Chapter 7 Disorders of Phosphorus
Hypophosphatemia and Hyperphosphatemia
Phosphorus plays an essential role in cellular structure and function.91 A constituent of structural phospholipids in cell membranes and hydroxyapatite in bone, phosphorus also is an integral component of nucleic acids and phosphoproteins involved in mitochondrial oxidative phosphorylation. Energy for essential metabolic processes (e.g., muscle contraction, neuronal impulse conduction, epithelial transport) is stored in high-energy phosphate bonds of adenosine triphosphate (ATP). The compound 2,3-diphosphoglycerate (2,3-DPG) decreases the affinity of hemoglobin for oxygen and facilitates the delivery of oxygen to tissues. Cyclic adenosine monophosphate (cAMP) is an intracellular second messenger for many polypeptide hormones. Phosphate is also an important urinary buffer, and urinary phosphate constitutes the majority of titratable acidity (see Chapter 9).
Phosphorus is important in the intermediary metabolism of protein, fat, and carbohydrate and as a component of glycogen. It stimulates glycolytic enzymes (e.g., hexokinase, phosphofructokinase) and participates in the phosphorylation of many glycolytic intermediates. Nicotinamide adenine dinucleotide phosphate (NADP+) is a coenzyme for important biochemical reactions. Phosphate regulates the activity of enzymes such as the glutaminase essential for ammoniagenesis (stimulated by increased phosphate concentrations) and the 1α-hydroxylase required for vitamin D activation (stimulated by decreased phosphate concentrations).
Physical chemistry
Phosphorus exists in organic (phospholipids and phosphate esters) and inorganic (orthophosphoric and pyrophosphoric acids) forms in the body. Almost all serum phosphorus is in the form of orthophosphate. Orthophosphoric acid is governed by the following set of equilibria:
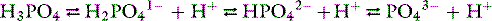
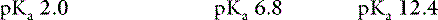
The pKa for the reaction between H2PO41− and HPO42− is 6.8 at the ionic strength and temperature of extracellular fluid (ECF), and these are the two prevailing ionic species at the normal ECF pH of 7.4. At this pH, H3PO4 and PO43− are present in negligible amounts, and plasma inorganic phosphorus principally consists of H2PO41− and HPO42−. At a pH of 7.4, the HPO42−:H2PO41− ratio is 4.0, and the average valence of phosphate in serum reflects this ratio. There are four times as many HPO42− as H2PO41− ions at a pH of 7.4, and therefore the average valence of phosphate at this pH is (4/5)(−2) + (1/5)(−1) = −1.8. Because the valence and number of milliequivalents (mEq) of phosphate in ECF are influenced by pH, it is easier to measure phosphate in millimoles (mmol) or milligrams (mg) of elemental phosphorus. Serum phosphorus concentrations typically are reported as elemental phosphorus and expressed as milligrams of elemental phosphorus per deciliter of serum. One millimole of phosphate contains 31 mg of elemental phosphorus. To convert milligrams per deciliter to millimoles per liter, divide milligrams per deciliter by 3.1. At a pH of 7.4, 1 mmol of phosphate equals 1.8 mEq, and conversion from millimoles per liter to milliequivalents per liter requires multiplication by 1.8.
Even though phosphorus circulates in organic and inorganic forms, clinical laboratories typically measure inorganic phosphate. Approximately 10% to 20% of the inorganic phosphate in serum is protein bound, and the remainder circulates as free anion or is complexed to sodium, magnesium, or calcium. The free and complexed fractions are available for ultrafiltration by the renal glomeruli.
Body stores and distribution
Phosphate is the body’s major intracellular anion, and translocation in and out of the intracellular compartment can rapidly change serum phosphorus concentration. Gradual changes in total body phosphate can be accommodated without noticeable changes in serum phosphorus concentration, resembling the situation with potassium (the major intracellular cation). Approximately 80% to 85% of total body phosphate is inorganic hydroxyapatite in bone, whereas 15% is in soft tissues such as muscle.59,88 Most soft tissue phosphorus is organic and can be readily converted to the inorganic form as needed. The ECF compartment contains less than 1% of total body phosphorus stores.
Normal serum concentrations
Normal serum phosphorus concentrations in adult dogs range from 2.5 to 6.0 mg/dL, but they are higher in dogs younger than 1 year.17,77,131,175 Serum phosphorus concentrations are highest in puppies less than 8 weeks of age (up to 10.8 mg/dL may be considered normal) and gradually decrease into the adult range after 1 year of age.73 Sex-related changes are not reported.134 The effect of age is less pronounced in cats, but immature cats have a tendency for higher serum concentrations.32 Bone growth and an increase in renal tubular reabsorption of phosphorus mediated by growth hormone presumably contribute to this age effect. Feeding also affects serum phosphorus concentration. A carbohydrate meal or infusion (e.g., 5% dextrose) decreases serum phosphorus concentration, because phosphate shifts into intracellular fluid as a result of stimulation of glycolysis and formation of phosphorylated glycolytic intermediates in muscle, liver, and adipose cells. In contrast, protein intake increases serum phosphorus concentration because of the relatively high phosphorus content of protein-rich diets.
Time of sampling affects the observed serum phosphorus concentration. People have substantial variations in serum phosphorus concentrations throughout the day.99 Acid-base balance also influences serum phosphorus concentration. Respiratory alkalosis stimulates glycolysis (by activating phosphofructokinase) and decreases serum phosphorus concentration. Thus, the measured serum phosphorus concentration is affected by several variables and does not accurately indicate total body phosphorus stores. Measuring serum phosphorus concentration after a 12-hour fast minimizes confounding factors, but the clinician must understand that the magnitude of hypophosphatemia or hyperphosphatemia may be incorrectly assessed if only one serum or plasma sample is analyzed.
Hemolysis may affect laboratory results because phosphate is present in erythrocytes. Human erythrocytes contain 8 μmol/dL red cells, whereas canine erythrocytes contain 35 μmol/dL and feline erythrocytes contain 26 μmol/dL.30 Hyperlipidemia and hyperproteinemia sometimes cause overestimation of serum phosphorus concentration, depending on the methodology used.27,68,97 This can become important when using drugs such as liposomal amphotericin B.95 Thrombocytosis and monoclonal gammopathy also may cause spurious increases in serum phosphorus concentration.92,103,108 Mannitol and other drugs may interfere with some assay systems, leading to erroneous measured values.62,179 Icterus and hemolysis were reported to result in artifactual hypophosphatemia in dogs with immune-mediated hemolytic anemia.72 Artifactual hypophosphatemia can occur in some automated systems but not in others. Thus, occurrence of hypophosphatemia in patients without known predisposing factors should prompt consideration of laboratory error.
Dietary intake
The average phosphorus content of commercial pet foods is approximately 1% on a dry matter basis. Dogs and cats need to ingest 0.5 to 3.0 g of phosphorus per day, depending on their body size and energy requirements. The source of dietary phosphorus markedly affects absorption and excretion of phosphorus in cats.56 The amount of phosphorus absorbed by the gastrointestinal tract, the amount excreted in the urine, and the extent of postprandial hyperphosphatemia were increased when monobasic and dibasic salts of phosphorus were fed but decreased when phosphorus originated from poultry, meat, and fish meal.
Intestinal absorption
Ingested organic phosphate is hydrolyzed in the gastrointestinal tract, liberating inorganic phosphate for absorption. Net intestinal phosphate absorption (i.e., the difference between dietary and fecal phosphate) is approximately 60% to 70% of the ingested load, and absorption is a linear function of phosphorus intake. In an animal in zero phosphorus balance, urinary phosphate excretion equals net intestinal phosphate absorption.
Intestinal phosphate absorption occurs via two mechanisms. Passive diffusion is the principal route and occurs primarily through the paracellular pathway. Active mucosal phosphate transport is a sodium-dependent, saturable carrier-mediated process. Calcitriol (1,25-dihydroxycholecalciferol) increases active intestinal mucosal phosphate transport, but this mechanism is probably important only during dietary phosphate deficiency. Both transport mechanisms function in the duodenum, whereas diffusion is the primary mechanism in the jejunum and ileum. Intestinal alkaline phosphatases may facilitate absorption by freeing inorganic phosphate for transport. Optimal phosphate transport occurs in an alkaline environment, and HPO4−2 is the main ionic species transported. Decreased intestinal phosphate absorption may occur with vitamin D deficiency and in malabsorptive states.
There is no evidence of a direct effect of parathyroid hormone (PTH) on intestinal phosphate absorption, and observed effects are probably mediated by the role of PTH in conversion of 25-hydroxycholecalciferol to calcitriol. High dietary ratios of calcium to phosphorus (>3 to 4) may suppress intestinal phosphate absorption, presumably through binding of phosphate by calcium and formation of poorly absorbed calcium phosphate complexes. During phosphate deprivation, the kidneys dramatically reduce phosphate excretion to negligible amounts in fewer than 3 days. Obligatory gastrointestinal loss continues for at least 3 weeks, but there is a diminution in the amount lost.99 This gastrointestinal loss may cause a cumulative negative phosphorus balance during phosphate deprivation.
Renal handling
The kidneys adjust tubular reabsorption of filtered phosphate to maintain zero balance. Normally, 80% to 90% of the filtered phosphate load is reabsorbed by the renal tubules, and renal dysfunction is the most common cause of hyperphosphatemia beside that found in young dogs.31,160
Phosphate crosses the luminal membranes of the proximal renal tubular cells by brush border sodium-phosphate cotransporters. The main transport protein in the proximal tubules (type IIa sodium-phosphate cotransporter) translocates three sodium ions and one divalent phosphate ion across the luminal membrane and thus promotes luminal electronegativity.168 Luminal entry is the rate-limiting step and the target for physiologic and pathophysiologic mechanisms that alter phosphate reabsorption.115 High dietary intake of phosphorus decreases proximal tubular reabsorption, whereas low dietary intake can result in nearly 100% proximal tubular reabsorption of phosphate. These dietary effects occur independently of changes in the plasma concentrations of phosphaturic hormones. PTH is the most important regulator of renal phosphate transport, and it decreases the tubular transport maximum for phosphate reabsorption (TmaxPi) in the proximal tubule where most phosphate reabsorption occurs. Apparently, no reabsorption occurs in the thin ascending limb or thick ascending limb of Henle’s loop, and the presence of a reabsorptive mechanism in the distal convoluted tubule is uncertain. Phosphate reabsorption is inhibited in the early proximal tubule by volume expansion with saline, but there may be a more distal reabsorptive site (at some point beyond the last portion of the proximal tubule accessible by micropuncture) that is sensitive to PTH and unaffected by saline volume expansion.
