Cancer Chemotherapy
General Principles of Cancer Chemotherapy
The use of chemical elixirs for the treatment of cancer can be traced through the medicinal customs and practices of a number of cultures.1 The modern use of pharmacologic agents to treat cancer began in the mid-1940s when Alfred Gilman and Louis Goodman showed the efficacy of nitrogen mustard in tumor-bearing mice, and these results were quickly translated and verified in human patients. These results and the efforts of others such as Sydney Farber with antifolates and George Hitchings and Gertrude Elion with purine analogs rapidly advanced the growing interest of treating cancer with drugs. The beginning of a systematic screening program for anticancer drugs at the National Cancer Institute (NCI) in 1955 set the framework for cancer chemotherapy development in both the public and private sectors and led to the characterization of many of the agents still in clinical use today.2,3
The basis of anticancer drug activity is the targeting of dividing cells through interference with processes involved in progression through the cell cycle. As shown in Figure 11-1, the major classes of drugs used to treat cancer work at various steps in the processes of DNA replication (S phase) and subsequent cell division (M phase). Another set of therapeutic agents, the signal transduction inhibitors, work by interfering with the signaling processes that trigger entry into the cell cycle and continuing cellular proliferation. This newer class of agents is discussed in Chapter 14, Section B, of this text. DNA synthesis is a complicated process involving anabolic processes to create the purine and pyrimidine nucleotide triphosphates required for replication, unwinding of the template DNA to provide access to the replication machinery, and the high fidelity process of creating complementary strands. Anticancer drugs work at all of these levels of DNA synthesis, including the antimetabolites that inhibit anabolic processes required for providing the nucleotide building blocks, topoisomerase inhibitors that interfere with the enzymatic process of DNA unwinding, cross-linking agents that through either interstrand or intrastrand interactions block the processes of strand separation and template processing, and the alkylating agents that interfere with the replication machinery through multiple mechanisms of altered binding and base recognition. The resulting effects of interacting at these levels of DNA replication can include the generation of DNA strand breaks, incomplete replication, and triggering of apoptotic signaling such that cell death is the ultimate result.
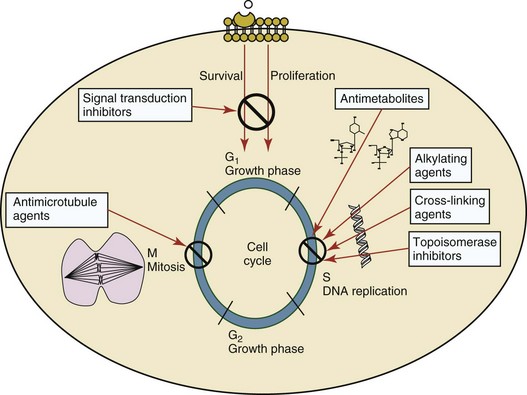
Figure 11-1 Cell cycle specificity of the major classes of drugs used in cancer chemotherapy. Although agents may have effects throughout the cell cycle, the phase where the major impact is realized is highlighted.
Processes in cell division not involving DNA replication are also targets for anticancer agents. The most prominent of these targets is tubulin with several classes of drugs having antitubulin activity. The mechanism of action of these agents involves either inhibiting the polymerization of tubulin or stabilizing the polymerized form so that depolymerization is blocked. The result of blocking either of these processes is the inhibition of microtubule function in the dividing cell. Microtubule function is critical to progression through mitosis via the spindle fiber formation and the separation of chromosome pairs into daughter cells. Blockade of this process by antitubulin agents has proved to be a very effective strategy of antiproliferation because cells blocked in this part of the cell cycle (M phase) can trigger apoptosis or undergo other mechanisms of cell death and loss of viability.
Terminology and Concepts
Terms that are related to the efficacy and toxicity of cancer chemotherapy are important concepts for understanding their pharmacologic activity. The therapeutic index for a given chemotherapeutic agent is the ratio between the toxic dose and the therapeutic dose for that drug. For most cytotoxic chemotherapy used to treat cancer, the therapeutic index is an abstract parameter because the administered dose is based on the maximum tolerated dose (MTD) rather than dose response. The MTD is an empirically derived value that represents the highest dose of a given drug that can be administered in the absence of unacceptable or irreversible side effects to a limited population sample. This is an important concept in cancer drug administration in that drug doses are generally based on this value rather than assessments of efficacy. A newer concept for drugs used to treat cancer is the biologically effective dose (BED), based on a measured response at a putative target or surrogate that is related to the mechanism of action of the agent. Determination of the BED is currently more related to the use of signal transduction inhibitors and molecularly targeted agents; however, the concept is not exclusive to these agents and this approach may be useful when applied to cytotoxic chemotherapy using dosing protocols not based on the MTD. Dose intensity (DI) is a measure of dose per unit of time and thus allows comparisons between protracted and compacted dosing schedules. Comparisons of DI between, for example, every 3 weeks and every week dosing allows for determining whether the total dose of the drug or the DI relates to toxicity or therapeutic outcome and the impact that altering dosing schedules can have on outcome. Therapeutic gain is often evaluated when combining two drugs or drug-radiation therapy combinations and quantitatively describes any improved tumor response relative to increased normal tissue toxicity when agents are used in a planned schedule. The basis for a positive therapeutic gain is the additive or synergistic tumor effects that exceed any summative toxicity patterns in normal tissues accomplished with combination therapy.
Indications and Goals of Therapy
The therapeutic intent and goals of a given chemotherapeutic regimen are important contributors to how a given drug is selected or assessed. Adjuvant therapy is the treatment with chemotherapeutic drugs following the surgical removal or radiation control of the primary tumor. The purpose is to treat occult disease and usually involves systemic drug exposure. Primary or neoadjuvant therapy is the utilization of chemotherapeutic drugs prior to treatment with other modalities, primarily surgical removal of the primary tumor, with the intent of decreasing tumor size for increased control and preventing possible postoperative growth of micrometastasis. Induction therapy is a similar concept to neoadjuvant therapy; however, this refers to the initial drug treatment phase with the intent of inducing remission in lymphoid or hematopoietic cancers. Maintenance therapy involves the use of chemotherapy in an ongoing basis to maintain remission. Consolidation therapy is intended to sustain an achieved remission. Rescue or salvage therapy is the use of chemotherapy after a tumor fails to respond to a previous therapy or after tumor recurrence. Palliative chemotherapy is delivered to decrease clinical signs in the case of unresectable or disseminated disease that is associated with functional disturbances or pain. The outcome of this therapy is based more on quality of life issues as opposed to other metrics of tumor response. The more subjective nature of assessing the effect of palliative chemotherapy, especially in terms of pain control, makes systematic testing of protocols difficult and treatment recommendations more at the discretion of the clinician and client. In the cases where organ function is impacted by tumor growth, more objective endpoints may exist in terms of functional improvements following treatment. Doses and scheduling of palliative intent therapies may also differ as strict adherence to schedules originating from trials where objective responses were measured may not be relevant and more patient-based endpoints employed. Radiosensitization is the enhancement of cytotoxicity when irradiation and chemotherapeutic agents are combined such that a therapeutic gain is obtained. The basis for chemotherapeutic exposure leading to enhanced radiosensitivity can be multifaceted and involve: (1) the enrichment of the tumor cell population in a more sensitive phase of the cell cycle, (2) increased tumor oxygenation through cytoreduction or alterations in tumor vascularization, and (3) selective killing of inherently radioresistant hypoxic cell fractions.
As a preliminary metric, the clinical measurements of the tumor response to cancer chemotherapy are useful for predicting the impact of treatment on the extent of disease or time interval of tumor control. Table 11-1 describes conventional measures of treatment response.
Table 11-1
Measures of Response in Cancer Therapy and Treatment
| Response Term | Abbreviation | Description |
| Complete remission/response | CR | Complete disappearance of tumor(s) and symptoms of disease. |
| Partial remission/response | PR | Decrease in tumor volume of ≥50% or decrease in tumor maximum diameter of >30%. |
| Stable disease | SD | Neither an increase nor a decrease in tumor size or disease symptoms (e.g., ±20% diameter changes). |
| Progressive disease | PD | Increase in tumor volume of >25% or increase of tumor maximum diameter of >20%; appearance of new lesions. |
| Median duration of response/median duration of survival | MDR/MDS | The median value for a group of individuals treated with a given therapy in terms of the length of time they achieved a complete or partial remission (MDR) or length of survival following implementation of therapy (MDS). |
| Progression-free interval/progression-free survival | PFI/PFS | The amount of time elapsed without evidence of progressive tumor growth (PFI) or survival without progressive growth of the tumor from treatment start (PFS). |
| Disease-free interval/disease-free survival | DFI/DFS | The amount of time that elapses without disease recurrence (DFI) or survival (DFS) of the patient following therapy. |
Tumor Susceptibility and Resistance
Individual cell sensitivity to anticancer agents has been addressed empirically through the screening of tumor cell panels associated with a given histotype. The NCI60 human tumor cell line panel is the most well characterized and studied compilation with more than 100,000 compounds and 50,000 extracts from natural products screened to date.4 Further, rich gene expression and other characterizations of these cell lines exist in public databases so that drug sensitivity and genotypic characteristics can be considered.5 The use of canine tumor cell line panels to screen drug sensitivity is becoming established6 as a viable way to identify potential drug combinations for further testing as well.
Chemosensitivity depends on a number of factors, including drug uptake into the cell, interaction with a cellular target, generation of lethal damage to important cellular macromolecules, repair of potentially lethal damage, and the cell’s response to generated damage as depicted in Figure 11-2. Uptake of some cancer chemotherapeutic agents occurs via passive diffusion due to their lipid-soluble properties; other compounds are actively transported into tumor cells. Melphalan is actively transported into cells by two amino acid transporters,7 and blocking transport with amino acid substrates or analogs can significantly reduce cytotoxicity.8 Other examples include nucleoside transporters used by Ara-C9 and gemcitabine10 and the reduced folate carrier system involved in methotrexate uptake.11 The intracellular target(s) for specific chemotherapeutic agents can play a role in determining sensitivity based on their levels and the nature of the interaction. For example, topoisomerase IIα levels can play a role in the sensitivity of tumor cells to doxorubicin12,13 as altered levels via decreased gene copy or transcriptional downregulation leads to a decrease in sensitivity (resistance). The opposite is true for thymidylate synthetase levels and 5-fluorouracil (5-FU) toxicity where increased levels of enzyme correlate to a decrease in sensitivity to 5-FU.14 Although the nature of the interaction with the target is different for doxorubicin and 5-FU, the fact that altered target levels can modulate response shows how quantitative interactions with the target can alter drug sensitivity.
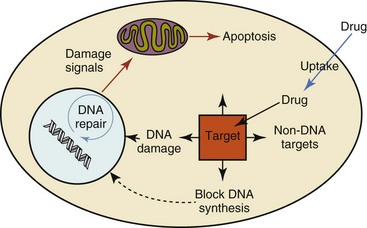
Figure 11-2 Processes involved in the pharmacologic activity and associated chemosensitivity of chemotherapeutic agents in tumor cells. Associated processes include drug uptake, interaction with drug target, impact on DNA and associated DNA repair, and the cellular response to these effects.
The extent of cellular damage, potential repair of that damage, and the cellular response occur in a tightly knit continuum that determines cellular fate. The generation of cellular damage is a consequence of interaction with a cellular target and can be either a primary or secondary event. In general for DNA-damaging agents, the resulting DNA lesions are caused by the interplay of DNA binding and DNA repair. For example, DNA strand breaks that result from O-6-methyl guanine lesions are due to aberrant mismatch repair processes and subsequent replication.15 DNA damage also triggers response pathways that can result in cell cycle arrest to allow for repair and subsequent survival or the triggering of apoptotic machinery that ultimately results in cell death. The definition of cellular response, whether mitotic catastrophe, apoptosis, necrosis, autophagy, or cellular stasis, depends on an intricate interplay of survival and death signaling and is often specific to the agent, dose at the critical target, and the cell lineage.16 Alterations in proapoptotic and antiapoptotic signaling clearly play a role in tumorigenesis and response to therapy,17 and the impact of antiapoptotic signaling in lymphoma by the mediators, bcl-2 and survivin, seem the most clear in regards to both chemosensitivity18-20 and response to therapy in humans and dogs.21-23 However, a clear understanding of the role of damage response and active cell death pathways in chemotherapeutic sensitivity and response in solid tumors is still lacking.
Tumor Cell Resistance
Acquired resistance, or selection of resistant cells, during the treatment process is thought to be one of the major mechanisms of therapeutic failure during cancer drug therapy. Resistance of tumor cells to chemotherapeutic agents can depend on the drug and its mechanism or be through a multidrug mechanism. In general, the development of acquired resistance to a specific agent can come via a variety of mechanisms associated with drug uptake, drug metabolism/detoxification, target modification, damage repair, or damage recognition and response. Changes in cellular drug levels can come about due to either a decrease in drug uptake or through increased efflux. Decreased expression of transporters known to play a role in drug uptake have been observed in response to treatment with melphalan in human breast cancer cells24 and acquired resistance to methotrexate in KB cells,25 with the resulting cells showing drug resistance that correlated with the lower intracellular drug levels. The induction of drug efflux pumps in response to drug treatment is a primary mechanism for multidrug resistance and will be discussed later in this section.
Alterations in metabolic or detoxification pathways within tumor cells are another mechanism by which acquired resistance occurs. Due to the fact that many chemotherapeutic agents are electrophilic based on their DNA-binding properties, enhancement of conjugation reactions with nucleophiles such as glutathione is a plausible mechanism of resistance.26 Induction of glutathione S-transferases has been shown to be a mechanism by which tumor cells can acquire resistance to nitrogen mustards.27,28 Although tumor cells themselves generally have limited drug metabolism capabilities, some metabolic pathways can play a role in the resistance phenotype. For example, the sensitivity of tumor cells to 5-FU is inversely correlated with the expression of dihydropyrimidine dehydrogenase,29-31 the enzyme predominantly responsible for the metabolism of 5-FU to the inactive 5-FUH2 metabolite.32 For prodrugs such as gemcitabine, which must be phosphorylated to the di- and tri-phosphate forms prior to eliciting an inhibitory effect on DNA synthesis,33,34 the enzyme responsible for this metabolic activation, deoxycytidine kinase,35,36 has been shown to be decreased in pancreatic tumor cells made resistant to this drug.37,38 This interplay between metabolic detoxification and activation, predominantly with drugs that are nucleotide analogs, leads to complex scenarios involving the upregulation of catabolic processes and the downregulation of anabolic processes regarding the cellular pharmacology of these cytotoxic agents.
Modifications in the cellular target of a given drug usually pertain to mutations in the target protein leading to a decrease in affinity or absence of drug interaction. These modifications can include a decrease in the levels of a specific target responsible for the generation of a toxic product, increases in target levels to ameliorate the effect of target inhibition, or target mutations such that the drug can no longer interact in a manner detrimental to the tumor cell. Decreased topoisomerase II gene expression and activity has been observed in human lung and colon cells with acquired resistance to the epipodophyllotoxins,39 etoposide, and teniposide, whose antitumor activity involves topoisomerase II–dependent DNA strand break formation.40,41 Target amplification as a mechanism of acquired resistance has been observed in methotrexate resistance where gene amplification and increased dihydrofolate reductase (DHFR) levels allow for cells to overcome DHFR inhibition by this agent.42 Mutations in targets such as β-tubulin in the case of paclitaxel43 and topoisomerase I for camptothecin44 affect binding of drug and interaction with the target, thus generating tumor cells resistant to the toxic mechanism of these agents. These examples of altered target levels and structures show that drug resistance can come about via either quantitative or qualitative change in the nature of the interaction of drug and target and will depend on the impact of these changes on tumor cell growth both in the absence and presence of the selective agent.
Damage repair in cancer cells treated with chemotherapy commonly refers to DNA repair processes since a majority of chemotherapy agents work at the level of the DNA. Resistance conferred through alteration in DNA repair include not only the induction of specific processes to repair discrete lesions but also more global DNA repair processes, such as postreplication and mismatch repair.45 Multiple studies have shown that enhanced removal of platinum adducts from tumor cell DNA correlate with acquired resistance46-48 to cisplatin, although the exact mechanism(s) and protein(s) responsible for repair of these lesions are unknown. The bulky DNA adducts generated by many cancer chemotherapeutic agents can cause replicative gaps in DNA that require postreplication surveillance and repair. The ability of cells to bypass these bulky lesions and interstrand cross-links during DNA replication has been found to be an important process in tolerance to agents causing these types of DNA damage (cisplatin, mitomycin C, melphalan); multiple DNA repair pathways can account for this release from DNA replication block.49 The fact that DNA repair pathways and processes are redundant and nondiscrete and that both lesion specific and global processes seem to play a role in determining drug resistance highlights the problems associated with attributing specific proteins or pathways to specific resistance phenotypes.
