Chapter 57 Pulmonary Hypertension in Non-Pulmonary Arterial Hypertension Patients
Overview of Pulmonary Hypertension
Definition and Nomenclature
Pulmonary hypertension (PH) is defined as a sustained mean pulmonary arterial blood pressure above 25 mmHg and pulmonary vascular resistance (PVR) of above 3 Wood units (240 dyne/s/cm− 5) at rest in the setting of a pulmonary capillary wedge pressure (PCWP) less than 15 mmHg.1 Pulmonary hypertension is a clinical syndrome typically characterized by hypoxemia, diminished exercise capacity, and dyspnea. In the vast majority of patients, PH develops as a consequence of hypoxic pulmonary vasoconstriction, vascular congestion, or impedance to pulmonary blood flow in the setting of primary lung, cardiac, or pulmonary vascular thromboembolic disease.
In contrast, mean pulmonary arterial hypertension (mPAH), a rare form of PH, results from an interplay between genetic and molecular factors that promotes pulmonary vascular endothelial dysfunction, vascular smooth muscle cell (VSMC) hypertrophy, and negative remodeling of small- and medium-sized pulmonary arteries in the absence of other cardiopulmonary disease.2 In PAH, pulmonary artery pressure (PAP) is often over 40 mmHg and may reach suprasystemic levels in severe cases; however, this occurs uncommonly in PH from non-PAH etiologies.3 Thus, PH and PAH are distinct pathophysiological entities, and although clinical signs and symptoms often overlap between these conditions, the terms are not synonymous.1
The contemporary PH classification system was created by an international panel of world experts at the 4th World Symposium on PH in 2008 (Dana Point, California) and broadly divides PH patients into two groups: those with PAH or PAH-associated conditions (i.e., formerly primary pulmonary hypertension) and PH that occurs in the setting of another cardiopulmonary disease (i.e., formerly secondary pulmonary hypertension).4
Chapter 56 is devoted to a discussion of PAH and PAH-associated conditions, whereas the current chapter provides an overview of the pathophysiology and treatment of disorders associated with secondary forms of PH. Specifically, this chapter will review primary diseases that modulate PH by causing (1) pulmonary venous hypertension, (2) chronic hypoxemia, (3) pulmonary thromboembolism, and (4) mechanical disruption to the normal pulmonary vasculature (i.e., World Health Organization [WHO] PH classification groups 2-5; Box 57-1).5 In clinical practice and throughout the published literature, the designation nonpulmonary arterial hypertension pulmonary hypertension is often invoked to describe these patients.
 Box 57-1 Clinical Classification of Pulmonary Hypertension by Etiology*
Box 57-1 Clinical Classification of Pulmonary Hypertension by Etiology*
1. Pulmonary arterial hypertension (PAH)
2. Pulmonary hypertension (PH) with left heart disease
3. PH associated with lung disease and/or hypoxemia
4. PH due to chronic thrombotic and/or embolic disease
5. Miscellaneous causes of PH (sarcoidosis, malignancy, fibrosing mediastinitis, Takayasu’s arteritis, iatrogenic)
Adapted from Simonneau G, Robbins IM, Beghetti M, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54(1 Suppl):S43–S54, 2009.
* Diseases in groups 2 to 5 are reviewed in the current chapter.
Epidemiology, Diagnosis, and Natural History
The prevalence of PH in the general population is not well established, and PH incidence varies substantially according to primary disease subtype. A recent report of 455 patients with an elevated left ventricular (LV) end-diastolic pressure (but without left-sided valvular disease) observed that over half had comorbid PH,6 whereas PH is present in more than 90% of selected chronic obstructive pulmonary disease (COPD) patients.7,8 Rates of PH vary significantly within a specific primary disease subpopulation as well. For example, Handa et al. reported a 5% PH prevalence rate in one cohort of asymptomatic or mildly symptomatic sarcoidosis patients despite abnormal chest radiography, restrictive pattern on pulmonary function testing (PF T), and decreased peripheral oxygen saturation levels. If persistent dyspnea is present in sarcoidosis, however, PH prevalence rates increase to over 50%.9,10
The likelihood of developing clinically evident PH is often tightly linked to comorbid cardiac or lung disease characteristics. For example, symptomatic PH due to impaired LV diastolic function from chronic systemic hypertension is an indolent process that progresses with respect to decline in myocardial compliance.11 In contrast, severe PH from acute altitude sickness occurs via sudden hypoxia-mediated pulmonary microvascular vasoconstriction and hyperemia, which may occur independently of pulmonary reserve.12
Pulmonary hypertension prognosis, treatment choice, and clinical response to therapy are strongly associated with PH disease subtype. At present, goal-directed medical therapy for restoration of pulmonary microvascular function with calcium channel blockers, endothelin receptor antagonists (ERAs), inhaled nitric oxide (iNO), selective phosphodiesterase type 5 (PDE5) inhibition, or prostanoid replacement therapy is approved by the U.S. Food and Drug Administration (FDA) for use only in PAH patients. As is discussed in greater detail later, conclusions from small clinical trials have demonstrated a favorable effect of various PAH therapies on pulmonary hemodynamics and exercise tolerance in some WHO Class 2 thorough 5 conditions,13 but inappropriate administration of advanced PAH therapies to non-PAH patients with PH is likely to be ineffective and possibly harmful.1,6 Therefore, the emphasis of contemporary diagnostic algorithms is on discrimination of PAH from non-PAH patients1 (Fig. 57-1A). Comprehensive clinical, radiographic, serological, echocardiographic, and/or invasive hemodynamic testing is often necessary to confirm the absence of disease states that predispose to PH prior to making the diagnosis of PAH7 (Fig. 57-1B).