The effects of calcitriol on renal phosphate transport are difficult to separate from the effects of calcitriol on PTH secretion and on phosphate transport in other organs (e.g., intestine, bone). Growth hormone increases proximal renal tubular phosphate reabsorption, which partially accounts for the increased serum phosphorus concentrations found in immature animals. Insulin and thyroxine also increase proximal tubular reabsorption of phosphate, whereas calcitonin and atrial natriuretic peptide inhibit proximal tubular phosphate reabsorption. High doses of adrenocorticotropic hormone (ACTH) or glucocorticoids increase renal phosphate excretion and may decrease serum phosphorus concentration.
The effects of acid-base balance on proximal tubular transport of phosphate are complex.115 Acute metabolic acidosis does not affect renal tubular reabsorption of phosphate, but chronic metabolic acidosis results in decreased proximal tubular transport, an effect possibly mediated by glucocorticoids. Respiratory acidosis decreases and respiratory alkalosis increases proximal tubular reabsorption of phosphate. Volume expansion increases urinary phosphate excretion and causes natriuresis because phosphate is cotransported with sodium in the proximal tubule.
Recently, additional factors that impact renal handling of inorganic phosphorus have been identified. Phosphatonins are circulating substances that increase renal loss of phosphorus. More than one has been identified. To date, they include fibroblast growth factor-23 (FGF-23), secreted frizzled-related protein (sFRP-4), fibroblast growth factor-7 (FGF-7), and matrix extracellular phosphoglycoprotein (MEPE).149 These substances decrease sodium phosphate transporters in proximal convoluted tubules, whereas FGF-23 and sFRP-4 are believed to also decrease formation of 1–25 dihydroxycholecalciferol. FGF-23 is increased in people with chronic renal failure, but the exact cause is unknown. Intestinal phosphatonins (factors released from the intestines due to increased intraluminal phosphate concentration) have also been suggested to exist. Although believed to be important in some pathologic conditions, their impact in normal phosphorus homeostasis is currently uncertain.
Hypophosphatemia
Clinical effects of hypophosphatemia
Hypophosphatemia can occur even when total body phosphorus is normal. Nonetheless, severe hypophosphatemia can have many detrimental effects. The most severe cellular damage seems to occur when there is concurrent phosphate depletion.99 Hypophosphatemia decreases erythrocyte concentrations of ATP, which increases erythrocyte fragility, leading to hemolysis. Hemolysis usually is not observed until serum phosphorus concentration decreases to 1.0 mg/dL or less. Hypophosphatemia also reduces erythrocyte 2,3-DPG concentrations, which impairs oxygen delivery to tissues. Leukocytes in hypophosphatemic patients have impaired chemotaxis, phagocytosis, and bacterial killing.38 This altered function may promote sepsis in hypophosphatemic patients receiving total parenteral nutrition. Platelet-associated abnormalities include shortened survival time, impaired clot retraction, megakaryocytosis in the bone marrow, and thrombocytopenia. Hemolytic anemia, thrombocytopenia, and impaired clot retraction occurred in starved dogs that were made hypophosphatemic by infusion of amino acids, ostensibly because of depletion of cellular ATP stores.178 Clinically, hemolysis has been reported in hypophosphatemic dogs and cats with diabetic ketoacidosis, hepatic lipidosis, and other disorders.2,80,173 Hemolysis was reported in four other hypophosphatemic diabetic cats, but cause and effect were obscured by the possibility of Heinz body anemia.23
Neuromuscular effects of hypophosphatemia include weakness and pain associated with rhabdomyolysis, as well as anorexia, vomiting, and nausea secondary to intestinal ileus.88,89 Decreased phosphate may impair central nervous system glucose use and ATP production, leading to metabolic encephalopathy, which has a wide range of manifestations in people (e.g., coma, seizure, confusion, irritability).99,177 Reversible impairment of cardiac contractility occurs in dogs with experimentally induced hypophosphatemia and in people with naturally occurring hypophosphatemia.65,66,180 Hypophosphatemia also causes proximal tubular bicarbonate wasting, reduction in titratable acidity, and impaired renal ammoniagenesis. However, serious acid-base disturbances do not arise in phosphate-deprived dogs.151 Phosphate deficiency produces bone demineralization via effects of PTH and calcitriol, and release of carbonate from bone may prevent serious metabolic acidosis. Hypomagnesemia frequently is found in hypophosphatemic people, but the reasons for this association are not clear.32
Causes of hypophosphatemia
Hypophosphatemia may be caused by translocation of phosphate from extracellular to intracellular fluid (maldistribution), increased loss (decreased renal reabsorption of phosphate), or decreased intake (decreased intestinal absorption of phosphate).99,136 Clinical conditions associated with hypophosphatemia are presented in Box 7-1. In confusing cases, one can measure fractional urinary phosphorus excretion to help determine if renal losses are responsible.
Box 7-1 Causes of Hypophosphatemia
Increased Loss (Reduced Renal Reabsorption)
• Renal tubular disorders (e.g., Fanconi syndrome)
• Proximally acting diuretics (e.g., carbonic anhydrase inhibitors) (?)*
* (?) Importance in veterinary medicine uncertain.
Translocation related to administration of a carbohydrate load (e.g., 5% dextrose infusion) is a common cause of hypophosphatemia in hospitalized people.15,79 Insulin facilitates entry of glucose and phosphate into cells, where glucose is phosphorylated to glycolytic intermediates. Interestingly, infusion of a higher concentration (e.g., 10% dextrose) for a shorter time seems to be less detrimental than infusing 4% glucose continuously.99 Malnourished patients receiving total parenteral nutrition are particularly susceptible to hypophosphatemia because of the accelerated rate of tissue repair as phosphate is incorporated into new cells and phosphate use during glycolysis.88,136 Hypophosphatemia as part of the “refeeding syndrome” (i.e., severe electrolyte changes in malnourished patients that are being fed parenterally or enterally) was more likely in patients that were more severely emaciated, had lower initial serum phosphate concentrations, and experienced more aggressive initial infusion of parenteral nutrition.107 Respiratory alkalosis likewise causes translocation because it stimulates glycolysis by activating phosphofructokinase.88 This effect has been demonstrated in experimental dogs but was marked only when hyperventilation was combined with glucose administration.19 Increased intracellular pH may be more important than increased extracellular pH for causing hypophosphatemia in respiratory alkalosis, which could explain why severe hypophosphatemia may occur in people with severe respiratory failure who are mechanically ventilated.99
Diabetic patients are especially at risk for hypophosphatemia. They often have total body phosphate deficits because of a loss of muscle mass, urinary phosphate losses, and impaired tissue use of phosphate related to insulin deficiency. Most diabetic cats in one study had mild hypophosphatemia at presentation, whereas 20 of 48 ketotic cats in another study were hypophosphatemic.23,146 Another study found only 7 of 104 diabetic cats to be hypophosphatemic. However, stratification of the cats into ketoacidotic and nonketoacidotic groups revealed that 5 of 38 ketoacidotic cats were hypophosphatemic and only 2 of 66 nonketotic cats were hypophosphatemic.39 Interestingly, serum phosphorus concentrations are often normal to increased at presentation in diabetic people, perhaps because of metabolic acidosis by organic acids (e.g., β-hydroxybutyrate), insulin deficiency, osmotic effects of hyperglycemia, or renal insufficiency.86,119
Administration of large doses of insulin makes hypophosphatemia even more likely in diabetic ketoacidotic patients. Severe hypophosphatemia has been reported in dogs and cats treated for diabetic ketoacidosis.2,23,173 Hypophosphatemia developed or worsened after insulin administration, and clinical signs (e.g., hemolysis, seizures) thought related to hypophosphatemia developed in 11 animals. Interestingly, four of these cats developed hemolytic anemia despite intravenous supplementation of potassium phosphate, and it is not clear whether the anemia was caused by inadequate phosphate supplementation or Heinz body formation.23
Although it is not documented in dogs and cats, hypophosphatemia may occur in people with certain rapidly growing tumors. Ostensibly, the rapidly dividing cells use phosphorus, removing it from the blood.99
Increased urinary loss of phosphorus often produces moderate hypophosphatemia in primary hyperparathyroidism, but clinical signs are caused by hypercalcemia.* If 2.5 mg/dL is considered the lower limit of normal, serum phosphorus concentration was decreased in approximately one third of reported cases associated with parathyroid adenoma, but in six of six cases associated with parathyroid hyperplasia.44 Hypophosphatemia is seen inconsistently in cats with primary hyperparathyroidism.43,82 The fractional excretion of phosphorus (FEPi) was increased in a few affected dogs.172 The normal FEPi was found to be 7.5% ± 4.6% in 10 normal dogs but 10% to 23% in a dog with primary hyperparathyroidism.29
Fanconi syndrome in basenjis is associated with decreased renal fractional reabsorption of phosphate, but serum phosphorus concentrations are normal.18 The renal tubular transport abnormality may be caused by metabolic or membrane defects affecting sodium transport, and the observed phosphaturia may be secondary to natriuresis.109 Loop diuretics (e.g., furosemide) and distally acting diuretics (e.g., thiazides) have little effect on renal phosphate excretion, but proximally acting diuretics (e.g., carbonic anhydrase inhibitors) may increase renal excretion of phosphate secondary to their effects on proximal tubular sodium reabsorption. In one study, acetazolamide (10 mg/kg intravenously three times daily) did not cause hypophosphatemia when administered to dogs over a 7-day period.137 Eclampsia in the bitch may be associated with hypophosphatemia and hypocalcemia.8,10 Presumably, increased PTH secretion in response to hypocalcemia leads to decreased renal reabsorption of phosphate.
Not reported in veterinary medicine, cranial trauma is associated with renal losses of phosphorus and hypophosphatemia.132 Acquired diabetes insipidus has been suggested as a possible reason.
