Chapter 17 Pathophysiology of Peripheral Artery Disease, Intermittent Claudication, and Critical Limb Ischemia
Peripheral artery disease (PAD) is a manifestation of systemic atherosclerosis that commonly coexists with coronary and carotid artery disease. This places the patient with PAD at high risk of cardiovascular events, including myocardial infarction (MI), ischemic stroke, and death.1,2 The pathobiology of atherosclerosis and atherothrombosis has been described previously (see Chapter 8).3,4 The focus of this chapter is on the pathophysiology of atherosclerotic disease in the arteries of the lower extremity that leads to the symptomatic manifestations of PAD, including claudication and critical limb ischemia (CLI). Understanding the pathophysiological mechanisms that underlie the development and progression of limb atherosclerosis and ischemic symptoms is critical in the overall management of the patient with PAD and for the development of potential new therapies. Table 17-1 summarizes the major pathophysiological mechanisms contributing to intermittent claudication and CLI that are reviewed in this chapter.
Table 17-1 Abnormalities Observed in Peripheral Artery Disease
| Changes in Pad | Possible Consequences |
|---|---|
| Hemodynamic | |
| Arterial stenosis/occlusion | Pressure drop across stenosis |
| Inability to increase flow relative to demand | |
| Collateral formation | Partial compensation for arterial stenosis |
| Increased blood viscosity | Reduced flow |
| Endothelial dysfunction | Altered arteriolar regulation of flow |
| Muscle capillary supply | Increased oxygen diffusion |
| Oxidant Stress | |
| Free radical generation | Endothelial and muscle injury |
| White cell activation | Contributes to oxidant injury |
| Mitochondrial DNA deletions | Indication of mitochondrial injury |
| Structural | |
| Distal axonal denervation | Muscle weakness |
| Reinnervation | Partial compensation |
| Type II fiber loss | Decreased muscle mass/strength |
| Metabolic | |
| Increased oxidative enzymes | Compensation for reduced oxygen delivery |
| Increased short-chain acylcarnitine | Reflects altered oxidative metabolism |
| Decreased electron transport | Accumulation related to performance |
| Chain activity | Reduced ATP production |
ATP, adenosine triphosphate; DNA, deoxyribonucleic acid; PAD peripheral artery disease.
Clinical Manifestations of Peripheral Artery Disease
The pathophysiology of PAD begins with progressive atherosclerosis, resulting in stenosis and occlusion of the major arteries supplying the lower extremities. Compared with the often acute nature of coronary and carotid atherothrombosis, clinical manifestations of PAD tend to be more chronic and progressive, with primarily functional consequences. Diagnosis of PAD can readily be established through noninvasive hemodynamic assessments. As noted in Chapter 16, the ankle-brachial index (ABI) is the ratio of the systolic blood pressure in the ankle to that in the arm. An ABI of 0.9 take all ABI values to second decimal place, i.e. 0.90 not 0.9 or less establishes a diagnosis of PAD with high sensitivity and specificity when compared with imaging modalities.5
Exercise Limitation and Systemic Risk
Patients with PAD suffer from exercise limitation secondary to impaired hemodynamics. The classic symptom of intermittent claudication is an exercise-induced discomfort in the calf associated with reversible muscle ischemia and relieved by rest. The term claudication is derived from the Latin word claudicato, meaning “to limp,” which is typical of the gait pattern of the patient who experiences claudication when walking. Claudication is characterized by a cramping and aching in the affected muscle. Discomfort develops only during exercise, steadily increases during walking activity to a point where the patient has to stop, and then is quickly relieved by rest without change of position. This sequence of exercise-induced progression and complete relief with rest are important clinical differentiators of claudication from other lower-extremity musculoskeletal conditions. Patients with claudication have severe limitations in exercise performance and walking ability. Compared to healthy individuals of the same age, patients with claudication have a 50% to 60% reduction in peak treadmill performance, reflecting a disability similar to that of patients with severe congestive heart failure (CHF).6 This exercise impairment is associated with a marked decrease in ambulatory activity and the physical dimension of several quality-of-life instruments.7
Although classic claudication symptoms occur in less than a third of patients with PAD, all patients with PAD have reductions in ambulatory activity and daily functional capacity.8 Even “asymptomatic” patients with PAD have a marked reduction in quality of life.9 Thus, major goals of treatment in ambulatory PAD patients are to prevent progression of systemic atherosclerosis and relieve the symptoms of claudication and enhance quality of life.
In addition to physical disability, PAD is a marker of systemic atherosclerotic disease and associated risk. Peripheral artery disease is associated with a three- to sixfold increased risk of coronary artery disease (CAD) and events, cerebral artery disease and stroke, and cardiovascular death.1,10 Thus, consensus guidelines recommend that patients with PAD be considered to have established atherosclerotic disease, and secondary prevention standards are applicable.11,12
Critical Limb Ischemia
Critical limb ischemia is the most severe of the limb manifestations of PAD. Critical limb ischemia is defined by chronic ischemic pain at rest and/or presence of ischemic skin lesions (gangrene or ulcerations). Although its epidemiology is not well known, a recent population study of 8000 individuals between 60 and 90 years of age found the prevalence of CLI to be 1.2%, and more women than men were affected.13 Prognosis for patients with CLI is poor.14,15 In clinical trials enrolling patients with CLI and an ischemic foot ulcer, the annualized risk of death or major amputation ranges from 33% to 50%.15 Independent predictors of worse outcomes include diabetes, end-stage renal disease, and cardiac dysfunction.16,17 Patients with CLI have more severe hemodynamic compromise than patients with claudication, owing to multiple and more distal levels of arterial occlusions. Tibial vessels are most often affected in CLI, usually in combination with disease in the popliteal and superficial femoral arteries (and other more proximal vessels), which leads to a severe compromise of blood flow and oxygen delivery to distal tissues.18 In CLI, ankle pressures typically are less than 50 mmHg. As CLI progresses, skin breakdown to ulceration and gangrene is inevitable. Resting energy expenditure is reduced in CLI compared to intermittent claudication, probably reflecting the fact that these patients become more inactive and sedentary.19 Revascularization to directly address the hemodynamic compromise in CLI remains the primary therapeutic approach.
Hemodynamics in Peripheral Artery Disease
Skeletal Muscle Oxygen Consumption
Muscle oxygen consumption both at rest and during exercise involves oxygen delivery (pulmonary oxygen uptake, oxygen content of hemoglobin (Hb), and regional blood flow) and oxygen extraction by skeletal muscle mitochondria. In healthy persons, maximal muscle oxygen consumption is determined primarily by maximal oxygen delivery, rather than mitochondrial metabolic rate.20 Muscle mitochondrial oxidative capacity remains tightly coupled with maximal exercise capacity and increases with exercise training.21 At the onset of submaximal exercise, skeletal muscle rapidly extracts oxygen from Hb, producing deoxyhemoglobin.22 The kinetics of the changes in tissue oxygen uptake are coupled to systemic oxygen consumption to maintain a balance between oxygen delivery and oxygen utilization.
Determinants of Limb Blood Flow in Healthy Individuals
At any given systemic blood pressure, the major determinant of blood flow in normal regional circulation is the peripheral resistance of the vascular bed supplied by major conduit vessels. This basic relationship can be expressed as:
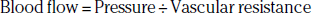
In healthy persons, exercise is a major stimulus for vasodilation, causing a decrease in peripheral resistance, which when combined with an increase in systemic pressure results in a large increase in arterial flow to skeletal muscle. Normal arteries have the capacity to support large volumetric increases in blood flow without a significant drop in pressure across the large and medium conduit vessels (Fig. 17-1).
