Chapter 8 Atherosclerosis
Knowledge of the pathobiology of atherosclerosis has continued to evolve at a rapid pace. Previously regarded as a mainly segmental disease, we now increasingly appreciate its diffuse nature. The traditional clinical focus on atherosclerosis has emphasized coronary artery disease (CAD). The attention of physicians in general, and of cardiovascular specialists in particular, now embraces other arterial beds, including the peripheral and cerebrovascular arterial beds.
Formerly considered an inevitable and relentlessly progressive degenerative process, we now recognize that quite to the contrary, atherogenesis progresses at varied paces. Increasing clinical and experimental evidence indicates that atheromatous plaques can evolve in vastly different fashions. Atheromata behave much more dynamically than traditionally conceived, from both structural and biological points of view. Plaques not only progress, but also may regress, and/or alter their qualitative characteristics in ways that decisively influence their clinical behavior.
Concepts of the pathobiology of atherosclerosis have likewise undergone perpetual revision. During much of the 20th century, most considered atherosclerosis a cholesterol storage disease. Recognition of the key role of interactions of vascular cells, blood cells (including leukocytes and platelets), and lipoproteins challenged this model later in the 20th century.1 Current thinking further broadens this schema, incorporating an appreciation of the global metabolic status of individuals, extending far beyond traditional risk factors as triggers to the atherogenic process.
This chapter will delineate the concepts of the widespread and diffuse distributions of atherosclerosis and its clinical manifestations, and also will describe progress in understanding its fundamental biology.
Risk Factors for Atherosclerosis: Traditional, Emerging, and Those on the Rise
Traditional Risk Factors for Atherosclerosis
Cholesterol
Experimental data have repeatedly shown a link between plasma cholesterol levels and the formation of atheromata. Pioneering work performed in Russia in the early 20th century showed that consumption by rabbits of a cholesterol-rich diet caused formation of arterial lesions that shared features with human atheromata.2 By mid-century, application of the ultracentrifuge to analysis of plasma proteins led to the recognition that various classes of lipoproteins transported cholesterol and other lipids through the aqueous medium of blood. Multiple epidemiological studies verified a link between one cholesterol-rich lipoprotein particle in particular—low-density lipoprotein (LDL)—and the risk for coronary heart disease.3 The characterization of familial hypercholesterolemia as a genetic disease provided further evidence linking LDL cholesterol levels with coronary heart disease. Heterozygotes for this condition had markedly elevated risk for atherosclerotic disease. Individuals homozygous for familial hypercholesterolemia commonly develop coronary heart disease within the first decade of life.
Elucidation of the LDL-receptor pathway and that mutations in the LDL receptor cause familial hypercholesterolemia provided proof positive of LDL’s role in atherogenesis.4 Yet the cholesterol hypothesis of atherogenesis still encountered skepticism. Many critics—some laypersons and some respected professionals—questioned aspects of the theory, pointing out that dietary cholesterol levels did not always correlate with cholesterolemia. Lack of proof that either dietary or drug intervention could modify outcomes dogged proponents of the cholesterol hypothesis of atherogenesis.5
Ultimately, controlled clinical trials that lowered LDL by interventions including partial intestinal bypass, bile acid–binding resins, and statin drugs showed reductions in coronary events and vindicated the cholesterol hypothesis. In appropriately powered trials conducted with sufficiently potent agents, lipid lowering also reduced overall mortality. Yet the very success of these interventions suggested there must be more to atherogenesis than cholesterol because a majority of events still occurred despite increasingly aggressive control of LDL cholesterol levels. Identification of proprotein convertase subtilisin/kexin type 9 (PCSK9) as the gene involved in autosomal dominant hypercholesterolemia has furnished new insight in this regard. Reduced function of this enzyme raises cellular LDL receptor numbers, and hence augments LDL clearance, leading to lower plasma LDL levels. Individuals with reduced function variants of PCSK9 who experience lifelong lower exposure to LDL show protection from atherosclerotic events even in the presence of other cardiovascular risk factors. These observations strengthen the case for involvement of LDL in atherogenesis, and for aggressive management of LDL in practice.6
Of course, aspects of the lipoprotein profile other than LDL can influence atherogenesis (see later discussion).3 Yet, because atherosclerotic events commonly occur in individuals with average levels of the major lipoprotein classes, a full understanding of atherogenesis requires consideration of factors other than blood lipids.
Systemic arterial hypertension
The relationship between arterial blood pressure and mortality emerged early from actuarial studies. Insurance underwriters had a major financial stake in mortality prediction. A simple measurement of blood pressure with a cuff sphygmomanometer powerfully predicted longevity. Data emerging from the Framingham Study and other observational cohorts verified a relationship between systemic arterial pressure and coronary heart disease events.7 Concordant observations from experimental animals and epidemiological studies bolstered the link between hypertension and atherosclerosis.
As in the case of high cholesterol, clinical evidence that pharmacological reduction in blood pressure could reduce coronary heart disease events proved fairly elusive. Early-intervention studies readily showed decreases in stroke and congestive heart failure endpoints following administration of antihypertensive drugs. Studies indicating clear-cut reductions in coronary heart disease events with antihypertensive treatment have accumulated much more recently.8
Mechanistically, antihypertensive drug therapy likely benefits atherosclerosis and its complications principally by lowering blood pressure, although some have posited other beneficial actions of various antihypertensive agents. A large randomized clinical trial, the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), showed no advantage over a 5-year period of a thiazide diuretic over an angiotensin receptor blocker, a calcium channel antagonist, or a β-adrenergic blocking agent.9,10
Clinical observations provide strong additional support for the concept that hypertension itself can promote atherogenesis. Atherosclerosis of the pulmonary arteries seldom occurs in individuals with normal pulmonary artery pressures, but even in relatively young patients with pulmonary hypertension, pulmonary arterial atheromata occur quite commonly. This “experiment of nature” supports the direct proatherogenic effect of hypertension in humans.
Cigarette smoking
Tobacco abuse, and cigarette smoking in particular, accentuates the risk of cardiovascular events.11 In the context of noncoronary arterial disease, cigarette smoking appears particularly important. The rapid return toward baseline rates of cardiovascular events after smoking cessation suggests that tobacco use alters the risk of thrombosis as much or more than it may accentuate atherogenesis per se. Classic studies in nonhuman primates have shown little effect of 2 to 3 years of cigarette smoke inhalation on experimental atherosclerosis in the presence of moderate hyperlipidemia.
Both first-hand and second-hand tobacco smoke impairs endothelial vasodilator functions—an index of arterial health.12 Smoking has many other adverse systemic effects, including eliciting a chronic inflammatory response implicated in atherothrombosis.13 Cigarette smoking seems to contribute particularly to abdominal aortic aneurysm formation. The mechanistic link between cigarette smoking and arterial aneurysm formation may resemble that invoked in the pathogenesis of smoking-related emphysema. Studies in genetically altered mice that inhale tobacco smoke have delineated a role for elastolytic enzymes such as matrix metalloproteinase (MMP)-12 in the destruction of lung extracellular matrix (ECM). Smoke-induced inflammation appears to release tumor necrosis factor (TNF)-α from macrophages that can elevate activity of elastolytic enzymes and promote pulmonary emphysema.14 A similar mechanism might well promote destruction of elastic laminae in the tunica media of the abdominal aorta, which characterizes aneurysm formation.
Age
Multiple observational studies have identified age as a potent risk factor for atherosclerotic events.15 Indeed, in the current cardiovascular risk algorithm based on the Framingham Study, age contributes substantially to risk calculation. Demographic trends portend a marked expansion in the elderly population, particularly women, in coming years. Although age-adjusted rates of cardiovascular disease may appear stable or even declining in men, the actual burden of disease in the elderly will increase because of their sheer number. In view of the expanding elderly population, evidence that supports the mutability of atherosclerosis assumes even greater importance (see later discussion).
Sex
Male sex contributes to heightened cardiovascular risk in numerous observational studies. Mechanisms for this increased burden of disease may reflect male-related proatherogenic factors and/or lack of protection conferred by female sex. Cardiovascular risk increases after menopause in women; many previously attributed the vascular protection enjoyed by premenopausal women to estrogen. But estrogen therapy in women (in more recent large-scale clinical trials) and in men (in the older Coronary Drug Project study) seems to confer hazard rather than benefit in the circumstances studied.16 Thus estrogen, certainly in combination with progesterone, does not provide a panacea for protection against cardiovascular events. Decreased in high-density lipoprotein (HDL) levels in blood after menopause might explain part of the apparent premenopausal protection from cardiovascular risk.
High-density lipoprotein
The Framingham risk algorithm recognizes lower strata of HDL—below 40 mg/dL in both women and men—as a risk factor. Numerous concordant population studies have pointed to the importance of HDL as an atheroprotective lipoprotein fraction. Indeed, in patients who undergo cardiac catheterization and display angiographically significant (CAD), low HDL is more common than high LDL.
Numerous animal studies have established the antiatherogenic effects of HDL or its major apolipoprotein (apo) AI, as have small biomarker studies in humans. Mechanisms by which HDL may protect against atherosclerosis include promotion of reverse lipid transport.17,18 Furthermore, nascent HDL particles can take up cholesterol from macrophages and other cells in a process that depends on the adenosine triphosphate (ATP) binding cassette transporter 1 (ABCA1). Mature HDL particles can also take up cholesterol through the related transporter ABCG1.19 In addition, HDL can have antiinflammatory effects.20 Mechanisms of HDL’s arterial protective effect may relate to its ability to bind and carry numerous proteins that may regulate lipid metabolism, oxidative stress, inflammation, and proteolysis.21
High-density lipoprotein’s metabolism is quite complex—more so in many respects than LDL.22 The steady-state level of HDL cholesterol, therefore, does not accurately predict cardiovascular protection. Although baseline levels of HDL cholesterol quite consistently track inversely with cardiovascular events, interventions that raise HDL cholesterol levels do not necessarily reduce event rates. Moreover, recent data challenge the extent to which HDL levels influence outcomes on individuals taking potent LDL-lowering treatment.23,24 The ability of HDL particles to effect cholesterol efflux from macrophages in vitro correlates with CAD status and the biomarker of risk, carotid intima media thickness.18 Yet the ability of pharmacologically induced increases in HDL to confer clinical benefit remains unproven. Several large-scale trials currently in progress should clarify whether drugs of several classes that elevate HDL can reduce clinical events.25,26
Emerging Risk Factors for Atherosclerosis
Homocysteine
Homocysteine, a product of amino acid metabolism, may contribute to atherothrombosis.27 Individuals with genetic defects that lead to elevated homocysteine levels (e.g., homocystinuria, commonly due to deficiency in cystathionine β-synthase) have a thrombotic diathesis. In vitro, treatment with homocysteine and related compounds can alter aspects of vascular cell function related to atherogenesis. Reliable clinical tests for hyperhomocysteinemia exist. Although clearly associated with elevated thrombotic risk in patients with homocystinuria, elevated levels of homocysteine in unselected populations only weakly predict cardiovascular risk (Fig. 8-1). Moreover, randomized trials that have used vitamin treatments to lower homocysteine levels have not documented improvements in cardiovascular outcomes.28–30 Enrichment of cereals and flour with folate in the United States has shifted dietary intake and should lower blood homocysteine levels in the American population.