Hypophosphatemia caused by dietary deficiency is unlikely in animals eating commercial diets with adequate protein content. A low-protein, low-phosphorus diet designed to dissolve struvite calculi (Prescription Diet S/D, Hill’s Pet Nutrition, Inc., Topeka, Kansas.) did not cause significant hypophosphatemia when fed to dogs over a 6-month period.1 Urinary phosphorus excretion decreased and calcium excretion increased in this study. Although vomiting and malabsorptive diseases potentially can cause phosphate loss, these disorders rarely cause hypophosphatemia in dogs or cats.31 Canine malabsorptive intestinal disorders often are characterized by hypocalcemia related to hypoalbuminemia, but serum phosphorus concentrations typically are normal.20,55
People have become hypophosphatemic after administration of magnesium- and aluminum-containing antacids.101 Whether phosphate depletion occurs depends on the patient’s phosphorus intake, dosage of the phosphate binding agent, duration of administration, and the preexisting phosphate balance of the patient. Vitamin D deficiency may cause hypophosphatemia because hypocalcemia increases PTH secretion, which increases renal phosphate excretion. Decreased intestinal phosphate absorption presumably also plays a role in this setting.
It has been stated that 38% of hyperadrenocortical dogs have hypophosphatemia, but actual serum phosphorus concentrations were not reported.125 In one study, an identifiable cause of hypophosphatemia could not be found in the majority of dogs with this serum biochemical abnormality.31 Hypophosphatemia, hypercalcemia, hyperglycemia, azotemia, hypokalemia, and acidosis have been reported in a dog and cat with hypothermia caused by exposure to low environmental temperature.138 The mechanisms responsible for these electrolyte and acid-base disturbances are uncertain, but translocation seems likely.
Renal transplantation has been associated with hypophosphatemia in people and cats.123 Ostensibly, the cause is decreased renal tubular reabsorption of phosphorus, which is currently believed to be due to phosphatonins. There was no obvious consequence of the hypophosphatemia in a report of 32 affected cats. Hemolysis occurred, but the incidence was not statistically different from normophosphatemic renal transplant cats. Changing the diet from a phosphorus-restricted diet to a maintenance diet plus supplementing sodium phosphate orally corrected the problem.
Disorders of renal tubular phosphate transport associated with hypophosphatemia in humans include X-linked hypophosphatemia, autosomal dominant hypophosphatemic rickets, oncogenic hypophosphatemic osteomalacia, and hereditary hypophosphatemic rickets with hypercalciuria.168 Naturally occurring mutations in the npt2 gene encoding the type IIa sodium-phosphate cotransporter have not been identified in these disorders, but rather mutations have been found in other phosphate-regulating genes. X-linked hypophosphatemia is caused by a mutation in the PHEX gene (i.e., phosphate-regulating gene with homology to endopeptidases on the X chromosome), which is expressed in bone, whereas autosomal dominant hypophosphatemic rickets is caused by a mutation in the FGF-23 gene, a member of the fibroblast growth factor family. Oncogenic hypophosphatemic osteomalacia occurs as a result of secretion of a humoral phosphaturic factor secreted by neoplastic cells. Hereditary hypophosphatemic rickets with hypercalciuria is similar to X-linked hypophosphatemia and autosomal dominant hypophosphatemic rickets except that it is associated with appropriately increased serum concentrations of calcitriol, whereas the other hereditary disorders are not. Renal tubular disorders of phosphate transport have not been conclusively identified in dogs and cats, but hypophosphatemia, increased urinary FEPi, low serum 25-hydroxycholecalciferol concentration, osteopenia, and pathologic fractures were reported in a young cat believed to have abnormal renal tubular phosphate transport and defective hepatic 25-hydroxylation of vitamin D.74
Cats with decreased serum concentrations of cobalamin and folate have been found to be at increased risk for hypophosphatemia.135 Most of the hypophosphatemic cats had gastrointestinal tract disease or pancreatitis. Therefore, it is not clear whether the changes in cobalamin and folate were epiphenomenon or cause-and-effect. However, as these patients frequently require nutritional supplementation (either enteral or parenteral), the need to avoid the refeeding syndrome was noted.
Postsurgical hypophosphatemia is a well-established problem in people. It is more common and can be particularly severe after hepatic surgery, especially major resection and transplantation.41 Although the mechanism was once suggested to be uptake of phosphorus by the regenerating liver, excessive renal losses are now considered to be responsible.116 “Phosphate diabetes” is the term used for increased urinary phosphate excretion due to decreased phosphate reabsorption, especially in hypophosphatemic patients.96 The occurrence of hypophosphatemia after hepatic surgery in particular is not surprising because acute hepatic damage due to any number of causes also is associated with hypophosphatemia. In fact, hypophosphatemia in people with hepatic damage has been suggested to reflect healing and regeneration of the liver.144 Hypophosphatemia has been seen in dogs experimentally intoxicated with xylitol,176 but it is uncertain whether hypophosphatemia is due to hepatic damage or to other metabolic effects of xylitol.
Hypophosphatemia previously has been associated with sepsis and gram-negative infections in people to the extent that some have suggested that it should be a diagnostic tool.171 Hypophosphatemia is believed to occur in these people due to redistribution of phosphorus into body cells. Recently, severe hypophosphatemia (<1 mg/dL) has been reported to be a major risk factor for mortality in these patients.154 Decreased ventricular stroke work with subsequent decreased arterial pressure is believed to be caused in part by the hypophosphatemia.
Intravenous administration of saccharated ferric oxide to treat iron deficiency has caused hypophosphatemia in people.155 Similar changes have not been reported in dogs or cats.
Treatment of hypophosphatemia
Prevention, when possible, is preferred to therapy. The clinician should anticipate potential hypophosphatemia and either administer supplemental phosphorus (e.g., patients receiving total parenteral nutrition or insulin treatment for diabetic ketoacidosis) or carefully monitor the patient for hypophosphatemia (e.g., patients receiving phosphate binders).
If hypophosphatemia occurs, one should seek to correct the underlying condition responsible for it. Whether phosphorus is administered depends on the magnitude of the hypophosphatemia and whether clinical signs are present. Asymptomatic animals with low serum phosphorus concentrations but without phosphorus depletion and those with serum phosphorus concentrations greater than 1.8 mg/dL and unlikely to decrease any lower (e.g., primary hyperparathyroidism) often do not require phosphate administration.
Phosphate supplementation seems appropriate for asymptomatic patients deemed at risk for developing symptomatic hypophosphatemia (e.g., diabetic ketoacidotic cat with serum phosphorus concentration of 1.6 mg/dL) and patients with clinical signs believed to result from hypophosphatemia. The clinician should keep in mind that oversupplementation (especially but not exclusively parenteral) can cause morbidity (e.g., hypocalcemia, soft tissue calcification, renal failure). Interestingly, treatment of asymptomatic hypophosphatemia in diabetic people is controversial and recommended only when hypophosphatemia is severe (<2.0 mg/dL).85 However, clinical experience in veterinary medicine suggests that anticipatory phosphorus supplementation is reasonable in some ketoacidotic cats.
If oral supplementation is suitable (which is uncommon in patients with severe hypophosphatemia), it may be safer and therefore preferable to parenteral supplementation.5,112 Oral phosphate administration is unacceptable in vomiting patients and perhaps in patients with diarrhea. If the enteral route is chosen, feeding skim or low-fat milk or a buffered laxative usually is effective. Patients symptomatic because of hypophosphatemia generally need parenteral replacement therapy. Administering phosphate intravenously is potentially dangerous because it may cause hypocalcemia, tetany, soft tissue mineralization, renal failure, or hyperphosphatemia.88 Therefore, phosphorus administration typically has consisted of injecting small amounts slowly over hours to days and monitoring the patient repeatedly (e.g., 0.01 to 0.06 mmol/kg/hr in dogs and cats with measurement of serum phosphorus concentration every 6 to 8 hours).80,173 Although such caution is wise, it is noteworthy that more aggressive phosphorus administration has been used in people (i.e., 0.16 to 0.64 mmol/kg over 4 to 12 hours in patients receiving total parenteral nutrition).32 Other groups have used similarly large doses over even shorter times (e.g., 0.4 to 0.8 mmol/kg depending on the degree of hypophosphatemia over 30 minutes in patients with cardiac disease), also without problems.180 Sodium phosphate and potassium phosphate are commonly used, but administration of glucose phosphate also has been reported.180 Selection of the particular form of phosphorus to administer is based on the patient’s serum electrolyte concentrations.
Currently, it seems safest to administer phosphate by constant-rate infusion at rates that have been used successfully in dogs and cats and to monitor the serum phosphorus concentration every 6 to 8 hours. Theoretically, adding phosphorus to fluids containing calcium may cause precipitation of calcium phosphate, but this appears to depend on relative concentrations of calcium and phosphorus. Phosphorus usually is administered after diluting it in physiologic saline solution. The volume of distribution for administered phosphate varies tremendously among hypophosphatemic people, and redistribution of phosphate can occur rapidly. Therefore, the dose necessary for repletion and the patient’s response to therapy cannot be predicted. In two studies of hypophosphatemic cats, total amounts of phosphorus infused intravenously ranged from 0.138 to 1.26 mmol/kg, indicating a wide range of total body phosphate deficits.2,80
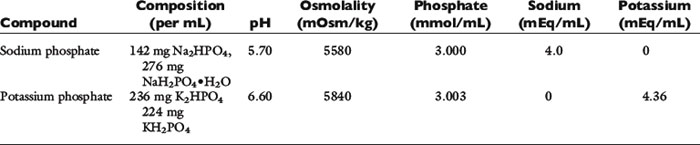
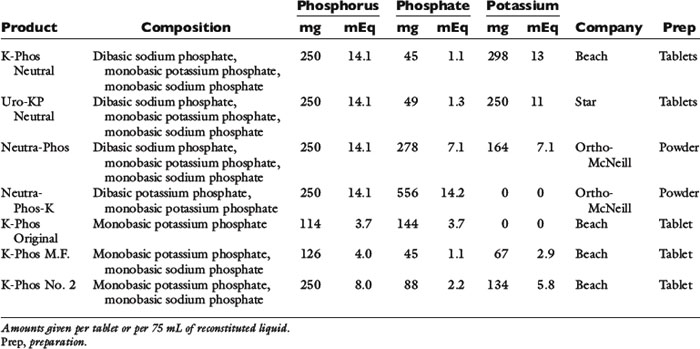
A conservative approach is to assume that intravenously administered phosphate remains in the ECF compartment (actually much of it enters the intracellular fluid). Development of hyperphosphatemia is unlikely with this approach. Prophylactic parenteral phosphate therapy (such as may be used for patients with diabetic ketoacidosis) may be reasonably estimated by giving one fourth to one half of the supplemented potassium as potassium phosphate and the rest as potassium chloride. However, decreased urinary phosphate excretion that develops during hypophosphatemia may persist during treatment and predispose to hyperphosphatemia. The products available for oral and parenteral use are summarized in Tables 7-1 through 7-3.