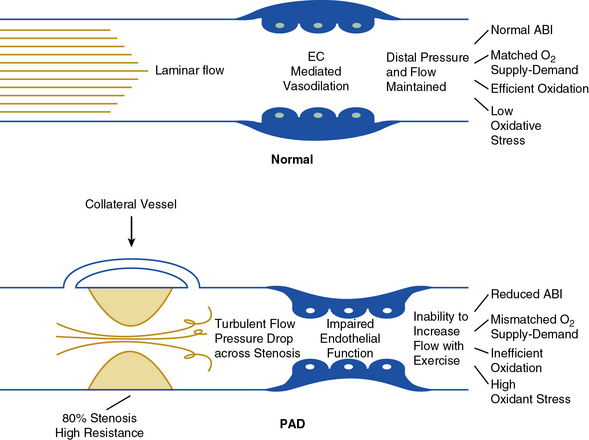
Figure 17-1 Normal arterial function. In healthy arteries (top) flow is laminar, and endothelial function is normal.
Therefore, blood flow and oxygen delivery match muscle metabolic demand at rest and exercise. Muscle metabolism is efficient, resulting in low oxidative stress. In contrast, in peripheral artery disease (bottom) arterial stenosis results in turbulent flow. Increased resistance associated with stenosis and loss of kinetic energy results in pressure drop across stenosis. Collateral vessels only partially compensate for arterial stenosis. In addition, endothelial function is impaired, resulting in further loss of vascular function. These changes limit blood flow response to exercise, resulting in mismatch of oxygen delivery to muscle metabolic demand. Changes in skeletal muscle metabolism further compromise efficient generation of high-energy phosphates. Oxidant stress—the result of inefficient oxidation—further impairs endothelial function and muscle metabolism. ABI, ankle-brachial index; EC, endothelial cell; PAD, peripheral artery disease.
Hemodynamic Abnormalities in Peripheral Artery Disease
The arterial occlusive disease process results in fixed-resistance elements in the circulation, and thus initiates multiple pathophysiological processes that manifest clinically as claudication, ischemic rest pain, or ulceration (see Fig. 17-1). Major factors that determine the pressure drop across an arterial stenosis include blood flow velocity and the resistance caused by the stenosis, which in turn is defined by the length and internal radius of the stenosis and blood viscosity. These parameters have been classically described by the Poiseuille equation, which defines the relationships between resistance, pressure, and flow:
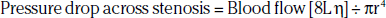
where L is length of stenosis, r is internal radius of the artery, and η is viscosity.
This equation makes clear that the radius or cross-sectional area of the stenosis is the primary factor in determining the drop in pressure and flow across a stenosis; a 50% reduction in cross-sectional diameter of the vessel results in a 16-fold increase in resistance. This relationship indicates that as a stenosis worsens, perfusion pressure (and thus the potential pressure that can be dissipated across the stenosis) and the maximal achieved blood flow will decrease dramatically. The dissipation of energy that occurs as blood flow traverses a stenosis is determined in part by the morphology of the stenosis and blood viscosity.23 Pressure drop across the stenosis manifests as reduced limb systolic pressure and decreased ABI as discussed earlier. In patients with PAD, arterial occlusions limit increase in blood flow to exercising muscle. Resting blood flow is usually preserved because of the pressure of arterial collaterals in most patients. In patients with CLI, however, even resting flow may be reduced below normal levels.
A common angiographic finding is multilevel occlusive disease, particularly in patients with severe symptoms of PAD. A patient with mild claudication may have stenosis at only a single site, such as the iliac artery, but a patient with moderate or severe claudication could have occlusive disease at multiple sites, including the iliac, femoral, and popliteal arteries. Patients with CLI often have diffuse disease affecting multiple arterial segments, such as the iliac, femoral, and tibial arteries. Tibial disease is quite common in CLI and predictive of a higher risk of ischemic ulceration and risk of amputation. Progression of the disease from intermittent claudication to chronic CLI is modulated by collateral vessel development and other compensatory mechanisms.
Based on the Poiseuille equation, the length of an individual stenosis has only mild impact on blood flow and the pressure gradient. Nevertheless, the hemodynamic effect of two equivalent lesions in series is double that of a single lesion.24 Thus, individual noncritical stenoses may become hemodynamically significant when combined in series in the same limb.25 In CLI, patients typically have disease in the inflow vessels (aorta, iliac arteries) and outflow vessels (superficial femoral, popliteal and tibial arteries).18 These lesions in series create more hemodynamic compromise than is typically seen in claudication.
Critical Artery Stenosis
The hemodynamic significance of an arterial stenosis is not only a function of the percent stenosis, but also linear flow velocity across the lesion, as reflected in the Poiseuille equation.26,27 The term critical artery stenosis is defined as the degree of stenosis that causes a decrease in distal blood flow. The concept integrates the relationship of a stenotic narrowing in an artery with arterial flow velocity and the resultant volumetric flow distal to the stenosis. Importantly, a critical artery stenosis may differ between resting and exercising states because flow velocity in these two conditions is different. Because the pressure gradient across any given stenosis is proportional to the flow velocity, states of higher flow velocity, as occurs with exercise, may result in a decrease in distal perfusion pressure, whereas states of lower velocity, as occurs at rest, may not. For example, resting blood flow velocity in the femoral artery may be only 10 to 20 cm/s, corresponding to a downstream calf blood flow of 1 to 2 mL/100 mL of tissue/min.28 When a large-vessel stenosis of 50% is imposed on the system at this resting flow velocity, loss of kinetic energy across the stenosis may cause no or only a minimal decrease in distal perfusion pressure. Distal flow will be maintained, since the mild reduction in perfusion pressure will be compensated by a reduction in downstream peripheral resistance. Once the stenosis becomes greater than 90%, there is a greater pressure gradient and fall in distal perfusion pressure, and changes in peripheral resistance can no longer compensate. Thus distal flow decreases. In this example, the critical arterial stenosis needed to reduce distal blood flow at rest is 90%.
During walking exercise, blood flow velocity increases—for example, to 150 cm/s. An exercise-induced increase in flow velocity across a 50% stenosis could significantly increase the pressure gradient and reduce distal perfusion pressure. The associated fall in peripheral resistance would be insufficient to compensate for the fall in pressure, and distal blood flow would decrease. Thus the critical arterial stenosis needed to reduce distal blood flow during exercise may be only 50%.28 The concept of critical artery stenosis has clinical significance. In a patient with a single iliac artery stenosis of 50%, the calf blood flow, pedal pulse examination, and ABI may be normal at rest. However, when flow velocity increases with exercise, the same iliac artery lesion becomes hemodynamically significant, resulting in a loss of pedal pulses due to the decrease in ankle pressure distal to the stenosis.
As in patients with CAD, the concept of fractional flow reserve describes the ratio of the blood flow through a diseased coronary artery to the maximal hyperemic flow through that artery in the absence of disease.29,30 This approach is applicable to PAD and provides a functional interpretation of any degree of percent stenosis and the associated critical arterial stenosis. For example, a functional flow reserve of 0.80 indicates a 20% reduction in maximal hyperemic blood flow due to a stenotic lesion. However, minimal luminal area (MLA, or degree of stenosis) correlates poorly with functional flow reserve until the artery disease results in a large reduction in MLA. Thus anatomy per se may not provide sufficient evidence of the functional significance of a particular degree of arterial stenosis.