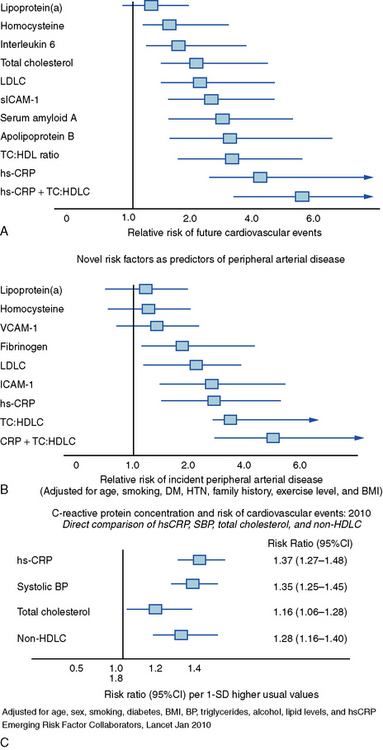
Figure 8-1 Predictive power of some established and emerging risk markers for coronary and peripheral atherosclerosis.
Relative risk for overall cardiovascular events (A) and incidence of peripheral artery disease (B) from the Women’s Health Study and the Physicians’ Health Study, respectively. Whisker plots show point estimates and confidence intervals (CIs) for various emerging and established risk factors for atherosclerotic complications. Rank, order, and magnitude of risk of peripheral artery disease in the Women’s Health Study and of cardiovascular events in the Physicians’ Health Study due to these established and emerging risk factors resembles that reported earlier (not shown). C, Direct comparison of hsCRP, systolic blood pressure, BP, total cholesterol, and non-HDLC adjusted for age, sex, smoking, diabetes, BMI, BP, triglycerides, alcohol, lipid levels, and hsCRP. Comparisons use data from 91,990 participants (5373 CHD events) from 31 studies. hs-CRP, C-reactive protein measured by the high-sensitivity assay; HTN, hypertension; LDLC, low-density lipoprotein cholesterol; sICAM-1, soluble intercellular adhesion molecule-1; TC:HDL ratio, total cholesterol/high-density lipoprotein ratio; TC:HDLC, total cholesterol/high-density lipoprotein cholesterol; VCAM-1, soluble vascular cell adhesion molecule.
(Reproduced with permission from Ridker PM: Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 107:363, 2003; Ridker PM, Stampfer MJ, Rifai N: Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral artery disease. JAMA 285:2481, 2001; and The Emerging Risk Factors Collaboration: C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet 375:132, 2010.)
Lipoprotein(a)
Lipoprotein(a) (Lp[a]—commonly pronounced “L P little A”) consists of a low-density lipoprotein particle with apolipoprotein a (apo a) covalently attached to apo B, the major apolipoprotein of LDL particles. Apo a should not be confused with apo A, the family that includes apolipoprotein A-I, the principal apolipoprotein of HDL. Lipoprotein(a) has considerable heterogeneity, determined genetically and related to the number of repeats of a structural motif known as a kringle in the apo a moiety of the special lipoprotein particle. The structural resemblance of apo a to plasminogen suggests that Lp(a) may inhibit fibrinolysis. Lipoprotein(a) levels in the general population have high skew. Most individuals lie in the lower range of distribution, with a few “outliers” in the higher levels of Lp(a). Black Americans have a higher frequency of elevated Lp(a). Those with Lp(a) levels substantially above normal appear to have increased cardiovascular risk. As in the case of homocysteine, Lp(a) in large populations only weakly predicts future cardiovascular disease,31 but genetic studies suggest a causal role of Lp(a) in provoking cardiovascular events32 (see Fig. 8-1).
Fibrinogen
Fibrinogen, the substrate of thrombin, provides the major meshwork of arterial thrombi. Levels of fibrinogen increase in inflammatory states as part of the acute-phase response. A consistent body of observational evidence links elevated levels of fibrinogen with cardiovascular risk.33,34 Standardization of assays for fibrinogen has proved difficult. Moreover, diurnal variation in plasma fibrinogen levels weakens its potential as a biomarker of cardiovascular risk, despite its obvious biological plausibility as a major participant in thrombosis. Fibrin deposition in plaques, first hypothesized by von Rokitansky in the mid-19th century, provides evidence of fibrinogen’s involvement in atherogenesis.35
Infection
The possibility that infectious agents or responses to infection may contribute to atherogenesis or precipitate atherosclerotic events has periodically captured the fancy of students of atherosclerosis. Many infectious agents could contribute to aspects of atherogenesis by direct cytopathic effect or through mediators they release or elicit as part of a host defense.36–41 The hemodynamic stresses of acute infection, and accentuated thrombotic risk or impaired fibrinolysis due to acute-phase reactants such as fibrinogen or plasminogen activator inhibitor (PAI)-1, might transiently heighten the risk for thrombotic complications of atherosclerosis.42 Some seroepidemiological studies have suggested links between exposure to viral and bacterial pathogens and various measures of atherosclerosis or risk of atherosclerotic events. Prospective studies properly controlled for confounding factors have shown weak, if any, correlation of antibody titers against various microbial or viral pathogens and cardiovascular events. Trials of various antibiotic regimens in patients with CAD have not shown reductions in recurrent events.43
Inflammation and atherosclerosis
Recognition that inflammation provides a unifying theme for many of the pathophysiological alterations that occur during atherogenesis has provoked both interest and controversy.44–46 A subsequent section of this chapter will discuss in detail the links between inflammatory processes and risk factors, and atherogenesis and complications of atherosclerosis. Emergence of C-reactive protein (CRP) as a validated marker of prospective cardiovascular risk has spawned countless studies proposing other inflammatory markers as potential predictors of atherosclerotic risk.47–49 Although some markers of inflammation (e.g., fibrinogen, PAI-1) have defined roles as mediators and markers, evidence regarding CRP as an effector rather than a marker remains unsettled. Despite some laboratory evidence that CRP can produce some effects that may promote atherothrombosis, genetic data do not support a causal role for CRP in cardiovascular events.50–52 Of course, biomarkers need not participate directly in pathogenic pathways to have utility as gauges of risk, as illustrated by the case of CRP.53–55
Genetic predisposition
The example of familial hypercholesterolemia recounted earlier irrefutably illustrates the link between gene mutation and atherosclerosis. The accelerated development of molecular genetic technology and the increasing ease of identifying and cataloging genetic polymorphisms have facilitated the search for genetic variants that predispose toward atherosclerosis or its complications.56 Monogenic conditions such as familial hypercholesterolemia do not appear to explain the majority of the burden or risk of atherosclerotic disease. The quest for genetic polymorphisms that predispose toward atherosclerosis has yielded many potential candidates. Genome-wide association studies have identified reproducible regions of the genome associated with increased cardiovascular risk.57–61 Some of the genes at locations so identified have well-established functions in pathogenic pathways for atherosclerosis.62,63 Other sites emerging from genome-wide association studies have unknown functions and have not been previously associated with cardiovascular disease. Notably, the chromosome 9p21 region concordantly associates with cardiovascular events in several independent large population studies. Yet even combinations of well-validated genetic markers may fail to improve estimation of cardiovascular risk beyond a simple query regarding family history of premature CAD (at < 60 years of age).64,65 Genetic markers of atherosclerotic risk should nonetheless prove useful in providing new understanding of disease mechanisms and may identify new avenues for intervention, even if they do not add substantially to risk stratification.
Risk Factors on the Rise
We are witnessing a transition in the pattern of atherosclerotic risk factors in the United States, and indeed worldwide.66 Certain traditional atherosclerotic risk factors are on the wane. For example, rates of smoking in the United States are declining, particularly in men. Dissemination of effective antihypertensive therapies has provided a means to reduce the degree or prevalence of this traditional atherosclerotic risk factor. Although many patients do not achieve the currently established targets for blood pressure, effective therapies have become much more widely implemented in recent decades.
We have made striking progress in combating high levels of LDL, a major traditional risk factor for atherosclerosis, as discussed earlier. In particular, the introduction of statins and accumulating evidence of their effectiveness as preventive therapies, combined with their relative ease of use and tolerability, should foster a secular trend toward lower LDL levels in the higher-risk segments of our population.
Although we justly derive considerable satisfaction from these pharmacological inroads into the traditional profile of cardiovascular risk, we are rapidly losing ground in other respects.66,67 The astounding increase in obesity in the U.S. population in the last decade alone represents a change in body habitus of substantial proportion in an instant on an evolutionary timescale. Short of major disasters or famines, this kind of rapid shift in body habitus may have no precedent in the history of our species. From the perspective of cardiovascular risk, the metabolic alterations that accompany this increased girth of our population should sound an alarm. Current data point to a significant increase in the prevalence of the components of the clustered risk factors often referred to as the metabolic syndrome.
The National Cholesterol Education Project Adult Treatment Panel III (ATP III) arbitrarily defined five components of the metabolic syndrome68 (Table 8-1); individuals with any three components have the syndrome, according to the ATP III criteria. There is disparity in the definitions of the metabolic syndrome among varied constituencies.69 Some have voiced cogent objections to the concept; for example, it is not clear that the sum of the risk of the metabolic syndrome components exceeds that of the individual factors combined, or that the components comprise a true syndrome, being linked mechanistically.70,71 But many clinicians find the construct practically useful insofar as it reflects the type of individuals seen more and more in practice.
Table 8-1 Criteria for the “Metabolic Syndrome”*
| Risk Factor | Defining Level |
|---|---|
| Abdominal Obesity† (Waist Circumference‡) | |
| Men | > 102 cm (> 40 in) |
| Women | > 88 cm (> 35 in) |
| Triglycerides | ≥ 150 mg/dL |
| HDLC | |
| Men | < 40 mg/dL |
| Women | < 50 mg/dL |
| Blood pressure | ≥ 130/≥85 mm Hg |
| Fasting glucose | ≥ 110 mg/dL |
HDLC, high-density lipoprotein cholesterol.
* Diagnosis is established when ≥ 3 of these risk factors are present.
† Abdominal obesity is more highly correlated with metabolic risk factors than is increased body mass index (BMI).
‡ Some men develop metabolic risk factors when circumference is only marginally increased.
From the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. JAMA 285:2486, 2001.