Some mechanisms of acquired resistance result in a phenotype in which the tumor is resistant to multiple chemotherapeutic agents or multidrug resistant (MDR). Some of the mechanisms discussed previously, including DNA repair, enhanced metabolism, or detoxification, and resistance to apoptosis can result in resistance to multiple agents; however, the MDR phenotype generally refers to tumor cells expressing individual or multiple members of the adenosine triphosphate (ATP)-binding cassette (ABC) transporter family who play a primary role in active efflux of drugs from cells. Forty-eight ABC genes have been identified in the human genome,50 and currently, fifteen members of the ABC transporter family have been recognized that include a cancer chemotherapeutic as a substrate for transport.51 These include the well-studied and characterized PGP/MDR1 (ABCB1), MXR/BCRP (ABCG2), MRP1 (ABCC1), and MRP2 (ABCC2). The basic function of the ABC transporters is conserved across the family and involves the ATP-dependent transport of xenobiotics and endogenous substrates from the inside of the cell to the extracellular space. The role of ABC transporters in multidrug resistance of canine and feline cancers is poorly explored; however, ABCB1 is expressed in canine lymphoma,52 canine mammary tumors,53 and canine and feline primary pulmonary carcinomas.54 ABCC1, ABCC2, ABCC5, ABCC10, and ABCG2 have all been shown to be expressed in canine mammary tumors as well.53,55 The normal tissue distribution of the ABC transporters is also beginning to be investigated in dogs, with initial studies showing similar tissue distributions and presumed function, although there appears to be some partial differences in relative expression in various tissues.56,57 A recent study has shown that feline ABCG2 has specific amino acid changes that lead to transporter dysfunction with regard to a number of substrates, suggesting that cats may have altered pharmacokinetic disposition for drugs that are ABCG2 substrates.58
Combination Therapies
The success of combination chemotherapy as compared to single-agent treatment is attributed to the overcoming of both natural and acquired resistance of tumor cells, as well as use of agents that differ in dose-limiting side effects. A premise of cancer chemotherapy is to kill the largest fraction of tumor cells possible with each dose, with the dose and timing of each therapy based on the normal tissue tolerance. Therefore a strategy that allows for more intensive cycles of fractional cell killing without exacerbating the recovery of normal tissue damage is preferred. Combination chemotherapy has been shown to be curative in humans with acute lymphocytic leukemia (ALL), Hodgkin’s disease, histiocytic lymphoma, and testicular carcinoma, whereas single-agent therapy was not.59 The success of combination therapies as opposed to single-agent therapy is best illustrated in veterinary oncology by treatment protocols for canine lymphoma. Doxorubicin is the most active single-agent therapy tested against canine lymphoma; other combination protocols that generally consist of cyclophosphamide, vincristine, and prednisone result in similar outcomes (Table 11-2). However, these same combination protocols with doxorubicin included empirically seem to increase both the median remission and median survival times over either doxorubicin alone or a combination protocol that excludes doxorubicin. It should be noted that the populations represented in Table 11-2 may not be homogeneous, and this data does not represent a formal reanalysis of these data sets.
Table 11-2
Response of Canine Lymphoma to Single-Agent Doxorubicin, Combination Protocols, and Combination Protocols Including Doxorubicin*
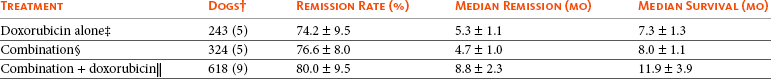
*Values represent the mean ± standard deviation (SD) from the cited studies. The combination protocols used included cyclophosphamide, vincristine, and prednisone, with methotrexate and actinomycin D included in some.
†Number represents the total number of dogs, with the number of individual studies in parentheses.
‡Data for doxorubicin alone studies were from references 60-64.
§Data for combination studies were from references 61, 65-68.
 Data for combination studies, including doxorubicin, were from references 68-76.
Data for combination studies, including doxorubicin, were from references 68-76.
Toxicities Associated with Drug Therapy of Cancer
Chemotherapy may fail to produce a positive clinical benefit for the reasons described previously but may also fail due to unacceptable toxicity. Anticipating and managing adverse events requires a thorough understanding of drug activity profiles and clinical experience modifying chemotherapeutic administration. The first step in the process of successfully managing cancer in companion animals is always a clear and frank discussion with the owner regarding the potential for benefit, toxicity, cost, and time commitment. A common understanding of the goals of therapy and committing to a continuing dialog as needs may change throughout treatment cannot be underestimated.
Dosing conventions have been developed from formal phase I studies for an increasing number of agents investigated specifically in companion animals. Nonetheless, suggested starting doses represent an estimate of the MTD from a small population of animals and safe individual patient dosing may vary substantially. There are numerous reasons for pharmacokinetic variability in cancer chemotherapy among a population of patients.77 Concurrent illness or organ dysfunction, extreme tumor burden, specific breed sensitivities (e.g., Collie with ABCB1 mut/mut) or idiosyncratic considerations (anticipated drug-drug interactions or drug allergies) will mandate modification of the protocol and dosing. Concurrent illness and organ dysfunction can also have profound effects on selection of anticancer agents and dosing. In general, predictable dose adjustments for pets with renal or hepatic disease have not been developed and treatment should be approached conservatively. Interestingly, in cats, the glomerular filtration rate (GFR) can be used to define an individual dose for carboplatin that will permit some patients with renal disease to be safely dosed that would not have been safe if dosed by conventional methods.78 Dose adjustments of 30% to 40% have been recommended for drugs that are ABCB1 substrates in Collie-type breeds in which an ABCB1 (mut/mut) phenotype is confirmed, and even in dogs with an ABCB1 (wt/mut) phenotype, dose adjustments may need to be made.79 Chemotherapeutic dosing in obese patients often raises questions about drug partitioning in lipid storage sites around the body. Distribution of many pharmaceutical agents may be affected in obese patients; however, there is no accepted scale for empiric dose adjustments in humans. Individual factors such as the specific drug, degree of obesity, and other comorbidities may convince a clinician to dose reduce or cap the dose of a chemotherapeutic agent.80 Some reviews suggest that dose reductions based on body mass may ultimately be detrimental to outcomes in obese patients.81 It is the initial chemotherapeutic intervention that is expected to result in the greatest opportunity to benefit the patient; therefore taking the time to assess the patient’s specific medical limitations and then proceeding with thoughtfully designing, administering, and completing a therapeutically robust protocol are highly desirable.
As individual patient tolerance and response to each compound in a multiagent protocol is observed, future modifications may be anticipated more accurately. The greatest benefit achievable with anticancer cytotoxic therapy requires a commitment to dose intensity. Optimal dose intensity demands therapeutic monitoring in order to either reduce or increase the dose based on the patient’s capacity to maintain a high quality lifestyle during effective therapy. The decision to increase the dose of an agent is conceptually challenging but important. In order to make a recommendation to increase dosing of a cytotoxic compound, owner understanding and monitoring of the patient’s white blood cell values and clinical events during the first treatment cycle are critical. A dose of a cytotoxic agent that does not result in any change in the target normal tissue (e.g., blood neutrophil count) is likely ineffective and could be increased 10% at the next infusion with continued follow-up to determine adequacy of dose adjustments (Figure 11-3). Dose reductions are deleterious to the optimum delivery of chemotherapy but are to be anticipated. Specific guidelines for dose adjustments of antineoplastic agents are not standardized. In general, a 20% to 25% reduction is recommended for the subsequent dose for patients experiencing a moderate or severe dose-limiting toxicity, such as neutropenia or emesis. Close monitoring and preemptive handling of signs may permit successful management of some potential future clinical signs, and clinical decisions are based on the extent and severity of the resulting signs as described in Table 11-3.
Table 11-3
Guidelines for Common Chemotherapy-Induced Toxicity
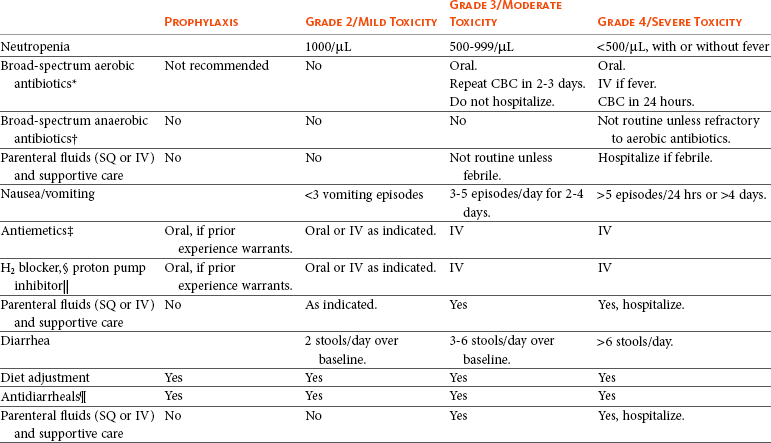
SQ, Subcutaneous; IV, intravenous; CBC, complete blood count.
*Fluoroquinolone (enrofloxacin 5-10 mg/kg once daily).
†Ampicillin 20 mg/kg oral (PO) or IV three times/day (TID) ± cephalosporin 20 mg/kg oral or IV TID.
‡Maropitant 2 mg/kg PO or SQ once daily × 3-5 days—dogs; 1 mg/kg PO or SQ once daily × 3-5 days—cats (not labeled for cats).
§Famotidine 0.5-1.0 mg/kg PO, SQ, or IV.
Pantoprazole 1 mg/kg IV or SQ as needed.
¶Loperamide 0.08 mg/kg PO TID; tylosin 10 mg/kg PO TID; metronidazole 15-25 mg/kg PO BID.
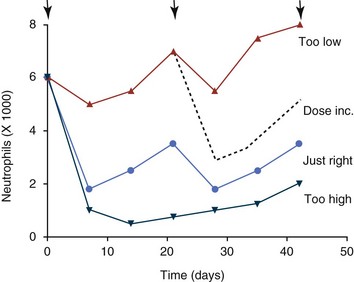
Figure 11-3 Blood neutrophil patterns following chemotherapy treatments (arrows). The appropriate dose (circles) results in noticeable nadirs with return to normal prior to the next dose. Doses that are too high or too low should prompt dose adjustments, including potential dose increases (dashed line).
The toxicity profile for anticancer agents may be categorized into immediately evident toxicities (at the time or within 24 to 48 hours after treatment), acute delayed effects (2 to 14 days), or cumulative/chronic toxicity (weeks, months, or years). Immediate toxicity may include infusion hypersensitivities due to histamine release associated with allergic reactions (l-asparaginase) or vehicle-induced mast cell degranulation (e.g., paclitaxel, etoposide). Routine management of these events with antihistamines and steroids may significantly reduce or eliminate this problem. Acute nausea and vomiting may occur with specific agents (e.g., cisplatin) or when the infusion is too rapid (e.g., doxorubicin). Preemptive antiemetic management is able to manage these situations well. Chemotherapeutics with vesicant properties can cause moderate-severe tissue necrosis if not administered safely through a suitable catheter. Vinca alkaloid and doxorubicin extravasations can be a very severe situation that should be avoided even if sedation is required or rescheduling must be recommended for safe catheter placement. Owners may need to be informed about this possibility prior to treatment and a management plan for this situation should be developed. Management recommendations for extravasations are included in the individual drug descriptions later.
Delayed acute effects from chemotherapy often include bone marrow suppression and nausea, vomiting, and diarrhea. In the majority of instances, these effects are self limiting and the incidence of hospitalization for such problems is low. Table 11-3 reviews the general therapeutic strategies for management of the most common types of adverse events experienced in companion animals following chemotherapy.82
Examples of potential cumulative and/or chronic toxicity include hepatic dysfunction after multiple doses of cyclohexylchloroethylnitrosourea (CCNU), cardiac abnormalities after exceeding a safe cumulative dose of doxorubicin, and renal disease after cisplatin use in dogs or doxorubicin use in cats. Screening recommendations and strategies to reduce the risks of such chronic effects have been developed and are incorporated into standard protocol procedures. It is critical to the success of treatment that owners be thoroughly informed about monitoring guidelines for the general signs and symptoms of chemotherapy-induced toxicity. Online educational resources for owners are readily available at www.csuanimalcancercenter.org. It is advisable to instruct the owner regarding monitoring and early responses when their pet experiences nausea and vomiting, diarrhea, or hematuria and it is important to inform the owner about how to obtain an accurate body temperature. These “at home” aids will allow the clinician to assess the management options should a concern arise.
Safety Concerns of Cancer Drug Therapy
In general, safety concerns for cancer therapy are only applied to the patient with regard to the impact of drug treatment. An issue with cytotoxic chemotherapy, however, is the preparation and distribution of the drugs, as well as active drug eliminated in the urine and feces of the patient and the potential exposure of health professionals, caregivers, and others in the home. The preparation of these drugs should be done under strict regulations and involves the use of protective clothing, gloves, masks, and chemical hoods. Studies have characterized the potential impact of secondary exposure to these agents on oncology health workers in human medicine with regard to cancer prevalence, reproductive risks, and acute toxicities, and the results show little risk.83 However, these results are a reflection of exposure in trained cohorts of individuals working in human medicine where fecal and urinary exposures are more limited. Clients with animals undergoing cancer therapy need to be informed of potential risks and safety precautions that need to be adhered to when dealing with oral medications and pet excrement. Simple precautions such as wearing gloves when handling oral medications and not opening capsules or splitting tablets are essential. Oral suspensions of these agents should be avoided. Urinary levels of some active drugs may remain high for days after treatment84 and fecal excretion may also be expected. Therefore careful avoidance, collection, and disposal of urine and feces must be recommended, as well as having pregnant women, small children, and immunosuppressed individuals in particular avoid any contact with pet wastes for a defined time period following treatments. Preparation of guidelines for minimizing exposure to individuals and the environment should be prepared and distributed to clients for general, as well as drug-specific, instructions.
Pharmacologic Principles in Cancer Therapy
Pharmacokinetic (PK) considerations in cancer drug therapy are important due to the relationship between drug exposure and pharmacodynamic (PD) response, whether efficacy or toxicity, that is more exact than the relationship between drug dose and PD response.85 PK considerations are also important with regard to interactions with other drugs,86 herbal products,87,88 and genetic differences among breeds and individuals89 that can cause changes in drug exposure at a given dose. Cytotoxic chemotherapy is usually dosed on an MTD-based schedule reflecting only acceptable toxicity and thus limits any informative role of drug half-life and effective therapeutic concentrations from initial dosing considerations. The most important PK parameters are those that have a relationship with either a response to therapy (efficacy) or toxicity, which is most often either the area under the plasma/serum concentration versus time curve (AUC) or the maximum drug concentration (Cmax) achieved, illustrated in Figure 11-4. The relationships of AUC and Cmax in the clinical pharmacology of doxorubicin illustrate the complex associations with PK considerations. The Cmax during doxorubicin infusion in humans is related to the incidence of cardiotoxicity both in adult90 and pediatric91 patients but is also associated with longer remissions in leukemia patients.92 A relationship between AUC values and decreased white blood cells has also been established with doxorubicin.93 However, no clear relationships between AUC and efficacy exist.94 These data have allowed for adjustments in doxorubicin dosing protocols so that intermediate infusion times (10 to 30 minutes) are utilized to decrease the Cmax and thus cardiotoxicity while still maintaining peak levels associated with effective therapy.
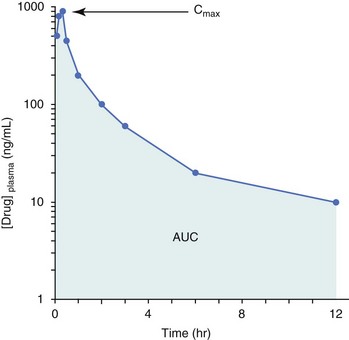
Figure 11-4 Illustration of pharmacokinetic parameters Cmax and AUC in a theoretical drug plasma concentration versus time plot.
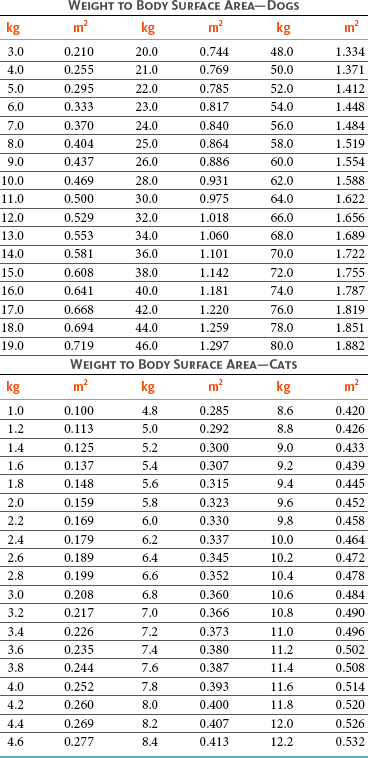
PK studies that relate drug exposure to responses are an important first step in establishing relationships that may be exploited for dose modification based on patient characteristics or therapeutic drug monitoring. These data are generally lacking for drugs used to treat cancer in companion animals with a few exceptions. Studies on the PK and myelotoxicity of carboplatin in cats have shown a clear relationship between drug exposure and the neutrophil nadir as well as drug clearance and GFR (Figure 11-5). The fact that PK parameters can be correlated both with a toxic endpoint and a physiologic function allows for the calculation of a dosing metric relating the GFR of an individual cat to a dose that produces a drug exposure (AUC) that results in acceptable toxicity.78 It remains to be determined whether such individualized dosing results in improved outcome in a heterogeneous population. Current drug-dosing convention for cancer chemotherapeutic agents is the use of body surface area (BSA) for dose normalization (mg/m2). Exceptions to this paradigm are the use of body weight (mg/kg) for dogs that weigh less than 15 kg and for cats with doxorubicin dosing based on empiric evidence showing a better toxicity profile for smaller dogs when mg/kg dosing is used.95 The approximate calculation for BSA in dogs and cats based on weight is as follows:

Figure 11-5 Relationship between (A) neutrophil nadir and carboplatin exposure and (B) platinum clearance and glomerular filtration rate (GFR) in cats being treated for cancer. Rights were not granted to include this figure in electronic media. Please refer to the printed book. (From Bailey DB, Rassnick KM, Erb HN, et al: Effect of glomerular filtration rate on clearance and myelotoxicity of carboplatin in cats with tumors, Am J Vet Res 65:1502, 2004.) Am J Vet Res
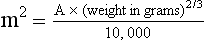
where A is equal to 10.1 for dogs and 10.0 for cats. The implementation of this equation relating body weight in kilograms to BSA in meters squared is shown for dogs and cats in Table 11-4.