Hyperphosphatemia
Clinical effects of hyperphosphatemia
Increased serum phosphorus concentration decreases serum calcium concentration so that the calcium phosphate solubility product ([Ca] × [Pi]) remains constant. Hypocalcemia (which may cause tetany) and soft tissue mineralization are the major clinical consequences of hyperphosphatemia.169 If hyperphosphatemia occurs in a hypercalcemic patient, systemic calciphylaxis (i.e., acute calcification of organs, including the heart and lungs166 may occur and can be fatal. Acute hyperphosphatemia (such as can occur after ingesting oral sodium phosphate solutions in preparation for colonoscopy) has been associated with acute renal failure in patients without prior hypercalcemia,118 and a direct toxic effect of phosphate on renal tubular cells has been hypothesized. In human end-stage renal disease patients, hyperphosphatemia is associated with increased cardiovascular mortality.36 After phosphate administration, deposition of calcium and phosphate in bone and soft tissue may contribute to hypocalcemia. The magnitude of hypocalcemia is related to the rate at which serum phosphorus concentration increases, but the exact relationship is unpredictable. The risk of soft tissue mineralization increases when the [Ca] × [Pi] solubility product exceeds 60 to 70.
Causes of hyperphosphatemia
Hyperphosphatemia in dogs and cats is primarily caused by decreased renal excretion, but increased intake and translocation also may be responsible (Box 7-2).169 Translocation occurring during treatment of hemolymphatic malignancies may cause tumor lysis syndrome (i.e., hyperphosphatemia, hypocalcemia, hyperkalemia, hyperuricemia, and oliguric acute renal failure). Myeloblasts and lymphoblasts may contain up to four times as much phosphate as normal cells, and destruction of these cells causes release of phosphate. This syndrome is uncommon in small animal practice. In one study of dogs with multicentric lymphosarcoma, serum phosphorus concentrations were normal before therapy and did not change after treatment.120 Urinary phosphorus excretion increased but probably because urine volume increased. There was no change in FEPi or renal function (as assessed by endogenous creatinine clearance). Chemotherapy in these dogs consisted of prednisone, vincristine, and L-asparaginase. However, acute tumor lysis syndrome has been reported in some animals with lymphosarcoma treated with chemotherapy with or without radiation therapy.28,93,94 Severe hyperphosphatemia (23.6 and 13.7 mg/dL) occurred in a dog and a cat (respectively), and mild hyperphosphatemia (7.4 and 7.7 mg/dL) occurred in two other affected dogs.28,93 Thus, it may be prudent to promote diuresis by intravenous administration of fluids before beginning chemotherapy in patients with lymphosarcoma suspected of having large tumor burdens (e.g., hepatosplenomegaly).
Box 7-2 Causes of Hyperphosphatemia

Decreased Excretion
Physiologic: Young Growing Animal
Laboratory Error (e.g., Lipemia, Hyperproteinemia) Depending on Methodology
* (?) Importance in veterinary medicine uncertain.
Massive tissue injury with rhabdomyolysis may cause hyperphosphatemia. Subsequent development of acute renal failure related to myoglobinuria further contributes to hyperphosphatemia.164 Hyperphosphatemia may occur after aortic thromboembolism in cats and was more common in nonsurvivors in one study.163 Hemolysis can produce hyperphosphatemia because of the phosphorus content of erythrocytes. Lactic acidosis and diabetic ketoacidosis can be associated with hyperphosphatemia because acidosis caused by organic acids apparently results in breakdown of ATP to AMP and inorganic phosphate by an unknown mechanism.119
Increased intake of phosphorus may occur with intravenous administration of phosphate-containing fluids, especially in immobilized patients in which bone resorption is occurring. Such therapy is uncommon in veterinary practice, except in the treatment of diabetic ketoacidosis and total parenteral nutrition.23,173 Increased absorption of phosphorus from the alimentary tract may occur with colonic infusion of hypertonic enema solutions or oral administration of sodium phosphate.47 Such enemas have caused severe hyperphosphatemia in small dogs and cats.9,78,147 Clinical signs in cats receiving phosphate enemas include lethargy, ataxia, vomiting, bloody diarrhea, mucous membrane pallor, and stupor. Laboratory abnormalities included marked hyperglycemia and hyperphosphatemia, mild hypernatremia, and lactic acidosis.9 Severe hyperphosphatemia, azotemia, and metabolic acidosis were reported in a cat treated with a phosphate-containing urinary acidifier (pHos-pHaid) at twice the recommended dosage.67
Vitamin D increases intestinal absorption of calcium and phosphorus and may produce hyperphosphatemia in addition to hypercalcemia. In one study, administration of vitamin D2 to dogs for 3 weeks caused hypercalcemia and azotemia, but serum phosphorus concentrations remained normal.165 However, intoxication with cholecalciferol-containing rodenticides causes azotemia, hypercalcemia, and hyperphosphatemia in dogs and cats.48,60,69,104,114 Topical medications containing calcipotriene, an analogue of calcitriol, also can cause hypercalcemia, hyperphosphatemia, metastatic soft tissue mineralization, and acute renal failure if ingested by dogs.51,71,124
Decreased urinary excretion is the main cause of hyperphosphatemia, and chronic renal failure is the most common cause of hyperphosphatemia in adult dogs and cats.31 Chronic renal disease causes a progressive decrease in the glomerular filtration rate (GFR), and the filtered load of phosphate (GFR × serum phosphorus concentration) decreases as GFR decreases. If phosphorus intake remains constant, phosphorus retention and transient hyperphosphatemia result. However, sustained hyperphosphatemia does not usually develop in early chronic renal failure because there is a compensatory increase in phosphate excretion by remnant nephrons. The effects of PTH on the kidneys mediate this increase in the FEPi. When the GFR decreases to 20% of normal or less (i.e., late chronic renal failure), this compensatory mechanism is exhausted and hyperphosphatemia develops.
Renal secondary hyperparathyroidism is a consistent finding in progressive renal disease.158,160 Hyperphosphatemia inhibits renal 1α-hydroxylase, which is present in the renal tubules (this inhibition impairs conversion of 25-hydroxycholecalciferol to calcitriol and thus reduces intestinal calcium absorption), and decreases serum ionized calcium concentration by the mass law effect ([Ca] × [Pi] = constant). The resultant hypocalcemia and the decreased serum calcitriol concentration stimulate PTH secretion. This increased PTH secretion increases renal excretion of phosphate and release of calcium and phosphate from bone. It also stimulates production of calcitriol. These actions normalize serum phosphorus and ionized calcium concentrations. Thus, calcium and phosphorus balance is maintained by a progressive increase in serum PTH concentration (in early chronic renal failure). However, as renal tubular destruction progresses, there are fewer proximal renal tubules and a decrease in the amount of 1α-hydroxylase enzyme present. This reduction in 1α-hydroxylase means that it is harder for increased concentrations of PTH to increase serum calcium concentration. It also means that calcitriol is not available to inhibit PTH secretion.117 As serum phosphate concentrations persistently remain increased, other changes also occur. Persistent hyperphosphatemia in rats increases the number and size of parathyroid cells. This is important because some percentage of each cell’s secretion is autonomous, and parathyroid hyperplasia means that there is a greater amount of nonsuppressible PTH secretion. Chronically increased PTH concentration leads to bone demineralization and other toxic effects of uremia (e.g., bone marrow suppression, uremic encephalopathy). In addition, uremia decreases the number of parathyroid gland calcitriol receptors, which subsequently decreases the responsiveness of parathyroid glands to the inhibitory effect of calcitriol on PTH release.21,90,110,162 Thus, both decreased calcitriol production and decreased numbers of parathyroid gland calcitriol receptors promote development of renal secondary hyperparathyroidism.
Renal secondary hyperparathyroidism can be prevented or reversed in dogs with experimentally induced chronic renal disease by reducing dietary phosphorus intake in proportion to the decrease in GFR.83,157,159 Early in the course of chronic renal disease, decreased phosphorus intake stimulates renal 1α-hydroxylase activity, which increases calcitriol production. Increased calcitriol enhances intestinal calcium absorption, increases serum ionized calcium concentration, and decreases PTH secretion. Late in the course of chronic renal disease, the kidneys are unable to produce sufficient calcitriol to promote normal intestinal absorption of calcium. Phosphorus restriction in advanced renal disease still decreases PTH secretion by unknown mechanisms independent of serum ionized calcium or calcitriol concentrations.162 These observations form the basis for restricting phosphorus in the medical management of chronic renal failure.