Blood Flow Response to Exercise in Intermittent Claudication
Most patients with PAD have no limb symptoms at rest (with the exception of those with CLI). This is because resting blood flow is sufficient to meet the relatively low metabolic needs of the tissue, and therefore there is no mismatch between supply and demand to maintain leg oxygen consumption.31,32 At the onset of leg exercise, patients with PAD have an initial rise in leg blood flow and leg oxygen consumption that is delayed.33 With a graded increase in exercise intensity, there is an initial linear increase in flow. However, as exercise intensity increases in PAD, blood flow reaches a plateau because of the limitation imposed by arterial obstructions. This plateau reflects dissipation of energy across the stenotic lesions, removing any additional driving force for increase in flow. Severity of arterial disease (defined by the ABI) correlates inversely with the maximal increase in flow.34 With cessation of exercise, the hyperemic phase (increased flow over resting levels) is prolonged in patients with PAD relative to healthy controls. Despite the plateau in oxygen delivery during exercise, further increases in oxidative work output are supported by increases in muscle oxygen extraction.35 Nonoxidative adenosine triphosphate (ATP) production also contributes to muscle energy metabolism.36 Importantly, muscle ischemia is not simply due to lack of increase in blood flow. The resultant mismatch between the demands for bioenergetics and the flow supply also contribute (see Fig. 17-1).
Other Contributors to Altered Blood Flow in Peripheral Artery Disease
Although arterial flow limitations are of critical importance in the pathophysiology of claudication, the hemodynamic status of the limb correlates poorly with exercise performance. Most studies have shown that resting ankle blood pressure (or ABI) and exercise blood flow do not predict treadmill walking time,37 whereas some studies have shown a weak positive correlation.38,39 This lack of consistent relationship between ABI and claudication-limited exercise capacity is surprising, especially given the relationship between ABI and exercise-induced peak blood flow. Thus, factors distal to the arterial obstruction likely contribute to functional limitations in PAD.
Endothelial Regulation of Flow
Blood flow and its distribution within skeletal muscle beds are determined by endothelial and microcirculatory factors (see Fig. 17-1). Endothelium-derived nitric oxide (NO) is central to the physiological regulation of arteriolar tone. Nitric oxide and prostaglandins (PGs) are major autocrine and paracrine mediators of local vascular resistance during exercise in normal individuals.40–42 Patients with atherosclerosis have a systemic abnormality in endothelial function that is associated with impaired vasodilation and enhanced platelet aggregation.43 A primary mediator of endothelial dysfunction is felt to be oxidant stress from the generation of superoxide anion.44 Consistent with the above, abnormalities in endothelium-dependent vasodilation have been observed in PAD.45 Amputation of an ischemic limb in CLI is associated with improvement of some markers of endothelial function, suggesting local generation of oxidative stress from an ischemic segment of limb.46 Thus, altered oxygen delivery to exercising skeletal muscle in PAD is related not only to the large-vessel occlusive process but also to endothelial dysfunction and impaired vasodilation.
Hemorheology in Peripheral Artery Disease
Peripheral artery disease is associated with altered hemorheology (flow properties of blood and its cellular components) that result in increased viscosity and altered flow as described by the Poiseuille equation (see earlier discussion). Patients with PAD have increased blood concentrations of fibrinogen, von Willebrand factor (vWF), and plasminogen activator inhibitor (PAI), as well as increased fibrin turnover.47 These changes also may affect blood flow characteristics in the microcirculation, but none of these factors has been directly correlated with claudication-limited exercise performance. Previous reports have noted that PAD patients have higher blood viscosity than age-matched controls; according to the Poiseuille law, this could be a contributing factor for exercise-induced ischemia.48 Red cell flexibility is reduced in patients with intermittent claudication, and thus the passage of erythrocytes through nutritive capillaries might be compromised by microcirculatory vessel plugging.49
Microcirculatory, Hemorheological, and Thrombophilic Abnormalities in Critical Limb Ischemia
A prominent feature of CLI is formation of cellular plugs and microthrombi in the microcirculation. Erythrocyte fluidity and erythrocyte volume fraction are reduced in patients with CLI compared with controls.50 These flow properties improve after amputation, suggesting that limb ischemia per se contributes to changes in red blood cell fluidity.51
In patients with CLI, a high peripheral white blood cell (WBC) count is associated with future amputation.52 However, it is unclear whether the increase in leukocytes is causative or simply reflects infection or other inflammatory process that predisposes to amputation. Leukocytes may play an important role in ischemic disease via formation of microemboli and induction of oxidative damage. Leukocyte adhesion is also increased in CLI.53 This may be due to increased endothelial expression of the adhesion molecules vascular cell adhesion molecule (VCAM)-1 and E-selectin. Adherent cells may further decrease lumen diameter in the microcirculation. Activated neutrophils may adhere to other leukocytes and blood cells, further narrowing the vessel lumen and, through release of mediators, increasing vessel wall damage. Activated leukocytes found in many vascular diseases are abnormally rigid, potentially exacerbating microvascular occlusion in CLI.
Platelet number and platelet activation also are increased in CLI.54 Activated platelets interact with endothelial receptors, releasing the potent vasoconstrictor thromboxane, further promoting vasoconstriction and platelet activation. In one study, P-selectin expression was significantly increased in patients with intermittent claudication and critical ischemia compared to controls.54
The vascular bed in PAD may be under increased vasoconstrictor tone. Decrease in the vasodilators NO and PGs was already discussed, as was increased exposure to the vasoconstrictor thromboxane. In CLI skeletal muscle resistance arterioles, α1- and α2-adrenergic receptor responses are increased.55 This finding has been confirmed in other studies, although the functional significance is unclear.56,57 Similarly, increases in endothelin messenger ribonucleic acid (mRNA) expression in CLI may cause vasoconstriction of the microcirculation.58
Edema in Critical Limb Ischemia
Microcirculatory abnormalities in CLI also predispose the patient to pedal edema.59 In a study of the rate of fluid filtration through the capillary wall of patients with CLI, the capillary filtration coefficient was increased compared with nonischemic and control limbs. These observations suggest a mechanism to explain the propensity to develop edema in CLI.60 Restoration of blood flow by surgical bypass grafting or angioplasty generates an increase in distal limb pressure, with associated increases in capillary hydrostatic pressure. This may lead to extravasation of fluid initially and tissue edema in patients who underwent revascularization for CLI.61
Inflammation and Oxidative Injury in Peripheral Artery Disease
In claudication, exercise is associated with an increase in plasma levels of thiobarbituric acid–reactive compounds, thromboxane, interleukin (IL)-8, soluble intercellular adhesion molecule (sICAM)-1, VCAM-1, vWF, E-selectin, and thrombomodulin.62–67 These observations suggest an acute inflammatory response to muscle ischemia during exercise (possibly reflecting reperfusion injury during recovery). After exercise-induced claudication, total neutrophil number and the proportion of activated neutrophils are higher in venous blood draining from the affected leg than in arterial blood.68 These venous-arterial differences are not observed in the circulation of the contralateral PAD-unaffected exercising leg. Furthermore, activated leukocytes release thromboxane A2 (TxA2), which is a vasoconstrictor and promotes platelet aggregation.69 In claudicants, P-selectin, which mediates platelet-endothelium interaction, may also contribute to platelet alterations in the microcirculation.70–72 Activated neutrophils also release elastase, which has been shown to exert harmful effects on the endothelium in vitro.73 Circulating elastase activities increase progressively from healthy individuals, to asymptomatic PAD patients, to symptomatic claudication.74 Furthermore, in patients with claudication, elastase activity increases further with exercise.75 These inflammatory responses to exercise may mediate adverse interactions with the microcirculation and skeletal muscle metabolism, which could further compromise exercise performance. Thus the generation of free radicals and oxidative stress can be mediators of tissue injury.