Pharmacodynamics
PD considerations for cytotoxic chemotherapy are generally related to standard measures of response (i.e., complete remission [CR], partial remission [PR], stable disease [SD]) and toxicity.96 A majority of the literature in veterinary oncology relates PD responses to specific drugs or combinations, doses, or schedules. Figure 11-6 shows the relationships between vinblastine doses and the incidence of grade III or IV neutropenia observed in a phase I cohort of dogs.97 These results relate a dose to a PD response with the absence of exposure PK data. PD endpoints can also be used as indicators of efficacy and potentially as targets of therapy. The proportion of dogs in remission following treatment for lymphoma is increased in the subgroup experiencing grade III or IV neutropenia compared to the group that did not show that level of observed toxicity (Figure 11-7).98 In this example, the therapeutic response was related to overall drug effects on normal tissues as indicated by the degree of neutropenia (PD response), whereas DI did not show a significant difference. Again, these data did not include exposure PK assessment and in this case only relate the therapeutic outcome to an observed drug response. A lack of complete PK/PD data relationships in veterinary medicine reduces the opportunities for therapeutic drug monitoring and the potential for optimizing efficacy.
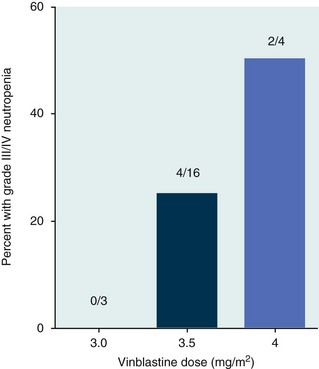
Figure 11-6 Relationship of prevalence of grade III/IV neutropenia with vinblastine dose in dogs being treated for cancer. (Data from Bailey DB, Rassnick KM, Kristal O, et al: Phase I dose escalation of single-agent vinblastine in dogs, J Vet Intern Med 22:1397, 2008.)
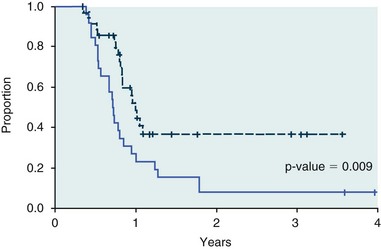
Figure 11-7 Proportion of dogs in remission following chemotherapy treatment for lymphoma. The dashed line represents those animals experiencing grade III/IV neutropenia, whereas the solid line represents those animals that did not show that level of toxicity. (From Ghan A, Johnson JL, Williams LE: Impact of chemotherapeutic dose intensity and hematologic toxicity on first remission duration in dogs with lymphoma treated with a chemoradiotherapy protocol, J Vet Intern Med 21:1332, 2007.)
Pharmaceutics
Pharmaceutics is the science associated with dosage form design with regard to formulation and optimizing drug delivery via a specific route. For example, improved formulations of clinical agents such as paclitaxel have made these compounds available for use in veterinary patients. The excipient (drug carrier) used in the original clinical formulation of paclitaxel (Taxol) was Cremophor EL, which causes histamine release and unacceptable toxicity when used in dogs and cats.99,100 A new water-soluble formulation of paclitaxel (Paccal Vet) has been tested and shown to be effective without associated hypersensitivity reactions in dogs with mast cell tumors. The ability of new formulations and delivery methods to alter the efficacy and toxicity profile of agents is a rapidly expanding field. It is expected that new technologies in drug formulation and targeting will be incorporated into veterinary medicine to alter drug delivery and distribution in a more favorable manner.
Specific Chemotherapeutic Agents
The alkylating agents are comprised of antitumor drugs whose mechanism of action involves the covalent binding of alkyl groups to cellular macromolecules. The cellular target of these agents is DNA in which they form monofunctional or bifunctional adducts that generate interstrand or intrastrand cross-links.
Nitrogen Mustards
Mechlorethamine
Basic Pharmacology: Mechlorethamine is frequently referred to as “nitrogen mustard” and was the first cytotoxic agent to show antineoplastic activity.101-103 Mechlorethamine undergoes spontaneous hydrolysis to 2-hydroxyethyl-2-chloroethylmethylamine and bis-2-hydroxyethylmethylamine, yielding nucleophilic reactive centers capable of forming DNA cross-links.104
Clinical Pharmacology: Mechlorethamine rapidly disappears from the plasma following intravenous (IV) administration primarily through spontaneous degradation, although some percentage of the drug is enzymatically metabolized.105 Mechlorethamine uptake into cells seems to be carrier mediated with decreased uptake as a mechanism for resistance.106
Clinical Use: Mechlorethamine is used predominantly in multiagent protocols for lymphoma in dogs.107-109 Experience with mechlorethamine as a single agent is not reported, although gastrointestinal (GI) and bone marrow toxicity are dose-limiting toxicities of conventional mustargen, vincristine, prednisone, and procarbazine (MOPP) protocols. Dosing of mechlorethamine in these protocols is reported as 3 mg/m2 IV on days 0 and 7 of a 21-day cycle.
Melphalan
Basic Pharmacology: Melphalan (l-phenylalanine mustard) is a nitrogen mustard containing DNA cross-linking agent with a similar structure and pharmacology to chlorambucil. The major difference is that melphalan is actively transported into tumor cells by amino acid transporters7 and its uptake can be blocked by the amino acid leucine. Melphalan has direct alkylating activity and does not require metabolic activation.
Clinical Pharmacology: Melphalan can be given orally with an oral bioavailability of approximately 30%. A relatively high percentage of melphalan (20% to 35%) is excreted unchanged in the urine with a majority of the remainder of the dose undergoing spontaneous chemical decomposition to inert products.110 The primary toxicity is myelosuppression.
Clinical Use: The primary indication for melphalan in companion animals is for management of myeloma. The initial dose of 0.1 mg/kg PO daily for 10 to 14 days should be reduced to 0.05 mg/kg PO daily based on control of the paraproteinemia and hematologic screening for both dogs and cats. Alternate dosing regimens have also been used for dogs: 7 mg/m2 PO daily for 5 days every 3 weeks or 2 mg/m2 PO daily for 10 days with a 10-days-off cycle and repeated as needed.
Cyclophosphamide
Basic Pharmacology: Cyclophosphamide (CP) is a nitrogen mustard–containing prodrug that is inactive in the absence of metabolic activation that occurs via microsomal mixed function oxidases predominantly in the liver.111 The activation of CP involves ring oxidation to 4-hydroxycyclophosphamide (4-OHCP), spontaneous and reversible ring opening to the amino aldehyde aldophosphamide, and the subsequent irreversible breakdown of aldophosphamide to phosphoramide mustard and acrolein. Phosphoramide mustard is considered the most active CP metabolite and is capable of bifunctional alkylation and cross-link production.112
Clinical Pharmacology: Recent studies have characterized the PK of CP and the 4-OHCP metabolite following both IV and oral dosing in dogs.113 The results of this study show that although exposure to CP is decreased following oral dosing, the overall exposure of 4-OHCP is similar when CP is dosed either intravenously or orally. The major dose-limiting side effects of CP are neutropenia and thrombocytopenia. GI toxicity (nausea and vomiting) is not common in dogs but has been observed in cats.114 Other common toxicities include alopecia in dog breeds with continually growing hair and hemorrhagic cystitis. Although hemorrhagic cystitis is uncommon at conventional doses, furosemide is often administered (1 mg/kg subcutaneous [SQ] or IV) prior to IV injection and precautions are taken at home to encourage vigorous hydration and frequent urination. CP should be discontinued permanently if hemorrhagic cystitis occurs and chlorambucil may be substituted in a multiagent protocol. Urine culture, antiinflammatory drugs, and antispasmodics should be initiated at the onset of clinical signs of cystitis. Aggressive intravesical therapy or even surgery may be necessary in severe instances.
Clinical Use: CP is commonly included in multiagent protocols for lymphoma in both dogs and cats. It is effectively administered as a bolus dose (250 mg/m2) by either an oral (PO) or IV route in the dog.113 A fractionated dosing schedule is also used in some protocols (50 mg/m2 for 3 to 4 consecutive days after doxorubicin).115 Metronomic therapy, the use of very low dosing for prolonged periods, has also been developed for CP. The rationale and application of metronomic therapy is covered extensively in Chapter 14, Section C, of this text. CP is administered IV (200 to 250 mg/m2) in many multiagent protocols for lymphoma in cats. The efficacy of oral dosing of CP has not been as carefully investigated in cats compared to dogs although it may be safely administered at doses of 300 mg/m2 PO every 3 weeks in multiagent protocols.116
Ifosfamide
Basic Pharmacology: Ifosfamide (IP) is a nitrogen mustard–containing prodrug that like CP requires metabolic activation by microsomal mixed function oxidases prior to generating the isofosforamide mustard metabolite capable of bifunctional alkylation.117
Clinical Pharmacology: The major difference between the clinical use of IP and CP is due to differences in the relative metabolism of the parent drugs, with dechloroethylation accounting for up to 25% of the metabolism of IP,118 whereas this number is much smaller for CP. This difference in metabolism accounts for an increase in the formation of the neurotoxic metabolite chloroacetaldehyde following IP dosing and potentially for the less favorable metabolism profile observed with IP following oral dosing.119 The primary toxicity associated with IP treatment is a dose-related myelosuppression, but nephrotoxicity and damage to the bladder epithelium are not uncommon. Vigorous hydration is required with IP administration; in addition, mesna, a urinary epithelial protectant, must be administered to avoid severe cystitis.
Clinical Use: Ifosfamide has been evaluated in dogs and cats with cancer and is primarily recommended for management of sarcomas. The recommended dose for dogs is 300 to 350 mg/m2 IV slow infusion with diuresis every 3 weeks, and for cats, the recommended dose is 900 mg/m2 IV slow infusion with diuresis every 3 weeks.120,121 The basis for such discrepancies in the MTD between species is not understood but reflects profound and interesting differences in metabolism pathways and most likely reduced generation of bioactive metabolites. A phase II study in feline vaccine sarcomas reported moderate objective response rates.122
Chlorambucil
Basic Pharmacology: Chlorambucil (p-bis[chloro-2-ethyl] amino-phenyl-4-butanoic acid) is a nitrogen mustard derivative that enters cells via passive diffusion123 and has direct bifunctional alkylating ability124 that is responsible for the cytotoxic activity.
Clinical Pharmacology: Chlorambucil is orally bioavailable with rapid absorption. Hepatic metabolism is extensive with the pharmacologically active phenylacetic acid being the primary metabolite and is presumably responsible for much of the clinical activity.125,126 The major dose-limiting toxicity is myelosuppression, including neutropenia and thrombocytopenia.
Clinical Use: Chlorambucil is used primarily for control of chronic lymphocytic leukemia (CLL) in dogs and for low-grade GI lymphoma in cats. Chronic oral dosing in dogs should begin with 3 to 6 mg/m2 PO every day for 1 to 2 weeks with a decrease to 3 to 6 mg/m2 PO every other day as determined by routine hematologic screening and control of the cancer. A convenient oral bolus dose of 20 mg/m2 PO every 2 weeks has been reported with excellent response in feline GI lymphoma.127
Nitrosoureas
Lomustine (Cyclohexylchloroethylnitrosourea)
Basic Pharmacology: Lomustine (CCNU, CeeNU) is a nitrosourea-based agent that is highly lipid soluble and enters cells by passive diffusion.128 Under aqueous conditions and at physiologic pH, lomustine will spontaneously decompose to a reactive center capable of DNA alkylation129,130 and DNA-DNA and DNA-protein cross-links.131
Clinical Pharmacology: The highly lipophilic properties of lomustine allows for rapid crossing of biologic membranes, including the blood-brain barrier. Lomustine undergoes extensive hepatic metabolism,132 predominantly by hydroxylation of the cyclohexyl ring, to metabolites with at least equivalent alkylating activity133 that presumably play an important role in the cytotoxic activity. This extensive hepatic metabolism is presumably responsible for the lack of oral bioavailability of the parent compound but rapid appearance of metabolites following oral dosing.134 The major dose-limiting toxicity is myelosuppression with acute neutropenia followed by a thrombocytopenia.135 Chronic administration may result in hepatic dysfunction requiring discontinuation of the drug temporarily or permanently.136 A recent report investigating coadministration of Denamarin, a product that increases glutathione levels and provides antioxidant properties, with CCNU reported reduced frequency of grade 4 hepatic toxicity.137 Further evaluation of whether this strategy should be routinely employed to reduce chronic hepatic toxicity is needed.
Clinical Use: CCNU (70 to 80 mg/m2 PO every 3 weeks) is most often used alone or in multiagent protocols for canine multicentric lymphoma, epitheliotropic lymphoma, mast cell tumors, and histiocytic sarcoma. In cats, CCNU (50 to 60 mg/m2 PO every 4 to 6 weeks) is used primarily for mast cell tumors and lymphoproliferative disorders.
Streptozotocin
Basic Pharmacology: Streptozotocin is a naturally occurring nitrosourea capable of DNA alkylation and inhibition of DNA synthesis in both bacteria and mammalian cells.138,139 Cellular uptake of streptozotocin depends on the glucose transporter 2 (GLUT2) transporter and expression of this transporter determines sensitivity of both insulinoma140 and pancreatic beta cells.141
Clinical Pharmacology: Streptozotocin is rapidly cleared from the blood following IV administration with reported half-life of 15 to 40 minutes in humans.142 Streptozotocin has unique activities for nitrosoureas, including inducing diabetes in animals143,144 and a lack of any significant bone marrow toxicity.145,146
Clinical Use: Streptozotocin is used to manage malignant insulinoma. Limited reports of efficacy have appeared in the literature, although transient normoglycemia occurred in the experience of these authors.147 The drug is dosed at 500 mg/m2 as an IV infusion with diuresis to avoid renal toxicity, similar to the protocol for cisplatin.
Other Alkylating Agents
Dacarbazine
Basic Pharmacology: Dacarbazine, or DTIC, is a prodrug that requires metabolic activation by the hepatic cytochrome P450 system148,149 to the resulting 5-aminoimidazole carboxamide and the active methylating intermediate methyldiazonium ion.150 Resulting DNA methylation products are 3-methyl adenine, 7-methyl guanine, and O-6-methyl guanine,151 which are presumably responsible for the cytotoxic activity.
Clinical Pharmacology: Dacarbazine has poor oral bioavailability and is administered intravenously. Use in cats is not recommended due to a lack of information regarding their ability to convert the parent drug to the active form. Dacarbazine is extensively metabolized in the liver and excreted in the urine. The major dose-limiting toxicity is GI toxicity, although occasional severe myelosuppression can be observed.
Clinical Use: In dogs, dacarbazine is used as a component of protocols for lymphoproliferative diseases in a relapse setting and historically for melanoma. As a single agent, an IV infusion dose of 800 to 1000 mg/m2 every 3 weeks has been used.152 When combined with other cytotoxics, the dose of 600 mg/m2 IV has been reported.153
Procarbazine
Basic Pharmacology: Procarbazine (PCB), like dacarbazine, is a prodrug requiring chemical or metabolic alteration for the generation of active, toxic metabolites.154,155 The mechanism of action of PCB could involve multiple interactions, including inhibition of DNA and RNA synthesis, but a predominant role for DNA methylation to form O-6-methyl guanine seems likely.156
Clinical Pharmacology: PCB is rapidly and completely absorbed after oral administration followed by rapid disappearance of the parent compound and subsequent appearance of metabolites.157 PCB and/or metabolites equilibrate rapidly between the blood and cerebrospinal fluid.158 IV delivery has been tested in humans with the appearance of neurotoxicity not seen with oral delivery, suggesting that first-pass metabolism associated with oral dosing significantly alters the spectrum of exposure to parent drug versus metabolites.159
Antitumor Antibiotics
The antitumor antibiotics consist of natural products from microbial fermentation including the anthracyclines, mitomycins, and actinomycins that have yielded clinically useful compounds with diverse mechanisms of action. Included in the discussion here are the anthracycline doxorubicin and a synthetic analog of the anthracenediones (mitoxantrone) and actinomycin D.
Doxorubicin
Basic Pharmacology
The cellular pharmacology of doxorubicin (DOX) is dominated by its ability to react with a number of cellular components and a multimodal mechanism of cellular toxicity. Its activities include DNA intercalation and inhibition of RNA and DNA polymerases160 and topoisomerase II,161 alkylation of DNA,162 reactive oxygen generation,163,164 perturbation of cellular Ca2+ homeostasis,165,166 inhibition of thioredoxin reductase,167 and interaction with plasma membrane components.168 These processes are involved in both the antitumor and dose-limiting side effects of DOX, with their relative contributions still open to some debate.