Phosphorus restriction also may prevent renal disease progression by minimizing renal interstitial mineralization.3 In rats with experimentally induced chronic renal failure, detrimental histologic changes (e.g., interstitial mineralization, inflammation, fibrosis) could be prevented and residual renal function maintained by dietary phosphorus restriction.76,84 In cats with experimentally induced renal disease, histologic changes were prevented by phosphorus restriction.139 In a study in rats with 80% nephrectomy, diet was carefully controlled so that only phosphorus intake differed between groups, and a beneficial effect of phosphorus restriction was clearly demonstrated with regard to mortality, proteinuria, histologic changes, creatinine clearance, and serum lipid concentrations over a period of 14 weeks.102 Similar beneficial effects were observed in dogs with 90% nephrectomy fed diets differing only in phosphorus content and followed for 12 months.22 A similar experiment using 48 dogs with experimentally induced renal failure found that the amount of dietary phosphorus was more important in clinical management than the amount of dietary protein.57 In studies of cats with naturally occurring chronic renal failure, renal secondary hyperparathyroidism was successfully managed using a combination of dietary restriction of phosphorus and administration of phosphate binders.11,49
In contrast to findings in early chronic renal failure, hyperphosphatemia is typical in acute renal failure because of insufficient time for compensatory mechanisms to develop. Hyperphosphatemia also occurs in uroabdomen or urethral obstruction because of urine reabsorption from the peritoneal cavity or decreased GFR caused by increased intratubular pressure resulting from urinary tract obstruction.26,54
Hypoparathyroidism in people causes mild hyperphosphatemia because renal reabsorption of phosphate is increased in the absence of PTH. Mild hyperphosphatemia also occurs in dogs with hypoparathyroidism but is overshadowed by the effects of hypocalcemia (e.g., muscle tremors, tetany, seizures, ataxia, behavioral aberrations).24,25,111,153 Hyperphosphatemia has also been reported in cats with hypoparathyroidism.130
Acromegalic people may develop hyperphosphatemia because of growth hormone’s effects on renal tubular phosphate reabsorption. Mild hyperphosphatemia has been reported in some acromegalic dogs and cats.52,126,128,129 Thyroxine increases renal tubular phosphate reabsorption, which contributes to the increased serum phosphorus concentrations observed in hyperthyroid cats.127,167,170 Hyperphosphatemia was reported in 21% of hyperthyroid cats in one study.127
Treatment of hyperphosphatemia
Volume expansion with saline dilutes ECF phosphate and enhances renal phosphate excretion in dehydrated patients. Increasing GFR by volume expansion increases the filtered load of phosphate, and natriuresis impairs proximal tubular phosphate reabsorption. Administration of glucose (and insulin if necessary) may temporarily decrease serum phosphorus concentration by promoting phosphorus entry into cells, although such therapy is rarely, if ever, necessary. All sources of phosphorus intake should be curtailed. In the diet, phosphorus restriction is accomplished primarily by protein restriction. As a rule, low-protein diets are also low in phosphorus. Calcium salts should not be administered to hyperphosphatemic patients because of the risk of metastatic soft tissue calcification. Iatrogenic calcinosis cutis has been reported in a dog and cat with hypoparathyroidism given calcium gluconate subcutaneously.140,148
In patients with severe, chronic renal failure, low-phosphorus diets are helpful but often insufficient. Dialysis is unpredictable because phosphate is a poorly diffusible ion. Therefore, the most practical and effective way to treat hyperphosphatemia in patients with stable chronic renal failure is to decrease intestinal phosphate absorption by orally administered phosphate binders. Such administration helps prevent ingested and endogenously secreted phosphate from being absorbed. Phosphate binders work because the cation in the binder combines with dietary phosphate, producing insoluble, nonabsorbable phosphate compounds. Adsorption of phosphate ions on the surface of binder particles may also contribute to their effect. The rate at which a binder dissolves depends on its water solubility, the pH of the environment, and the dosage.152
The most widely used oral phosphate-binding agents contain aluminum or calcium and hydroxide, carbonate, or acetate (see Table 7-1).35,75,160 The appropriate dosage must be determined empirically, but 90 to 100 mg/kg/day divided two or three times daily is a reasonable starting point. Lower dosages of calcium acetate (50 to 60 mg/kg/day) may be sufficient because it has a greater capacity to bind phosphate than does calcium carbonate.105 Magnesium-containing compounds are not useful as phosphate binders because they cause diarrhea, and limited ability to excrete magnesium in renal failure patients increases the risk of hypermagnesemia42.
Aluminum hydroxide and aluminum carbonate are commonly used phosphate binders. Aluminum hydroxide reduces intestinal phosphorus absorption in normal and uremic people.34 Aluminum is a better binding agent for phosphate than calcium or magnesium in the acidic gastric environment.152 This effect is less important at the higher intestinal pH. Aluminum-containing gels are better tolerated by many dogs and cats when given as tablets or capsules, but the desiccated form has a lower phosphate-binding capacity than the liquid gel.142 Aluminum oxide gel prepared to maximize phosphate binding has been studied in dogs.142,143 Constipation is a common side effect of aluminum-containing phosphate binders.
In people undergoing hemodialysis, osteomalacia and dialysis encephalopathy have been correlated with the aluminum content of dialysis water.121 In one study, encephalopathy occurred in dialysis patients receiving aluminum hydroxide despite a negligible aluminum content of dialysis water.4 Aluminum can be absorbed from the intestinal tract in normal people81 and uremic people,14,34 and aluminum-induced bone disease can occur in nondialyzed patients after oral administration of aluminum hydroxide.6 The toxicity of aluminum-containing phosphate binders in human patients with renal failure is now well established, and they have been replaced by calcium-containing phosphate binders.50 It still is unclear whether aluminum-containing phosphate binders represent a hazard to dogs with chronic renal failure.
Calcium salts such as calcium carbonate and calcium acetate also have been used as phosphate binders. Calcium carbonate decreases intestinal phosphate absorption in normal and uremic people.* Calcium citrate also has been advocated as a phosphate binder but should not be given with aluminum-containing compounds because citrate enhances aluminum absorption.40,64,113,122,156 Nausea, constipation, and hypercalcemia are potential side effects of calcium-containing phosphate binders. Simultaneous use of calcitriol and calcium-containing phosphate binders to manage renal secondary hyperparathyroidism increases the risk of hypercalcemia. Calcium acetate binds more phosphate than either calcium citrate or calcium carbonate, and less calcium is absorbed from the intestine during its use.152 Calcium acetate binds phosphate better than aluminum carbonate at the neutral pH found in the small intestine, but aluminum carbonate is better at the lower gastric pH.152 In vivo, both were about equally effective.
Phosphate binders are most effective when given with meals. In one study, calcium acetate reduced intestinal absorption of phosphate best when ingested just before or after a meal but was much less effective if given 2 hours after eating.150 Approximately one third as much phosphate was removed from the body when calcium acetate was given during fasting compared with when it was given with a meal. The endogenous phosphate removed probably originated from basal intestinal secretions or passive diffusion into the intestine. Ingestion of a meal also decreased the absorption of calcium from the calcium acetate. Thus, calcium-containing phosphate binders should be given with meals to reduce the risk of hypercalcemia.
The search for new phosphorus binders has continued because of the bone toxicity and encephalopathy associated with use of aluminum-containing compounds and the hypercalcemia and soft tissue (including cardiovascular) calcification associated with use of calcium-containing compounds.50 Sevelamer hydrochloride is a cross-linked polymeric resin that binds phosphorus and releases chloride. It does not contain aluminum or calcium. Sevelamer is believed to adhere to the mucosa of the intestines, thus slowing its transit time and allowing for extended periods of phosphate binding 58 It reduces the risk of vascular and renal calcification that occurs in human patients with chronic renal failure treated with calcium-containing compounds.37 It is very expensive, causes some adverse gastrointestinal effects, and has the potential to bind other substances (e.g., bile acids, cholesterol, vitamins) in addition to phosphorus. Initial reports suggested that sevelamer was similar in effectiveness to calcium acetate in binding phosphorus but with less risk of hypercalcemia.16 However, a recent study found calcium acetate superior to sevelamer in control of hyperphosphatemia and calcium-phosphorus product.133 Sevelamer decreased serum bicarbonate concentrations in this study, presumably as a result of the release and absorption of the hydrochloride moiety. In another study,58 once-daily administration was as effective as administration three times per day in some patients.
Lanthanum carbonate also contains no aluminum and no calcium, is not absorbed from the gastrointestinal tract, and acts as an efficient phosphorus binder.50 Its effects are similar to those of calcium carbonate but without risk of bone toxicity or hypercalcemia.45 It binds to phosphate at low and high pH, making it effective in both the stomach and small intestine.63 Lanthanum is excreted primarily in bile and should not accumulate in patients with renal failure, but its long-term safety is unknown.
Phosphate binder effectiveness is monitored by measuring fasting serum phosphorus concentration. The goal is to maintain the serum phosphorus concentration in the normal range. In normophosphatemic patients with early renal insufficiency, one may monitor fasting FEPi to determine the efficacy of phosphate restriction. Dogs with spontaneous chronic renal failure (mean serum creatinine concentration, 2.3 mg/dL) had significantly higher FEPi values than control dogs (23% vs. 5%), respectively, and FEPi decreased in both groups after feeding of Prescription Diet K/D.70 In one dog with chronic renal failure, FEPi was below the mean value for the chronic renal failure group despite increased serum PTH concentration. It has been suggested that FEPi values less than 30% are indicative of adequate phosphate restriction.53 This method is limited by the wide range of normal values for FEPi.46,141 The response to phosphate binders may be relatively slow because the pool of accumulated phosphate is large and the persistent osteolytic effects of PTH provide a large endogenous phosphate load. Thus, the clinician should not be discouraged if the patient responds slowly to phosphate binder therapy. Recently, increased salivary secretion of phosphate in hyperphosphatemic people with renal failure has been identified.12,145 This route is currently being examined as a possible means by which phosphorus can be chelated and removed from the body.
Appendix
Calculation of Amount of PO43- and H3PO4 Present in Extracellular Fluid at a pH of 7.4
The Henderson-Hasselbalch equation is derived from the formula for the dissociation constant of an acid. For the ionic species of phosphate of interest:
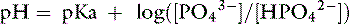
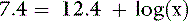
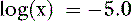
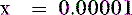
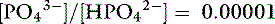
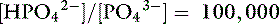
Thus, at a pH of 7.4, there are 100,000 molecules of HPO42− for every molecule of PO43−.
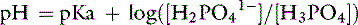
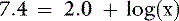
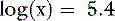
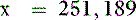
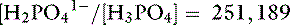
Thus, at a pH of 7.4, there are 251,189 molecules of H2PO41− for every molecule of H3PO4.
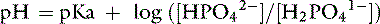
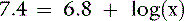
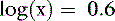
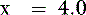
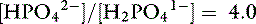
From these calculations, it can be determined that, at a pH of 7.4, there will be 1,004,756 molecules of HPO42−, 251,189 molecules of H2PO41−, and 10 molecules of PO43− for every molecule of H3PO4. Therefore, it can be seen that the amounts of H3PO4 and PO43− present in ECF at a pH of 7.4 can be safely ignored.
1 Abdullahi S.U., Osborne C.A., Leininger J.R., et al. Evaluation of a calculolytic diet in female dogs with induced struvite urolithiasis. Am J Vet Res. 1984;45:1508-1519.
2 Adams L.G., Hardy R.M., Weiss D.J., et al. Hypophosphatemia and hemolytic anemia associated with diabetes mellitus and hepatic lipidosis in cats. J Vet Intern Med. 1993;7:266-271.
3 Alfrey A.C. Effect of dietary phosphate restriction on renal function and deterioration. Am J Clin Nutr. 1988;47:153-156.
4 Alfrey A.C., LeGendre G.R., Kaehny W.D. The dialysis encephalopathy syndrome: possible aluminum intoxication. N Engl J Med. 1976;294:184-188.
5 Amanzadeh J., Reilly R. Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nature Clinical Practice. 2006;2:136-148.
6 Andreoli S.P., Bergstein J.M., Sherrard D.J. Aluminum intoxication from aluminum-containing phosphate binders in children with azotemia not undergoing dialysis. N Engl J Med. 1984;310:1079-1084.
7 Andreoli S.P., Dunson J., Bergstein J.M. Calcium carbonate is an effective phosphorus binder in children with chronic renal failure. Am J Kidney Dis. 1987;9:206-210.
