology, and management. Lancet Neurology. 2009;8:857–868.
Klit H., Finnerup N.B., Overvad K., et al. Pain following stroke: a population-based follow-up study. PLoS One. 2011;6:e27607.
Koltzenburg M., Scadding J. Neuropathic pain. Current Opinion in Neurology. 2001;14:641–647.
Kumar G., Soni C.R. Central post-stroke pain: current evidence. Journal of Neurological Sciences. 2009;284:10–17.
Kurland L.T. Descriptive epidemiology of selected neurologic and myopathic disorders with a particular reference to a survey in Rochester, Minnesota. Journal of Chronic Diseases. 1958;8:378–415.
Lampl C., Yazdi K., Roper C. Amitriptyline in the prophylaxis of central poststroke pain. Preliminary results of 39 patients in a placebo-controlled, long-term study. Stroke. 2002;33:3030–3032.
Leijon G., Boivie J. Central post-stroke pain—a controlled trial of amitriptyline and carbamazepine. Pain. 1989;36:27–36.
Leijon G., Boivie J., Johansson I. Central post-stroke pain: neurological symptoms and pain characteristics. Pain. 1989;36:13–25.
Lenz F.A., Gracely R.H., Baker F.H., et al. Reorganization of sensory modalities evoked by microstimulation in region of the thalamic principal sensory nucleus in patients with pain due to nervous system injury. Journal of Comparative Neurology. 1998;399:125–138.
Lenz F.A., Gracely R.H., Rowland L.H., et al. A population of cells in the human thalamic principal sensory nucleus respond to painful mechanical stimuli. Neuroscience Letters. 1994;180:46–50.
Lenz F.A., Kwan H.C., Dostrovsky J.O., et al. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Research. 1989;496:357–360.
Lenz F.A., Weiss N., Ohara S., et al. The role of the thalamus in pain. Supplements to Clinical Neurophysiology. 2004;57:50–61.
Levendoglu F., Ogun C.O., Ozerbil O., et al. Gabapentin is a first line drug for the treatment of neuropathic pain in spinal cord injury. Spine. 2004;29:743–751.
MacGowan D.J.L., Janal M.N., Clark W.C., et al. Central post stroke pain and Wallenberg’s lateral medullary infarction: frequency, character, and determinants in 63 patients. Neurology. 1997;49:120–125.
Merskey H., Bogduk N. Classification of chronic pain: descriptions of chronic pain syndromes and definitions of pain terms. Seattle: IASP Press; 1994.
Moseley G.L. Using visual illusion to reduce at-level neuropathic pain in paraplegia. Pain. 2007;130:294–298.
Moulin D.E., Foley K.M., Ebers G.C. Pain syndromes in multiple sclerosis. Neurology. 1988;38:1830–1834.
Mueller M.E., Gruenthal M., Olson W.L., et al. Gabapentin for relief of upper motor neuron symptoms in multiple sclerosis. Archives of Physical Medicine and Rehabilitation. 1997;78:521–524.
Nathan P.W. Reference of sensation at the spinal level. Journal of Neurology, Neurosurgery, and Psychiatry. 1956;19:88–100.
Nathan P.W., Smith M.C. Clinico-anatomical correlation in anterolateral cordotomy. In: Bonica J., ed. Advances in pain research and therapy. New York: Raven Press; 1979:921–926.
Norrbrink B.C., Kowalski J., Lundeberg T. A comprehensive pain management programme comprising educational, cognitive and behavioural interventions for neuropathic pain following spinal cord injury. Journal of Rehabilitation Medicine. 2006;38:172–180.
Norrbrink C., Lundeberg T. Tramadol in neuropathic pain after spinal cord injury: a randomized, double-blind, placebo-controlled trial. Clinical Journal of Pain. 2009;25:177–184.
Nurmikko T.J., Gupta S., Maclver K. Multiple sclerosis–related central pain disorders. Current Pain and Headache Reports. 2010;14:189–195.