Clinical Pharmacology
Following intravenous dosing, DOX is extensively distributed to tissues, with binding to cellular DNA169 and anionic lipids170,171 determining the magnitude of tissue uptake.172 The elimination of DOX occurs through renal and biliary elimination of parent drug, as well as metabolism to doxorubicinol and the 7-hydroxy aglycone. Metabolism to doxorubicinol is via side chain reduction mediated by aldo-keto reductases173 and 7-hydroxy aglycone by reductive cleavage of the sugar moiety both by the liver and extrahepatic tissues.174 The dose-limiting toxicities associated with DOX treatment are infusion-rate–dependent hypersensitivity, myelosuppression, GI toxicity, and a well-established cumulative dose-related cardiotoxicity.175 In addition, cats may develop renal tubular damage following repeated dosing.176
Clinical Use
DOX is the most active single agent available for a wide variety of cancers in companion animals. The drug may be used alone or in combination protocols for lymphoma, osteosarcoma (OSA), and most mesenchymal and epithelial neoplasms. Conventional dosing regimens are 30 mg/m2 slow IV bolus or infusion (10 to 30 minutes) every 3 weeks in dogs larger than 15 kg, 1 mg/kg for dogs smaller than 15 kg, and for all cats. This nonuniform dosing formula reflects the cumulative experience of specialists realizing that small dogs and cats are often overdosed with DOX using the BSA prescription base. Unfortunately, a uniform prescription model has not been developed, and thus dose adjustments should be anticipated following the initial DOX administration. The drug is diluted in saline and administered as a slow bolus or infusion over 10 to 30 minutes. Vigilant observation during the infusion is required to ensure proper IV delivery. Any concern about catheter placement should result in replacement using an alternate vein before beginning the infusion. Sedation may be required. If it is suspected that the drug has been delivered external to the vein, stop the infusion, aspirate remaining product out of the catheter, and remove it. Dexrazoxane (Zinecard) is used to reduce cardiac toxicity in humans, and infusion within a 3-hour period at 10 times the prescribed DOX dose may be useful for DOX extravasation.177 Additional infusions at 24 and 48 hours may be useful as well. If DOX is being administered, a source for dexrazoxane should be identified through a local hospital pharmacy because it is expensive and sometimes difficult to obtain rapidly. Despite active management, DOX extravasations may progress to extensive tissue necrosis requiring surgical intervention, including potential amputation.
Cardiac performance should be carefully evaluated prior to each DOX infusion to detect any new murmurs, arrhythmias, or pulse deficits. Routine electrocardiography or echocardiography is not necessary due to limited sensitivity and specificity of these monitoring procedures for DOX cardiotoxicity in dogs and cats. However, any abnormalities that develop during treatment should be pursued with full diagnostic evaluation. Most protocols ostensibly limit the DOX cumulative dose to 120 to 150 mg/m2 (4 to 5 doses) in order to limit potential toxicity in the general population and particularly in certain at-risk breeds, such as the Boxer and Doberman Pinscher. The cumulative dose of DOX may be increased as dictated by the need for additional treatment, such as in relapsed lymphoma following a formal cardiac evaluation. A complete blood count (CBC) is recommended prior to each DOX infusion, and in cats, a serum creatinine and urine specific gravity are recommended to identify any changes in renal function.176
Mitoxantrone
Basic Pharmacology
Mitoxantrone is a synthetic DOX analog and maintains similar activity as DOX in terms of DNA intercalation and the inhibition of RNA and DNA polymerases and topoisomerase II.178,179 However, mitoxantrone does not cause oxidative damage to cells180 and has a reduced potential to undergo one-electron reduction and generate reactive oxygen species.181
Clinical Pharmacology
Following IV administration, mitoxantrone is extensively distributed to tissues with residual levels being long lasting. Mitoxantrone is not extensively metabolized and a fraction of the drug (<30%) is excreted unchanged in the urine and feces.182-184 Dose-limiting toxicities include GI disturbances and myelosuppression. Cardiotoxicity has not been reported in dogs and only rarely in humans.
Clinical Use
Mitoxantrone (5 to 6 mg/m2 IV slow bolus every 3 weeks) is used as a cardiac-sparing anthracycline in dogs that have reached the cumulative level of DOX or with evidence of cardiomyopathy and is at risk of further damage with doxorubicin administration. The clinical indications for mitoxantrone include lymphoproliferative disorders and, most recently, transitional cell carcinoma (TCC) of the bladder and urethra.185
Actinomycin D (Dactinomycin)
Basic Pharmacology
Actinomycin D, or dactinomycin (DACT), consists of two symmetric polypeptide chains attached to a central phenoxazone ring. DACT has been shown to interact with double-stranded DNA in multiple ways in a sequence-dependent manner,186-188 and also bind to single-stranded DNA.189 The resulting interactions of DACT with both double- and single-stranded DNA results in a potent inhibition of transcription, thus inhibiting RNA and protein synthesis.190,191 DACT is taken up into cells by passive diffusion,192 and the sensitivity of cells may depend on uptake and retention193 with ABCB1 playing a role in DACT efflux.194
Clinical Pharmacology
Following IV administration, DACT is rapidly distributed to tissues and then slowly eliminated from tissues. Metabolism is minimal with 20% of DACT excreted unchanged in the urine and 14% in the feces.195 The major dose-limiting toxicities of DACT are myelosuppression and GI toxicity.195
Clinical Use
Actinomycin D is used in multi-agent protocols for dogs with lymphoproliferative diseases in the relapse setting or as a doxorubicin substitute in dogs with cardiac abnormalities. It is administered IV at 0.5 to 0.75 mg/m2 every 3 weeks. There is risk of perivascular damage following extravasation.
Antimetabolites
The antimetabolites are comprised of agents that inhibit the use of cellular metabolites in the course of cell growth and division. Therefore these agents are generally analogs of compounds used in the normal course of metabolism and in the case of cancer chemotherapeutics, specifically anabolic processes associated with DNA replication.
Cytosine Arabinoside (Cytarabine)
Basic Pharmacology
Cytosine arabinoside (Ara-C), or cytarabine, acts as an analog to deoxycytidine and is phosphorylated in cells to generate arabinosylcytosine triphosphate (ara-CTP), which acts as a competitive inhibitor of DNA polymerase α.196 Ara-CTP is also incorporated into DNA, which correlates with cytotoxicity197 and thus presumably is the primary mechanism of action. Once incorporated into DNA, it cannot be excised198 and inhibits both the function of the DNA template and subsequent synthesis.199 Ara-C has also been reported to have a differentiating function in leukemic cells through decreased c-myc expression.200 Ara-C is actively transported into tumor cells via nucleoside transporters201 and is phosphorylated sequentially by deoxycytidine kinase, deoxycytidine monophosphate (dCMP) kinase, and nucleoside diphosphate kinase.202
Clinical Pharmacology
Ara-C is water-soluble and dosed by IV infusion or SQ bolus injection. It distributes rapidly in total body water and crosses into the central nervous system (CNS), reaching levels 20% to 40% of those observed in the plasma.203 The primary mode of metabolism is deamination by the liver and extrahepatic tissues. Observed dose-limiting toxicities are myelosuppression and occasionally GI disturbances.
Clinical Use
Ara-C is an infrequent component of combination protocols for leukemias and lymphomas in dogs and cats. It is more often incorporated into treatment protocols for patients with a potential for CNS involvement. Ara-C is ideally administered as a constant rate infusion over a 4 to 5 day period. However, a more convenient method of administration in dogs and cats is SQ injection twice daily for 2 consecutive days at 150 mg/m2 (total dose = 600 mg/m2). Low-dose SQ Ara-C (50 mg/m2 twice daily for 2 days or 100 mg/m2 as a constant rate infusion for 1 day) has been reported to improve clinical signs in dogs with meningoencephalitis of undetermined origin when combined with prednisone.204,205 The collective reported data regarding efficacy of Ara-C for this condition are not sufficient to make a treatment recommendation at this time to use Ara-C in addition to prednisone. Results of a recent small study suggest improved responses in dogs with naïve stage V multicentric lymphoma when Ara-C (150 mg/m2/day as a continuous rate infusion) was infused over 5 days following the first and second cycle of a conventional cyclophosphamide, hydroxydaunorubicin (DOX), vincristine (Oncovin), prednisone (CHOP)-based protocol compared to the CHOP protocol alone.206 Bone marrow support with human granulocyte colony-stimulating factor (G-CSF) and erythropoietin were co-administered with Ara-C, and patients in this treatment group did not experience increased adverse events. Further investigation of this protocol is certainly of interest.
Methotrexate
Basic Pharmacology
Methotrexate (MTX) is a folate analog that inhibits the enzyme dihydrofolate reductase, thus depleting reduced folate pools required for purine and thymidylate biosynthesis.207 MTX is also converted to polyglutamates that act as direct inhibitors of folate-dependent enzymes that play a role in de novo purine and thymidylate synthesis.208,209 MTX enters cells via active transport through the reduced folate carrier.11
Clinical Pharmacology
The oral bioavailability of MTX is high at lower doses but becomes variable as doses increase210; thus it is usually dosed orally at lower doses and intravenously at higher doses. The PK of MTX is well understood across species211 and is dominated by enterohepatic recycling that accounts for the observed GI side effects at doses that do not cause hematopoietic toxicities. At higher doses, both GI toxicity and myelosuppression are observed. MTX does not undergo substantial hepatic metabolism except when administered at high doses and is primarily excreted unchanged in the urine.211
Gemcitabine
Basic Pharmacology
Gemcitabine, or 2,2-difluorodeoxycytidine (dFdC), is actively transported into cells by nucleoside transporters212 and metabolized by phosphorylation to dFdC monophosphorylated (dFdCMP), dFdC diphosphorylated (dFdCDP), and dFdC triphosphorylated (dFdCTP) species.202 The effect of dFdC treatment on cells is the inhibition of DNA synthesis through dFdCTP inhibition of DNA polymerase,213 dFdCDP inhibition of ribonucleotide reductase and subsequent depletion of deoxyribonucleotide pools,33 and dFdCTP incorporation into DNA leading to strand termination.34 The dFdCTP incorporated into newly synthesized DNA appears resistant to normal DNA repair,214 and its presence is critical for triggering apoptosis by this agent.215 Recent studies suggest that the primary deamination metabolite of dFdC, difluorodeoxyuridine (dFdU), may also play a role in cytotoxicity.216
Clinical Pharmacology
Gemcitabine is dosed intravenously because oral dosing leads to low systemic exposure217 presumably due to extensive first-pass metabolism in the liver through deamination to the dFdU metabolite.218 The length of the infusion also seems to be a potentially important variable as longer, constant rate infusions have been shown to lead to increased intracellular dFdCTP levels and enhanced response as opposed to shorter infusions.219 The dose-limiting toxicity of dFdC is hematologic in both humans and dogs.202,220
Clinical Use
Gemcitabine use has been reported infrequently in clinical studies in dogs and cats. Recent reports have used gemcitabine as a single agent or in combination with other antineoplastic agents or combined with radiation therapy. Dosing regimens employed in dogs involve either high dose (800 mg/m2 IV over 20 to 30 minutes every week for 4 weeks) or low dose (25 to 50 mg/m2 IV once or twice a week per protocol) options, depending on the use of other cytotoxics in the protocol. Cats have been treated with low-dose regimens (20 to 25 mg/m2 or 2 mg/kg weekly to biweekly) in combination with full-dose carboplatin or radiation therapy.221-223 All reports indicate that bone marrow and GI toxicity is moderate to significant with high-dose gemcitabine but manageable with routine prophylaxis. Results to date with current administration schedules support only limited, if any, gemcitabine-specific antitumor activity.220,221,224-227
5-Fluorouracil
Basic Pharmacology
5-Fluororacil (5-FU) is a halogenated analog of uracil that enters cells using a facilitated-transport system shared by adenine, uracil, and hypoxanthine.228 5-FU is converted to active nucleotide forms intracellularly by a series of phosphorylase and kinase reactions to yield monophosphate, diphosphate, and triphosphate forms of both fluorouridine and fluorodeoxyuridine,229,230 which are incorporated into RNA and DNA interfering231 with synthesis and function.232-234 The 5-FU metabolite, FdUMP, is an inhibitor of thymidylate synthetase leading to depletion of thymidine 5′ monophosphate and thymidine 5′ triphosphate.235 The alterations in thymidine and deoxyuridine phosphate pools caused by thymidylate synthetase inhibition, effects on DNA synthesis and integrity, as well as effects on RNA synthesis and processing, are all thought to play a role in cytotoxicity induced by 5-FU.
Clinical Pharmacology
5-FU is dosed intravenously and is extensively metabolized in many tissues by dihydropyrimidine dehydrogenase to dihydrofluorouracil, which is further catabolized to α-fluoro-β-alanine, ammonia, and carbon dioxide (CO2).236,237 Approximately 90% of an administered dose is metabolized, and both 5-FU and its catabolites undergo biliary excretion with less than 5% of the parent drug renally excreted. 5-FU causes a dose-dependent myelosuppression, GI toxicity, and neurotoxicity in dogs. Inadvertent ingestion of a topical 5-FU cream is toxic and/or fatal.231 5-FU is contraindicated in cats due to severe CNS toxicity.
Antimicrotubule Agents
The antimicrotubule agents currently used in veterinary medicine are structurally complex agents belonging to the taxane or vinca alkaloid classes of compounds. These agents have a mechanism of action involving interference with the polymerization or depolymerization of the microtubules that play critical roles in cell function and division.
Taxanes (Paclitaxel and Docetaxel)
Basic Pharmacology
The clinically used taxanes (paclitaxel and docetaxel) both act by stabilizing microtubules against depolymerization and thus inhibit reorganization dynamics required for carrying out cellular functions.238-240 This alteration in microtubule function causes an abnormal organization of spindle microtubules involved in chromosome segregation during mitosis, leading to mitotic arrest.241 Paclitaxel and docetaxel share identical mechanisms of action, with the increased potency of docetaxel242 attributable to an approximately twofold higher affinity for tubulin binding as compared to paclitaxel.243
Clinical Pharmacology
The clinical use of the taxanes is complicated by their poor solubility and the use of excipients, including Cremophor EL (paclitaxel) and polysorbate 80 (docetaxel) to allow for IV administration. Both paclitaxel and docetaxel are rapidly distributed throughout the body and eliminated slowly primarily by hepatic metabolism and biliary excretion. Renal elimination is 10% or less for both compounds. Toxicities associated with taxanes include hypersensitivity reactions that are attributable to the Cremophor EL and polysorbate 80 utilized in formulation. Diarrhea and neutropenia are the major dose-limiting, taxane-specific toxicities observed.
Clinical Use
The use of paclitaxel has not been frequently described in either dogs or cats. This is likely due to the requirement for significant pretreatment with antihistamines and steroids followed by a prolonged infusion with continued monitoring for acute hypersensitivity. One report documented several responses in dogs treated with paclitaxel at a dose of 165 mg/m2 slow IV infusion every 3 weeks.100 Hypersensitivity was frequent despite pretreatment, and significant bone marrow toxicity was observed, leading to the conclusion that the recommended dose for further evaluation is 132 mg/m2 as a slow IV infusion every 3 weeks in dogs. Paclitaxel in cats has been anecdotally used at 80 mg/m2 slow IV infusion every 3 weeks in anecdotal reports with similar need for pretreatment. As discussed earlier in the section on pharmaceutics, a water-soluble formulation of paclitaxel (Paccal Vet) is currently under development for veterinary use, with dose regimens and safety profiles still to be determined.
Docetaxel has been investigated in dogs and cats. In order to overcome hypersensitivity reactions, a strategy was developed to administer oral docetaxel with cyclosporine as an absorption aid. Docetaxel and cyclosporine compete with ABCB1-mediated excretion mechanisms on the enterocytes, and both are substrates for CYP3A—the phase I enzymes found in enterocytes and the liver. This was initially studied in normal beagle dogs, confirming acceptable docetaxel bioavailability.244 This strategy was subsequently investigated in phase I studies in dogs and cats with cancer in which the MTD of docetaxel is 1.63 mg/kg and 1.75 mg/kg PO (by gavage) every 2 to 3 weeks, respectively, when combined with cyclosporine (5 mg/kg PO).245,246 Although no hypersensitivities were reported, diarrhea was the dose-limiting adverse event. A recent report of IV docetaxel in cats indicates that hypersensitivity in cats is less difficult to manage compared to hypersensitivity in dogs.247 The MTD for docetaxel in cats is 2.25 mg/kg IV infused over 1 hour with routine pretreatment (antihistamine, steroids, famotidine). Tumor response data following oral or IV docetaxel have not been reported to date.
Vinca Alkaloids (Vinblastine and Vincristine)
The vinca alkaloids as a class of antitumor agents consist of the naturally occurring vincristine (VCR) and vinblastine (VBL), as well as a semisynthetic derivative and metabolite of VBL, vindesine (VDS), and the semisynthetic derivative of VBL, vinorelbine (VRL). These agents all share a similar mechanism of action; however, focus will be on VCR and VBL in this section due to their use in veterinary medicine.
Basic Pharmacology
The vinca alkaloids bind to a distinct site on tubulin248 and inhibit microtubule assembly.249 This inhibition of microtubule function leads to a disruption in the mitotic spindle apparatus resulting in metaphase arrest and cytotoxicity.250,251 The vinca alkaloids enter cells by a simple diffusion process. Exposure time and concentration seem to be important variables in determining cytotoxicity.