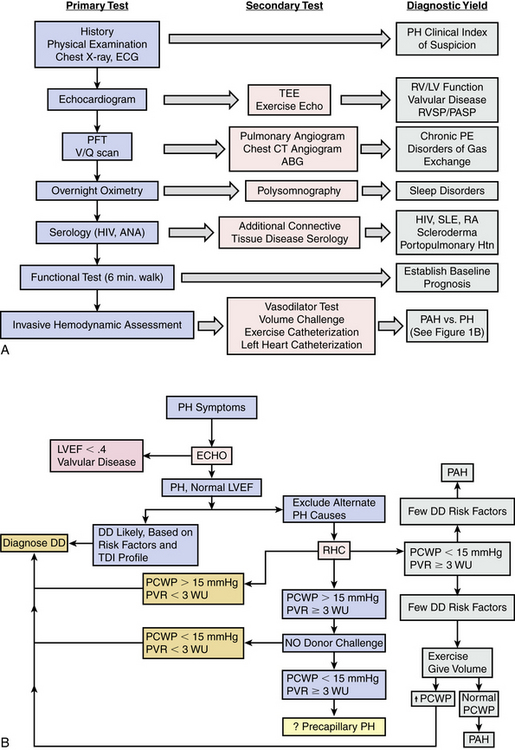
Figure 57-1 Pulmonary hypertension (PH) diagnostic algorithm.
A, Low clinical index of suspicion initiates clinical evaluation for PH. Primary test results aid the clinician in establishing the diagnosis of PH and determining the most likely PH classification. Secondary tests are based on results from primary tests and provide additional information for determination of PH etiology. B, One possible diagnostic algorithm for discriminating pulmonary arterial hypertension (PAH) vs. PH from primary left-heart disease. ABG, arterial blood gas; ANA, antinuclear antibody; CT, computed tomography; CXR, chest x-ray; DD, diastolic dysfunction; ECG, electrocardiogram; ECHO, echocardiogram; HIV, human immunodeficiency virus; LV, left ventricle; LVEF, left ventricular ejection fraction; NO, nitric oxide; PASP, pulmonary artery systolic pressure; PCWP, pulmonary capillary wedge pressure; PE, pulmonary embolism; PFT, pulmonary function test; PVR, pulmonary vascular resistance; RHC, right heart catheterization; RV, right ventricle; RVSP, right ventricular systolic pressure; SLE, systemic lupus erythematosus; TEE, transesophageal echocardiogram; V/Q, ventilation/perfusion; WU, Wood units.
(A adapted from McLaughlin VV, Archer SL, Badesch DB, et al: ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. J Am Coll Cardiol 53:1573–1619, 2009. B adapted from Hoeper MM, Barbera JA, Channick RN, et al: Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol 54[1 Suppl]:S85–S96, 2009.)
Pulmonary hypertension is underrecognized in clinical practice.14 Initiation of diagnostic testing for PH therefore requires a low index of clinical suspicion among practitioners who must recognize clues that suggest PH pathophysiology, such as familial or genetic risk factors for PAH, or comorbid conditions known to promote elevations in PAP.
Pulmonary Venous Hypertension
Pathophysiology of Pulmonary Hypertension due to Left-Sided Heart Disease
Acute left heart failure
Among the most common causes of PH is left-sided cardiac disease from LV systolic or diastolic dysfunction, or left-sided valvular disease (Table 57-1). In nonvalvular forms of left-sided heart disease, increased LV end-diastolic filling pressure is transmitted retrograde to the pulmonary venous and arterial circulatory beds. Acute changes to normal LV pressure-volume hemodynamics, such as occurs during an acute myocardial infarction (MI) or as a consequence of mitral valve leaflet rupture, predispose to sudden and dramatic increases in left atrial pressure and PAP15 (Fig. 57-2). Owing to the noncompacted, thin-walled architecture of the right ventricle (RV), acute pressure loading is poorly tolerated and results in RV systolic dysfunction, a major determinant of outcome in PH.16 Acute increases in PAP results in a congestive vasculopathy characterized by decreased pulmonary arteriolar compliance and loss of normal autoregulation of pulmonary vasomotor tone.17 These pathophysiological changes are generally reversible with pharmacotherapies that promote pulmonary vasodilation, reduce cardiac preload (e.g., nitric oxide [NO] donors), or directly alleviate pulmonary vascular congestion (e.g., loop diuretics).
Table 57-1 Cardiovascular Diseases That Predispose to Pulmonary Hypertension
| Clinical Feature | Mechanism |
|---|---|
| Chronic systemic hypertension | ↑ Left ventricular (LV) afterload → LV hypertrophy → ↓ LV compliance → ↑ LV end-diastolic filling pressure → pulmonary venous hypertension |
| Diabetes mellitus | Intramyocardial microcirculatory and epicardial coronary vascular dysfunction → LV systolic or diastolic dysfunction → pulmonary venous hypertension |
| Coronary artery disease (CAD) | Myocardial ischemia → LV systolic or diastolic dysfunction → pulmonary venous hypertension |
| Atrial fibrillation | Loss of “atrial kick” → ↑ left atrial and pulmonary venous congestion |
| Impaired diastolic function | ↑ End-diastolic filling pressure secondary to restrictive, infiltrative, or genetic cardiomyopathy → pulmonary venous hypertension |
| Mitral stenosis | ↑ Transmitral valve pressure ± atrial fibrillation → ↑ pulmonary venous hypertension |
| Mitral regurgitation (MR) | Chronic LV volume overload → LV cavitary dilation → ↑ LV end-diastolic filling pressure → pulmonary venous hypertension With elevated mitral regurgitant fraction, pulmonary artery pressure (PAP) is elevated secondary to pulmonary circulatory volume and pressure overload, particularly during exercise |
| Aortic insufficiency | Chronic LV volume overload → LV cavitary dilation → ↑ LV end-diastolic filling pressure → pulmonary venous hypertension |
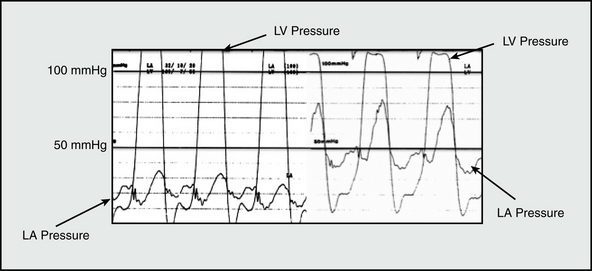
Figure 57-2 Effect of acute mitral regurgitation (MR) on left atrial pressure.