O’Connor A.B., Schwid S.R., Herrmann D.N., et al. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. 2008;137:96–111.
Ofek H., Defrin R. The characteristics of chronic central pain after traumatic brain injury. Pain. 2007;131:330–340.
Osterberg A., Boivie J. Central pain in multiple sclerosis—a double-blind placebo-controlled trial of amitriptyline and carbamazepine. In: Österberg A., ed. Central pain in multiple sclerosis—clinical characteristics, sensory abnormalities, and treatment. Linköpings Universitet, thesis, 2005.
Osterberg A., Boivie J. Central pain in multiple sclerosis—sensory abnormalities. European Journal of Pain. 2010;14:104–110.
Osterberg A., Boivie J., Thuomas K.A. Central pain in multiple sclerosis—prevalence and clinical characteristics. European Journal of Pain. 2005;9:531–542.
Pagni C.A. Central pain due to spinal cord and brain damage. In: Wall P.D., Melzack R., eds. Textbook of pain. ed 2. Edinburgh: Churchill Livingstone; 1989:634–655.
Pertovaara A., Wei H. Dual influence of the striatum on neuropathic hypersensitivity. Pain. 2008;137:50–59.
Peyron R., Garcia-Larrea L., Gregoire M.C., et al. Allodynia after lateral-medullary (Wallenberg) infarct. A PET study. Brain. 1998;121:345–356.
Peyron R., Garcia-Larrea L., Gregoire M.C., et al. Parietal and cingulate processes in central pain. A combined positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) study of an unusual case. Pain. 2000;84:77–87.
Ralston H.J., III. Pain and the primate thalamus. Progress in Brain Research. 2005;149:1–10.
Riddoch G. The clinical features of central pain I. Lancet. 1938;231:1093–1098.
Riddoch G. The clinical features of central pain II. Lancet. 1938;231:1150–1156.
Riddoch G. The clinical features of central pain III. Lancet. 1938;231:1205–1209.
Rinaldi P.C., Young R.F., Albe-Fessard D., et al. Spontaneous neuronal hyperactivity in the medial and intralaminar thalamic nuclei of patients with deafferentation pain. Journal of Neurosurgery. 1991;74:415–421.
Rintala D.H., Fiess R.N., Tan G., et al. Effect of dronabinol on central neuropathic pain after spinal cord injury: a pilot study. American Journal of Physical Medicine & Rehabilitation. 2010;89:840–848.
Rintala D.H., Holmes S.A., Courtade D., et al. Comparison of the effectiveness of amitriptyline and gabapentin on chronic neuropathic pain in persons with spinal cord injury. Archives of Physical Medicine and Rehabilitation. 2007;88:1547–1560.
Rog D.J., Nurmikko T.J., Friede T., et al. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology. 2005;65:812–819.
Rowbotham M.C., Twilling L., Davies P.S., et al. Oral opioid therapy for chronic peripheral and central neuropathic pain. New England Journal of Medicine. 2003;348:1223–1232.
Schestatsky P., Kumru H., Valls-Sole J., et al. Neurophysiologic study of central pain in patients with Parkinson disease. Neurology. 2007;69:2162–2169.
Sherrington C.S. The integrative action of the nervous system. Cambridge: Cambridge University Press; 1948.
Siddall P.J., Cousins M.J., Otte A., et al. Pregabalin in central neuropathic pain associated with spinal cord injury: a placebo-controlled trial. Neurology. 2006;67:1792–1800.
Siddall P.J., McClelland J.M., Rutkowski S.B., et al. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257.
Siddall P.J., Molloy A.R., Walker S., et al. The efficacy of intrathecal morphine and clonidine in the treatment of pain after spinal cord injury. Anesthesia and Analgesia. 2000;91:1–6.
Sindrup S.H., Otto M., Finnerup N.B., et al. Antidepressants in the treatment of neuropathic pain. Basic & Clinical Pharmacology & Toxicology. 2005;96:399–409.
Solaro C., Brichetto G., Amato M.P., et al. The prevalence of pain in multiple sclerosis: a multicenter, cross-sectional study. Neurology. 2004;63:919–921.