Clinical Pharmacology
The vinca alkaloids are administered by IV infusion, rapidly distribute to tissues, and are slowly eliminated primarily by hepatic metabolism and biliary excretion of parent drug and metabolites. Urinary excretion of parent drug and metabolites is relatively low: 10% to 20%. One of the metabolites of VBL is desacetylvinblastine (vindesine), which is active and has been identified in dogs.252 VBL and VCR differ in their respective toxicities with VCR being less myelosuppressive than VBL but causing more peripheral neurotoxic and GI effects, including significant ileus.
Clinical Use
Vincristine is used predominantly as a component in multiagent protocols for dogs and cats with lymphoma. It is also used as a single agent for dogs with transmissible venereal tumor. The dose for vincristine is 0.5 to 0.75 mg/m2 IV bolus weekly in both dogs and cats or as defined in the protocol. All vinca alkaloids are tissue vesicants if delivered extravascularly, although not as serious as doxorubicin. Extreme care should be taken with the injection.
Vinblastine is most often used to manage canine mast cell tumors, either as a single agent or in combination with other agents. Several dose-schedule variations have been developed in the last 5 years. Vinblastine as a single agent or when appropriately combined with other cytotoxic agents may be administered at 2.5 mg/m2 IV every 1 to 2 weeks253 or 3.0 to 3.5 mg/m2 IV every 2 to 3 weeks.97
Vinorelbine is a synthetic vinca alkaloid and has been used in dogs with a variety of tumors at a starting dose of 15 mg/m2 IV over 5 minutes once weekly. There are insufficient numbers of dogs evaluated to accurately quantify tumor response at this time.254,255
Topoisomerase Inhibitors
The topoisomerase inhibitors represent classes of drugs that inhibit either the type I or type II topoisomerase enzymes that are involved in the unlinking and unwinding of the DNA strand for replication and transcription. The major classes of topoisomerase II inhibitors used in veterinary oncology are the anthracyclines, which have already been discussed, and the epipodophyllotoxins, of which etoposide and teniposide are the clinically relevant members. The major class of topoisomerase I inhibitors used in human oncology are the camptothecins, which have found little use so far in veterinary medicine and will not be discussed here.
Epipodophyllotoxins (Etoposide and Teniposide)
Basic Pharmacology
Etoposide (VP-16) and teniposide (VM-26) both inhibit the catalytic activity of topoisomerase II256 by stabilizing a protein-DNA cleavage complex257 that ultimately results in the generation of single- and double-strand DNA breaks.258 These compounds enter tumor cells by simple diffusion across the cell membrane, and increased levels of topoisomerase II in proliferating tumor cells increases selectivity.259
Clinical Pharmacology
Etoposide has been evaluated in dogs both intravenously and orally. Etoposide administered IV is associated with severe histamine release in dogs associated with the polysorbate 80 vehicle as described previously with the use of IV docetaxel. Oral dosing has shown low and highly variable bioavailability in dogs, making this route of delivery difficult to use.260 Etoposide is eliminated following dosing by hepatic metabolism and renal elimination of both parent drugs (30% to 40% of the dose) and glucuronide metabolites. The major dose-limiting toxicity of IV etoposide in the dog is hypersensitivity.261
Clinical Use
Based on the hypersensitivity reactions experienced in dogs following IV etoposide and the low bioavailability of orally administered etoposide, it is not recommended for use. Strategies to overcome the vehicle-induced hypersensitivity by reformulation or to improve bioavailability are required to continue evaluating etoposide. No studies have been reported in cats.
Steroids
Basic Pharmacology
Prednisone, or prednisolone, is a corticosteroid that presumably induces killing of hematopoietic cancer cells through interaction with the glucocorticoid receptor262 and the induction of apoptosis.263 Mechanisms of apoptosis induction by corticosteroids in hematologic cancers is still not completely understood, and multiple mechanisms exist whereby tumor cells of hematopoietic origin resist steroid-induced killing.264
Clinical Pharmacology
Prednisone is generally well tolerated in dogs over short time periods (weeks) when administered on a tapering schedule to a tolerable baseline dose dependent on the response of the patient and the cancer. The adrenal-pituitary axis can become suppressed, with signs of iatrogenic hyperadrenocorticism occurring if prednisone is continued at immunosuppressive doses.
Clinical Use
Prednisone is widely used for management of lymphoid malignancies, mast cell tumors, and brain tumors in dogs and cats. Dogs are often dosed at 2 mg/kg (or 40 mg/m2) PO daily at the beginning of multiagent protocols for lymphoma and are weaned off the drug over 3 to 4 weeks. Cats are tolerant of prednisone or prednisolone and are maintained at 5 mg PO every 24 hours or twice a day as needed. Prednisone is also used to manage signs and side effects of chemotherapy-induced toxicity, such as hypersensitivities or hemorrhagic cystitis. Antiinflammatory doses are used in dogs (0.5 to 1.0 mg/kg PO once daily and reduced as indicated by signs).
Others
Platinum (Carboplatin and Cisplatin)
Basic Pharmacology
The activity of platinum-containing antitumor agents is through covalent binding to DNA through displacement reactions resulting in bifunctional lesions and interstrand or intrastrand cross-links.265 The formation of interstrand cross-links has been shown to correlate to cytotoxicity,266 presumably by blocking strand separation required for replication and transcription. Reactions with water are an important component of the pharmacology of cisplatin due to some of the aquatic species potentially crossing cell membranes more rapidly.267
Clinical Pharmacology
Both cisplatin and carboplatin are administered intravenously. Metabolism of both cisplatin and carboplatin occurs primarily through reactions with water and elimination by binding to plasma and tissue proteins. Urinary elimination of unbound and bound forms accounts for nearly 50% of the cisplatin dose 5 days after administration. Carboplatin is predominantly excreted in the urine with approximately 65% of the dose recovered in the urine 24 hours after administration.267 Strong correlations exist between carboplatin exposure and renal function such that simple formulas have been derived in both humans268 and cats269 for dosing calculations based on renal function. The most common toxicities associated with platinum therapies are vomiting and myelosuppression. Nephrotoxicity can occur with cisplatin therapy, and vigorous diuresis is required to avoid or decrease renal toxicity.
Clinical Use
Cisplatin (50 to 70 mg/m2 IV infusion administered with diuresis and antiemetics every 3 weeks) is indicated primarily for canine OSA. A variety of other tumor types have been reported to be marginally sensitive to cisplatin. Cisplatin is contraindicated in cats.
Carboplatin (300 mg/m2 IV over 10 to 15 minutes every 3 weeks) is preferred to cisplatin because of the reduced incidence of nausea/vomiting, the absence of nephrotoxicity, and ease of administration. Myelosuppression is the dose-limiting toxicity. It is used for management of OSA in dogs, as well as a variety of sarcomas and carcinomas. The traditional dose of carboplatin in cats is 240 mg/m2 IV over 10 minutes every 3 weeks.270 It has a reported indication for sarcomas and carcinomas. As in humans, an individualized dose of carboplatin may be calculated based on GFR.78 This provides a uniform systemic dose exposure in cats that may have impaired but subclinical renal disease, as well as makes treatment potentially feasible in cats with overt renal disease. Measurement of GFR requires some extra time and expense but increases the dose intensity and may thus improve response, although no response data using this dosing strategy have been reported to date.
Hydroxyurea
Basic Pharmacology
Hydroxyurea enters cells via passive diffusion271 and is an inhibitor of ribonucleotide reductase272 resulting in depletion of deoxyribonucleotide pools.273 This interaction with ribonucleotide reductase can also lead to the allosteric inhibition of other enzymes in the DNA precursor synthesis pathway that make up the replitase complex.274 The magnitude of decrease in cellular deoxyribonucleotide pools induced by hydroxyurea treatment correlates with inhibition of DNA synthesis observed.275
Clinical Pharmacology
Hydroxyurea is dosed orally and distributes rapidly to all tissues. Elimination is through hepatic metabolism, as well as urinary elimination of the parent compound. Toxicities associated with hydroxyurea treatment include GI effects, myelosuppression, onycholysis, and pulmonary fibrosis; cats are more susceptible to myelosuppressive effects and potentially methemoglobinemia at higher doses.
Clinical Use
Hydroxyurea is used primarily for management of bone marrow disorders such as polycythemia vera and granulocytic leukemias. Hydroxyurea is safe to use at 50 to 60 mg/kg orally once daily initially for several weeks followed by a decreasing dose adjustment as needed to maintain a low red blood cell count in the case of polycythemia vera. It has been recently evaluated as an alternative agent for advanced mast cell tumors in dogs.276
L-Asparaginase
Basic Pharmacology
The enzymatic function of l-asparaginase is the hydrolysis of l-asparagine to l-aspartic acid. This depletion of circulating l-asparagine leads to inhibition of protein synthesis in tumor cells lacking l-asparagine synthetase, causing induction of apoptosis.277
Clinical Pharmacology
l-Asparaginase can be dosed subcutaneously, intramuscularly, or intraperitoneally, and blood levels of the protein remain for weeks. Hypersensitivity reactions can lead to a shorter half-life through enhanced clearance. The toxicities associated with l-asparaginase treatment are due to either hypersensitivity reactions, which may be accentuated following repeated exposures, or to decreased protein synthesis from depleted l-asparagine pools.278 Hypersensitivity may be managed through pretreatment with antihistamines and dexamethasone, although owners should be advised to observe the dog for 1 to 4 hours after treatment for signs of hypersensitivity.
Future Directions in Drug Therapies for Cancer
Population Pharmacokinetics
The convention for drug dosing is to normalize the dose to the weight or surface area of the patient. This is done even though there is often no data to support a relationship between a given drug’s exposure in the patient and either of these parameters. These conventions are based on the idea that body weight or BSA is related to drug distribution and/or elimination in a manner that allows for consistent drug exposure in treated individuals. Dosing in mg/kg or mg/m2 is a crude attempt at individualized dosing, and for some drugs, substantial variability is expected. Studies specifically aimed at determining what demographic characteristics in the patient population determine variability in drug exposure are termed population pharmacokinetic studies.
Molecular Profiling
Tumors have traditionally been classified by descriptive characteristics, such as tissue or organ of origin, histology, aggressiveness, and extent of spread. That empiric rubric is being challenged, as molecular classifications made possible by microarrays and other profiling technologies become increasingly common and persuasive.279,280 The reductionist program would suggest that eventually all differences among traditional tumor types will be reduced to statements about molecules in the tumors and about the interactions among those molecules. Hence it might then be possible to study physiologic processes in one type of cancer and extrapolate the results in a predictive manner to another type through commonalities in their molecular constitutions. But what if we want to do the same thing at the pharmacologic level—to extrapolate and predict drug sensitivity based on molecular characteristics of the tumor? These types of decisions are already being used in human medicine at a discrete level for the use of antiestrogens in estrogen receptor–positive breast cancers,281 and the use of select molecularly targeted agents based on the mutation status of target molecules.282,283 However, responses to traditional chemotherapy agents, which still make up the backbone of available therapies both in human and veterinary medicine, are more complex and do not generally sort as responders and nonresponders based on single, or even a few, molecular characteristics. Examples do exist in which this is the case such as overexpression of ABCB1 and the multidrug resistance phenotype,284 but generally, a multitude of genes involved in drug activation, detoxification, DNA repair, stress responses, and a myriad of other known and unknown pathways play a role in determining tumor cell chemosensitivity. Therefore a mechanism that can evaluate multiple factors in a tumor indiscriminately and determine whether it is sensitive or insensitive to a given chemotherapeutic agent would be an invaluable adjunct in determining which drugs to utilize for which individual tumor.
Prognostic Evaluation
Current clinical practice in both human and veterinary oncology bases the choice of cytotoxic chemotherapy on descriptive histopathology characteristics. For example, a diagnosis of OSA in a veterinary patient would lead to the use of adjuvant DOX and/or a platinum-based drug (carboplatin or cisplatin) following surgical resection of the tumor. Why are these drugs used? The easy answer is that studies have shown that dogs receiving either of these drugs following surgery live significantly longer than dogs receiving surgery alone.285 Studies in human patients have shown in a variety of tumor types that in vitro chemosensitivity testing of tumor biopsies and tailoring therapy can lead to increases in antitumor response.286-289 Therefore basing therapy on an empiric assessment of drug sensitivity rather than on tumor type alone is a strategy that can lead to preferred outcomes. Issues with the use of chemosensitivity assessment in clinical practice are the technical difficulties associated with tissue procurement and culturing and measures of drug response. Another approach to predicting the chemosensitivity of tumors has evolved around gene expression profiling and informatics.290,291 Although much attention has been focused on recent evidence of research impropriety and improper validation of predictors used for clinical trials in human lung and breast cancers292,293 and this has dampened some enthusiasm for using genomic predictors in chemosensitivity profiling, it should be noted that extensive review of these data by statisticians294 and NCI review panels has found that the errors made were in data handling and consistency in analysis. Thus these unfortunate events should not be an indictment of “omics” approaches in making clinical decisions but rather a stark reminder that correct and careful research approaches and data analysis must be adhered to.
Novel Combinations
The approval of the first targeted agent for veterinary applications in the United States (Palladia) has provided access to a multitargeted tyrosine kinase inhibitor. Biologic agents, including species-specific cytokines, peptides, monoclonal antibodies, chimeric molecules, and targeted toxins will also invariably become more prevalent as experimental therapies in veterinary medicine. The use of these novel agents in combination with traditional cytotoxic chemotherapy will likely follow the development pathway seen in human oncology, which includes adding these agents to standard protocols in a disease-specific manner. Thus changes to current standards of practice and care should be expected as these newer agents are incorporated and tested.
References
1. Morrison, WB. Cancer chemotherapy: an annotated history. J Vet Intern Med. 2010;24:1249.
2. Chabner, BA, Roberts, TG, Jr. Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65.
3. DeVita, VT, Jr., Chu, E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643.
4. Shoemaker, RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813.
5. Sharma, SV, Haber, DA, Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10:241.
6. Thamm, DH, Rose, B, Kow, K, et al. Masitinib as a chemosensitizer of canine tumor cell lines: A proof of concept study. Vet J. 2011;191(1):131–134.
7. Begleiter, A, Lam, H-YP, Grover, J, et al. Evidence for active transport of melphalan by two amino acid carriers in L5178Y lymphoblasts in vitro. Cancer Res. 1979;39:353.
8. Vistica, D. Cytotoxicity as an indicator for transport mechanism: evidence that murine bone marrow progenitor cells lack a high-affinity leucine carrier that transports melphalan in murine L1210 leukemia cells. Blood. 1980;56:427.
9. Wiley, JS, Jones, SP, Sawyer, WH, et al. Cytosine arabinoside influx and nucleoside transport sites in acute leukemia. J Clin Invest. 1982;69:479.
10. Mackey, JR, Mani, RS, Selner, M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349.
11. Goldman, ID, Lichtenstein, NS, Oliverio, VT. Carrier-mediated transport of the folic acid analogue, methotrexate, in the L1210 leukemia cell. J Biol Chem. 1968;243:5007.
12. Withoff, S, Keith, WN, Knol, AJ, et al. Selection of a subpopulation with fewer DNA topoisomerase II alpha gene copies in a doxorubicin-resistant cell line panel. Br J Cancer. 1996;74:502.
13. Wang, H, Jiang, Z, Wong, YW, et al. Decreased CP-1 (NF-Y) activity results in transcriptional down-regulation of topoisomerase IIalpha in a doxorubicin-resistant variant of human multiple myeloma RPMI 8226. Biochem Biophys Res Commun. 1997;237:217.
14. Moran, RG, Spears, CP, Heidelberger, C. Biochemical determinants of tumor sensitivity to 5-fluorouracil: ultrasensitive methods for the determination of 5-fluoro-2′-deoxyuridylate, 2′-deoxyuridylate, and thymidylate synthetase. Proc Natl Acad Sci U S A. 1979;76:1456.
15. Karran, P, Bignami, M. DNA damage tolerance, mismatch repair and genome instability. BioEssays. 1994;16:833.
16. Brown, JM, Attardi, LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231.
17. Igney, FH, Krammer, PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277.
18. Reed, JC, Kitada, S, Takayama, S, et al. Regulation of chemoresistance by the bcl-2 oncoprotein in non-Hodgkin’s lymphoma and lymphocytic leukemia cell lines. Ann Oncol. 1994;5(Suppl 1):61.
19. Schmitt, CA, Rosenthal, CT, Lowe, SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6:1029.
20. Ambrosini, G, Adida, C, Altieri, DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917.
21. Kramer, M, Hermans, J, Parker, J, et al. Clinical significance of bcl2 and p53 protein expression in diffuse large B-cell lymphoma: a population-based study. J Clin Oncol. 1996;14:2131.
22. Sohn, SK, Jung, JT, Kim, DH, et al. Prognostic significance of bcl-2, bax, and p53 expression in diffuse large B-cell lymphoma. Am J Hematol. 2003;73:101.
23. Rebhun, RB, Lana, SE, Ehrhart, EJ, et al. Comparative analysis of survivin expression in untreated and relapsed canine lymphoma. J Vet Intern Med. 2008;22:989.
24. Moscow, JA, Swanson, CA, Cowan, KH. Decreased melphalan accumulation in a human breast cancer cell line selected for resistance to melphalan. Br J Cancer. 1993;68:732.