Intraprocedural hemodynamic tracing obtained during mitral balloon valvotomy captures dramatic increase in mean left atrial (LA) pressure from 20 to 48 mmHg due to acute mitral insufficiency. Sudden changes in LA or pulmonary pressure are poorly tolerated by right ventricle and may induce cor pulmonale. LV, left ventricle.
(Adapted from Ha JW, Chung N, Chang BC, et al: Acute mitral regurgitation due to leaflet tear after balloon valvotomy. Circulation 98:2095–2097, 1998.)
Chronic nonvalvular left-sided heart failure
In addition to passive pulmonary vascular congestion, circulating levels of the vasoactive peptide endothelin-1 (ET-1) positively correlate with PH severity in chronic left-sided heart failure.18 Pathophysiological concentrations of ET-1 disrupt normal vasomotor tone via activation of ETA/B receptors on VSMCs that increases intracellular [Ca+ 2]i levels. Endothelin-1 also promotes release of catecholamines (e.g., norepinephrine).19,20 Together, these processes are linked to VSMC contraction and in PH offset vasodilatory cell signaling pathways, resulting in pulmonary VSMC vasoconstriction.
Chronically elevated pulmonary venous pressure induces a cellular environment in pulmonary arterioles characterized by inflammation and increased levels of reactive oxygen species (ROS) generation. Over time, these maladaptive molecular processes are implicated in the development of irreversible pathological changes to normal pulmonary blood vessel architecture, including intimal fibrosis and VSMC hypertrophy and proliferation. Chronic RV pressure overload is also linked to the propagation of worsening left-heart failure by promoting abnormal changes in RV chamber deformation that adversely influences LV geometry.21
Diagnosis, Treatment, and Natural History
The diagnosis of PH from left-sided heart failure is often evident on clinical grounds alone. Complaints of decreased exercise tolerance, dyspnea, and lower-extremity edema are common in PH but do not necessarily discriminate right- from left-sided congestive heart failure. Therefore, echocardiography is used to estimate PA systolic pressure and evaluate RV size and function. Invasive hemodynamic monitoring with right heart catheterization confirms the diagnosis of PH and excludes alternate etiologies of PH-like symptoms, such as constrictive pericardial disease, in which PVR is usually normal.
Pulmonary artery pressure, PVR, and RV systolic function are independent predictors of outcome in patients with chronic left-sided heart failure. In one study of 377 patients undergoing right heart catheterization with low LV ejection fraction and a history of congestive heart failure, mean pulmonary artery pressure (mPAP) over 29 mmHg portended about a threefold higher 36-month mortality rate (irrespective of RV function) compared to patients with normal mPAP. 22 Interpretation of pulmonary hemodynamics, however, must occur according to an individual patient’s specific clinical scenario. Although typically, PAP positively correlates with PH severity in chronic left-sided heart failure, this is not always the case. Since the generation of PAP is dependent on RV systolic function, abnormally low PAP may be observed in severe PH with RV failure. In this scenario, left-sided heart failure–mediated pulmonary vascular congestion results in an increased PVR even if PAP is normal or low.18 Pulmonary hemodynamic indices commonly used in clinical practice are provided in Table 57-2.
Table 57-2 Pulmonary Hemodynamic Measurements and Normal Ranges
| Measurement | Equation | Normal Range |
|---|---|---|
| Mean RA BP | Directly measured (PA catheter) | 0-8 mmHg |
| Pulmonary artery (PA) BP | Directly measured (PA catheter) | Systolic (PASP): 15-25 mmHg Diastolic (PADP): 4-12 mmHg mPAP 10-20 mmHg |
| PA capillary wedge pressure | Directly measured (PA catheter) PASP + (2 × PADP)]/3 |
6-12 mmHg |
| Cardiac output | Heart rate × Stroke volume/1000 | 4-7 L/min |
| PVR | 80 × (mPAP − PCWP)/cardiac output | 20-130 dyne/s/cm− 5 or 0.25-1.6 Wood units |
| Transpulmonary gradient | Mean SBP − Mean RABP | 5-8 mmHg |
BP, blood pressure; mPAP, mean pulmonary artery pressure; PA, pulmonary artery; PADP, pulmonary artery diastolic pressure; PASP, pulmonary artery systolic pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RA, right atrial; SBP, systolic blood pressure.
Conventional heart failure pharmacotherapy is the cornerstone treatment strategy for PH from nonvalvular left-sided cardiac disease. Angiotensin-converting enzyme (ACE) inhibitors, β-adrenergic receptor antagonists, loop diuretics, and vasodilators (e.g., hydralazine) often effectively decrease PVR and PAP, thereby promoting favorable responses in RV systolic function. A subset of chronic left-sided heart failure patients tends to exhibit a degree of PH that seems clinically and hemodynamically out of proportion to the severity of LV dysfunction, raising suspicion that alternative processes beyond passive vascular congestion alone are in play.1,7 Overall, sufficiently powered randomized clinical trials evaluating the effect of iNO, prostacyclin replacement, and ERAs for PH due to chronic left-sided heart failure have either failed to demonstrate a beneficial effect on pulmonary vascular hemodynamics or did so, but at the cost of significant adverse clinical events, including increased early mortality in one large trial of intravenous epoprostenol.23 Sildenafil, which promotes vasodilation by inhibiting PDE5 in lung VSMCs that consume cyclic guanosine monophosphate (cGMP), appears to decrease PAP and PVR safely without compromising cardiac output13 (Fig. 57-3). The effect of sildenafil on long-term outcome and survival in chronic heart failure patients is not yet known.