Soler M.D., Kumru H., Pelayo R., et al. Effectiveness of transcranial direct current stimulation and visual illusion on neuropathic pain in spinal cord injury. Brain. 2010;133:2565–2577.
Svendsen K., Jensen T.S., Overvad K., et al. Pain in patients with multiple sclerosis. A population based study. Archives of Neurology. 2003;60:1089–1094.
Svendsen K.B., Jensen T.S., Bach F.W. Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. British Medical Journal. 2004;329:253–261.
Svendsen K.B., Jensen T.S., Hansen H.J., et al. Sensory function and quality of life in patients with multiple sclerosis and pain. Pain. 2005;114:473–481.
Svendsen K.B., Sørensen L., Jensen T.S., et al. MRI of the central nervous system in MS patients with and without pain. European Journal of Pain. 2011;15:395–401.
Tai Q., Kirshblum S., Chen B., et al. Gabapentin in the treatment of neuropathic pain after spinal cord injury: a prospective, randomized, double-blind, crossover trial. Journal of Spinal Cord Medicine. 2002;25:100–105.
Tasker R.R., de Carvalho G., Dostrovsky J.O. The history of central pain syndromes, with observations concerning pathophysiology and treatment. In: Casey K.L., ed. Pain and central nervous system disease: the central pain syndromes. New York: Raven Press; 1991:31–58.
Treede R.D., Jensen T.S., Campbell J.N., et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–1635.
Vestergaard K., Andersen G., Gottrup H., et al. Lamotrigine for central post-stroke pain: a randomised controlled trial. Neurology. 2001;56:184–190.
Vestergaard K., Andersen G., Jensen T.S. Treatment of central post-stroke pain with a selective serotonin reuptake inhibitor. European Journal of Neurology. 1996;3(Suppl 5):169.
Vestergaard K., Nielsen J., Andersen G., et al. Sensory abnormalities in consecutive unselected patients with central post-stroke pain. Pain. 1995;61:177–186.
Vranken J.H., Dijkgraaf M.G., Kruis M.R., et al. Iontophoretic administration of S(+)-ketamine in patients with intractable central pain: a placebo-controlled trial. Pain. 2005;118:224–231.
Vranken J.H., Dijkgraaf M.G., Kruis M.R., et al. Pregabalin in patients with central neuropathic pain: a randomized, double-blind, placebo-controlled trial of a flexible-dose regimen. Pain. 2008;136:150–157.
Vranken J.H., Hollmann M.W., van der Vegt M.H., et al. Duloxetine in patients with central neuropathic pain caused by spinal cord injury or stroke: a randomized, double-blind, placebo-controlled trial. Pain. 2011;152:267–273.
Wallace M.S., Rauck R.L., Deer T. Ziconotide combination intrathecal therapy: rationale and evidence. Clinical Journal of Pain. 2010;26:635–644.
Wang G., Thompson S.M. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. Journal of Neuroscience. 2008;28:11959–11969.
Wasner G., Deuschl G. Pain in Parkinson’s disease. In: Cervero F., Jensen T.S., eds. Handbook of Clinical Neurology: PAIN, vol 81. Edinburgh: Elsevier; 2006:747–760.
White J.C., Sweet W.H. Pain and the neurosurgeon. A forty year experience. Springfield, IL: Charles C Thomas; 1969.
Widar M., Ahlstrom G. Disability after a stroke and the influence of long-term pain on everyday life. Scandinavian Journal of Caring Sciences. 2002;16:302–310.
Willis W.D., Westlund K.N. Neuroanatomy of the pain system and of the pathways that modulate pain. Journal of Clinical Neurophysiology. 1997;14:2–31.
Woolf C.J. Pain: moving from symptom control toward mechanism-specific pharmacologic treatment. Annals of Internal Medicine. 2004;140:441–451.
Young G.B., Blume W.T. Pain epileptic seizures. Brain. 1983;106:537–554.