8 Aroch I., Srebro H., Shpigel N.Y. Serum electrolyte concentrations in bitches with eclampsia. Vet Rec. 1999;145:318-320.
9 Atkins C.E., Tyler R., Greenlee P. Clinical, biochemical, acid-base, and electrolyte abnormalities in cats after hypertonic sodium phosphate enema administration. Am J Vet Res. 1985;46:980-988.
10 Austad R., Bjerkas E. Eclampsia in the bitch. J Small Anim Pract. 1976;17:795-798.
11 Barber P.J., Rawlings J.M., Markwell P.J., et al. Effect of dietary restriction on renal secondary hyperparathyroidism in the cat. J Small Anim Pract. 1999;40:62-70.
12 Bellinghieri G., Santoro D., Savica V. Emerging drugs for hyperphosphatemia. Expert Opin Emerg Drugs. 2007;12:355-365.
13 Berger B., Feldman E.C. Primary hyperparathyroidism in dogs: 21 cases (1976–1986). J Am Vet Med Assoc. 1987;191:350-356.
14 Berlyne G.M., Ben-Ari J., Pest D., et al. Hyperaluminaemia from aluminum resins in renal failure. Lancet. 1970;2:494-496.
15 Betro M.G., Pain R.W. Hypophosphatemia and hyperphosphatemia in a hospital population. Lancet. 1972;1:273-276.
16 Bleyer A.J., Burke S.K., Dillon M., et al. A comparison of the calcium-free phosphate binder sevelamer hydrochloride with calcium acetate in the treatment of hyperphosphatemia in hemodialysis patients. Am J Kidney Dis. 1999;33:694-701.
17 Bloom F. The blood chemistry of the dog and cat. New York: Gamma Publications; 1960.
18 Bovee K.C., Joyce T., Blazer-Yost B., et al. Characterization of renal defects in dogs with a syndrome similar to Fanconi syndrome in man. J Am Vet Med Assoc. 1979;174:1094-1099.
19 Brautbar N., Leibovici H., Massry S.G. On the mechanism of hypophosphatemia during acute hyperventilation: evidence for increased muscle glycolysis. Miner Electrolyte Metab. 1983;9:45-50.
20 Breitschwerdt E.B., Halliwell W.H., Foley C.W., et al. A hereditary diarrhetic syndrome in the basenji characterized by malabsorption, protein losing enteropathy, and hypergammaglobulinemia. J Am Anim Hosp Assoc. 1980;16:551-560.
21 Brown A.J., Dusso A., Lopez-Hilker S., et al. 1,25-(OH)2D receptors are decreased in parathyroid glands from chronically uremic dogs. Kidney Int. 1989;35:19-23.
22 Brown S., Finco D., Crowell W.A., et al. Beneficial effect of moderate phosphate restriction in partially nephrectomized dogs on a low protein diet (abstract). Kidney Int. 1987;31:380.
23 Bruskiewicz K.A., Nelson R.W., Feldman E.C., et al. Diabetic ketosis and ketoacidosis in cats: 42 cases (1980–1995). J Am Vet Med Assoc. 1997;211:188-192.
24 Bruyette D.S., Feldman E.C. Primary hypoparathyroidism in the dog. J Vet Intern Med. 1988;2:7-14.
25 Burk R.L., Schaubhut C.W. Spontaneous primary hypoparathyroidism in a dog. J Am Anim Hosp Assoc. 1975;11:784-785.
26 Burrows C.F., Bovee K.C. Metabolic changes due to experimentally-induced rupture of the canine urinary bladder. Am J Vet Res. 1974;35:1083-1088.
27 Busse J.C., Gelbard M.A., Byrnes J.J., et al. Pseudohyperphosphatemia and dysproteinemia. Arch Intern Med. 1987;147:2045-2046.
28 Calia C.M., Hohenhaus A.E., Fox P.R., et al. Acute tumor lysis syndrome in a cat with lymphoma. J Vet Intern Med. 1996;10:409-411.
29 Carillo J.M., Burk R.L., Bode C. Primary hyperparathyroidism in a dog. J Am Vet Med Assoc. 1979;174:67-71.
30 Carlson G.P. Fluid, electrolyte, and acid base balance. In: Kaneko J.J., editor. Clinical biochemistry of domestic animals. 4th ed. New York: Academic Press; 1989:543-575.
31 Chew D.J., Meuten D.J. Disorders of calcium and phosphorus metabolism. Vet Clin North Am. 1982;12:411-438.
32 Clark C.L., Sacks G.S., Dickerson R.N., et al. Treatment of hypophosphatemia in patients receiving specialized nutrition support using a graduated dosing scheme. Results from a prospective clinical trial. Crit Care Med. 1995;23:1504-1511.
33 Clarkson E.M., McDonald S.J., deWardener H.E. The effect of a high intake of calcium carbonate in normal subjects and patients with chronic renal failure. Clin Sci. 1966;30:425-438.
34 Clarkson E.M., Luck V.A., Hynson W.V., et al. The effect of aluminum hydroxide on calcium, phosphorus, and aluminum balances, the serum parathyroid hormone concentration and the aluminum content of bone in patients with chronic renal failure. Clin Sci. 1972;43:519-531.
35 Coburn J.W., Salusky I.B. Control of serum phosphorus in uremia. N Engl J Med. 1989;320:1140-1142.
36 Coladonato J. Control of hyperphosphatemia among patients with ESRD. J Am Soc Nephrol. 2005;16:S107-S114.
37 Cozzolino M., Staniforth M.E., Liapis H., et al. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int. 2003;64:1653-1661.
38 Craddock P.R., Yawata Y., VanSanten L., et al. Acquired phagocyte dysfunction. N Engl J Med. 1974;290:1403-1407.
39 Crenshaw K.L., Peterson M.E. Pretreatment of clinical and laboratory evaluation of cats with diabetes mellitus: 104 cases (1992–1994). J Am Vet Med Assoc. 1996;209:943-949.
40 Cushner H.M., Copley J.B., Lindberg J.S., et al. Calcium citrate, a non-aluminum-containing phosphate-binding agent for treatment of CRF. Kidney Int. 1988;33:95-99.
41 Datta H., Malik M., Neely R. Hepatic surgery-related hypophosphatemia. Clin Chim Acta. 2007;380:13-23.
42 Delmez J.A., Kelber J., Norword K.Y., et al. Magnesium carbonate as a phosphorus binder: a prospective controlled crossover study. Kidney Int. 1996;49:163-167.
43 den Hertog E., Goossens M.M., van der Linde Sipman J.S., et al. Primary hyperparathyroidism in two cats. Vet Q. 1997;19:81-84.
44 DeVries S.E., Feldman E.C., Nelson R.W., et al. Primary parathyroid gland hyperplasia in dogs: six cases (1982–1991). J Am Vet Med Assoc. 1993;202:1132-1136.
45 D’Haese P.C., Spasovski G.B., Sikole A., et al. A multicenter study on the effects of lanthanum carbonate (Fosrenol™) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int. 2003;63:S73-S78.
46 DiBartola S.P., Chew D.J., Jacobs G. Quantitative urinalysis including 24-hour urinary protein excretion in the dog. J Am Anim Hosp Assoc. 1980;16:537-546.
47 DiPalma J.A., Buckley S.E., Warner B.A., et al. Biochemical effects of oral sodium phosphate. Dig Dis Sci. 1996;41:749-753.
48 Dougherty S.A., Center S.A., Dzanis D.A. Salmon calcitonin as adjunct treatment for vitamin D toxicosis in a dog. J Am Vet Med Assoc. 1990;196:1269-1272.
49 Elliott J., Rawlings J.M., Markwell P.J., et al. Survival of cats with naturally-occurring chronic renal failure: effect of dietary management. J Small Anim Pract. 2000;41:235-242.
50 Emmett M. A comparison of clinically useful phosphorus binders for patients with chronic renal failure. Kidney Int. 2004;66:S25-S32.
51 Fan T.M., Simpson K.W., Trasti S., et al. Calcipotriol toxicity in a dog. J Small Anim Pract. 1998;39:581-586.
52 Feldman E.C., Nelson R.W. Disorders of growth hormone. In Canine and feline endocrinology and reproduction, 2nd ed, Philadelphia: WB Saunders; 1996:60.
53 Finco D.R. The role of phosphorus restriction in the management of chronic renal failure in the dog and cat. Proc 7th Kal Kan Symposium. 1983:131-134.
54 Finco D.R., Cornelius L.M. Characterization and treatment of water, electrolyte, and acid-base imbalances of induced urethral obstruction in the cat. Am J Vet Res. 1977;38:823-830.
55 Finco D.R., Duncan J.R., Schall W.D., et al. Chronic enteric disease and hypoproteinemia in 9 dogs. J Am Vet Med Assoc. 1973;163:262-271.
56 Finco D.R., Barsanti J.A., Brown S.A. Influence of dietary source of phosphorus on fecal and urinary excretion of phosphorus and other minerals by male cats. Am J Vet Res. 1989;50:263-266.
57 Finco D.R., Brown S.A., Crowell W.A., et al. Effects of dietary phosphorus and protein in dogs with chronic renal failure. Am J Vet Res. 1992;53:2264-2271.
58 Fischer D., Cline K., Plone M., et al. Results of a randomized crossover study comparing once-daily and thrice-daily sevelamer dosing. Am J Kid Dis. 2006;48:437-444.
59 Fitzgerald F. Clinical hypophosphatemia. Annu Rev Med. 1978;29:177-189.
60 Fooshee S.K., Forrester S.D. Hypercalcemia secondary to cholecalciferol rodenticide toxicosis in two dogs. J Am Vet Med Assoc. 1990;196:1265-1268.
61 Fournier A., Moriniere P., Sebert J.L., et al. Calcium carbonate, an aluminum-free agent for control of hyperphosphatemia, hypocalcemia, and hyperparathyroidism in uremia. Kidney Int. 1986;29:S114-S119.
62 Fraser D., Hones G., Kooh S.W., et al. Calcium and phosphate metabolism. In: Tietz N.W., editor. Fundamentals of clinical chemistry. Philadelphia: WB Saunders; 1987:705-728.
63 Fricker S. The therapeutic application of lanthanides. Chem Soc Rev. 2006;35:524-533.
64 Froment D.H., Molitoris B.A., Buddington B., et al. Site and mechanism of enhanced gastrointestinal absorption of aluminum by citrate. Kidney Int. 1989;36:978-984.
65 Fuller T.J., Carter N.W., Barcenas C., et al. Reversible changes of the muscle cell in experimental phosphorus deficiency. J Clin Invest. 1976;57:1019-1024.