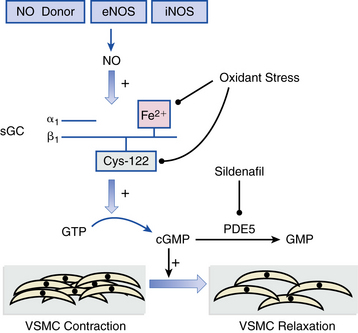
Figure 57-3 Nitric oxide (NO)-soluble guanylyl cyclase signaling in vascular smooth muscle cells (VSMCs).
NO generated from nitric oxide synthase (NOS) or pharmacological sources (e.g., nitroglycerin, inhaled NO [iNO]) activate the heterodimer soluble guanylyl cyclase (sGC) to catalyze conversion of cytosolic guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which results in VSMC relaxation. In pulmonary vasculature, phosphodiesterase-5 (PDE5) hydrolyzes cGMP to form guanosine monophosphate (GMP), thereby decreasing bioactive cGMP levels. Sildenafil is a selective PDE5 inhibitor that increases cGMP levels to promote VSMC dilation. Increases in pulmonary vascular reactive oxygen species (ROS) formation, which occurs in various forms of pulmonary hypertension (PH), may also impair sGC activation via oxidation of the prosthetic heme group or cysteine-122 located in the catalytically active β1-subunit of sGC. eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase.
Pulmonary Hypertension due to Left-Sided Valvular Disease
Pulmonary hypertension due to left-sided valvular disease most often occurs as a consequence of mitral regurgitation (MR) or mitral stenosis and less commonly from severe aortic regurgitation. In aortic stenosis, initial pressure loading–induced LV hypertrophy is protective against PH. However, in decompensated aortic stenosis, LV cavitary dilation due to volume overload is associated with progressive PH.24 The final common pathway in the pathophysiology of PH, irrespective of valve lesion, is pulmonary venous hypertension. In only MR, however, PH is a key determinate for the timing of surgical valve therapy. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend mitral valve surgery (class IIa, level of evidence C) in asymptomatic patients with severe MR, preserved LV function, and PH (systolic PAP >50 mmHg).25 Magne et al. reported that in a cohort of 78 asymptomatic patients with at least moderate MR from degenerative mitral valve disease, resting PH (systolic PAP >60 mmHg) and exercise-induced PH were associated with a significantly lower 2-year symptom free survival rate (36 ± 14% vs. 59 ± 7% and 35 ± 8% vs. 75 ± 7%, respectively). Exercise-induced PH (particularly when systolic PAP >56 mmHg) was also an independent risk factor for development of symptoms.26 These data support exercise-induced PH as one potentially useful clinical marker for estimating the timing of surgical intervention for MR.
Considerations for Cardiac Surgery, Orthotopic Heart Transplantation, and Heart-Lung Transplantation in the Pulmonary Hypertension Patient
In contrast to select end-stage PAH patients for whom bilateral lung transplantation may be indicated, severe preoperative PH in non-PAH patients is associated with an increased rate of adverse outcome in cardiac surgery patients who require cardiopulmonary bypass (CPB).27 In the case of orthotopic heart transplant candidates, a transpulmonary gradient greater than 15 mmHg or PVR greater than 5 Wood units that is unchanged despite preoperative NO donor or selective PDE5 inhibitor pharmacotherapy is an absolute contraindication to surgery, owing to high rates of premature graft failure and early mortality.28
Pulmonary hypertension reversibility in response to pulmonary vasodilator therapy appears to improve 30-day posttransplant mortality rates compared to patients with fixed PH.29 Although universally accepted pulmonary hemodynamic thresholds do not exist, a target PVR of less than 2.5 Wood units or transpulmonary gradient less than 12 mmHg is often used in clinical practice to define preoperative optimization for CPB-requiring surgery. For patients with fixed PH requiring cardiac transplantation, left ventricular assist device (LVAD) therapy may improve operative candidacy. Zimpfer and colleagues reported that in 35 consecutive cardiac transplant candidates with fixed PH receiving LVAD therapy, PVR decreased from 5.1 Wood units at baseline to 3.2 Wood units 3 days following device implantation, an effect sustained over 6 weeks.30 Larger clinical trials are necessary to determine the complete risk/benefit profile (including financial costs) of LVAD implantation under these circumstances. The role of right ventricular assist device (RVAD) therapy in improving PH is unknown.
Pulmonary Hypertension Under Conditions of Hypoxemia
Pulmonary hypertension is a potential component of most chronic lung disease syndromes that cause hypoxemia. It has long been established that PH severity in COPD, interstitial lung disease (ILD), cystic fibrosis, and bronchiectasis is predictive of morbidity and mortality.1,31 A stronger appreciation developed over the previous decade regarding the central role of PH in the natural history of sleep-disordered breathing (SDB) and alveolar hypoventilation disorders.
Hypoxic pulmonary vasoconstriction is the cornerstone pathophysiological process common to lung disease–associated forms of PH. Small and precapillary blood vessel vasoconstriction in response to hypoxia distinguishes pulmonary from coronary, cerebral, and skeletal circulations; a fall in alveolar partial pressure of oxygen (PO2) to less than 50 mmHg results in an increase in PVR by 50%.32,33
A multitude of cell signaling mechanisms modulated by hypoxia have been implicated in pulmonary vasoconstriction, including increased ET-1 production, intracellular [Ca+ 2]i-mediated activation of oxygen-sensitive Ca+ 2 channels in VSMC, and local activation of α-adrenergic receptors. In addition, hypoxia inducible factor (HIF)-1α, a “master” transcription factor activated in response to hypoxia, may modulate the pathogenesis of PH in chronic lung disease via nuclear factor (NF)-κB-mediated pulmonary VSMC proliferation, among other pathological cell signaling pathways that promote negative pulmonary vascular remodeling.34 Alternatively, suppression of HIF-1α requires iron (Fe[II]), raising speculation that iron supplementation is a potential therapeutic target for attenuation of pathogenic HIF-1α-dependent cell signaling in PH. For example, Smith et al. recently reported that iron infusion (Fe[III]-hydroxide sucrose, 200 mg) administered to healthy individuals who developed PH following exposure to hypoxic conditions at elevated altitudes decreased PA systolic from a mean of 37 to 31 mmHg (P = 0.01).35
Late-stage findings in hypoxia-mediated PH include arteriole VSMC hypertrophy and hyperplasia, which lead to medial thickening and may contribute to PH via a reduction in the pulmonary vessel lumen area.
Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease is the most common cause of hypoxemia-induced PH and cor pulmonale, accounting for more than 80% of cases.36 A mPAP “cutoff” of 20 mmHg captured 91% of 120 severe emphysema patients in one widely cited retrospective analysis.1,8 It is uncommon for PAP to exceed 40 mmHg in patients with COPD, and if this value is used to define PH, disease prevalence drops substantially.1
Diagnosis
Jugular venous pressure assessment or detection of a loud pulmonic component of the second heart sound may be obscured in COPD patients with PH due to chest hyperinflation. Echocardiography is generally sufficient for diagnosis of PH in COPD patients, but rotational changes to normal heart anatomy in severely hyperinflated patients may distort acoustic windows and limit detection of the tricuspid regurgitant jet envelope required for PA systolic pressure estimation. In these patients, right heart catheterization is indicated.
Natural history
Progression of PH in COPD patients is indolent, with an average change in mPAP of 0.5 mmHg/year.37 In one case series of 84 patients undergoing right heart catheterization after initiation of long-term oxygen therapy for severe COPD, the 5-year mortality rate for patients with a PAP above 25 mmHg was 36%, vs. 62% for the remainder of the study population.38 Identification of COPD patients at risk for RV dysfunction does not, however, necessarily require echocardiography or invasive hemodynamic monitoring. The presence of one or more electrocardiographic signs of cor pulmonale in COPD patients is associated with a significantly decreased lifespan.39
Interstitial Lung Disease
Interstitial lung disease is characterized by impaired gas exchange, patchy collagen fibrosis, fibroblastic foci deposition, and in certain instances, noncaseating granulomas on lung histopathological analysis.40 Owing to pulmonary blood vessel compression from fibrosis, PH is a potential complication in most forms of ILD. In idiopathic pulmonary fibrosis (IPF), the most aggressive ILD subtype, histopathological findings include adverse vascular remodeling of small muscular pulmonary arteries, destruction of capillary beds, peripulmonary vessel adventitial thickening, and VSMC hypertrophy with increased collagen and elastin accumulation.41 Together, these changes contribute to low diffusion capacity (DLCO) and development of PH that are classic features of this condition.
Diagnosis and treatment
A mean PA diameter greater than 29 mm on computed tomography (CT) scan is suspicious for PH, but echocardiography or right heart catheterization remain the gold standard diagnostic tests for PH in ILD.42 Universal guidelines for iNO, PDE5 inhibitors, ERAs, or prostacyclin replacement therapy in ILD do not exist; current management emphasizes alleviation of hypoxemia with supplemental oxygen and improvement in alveolar gas exchange (e.g., corticosteroids if appropriate). A randomized clinical tria l to evaluate iloprost in IPF patients with PH is complete, but results have not yet been published (clinicaltrials.gov; NCT00109681).
Pulmonary Hypertension in Obstructive Sleep Apnea
Obstructive sleep apnea (OSA) is a clinical syndrome resulting from involuntary collapse of pharyngeal muscles during sleep. Decreases in the arterial partial pressure of oxygen (PaO2) and increased sleep arousal and sympathetic tone adversely affect cardiovascular function by increasing LV afterload, systemic blood pressure, and heart rate. The aggregate effect of these pathological adaptations results in increased rates of OSA-associated ischemic heart disease, congestive heart failure, and arrhythmias. Furthermore, these processes in turn promote PH via left-sided heart failure–induced passive pulmonary vascular congestion. Obesity, which is associated with increases in cardiac output compared to lean individuals, correlates positively with PAP, irrespective of OSA status.43 This observation suggests that high cardiac output, perhaps by inducing reactive pulmonary vasoconstriction from increased circulating blood volume, may contribute to elevated rates of PH observed in patients with obesity, OSA, or both.
Continuous positive pressure airway pressure (CPAP), which in OSA patients is associated with improvement in LV diastolic function, decreases PAP (≈︀ 5 mmHg over 4 months of CPAP use) and is associated with decreased mortality and hospitalization rates in OSA patients.43–45
Chronic High-Altitude Exposure
Over 140 million people live at high altitude (HA) (> 2500 m above sea level).46 A parabolic inverse relationship between PAP and peripheral oxygen saturation (SaO2) exists in populations of native HA dwellers.46 It is believed that chronic hypoxia-mediated vasoconstriction from low oxygen tension at HA results in several cardiopulmonary adaptations, which include RV hypertrophy, VSMC hypertrophy of small and distal pulmonary arterioles, increased oxygen carrying capacity due to reactive erythrocytosis, and increased heart rate at baseline and on exertion.46 If these protective responses are disrupted, typically from hypoventilation-mediated worsening of hypoxia, a cascade of maladaptive responses may ensue, resulting in chronic mountain sickness syndrome (CMS). Severe PH, cor pulmonale, and excessive polycythemia are hallmark clinical features of CMS. In contrast to acute altitude sickness, pulmonary edema from severe PH is not typical. Altitude descent is usually required to cure CMS.47
Pulmonary Hypertension Secondary to Pulmonary Thromboembolic Disease
Chronic Thromboembolic Pulmonary Hypertension
Chronic thromboembolic pulmonary hypertension (CTEPH) is characterized by luminal obliteration of central and distal pulmonary arteries, which results in PH and RV failure. Epidemiological data describing the frequency of CTEPH in the general population is limited; however, 0.5% to 5% of patients surviving an acute pulmonary embolism (PE) tend to develop this condition.48 Histopathological analysis demonstrates intraluminal and in situ thrombus, and stenosis from associated fibrous deposition in the wall of pulmonary arteries.49,50 Pulmonary hypertensive arteriopathy is present in vessels that are distinct from the site of thrombus, suggesting thrombus potentiation of diffuse injury to the pulmonary vasculature.