Oxidative Injury in Peripheral Artery Disease
Animal models have shown that both ischemia and ischemia-reperfusion are associated with oxidative stress due to production of free radicals.76,77 Patients with claudication do not deliver sufficient oxygen to exercising muscle and have a prolonged hyperemic oxygen-rich phase during recovery from exercise.78 Muscle ischemia during exercise and reperfusion after claudication-limited exercise are associated with an increase in oxidant stress.79,80 Blood levels of malondialdehyde (a marker of free radical generation) are elevated at rest in PAD and increase further with exercise.80 Peripheral artery disease patients also have systemic evidence of neutrophil and platelet activation and endothelial injury.62
The oxidative stress observed in PAD may be part of a broad spectrum of inflammatory responses to systemic atherosclerosis that is enhanced by exercise.66 Production of oxygen free radicals may be a unifying mechanism of skeletal muscle injury in PAD (Fig. 17-2). Repeated episodes of ischemia during exercise and reperfusion during recovery may promote oxidant injury to endothelial cells (ECs), muscle mitochondria, muscle fibers, and distal motor axons. Oxidative injury to these tissues may in turn promote chronic changes in muscle structure and metabolism, leading to loss of function in PAD that cannot be explained simply by a reduction in blood flow and oxygen delivery. Mitochondria are the major source of free radicals within the cell, so mitochondrial deoxyribonucleic acid (DNA) may be a useful marker of oxidant injury.81
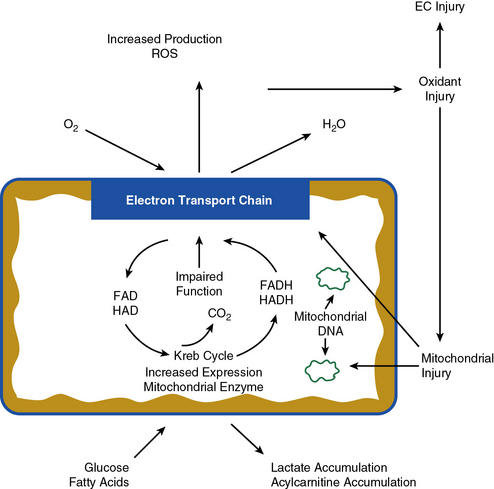
Figure 17-2 Alterations in muscle metabolism in peripheral artery disease (PAD).
Oxidant stress results in endothelial and mitochondrial injuries that cause mitochondrial deoxyribonucleic acid (DNA) deletions and impairment of electron transport chain function. Sequelae are increase in expression of mitochondrial enzymes and accumulation of lactate and acylcarnitines. EC, endothelial cell.
Muscle mitochondrial oxidative damage in patients with PAD is readily demonstrated as accumulation of somatic mutations in mitochondrial DNA. For example, patients with PAD have an increased frequency of a mitochondrial DNA 4977 bp deletion mutation.82 This is a common finding in other tissues under conditions of oxidative stress. More importantly, muscle mitochondria from patients with PAD have specific defects in key steps of the electron transport chain (see Fig. 17-2). These steps have been previously identified as targets of oxidative injury in myocardial perfusion-reperfusion models.83 Mitochondrial oxidative injury may represent a positive feedback system because electron transport chain impairment increases mitochondrial free radical production, which results in more electron transport dysfunction. Such mechanisms may eventually result in cell loss due to apoptosis.84,85
Strategies to reduce or modulate oxidant stress may be important to prevent not only atherosclerotic disease progression, but also to protect skeletal muscle from oxidant injury. Supplementation with the antioxidant vitamin C improves endothelial function in patients with diabetes.86 However, long-term administration of vitamins C and E does not improve endothelial function in patients with cardiovascular disease.87 In an animal model, ischemia-reperfusion injury of skeletal muscle microcirculation (defined as microvascular vasoconstriction and plugging and inhibition of NO production) is prevented by a combination of antioxidant vitamins and l-arginine.88 Important challenges remain concerning the development of antioxidant therapy. A relevant antioxidant should target specific subcellular locations (e.g., mitochondria) and not be capable of propagating oxidative injury. Thus it remains unclear how oxidant stress can be optimally modulated in PAD, or if antioxidants will favorably alter the pathophysiology of claudication.
Muscle Structure and Function in Peripheral Artery Disease
In healthy humans, exercise requires coordinated recruitment of appropriate muscle fiber types to meet the demands of specific exercise conditions. There is recruitment of type I oxidative slow-twitch fibers that have high mitochondrial content with low-intensity repetitive contractions. Depending on the exercise intensity of these contractions, the fuel is a balance of fat and carbohydrate oxidation. In contrast, rapid forceful muscle contractions require recruitment of type II glycolytic fast-twitch fibers. These fibers have fewer mitochondria than type I fibers and have easy fatigability. Type II fibers include two subtypes: type IIa fibers have intermediate oxidative and contractile properties, and type IIb fibers have the greatest capacity for force generation.
Patients with PAD develop several histological abnormalities in their skeletal muscle. These changes reflect a complex combination of changes associated with disuse due to exercise limitation and direct injury from ischemia, ischemia-reperfusion, and chronic inflammatory mechanisms. Muscle biopsy studies have shown a decrease in type II fast-twitch fiber area that is associated with muscle weakness.89 These observations have been extended to patients with CLI, where decreases in skeletal muscle myosin isoforms of types IIa and IIb fibers were observed.90 Various morphological alterations have been identified in the skeletal muscle of PAD patients, including muscle apoptosis and atrophy, increased fiber type switching from oxidative type I fibers to glycolytic type II fibers, muscle fiber denervation, altered myosin heavy chain expression, and mitochondrial DNA injury.82,89,91–93 Increasing evidence indicates that inflammatory mediators play an important role in skeletal muscle wasting and fatigue. Tumor necrosis factor (TNF)-α and IL-6, which are increased in PAD, induce skeletal muscle protein breakdown in rats and are negatively related to muscle mass and muscle strength in elderly individuals.66,94–96 Furthermore, TNF-α may provoke apoptosis in skeletal myocytes.97 McDermott et al. found that in PAD, higher levels of inflammatory markers (CRP, IL-6, and sVCAM-1) were associated with a small calf area. IL-6 and sVCAM-1 also were associated with a higher percent of calf muscle fat.98 However, the impact of these observations on muscle function and exercise performance in PAD has not been established.