Note that LDL, the traditional focus of guidelines and therapies, does not figure among the metabolic syndrome criteria; instead, lower ranges of HDL and higher levels of triglycerides characterize the syndrome. Thus, we may witness a shift in lipid risk factor burden from primarily LDL to the dyslipidemia associated with obesity and/or insulin resistance, characterized by lower HDL and higher triglycerides. The success of statins in reducing LDL cholesterol will likely contribute to the increased importance of such dyslipidemia in the future. Although individuals with the metabolic syndrome cluster commonly have dyslipidemia, their levels of LDL cholesterol may be average or even below average. This finding may provide false reassurance to physicians and patients alike. Although LDL cholesterol levels in such patients may not be especially elevated, the quality of the lipoprotein particles may prove particularly atherogenic.72 Low-density lipoprotein particles in those with high levels of triglycerides and low levels of HDL tend to be small and dense. Such small, dense LDL particles appear to bind to proteoglycans in the arterial intima with more avidity. Their retention in the intima may facilitate their oxidative modification, so small, dense LDL particles may have particularly atherogenic properties.73 Prolonged retention and increased entry may promote lipoprotein accumulation in the artery wall.74
Hyperglycemia
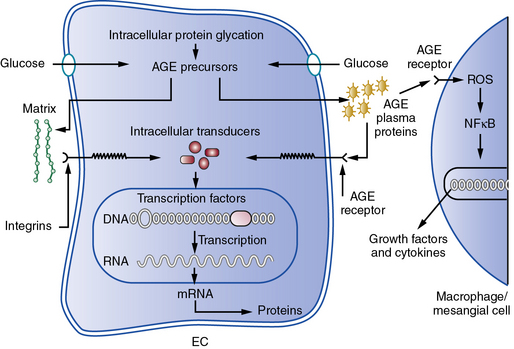
Hyperglycemia may contribute independently to the pathogenesis of atherosclerosis.75 In the presence of higher levels of glucose, nonenzymatic glycation and other types of oxidative posttranslational modification of various macromolecules increases. Hemoglobin A1c, a glycated form of this oxygen-carrying pigment, reflects this biochemical process. Accumulation of glycated macromolecules ultimately leads to the formation of complex condensates known as advanced glycation end products (AGEs) that may trigger inflammatory and oxidative responses implicated in atherogenesis.76 A cell surface receptor for AGEs known as RAGE may transduce proinflammatory signals when occupied by AGE-modified ligands77 (Fig. 8-2). This and other mechanisms link hyperglycemia and insulin resistance to aspects of host defenses considered essential for the atherogenic process, but strict glycemic control does not necessarily improve cardiovascular outcomes, as shown in several recent large clinical trials.78–81
Interactions of risk factors with cells in the arterial wall and leukocytes during atherogenesis
We increasingly understand atherosclerosis as a process that involves cellular interactions with risk factors such as high levels of LDL. This contemporary view contrasts with previous notions that the arterial wall passively accumulated cholesterol. This crosstalk among cells of varying types during atherogenesis involves more than just the intrinsic cells of the arterial wall, endothelium, and vascular smooth muscle cells (VSMCs) (see Chapters 2 and 3).44,82 Indeed, the mononuclear phagocyte also contributes importantly to atherogenesis. Normal endothelium resists prolonged contact with leukocytes, including blood monocytes, precursors of the tissue macrophages that accumulate in atheromata. A mechanism involving expression of particular leukocyte adhesion molecules on the endothelial surface likely mediates recruitment of blood monocytes to sites of formation of the earliest atherosclerotic lesions.83
The heterogeneity of monocytes and the macrophages to which they give rise has generated considerable recent interest.84–87 A particularly proinflammatory subset of monocytes accumulate in the blood of hypercholesterolemic mice. Macrophages exhibiting atherogenic functions also appear to accumulate in atherosclerotic lesions, and therapeutic interventions may modulate these functions.88
Adhesion molecules considered important in this process include members of the selectin superfamily, such as P-selectin. Leukocytes passing through the arterial circulation can bind to patches of endothelial cells (ECs) expressing P-selectin, which mediates a rolling or saltatory slowing of leukocytes. The more permanent adhesion of the tethered white cell depends on expression of another category of leukocyte adhesion molecule expressed on the endothelial surface at sites prone to lesion formation: members of the immunoglobulin (Ig)G superfamily, including vascular cell adhesion molecule-1 (VCAM-1). Both P-selectin and VCAM-1, among other adhesion molecules, show increased expression in regions of human atherosclerotic plaques and on the macrovascular endothelium overlying nascent atherosclerotic plaques in experimental animals. Other leukocyte adhesion molecules certainly participate in this capture of blood leukocytes, but considerable evidence from genetically altered mice supports the essential involvement of P-selectin and VCAM-1 in lesion formation.
Once firmly bound to the endothelial surface, white blood cells must receive chemoattractant stimuli to penetrate into the intima. Among such signals, monocyte chemoattractant protein-1 (MCP-1) may be particularly important. Again, experiments in genetically altered mice support the involvement of MCP-1 in the formation of experimental atherosclerotic lesions. Other chemokines, such as the cell surface–associated molecule fractalkine, also may contribute to this process. In addition to mononuclear phagocytes, T lymphocytes accumulate in human atherosclerotic plaques, where they may play important regulatory roles. Adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1), overexpressed by ECs overlying atheromata, may participate with VCAM-1 in the adhesion of T lymphocytes. Moreover a trio of chemokines induced by the T-cell activator interferon (IFN)-γ may promote chemoattraction of adherent T cells into the arterial intima.89 Mast cells, long recognized in the leukocyte population of the arterial adventitia, also localize within the intimal lesions of atherosclerosis. Although vastly outnumbered by macrophages, mast cells may also contribute to lesion formation or complication.82,90–92 The chemokine exotaxin may participate in recruiting mast cells to the arterial intima.
Once present in the arterial intima, these various classes of leukocytes undergo diverse activation reactions that may potentiate atherogenesis. For example, monocytes mature into macrophages in the atherosclerotic plaque, where they overexpress a series of scavenger receptors that can capture modified lipoproteins that accumulate in the atherosclerotic intima. Because their levels do not decrease as cells accumulate cholesterol, these scavenger receptors permit formation of foam cells, a hallmark of the atheromatous plaque. Macrophages within the atherosclerotic intima proliferate and become a rich source of mediators, including reactive oxygen species and proinflammatory cytokines, that may contribute to progression of atherosclerosis.93
One of the key signals for macrophage activation, macrophage colony-stimulating factor (M-CSF), can enhance scavenger receptor expression and promote replication of macrophages and their production of proinflammatory cytokines. Experiments in mutant mice have shown an important role for M-CSF in the formation of atheromata.
The T cells in the atherosclerotic plaque also appear to modulate aspects of atherogenesis.45,94 Interferon-γ, a strong activating stimulus for macrophages produced by activated type 1 helper T (TH1) cells, localizes in plaques. Indicators of the action of IFN-γ, such as induction of the class II major histocompatibility antigen molecules, provide evidence for the biological activity of IFN-γ in atherosclerotic plaques.
Once recruited to the intima, white blood cells can perpetuate, amplify, or mollify the ongoing inflammatory response that led to their recruitment. The function of the “professional phagocytes” adds to the proinflammatory mediators elaborated by the intrinsic vascular wall cells, and perpetuates and amplifies the local inflammatory response. T lymphocyte subsets may also quell inflammation. T helper 2 cells that produce interleukin (IL)-10, a putative antiatherosclerotic cytokine, also localize in plaques. Regulatory T cells (Treg) produce transforming growth factor (TGF)-β, another antiinflammatory and fibrogenic mediator that may modulate plaque biology.95 The role in atherogenesis of another T-cell subset, TH17, remains unsettled.96–98 Dendritic cells specialized in surveying the environment and presenting antigens to T cells arrive early in the arterial intima of mice subjected to hyperlipidemia.99–101 The nature of the antigenic stimulus to T-cell activation remains speculative, although animal experiments have suggested some candidates.102
Atherosclerosis progression
Recruitment of blood leukocytes and their activation in the arterial intima sets the stage for progression of atherosclerosis. Proinflammatory mediators produced by these various cell types lead to elaboration of factors that can stimulate migration of smooth muscle cells (SMCs) from the tunica media into the intima.103 The normal human tunica intima contains resident SMCs. Growth factors produced locally by activated leukocytes provide a paracrine stimulus to SMC proliferation. Activated SMCs also appear capable of producing growth factors that can stimulate their own proliferation or that of their neighbors, an autocrine pathway of proliferation.104 Smooth muscle cells also die in atheromata.105,106 Depletion of SMCs may influence the biology of plaques by disturbing repair and maintenance of the fibrous cap.107,108
Other mediators present in atheromatous plaques, such as TGF-β, can augment production of macromolecules of the ECM, including interstitial collagen.109,110 Thus, “maturing” atherosclerotic lesions assume fibrous as well as fatty characteristics. Ultimately, the established atherosclerotic plaque develops a central lipid core encapsulated in fibrous ECM. In particular, the fibrous cap—the layer of connective tissue overlying the lipid core and separating it from the arterial lumen—forms during this phase of lesion progression.
Calcification, another characteristic of the advancing atherosclerotic plaque, also involves tightly regulated biological functions. The expression of certain calcium-binding proteins within the plaque may sequester calcium hydroxyapatite. Such deposits, far from fixed, can undergo resorption as well as deposition. Inflammatory pathways participate in regulating mineralization of atheromata.111 Reminiscent of bone metabolism, activated macrophages within the plaque appear to function as osteoclasts. Indeed, mice deficient in M-CSF, the macrophage activator, show increased accumulation of calcified deposits.112 This observation supports the dynamic nature of calcium accretion in the plaque.
The atheroma eventually develops a central region filled with lipid, inflammatory cells, and cellular debris. Macrophage death, including apoptotic (programmed) death, contributes to formation of this lipid-rich core.105,113 Indeed, classical pathologists often referred to this region of the plaque as the necrotic core of atheroma. Defective clearance of apoptotic cells, a process denoted efferocytosis, may contribute to the formation of the plaque’s lipid core.114
During the progression of atherosclerotic plaques, migration and proliferation of SMCs, accumulation of ECM, and calcification lead to the transition from the fatty streak, dominated by the lipid-laden macrophages known as foam cells, to the fibrocalcific plaque that can produce arterial stenoses and other complications. Although this phase of lesion progression in humans may begin in youth, it often continues for many decades. Notably, atherosclerotic plaques often produce no symptoms during this generally prolonged phase of lesion evolution. Although traditionally viewed as a disease of middle and later life, the seeds of atherosclerosis are sown much earlier. This recognition highlights the importance of early and aggressive reduction of risk factors, best accomplished by lifestyle modification rather than pharmacological intervention during the formative phase of the disease process.
The Diversity of Atherosclerosis
Heterogeneity of Atherosclerosis Lesions
Although in the past we have focused on atherosclerosis of the coronary arteries, we now recognize increasingly that atherosclerosis reaches beyond the coronary bed. For example, atherosclerosis underlies many ischemic strokes. Although peripheral artery disease jeopardizes limb more than life, the limitation of exercise capacity and the considerable burden of nonhealing ulcers and other complications render this manifestation of atherosclerosis important from both medical and economical points of view (see Chapter 17). In addition, atherosclerotic involvement of the renal arteries contributes to end-stage renal disease and refractory hypertension in many instances (see Chapter 22).