25. Saikawa, Y, Knight, CB, Saikawa, T, et al. Decreased expression of the human folate receptor mediates transport-defective methotrexate resistance in KB cells. J Biol Chem. 1993;268:5293.
26. Tew, KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54:4313.
27. Wang, AL, Tew, KD. Increased glutathione-S-transferase activity in a cell line with acquired resistance to nitrogen mustards. Cancer Treat Rep. 1985;69:677.
28. Lewis, AD, Hickson, ID, Robson, CN, et al. Amplification and increased expression of alpha class glutathione S-transferase-encoding genes associated with resistance to nitrogen mustards. Proc Natl Acad Sci U S A. 1988;85:8511.
29. Beck, A, Etienne, MC, Cheradame, S, et al. A role for dihydropyrimidine dehydrogenase and thymidylate synthase in tumour sensitivity to fluorouracil. Eur J Cancer. 1994;30A:1517.
30. Ishikawa, Y, Kubota, T, Otani, Y, et al. Dihydropyrimidine dehydrogenase activity and messenger RNA level may be related to the antitumor effect of 5-fluorouracil on human tumor xenografts in nude mice. Clin Cancer Res. 1999;5:883.
31. Scherf, U, Ross, DT, Waltham, M, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24:236.
32. Heggie, GD, Sommadossi, JP, Cross, DS, et al. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987;47:2203.
33. Heinemann, V, Xu, YZ, Chubb, S, et al. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567.
34. Huang, P, Chubb, S, Hertel, LW, et al. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110.
35. Heinemann, V, Hertel, LW, Grindey, GB, et al. Comparison of the cellular pharmacokinetics and toxicity of 2′,2′-difluorodeoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1988;48:4024.
36. Bouffard, DY, Laliberte, J, Momparler, RL. Kinetic studies on 2′,2′-difluorodeoxycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem Pharmacol. 1993;45:1857.
37. Ohhashi, S, Ohuchida, K, Mizumoto, K, et al. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008;28:2205.
38. Nakano, Y, Tanno, S, Koizumi, K, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457.
39. Long, BH, Wang, L, Lorico, A, et al. Mechanisms of resistance to etoposide and teniposide in acquired resistant human colon and lung carcinoma cell lines. Cancer Res. 1991;51:5275.
40. Minocha, A, Long, BH. Inhibition of the DNA catenation activity of type II topoisomerase by VP16–213 and VM26. Biochem Biophys Res Commun. 1984;122:165.
41. Chen, GL, Yang, L, Rowe, TC, et al. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem. 1984;259:13560.
42. Alt, FW, Kellems, RE, Bertino, JR, et al. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem. 1978;253:1357.
43. Giannakakou, P, Sackett, DL, Kang, Y-K, et al. Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem. 1997;272:17118.
44. Andoh, T, Ishii, K, Suzuki, Y, et al. Characterization of a mammalian mutant with a camptothecin-resistant DNA topoisomerase I. Proc Natl Acad Sci U S A. 1987;84:5565.
45. Chaney, SG, Sancar, A. DNA repair: Enzymatic mechanisms and relevance to drug response. J Natl Cancer Inst. 1996;88:1346.
46. Behrens, BC, Hamilton, TC, Masuda, H, et al. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 1987;47:414.
47. Masuda, H, Tanaka, T, Matsuda, H, et al. Increased removal of DNA-bound platinum in a human ovarian cancer cell line resistant to cis-diamminedichloroplatinum(II). Cancer Res. 1990;50:1863.
48. Parker, RJ, Eastman, A, Bostick-Bruton, F, et al. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:772.
49. Nojima, K, Hochegger, H, Saberi, A, et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005;65:11704.
50. Borst, P, Elferink, RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537.
51. Fletcher, JI, Haber, M, Henderson, MJ, et al. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer. 2010;10:147.
52. Lee, JJ, Hughes, CS, Fine, RL, et al. P-glycoprotein expression in canine lymphoma: a relevant, intermediate model of multidrug resistance. Cancer. 1996;77:1892.
53. Honscha, KU, Schirmer, A, Reischauer, A, et al. Expression of ABC-transport proteins in canine mammary cancer: Consequences for chemotherapy. Reprod Domest Anim. 2009;44(Suppl 2):218.
54. Hifumi, T, Miyoshi, N, Kawaguchi, H, et al. Immunohistochemical detection of proteins associated with multidrug resistance to anti-cancer drugs in canine and feline primary pulmonary carcinoma. J Vet Med Sci. 2010;72:665.
55. Nowak, M, Madej, JA, Dziegiel, P. Expression of breast cancer resistance protein (BCRP-1) in canine mammary adenocarcinomas and adenomas. In Vivo. 2009;23:705.
56. Conrad, S, Viertelhaus, A, Orzechowski, A, et al. Sequencing and tissue distribution of the canine MRP2 gene compared with MRP1 and MDR1. Toxicology. 2001;156:81.
57. Yabuuchi, H, Tanaka, K, Maeda, M, et al. Cloning of the dog bile salt export pump (BSEP; ABCB11) and functional comparison with the human and rat proteins. Biopharm Drug Dispos. 2008;29:441.
58. Ramirez, CJ, Minch, JD, Gay, JM, et al. Molecular genetic basis for fluoroquinolone-induced retinal degeneration in cats. Pharmacogenet Genomics. 2011;21:66.
59. Chabner, BA. Clinical strategies for cancer treatment: the role of drugs. In: Chabner BA, Collins JM, eds. Cancer chemotherapy: Principles and practice. Philadelphia: JB Lippincott, 1990.
60. Page, RL, Macy, DW, Ogilvie, GK, et al. Phase III evaluation of doxorubicin and whole-body hyperthermia in dogs with lymphoma. Int J Hyperthermia. 1992;8:187.
61. Carter, RF, Harris, CK, Withrow, SJ, et al. Chemotherapy of canine lymphoma with histopathological correlation- doxorubicin alone compared to COP as 1st treatment regimen. J Am Anim Hosp Assoc. 1987;23:587.
62. Postorino, NC, Susaneck, SJ, Withrow, SJ, et al. Single agent therapy with adriamycin for canine lymphosarcoma. J Am Anim Hosp Assoc. 1989;25:221.
63. Valerius, KD, Ogilvie, GK, Mallinckrodt, CH, et al. Doxorubicin alone or in combination with asparaginase, followed by cyclophosphamide, vincristine, and prednisone for treatment of multicentric lymphoma in dogs: 121 cases (1987–1995). J Am Vet Med Assoc. 1997;210:512.
64. Mutsaers, AJ, Glickman, NW, DeNicola, DB, et al. Evaluation of treatment with doxorubicin and piroxicam or doxorubicin alone for multicentric lymphoma in dogs. J Am Vet Med Assoc. 2002;220:1813.
65. Cotter, SM. Treatment of lymphoma and leukemia with cyclophosphamide, vincristine, and prednisone. 1. Treatment of Dogs. J Am Anim Hosp Assoc. 1983;19:159.
66. MacEwen, EG, Brown, NO, Patnaik, AK, et al. Cyclic combination chemotherapy of canine lymphosarcoma. J Am Vet Med Assoc. 1981;178:1178.
67. MacEwen, EG, Hayes, AA, Matus, RE, et al. Evaluation of some prognostic factors for advanced multicentric lymphosarcoma in the dog—147 Cases (1978–1981). J Am Vet Med Assoc. 1987;190:564.
68. Khanna, C, Lund, EM, Redic, KA, et al. Randomized controlled trial of doxorubicin versus dactinomycin in a multiagent protocol for treatment of dogs with malignant lymphoma. J Am Vet Med Assoc. 1998;213:985.
69. Greenlee, PG, Filippa, DA, Quimby, FW, et al. Lymphomas in dogs. A morphologic, immunologic, and clinical study. Cancer. 1990;66:480.
70. Stone, MS, Goldstein, MA, Cotter, SM. Comparison of 2 protocols for induction of remission in dogs with lymphoma. J Am Anim Hosp Assoc. 1991;27:315.
71. Myers, NC, 3rd., Moore, AS, Rand, WM, et al. Evaluation of a multidrug chemotherapy protocol (ACOPA II) in dogs with lymphoma. J Vet Intern Med. 1997;11:333.
72. Boyce, KL, Kitchell, BE. Treatment of canine lymphoma with COPLA/LVP. J Am Anim Hosp Assoc. 2000;36:395.
73. Morrison-Collister, KE, Rassnick, KM, Northrup, NC, et al. A combination chemotherapy protocol with MOPP and CCNU consolidation (Tufts VELCAP-SC) for the treatment of canine lymphoma. Vet Comp Oncol. 2003;1:180.
74. Zemann, BI, Moore, AS, Rand, WM, et al. A combination chemotherapy protocol (VELCAP-L) for dogs with lymphoma. J Vet Intern Med. 1998;12:465.
75. Keller, ET, MacEwen, EG, Rosenthal, RC, et al. Evaluation of prognostic factors and sequential combination chemotherapy with doxorubicin for canine lymphoma. J Vet Intern Med. 1993;7:289.
76. Garrett, LD, Thamm, DH, Chun, R, et al. Evaluation of a 6-month chemotherapy protocol with no maintenance therapy for dogs with lymphoma. J Vet Intern Med. 2002;16:704.
77. Undevia, SD, Gomez-Abuin, G, Ratain, MJ. Pharmacokinetic variability of anticancer agents. Nat Rev Cancer. 2005;5:447.
78. Bailey, DB, Rassnick, KM, Erb, HN, et al. Effect of glomerular filtration rate on clearance and myelotoxicity of carboplatin in cats with tumors. Am J Vet Res. 2004;65:1502.
79. Mealey, KL, Fidel, J, Gay, JM, et al. ABCB1–1Delta polymorphism can predict hematologic toxicity in dogs treated with vincristine. J Vet Intern Med. 2008;22:996.
80. Thompson, LA, Lawson, AP, Sutphin, SD, et al. Description of current practices of empiric chemotherapy dose adjustment in obese adult patients. J Oncol Pract. 2010;6:141.
81. Hunter, RJ, Navo, MA, Thaker, PH, et al. Dosing chemotherapy in obese patients: actual versus assigned body surface area (BSA). Cancer Treat Rev. 2009;35:69.
82. Vail, DM. Supporting the veterinary cancer patient on chemotherapy: neutropenia and gastrointestinal toxicity. Top Companion Anim Med. 2009;24:122.
83. Dranitsaris, G, Johnston, M, Poirier, S, et al. Are health care providers who work with cancer drugs at an increased risk for toxic events? A systematic review and meta-analysis of the literature. J Oncol Pharm Pract. 2005;11:69.
84. Hamscher, G, Mohring, SA, Knobloch, A, et al. Determination of drug residues in urine of dogs receiving anti-cancer chemotherapy by liquid chromatography-electrospray ionization- tandem mass spectrometry: is there an environmental or occupational risk? J Anal Toxicol. 2010;34:142.
85. Evans, WE, Relling, MV. Clinical pharmacokinetics-pharmacodynamics of anticancer drugs. Clin Pharmacokinet. 1989;16:327.
86. Eckhoff, GA. Mechanisms of adverse drug-reactions and interactions in veterinary-medicine. J Am Vet Med Assoc. 1980;176:1131.
87. He, SM, Yang, AK, Li, XT, et al. Effects of herbal products on the metabolism and transport of anticancer agents. Expert Opin Drug Metab Toxicol. 2010;6:1195.
88. Tarirai, C, Viljoen, AM, Hamman, JH. Herb-drug pharmacokinetic interactions reviewed. Expert Opin Drug Metab Toxicol. 2010;6:1515.
89. Mealey, KL. Therapeutic implications of the MDR-1 gene. J Vet Pharmacol Ther. 2004;27:257.
90. Legha, SS, Benjamin, RS, Mackay, B, et al. Reduction of doxorubicin cardiotoxicity by prolonged continuous intravenous infusion. Ann Intern Med. 1982;96:133.
91. Berrak, SG, Ewer, MS, Jaffe, N, et al. Doxorubicin cardiotoxicity in children: reduced incidence of cardiac dysfunction associated with continuous-infusion schedules. Oncol Rep. 2001;8:611.
92. Preisler, HD, Gessner, T, Azarnia, N, et al. Relationship between plasma adriamycin levels and the outcome of remission induction therapy for acute nonlymphocytic leukemia. Cancer Chemother Pharmacol. 1984;12:125.
93. Piscitelli, SC, Rodvold, KA, Rushing, DA, et al. Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small cell lung cancer. Clin Pharmacol Ther. 1993;53:555.
94. Danesi, R, Fogli, S, Gennari, A, et al. Pharmacokinetic-pharmacodynamic relationships of the anthracycline anticancer drugs. Clin Pharmacokinet. 2002;41:431.
95. Arrington, KA, Legendre, AM, Tabeling, GS, et al. Comparison of body surface area-based and weight-based dosage protocols for doxorubicin administration in dogs. Am J Vet Res. 1994;55:1587.
96. Veterinary co-operative oncology group—common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.0. Vet Comp Oncol. 2004;2:195.
97. Bailey, DB, Rassnick, KM, Kristal, O, et al. Phase I dose escalation of single-agent vinblastine in dogs. J Vet Intern Med. 2008;22:1397.
98. Vaughan, A, Johnson, JL, Williams, LE. Impact of chemotherapeutic dose intensity and hematologic toxicity on first remission duration in dogs with lymphoma treated with a chemoradiotherapy protocol. J Vet Intern Med. 2007;21:1332.
99. Eschalier, A, Lavarenne, J, Burtin, C, et al. Study of histamine release induced by acute administration of antitumor agents in dogs. Cancer Chemother Pharmacol. 1988;21:246.
100. Poirier, VJ, Hershey, AE, Burgess, KE, et al. Efficacy and toxicity of paclitaxel (Taxol) for the treatment of canine malignant tumors. J Vet Intern Med. 2004;18:219.
101. Goodman, LS, Wintrobe, MM, et al. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J Am Med Assoc. 1946;132:126.
102. Jacobson, LO, Spurr, CL, et al. Studies on the effect of methyl bis (beta-chloroethyl) amine hydrochloride on diseases of the hemopoietic system. J Clin Invest. 1946;25:909.
103. Rhoads, CP. Nitrogen mustards in the treatment of neoplastic disease; official statement. J Am Med Assoc. 1946;131:656.
104. Kohn, KW, Spears, CL, Doty, P. Inter-strand crosslinking of DNA by nitrogen mustard. J Mol Biol. 1966;19:266.
105. Skipper, HE, Bennett, LL, Jr., Langham, WH. Over-all tracer studies with C14 labeled nitrogen mustard in normal and leukemic mice. Cancer. 1951;4:1025.
106. Goldenberg, GJ, Vanstone, CL, Israels, LG, et al. Evidence for a transport carrier of nitrogen mustard in nitrogen mustard-sensitive and -resistant L5178Y lymphoblasts. Cancer Res. 1970;30:2285.
107. Rassnick, KM, Mauldin, GE, Al-Sarraf, R, et al. MOPP chemotherapy for treatment of resistant lymphoma in dogs: a retrospective study of 117 cases. J Vet Intern Med. 2002;16:1989–2000.
108. Rassnick, KM, Bailey, DB, Malone, EK, et al. Comparison between L-CHOP and an L-CHOP protocol with interposed treatments of CCNU and MOPP (L-CHOP-CCNU-MOPP) for lymphoma in dogs. Vet Comp Oncol. 2010;8:243.
109. Brodsky, EM, Maudlin, GN, Lachowicz, JL, et al. Asparaginase and MOPP treatment of dogs with lymphoma. J Vet Intern Med. 2009;23:578.
110. Tew, KD, Colvin, OM, Chabner, BA. Alkylating agents. In: Chabner BA, Longo DL, eds. Cancer chemotherapy & biotherapy: principles and practice. Philadelphia: Lippincott Williams & Wilkins, 2001.
111. Cohen, JL, Jao, JY. Enzymatic basis of cyclophosphamide activation by hepatic microsomes of the rat. J Pharmacol Exp Ther. 1970;174:206.
112. Colvin, M, Brundrett, RB, Kan, MN, et al. Alkylating properties of phosphoramide mustard. Cancer Res. 1976;36:1121.
113. Warry, E, Hansen, RJ, Gustafson, DL, et al. Pharmacokinetics of cyclophosphamide after oral and intravenous administration to dogs with lymphoma. J Vet Intern Med. 2011;25:903.
114. Fetting, JH, McCarthy, LE, Borison, HL, et al. Vomiting induced by cyclophosphamide and phosphoramide mustard in cats. Cancer Treat Rep. 1982;66:1625.
115. Lori, JC, Stein, TJ, Thamm, DH. Doxorubicin and cyclophosphamide for the treatment of canine lymphoma: a randomized, placebo-controlled study. Vet Comp Oncol. 2010;8:188.
116. Hadden, AG, Cotter, SM, Rand, W, et al. Efficacy and toxicosis of VELCAP-C treatment of lymphoma in cats. J Vet Intern Med. 2008;22:153.
117. Creaven, PJ, Allen, LM, Alford, DA, et al. Clinical pharmacology of isophosphamide. Clin Pharmacol Ther. 1974;16:77.
118. Norpoth, K. Studies on the metabolism of isopnosphamide (NSC-109724) in man. Cancer Treat Rep. 1976;60:437.
119. Lind, MJ, Roberts, HL, Thatcher, N, et al. The effect of route of administration and fractionation of dose on the metabolism of ifosfamide. Cancer Chemother Pharmacol. 1990;26:105.