It is parsimonious to speculate that this syndrome is merely a consequence of incompletely recanalized PE but up to 50% of CTEPH patients have not been previously diagnosed with a pulmonary embolism. Furthermore, a surprisingly strong association between CTEPH and asplenism exists. In one case-controlled analysis of 257 patients, splenectomy was present in 8.5% of CTEPH patients, compared with 2.5% of PAH patients and 0.5% of patients with chronic parenchymal lung disease.51 The precise mechanisms to account for this association remain speculative. Loss of hematopoietic filtering in asplenic patients may generate a prothrombotic environment in vascular tissue, owing to elevated circulating levels of platelets and abnormal erythrocytes. One hypothesis suggests that the platelet-derived vascular effector serotonin, implicated in microthrombus formation and VSMC constriction in PAH-associated conditions, may contribute to PH in asplenic patients. Alternatively, in asplenic patients, unfiltered structurally abnormal red blood cells may function as procoagulant intermediaries because of expression of the negatively charged phospholipid phosphatidylserine on the outer cell membrane, which interacts with thrombin (see Hemoglobinopathy, later).52,53 Additional risk factors include a history of venothromboembolism, positive anticardiolipin antibody status, osteomyelitis, surgically placed ventriculoatrial shunt, inflammatory bowel disease, and malignancy. 54
Diagnosis and medical treatment
CTEPH often presents with progressive exertional dyspnea. Late in the disease course, signs and symptoms of decreased right-sided cardiac output, such as exertional chest pain, increased abdominal girth and lower-extremity edema, and syncope or near-syncope may be present. A bruit auscultated over the lung fields, present in up to 30% of patients, reflects turbulent flow through partially occluded pulmonary vessels.55
In contrast to PAH and most other forms of PH, CTEPH often involves proximal pulmonary arteries. Thus, the presence of one or more ventilation/perfusion (V/Q) mismatched segmental (or larger) defects detected by V/Q scintigraphy is often a key finding that distinguishes CTEPH from PAH. In up to 45% of patients with CTEPH, lower-extremity venous duplex ultrasound examination suggests venothromboembolism.55 Although pulmonary angiography has traditionally been the gold standard diagnostic imaging modality in this condition, multidimensional CT or magnetic resonance angiography (MRA) is at least equally effective in determining proximal, segmental, and subsegmental clot burden, and is superior for detecting alternative PH-associated lung disease (e.g., fibrosing mediastinitis, adenopathy, tumors of the pulmonary artery)56 (Fig. 57-4). Nevertheless, pulmonary angiography is necessary for defining CTEPH type, which predicts operative success, and is therefore recommended for all surgical intervention candidates (see later discussion).
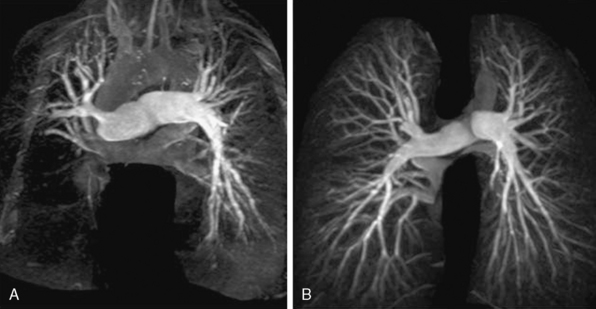
Figure 57-4 A, Preoperative pulmonary magnetic resonance angiography (MRA) in patient with chronic thromboembolic pulmonary hypertension (CTEPH). B, Images acquired from same patient following surgical endarterectomy.
(Adapted from Hamilton-Craig C, Kermeen F, Dunning JJ, et al: Cardiovascular magnetic resonance prior to surgical treatment of chronic thromboembolic pulmonary hypertension. Eur Heart J 31:1040, 2010.)
For inoperable CTEPH patients, a systolic PAP over 40 mmHg, elevated PVR (> 584 dyne/s/cm− 5), and/or elevated right atrial pressure (> 12 mmHg) is associated with a poor prognosis. One study conducted prior to the era of modern PAH pharmacotherapy demonstrated that in CTEPH patients with mPAP over 50 mmHg treated only with anticoagulation, the 2-year survival rate was below 20%.57 In contrast, data from contemporary trials in nonoperative CTEPH patients suggest that therapy with PDE5 inhibitors, ERAs, or prostacyclin replacement therapy improves outcome. For example, one recently published retrospective analysis of 84 inoperable CTEPH patients on maximal medical therapy reported a survival rate of 68% at 5 years.58 In the BENEFiT trial, 157 patients with inoperable or surgically refractory CTEPH were randomized to receive ERA therapy with the ETA/B receptor antagonist bosentan or placebo. Bosentan was well tolerated and resulted in a statistically significant 24% decrease in PVR from baseline, but did not influence exercise tolerance.59 Pilot studies evaluating the effects of iNO and/or selective PDE5 inhibition have demonstrated similar beneficial effects on pulmonary vascular hemodynamics.60 At present, initiation of PAH therapies in CTEPH is generally recommended for poor surgical candidates or those with disease refractory to surgery awaiting lung transplantation.