From a pathophysiological perspective, atherosclerosis in different distributions of the arterial tree overlap considerably. Although the fundamental cellular and molecular events that underlie atherosclerosis in various arterial trees seem similar, certain complications appear distinct. For example, ectasia and eventual aneurysm formation affect the atherosclerotic abdominal aorta more commonly than do stenosis and thrombosis leading to total aortic occlusion. In addition, the aorta, and particularly the proximal portions of its trunk, appears especially important as a source for atheroemboli that may cause cerebral or renal infarctions.115 Atherosclerosis of the extracranial vessels that perfuse the brain often develop stenosis, but ulceration of carotid plaques with embolization of atheromatous material commonly causes transient ischemic attacks or monocular blindness.116
Some of the regional variations in the expressions of atherosclerosis may depend on embryological factors. Endothelial cells in different regions of the arterial tree can display considerable heterogeneity, as determined by a variety of markers.117 Developmental biologists have long recognized that SMCs found in various segments of the arterial tree may have distinct embryological origins. For example, SMCs in the ascending aorta and other arteries of the upper body derive from neural crest cells rather than mesenchyme.118 Thus, SMCs can arise even from different germ layers in the lower body and neurectoderm in certain upper body arteries. The developmental biology of arteriogenesis and the determination of smooth muscle and EC lineages constitute a frontier of contemporary vascular biology research. For example, some but not all recent work suggests that both endothelial cells and smooth muscle cells, for example, may derive from bone marrow postnatally in injured, diseased, or transplanted arteries.119–123 This recognition not only has intrinsic scientific interest but also may have therapeutic implications for regenerative medicine.
Atherosclerosis: a Focal or Diffuse Disease?
We classically understand atherosclerosis as a segmental process. Much of our traditional diagnostic armamentarium and treatment modalities aim to identify stenoses and restore flow by revascularization. Nonetheless, we recognize increasingly the diffuse nature of atherosclerosis. Classic comparison of histopathological examination with angiograms showed that the arteriogram vastly underestimates involvement of coronary arteries by atherosclerosis. More recently, the application of intravascular ultrasound has renewed our appreciation of the diffuse nature of coronary atherosclerosis. Arterial stenoses often cause ischemia and bring the patient to the attention of clinicians. Various noninvasive modalities can disclose ischemia. Contrast angiography readily localizes the focal stenoses that most often cause demand ischemia. Yet cross-sectional images obtained by intravascular ultrasound reveal that segments of arteries that appear absolutely normal by angiogram may nonetheless harbor a substantial burden of atherosclerotic disease. 124
The process of arterial remodeling during atherogenesis explains this apparent paradox. During much of its life history, an atherosclerotic plaque grows in an outward, or abluminal, direction. Thus the plaque can grow silently without producing stenosis. Morphometric studies in nonhuman primates by Clarkson et al. first called attention to this compensatory enlargement of arteries that preserves the lumen during atherogenesis.125 Oft-cited studies by Glagov et al. established the relevance of this process to human coronary atherosclerosis.126 Remodeling also occurs in atherosclerotic peripheral arteries.127 Expansive arterial remodeling can influence the clinical manifestations of atheromata, with those with increased compensatory enlargement more prone to cause clinical events.128 Luminal encroachment usually occurs relatively late in the life history of an atheromatous plaque.
Well-performed and systematic histopathological studies have shown that atherosclerotic disease begins early in life. In the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study, the aortas and coronary arteries of Americans 34 years of age or younger who died of noncardiac causes revealed consistent involvement of the dorsal surface of the abdominal aorta by both fatty and raised arterial plaques.129 The coronary arteries, including the proximal portion of the left anterior descending (LAD) coronary artery, also disclosed involvement, even in this young population. The Bogalusa Heart Study also showed a correlation between risk factors during life and the degree of atherosclerotic involvement at autopsy.130 These systematic observations agree with reports of a substantial burden of coronary arterial atherosclerosis in young American male casualties during the Korean and Vietnam wars.131 Indeed, maternal hypercholesterolemia associates with fatty streak formation in fetuses.132
These various data indicate that atherosclerosis affects arteries far more diffusely than we believed only a few years ago. The process begins much earlier in life than generally acknowledged. Indeed, intravascular ultrasound studies have shown that 1 in 6 American teenagers has significant atherosclerotic involvement of the coronary arteries.133 These findings have considerable importance for understanding the pathophysiology of the clinical manifestations of atherosclerosis. They also have important implications for the management of this disease (see later discussion).
Shear Stress and Atheroprotection: Why Atherosclerosis Begins Where It Does
The foregoing section emphasizes the diffuse nature of atherosclerosis in adults. Yet in both humans and experimental animals, atherosclerosis begins in certain stereotyped locales. The predilection of atherosclerosis for branch points and flow dividers appears quite consistent across species.
Why do these sites have a predisposition to early atherogenesis? Decades of sophisticated biomechanical analysis have established that atheromata tend to form at sites of disturbed blood flow, particularly areas of low shear stress.134,135 Endothelial cells somehow can sense shear stress; in areas of laminar shear stress in vivo and in monolayers of cultured cells in vitro, for example, ECs align their long axes parallel to the direction of flow. At branch points and dividers in the arterial tree, the well-ordered cobblestone array of the endothelial monolayer changes—cells appear more polygonally and irregularly shaped. Areas of low shear stress show heightened EC turnover, increased permeability, and prolonged retention of lipoprotein particles in the subendothelial regions of the intima. Such data, accumulated over many decades, provide answers to the question of what goes awry at sites of lesion predilection.
More recent data have inspired a different and complementary hypothesis to explain the focality of atherosclerosis initiation. Transcriptional profiling provides a “snapshot” of the expression of a large number of genes in a single experiment. The pattern of genes expressed by ECs subjected to controlled physiological levels of laminar shear stress in vitro differs strikingly from that of resting ECs in vitro. Several of genes differentially expressed by ECs experiencing laminar shear stress appear to have “atheroprotective” functions. A number of putative atheroprotective genes rise selectively under conditions of physiological laminar shear stress. These findings suggest that regions of undisturbed laminar shear stress enjoy tonic endogenous antioxidant, vasodilatory, and antiinflammatory properties conferred by the function of these putative “atheroprotective” genes.136
For example, superoxide dismutase can catabolize the highly reactive superoxide anion (O2−). Cyclooxygenase (COX)-2 can give rise to the vasodilatory and antiaggregatory arachidonic acid metabolite prostacyclin. The endothelial isoform of nitric oxide synthase (eNOS) produces the endogenous vasodilator, antithrombotic, and antiinflammatory mediator nitric oxide (NO), which exerts antiinflammatory actions by combating leukocyte adhesion to the activated EC. The transcription factor Krüppel-like factor 2 (KLF-2) has emerged as an integrator of shear stress and altered endothelial functions implicated in “atheroprotection.”137 Flow-mediated stimulation of adenosine monophosphate (AMP)-dependent protein kinase may in turn signal activation of KLF-2.138
At regions of disturbed flow—for example, near branch points and flow dividers—expression of these endogenous atheroprotective genes should decline. Indeed, regions predisposed to early lesion formation show activation of nuclear factor (NF)κB, the master regulator of inflammatory gene expression.139 Because NO can antagonize activation of NFκB, absence of laminar flow in these regions may explain, at least in part, the tendency of nascent atheromata to form at such sites.140
Thus, atherosclerosis is both a focal and diffuse disease. It begins focally for reasons we understand in increasing detail. The stenoses that cause flow-limiting lesions tend to localize in similar regions. Much of our diagnostic and therapeutic activity in contemporary cardiology and vascular medicine has traditionally focused on these stenoses. Advances in both arterial biology and clinical science heighten our appreciation of the diffuse distribution of atherosclerosis and the systemic nature of the risk factors that promote its development. These considerations help us understand how optimum medical management with systemic therapies can confer benefits on par with those derived from revascularization procedures in many patients.141
Pathophysiology of Thrombotic Complications of Atherosclerosis
As noted, flow-limiting stenoses have driven much of the diagnostic and therapeutic activity in clinical atherosclerosis for many decades. Patients with flow-limiting lesions often experience symptoms due to ischemia (e.g., angina pectoris in the coronary circulation, intermittent claudication in peripheral artery disease). We can readily diagnose ischemia by various noninvasive modalities. We can localize stenoses through invasive and noninvasive angiographic techniques. Percutaneous and surgical approaches can effectively relieve ischemia due to focal stenoses, but do not address lesions that do not limit flow.
We recognize increasingly that in many cases, acute thrombosis does not occur in the most tightly narrowed segments of an artery. The best illustration of this counterintuitive observation arises from analyses of fatal coronary thromboses. Clinical evidence suggests that a minority of fatal coronary thrombi occur where stenoses exceed 60%.142,143 Thus, many occlusive thrombi complicate lesions that neither limit flow nor meet traditional angiographic criteria for “significance.” Fewer data exist regarding the substrates for thrombosis in noncoronary arteries. But in peripheral and carotid arteries, inflammation—tightly linked to mechanisms of thrombosis—characterizes atherosclerotic lesions. Intraplaque hemorrhage appears quite common in carotid plaques that cause cerebral ischemic disease as well.
Although most fatal myocardial infarctions (MIs) occur at sites of noncritical stenosis, it does not follow that high-grade stenoses cause fewer heart attacks than less obstructive lesions. Indeed, on a “per-lesion basis,” tighter stenoses are more likely to give rise to acute MIs.144 Because the noncritically stenotic lesions outnumber “tight” stenoses, the total risk attributable to less stenotic lesions exceeds the risk due to the less numerous lesions that cause higher-grade stenoses.
A common confusion surrounds the distinction between lesion size and degree of stenosis. Based on our traditional angiographically centered view of atherosclerosis, many assume that lesions that cause high-grade stenoses are larger than those that cause less obstruction. This fallacy fails to consider the importance of outward remodeling or compensatory enlargement. The outward growth of most atherosclerotic plaques before they begin to encroach on the lumen protects the lumen from obstruction and conceals the growing lesion from visualization by angiography until the latter stages of its evolution. Thus, low-grade stenoses do not equate with smaller plaques. Indeed, larger and eccentrically remodeled plaques may cause acute coronary syndromes (ACSs) more frequently than smaller plaques that do not exhibit compensatory enlargement and/or produce greater degrees of stenosis.145
Concordant evidence from several avenues of investigation suggests that a physical disruption of the atherosclerotic plaque, rather than a critical degree of stenosis, commonly precipitates arterial thromboses. Four mechanisms of plaque disruption may cause thrombosis or rapid plaque expansion146,147 (Fig. 8-3). A through-and-through fracture of the plaque’s fibrous cap causes most fatal coronary thromboses (see Fig. 8-3). Our group hypothesized a model of the pathophysiology of this common mechanism of atherosclerotic plaque disruption that focused on the metabolism of interstitial collagen. This ECM macromolecule accounts for much of the biomechanical strength of the plaque’s fibrous cap. Further hypothesizing that inflammation regulates the metabolism of interstitial forms of collagen and might regulate the stability of an atherosclerotic plaque (Fig. 8-4), we found that certain proinflammatory cytokines expressed in the atherosclerotic plaque can inhibit the ability of the SMC to synthesize new collagen required to repair and maintain the integrity of the plaque’s fibrous cap. Notably, the signature TH1 cytokine IFN-γ can inhibit interstitial collagen gene expression in human VSMCs.109
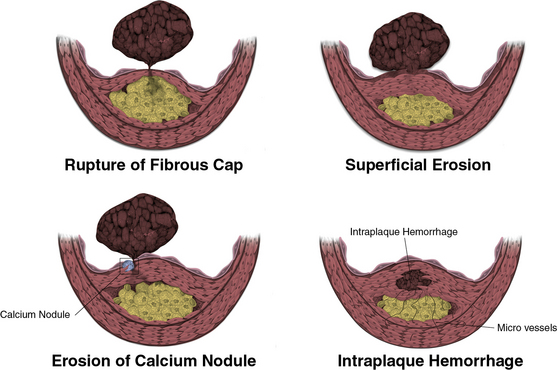
Figure 8-3 Mechanisms of plaque disruption.