Patients with claudication also demonstrate extensive skeletal muscle denervation by histological criteria. Denervation injury has been confirmed by electrophysiological testing, and these abnormalities are progressive over time.99 Changes in skeletal muscle fiber type and neurological function correlate with a decrease in muscle strength.89 Sensory nerve function is impaired in PAD, particularly in patients with CLI.100 Neuropathic symptoms are often obscured by the effects of ischemia on other tissues. The neurophysiological changes suggest that the underlying pathophysiology is a distal axonopathy affecting nerve fibers of all sizes. Measures of blood flow in the leg correlate with neurological symptom scores, examination scores, and electrophysiological testing.101
In addition to changes in muscle fibers, muscle capillarity is increased in skeletal muscle from patients with PAD.102 If capillary architecture is normal, this suggests that distal diffusion distances are not a limiting factor for oxygen delivery in PAD. Increased capillarity may be in compensation for the reduction in large-vessel blood flow, and these changes in peripheral diffusion (higher conductance) may have functional relevance.103
Several gait abnormalities also have been described in claudication.104 The findings are primarily slowed walking speed due to decreased step length and cadence. Gait stability is favored over walking speed. Whether these gait abnormalities are related to muscle denervation and weakness or are adaptations to minimize development of pain is unknown.105 These observations may explain in part why the reduced exercise performance of a patient with claudication cannot be entirely explained by alterations in limb blood flow and pressure.
Alterations in Skeletal Muscle Metabolism
When patients with PAD exercise, skeletal muscle blood flow is insufficient to meet metabolic demand, as described earlier in this chapter. This limitation in the blood flow response to exercise has metabolic consequences. In patients with PAD, muscle oxygen saturation and phosphocreatine levels are normal at rest. At the onset of exercise, however, there is a marked delay in systemic uptake of oxygen that parallels a slowed response in skeletal muscle uptake of oxygen.6,106 Phosphocreatine is used preferentially for energy creation in patients with PAD compared with control subjects at equivalent exercise work loads.78 These observations would suggest that a block occurs in early utilization of oxygen at the onset of exercise and prior to limitation of oxygen delivery due to large-vessel occlusions.
Patients with PAD also have changes in oxidative metabolism that appear intrinsic to skeletal muscle. A potential site of impairment of oxidative metabolism in PAD is the electron transport chain, which is vulnerable to free radical injury, as noted earlier.107 Skeletal muscle from legs affected by PAD has reduced activity of mitochondrial NADH dehydrogenase of complex I and ubiquinol–cytochrome C oxidoreductase (complex III).108 These observations suggest that electron transport chain activity is impaired, likely secondary to ischemia-reperfusion injury, and may contribute to metabolic dysfunction in PAD.
Altered mitochondrial respiration may have functional consequences. For example, pulmonary oxygen uptake kinetics are slowed at the onset of exercise in patients with PAD, as described earlier. The kinetic changes are independent of the hemodynamic severity of the vascular disease and may thus relate to muscle metabolic abnormalities. Consistent with slowed ventilatory oxygen kinetics, patients with PAD have altered mitochondrial respiration. A number of investigators have used phosphorus-31 (31P) magnetic resonance spectroscopy (MRS) to evaluate mitochondrial respiration in the muscle of control subjects and patients with PAD.109 Using muscle adenosine diphosphate (ADP) concentration as a marker of the state of mitochondrial respiration, mitochondrial function in PAD is characterized by an increased level of ADP to maintain cellular respiration. An altered ADP–respiratory control relationship is unusual in human chronic diseases, but is characteristic of inherited disorders of the electron transport chain. Given these observations, PAD muscle energetics cannot entirely be explained by reduced blood flow.
Muscle mitochondrial content and mitochondrial enzyme activities reflect the functional state of the individual. Skeletal muscle mitochondrial oxidative enzyme activities increase with exercise training and decrease with prolonged bed rest or inactivity.110 In healthy individuals, muscle mitochondrial content is positively correlated with peak oxygen uptake, indicating the importance of muscle oxidative capacity in determining exercise performance.111 In PAD, marked limitation in walking activity and resultant sedentary behavior would be expected to result in a decrease in muscle mitochondrial enzyme content and activity (detraining). In contrast, several studies have shown an increased mitochondrial content in muscle of patients with PAD.112,113 This increased mitochondrial expression appears to be a direct consequence of, and is proportionate to, the severity of occlusive disease as assessed by leg hemodynamics.114 Thus, alterations in skeletal muscle mitochondria in PAD appear to reflect the severity of the underlying occlusive disease process. An increased mitochondrial content might improve oxygen extraction under ischemic conditions and could reflect a compensatory mechanism for any intrinsic abnormality in mitochondrial oxidative capacity. Interestingly, increased mitochondrial expression is also associated with inherited disorders of mitochondrial electron transport, suggesting a mechanistic and functional link with the acquired disorder in PAD discussed earlier.
During normal metabolic conditions, fuel substrates such as fatty acids, protein, and carbohydrates are converted to acyl-coenzyme A (CoA) intermediates for oxidative metabolism in the Krebs cycle. These coA–coupled intermediates are linked to the cellular carnitine pool through reversible transfer of acyl groups between carnitine and CoA.115 One of the functions of carnitine is to serve as a buffer for the acyl-CoA pool by the formation of acylcarnitines. Thus, during conditions of metabolic stress, incomplete oxidation or utilization of an acyl-CoA will lead to their accumulation. Transfer of the acyl group to carnitine will result in accumulation of the corresponding acylcarnitine.
Patients with PAD have alterations in carnitine metabolism, as evidenced by accumulation of short-chain acylcarnitines in plasma and skeletal muscle from the legs affected by arterial disease.116,117 This accumulation of acylcarnitines implies that acyl-CoA is not being efficiently oxidized, given that the acyl-CoA pool is in equilibrium with the acylcarnitine pool. Importantly, acylcarnitine accumulation may have functional significance in that patients with the greatest accumulation have the most reduced treadmill exercise performance. The degree of metabolic abnormality (as defined by acylcarnitine accumulation) is a better predictor of treadmill exercise performance than the ABI, emphasizing the importance of altered skeletal muscle metabolism in the pathophysiology of claudication.
Conclusions
Patients with PAD and claudication have profound limitation in their exercise performance. Large-vessel obstruction impairs delivery of oxygenated blood to skeletal muscle during exercise, resulting in a supply/demand mismatch. Arterial hemodynamics and large-vessel blood flow, however, do not fully account for the exercise limitations observed in patients with claudication. Changes in microcirculation and skeletal muscle structure and metabolic function significantly contribute to disease pathophysiology. Understanding these multiple components of exercise limitation provides insight into treatment approaches that address the spectrum of abnormalities seen in patients with claudication.
Critical limb ischemia is a state characterized by severe impairment of blood flow to the limb whereby the metabolic requirements of the tissue at rest are not met. Multiple occlusive lesions of the limb arteries, coupled with functional and structural changes in the microcirculation, are responsible for inadequate tissue perfusion and formation of skin ulcers and necrosis. Age, smoking, and diabetes are major risk factors for CLI. Inflammatory mediators and endogenous procoagulants contribute to development and progression of CLI. Blood components such as red cells, white cells, and platelets aggregate and perturb blood flow in the microcirculation.
Revascularization procedures are the mainstay of treatment for CLI. Further understanding of the pathophysiological disturbances that occur in CLI may lead to additional strategies to preserve limb viability and improve symptoms.
1 Criqui M.H., Langer R.D., Fronek A., et al. Mortality over a period of 10 years in patients with peripheral arterial disease. N Engl J Med. 1992;326:381–386.
2 Pande R.L., Perlstein T.S., Beckman J.A., et al. Secondary prevention and mortality in peripheral artery disease: National Health and Nutrition Examination Study, 1999 to 2004. Circulation. 2011;124:17–23.