120. Rassnick, KM, Frimberger, AE, Wood, CA, et al. Evaluation of ifosfamide for treatment of various canine neoplasms. J Vet Intern Med. 2000;14:271.
121. Rassnick, KM, Moore, AS, Northrup, NC, et al. Phase I trial and pharmacokinetic analysis of ifosfamide in cats with sarcomas. Am J Vet Res. 2006;67:510.
122. Rassnick, KM, Rodriguez, CO, Khanna, C, et al. Results of a phase II clinical trial on the use of ifosfamide for treatment of cats with vaccine-associated sarcomas. Am J Vet Res. 2006;67:517.
123. Begleiter, A, Goldenberg, GJ. Uptake and decomposition of chlorambucil by L5178Y lymphoblasts in vitro. Biochem Pharmacol. 1983;32:535.
124. Jiang, BZ, Bank, BB, Hsiang, YH, et al. Lack of drug-induced DNA cross-links in chlorambucil-resistant Chinese hamster ovary cells. Cancer Res. 1989;49:5514.
125. Mitoma, C, Onodera, T, Takegoshi, T, et al. Metabolic disposition of chlorambucil in rats. Xenobiotica. 1977;7:205.
126. Goodman, GE, McLean, A, Alberts, DS, et al. Inhibition of human tumour clonogenicity by chlorambucil and its metabolites. Br J Cancer. 1982;45:621.
127. Stein, TJ, Pellin, M, Steinberg, H, et al. Treatment of feline gastrointestinal small-cell lymphoma with chlorambucil and glucocorticoids. J Am Anim Hosp Assoc. 2010;46:413.
128. Begleiter, A, Lam, HP, Goldenberg, GJ. Mechanism of uptake of nitrosoureas by L5178Y lymphoblasts in vitro. Cancer Res. 1977;37:1022.
129. Montgomery, JA, James, R, McCaleb, GS, et al. The modes of decomposition of 1,3-bis(2-chloroethyl)-1-nitrosourea and related compounds. J Med Chem. 1967;10:668.
130. Colvin, M, Brundrett, RB, Cowens, W, et al. A chemical basis for the antitumor activity of chloroethylnitrosoureas. Biochem Pharmacol. 1976;25:695.
131. Kohn, KW. Interstrand cross-linking of DNA by 1,3-bis(2-chloroethyl)-1-nitrosourea and other 1-(2-haloethyl)-1-nitrosoureas. Cancer Res. 1977;37:1450.
132. Hill, DL, Kirk, MC, Struck, RF. Microsomal metabolism of nitrosoureas. Cancer Res. 1975;35:296.
133. Wheeler, GP, Johnston, TP, Bowdon, BJ, et al. Comparison of the properties of metabolites of CCNU. Biochem Pharmacol. 1977;26:2331.
134. Lee, FY, Workman, P, Roberts, JT, et al. Clinical pharmacokinetics of oral CCNU (lomustine). Cancer Chemother Pharmacol. 1985;14:125.
135. Heading, KL, Brockley, LK, Bennett, PF. CCNU (lomustine) toxicity in dogs: a retrospective study (2002–07). Aust Vet J. 2011;89:109.
136. Hosoya, K, Lord, LK, Lara-Garcia, A, et al. Prevalence of elevated alanine transaminase activity in dogs treated with CCNU (Lomustine). Vet Comp Oncol. 2009;7:244.
137. Skorupski, KA, Hammond, GM, Irish, AM, et al. Prospective randomized clinical trial assessing the efficacy of denamarin for prevention of CCNU-induced hepatopathy in tumor-bearing dogs. J Vet Intern Med. 2011;25:838.
138. Reusser, F. Mode of action of streptozotocin. J Bacteriol. 1971;105:580.
139. Bhuyan, BK. The action of streptozotocin on mammalian cells. Cancer Res. 1970;30:2017.
140. Schnedl, WJ, Ferber, S, Johnson, JH, et al. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes. 1994;43:1326.
141. Hosokawa, M, Dolci, W, Thorens, B. Differential sensitivity of GLUT1- and GLUT2-expressing beta cells to streptozotocin. Biochem Biophys Res Commun. 2001;289:1114.
142. Adolphe, AB, Glasofer, ED, Troetel, WM, et al. Preliminary pharmacokinetics of streptozotocin, an antineoplastic antibiotic. J Clin Pharmacol. 1977;17:379.
143. Schein, PS, Cooney, DA, Vernon, ML. The use of nicotinamide to modify the toxicity of streptozotocin diabetes without loss of antitumor activity. Cancer Res. 1967;27:2324.
144. Schein, PS, Rakieten, N, Cooney, DA, et al. Streptozotocin diabetes in monkeys and dogs, and its prevention by nicotinamide. Proc Soc Exp Biol Med. 1973;143:514.
145. Schein, PS. 1-methyl-1-nitrosourea and dialkylnitrosamine depression of nicotinamide adenine dinucleotide. Cancer Res. 1969;29:1226.
146. Panasci, LC, Fox, PA, Schein, PS. Structure-activity studies of methylnitrosourea antitumor agents with reduced murine bone marrow toxicity. Cancer Res. 1977;37:3321.
147. Moore, AS, Nelson, RW, Henry, CJ, et al. Streptozocin for treatment of pancreatic islet cell tumors in dogs: 17 cases (1989–1999). J Am Vet Med Assoc. 2002;221:811.
148. Audette, RC, Connors, TA, Mandel, HG, et al. Studies on the mechanism of action of the tumour inhibitory triazenes. Biochem Pharmacol. 1973;22:1855.
149. Reid, JM, Kuffel, MJ, Miller, JK, et al. Metabolic activation of dacarbazine by human cytochromes P450: the role of CYP1A1, CYP1A2, and CYP2E1. Clin Cancer Res. 1999;5:2192.
150. Nagasawa, HT, Shirota, FN, Mizuno, NS. The mechanism of alkylation of DNA by 5-(3-methyl-1-triazeno)imidazole-4-carboxamide (MIC), a metabolite of DIC (NSC-45388). Non-involvement of diazomethane. Chem Biol Interact. 1974;8:403.
151. Kleihues, P, Kolar, GF, Margison, GP. Interaction of the carcinogen 3,3-dimethyl-1-phenyltriazene with nucleic acids of various rat tissues and the effect of a protein-free diet. Cancer Res. 1976;36:2189.
152. Griessmayr, PC, Payne, SE, Winter, JE, et al. Dacarbazine as single-agent therapy for relapsed lymphoma in dogs. J Vet Intern Med. 2009;23:1227.
153. Flory, AB, Rassnick, KM, Al-Sarraf, R, et al. Combination of CCNU and DTIC chemotherapy for treatment of resistant lymphoma in dogs. J Vet Intern Med. 2008;22:164.
154. Gale, GR, Simpson, JG, Smith, AB. Studies of the mode of action of N-isopropyl-alpha-(2-methylhydrazino)-p-toluamide. Cancer Res. 1967;27:1186.
155. Moloney, SJ, Wiebkin, P, Cummings, SW, et al. Metabolic activation of the terminal N-methyl group of N-isopropyl-alpha-(2-methylhydrazino)-p-toluamide hydrochloride (procarbazine). Carcinogenesis. 1985;6:397.
156. Schold, SC, Jr., Brent, TP, von Hofe, E, et al. O6-alkylguanine-DNA alkyltransferase and sensitivity to procarbazine in human brain-tumor xenografts. J Neurosurg. 1989;70:573.
157. Shiba, DA, Weinkam, RJ. Quantitative analysis of procarbazine, procarbazine metabolites and chemical degradation products with application to pharmacokinetic studies. J Chromatogr. 1982;229:397.
158. Oliverio, VT, Denham, C, Devita, VT, et al. Some pharmacologic properties of a new antitumor agent, N-isopropyl-alpha-(2-methylhydrazino)-p-toluamide, hydrochloride (Nsc-77213). Cancer Chemother Rep. 1964;42:1.
159. Chabner, BA, Sponzo, R, Hubbard, S, et al. High-dose intermittent intravenous infusion of procarbazine (NSC-77213). Cancer Chemother Rep. 1973;57:361.
160. Zunino, F, Gambetta, R, Di Marco, A. The inhibition in vitro of DNA polymerase and RNA polymerase by daunomycin and Adriamycin. Biochem Pharmacol. 1975;24:309.
161. Tewey, KM, Chen, GI, Nelson, EM, et al. Intercalative anti-tumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase. J Biol Chem. 1984;259:9182.
162. Taatjes, DJ, Gaudiano, G, Resing, K, et al. Alkylation of DNA by the anthracycline, antitumor drugs adriamycin and daunomycin. J Med Chem. 1996;39:4135.
163. Doroshow, JH. Role of hydrogen peroxide and hydroxyl radical in the killing of ehrlich tumor cells by anticancer quinones. Proc Natl Acad Sci U S A. 1985;83:4514.
164. Bachur, NR, Gordon, SL, Gee, MV. A general mechanism for microsomal activation of quinone anticancer agents to free radicals. Cancer Res. 1978;38:1745.
165. Pessah, IN, Durie, EL, Schiedt, MJ, et al. Anthraquinone-sensitized Ca + release channel from rat cardiac sarcoplasmic reticulum: possible receptor-mediated mechanism of doxorubicin cardiomyopathy. Mol Pharmacol. 1990;37:503.
166. Oakes, SG, Schlager, JJ, Santone, KS, et al. Doxorubicin blocks the increase in intracellular Ca ++, part of a second messenger system in N1E-115 murine neuroblastoma cells. J Pharmacol Exp Ther. 1990;252:979.
167. Mau, BL, Powis, G. Inhibition of cellular thioredoxin reductase by diaziquone and doxorubicin: relationship to the inhibition of cell proliferation and decreased ribonucleotide reductase activity. Biochem Pharmacol. 1992;43:1621.
168. Morre, DJ, Kim, C, Paulik, M, et al. Is the drug-responsive NADH oxidase of the cancer cell plasma membrane a molecular target for adriamycin? J Bioenerg Biomembr. 1997;29:269.
169. Terasaki, T, Iga, T, Sugiyama, Y, et al. Experimental evidence of characteristic tissue distribution of Adriamycin: tissue DNA concentration as a determinant. J Pharm Pharmacol. 1982;34:597.
170. Nicolay, K, Timmers, RJM, Spoelstra, E, et al. The interaction of adriamycin with cardiolipin in model and rat liver mitochondrial membranes. Biochim Biophys Acta. 1984;778:359.
171. Goormaghtigh, E, Chatelain, P, Caspers, J, et al. Evidence of a specific complex between adriamycin and negatively-charged phospholipids. Biochim Biophys Acta. 1980;597:1.
172. Gustafson, DL, Rastatter, JC, Colombo, T, et al. Doxorubicin pharmacokinetics: macromolecule binding, metabolism and elimination in the context of a physiological model. J Pharm Sci. 2002;91:1488.
173. Ahmed, NK, Felsted, RL, Bachur, NR. Daunorubicin reduction mediated by aldehyde and ketone reductases. Xenobiotica. 1981;11:131.
174. Pan, SS, Bachur, NR. Xanthine oxidase catalyzed reductive cleavage of anthracycline antibiotics and free radical formation. Mol Pharmacol. 1980;17:95.
175. Young, RC, Ozols, RF, Myers, CE. The anthracycline neoplastic drugs. N Engl J Med. 1981;305:139.
176. O’Keefe, DA, Sisson, DD, Gelberg, HB, et al. Systemic toxicity associated with doxorubicin administration in cats. J Vet Intern Med. 1993;7:309.
177. Thamm, DH, Vail, DM. Aftershocks of cancer chemotherapy: managing adverse effects. J Am Anim Hosp Assoc. 2007;43:1.
178. Foye, WO, Vajragupta, O, Sengupta, SK. DNA-binding specificity and RNA polymerase inhibitory activity of bis(aminoalkyl)anthraquinones and bis(methylthio)vinylquinolinium iodides. J Pharm Sci. 1982;71:253.
179. Crespi, MD, Ivanier, SE, Genovese, J, et al. Mitoxantrone affects topoisomerase activities in human breast cancer cells. Biochem Biophys Res Commun. 1986;136:521.
180. Patterson, LH, Gandecha, BM, Brown, JR. 1,4-Bis(2-[(2-hydroxyethyl)amino]ethylamino)-9,10-anthracenedione, an anthraquinone antitumour agent that does not cause lipid peroxidation in vivo; comparison with daunorubicin. Biochem Biophys Res Commun. 1983;110:399.
181. Nguyen, B, Gutierrez, PL. Mechanism(s) for the metabolism of mitoxantrone: electron spin resonance and electrochemical studies. Chem Biol Interact. 1990;74:139.
182. Alberts, DS, Peng, YM, Leigh, S, et al. Disposition of mitoxantrone in cancer patients. Cancer Res. 1985;45:1879.
183. Lu, K, Savaraj, N, Loo, TL. Pharmacological disposition of 1,4-dihydroxy-5–8-bis[[2 [(2-hydroxyethyl)amino]ethyl]amino]-9,10-anthracenedione dihydrochloride in the dog. Cancer Chemother Pharmacol. 1984;13:63.
184. Chiccarelli, FS, Morrison, JA, Cosulich, DB, et al. Identification of human urinary mitoxantrone metabolites. Cancer Res. 1986;46:4858.
185. Henry, CJ, McCaw, DL, Turnquist, SE, et al. Clinical evaluation of mitoxantrone and piroxicam in a canine model of human invasive urinary bladder carcinoma. Clin Cancer Res. 2003;9:906.
186. Takusagawa, F, Dabrow, M, Neidle, S, et al. The structure of a pseudo intercalated complex between actinomycin and the DNA binding sequence d(GpC). Nature. 1982;296:466.
187. Takusagawa, F, Goldstein, BM, Youngster, S, et al. Crystallization and preliminary X-ray study of a complex between d(ATGCAT) and actinomycin D. J Biol Chem. 1984;259:4714.
188. Brown, SC, Mullis, K, Levenson, C, et al. Aqueous solution structure of an intercalated actinomycin D-dATGCAT complex by two-dimensional and one-dimensional proton NMR. Biochemistry. 1984;23:403.
189. Wadkins, RM, Jovin, TM. Actinomycin D and 7-aminoactinomycin D binding to single-stranded DNA. Biochemistry. 1991;30:9469.
190. Goldberg, IH, Rabinowitz, M, Reich, E. Basis of actinomycin action. I. DNA binding and inhibition of RNA-polymerase synthetic reactions by actinomycin. Proc Natl Acad Sci U S A. 1962;48:2094.
191. Reich, E, Franklin, RM, Shatkin, AJ, et al. Action of actinomycin D on animal cells and viruses. Proc Natl Acad Sci U S A. 1962;48:1238.
192. Kessel, D, Wodinsky, I. Uptake in vivo and in vitro of actinomycin D by mouse leukemias as factors in survival. Biochem Pharmacol. 1968;17:161.
193. Inaba, M, Johnson, RK. Decreased retention of actinomycin D as the basis for cross-resistance in anthracycline-resistant sublines of P388 leukemia. Cancer Res. 1977;37:4629.
194. Diddens, H, Gekeler, V, Neumann, M, et al. Characterization of actinomycin-D-resistant CHO cell lines exhibiting a multidrug-resistance phenotype and amplified DNA sequences. Int J Cancer. 1987;40:635.
195. Galbraith, WM, Mellett, LB. Tissue disposition of 3H-actinomycin D (NSC-3053) in the rat, monkey, and dog. Cancer Chemother Rep. 1975;59:1601.
196. Furth, JJ, Cohen, SS. Inhibition of mammalian DNA polymerase by the 5′-triphosphate of 1-beta-d-arabinofuranosylcytosine and the 5′-triphosphate of 9-beta-d-arabinofuranoxyladenine. Cancer Res. 1968;28:2061.
197. Kufe, DW, Major, PP, Egan, EM, et al. Correlation of cytotoxicity with incorporation of ara-C into DNA. J Biol Chem. 1980;255:8997.
198. Major, PP, Egan, EM, Herrick, DJ, et al. Effect of ARA-C incorporation on deoxyribonucleic acid synthesis in cells. Biochem Pharmacol. 1982;31:2937.
199. Mikita, T, Beardsley, GP. Functional consequences of the arabinosylcytosine structural lesion in DNA. Biochemistry. 1988;27:4698.
200. Bianchi Scarra, GL, Romani, M, Coviello, DA, et al. Terminal erythroid differentiation in the K-562 cell line by 1-beta-D-arabinofuranosylcytosine: accompaniment by c-myc messenger RNA decrease. Cancer Res. 1986;46:6327.
201. Plagemann, PG, Marz, R, Wohlhueter, RM. Transport and metabolism of deoxycytidine and 1-beta-D-arabinofuranosylcytosine into cultured Novikoff rat hepatoma cells, relationship to phosphorylation, and regulation of triphosphate synthesis. Cancer Res. 1978;38:978.
202. Garcia-Carbonero, R, Ryan, DP, Chabner, BA. Cytidine analogs. In: Chabner BA, Longo DL, eds. Cancer chemotherapy & biotherapy: Principles and practice. Philadelphia: Lippincott Williams & Wilkins, 2001.