Andersen G., Vestergaard K., Ingeman-Nielsen M., et al. Incidence of central post-stroke pain. Pain. 1995;61:187–193.
Attal N., Cruccu G., Baron R., et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2009 revision. European Journal of Neurology. 17, 2010. 1113–e88
Baron R., Binder A., Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurology. 2010;9:807–819.
Bogousslavsky J., Regli F., Uske A. Thalamic infarcts: clinical syndromes, etiology, and prognosis. Neurology. 1988;38:837–848.
Boivie J., Leijon G., Johansson I. Central post-stroke pain—a study of the mechanisms through analysis of the sensory abnormalities. Pain. 1989;37:173–185.
Bowsher D. Central pain: clinical and physiological characteristics. Journal of Neurology, Neurosurgery, and Psychiatry. 1996;61:62–69.
Casey K.L., Beydoun A., Boivie J., et al. Laser-evoked cerebral potentials and sensory function in patients with central pain. Pain. 1996;64:485–491.
Costigan M., Scholz J., Woolf C.J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual Review of Neuroscience. 2009;32:1–32.
Craig A.D. How do you feel? Interoception: the sense of the physiological condition of the body. Nature Reviews. Neuroscience. 2002;3:655–666.
Cruccu G., Biasiotta A., Di Rezze S., et al. Trigeminal neuralgia and pain related to multiple sclerosis. Pain. 2009;143:186–191.
Cruccu G., Sommer C., Anand P., et al. EFNS guidelines on neuropathic pain assessment: revised 2009. European Journal of Neurology. 2010;17:1010–1018.
Déjerine J., Roussy G. La syndrome thalamique. Revue Neurologique. 1906;14:521–532.
Dostrovsky J.O. Brainstem and thalamic relays. In: Cervero F., Jensen T.S., eds. Handbook of Clinical Neurology: PAIN, vol 81. Edinburgh: Elsevier; 2006:127–139.
Ducreux D., Attal N., Parker F., et al. Mechanisms of central neuropathic pain: a combined psychophysical and fMRI study in syringomyelia. Brain. 2006;129:963–976.
Finnerup N.B. A review of central neuropathic pain states. Current Opinion in Anaesthesiology. 2008;21:586–589.
Finnerup N.B., Jensen T.S. Spinal cord injury pain—mechanisms and treatment. European Journal of Neurology. 2004;11:73–82.
Finnerup N.B., Johannesen I.L., Fuglsang-Frederiksen A., et al. Sensory function in spinal cord injury patients with and without central pain. Brain. 2003;126:57–70.
Garcin R. La douleur dans les affections organiques du systeme nerveux central. Revue Neurologique. 1937;68:105–153.
Greenspan J.D., Ohara S., Sarlani E., et al. Allodynia in patients with post-stroke central pain (CPSP) studied by statistical quantitative sensory testing within individuals. Pain. 2004;109:357–366.
Hains B.C., Saab C.Y., Waxman S.G. Changes in electrophysiological properties and sodium channel NaV 1.3 expression in thalamic neurons after spinal cord injury. Brain. 2005;128:2359–2371.
Head H., Holmes G. Sensory disturbances from cerebral lesions. Brain. 1911;34:102–254.
Jensen T.S., Baron R. Translation of symptoms and signs into mechanisms in neuropathic pain. Pain. 2003;102:1–8.
Jensen T.S., Lenz F.A. Central post-stroke pain: a challenge for the scientist and the clinician. Pain. 1995;61:161–164.
Kim J.H., Greenspan J.D., Coghill R.C., et al. Lesions limited to the human thalamic principal somatosensory nucleus (ventral caudal) are associated with loss of cold sensations and central pain. Journal of Neuroscience. 2007;27:4995–5004.
Kim J.S., Choi-Kwon S. Sensory sequelae of medullary infarction: differences between lateral and medial medullary syndrome. Stroke. 1999;32:2697–2703.