3 Libby P., Ridker P.M., Hansson G.K. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325.
4 Rocha V.Z., Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. 2009;6:399–409.
5 Dachun X., Jue L., Liling Z., et al. Sensitivity and specificity of the ankle-brachial index to diagnose peripheral artery disease: a structured review. Vasc Med. 2010;15:361–369.
6 Bauer T.A., Regensteiner J.G., Brass E.P., et al. Oxygen uptake kinetics during exercise are slowed in patients with peripheral arterial disease. J Appl Physiol. 1999;87:809–816.
7 McDermott M.M., Greenland P., Liu K., et al. Leg symptoms in peripheral arterial disease: associated clinical characteristics and functional impairment. JAMA. 2001;286:1599–1606.
8 McDermott M.M., Mehta S., Liu K., et al. Leg symptoms, the ankle-brachial index, and walking ability in patients with peripheral arterial disease. J Gen Intern Med. 1999;14:173–181.
9 Vogt M.T., Cauley J.A., Kuller L.H., et al. Functional status and mobility among elderly women with lower extremity arterial disease: the study of osteoporotic fractures. J Am Geriatr Soc. 1994;42:923–929.
10 Fowkes F.G., Murray G.D., Butcher I., et al. Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: a meta-analysis. JAMA. 2008;300:197–208.
11 Hirsch A.T., Haskal Z.J., Hertzer N.R., et al: ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease) endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation, J Am Coll Cardiol 47:1239–1312, 2006.
12 Norgren L., Hiatt W.R., Dormandy J.A., et al. Inter-society consensus for the management of peripheral arterial disease (TASC II). J Vasc Surg. 2007;45:S5–S67.
13 Sigvant B., Wiberg-Hedman K., Bergqvist D., et al. A population-based study of peripheral arterial disease prevalence with special focus on critical limb ischemia and sex differences. J Vasc Surg. 2007;45:1185–1191.
14 Dormandy J., Mahir M., Ascady G., et al. Fate of the patient with chronic leg ischaemia. A review article. J Cardiovasc Surg (Torino). 1989;30:50–57.
15 Belch J., Hiatt W.R., Baumgartner I., et al. Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet. 2011;377:1929–1937.
16 Gray B.H., Grant A.A., Kalbaugh C.A., et al. The impact of isolated tibial disease on outcomes in the critical limb ischemic population. Ann Vasc Surg. 2010;24:349–359.
17 Faglia E., Clerici G., Clerissi J., et al. Long-term prognosis of diabetic patients with critical limb ischemia: a population-based cohort study. Diabetes Care. 2009;32:822–827.
18 Van B.E., Nikol S., Norgren L., et al. Insights on the role of diabetes and geographic variation in patients with critical limb ischaemia. Eur J Vasc Endovasc Surg. 2011;42:365–373.
19 Gardner A.W., Montgomery P.S. Resting energy expenditure in patients with intermittent claudication and critical limb ischemia. J Vasc Surg. 2010;51:1436–1441.
20 Richardson R.S., Grassi B., Gavin T.P., et al. Evidence of O2 supply-dependent VO2 max in the exercise-trained human quadriceps. J Appl Physiol. 1999;86:1048–1053.
21 Richardson R.S., Leigh J.S., Wagner P.D., et al. Cellular PO2 as a determinant of maximal mitochondrial O(2) consumption in trained human skeletal muscle. J Appl Physiol. 1999;87:325–331.
22 Grassi B., Pogliaghi S., Rampichini S., et al. Muscle oxygenation and pulmonary gas exchange kinetics during cycling exercise on-transitions in humans. J Appl Physiol. 2003;95:149–158.
23 Young D.F., Tsai F.Y. Flow characteristics in models of arterial stenoses. II. Unsteady flow. J Biomech. 1973;6:547–559.
24 Flanigan D.P., Tullis J.P., Streeter V.L., et al. Multiple subcritical arterial stenoses: effect on poststenotic pressure and flow. Ann Surg. 1977;186:663–668.
25 Karayannacos P.E., Talukder N., Nerem R.M., et al. The role of multiple noncritical arterial stenoses in the pathogenesis of ischemia. J Thorac Cardiovasc Surg. 1977;73:458–469.
26 Young D.F., Cholvin N.R., Kirkeeide R.L., et al. Hemodynamics of arterial stenoses at elevated flow rates. Circ Res. 1977;41:99–107.
27 Demer L., Gould K.L., Kirkeeide R. Assessing stenosis severity: coronary flow reserve, collateral function, quantitative coronary arteriography, positron imaging, and digital subtraction angiography. A review and analysis. Prog Cardiovasc Dis. 1988;30:307–322.
28 Lewis P., Psaila J.V., Morgan R.H., et al. Common femoral artery volume flow in peripheral vascular disease. Br J Surg. 1990;77:183–187.
29 Park S.J., Ahn J.M., Kang S.J. Paradigm shift to functional angioplasty: new insights for fractional flow reserve- and intravascular ultrasound-guided percutaneous coronary intervention. Circulation. 2011;124:951–957.
30 Pijls N.H., Van Gelder B., Van der Voort P., et al. Fractional flow reserve. A useful index to evaluate the influence of an epicardial coronary stenosis on myocardial blood flow. Circulation. 1995;92:3183–3193.
31 Pentecost B.L. The effect of exercise on the external iliac vein blood flow and local oxygen consumption in normal subjects, and in those with occlusive arterial disease. Clin Sci. 1964;27:437–445.
32 Hillestad LK: The peripheral blood flow in intermittent claudication IV. The significance of the claudication distance, Acta Med Scand 173:467–478, 1963.
33 Bauer T.A., Brass E.P., Hiatt W.R. Impaired muscle oxygen use at onset of exercise in peripheral arterial disease. J Vasc Surg. 2004;40:488–493.
34 Sumner D.S., Strandness D.E.Jr. The relationship between calf blood flow and ankle blood pressure in patients with intermittent claudication. Surgery. 1969;65:763–771.
35 Maass U., Alexander K. Effect of treadmill exercise on blood gases and acid-base balance in patients with intermittent claudication. Z Kardiol. 1983;72:537–542.
36 Hansen J.E., Sue D.Y., Oren A., et al. Relation of oxygen uptake to work rate in normal men and men with circulatory disorders. Am J Cardiol. 1987;59:669–674.
37 Szuba A., Oka R.K., Harada R., et al. Limb hemodynamics are not predictive of functional capacity in patients with PAD. Vasc Med. 2006;11:155–163.
38 Gardner A.W., Ricci M.A., Case T.D., et al. Practical equations to predict claudication pain distances from a graded treadmill test. Vasc Med. 1996;1:91–96.
39 Gardner A.W., Skinner J.S., Cantwell B.W., et al. Prediction of claudication pain from clinical measurements obtained at rest. Med Sci Sports Exerc. 1992;24:163–170.
40 Gordon M.B., Jain R., Beckman J.A., et al. The contribution of nitric oxide to exercise hyperemia in the human forearm. Vasc Med. 2002;7:163–168.
41 Kinlay S., Creager M.A., Fukumoto M., et al. Endothelium-derived nitric oxide regulates arterial elasticity in human arteries in vivo. Hypertension. 2001;38:1049–1053.