Rupture of fibrous cap (upper left) causes two thirds to three quarters of fatal coronary thrombosis. Superficial erosion (upper right) occurs in one fifth to a one quarter of all cases of fatal coronary thrombosis. Certain populations, such as diabetic individuals and women, appear more susceptible to superficial erosion as a mechanism of plaque disruption and thrombosis. Erosion of a calcium nodule may also cause plaque disruption and thrombosis (lower left). In addition, friable microvessels in the base of an atherosclerotic plaque may rupture and cause intraplaque hemorrhage (lower right). Consequent local generation of thrombin may stimulate smooth muscle proliferation, migration, and collagen synthesis, promoting fibrosis and plaque expansion on a subacute basis. Severe intraplaque hemorrhage also can cause sudden lesion expansion by a mass effect acutely.
(Reproduced with permission from Libby P, Theroux P: Pathophysiology of coronary artery disease. Circulation 111:3481, 2005).
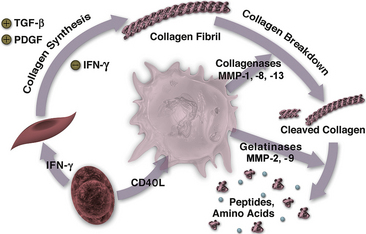
Figure 8-4 Relationship between inflammation and collagen metabolism in the atherosclerotic plaque.
T lymphocytes can elaborate interferon (IFN)-γ, which reduces ability of smooth muscle cell (SMC) to synthesize new collagen. T cell also can stimulate the macrophage through CD40 ligand to elaborate interstitial collagenases, matrix metalloproteinase MMP-1, -8, -13—enzymes that affect initial cleavage in the collagen fibril. Elaboration of gelatinases such as MMP-2, -9 can contribute to further cleavage of collagen fragments to peptides and amino acids. Transforming growth factor (TGF)-β and platelet-derived growth factor (PDGF) from autocrine or paracrine sources, including degranulating platelets, can promote collagen synthesis by SMCs.
The interstitial collagen triple helix resists breakdown by most proteinases. We described the overexpression of proteolytic enzymes specialized in the catabolism of collagen in the atherosclerotic plaque, and further demonstrated that inflammatory mediators found in the atheroma can enhance the expression of these collagenolytic enzymes, members of the MMP family. Cells in human atheromata overexpress all three members of the human interstitial collagenase family (MMP-1, MMP-13, and MMP-8). We have furnished evidence that collagen breakdown occurs in situ in human atherosclerotic plaques.148,149 These findings indicate that the interstitial collagenase MMPs exist in their active forms rather than their precursor zymogen forms. Furthermore, demonstration of collagenolysis in situ indicates that these collagenases overwhelm their endogenous inhibitors, including tissue inhibitors of MMPs (TIMPs).150 We also ascribed a hitherto unknown function to tissue factor pathway inhibitor (TFPI)-2 as a potent collagenase inhibitor.151 Although we find little regulation of TIMPs in atherosclerotic lesions, TFPI-2, abundant in normal arteries, shows decreased levels in atherosclerotic lesions. Thus, a preponderance of proteinases over their inhibitors prevails in the atherosclerotic plaque.
Colocalization of proteinases with inflammatory cells, and regulation of their expression by products of inflammatory cells, strongly implicate disordered collagen metabolism as a key mechanism for atherosclerotic plaque destabilization. Experiments using genetically altered mice have demonstrated the importance of collagen catabolism due to MMP collagenases in the regulation of the steady-state level of this ECM macromolecule in experimental atheromata using both loss-of-function and gain-of-function strategies.152–154 The finding that inflammatory mediators elicit overexpression of MMP collagenases from macrophages supports the view that inflammation promotes the thrombotic complications of atherosclerosis. This mechanistic insight aids the understanding of how biomarkers of inflammation can help predict such events.
Superficial erosion of the endothelial monolayer constitutes an important cause of a minority of coronary thromboses155,156 (see Fig. 8-3). Women, the elderly, patients with diabetes, and patients with hypertriglyceridemia appear to have a higher frequency of superficial erosion as a cause of fatal thrombosis than do younger, male hypercholesterolemic individuals. Various molecular and cellular mechanisms may underlie superficial erosion. Excessive proteolysis of ECM macromolecules that make up the subendothelial basement membrane may predispose toward endothelial desquamation and superficial erosion. Apoptosis of ECs may also promote superficial erosion. Various proinflammatory stimuli can sensitize ECs to apoptosis. In addition, hypochlorous acid, a product of myeloperoxidase—an enzyme found in leukocytes in atherosclerotic plaques—can provoke EC apoptosis.157 Local generation of tissue factor from dying ECs may also contribute to thrombosis at sites of superficial erosion.
Atherosclerotic plaques often harbor rich plexi of microvessels. Neovascularization of plaques provides an additional portal for trafficking of leukocytes that may promote the inflammatory process.158 Neovessels in the plaque, like those in the diabetic retina, may be friable and fragile. Intraplaque hemorrhage due to disruption of microvessels may cause sudden plaque expansion (see Fig. 8-3). Local generation of thrombin and other mediators associated with coagulation in situ may promote lesion growth. For example, platelet-derived growth factor (PDGF), TGF-β, and platelet factor 4 (PF4), released by platelets at sites of microvascular hemorrhage and intramural thrombosis, may hasten local fibrosis. Thrombin can stimulate SMC migration, division, and collagen synthesis. Thus, microvascular disruption, while not provoking an occlusive thrombus, may promote lesion evolution nonetheless.
Erosion through the intima of a calcified nodule represents another less common form of atherosclerotic plaque disruption associated with thrombosis (see Fig. 8-3). The active metabolism of calcium hydroxyapatite, with its accretion and removal as described earlier, can contribute to calcium accumulation in regions of atherosclerotic plaques. The tendency of atheromata to accumulate calcium has given rise to clinical testing. Increasing evidence supports the contention that the amount of coronary artery calcification correlates with the burden of atherosclerotic disease. Moreover, emerging data suggest a correlation between calcium score and risk of future cardiovascular events.
The degree to which calcium scoring or any other imaging modality (such as computed tomographic coronary angiography) can provide prognostic information beyond that available from risk algorithms based on traditional risk factors and selected emerging biomarkers, such as CRP measured with the high-sensitivity assay (hsCRP), remains uncertain. The notion that arterial calcification bears a relationship to atherosclerosis complication holds considerable sway. Yet, current biological understanding and preliminary clinical evidence suggest that coronary lesions most likely to cause thrombosis contain less calcium than lesions that cause flow-limiting stenoses and ischemia.159,160 Thus, the calcified lesions may have less propensity to disrupt and provoke coronary thrombosis than those with little calcium.
Mutability of the Atherosclerotic Plaque
The classic concept of atherosclerosis assumed a steady, relentless, and continuous progression of the disease. But serial angiographic studies suggest that stenoses evolve in a discontinuous fashion, with periods of relative quiescence punctuated by spurts in growth. Our current understanding of plaque pathobiology suggests a plausible mechanism to explain this angiographically discontinuous evolution of arterial stenoses. Careful study of the coronary arteries of individuals who succumb to noncardiac death, and of coronary arteries perfusion-fixed in the operating room from hearts of transplantation recipients with ischemic cardiomyopathy, as well as observations of cholesterol-fed nonhuman primates, all indicate that areas of plaque disruption or superficial erosion with nonocclusive mural thrombus formation occur frequently. The picture that arises from an amalgamation of these human and experimental observations indicates that most plaque disruptions with thrombosis in situ do not proceed to a total occlusion and, indeed, usually pass unnoticed by the patient or physician (Fig. 8-5). For various reasons, plaque disruptions with mural thrombosis may not proceed to a disastrous total thrombosis in all events. In many patients, the endogenous fibrinolytic mechanisms or antithrombotic effects of dietary or pharmacological intervention may prevent thrombi from propagating. Larger arteries such as the aorta have sufficient flow to prevent mural thrombi from progressing to total occlusion in most instances. Indeed, although inspection of the aortas of many patients with atherosclerosis discloses many ulcerated lesions with mural thrombi, aortic occlusion due to thrombosis fortunately occurs relatively rarely (Fig. 8-6).
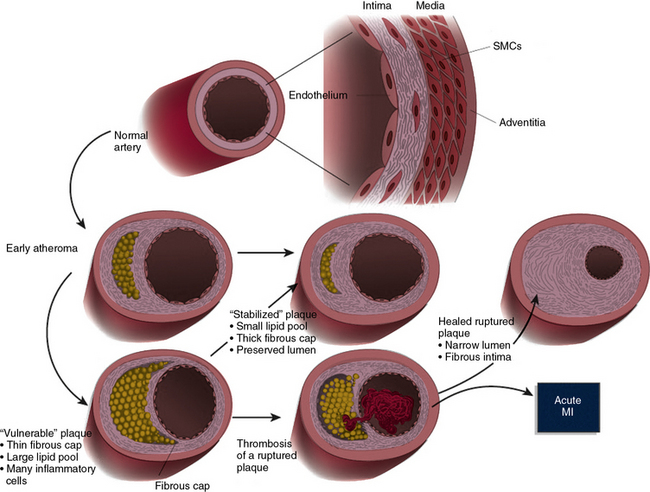
Figure 8-5 Schematic of the life history of an atheroma.
A normal human coronary artery has a typical trilaminar structure. Endothelial cells (ECs) in contact with blood in arterial lumen rest on a basement membrane. Intimal layer in adult humans generally contains a small amount of smooth muscle cells (SMCs) scattered within intimal extracellular matrix (ECM). Internal elastic lamina forms a barrier between the tunica intima and underlying tunica media. Tunica media consists of multiple layers of SMCs much more tightly packed than in diffusely thickened intima, and embedded in a matrix rich in elastin and collagen. In early atherogenesis, recruitment of inflammatory cells and accumulation of lipids leads to formation of a lipid-rich core, as artery enlarges in an outward (abluminal) direction to accommodate intimal expansion. If inflammatory conditions prevail and risk factors such as dyslipidemia persist, lipid core can grow, and proteinases secreted by activated leukocytes can degrade extracellular matrix; whereas proinflammatory cytokines such as interferon (IFN)-γ can limit synthesis of new collagen. These changes can thin the fibrous cap and render it friable and susceptible to rupture. When a plaque ruptures, blood coming in contact with tissue factor in plaque coagulates; platelets activated by thrombin generated from coagulation cascade, and by contact with collagen in intimal compartment, instigate thrombus formation. If thrombus occludes the vessel persistently, acute myocardial infarction (MI) can result (dusky blue area in anterior wall of left ventricle [lower right]). Thrombus may eventually resorb secondary to endogenous or therapeutic thrombolysis, but a wound-healing response triggered by thrombin generated during blood coagulation can stimulate smooth muscle proliferation. Platelet-derived growth factor (PDGF) released from activated platelets stimulates SMC migration. Transforming growth factor (TGF)-β, also released from activated platelets, stimulates interstitial collagen production. This increased migration, proliferation, and ECM synthesis by SMCs thickens fibrous cap and causes further intimal expansion, often in an inward direction, constricting lumen. Such stenotic lesions produced by luminal encroachment of fibrosed plaque may restrict flow—particularly under situations of increased cardiac demand—leading to ischemia, commonly provoking symptoms such as angina pectoris. Such advanced stenotic plaques, being more fibrous, may prove less susceptible to rupture and renewed thrombosis. Lipid lowering can reduce lipid content and calm intimal inflammatory response, yielding a more stable plaque with a thick fibrous cap and preserved lumen (center).