203. Ho, DH, Frei, E, 3rd. Clinical pharmacology of 1-beta-d-arabinofuranosyl cytosine. Clin Pharmacol Ther. 1971;12:944.
204. Menaut, P, Landart, J, Behr, S, et al. Treatment of 11 dogs with meningoencephalomyelitis of unknown origin with a combination of prednisolone and cytosine arabinoside. Vet Rec. 2008;162:241.
205. Smith, PM, Stalin, CE, Shaw, D, et al. Comparison of two regimens for the treatment of meningoencephalomyelitis of unknown etiology. J Vet Intern Med. 2009;23:520.
206. Marconato, L, Bonfanti, U, Stefanello, D, et al. Cytosine arabinoside in addition to VCAA-based protocols for the treatment of canine lymphoma with bone marrow involvement: does it make the difference? Vet Comp Oncol. 2008;6:80.
207. Allegra, CJ, Fine, RL, Drake, JC, et al. The effect of methotrexate on intracellular folate pools in human MCF-7 breast cancer cells. Evidence for direct inhibition of purine synthesis. J Biol Chem. 1986;261:6478.
208. Allegra, CJ, Chabner, BA, Drake, JC, et al. Enhanced inhibition of thymidylate synthase by methotrexate polyglutamates. J Biol Chem. 1985;260:9720.
209. Fabre, I, Fabre, G, Goldman, ID. Polyglutamylation, an important element in methotrexate cytotoxicity and selectivity in tumor versus murine granulocytic progenitor cells in vitro. Cancer Res. 1984;44:3190.
210. Wan, SH, Huffman, DH, Azarnoff, DL, et al. Effect of route of administration and effusions on methotrexate pharmacokinetics. Cancer Res. 1974;34:3487.
211. Bischoff, KB, Dedrick, RL, Zaharko, DS, et al. Methotrexate pharmacokinetics. J Pharm Sci. 1971;60:1128.
212. Mackey, JR, Mani, RS, Selner, M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349.
213. Gandhi, V, Plunkett, W. Modulatory activity of 2′,2′-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res. 1990;50:3675.
214. Gandhi, V, Legha, J, Chen, F, et al. Excision of 2′,2′-difluorodeoxycytidine (gemcitabine) monophosphate residues from DNA. Cancer Res. 1996;56:4453.
215. Huang, P, Plunkett, W. Fludarabine- and gemcitabine-induced apoptosis: incorporation of analogs into DNA is a critical event. Cancer Chemother Pharmacol. 1995;36:181.
216. Veltkamp, SA, Pluim, D, van Eijndhoven, MA, et al. New insights into the pharmacology and cytotoxicity of gemcitabine and 2′,2′-difluorodeoxyuridine. Mol Cancer Ther. 2008;7:2415.
217. Veltkamp, SA, Jansen, RS, Callies, S, et al. Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study. Clin Cancer Res. 2008;14:3477.
218. Veltkamp, SA, Pluim, D, van Tellingen, O, et al. Extensive metabolism and hepatic accumulation of gemcitabine after multiple oral and intravenous administration in mice. Drug Metab Dispos. 2008;36:1606.
219. Tempero, M, Plunkett, W, Ruiz Van Haperen, V, et al. Randomized phase II comparison of dose-intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol. 2003;21:3402.
220. Turner, AI, Hahn, KA, Rusk, A, et al. Single agent gemcitabine chemotherapy in dogs with spontaneously occurring lymphoma. J Vet Intern Med. 2006;20:1384.
221. LeBlanc, AK, LaDue, TA, Turrel, JM, et al. Unexpected toxicity following use of gemcitabine as a radiosensitizer in head and neck carcinomas: a veterinary radiation therapy oncology group pilot study. Vet Radiol Ultrasound. 2004;45:466.
222. Martinez-Ruzafa, I, Dominguez, PA, Dervisis, NG, et al. Tolerability of gemcitabine and carboplatin doublet therapy in cats with carcinomas. J Vet Intern Med. 2009;23:570.
223. Jones, PD, de Lorimier, LP, Kitchell, BE, et al. Gemcitabine as a radiosensitizer for nonresectable feline oral squamous cell carcinoma. J Am Anim Hosp Assoc. 2003;39:463.
224. Dominguez, PA, Dervisis, NG, Cadile, CD, et al. Combined gemcitabine and carboplatin therapy for carcinomas in dogs. J Vet Intern Med. 2009;23:130.
225. Marconato, L, Lorenzo, RM, Abramo, F, et al. Adjuvant gemcitabine after surgical removal of aggressive malignant mammary tumours in dogs. Vet Comp Oncol. 2008;6:90.
226. Marconato, L, Zini, E, Lindner, D, et al. Toxic effects and antitumor response of gemcitabine in combination with piroxicam treatment in dogs with transitional cell carcinoma of the urinary bladder. J Am Vet Med Assoc. 2011;238:1004.
227. McMahon, M, Mathie, T, Stingle, N, et al. Adjuvant carboplatin and gemcitabine combination chemotherapy postamputation in canine appendicular osteosarcoma. J Vet Intern Med. 2011;25:511.
228. Wohlhueter, RM, McIvor, RS, Plagemann, PG. Facilitated transport of uracil and 5-fluorouracil, and permeation of orotic acid into cultured mammalian cells. J Cell Physiol. 1980;104:309.
229. Reyes, P. The synthesis of 5-fluorouridine 5′-phosphate by a pyrimidine phosphoribosyltransferase of mammalian origin. I. Some properties of the enzyme from P1534J mouse leukemic cells. Biochemistry. 1969;8:2057.
230. Houghton, JA, Houghton, PJ. Elucidation of pathways of 5-fluorouracil metabolism in xenografts of human colorectal adenocarcinoma. Eur J Cancer Clin Oncol. 1983;19:807.
231. Snavely, NR, Snavely, DA, Wilson, BB. Toxic effects of fluorouracil cream ingestion on dogs and cats. Arch Dermatol. 2010;146:1195.
232. Kufe, DW, Major, PP. 5-Fluorouracil incorporation into human breast carcinoma RNA correlates with cytotoxicity. J Biol Chem. 1981;256:9802.
233. Kufe, DW, Major, PP, Egan, EM, et al. 5-Fluoro-2′-deoxyuridine incorporation in L1210 DNA. J Biol Chem. 1981;256:8885.
234. Tanaka, M, Yoshida, S, Saneyoshi, M, et al. Utilization of 5-fluoro-2′-deoxyuridine triphosphate and 5-fluoro-2′-deoxycytidine triphosphate in DNA synthesis by DNA polymerases alpha and beta from calf thymus. Cancer Res. 1981;41:4132.
235. Santi, DV, McHenry, CS, Sommer, H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry. 1974;13:471.
236. Naguib, FN, el Kouni, MH, Cha, S. Enzymes of uracil catabolism in normal and neoplastic human tissues. Cancer Res. 1985;45:5405.
237. Grem, JL. 5-fluoropyrimidines. In: Chabner BA, Longo DL, eds. Cancer chemotherapy & biotherapy: principles and practice. Philadelphia: Lippincott Williams & Wilkins, 2001.
238. Schiff, PB, Fant, J, Horwitz, SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665.
239. Schiff, PB, Horwitz, SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1980;77:1561.
240. Ringel, I, Horwitz, SB. Studies with RP 56976 (taxotere): a semisynthetic analogue of taxol. J Natl Cancer Inst. 1991;83:288.
241. Jordan, MA, Toso, RJ, Thrower, D, et al. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci U S A. 1993;90:9552.
242. Bissery, MC, Guenard, D, Gueritte-Voegelein, F, et al. Experimental antitumor activity of taxotere (RP 56976, NSC 628503), a taxol analogue. Cancer Res. 1991;51:4845.
243. Diaz, JF, Andreu, JM. Assembly of purified GDP-tubulin into microtubules induced by taxol and taxotere: reversibility, ligand stoichiometry, and competition. Biochemistry. 1993;32:2747.
244. McEntee, M, Silverman, JA, Rassnick, K, et al. Enhanced bioavailability of oral docetaxel by co-administration of cyclosporin A in dogs and rats. Vet Comp Oncol. 2003;1:105.
245. McEntee, MC, Rassnick, KM, Bailey, DB, et al. Phase I and pharmacokinetic evaluation of the combination of orally administered docetaxel and cyclosporin A in tumor-bearing cats. J Vet Intern Med. 2006;20:1370.
246. McEntee, MC, Rassnick, KM, Lewis, LD, et al. Phase I and pharmacokinetic evaluation of the combination of orally administered docetaxel and cyclosporin A in tumor-bearing dogs. Am J Vet Res. 2006;67:1057.
247. Shiu, KB, McCartan, L, Kubicek, L, et al. Intravenous administration of docetaxel to cats with cancer. J Vet Intern Med. 2011;25:916.
248. Correia, JJ. Effects of antimitotic agents on tubulin-nucleotide interactions. Pharmacol Ther. 1991;52:127.
249. Wilson, L, Jordan, MA, Morse, A, et al. Interaction of vinblastine with steady-state microtubules in vitro. J Mol Biol. 1982;159:125.
250. Bruchovsky, N, Owen, AA, Becker, AJ, et al. Effects of vinblastine on the proliferative capacity of L cells and their progress through the division cycle. Cancer Res. 1965;25:1232.
251. Tucker, RW, Owellen, RJ, Harris, SB. Correlation of cytotoxicity and mitotic spindle dissolution by vinblastine in mammalian cells. Cancer Res. 1977;37:4346.
252. Creasey, WA, Marsh, JC. Metabolism of vinblastine (VBL) in the dog. Proceedings of the American Association for Cancer Research. 1973;14:57. [(abstract)].
253. Vickery, KR, Wilson, H, Vail, DM, et al. Dose-escalating vinblastine for the treatment of canine mast cell tumour. Vet Comp Oncol. 2008;6:111.
254. Grant, IA, Rodriguez, CO, Kent, MS, et al. A phase II clinical trial of vinorelbine in dogs with cutaneous mast cell tumors. J Vet Intern Med. 2008;22:388.
255. Poirier, VJ, Burgess, KE, Adams, WM, et al. Toxicity, dosage, and efficacy of vinorelbine (Navelbine) in dogs with spontaneous neoplasia. J Vet Intern Med. 2004;18:536.
256. Minocha, A, Long, BH. Inhibition of the DNA catenation activity of type II topoisomerase by VP16–213 and VM26. Biochem Biophys Res Commun. 1984;122:165.
257. Chen, GL, Yang, L, Rowe, TC, et al. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J Biol Chem. 1984;259:13560.
258. Long, BH, Musial, ST, Brattain, MG. Single- and double-strand DNA breakage and repair in human lung adenocarcinoma cells exposed to etoposide and teniposide. Cancer Res. 1985;45:3106.
259. Sullivan, DM, Latham, MD, Ross, WE. Proliferation-dependent topoisomerase II content as a determinant of antineoplastic drug action in human, mouse, and Chinese hamster ovary cells. Cancer Res. 1987;47:3973.
260. Flory, AB, Rassnick, KM, Balkman, CE, et al. Oral bioavailability of etoposide after administration of a single dose to tumor-bearing dogs. Am J Vet Res. 2008;69:1316.
261. Pommier, YG, Goldwasser, F, Strumberg, D. Topoisomerase II inhibitors: epipodophyllotoxins, acridines, ellipticines, and bisdioxopiperazines. In: Chabner BA, Longo DL, eds. Cancer chemotherapy & biotherapy: principles and practice. Philadelphia: Lippincott Williams & Wilkins, 2001.
262. Baxter, JD, Harris, AW, Tomkins, GM, et al. Glucocorticoid receptors in lymphoma cells in culture: relationship to glucocorticoid killing activity. Science. 1971;171:189.
263. Greenstein, S, Ghias, K, Krett, NL, et al. Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clin Cancer Res. 2002;8:1681.
264. Moalli, PA, Rosen, ST. Glucocorticoid receptors and resistance to glucocorticoids in hematologic malignancies. Leuk Lymphoma. 1994;15:363.
265. Fichtinger-Schepman, AM, van der Veer, JL, den Hartog, JH, et al. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry. 1985;24:707.
266. Zwelling, LA, Anderson, T, Kohn, KW. DNA-protein and DNA interstrand cross-linking by cis- and trans-platinum(II) diamminedichloride in L1210 mouse leukemia cells and relation to cytotoxicity. Cancer Res. 1979;39:365.
267. Reed, E, Kohn, KW, Chabner, BA, et al. Platinum analogues. In: Cancer chemotherapy: principles and practice. Philadelphia: JB Lippincott; 1990.
268. Calvert, AH, Newell, DR, Gumbrell, LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol. 1989;7:1748.
269. Bailey, DB, Rassnick, KM, Erb, HN, et al. Effect of glomerular filtration rate on clearance and myelotoxicity of carboplatin in cats with tumors. Am J Vet Res. 2004;65:1502.
270. Kisseberth, WC, Vail, DM, Yaissle, J, et al. Phase I clinical evaluation of carboplatin in tumor-bearing cats: a Veterinary Cooperative Oncology Group study. J Vet Intern Med. 2008;22:83.
271. Morgan, JS, Creasey, DC, Wright, JA. Evidence that the antitumor agent hydroxyurea enters mammalian cells by a diffusion mechanism. Biochem Biophys Res Commun. 1986;134:1254.
272. Turner, MK, Abrams, R, Lieberman, I. Meso-alpha, beta-diphenylsuccinate and hydroxyurea as inhibitors of deoxycytidylate synthesis in extracts of Ehrlich ascites and L cells. J Biol Chem. 1966;241:5777.
273. Skoog, L, Nordenskjold, B. Effects of hydroxyurea and 1-beta-D-arabinofuranosyl-cytosine on deoxyribonucleotide pools in mouse embryo cells. Eur J Biochem. 1971;19:81.
274. veer Reddy, GP, Pardee, AB. Inhibitor evidence for allosteric interaction in the replitase multienzyme complex. Nature. 1983;304:86.
275. Bianchi, V, Pontis, E, Reichard, P. Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J Biol Chem. 1986;261:16037.
276. Rassnick, KM, Al-Sarraf, R, Bailey, DB, et al. Phase II open-label study of single-agent hydroxyurea for treatment of mast cell tumours in dogs. Vet Comp Oncol. 2010;8:103.
277. Story, MD, Voehringer, DW, Stephens, LC, et al. L-asparaginase kills lymphoma cells by apoptosis. Cancer Chemother Pharmacol. 1993;32:129.
278. Chabner, BA, Sallan, SE. Enzyme therapy: L-asparaginase. In: Chabner BA, Longo DL, eds. Cancer chemotherapy & biotherapy: principles and practice. Philadelphia: Lippincott Williams & Wilkins, 2001.
279. Su, AI, Welsh, JB, Sapinoso, LM, et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61:7388.
280. Golub, TR, Slonim, DK, Tamayo, P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531.
281. Moseson, DL, Sasaki, GH, Kraybill, WG, et al. The use of antiestrogens tamoxifen and nafoxidine in the treatment of human breast cancer in correlation with estrogen receptor values. A phase II study. Cancer. 1978;41:797.
282. Flaherty, KT, Puzanov, I, Kim, KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809.
283. Paez, JG, Janne, PA, Lee, JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497.
284. Ueda, K, Cornwell, MM, Gottesman, MM, et al. The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem Biophys Res Commun. 1986;141:956.
285. Mauldin, G, Matus, R, Patnaik, A, et al. Efficacy and toxicity of doxorubicin and cyclophosphamide used in the treatment of selected malignant tumors in 23 cats. J Vet Intern Med. 1988;2:60.
286. Herzog, TJ, Krivak, TC, Fader, AN, et al. Chemosensitivity testing with ChemoFx and overall survival in primary ovarian cancer. Am J Obstet Gynecol. 2010;203:68. [e1].
287. Wakatsuki, T, Irisawa, A, Imamura, H, et al. Complete response of anaplastic pancreatic carcinoma to paclitaxel treatment selected by chemosensitivity testing. Int J Clin Oncol. 2010;15:310.
288. Ugurel, S, Schadendorf, D, Pfohler, C, et al. In vitro drug sensitivity predicts response and survival after individualized sensitivity-directed chemotherapy in metastatic melanoma: a multicenter phase II trial of the Dermatologic Cooperative Oncology Group. Clin Cancer Res. 2006;12:5454.
289. Staib, P, Staltmeier, E, Neurohr, K, et al. Prediction of individual response to chemotherapy in patients with acute myeloid leukaemia using the chemosensitivity index Ci. Br J Haematol. 2005;128:783.
290. Lee, JK, Havaleshko, DM, Cho, H, et al. A strategy for predicting the chemosensitivity of human cancers and its application to drug discovery. Proc Natl Acad Sci U S A. 2007;104:13086.
291. Staunton, JE, Slonim, DK, Coller, HA, et al. Chemosensitivity prediction by transcriptional profiling. Proc Natl Acad Sci U S A. 2001;98:10787.
292. Potti, A, Dressman, HK, Bild, A, et al. Retraction: Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2011;17:135.
293. Bonnefoi, H, Potti, A, Delorenzi, M, et al. Retraction–Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: a substudy of the EORTC 10994/BIG 00-01 clinical trial. Lancet Oncol. 2011;12:116.
294. Baggerly, KA, Coombes, KR. Deriving chemosensitivity from cell lines: Forensic bioinformatics and reproducible research in high-throughput biology. Ann Appl Stat. 2009;3:1309.