Klit H., Andersen G., Jensen T.S., et al. Central poststroke pain: a population-based study. Pain. 152, 2011. 828–824
Klit H., Finnerup N.B., Jensen T.S. Central post stroke pain: clinical characteristics, pathophysiology, and management. Lancet Neurology. 2009;8:857–868.
Klit H., Finnerup N.B., Overvad K., et al. Pain following stroke: a population-based follow-up study. PLoS One. 2011;6:e27607.
Koltzenburg M., Scadding J. Neuropathic pain. Current Opinion in Neurology. 2001;14:641–647.
Kumar G., Soni C.R. Central post-stroke pain: current evidence. Journal of Neurological Sciences. 2009;284:10–17.
Leijon G., Boivie J. Central post-stroke pain—a controlled trial of amitriptyline and carbamazepine. Pain. 1989;36:27–36.
Lenz F.A., Weiss N., Ohara S., et al. The role of the thalamus in pain. Supplements to Clinical Neurophysiology. 2004;57:50–61.
MacGowan D.J.L., Janal M.N., Clark W.C., et al. Central post stroke pain and Wallenberg’s lateral medullary infarction: frequency, character, and determinants in 63 patients. Neurology. 1997;49:120–125.
Nathan P.W. Reference of sensation at the spinal level. Journal of Neurology, Neurosurgery, and Psychiatry. 1956;19:88–100.
Nurmikko T.J., Gupta S., MacIver K. Multiple sclerosis–related central pain disorders. Current Pain and Headache Reports. 2010;14:189–195.
Osterberg A., Boivie J. Central pain in multiple sclerosis—sensory abnormalities. European Journal of Pain. 2010;14:104–110.
Peyron R., Garcia-Larrea L., Gregoire M.C., et al. Allodynia after lateral-medullary (Wallenberg) infarct. A PET study. Brain. 1998;121:345–356.
Ralston H.J., III. Pain and the primate thalamus. Progress in Brain Research. 2005;149:1–10.
Rog D.J., Nurmikko T.J., Friede T., et al. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology. 2005;65:812–819.
Rowbotham M.C., Twilling L., Davies P.S., et al. Oral opioid therapy for chronic peripheral and central neuropathic pain. New England Journal of Medicine. 2003;348:1223–1232.
Sherrington C.S. The integrative action of the nervous system. Cambridge: Cambridge University Press; 1948.
Siddall P.J., Cousins M.J., Otte A., et al. Pregabalin in central neuropathic pain associated with spinal cord injury: a placebo-controlled trial. Neurology. 2006;67:1792–1800.
Svendsen K.B., Jensen T.S., Hansen H.J., et al. Sensory function and quality of life in patients with multiple sclerosis and pain. Pain. 2005;114:473–481.
Svendsen K.B., Sørensen L., Jensen T.S., et al. MRI of the central nervous system in MS patients with and without pain. European Journal of Pain. 2011;15:395–401.
Treede R.D., Jensen T.S., Campbell J.N., et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–1635.
Vestergaard K., Andersen G., Gottrup H., et al. Lamotrigine for central post-stroke pain: a randomised controlled trial. Neurology. 2001;56:184–190.
Vestergaard K., Nielsen J., Andersen G., et al. Sensory abnormalities in consecutive unselected patients with central post-stroke pain. Pain. 1995;61:177–186.
Vranken J.H., Dijkgraaf M.G., Kruis M.R., et al. Pregabalin in patients with central neuropathic pain: a randomized, double-blind, placebo-controlled trial of a flexible-dose regimen. Pain. 2008;136:150–157.
Vranken J.H., Hollmann M.W., van der Vegt M.H., et al. Duloxetine in patients with central neuropathic pain caused by spinal cord injury or stroke: a randomized, double-blind, placebo-controlled trial. Pain. 2011;152:267–273.
Wang G., Thompson S.M. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. Journal of Neuroscience. 2008;28:11959–11969.
Woolf C.J. Pain: moving from symptom control toward mechanism-specific pharmacologic treatment. Annals of Internal Medicine. 2004;140:441–451.