(Reproduced with permission from Libby P: Inflammation in atherosclerosis. Nature 420:868, 2002.)
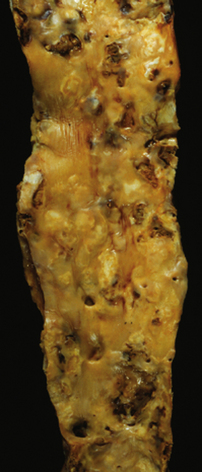
Figure 8-6 Widespread atheromatous involvement of abdominal aorta in patient with atherosclerosis.
Note the variety of atheromata in different stages of evolution within a few centimeters of one another. There are ulcerated plaques, raised lesions, and fatty streaks among other types of lesions demonstrated in this example.
The failure of most mural thrombi to progress to total occlusion does not imply that such events have a benign course. Although subclinical at the time that they occur, the mural thrombi elicit a local “wound-healing” response that tends to promote plaque progression. Indeed, one of the types of “crisis” in the history of a plaque that can lead to its sudden evolution, as disclosed by serial angiographic studies, likely reflects such a scenario (see Fig. 8-5). A localized plaque disruption with mural thrombus can engender a healing response induced by platelet products released at short range, such as TGF-β, a potent stimulus to collagen gene expression by SMCs. Platelet-derived growth factor, also released during platelet aggregation, stimulates SMC migration. Thrombin locally generated at sites of thrombosis can stimulate SMC proliferation, migration, and collagen gene expression.
All these molecular and cellular mechanisms conspire to promote a cycle of SMC migration, proliferation, and local collagen synthesis. Quite plausibly, this scenario leads to evolution from a lipid-rich plaque with abundant inflammatory cells to a more fibrous lesion, rich in connective tissue, often with a paucity of inflammatory cells due to their death or departure, accretion of calcium deposits, and a relative lack of lipid accumulation. Careful study of atheromata obtained at autopsy shows signs of healed plaque disruption in some fibrous lesions161,162 (Fig. 8-7).
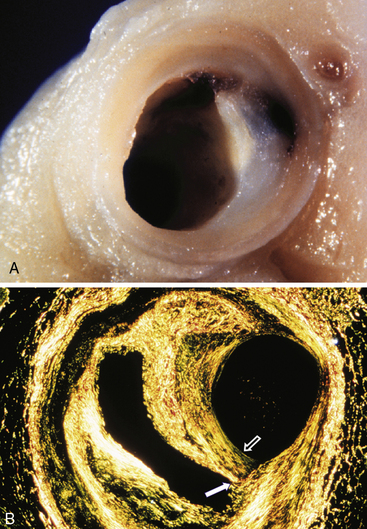
Figure 8-7 Hemorrhage, thrombosis, and plaque healing as a mechanism of atheroma progression.
A, Photograph of coronary artery with plaque rupture that has led to an intraplaque hematoma without an occlusive thrombus. As explained in text, thrombin and platelet products such as platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β, elaborated locally at the site of microthrombosis and hematoma formation, can stimulate fibrosis. B, This Sirius red–stained preparation of cross-section of coronary artery shows an area of plaque rupture (solid arrow) that healed to cause further accretion of a layer of collagen (open arrow) and luminal encroachment. This example illustrates the “archaeology” of the atherosclerotic plaque: distant plaque rupture, followed by healing and fibrosis, with progression of the lesion to stenosis. Note that the arterial lumen at the time of the original plaque rupture would not have shown a critical narrowing.
(These figures were kindly provided by the late Prof. Michael J. Davies.)
The biological scenario just described depicts a more dynamic life history of the atherosclerotic plaque than that heretofore recognized (see Fig. 8-5). The heterogeneity of human atherosclerotic plaques, a concept now gaining considerable currency, highlights the importance of the qualitative characteristics of a lesion, not just its size. Whereas previous concepts emphasized the structure of atherosclerotic plaques, contemporary thinking accords a greater contribution to the biological characteristics of the plaque.
“Stable” vs. “Vulnerable” Plaques
Recognition of the heterogeneity of atherosclerotic plaques has fostered adoption of a dichotomous view of atheromata: “stable” versus “vulnerable” plaques. The notion of the vulnerable plaque has engendered considerable interest from pathologists and practitioners alike. Current cardiologic parlance uses the term “vulnerable plaque” to designate a lesion characterized by a thin fibrous cap, a large lipid core, and a surfeit of inflammatory cells. Use of this term and its opposite, the “stable plaque,” extends findings largely obtained at postmortem examination of humans who succumbed to acute MI. Although the dichotomization of plaques into vulnerable and stable provides a useful shorthand, we should exercise care in uncritically extrapolating morphological findings to foretell clinical complications.147
This pigeon-holing of plaques has engendered considerable effort to develop methods for identifying vulnerable or high-risk lesions, but such a view of atherosclerosis oversimplifies a disease of staggering complexity. For example, considerable evidence suggests that a given arterial tree has not one but many vulnerable plaques. Angioscopic and intravascular ultrasonographic study of coronary arteries, as well as interpretation of angiographic observations, support the multiplicity of disrupted plaques in patients with ACSs.163
Inflammation may provide a mechanistic link between “instability” of coronary and carotid plaques in the same individuals. As previously mentioned, the aorta in an atherosclerotic individual often shows multiple ulcerated lesions, often within millimeters of fatty streaks, raised fibrous lesions, and resorbing healing thrombi (see Fig. 8-5). Thus, the quest to identify a single vulnerable plaque underestimates the complexity of the clinical challenge. Our broader view of the risk of atherosclerotic complications seeks the “vulnerable patient” and targets intervention more widely than to a single vulnerable plaque.
How could one approach such a vulnerable patient? Experimental and clinical consideration provide grounds for considerable optimism in this regard. Atherosclerosis exhibits considerable mutability; lipid lowering and other manipulations can alter features of plaques paramount to the clinical expression of the disease. Preclinical studies have shown that lipid lowering by diet or by treatment with statins can alter features of plaques associated with vulnerability in humans, such as proteinase activity or collagen content.164–166 Likewise, treatment with angiotensin-converting enzyme (ACE) inhibitors can confer characteristics of stability on experimentally produced plaques in rabbits.167 Studies in atherosclerosis-prone mice have demonstrated that interruption of inflammatory signaling pathways or manipulation of TGF-β can alter features of plaques associated with their propensity to rupture and provoke thrombosis. In addition to structural changes relating to the collagen content of plaques that may determine their biomechanical integrity, interventions such as lipid lowering can reduce expression of tissue factor, hence lowering the thrombogenic potential of the atherosclerotic plaque.
Clinical studies also support the mutability of atherosclerosis, abundantly demonstrated in animals by the experiments described earlier. Infusion of forms of apo A-I can cause modest reductions in the volume of atherosclerotic plaques, as monitored by intravascular ultrasound.168,169 Aggressive lipid-lowering therapy with a statin can arrest or even reverse accumulation of atherosclerotic plaque, as determined by serial ultrasonographic study.170 Careful histological correlations with magnetic resonance imaging (MRI) disclose evidence of the mutability of atherosclerotic plaques in intact humans.171 These preclinical and clinical observations inspire considerable optimism regarding our ability to manipulate atherosclerotic plaques to benefit patient outcomes. The disease appears much more mutable than generally appreciated in the past.
Atherosclerosis: a Systemic Disease
Our traditional medical focus on atherosclerosis as a segmental disease caused by cholesterol accumulation has undergone an accelerated revision. We increasingly understand the global nature of factors that encompass the entire metabolic state of the patient, not just the serum cholesterol level. As the spectrum of risk factors in our population shifts, our attention must likewise broaden to encompass not just hypercholesterolemia but also the dyslipidemia associated with the metabolic syndrome and diabetes. As elevated LDL cholesterol and smoking recede as risk factors in our society, we must acknowledge the increasing contribution of obesity and insulin resistance.
We also recognize increasingly the importance of atherosclerosis beyond the coronary arteries. Our purview should now embrace the entire arterial tree with all its beds. We must strive to fill knowledge gaps about regional differences in human atherosclerosis, as well as differences in the biology and clinical manifestations of atherosclerosis associated with the shifting pattern of risk factors.
Our comprehension of the pathophysiology of atherosclerosis has undergone a revolution in recent decades. Emerging data and deeper understanding of this process will continue to change our concepts of this disease in the future. We should take immense satisfaction in the therapeutic inroads furnished by contemporary pharmacological tools. Reduction in coronary heart disease and cerebrovascular events accruing from treatments like statins and ACE inhibitors has changed medicine irrevocably, and provides striking benefits to patients. Therapeutic advances that have emerged from the application of progress in basic science, and have proved effective in randomized clinical trials, furnish cardiovascular medicine with an enviable and unparalleled database for the practice of evidence-based medicine.
Yet despite the victory of clinical science, much remains undone. The majority of cardiovascular events still occur despite optimal therapy that addresses multiple facets of our current understanding of the pathophysiology of this disease. Our challenge for the future is clear. We must strive to turn the tide on the alarming trends toward increased cardiovascular risk due to obesity and physical inactivity. We must not relent in our quest to advance the scientific understanding of atherogenesis and the translation to innovative therapies that address the residual and ever-growing burden of disease.
Many of the current landmark studies in the cardiovascular arena have focused on death and ACSs as “major adverse coronary events.” These endpoints offer relative ease of study, adjudication, and quantification. But we must not lose sight of the enormous impediment to quality of life due to intermittent claudication caused by peripheral artery disease, and the loss of ability to communicate and live independently due to cerebrovascular disease. In addition to the human costs of these manifestations of noncoronary atherosclerosis, our society shoulders a substantial economic burden owing to the ravages of peripheral artery and cerebrovascular disease. We must pursue these issues with the same fervor we have accorded to CAD. This goal will become even more important as elder segments of our population continue to increase in numbers. The powerful tools available to investigators today provide grounds for optimism. Further inroads against the residual morbidity and mortality from atherosclerosis will emerge from future application of basic science to alter the biology of this disease, regarded not so long ago as an inevitable companion of the aging process.
1 Libby P., Ridker P.M., Hansson G.K. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325.
2 Anitschkow N., Chalatow S. On experimental cholesterin steatosis and its significance in the origin of some pathological processes (1913). Reprinted in Arteriosclerosis. 1983;3:178–182.