Surgical treatment
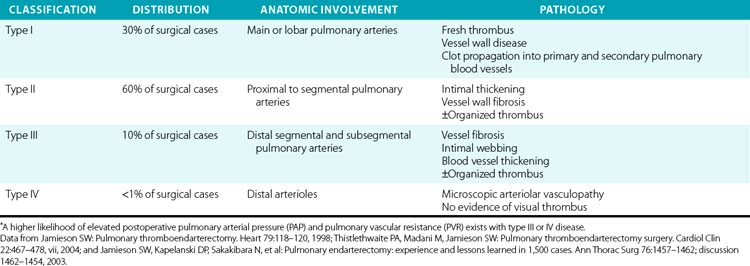
Primary treatment for CTEPH is pulmonary thromboendarterectomy for excision of thromboembolic and fibrotic tissue adherent to the lung vessel wall61,62 (Table 57-3). Deep hypothermic circulatory arrest with CPB is a strategy used to minimize bleeding in the surgical field, and at experienced centers has been met with low neurological morbidity rates and favorable outcomes.63 Factors favoring operative success include a proximal clot burden and a decrease in mPAP in response to preoperative iNO treatment.64 When successful, surgery results in a substantial reduction in clot burden and PVR65 (see Fig. 57-4). In one prospective analysis of 181 CTEPH patients, compared with nonsurgical treatment, pulmonary thromboendarterectomy was associated with a significantly lower PVR (586 ± 248 vs. 269 ± 201 dyne/s/cm− 5) and mPAP, (45 ± 12 vs. 25 ± 11 mmHg).66 In this study, surgeries performed within the last decade were associated with a 6% or lower mortality rate. Lung transplantation should be considered for those with inoperable disease or an incomplete response to thromboendarterectomy.55 Lifelong anticoagulation is recommended for all patients irrespective of operative candidacy.
Pulmonary Hypertension with Hemoglobinopathies
Pulmonary hypertension from primary blood dyscrasias that are characterized by abnormal erythrocyte structure or hemolysis occurs as a consequence of decreased bioavailable NO, increased microthrombi formation, and possibly, increased vascular endothelial ET-1 synthesis (see mechanism details later). Examples include sickle cell disease (SCD), hereditary spherocytosis, β-thalassemia (particularly in patients with splenectomy), and stomatocytosis.
Pulmonary hypertension is a part of the SCD syndrome in about 40% of patients. The 4-year mortality rate is 40% in SCD patients with PH; when RV systolic pressure is above 30 mmHg, there is a 10-fold increase in mortality compared with non-PH SCD patients.67 Histopathological findings in SCD and hemolysis-associated PH demonstrate a pulmonary arteriopathy similar to that observed in PAH. Specifically, the presence of pulmonary VSMC hypertrophy, neomuscularization of small- and medium-sized pulmonary arteries, luminal microthrombi, and plexiform lesions are described at autopsy in patients with these various primary disorders of erythrocyte structure68 (Fig. 57-5).
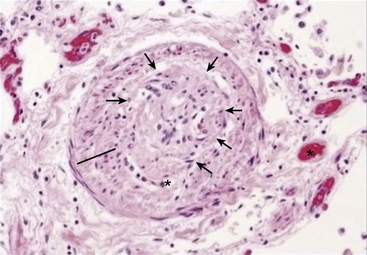
Figure 57-5 Histopathological findings in sickle cell disease (SCD)–associated pulmonary hypertension (PH).
Hematoxylin and eosin preparation of distal pulmonary blood vessel in patient with sickle cell anemia reveals vascular smooth muscle cell (VSMC) hypertrophy, intimal thickening (line), and subtotal obliteration of vessel lumen due to a plexiform lesion (outlined by arrows). Microvessel thrombus is also noted (star).
(From Machado RF, Gladwin MT: pulmonary hypertension in hemolytic disorders: pulmonary vascular disease: the global perspective. Chest 137[6 Suppl]:30 S–38 S, 2010.)
Potential Mechanisms Linking Pulmonary Hypertension and Sickle Cell Disease
Decreased bioavailable nitric oxide
In the pulmonary vasculature, NO is a critical intermediary that promotes vasodilation, decreases platelet aggregation, and is a potent antagonist of vascular inflammation. In SCD and hemolytic anemias, erythrocyte deformity or rupture results in release of free hemoglobin (Hb) into plasma. In the microcirculation, free Hb interacts with NO to form methemoglobin and nitrate (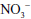 ) [
) [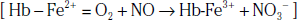 ], thereby decreasing bioavailable levels of NO, which increases pulmonary vascular reactivity.
], thereby decreasing bioavailable levels of NO, which increases pulmonary vascular reactivity.
Ischemia-reperfusion injury in small pulmonary arterioles and capillaries is a hallmark feature in the pathophysiology of SCD occlusive crises.69,70 The downstream molecular effect of these events includes scavenging of bioavailable NO by superoxide 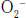 in a reaction that generates peroxynitrite anion (ONOO−).71 Superoxide is generated during ischemia-reperfusion injury via (1) direct activation of ROS generating enzymes (e.g., xanthin oxidase), (2) increased NADPH oxidase activity modulated by enhanced leukocyte recruitment to the vascular endothelium, (3) uncoupling of endothelial nitric oxide synthase (eNOS) due to enzyme cofactor depletion (e.g., BH4), and (4) inducible nitric oxide synthase (iNOS)-dependent NO-mediated ROS formation.70 In addition to scavenging NO,
in a reaction that generates peroxynitrite anion (ONOO−).71 Superoxide is generated during ischemia-reperfusion injury via (1) direct activation of ROS generating enzymes (e.g., xanthin oxidase), (2) increased NADPH oxidase activity modulated by enhanced leukocyte recruitment to the vascular endothelium, (3) uncoupling of endothelial nitric oxide synthase (eNOS) due to enzyme cofactor depletion (e.g., BH4), and (4) inducible nitric oxide synthase (iNOS)-dependent NO-mediated ROS formation.70 In addition to scavenging NO, 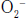 is converted to hydrogen peroxide (H2O2), which has been shown in vitro to disrupt endothelium-independent vasodilatory signaling pathways by oxidatively modifying a key cysteinyl thiol involved in normal NO sensing by sGC72 (see Fig. 57-3).
is converted to hydrogen peroxide (H2O2), which has been shown in vitro to disrupt endothelium-independent vasodilatory signaling pathways by oxidatively modifying a key cysteinyl thiol involved in normal NO sensing by sGC72 (see Fig. 57-3).