66 Fuller T.J., Nichols W.W., Brenner B.J., et al. Reversible depression in myocardial performance in dogs with experimental phosphorus deficiency. J Clin Invest. 1978;62:1194-1200.
67 Fulton R.B., Fruechte L.K. Poisoning induced by administration of a phosphate-containing urinary acidifier in a cat. J Am Vet Med Assoc. 1991;198:883-885.
68 Glick M.R., Ryder K.W., Glick S.J. Interferographs. Indianapolis: Science Enterprises Inc; 1991.
69 Gunther R., Felice L.J., Nelson R.K. Toxicity of a vitamin D3 rodenticide to dogs. J Am Vet Med Assoc. 1988;193:211-214.
70 Hansen B., DiBartola S.P., Chew D.J., et al. Clinical and metabolic findings in dogs with spontaneous chronic renal failure fed two different diets. Am J Vet Res. 1992;53:326-334.
71 Hare R., Dobbs C., Sayman K., et al. Calcipotriene poisoning in dogs. Vet Med. 2000;95:770-778.
72 Harkin K.R., Braselton W.E., Tvedten H. Pseudohypophosphatemia in two dogs with immune-mediated hemolytic anemia. J Vet Intern Med. 1998;12:178-181.
73 Harper E.J., Hackett R.M., Wilkinson J., et al. Age-related variations in hematologic and plasma biochemical test results in beagles and Labrador retrievers. J Am Vet Med Assoc. 2003;223:1436-1442.
74 Henik R.A., Forrest L.J., Friedman A.L. Rickets caused by excessive renal phosphate loss and apparent abnormal vitamin D metabolism in a cat. J Am Vet Med Assoc. 1999;215:1644-1649.
75 Hercz G., Coburn J.W. Prevention of phosphate retention and hyperphosphatemia in uremia. Kidney Int. 1987;32:S215-S220.
76 Ibels L.S., Alfrey A.C., Haut L., et al. Preservation of function in experimental renal disease by dietary restriction of phosphate. N Engl J Med. 1978;298:122.
77 Ikeuchi J., Yoshizaki T., Hirata M. Plasma biochemistry values of young beagle dogs. J Toxicol Sci. 1991;16:49-59.
78 Jorgensen L.S., Center S.A., Randolph J.F., et al. Electrolyte abnormalities induced by hypertonic phosphate enemas in two cats. J Am Vet Med Assoc. 1985;187:1367-1368.
79 Juan D., Elrazak M.A. Hypophosphatemia in hospitalized patients. JAMA. 1979;242:163-164.
80 Justin R.B., Hohenhaus A.E. Hypophosphatemia associated with enteral alimentation in cats. J Vet Intern Med. 1995;9:228-233.
81 Kaehny W.D., Hegg A.P., Alfrey A.C. Gastrointestinal absorption of aluminum from aluminum-containing antacids. N Engl J Med. 1977;296:1389-1390.
82 Kallet A.J., Richter K.P., Feldman E.C., et al. Primary hyperparathyroidism in cats: seven cases (1984–1989). J Am Vet Med Assoc. 1991;199:1767-1771.
83 Kaplan M.A., Canterbury J.M., Bourgoignie J.J., et al. Reversal of hyperparathyroidism in response to dietary phosphorus restriction in the uremic dog. Kidney Int. 1979;15:43.
84 Karlinsky M.L., Haut L., Buddington B., et al. Preservation of renal function in experimental glomerulonephritis. Kidney Int. 1980;17:293.
85 Kassirer J.P., Hricik D.E., Cohen J.J. Phosphate. In: Repairing body fluids: principles and practice. Philadelphia: WB Saunders; 1989:110-117.
86 Kebler R., McDonald F.D., Cadnapaphornchai P. Dynamic changes in serum phosphorus level in diabetic ketoacidosis. Am J Med. 1985;79:571-576.
87 Klausner J.S., O’Leary T.P., Osborne C.A. Calcium urolithiasis in two dogs with parathyroid adenomas. J Am Vet Med Assoc. 1987;191:1423-1426.
88 Knochel J.P. The pathophysiology and clinical characteristics of severe hypophosphatemia. Arch Intern Med. 1977;137:203-220.
89 Knochel J.P. Skeletal muscle in hypophosphatemia and phosphorus deficiency. Adv Exp Med Biol. 1978;103:357-366.
90 Korkor A.B. Reduced binding of [3H]1,25-dihydroxyvitamin D3 in the parathyroid glands of patients with renal failure. N Engl J Med. 1987;316:1573.
91 Kreisberg R.A. Phosphorus deficiency and hypophosphatemia. Hosp Pract. 1977;12:121-128.
92 Kristensen A.T., Klausner J.S., Weiss D.J., et al. Spurious hyperphosphatemia in a dog with chronic lymphocytic leukemia and an IgM monoclonal gammopathy. Vet Clin Pathol. 1991;20:45-48.
93 Laing E.J., Carter R.F. Acute tumor lysis syndrome following treatment of canine lymphoma. J Am Anim Hosp Assoc. 1988;24:691-696.
94 Laing E.J., Fitzpatrick P.J., Norris A.M., et al. Half-body radiotherapy in the treatment of canine lymphoma. J Vet Intern Med. 1989;3:102-108.
95 Lane J., Rehak N., Hortin G., et al. Pseudohyperphosphatemia associated with high-dose liposomal amphotericin B therapy. Clin Chim Acta. 2008;387:145-149.
96 Laroche M., Boyer J. Phosphate diabetes, tubular phosphate reabsorption and phosphatonins. Joint Bone Spine. 2005;72:376-381.
97 Leehey D.J., Daugirdas J.T., Ing T.S., et al. Spurious hyperphosphatemia due to hyperlipidemia. Arch Intern Med. 1985;145:743-744.
98 Legendre A.M., Merkley D.F., Carrig C.B., et al. Primary hyperparathyroidism in a dog. J Am Vet Med Assoc. 1976;168:694-696.
99 Levine B.S., Kleeman C.R. Hypophosphatemia and hyperphosphatemia: clinical and pathophysiologic aspects. In: Narins R.G., editor. Clinical disorders of fluid and electrolyte metabolism. 5th ed. New York: McGraw-Hill; 1994:1045-1090.
100 Lopez S., Galceran T., Slatopolsky E. Evaluation of calcium carbonate as an effective phosphorus-binding agent in the dog. Clin Res. 1984;32:452A.
101 Lotz M., Zisman E., Bartter F.C. Evidence for a phosphorus depletion syndrome in man. N Engl J Med. 1968;278:409-415.
102 Lumlertgul D., Burke T.J., Gillum D.M., et al. Phosphate depletion arrests progression of chronic renal failure independent of protein intake. Kidney Int. 1986;29:658.
103 Lutomski D.M., Bower R.H. The effect of thrombocytosis on serum potassium and phosphorus concentrations. Am J Med Sci. 1994;307:255-258.
104 MacKenzie C.P., Burnie A.G., Head K.W. Poisoning in four dogs by a compound containing warfarin and calciferol. J Small Anim Pract. 1987;28:433-445.
105 Mai M.L., Emmett M.E., Sheikh M.S., et al. Calcium acetate, an effective phosphorus binder in patients with renal failure. Kidney Int. 1989;36:690-695.
106 Malberti F., Surian M., Colussi G., et al. Calcium carbonate: a suitable alternative to aluminum hydroxide as phosphate binder. Kidney Int. 1988;33:S184-S185.
107 Marvin V., Brown D., Portlock J., et al. Factors contributing to the development of hypophosphatemia when refeeding using parenteral nutrition. Pharm World Sci. 2008;30:329-335.
108 Mavrikakis M., Vaiopoulos G., Athanassiades P., et al. Pseudohyperphosphatemia in multiple myeloma. Am J Hematol. 1996;51:178-179.
109 McNamara P.D., Rea C.T., Bovee K.C., et al. Cystinuria in dogs: comparison of the cystinuric component of the Fanconi syndrome in basenji dogs to isolated cystinuria. Metabolism. 1989;38:8-15.
110 Merke J., Hugel U., Zlotkowski A., et al. Diminished parathyroid 1,25-dihydroxycholecalciferol receptors in experimental uremia. Kidney Int. 1987;32:350-353.
111 Meyer D.J., Terrell T.G. Idiopathic hypoparathyroidism in a dog. J Am Vet Med Assoc. 1976;168:858-860.
112 Moe S. Disorders involving calcium, phosphorus and magnesium. Prim Care Clin Office Pract. 2008;35:215-237.
113 Molitoris B.A., Froment D.H., MacKenzie T.A., et al. Citrate: a major factor in the toxicity of orally administered aluminum compounds. Kidney Int. 1989;36:949-953.
114 Moore F.M., Kudisch M., Richter K., et al. Hypercalcemia associated with rodenticide poisoning in three cats. J Am Vet Med Assoc. 1988;193:1099-1100.
115 Murer H., Hernando N., Forster I., et al. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev. 2000;80:1373-1409.
116 Nafidi O., Lapointe R., Lepage R., et al. Mechanisms of renal phosphate loss in liver resection-associated hypophosphatemia. Ann Surg. 2009;249:824-827.
117 Nagode L.A., Chew D.J., Podell M. Benefits of calcitriol therapy and serum phosphorus control in dogs and cats with renal failure. Vet Clin North Am. 1996;26:1293-13330.
118 Ori Y., Herman M., Tobar A., et al. Acute phosphate nephropathy: an emerging threat. Am J Med Sci. 2008;336:309-314.
119 Oster J.R., Perez G.O., Vaamonde C.A. Relationship between blood pH and potassium and phosphorus during acute metabolic acidosis. Am J Physiol. 1978;235:F345-F351.
120 Page R.L., Leifer C.E., Matus R.E. Uric acid and phosphorus excretion in dogs with lymphosarcoma. Am J Vet Res. 1986;47:910-912.
121 Parkinson I.S., Ward M.K., Kerr D.N.S. Dialysis encephalopathy, bone disease, and anemia: the aluminum intoxication syndrome during regular hemodialysis. J Clin Pathol. 1981;34:1285-1294.
122 Partridge N.A., Regnier F.E., White J.L., et al. Influence of dietary constituents on intestinal absorption of aluminum. Kidney Int. 1989;35:1413-1417.
123 Paster E., Mehl M., Kass P., et al. Hypophosphatemia in cats after renal transplantation. Vet Surg. 2009;38:983-989.
124 Pesillo S.A., Khan S.A., Rozanski E.A., et al. Calcipotriene toxicosis in a dog successfully treated with pamidronate disodium. J Vet Emerg Crit Care. 2002;12:177-181.