3 Di Angelantonio E., Sarwar N., Perry P., et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000.
4 Goldstein J.L., Brown M.S. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–438.
5 Steinberg D. The cholesterol wars: the skeptics vs. the preponderance of evidence, ed 1. San Diego, CA: Elsevier Academic Press; 2007.
6 Horton J.D., Cohen J.C., Hobbs H.H. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 50 Suppl. 2009:S172–S177.
7 Lloyd-Jones D.M., Evans J.C., Levy D. Hypertension in adults across the age spectrum: current outcomes and control in the community. JAMA. 2005;294(4):466–472.
8 Wang J.G., Staessen J.A., Franklin S.S., et al. Systolic and diastolic blood pressure lowering as determinants of cardiovascular outcome. Hypertension. 2005;45(5):907–913.
9 Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs. diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA. 2002;288(23):2981–2997.
10 Onuigbo M.A. ALLHAT findings revisited in the context of subsequent analyses, other trials, and meta-analyses. Arch Intern Med. 169(19), 1810, 2009. author reply 1810–1811
11 Smoking-attributable mortality, years of potential life lost, and productivity losses–United States, 2000–2004. MMWR Morb Mortal Wkly Rep. 2008;57(45):1226–1228.
12 Thomas G.N., Chook P., Yip T.W., et al. Smoking without exception adversely affects vascular structure and function in apparently healthy Chinese: implications in global atherosclerosis prevention. Int J Cardiol. 2008;128(2):172–177.
13 Yanbaeva D.G., Dentener M.A., Creutzberg E.C., et al. Systemic effects of smoking. Chest. 2007;131(5):1557–1566.
14 Churg A., Wang R.D., Tai H., et al. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med. 2003;167(8):1083–1089.
15 Marma A.K., Berry J.D., Ning H., et al. Distribution of 10-year and lifetime predicted risks for cardiovascular disease in US adults: findings from the National Health and Nutrition Examination Survey 2003 to 2006. Circ Cardiovasc Qual Outcomes. 2010;3(1):8–14.
16 Magliano D.J., Rogers S.L., Abramson M.J., et al. Hormone therapy and cardiovascular disease: a systematic review and meta-analysis. BJOG. 2006;113(1):5–14.
17 Rye K.A., Bursill C.A., Lambert G., et al. The metabolism and anti-atherogenic properties of HDL. J Lipid Res. 50 Suppl. 2009:S195–S200.
18 Khera A.V., Cuchel M., de la Llera-Moya M., et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364(2):127–135.
19 Tall A.R., Yvan-Charvet L., Terasaka N., et al. ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7(5):365–375.
20 Rye K.A., Barter P.J. Antiinflammatory actions of HDL: a new insight. Arterioscler Thromb Vasc Biol. 2008;28(11):1890–1891.
21 Vaisar T., Pennathur S., Green P.S., et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117(3):746–756.
22 Brewer H.B.Jr. HDL metabolism and the role of HDL in the treatment of high-risk patients with cardiovascular disease. Curr Cardiol Rep. 2007;9(6):486–492.
23 Barter P., Gotto A.M., LaRosa J.C., et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357(13):1301–1310.
24 Ridker P.M., Genest J., Boekholdt M., et al. HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial. Lancet. 2010;376(9738):333–339.
25 Duffy D., Rader D.J. Update on strategies to increase HDL quantity and function. Nat Rev Cardiol. 2009;6(7):455–463.
26 Cannon C.P., Shah S., Dansky H.M., et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363(25):2406–2415.
27 Handy D.E., Loscalzo J. Homocysteine and atherothrombosis: diagnosis and treatment. Curr Atheroscler Rep. 2003;5(4):276–283.
28 Lonn E., Yusuf S., Arnold M.J., et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med. 2006;354(15):1567–1577.
29 Bonaa K.H., Njolstad I., Ueland P.M., et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med. 2006;354(15):1578–1588.
30 Armitage J.M., Bowman L., Clarke R.J., et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA. 2010;303(24):2486–2494.
31 Erqou S., Kaptoge S., Perry P.L., et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–423.
32 Clarke R., Peden J.F., Hopewell J.C., et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–2528.
33 Danesh J., Lewington S., Thompson S.G., et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294(14):1799–1809.
34 Mora S., Rifai N., Buring J.E., et al. Additive value of immunoassay-measured fibrinogen and high-sensitivity C-reactive protein levels for predicting incident cardiovascular events. Circulation. 2006;114(5):381–387.
35 Bini A., Kudryk B.J. Fibrinogen in human atherosclerosis. Ann N Y Acad Sci. 1995;748:461–471. discussion 471–463
36 Libby P., Egan D., Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research, [Review] [91 refs]. Circulation, 96;11. 1997:4095–4103.
37 Muhlestein J.B. Bacterial infections and atherosclerosis, [Review] [31 refs]. J Invest Med, 46;8. 1998:396–402.
38 Ridker P.M. Are associations between infection and coronary disease causal or due to confounding? Am J Med. 1999;106(3):376–377. (editorial, comment)
39 Zhu J., Nieto F.J., Horne B.D., et al. Prospective study of pathogen burden and risk of myocardial infarction or death. Circulation. 2001;103(1):45–51.
40 Danesh J., Whincup P., Lewington S., et al. Chlamydia pneumoniae IgA titres and coronary heart disease; prospective study and meta-analysis. Eur Heart J. 2002;23(5):371–375.
41 Kalayoglu M.V., Libby P., Byrne G.I. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA. 2002;288(21):2724–2731.
42 Corrales-Medina V.F., Madjid M., Musher D.M. Role of acute infection in triggering acute coronary syndromes. Lancet Infect Dis. 2010;10(2):83–92.
43 Andraws R., Berger J.S., Brown D.L. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: a meta-analysis of randomized controlled trials. JAMA. 2005;293(21):2641–2647.
44 Libby P., Ridker P.M., Hansson G.K. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129–2138.
45 Andersson J., Libby P., Hansson G.K. Adaptive immunity and atherosclerosis. Clin Immunol. 2010;134(1):33–46.
46 Libby P., Crea F. Clinical implications of inflammation for cardiovascular primary prevention. Eur Heart J. 2010;31(7):777–783.
47 Ridker P.M. C-reactive protein: eighty years from discovery to emergence as a major risk marker for cardiovascular disease. Clin Chem. 2009;55(2):209–215.
48 Ridker P.M., MacFadyen J.G., Libby P., et al. Relation of baseline high-sensitivity C-reactive protein level to cardiovascular outcomes with rosuvastatin in the Justification for Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER). Am J Cardiol. 2010;106(2):204–209.
49 Kaptoge S., Di Angelantonio E., Lowe G., et al. Emerging risk factors collaboration. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375(9709):132–140.
50 Zacho J., Tybjaerg-Hansen A., Jensen J.S., et al. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359(18):1897–1908.
51 Elliott P., Chambers J.C., Zhang W., et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302(1):37–48.
52 Nordestgaard B.G. Does elevated C-reactive protein cause human atherothrombosis? Novel insights from genetics, intervention trials, and elsewhere. Curr Opin Lipidol. 2009.
53 Ridker P.M., Buring J.E., Rifai N., et al. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611–619.
54 Ridker P.M., Paynter N.P., Rifai N., et al. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118(22):2243–2251.
55 Wilson P.W., Pencina M., Jacques P., et al. C-reactive protein and reclassification of cardiovascular risk in the Framingham heart study. Circ Cardiovasc Qual Outcomes. 2008;1(2):92–97.
56 Hamsten A., Eriksson P. Identifying the susceptibility genes for coronary artery disease: from hyperbole through doubt to cautious optimism. J Intern Med. 2008;263(5):538–552.
57 Samani N.J., Erdmann J., Hall A.S., et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–453.
58 Schunkert H., Gotz A., Braund P., et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation. 2008;117(13):1675–1684.
59 Kathiresan S., Melander O., Guiducci C., et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40(2):189–197.
60 Palomaki G.E., Melillo S., Bradley L.A. Association between 9p21 genomic markers and heart disease: a meta-analysis. JAMA. 2010;303(7):648–656.
61 Teslovich T.M., Musunuru K., Smith A.V., et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713.
62 Burkhardt R., Toh S.A., Lagor W.R., et al. Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J Clin Invest. 2010;120(12):4410–4414.
63 Musunuru K., Pirruccello J.P., Do R., et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363(23):2220–2227.
64 Ripatti S., Tikkanen E., Orho-Melander M., et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376(9750):1393–1400.
65 Paynter N.P., Chasman D.I., Pare G., et al. Association between a literature-based genetic risk score and cardiovascular events in women. JAMA. 2010;303(7):631–637.
66 Gaziano J.M. Fifth phase of the epidemiologic transition: the age of obesity and inactivity. JAMA. 2010;303(3):275–276.
67 Lloyd-Jones D., Adams R.J., Brown T.M., et al. Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010;121(7):e46–e215.
68 Grundy S.M., Cleeman J.I., Daniels S.R., et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112(17):2735–2752.
69 Alberti K.G., Eckel R.H., Grundy S.M., et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645.
70 Kahn R., Buse J., Ferrannini E., et al. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28(9):2289–2304.
71 Mente A., Yusuf S., Islam S., et al. Metabolic syndrome and risk of acute myocardial infarction a case-control study of 26,903 subjects from 52 countries. J Am Coll Cardiol. 2010;55(21):2390–2398.
72 Berneis K.K., Krauss R.M. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43(9):1363–1379.
73 Ai M., Otokozawa S., Asztalos B.F., et al. Small dense LDL cholesterol and coronary heart disease: results from the Framingham Offspring Study. Clin Chem. 2010;56(6):967–976.
74 Tabas I., Williams K.J., Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844.
75 Selvin E., Steffes M.W., Zhu H., et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med. 2010;362(9):800–811.
76 Giacco F., Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–1070.
77 Yan S.F., Ramasamy R., Schmidt A.M. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106(5):842–853.
78 Gerstein H.C., Miller M.E., Byington R.P., et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545–2559.
79 Patel A., MacMahon S., Chalmers J., et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560–2572.
80 Duckworth W., Abraira C., Moritz T., et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360(2):129–139.
81 Holman R.R., Paul S.K., Bethel M.A., et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577–1589.
82 Libby P., Shi G.P. Mast cells as mediators and modulators of atherogenesis. Circulation. 2007;115(19):2471–2473.
83 Mestas J., Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18(6):228–232.
84 Swirski F.K., Libby P., Aikawa E., et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205.
85 Tacke F., Alvarez D., Kaplan T.J., et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117(1):185–194.
86 Libby P., Nahrendorf M., Pittet M.J., et al. Diversity of denizens of the atherosclerotic plaque: not all monocytes are created equal. Circulation. 2008;117:3168–3170.
87 Woollard K.J., Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol. 2010;7(2):77–86.
88 Bouhlel M.A., Derudas B., Rigamonti E., et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137–143.
89 Bromley S.K., Mempel T.R., Luster A.D. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9(9):970–980.
90 Kovanen P.T. Mast cells: multipotent local effector cells in atherothrombosis. Immunol Rev. 2007;217:105–122.
91 Bot I., de Jager S.C., Zernecke A., et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115(19):2516–2525.
92 Sun J., Sukhova G.K., Wolters P.J., et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13(6):719–724.
93 Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874.