42 Boushel R., Langberg H., Gemmer C., et al. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol. 2002;543:691–698.
43 Anderson T.J., Gerhard M.D., Meredith I.T., et al. Systemic nature of endothelial dysfunction in atherosclerosis. Am J Cardiol. 1995;75:71B–74B.
44 Tsao P.S., Buitrago R., Chang H., et al. Effects of diabetes on monocyte-endothelial interactions and endothelial superoxide production in fructose-induced insulin-resistant and hypertensive rats. Circulation. 1995;92:A2666.
45 Liao J.K., Bettmann M.A., Sandor T., et al. Differential impairment of vasodilator responsiveness of peripheral resistance and conduit vessels in humans with atherosclerosis. Circ Res. 1991;68:1027–1034.
46 Newton D.J., Khan F., Kennedy G., et al. Improvement in systemic endothelial condition following amputation in patients with critical limb ischemia. Int Angiol. 2008;27:408–412.
47 Woodburn K.R., Lowe G.D., Rumley A., et al. Relation of haemostatic, fibrinolytic, and rheological variables to the angiographic extent of peripheral arterial occlusive disease. Int Angiol. 1995;14:346–352.
48 Dormandy J.A., Hoare E., Colley J., et al. Clinical, haemodynamic, rheological, and biochemical findings in 126 patients with intermittent claudication. Br Med J. 1973;4:576–581.
49 Reid H.L., Dormandy J.A., Barnes A.J., et al. Impaired red cell deformability in peripheral vascular disease. Lancet. 1976;1:666–668.
50 Holmberg A., Sandhagen B., Bergqvist D. Hemorheologic variables in critical limb ischemia before and after infrainguinal reconstruction. J Vasc Surg. 2000;31:691–695.
51 Nash G.B., Thomas P.R., Dormandy J.A. Abnormal flow properties of white blood cells in patients with severe ischaemia of the leg. Br Med J (Clin Res Ed). 1988;296:1699–1701.
52 Belch J.J., Sohngen M., Robb R., et al. Neutrophil count and amputation in critical limb ischaemia. Int Angiol. 1999;18:140–144.
53 Anderson S.I., Shiner R., Brown M.D., et al. ICAM-1 expression and leukocyte behavior in the microcirculation of chronically ischemic rat skeletal muscles. Microvasc Res. 2006;71:205–211.
54 Cassar K., Bachoo P., Ford I., et al. Platelet activation is increased in peripheral arterial disease. J Vasc Surg. 2003;38:99–103.
55 Jarajapu Y.P., Coats P., McGrath J.C., et al. Increased alpha(1)- and alpha(2)-adrenoceptor-mediated contractile responses of human skeletal muscle resistance arteries in chronic limb ischemia. Cardiovasc Res. 2001;49:218–225.
56 Coats P., Hillier C. Differential responses in human subcutaneous and skeletal muscle vascular beds to critical limb ischaemia. Eur J Vasc Endovasc Surg. 2000;19:387–395.
57 Jarajapu Y.P., McGrath J.C., Hillier C., et al. The alpha 1-adrenoceptor profile in human skeletal muscle resistance arteries in critical limb ischaemia. Cardiovasc Res. 2003;57:554–562.
58 Tsui J.C., Baker D.M., Biecker E., et al. Evidence for the involvement of endothelin-1 but not urotensin-II in chronic lower limb ischaemia in man. Eur J Vasc Endovasc Surg. 2003;25:443–450.
59 Jacobs M.J., Ubbink D.T., Kitslaar P.J., et al. Assessment of the microcirculation provides additional information in critical limb ischaemia. Eur J Vasc Surg. 1992;6:135–141.
60 Anvar M.D., Khiabani H.Z., Kroese A.J., et al. Alterations in capillary permeability in the lower limb of patients with chronic critical limb ischaemia and oedema. Vasa. 2000;29:106–111.
61 Coats P., Wadsworth R. Marriage of resistance and conduit arteries breeds critical limb ischemia. Am J Physiol Heart Circ Physiol. 2005;288:H1044–H1050.
62 Edwards A.T., Blann A.D., Suarez-Mendez V.J., et al. Systemic responses in patients with intermittent claudication after treadmill exercise. Br J Surg. 1994;81:1738–1741.
63 Belch J.J., McLaren M., Khan F., et al. The inflammatory process in intermittent claudication. Eur Heart J. 2002;4:B31–B34.
64 Belch J.J., Mackay I.R., Hill A., et al. Oxidative stress is present in atherosclerotic peripheral arterial disease and further increased by diabetes mellitus. Int Angiol. 1995;14:385–388.
65 Brevetti G., De Caterina M., Martone V.D., et al. Exercise increases soluble adhesion molecules ICAM-1 and VCAM-1 in patients with intermittent claudication. Clin Hemorheol Microcirc. 2001;24:193–199.
66 Signorelli S.S., Mazzarino M.C., Di Pino L., et al. High circulating levels of cytokines (IL-6 and TNFalpha), adhesion molecules (VCAM-1 and ICAM-1) and selectins in patients with peripheral arterial disease at rest and after a treadmill test. Vasc Med. 2003;8:15–19.
67 Blann A.D., Dobrotova M., Kubisz P., et al. von Willebrand factor, soluble P-selectin, tissue plasminogen activator and plasminogen activator inhibitor in atherosclerosis. Thromb Haemost. 1995;74:626–630.
68 Neumann F.J., Waas W., Diehm C., et al. Activation and decreased deformability of neutrophils after intermittent claudication. Circulation. 1990;82:922–929.
69 Paul B.Z., Jin J., Kunapuli S.P. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Biol Chem. 1999;274:29108–29114.
70 Kirkpatrick U.J., Mossa M., Blann A.D., et al. Repeated exercise induces release of soluble P-selectin in patients with intermittent claudication. Thromb Haemost. 1997;78:1338–1342.
71 Cassar K., Bachoo P., Ford I., et al. Platelet activation is increased in peripheral arterial disease. J Vasc Surg. 2003;38:99–103.
72 Hope S.A., Meredith I.T. Cellular adhesion molecules and cardiovascular disease. Part I. Their expression and role in atherogenesis. Intern Med J. 2003;33:380–386.
73 Mehta J., Dinerman J., Mehta P., et al. Neutrophil function in ischemic heart disease. Circulation. 1989;79:549–556.
74 Lowe G.D.O., Fowkes F.G.R., Dawes J., et al. Blood viscosity, fibrinogen, and activation of coagulation and leukocytes in peripheral arterial disease and the normal population in the Edinburgh Artery Study. Circulation. 1993;87:1915–1920.
75 Turton E.P., Coughlin P.A., Kester R.C., et al. Exercise training reduces the acute inflammatory response associated with claudication. Eur J Vasc Endovasc Surg. 2002;23:309–316.
76 Karmazyn M. Ischemic and reperfusion injury in the heart: cellular mechanisms and pharmacologic interventions. Can J Physiol Pharmacol. 1991;69:719–730.
77 Turrens J.F., Beconi M., Barilla J., et al. Mitochondrial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic Res Commun. 1991;12–13(Pt 2):681–689.
78 Kemp G.J., Roberts N., Bimson W.E., et al. Mitochondrial function and oxygen supply in normal and in chronically ischemic muscle: a combined 31P magnetic resonance spectroscopy and near infrared spectroscopy study in vivo. J Vasc Surg. 2001;34:1103–1110.
79 Hickman P., Harrison D.K., Hill A., et al. Exercise in patients with intermittent claudication results in the generation of oxygen derived free radicals and endothelial damage. Adv Exp Med Biol. 1994;361:565–570.