125 Peterson M.E. Hyperadrenocorticism. Vet Clin North Am. 1984;14:731-749.
126 Peterson M.E. Endocrine disorders in cats: four emerging diseases. Comp Contin Educ Pract Vet. 1988;10:1353-1362.
127 Peterson M.E., Kintzer P.P., Cavanagh P.G., et al. Feline hyperthyroidism: pretreatment clinical and laboratory evaluation of 131 cases. J Am Vet Med Assoc. 1983;183:103-110.
128 Peterson M.E., Taylor R.S., Greco D.S., et al. Spontaneous acromegaly in the cat. Proc Am Coll Vet Intern Med. 1986:14-43. Washington, DC
129 Peterson M.E., Taylor R.S., Greco D.S., et al. Acromegaly in 14 cats. J Vet Intern Med. 1990;4:192-201.
130 Peterson M.E., James K.M., Wallace M., et al. Idiopathic hypoparathyroidism in five cats. J Vet Intern Med. 1991;5:47-51.
131 Pickrell J.A., Schluter S.J., Belasich J.J., et al. Relationship of age of normal dogs to blood serum constituents and reliability of measured single values. Am J Vet Res. 1974;35:897-903.
132 Polderman K., Bloemers F., Peerdeman S., et al. Hypomagnesemia and hypophosphatemia at admission in patients with severe head injury. Crit Care Med. 2000;28:2022-2025.
133 Qunibi W.Y., Hootkins R.E., McDowell L.L., et al. Treatment of hyperphosphatemia in hemodialysis patients: the Calcium Acetate Renagel Evaluation (CARE Study). Kidney Int. 2004;65:1914-1926.
134 Ralston Purina Company. Normal blood values for cats and normal blood values for dogs. St. Louis: Ralston Purina Company; 1975.
135 Reed N., GunnMoore D., Simpson K. Cobalamin, folate and inorganic phosphate abnormalities in ill cats. J Feline Med Surg. 2007;9:278-288.
136 Ritz E. Acute hypophosphatemia. Kidney Int. 1982;22:84-94.
137 Rose R.J., Carter J. Some physiological and biochemical effects of acetazolamide in the dog. J Vet Pharmacol Ther. 1979;2:215-221.
138 Ross L.A., Goldstein M. Biochemical abnormalities associated with accidental hypothermia in a dog and cat. Abstract. Proc Am Coll Vet Intern Med. 1981:66. St. Louis
139 Ross L.A., Finco D.R., Crowell W.A., et al. Effect of dietary phosphorus restriction on the kidneys of cats with reduced renal mass. Am J Vet Res. 1982;43:1023.
140 Ruopp J.L. Primary hypoparathyroidism in a cat complicated by suspect iatrogenic calcinosis cutis. J Am Anim Hosp Assoc. 2001;37:370-373.
141 Russo E.A., Lees G.E., Hightower D. Evaluation of renal function in cats, using quantitative urinalysis. Am J Vet Res. 1986;47:1308-1312.
142 Rutherford E., Mercado A., Hruska K., et al. An evaluation of a new and effective phosphorus binding agent. Trans Am Soc Artif Intern Organs. 1973;19:446-449.
143 Rutherford E., King S., Perry B., et al. Use of a new phosphate binder in chronic renal insufficiency. Kidney Int. 1980;17:528-534.
144 Saito T., Tojo K., Miyashita Y., et al. Acute liver damage and subsequent hypophosphatemia in malnourished patients: case reports and review of literature. Int J Eat Disord. 2008;41:188-192.
145 Savica V., Calo L., Monardo P., et al. Salivary phosphorus and phosphate content of beverages: Implications for the treatment of uremic hyperphosphatemia. J Ren Nutr. 2009;19:69-72.
146 Schaer M. A clinical survey of 30 cats with diabetes mellitus. J Am Anim Hosp Assoc. 1977;13:23-27.
147 Schaer M., Cavanaugh P., Hause W., et al. Iatrogenic hyperphosphatemia, hypocalcemia and hypernatremia in a cat. J Am Anim Hosp Assoc. 1977;13:39-41.
148 Schaer M., Gin P.E., Fox L.E., et al. Severe calcinosis cutis associated with treatment of hypoparathyroidism in a dog. J Am Anim Hosp Assoc. 2001;37:364-369.
149 Shaikh A., Berndt T., Kumar R. Regulation of phosphate homeostasis by the phosphatonins and other novel mediators. Pediatr Nephrol. 2008;23:1203-1210.
150 Schiller L.R., Santa Ana C.A., Sheikh M.S., et al. Effect of the time of administration of calcium acetate on phosphorus binding. N Engl J Med. 1989;320:1110-1113.
151 Schmidt R.W. Effects of phosphate depletion on acid-base status in dogs. Metabolism. 1978;27:943-952.
152 Sheikh M.S., Maguire J.A., Emmett M., et al. Reduction of dietary phosphorus absorption by phosphorus binders: a theoretical, in vitro, and in vivo study. J Clin Invest. 1989;83:66-73.
153 Sherding R.F., Meuten D.J., Chew D.J., et al. Primary hypoparathyroidism in the dog. J Am Vet Med Assoc. 1980;176:439-444.
154 Shor R., Halabe A., Rishver S., et al. Severe hypophosphatemia in sepsis as a mortality predictor. Ann Clin Lab Sci. 2006;36:67-72.
155 Shimizu Y., Tada Y., Yamauchi M., et al. Hypophosphatemia induced by intravenous administration of saccharated ferric oxide Another form of FGF23-related hypophosphatemia. Bone. 2009;45:814-816.
156 Slanina P., Falkeborn Y., Frech W., et al. Aluminum concentration in the brain and bone of rats fed citric acid, aluminum citrate or aluminum hydroxide. Food Chem Toxicol. 1984;27:391-397.
157 Slatopolsky E., Bricker N.S. The role of phosphorus restriction in the prevention of secondary hyperparathyroidism in chronic renal disease. Kidney Int. 1973;4:141.
158 Slatopolsky E., Caglar S., Pennell J.P., et al. On the pathogenesis of hyperparathyroidism in chronic renal insufficiency in the dog. J Clin Invest. 1971;50:492.
159 Slatopolsky E., Caglar S., Gradowska L., et al. On the prevention of secondary hyperparathyroidism in experimental chronic renal disease using “proportional reduction” of dietary phosphorus intake. Kidney Int. 1972;2:147.
160 Slatopolsky E., Rutherford W.E., Rosenbaum R., et al. Hyperphosphatemia. Clin Nephrol. 1977;7:138-146.
161 Slatopolsky E., Weerts C., Lopez-Hilker S., et al. Calcium carbonate as a phosphate binder in patients with chronic renal failure undergoing dialysis. N Engl J Med. 1986;315:157-161.
162 Slatopolsky E., Lopez-Hilker S., Delmez J., et al. The parathyroid-calcitriol axis in health and chronic renal failure. Kidney Int. 1990;38:S41-S47.
163 Smith S.A., Tobias A.H., Jacob K.A., et al. Arterial thromboembolism in cats: acute crisis in 127 cases (1992–2001) and long-term management with low-dose aspirin in 24 cases. J Vet Int Med. 2003;17:73-83.
164 Spangler W.L., Muggli F.M. Seizure-induced rhabdomyolysis accompanied by renal failure in a dog. J Am Vet Med Assoc. 1978;172:1190-1194.
165 Spangler W.L., Gribble D.H., Lee T.C. Vitamin D intoxication and the pathogenesis of vitamin D nephropathy in the dog. Am J Vet Res. 1979;40:73-83.
166 Suryadevara M., Schurman S.J., Landas S.K., et al. Systemic calciphylaxis. Pediatr Blood Cancer. 2008;51:548-550.
167 Taylor J.A., Jacobs R.M., Lumsden J.H., et al. Perspectives on the diagnosis of feline hyperthyroidism. Can Vet J. 1989;30:477-481.
168 Tenenhouse H.S., Murer H. Disorders of renal tubular phosphate transport. J Am Soc Nephrol. 2003;14:240-247.
169 Thatte L., Oster J.R., Singer I., et al. Review of the literature: severe hyperphosphatemia. Am J Med Sci. 1995;310:167-174.
170 Turrel J.M., Feldman E.C., Nelson R.W., et al. Thyroid carcinoma causing hyperthyroidism in cats: 14 cases (1981–1986). J Am Vet Med Assoc. 1988;193:359-364.
171 von Landenberg P., Shoefeld Y. New approaches in the diagnosis of sepsis. Isr Med Assoc J. 2001;3:439-442.
172 Weir E.C., Morrdin R.W., Barthold S.W., et al. Primary hyperparathyroidism in a dog: biochemical, bone, histomorphometric, and pathologic findings. J Am Vet Med Assoc. 1986;189:1471-1474.
173 Willard M.D., Zerbe C.A., Schall W.D., et al. Severe hypophosphatemia associated with diabetes mellitus in six dogs and one cat. J Am Vet Med Assoc. 1987;190:1007-1010.
174 Wilson J.W., Harris S.G., Moor W.D., et al. Primary hyperparathyroidism in a dog. J Am Vet Med Assoc. 1974;164:942-946.
175 Wolford S.T., Schroer R.A., Gohs F.X., et al. Effect of age on serum chemistry profile, electrophoresis and thyroid hormones in beagle dogs two weeks to one year of age. Vet Clin Pathol. 1988;17:35-42.
176 Xia Z., He Y., Yu J. Experimental acute toxicity of xylitol in dogs. J Vet Pharmacol Ther. 2009;32:465-469.
177 Yawata Y., Craddock P., Hebbel R., et al. Hyperalimentation hypophosphatemia: hematologic and neurologic dysfunction due to ATP depletion. Clin Res. 1973;31:729A.
178 Yawata Y., Hebbel R.P., Silvis S., et al. Blood cell abnormalities complicating the hypophosphatemia of hyperalimentation: erythrocyte and platelet ATP deficiency associated with hemolytic anemia and bleeding in hyperalimented dogs. J Lab Clin Med. 1974;84:643-653.
179 Young D.S. Effects of drugs on clinical laboratory tests. Washington, DC: AACC Press; 1995.
180 Zazzo J.F., Troche G., Ruel P., et al. High incidence of hypophosphatemia in surgical intensive care patients. Efficacy of phosphorus therapy on myocardial function. Intensive Care Med. 1995;21:826-831.