94 Mallat Z., Taleb S., Ait-Oufella H., et al. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2008.
95 Taleb S., Tedgui A., Mallat Z. Regulatory T-cell immunity and its relevance to atherosclerosis. J Intern Med. 2008;263(5):489–499.
96 van Es T., van Puijvelde G.H., Ramos O.H., et al. Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun. 2009;388(2):261–265.
97 Smith E., Prasad K.M., Butcher M., et al. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2010;121(15):1746–1755.
98 Taleb S., Tedgui A., Mallat Z. Interleukin-17: friend or foe in atherosclerosis? Curr Opin Lipidol. 2010;21(5):404–408.
99 Packard R.R.S., Maganto-Garcia E., Gotsman I., et al. CD11c + dendritic cells maintain antigen processing, presentation capabilities, and CD4 + T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ Res. 2008;103:965–973.
100 Randolph G.J., Potteaux S. Vascular dendritic cells as gatekeepers of lipid accumulation within nascent atherosclerotic plaques. Circ Res. 2010;106(2):227–229.
101 Paulson K.E., Zhu S.N., Chen M., et al. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106(2):383–390.
102 Hermansson A., Ketelhuth D.F., Strodthoff D., et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. 2010;207(5):1081–1093.
103 Bentzon J.F., Weile C., Sondergaard C.S., et al. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26(12):2696–2702.
104 Orlandi A., Bochaton-Piallat M.L., Gabbiani G., et al. Aging, smooth muscle cells and vascular pathobiology: implications for atherosclerosis. Atherosclerosis. 2006;188(2):221–230.
105 Geng Y.J., Libby P. Progression of atheroma: a struggle between death and procreation. Arterioscler Thromb Vasc Biol. 2002;22(9):1370–1380.
106 Kavurma M.M., Tan N.Y., Bennett M.R. Death receptors and their ligands in atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(10):1694–1702.
107 Clarke M.C., Figg N., Maguire J.J., et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12(9):1075–1080.
108 Clarke M.C., Littlewood T.D., Figg N., et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102(12):1529–1538.
109 Amento E.P., Ehsani N., Palmer H., et al. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1991;11:1223–1230.
110 Robertson A.K., Rudling M., Zhou X., et al. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112(9):1342–1350.
111 Hjortnaes J., Butcher J., Figueiredo J.L., et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J. 2010;31(16):1975–1984.
112 Rajavashisth T., Qiao J.H., Tripathi S., et al. Heterozygous osteopetrotic (op) mutation reduces atherosclerosis in LDL receptor- deficient mice. J Clin Invest. 1998;101(12):2702–2710.
113 Gautier E.L., Huby T., Witztum J.L., et al. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation. 2009;119(13):1795–1804.
114 Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46.
115 Russo C., Jin Z., Rundek T., et al. Atherosclerotic disease of the proximal aorta and the risk of vascular events in a population-based cohort: the Aortic Plaques and Risk of Ischemic Stroke (APRIS) study. Stroke. 2009;40(7):2313–2318.
116 Takaya N., Yuan C., Chu B., et al. Association between carotid plaque characteristics and subsequent ischemic cerebrovascular events: a prospective assessment with MRI–initial results. Stroke. 2006;37(3):818–823.
117 Aird W.C. Mechanisms of endothelial cell heterogeneity in health and disease. Circ Res. 2006;98(2):159–162.
118 Majesky M.W. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27(6):1248–1258.
119 Dong C., Crawford L.E., Goldschmidt-Clermont P.J. Endothelial progenitor obsolescence and atherosclerotic inflammation. J Am Coll Cardiol. 2005;45(9):1458–1460.
120 Werner N., Kosiol S., Schiegl T., et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353(10):999–1007.
121 Shantsila E., Watson T., Lip G.Y. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol. 2007;49(7):741–752.
122 Bentzon J.F., Falk E. Circulating smooth muscle progenitor cells in atherosclerosis and plaque rupture: current perspective and methods of analysis. Vasc Pharmacol. 2010;52(1–2):11–20.
123 Hagensen M.K., Shim J., Thim T., et al. Circulating endothelial progenitor cells do not contribute to plaque endothelium in murine atherosclerosis. Circulation. 2010;121(7):898–905.
124 Lavoie A.J., Bayturan O., Uno K., et al. Plaque progression in coronary arteries with minimal luminal obstruction in intravascular ultrasound atherosclerosis trials. Am J Cardiol. 2010;105(12):1679–1683.
125 Clarkson T.B., Prichard R.W., Morgan T.M., et al. Remodeling of coronary arteries in human and nonhuman primates. JAMA. 1994;271(4):289–294.
126 Glagov S., Weisenberg E., Zarins C., et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:371–375.
127 Vink A., Schoneveld A.H., Borst C., et al. The contribution of plaque and arterial remodeling to de novo atherosclerotic luminal narrowing in the femoral artery. J Vasc Surg. 2002;36(6):1194–1198.
128 Schoenhagen P., Sipahi I. Arterial remodelling: an independent pathophysiological component of atherosclerotic disease progression and regression. Insights from serial pharmacological intervention trials. Eur Heart J. 2007;28(19):2299–2300.
129 McGill H.C.Jr, McMahan C.A., Zieske A.W., et al. Associations of coronary heart disease risk factors with the intermediate lesion of atherosclerosis in youth. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. Arterioscler Thromb Vasc Biol. 2000;20(8):1998–2004.
130 Li S., Chen W., Srinivasan S.R., et al. Childhood cardiovascular risk factors and carotid vascular changes in adulthood: the Bogalusa Heart Study. JAMA. 2003;290(17):2271–2276.
131 Virmani R., Robinowitz M., Geer J.C., et al. Coronary artery atherosclerosis revisited in Korean war combat casualties. Arch Pathol Lab Med. 1987;111(10):972–976.
132 Palinski W. Maternal-fetal cholesterol transport in the placenta: good, bad, and target for modulation. Circ Res. 2009;104(5):569–571.
133 Tuzcu E.M., Kapadia S.R., Tutar E., et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation. 2001;103(22):2705–2710.
134 Gimbrone M.A.Jr, Topper J.N., Nagel T., et al. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann N Y Acad Sci. 2000;902:230–239. discussion 239–240
135 Libby P., Aikawa M., Jain M.K. Vascular endothelium and atherosclerosis. Handb Exp Pharmacol. 2006;176 Pt 2:285–306.
136 Dai G., Vaughn S., Zhang Y., et al. Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101(7):723–733.
137 Parmar K.M., Nambudiri V., Dai G., et al. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;289:26714–26719.
138 Young A., Wu W., Sun W., et al. Flow activation of AMP-activated protein kinase in vascular endothelium leads to Kruppel-like factor 2 expression. Arterioscler Thromb Vasc Biol. 2009;29(11):1902–1908.
139 Hajra L., Evans A.I., Chen M., et al. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;97(16):9052–9057.
140 Peng H.B., Libby P., Liao J.K. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem. 1995;270(23):14214–14219.
141 Boden W.E., O’Rourke R.A., Teo K.K., et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356(15):1503–1516.
142 Corti R., Fuster V., Badimon J.J. Pathogenetic concepts of acute coronary syndromes. J Am Coll Cardiol. 2003;41(4 Suppl S):7S–14S.
143 Libby P. The molecular mechanisms of the thrombotic complications of atherosclerosis. J Int Med. 2008;263:517–527.
144 Fuster V. Mechanisms leading to myocardial infarction: insights from vascular biology. Circulation. 1994;90(4):2126–2146.
145 Schoenhagen P., Ziada K.M., Kapadia S.R., et al. Extent and direction of arterial remodeling in stable versus unstable coronary syndromes: an intravascular ultrasound study. Circulation. 2000;101(6):598–603.
146 Libby P., Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488.
147 Finn A.V., Nakano M., Narula J., et al. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30(7):1282–1292.
148 Galis Z., Sukhova G., Lark M., et al. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503.
149 Sukhova G.K., Schonbeck U., Rabkin E., et al. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99(19):2503–2509.
150 Dollery C.M., Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69(3):625–635.
151 Herman M.P., Sukhova G.K., Kisiel W., et al. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix metalloproteinases with implications for atherosclerosis. J Clin Invest. 2001;107(9):1117–1126.
152 Fukumoto Y., Deguchi J.O., Libby P., et al. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110(14):1953–1959.
153 Deguchi J.O., Aikawa E., Libby P., et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112(17):2708–2715.
154 Schneider F., Sukhova G.K., Aikawa M., et al. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117(7):931–939.
155 van der Wal A.C., Becker A.E., van der Loos C.M., et al. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44.
156 Virmani R., Burke A.P., Farb A., et al. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13–C18.
157 Sugiyama S., Kugiyama K., Aikawa M., et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24(7):1309–1314.
158 Moulton K.S. Angiogenesis in atherosclerosis: gathering evidence beyond speculation. Curr Opin Lipidol. 2006;17(5):548–555.
159 Beckman J.A., Ganz J., Creager M.A., et al. Relationship of clinical presentation and calcification of culprit coronary artery stenoses. Arterioscler Thromb Vasc Biol. 2001;21(10):1618–1622.
160 Bayturan O., Tuzcu E.M., Nicholls S.J., et al. Attenuated plaque at nonculprit lesions in patients enrolled in intravascular ultrasound atherosclerosis progression trials. JACC Cardiovasc Interv. 2009;2(7):672–678.
161 Mann J., Davies M.J. Mechanisms of progression in native coronary artery disease: role of healed plaque disruption. Heart. 1999;82(3):265–268.
162 Burke A.P., Kolodgie F.D., Farb A., et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103(7):934–940.
163 Libby P. Act local, act global: inflammation and the multiplicity of "vulnerable" coronary plaques. J Am Coll Cardiol. 2005;45(10):1600–1602.
164 Aikawa M., Rabkin E., Okada Y., et al. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998;97:2433–2444.
165 Libby P., Aikawa M. Stabilization of atherosclerotic plaques: New mechanisms and clinical targets. Nat Med. 2002;8(11):1257–1262.
166 Libby P., Sasiela W. Plaque stabilization: can we turn theory into evidence? Am J Cardiol. 2006;98:S26–S33.
167 Hernandez-Presa M.A., Bustos C., Ortego M., et al. ACE inhibitor quinapril reduces the arterial expression of NF-kappaB-dependent proinflammatory factors but not of collagen I in a rabbit model of atherosclerosis. Am J Pathol. 1998;153(6):1825–1837.
168 Nissen S.E., Tsunoda T., Tuzcu E.M., et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290(17):2292–2300.
169 Tardif J.C., Gregoire J., L’Allier P.L., et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: a randomized controlled trial. JAMA. 2007;297(15):1675–1682.
170 Nissen S.E., Nicholls S.J., Sipahi I., et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295(13):1556–1565.
171 Nicholls S.J., Ballantyne C.M., Barter P.J., et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078–2087.
172 Zhao X.Q., Yuan C., Hatsukami T.S., et al. Effects of prolonged intensive lipid-lowering therapy on the characteristics of carotid atherosclerotic plaques in vivo by MRI: a case-control study. Arterioscler Thromb Vasc Biol. 2001;21(10):1623–1629.