80 Ciuffetti G., Mercuri M., Mannarino E., et al. Free radical production in peripheral vascular disease. A risk for critical ischaemia? Int Angiol. 1991;10:81–87.
81 Melov S., Shoffner J.M., Kaufman A., et al. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995;23:4122–4126.
82 Bhat H.K., Hiatt W.R., Hoppel C.L., et al. Skeletal muscle mitochondrial DNA injury in patients with unilateral peripheral arterial disease. Circulation. 1999;99:807–812.
83 Rouslin W., Ranganathan S. Impaired function of mitochondrial electron transfer complex I in canine myocardial ischemia: loss of flavin mononucleotide. J Mol Cell Cardiol. 1983;15:537–542.
84 Kumar D., Jugdutt B.I. Apoptosis and oxidants in the heart. J Lab Clin Med. 2003;142:288–297.
85 Sastre J., Pallardo F.V., Vina J. The role of mitochondrial oxidative stress in aging. Free Radic Biol Med. 2003;35:1–8.
86 Ting H.H., Timimi F.K., Boles K.S., et al. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1996;97:22–28.
87 Kinlay S., Behrendt D., Fang J.C., et al. Long-term effect of combined vitamins E and C on coronary and peripheral endothelial function. J Am Coll Cardiol. 2004;43:629–634.
88 Nanobashvili J., Neumayer C., Fuegl A., et al. Combined L-arginine and antioxidative vitamin treatment mollifies ischemia-reperfusion injury of skeletal muscle. J Vasc Surg. 2004;39:868–877.
89 Regensteiner J.G., Wolfel E.E., Brass E.P., et al. Chronic changes in skeletal muscle histology and function in peripheral arterial disease. Circulation. 1993;87:413–421.
90 Steinacker J.M., Opitz-Gress A., Baur S., et al. Expression of myosin heavy chain isoforms in skeletal muscle of patients with peripheral arterial occlusive disease. J Vasc Surg. 2000;31:443–449.
91 Askew C.D., Green S., Walker P.J., et al. Skeletal muscle phenotype is associated with exercise tolerance in patients with peripheral arterial disease. J Vasc Surg. 2005;41:802–807.
92 McGuigan M.R., Bronks R., Newton R.U., et al. Muscle fiber characteristics in patients with peripheral arterial disease. Med Sci Sports Exerc. 2001;33:2016–2021.
93 Mitchell R.G., Duscha B.D., Robbins J.L., et al. Increased levels of apoptosis in gastrocnemius skeletal muscle in patients with peripheral arterial disease. Vasc Med. 2007;12:285–290.
94 Goodman M.N. Tumor necrosis factor induces skeletal muscle protein breakdown in rats. Am J Physiol. 1991;260:E727–E730.
95 Goodman M.N. Interleukin-6 induces skeletal muscle protein breakdown in rats. Proc Soc Exp Biol Med. 1994;205:182–185.
96 Visser M., Pahor M., Taaffe D.R., et al. Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study. J Gerontol A Biol Sci Med Sci. 2002;57:M326–M332.
97 Meadows K.A., Holly J.M., Stewart C.E. Tumor necrosis factor-alpha-induced apoptosis is associated with suppression of insulin-like growth factor binding protein-5 secretion in differentiating murine skeletal myoblasts. J Cell Physiol. 2000;183:330–337.
98 McDermott M.M., Ferrucci L., Guralnik J.M., et al. Elevated levels of inflammation, D-dimer, and homocysteine are associated with adverse calf muscle characteristics and reduced calf strength in peripheral arterial disease. J Am Coll Cardiol. 2007;50:897–905.
99 England J.D., Ferguson M.A., Hiatt W.R., et al. Progression of neuropathy in peripheral arterial disease. Muscle Nerve. 1995;18:380–387.
100 Laghi P.F., Pastorelli M., Beermann U., et al. Peripheral neuropathy associated with ischemic vascular disease of the lower limbs. Angiology. 1996;47:569–577.
101 Weinberg D.H., Simovic D., Isner J., et al. Chronic ischemic monomelic neuropathy from critical limb ischemia. Neurology. 2001;57:1008–1012.
102 McGuigan M.R., Bronks R., Newton R.U., et al. Muscle fiber characteristics in patients with peripheral arterial disease. Med Sci Sports Exerc. 2001;33:2016–2021.
103 Sala E., Noyszewski E.A., Campistol J.M., et al. Impaired muscle oxygen transfer in patients with chronic renal failure. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1240–R1248.
104 Scherer S.A., Bainbridge J.S., Hiatt W.R., et al. Gait characteristics of patients with claudication. Arch Phys Med Rehabil. 1998;79:529–531.
105 Scherer S.A., Hiatt W.R., Regensteiner J.G. Lack of relationship between gait parameters and physical function in peripheral arterial disease. J Vasc Surg. 2006;44:782–788.
106 Bauer T.A., Brass E.P., Barstow T.J., et al. Skeletal muscle StO(2) kinetics are slowed during low work rate calf exercise in peripheral arterial disease. Eur J Appl Physiol. 2007;100:143–151.
107 Rouslin W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am J Physiol. 1983;244:H743–H748.
108 Brass E.P., Hiatt W.R., Gardner A.W., et al. Decreased NADH dehydrogenase and ubiquinol-cytochrome c oxidoreductase in peripheral arterial disease. Am J Physiol. 2001;280:H603–H609.
109 Kemp G.J., Taylor D.J., Thompson C.H., et al. Quantitative analysis by 31P magnetic resonance spectroscopy of abnormal mitochondrial oxidation in skeletal muscle during recovery from exercise. NMR Biomed. 1993;6:302–310.
110 Wibom R., Hultman E., Johansson M., et al. Adaptation of mitochondrial ATP production in human skeletal muscle to endurance training and detraining. J Appl Physiol. 1992;73:2004–2010.
111 Wang H., Hiatt W.R., Barstow T.J., et al. Relationships between muscle mitochondrial DNA content, mitochondrial enzyme activity and oxidative capacity in man: Alterations with disease. Eur J Appl Physiol. 1999;80:22–27.
112 Lundgren F., Dahllof A.G., Schersten T., et al. Muscle enzyme adaptation in patients with peripheral arterial insufficiency: Spontaneous adaptation, effect of different treatments and consequences on walking performance. Clin Sci. 1989;77:485–493.
113 Hiatt W.R., Regensteiner J.G., Wolfel E.E., et al. Effect of exercise training on skeletal muscle histology and metabolism in peripheral arterial disease. J Appl Physiol. 1996;81:780–788.
114 Jansson E., Johansson J., Sylven C., et al. Calf muscle adaptation in intermittent claudication. Side-differences in muscle metabolic characteristics in patients with unilateral arterial disease. Clin Physiol. 1988;8:17–29.
115 Brass E.P., Hoppel C.L. Relationship between acid-soluble carnitine and coenzyme A pools in vivo. Biochem J. 1980;190:495–504.
116 Hiatt W.R., Nawaz D., Brass E.P. Carnitine metabolism during exercise in patients with peripheral vascular disease. J Appl Physiol. 1987;62:2383–2387.
117 Hiatt W.R., Wolfel E.E., Regensteiner J.G., et al. Skeletal muscle carnitine metabolism in patients with unilateral peripheral arterial disease. J Appl Physiol. 1992;73:346–353.