Intracellular levels of arginase, which converts L-arginine to ornithine, are elevated in sickled erythrocytes and released into plasma upon hemolysis.73 It has been postulated, therefore, that since L-arginine is a substrate for NO synthesis, this may represent an additional mechanism by which bioavailable NO is decreased in SCD. Data examining arginine replacement therapy, PDE5 inhibition with sildenafil, or iNO to treat PH in SCD, although encouraging, are at this time limited to case reports or very small patients series.74
The role of arginase in modulating PH in other primary blood dyscrasias has been evaluated. In paroxysmal nocturnal hemoglobinuria (PNH), an acquired mutation results in the absence of erythrocyte membrane-bound glycosylphosphatidylinositol-anchored proteins, which predisposes to compliment-mediated hemolysis and free Hb scavenging of NO. Patients with PNH express a low bioactive NO state in several ways, including PH, gastrointestinal smooth muscle cell (SMC) hyperactivity, and erectile dysfunction (ED).75 Data extracted from patients enrolled in the Transfusion Reduction Efficacy and Safety Clinical Investigation Using Eculizumab in Paroxysmal Nocturnal Hemoglobinuria (TRIUMPH) study demonstrated that plasma arginase activity levels were 10-fold higher compared with the normal range, a finding associated with a roughly 30-fold increase in NO consumption. Interestingly, by blocking compliment C5 and thereby decreasing erythrocyte hemolysis, eculizumab treatment resulted in a significant decrease in hemolysis, N-terminal pro-brain natriuretic peptide levels, and patient-reported dyspnea.76
Less well-established mechanisms that may influence NO bioavailability may involve asymmetrical dimethylarginine (ADMA), a NOS inhibitor, which is present at elevated concentrations in plasma of SCD patients.77
Hypercoagulable state
In SCD, stroke, veno-occlusive crises, and PH are believed to occur in part from thrombus formation in the cerebral and pulmonary microcirculations. Decreased bioavailable NO (through loss of platelet aggregation antagonism), increased whole blood tissue factor expression and thrombin formation, and decreased levels of the coagulation pathway inhibitors activated protein C (APC) and its cofactor protein S contribute to a hypercoagulable state. In SCD, β-thalassemia, and/or asplenic patients, erythrocyte membrane dyssymmetry results in abundant exposure of phosphatidyl choline, which is linked to increased erythrocyte vascular endothelial cell (ED) adhesion and thrombin activation.78 Antiplatelet therapy with aspirin is recommended in selected high-risk SCD patients for primary prevention of ischemic stroke; however, the effect of these agents in modulating a decrease in PAP has not been adequately tested.
Despite these observations, the attributable contribution to the pathophysiology of PH by free Hb scavenging of NO remains unresolved. A surprisingly low difference in reticulocyte count and lactate dehydrogenase (LDH) levels between non-PH and PH patients with SCD; lack of a positive association between extent of hemolysis and PH disease severity; and an absence of extrahematological clinical signs or symptoms indicative of decreased bioactive NO, such as esophageal spasm or ED (i.e., impotence but not priapism), has led some to suggest that alternative mechanisms may play an underrecognized role in promoting PH in SCD.52
Increased endothelin-1 synthesis
A threefold increase in circulating ET-1 levels are observed in patients with SCD compared to age-matched normal controls.79 This presumably occurs as a consequence of microvascular hypoxia and increased sheer stress in SCD that triggers pulmonary vascular endothelial secretion of ET-1. In vitro, exposure of vascular ECs to sickled erythrocytes significantly increases ET-1 gene transcription, suggesting that ET-1 may act as a vasoconstrictor intermediary in SCD.80 Because of the central role of ET-1 in the pathophysiology of PAH, investigators have tested the hypothesis that ETA/B receptor antagonism may attenuate PH disease severity in SCD. Early data from small clinical trials have demonstrated about a 10% increase in distance performed during a 6-minute walk test in SCD patients with PH treated with bosentan therapy for 6 months. In the same study, a mean fall of 6 mmHg in estimated systolic PAP (by echocardiography) was also observed.81 Larger randomized double-blinded placebo-controlled clinical trials to investigate further bosentan therapy in a similar patient cohort are ongoing (ASSET-1 and -2 at clinicaltrials.gov).82
Other Secondary Causes of Pulmonary Hypertension
Numerous other less common disease states are associated with PH by causing direct compression of the pulmonary vasculature via mass effect or vessel wall infiltration. Fibrosing mediastinitis, which is associated with granulomatous diseases such as histoplasmosis, is an immunologically mediated response to caseous nodes. Fibrotic encroachment of large and small pulmonary arteries and veins has been observed at necroscopy of patients when this condition includes severe PH. A similar PH pathophysiology has been implicated in sarcoidosis, where impedance to pulmonary blood flow occurs secondary to fibrosis of the pulmonary vasculature in the setting of extensive parenchymal lung disease or direct invasion of the intima and media of the pulmonary arteries with noncaseating granulomas, resulting in blood vessel encroachment.
Primary inflammatory vascular diseases including pulmonary capillary hemangiomatosis, characterized by uncontrolled proliferation of capillaries infiltrating vascular, bronchial, and interstitial pulmonary structures, and Takayasu’s arteritis (see Chapter 42) are rare causes of PH. An important iatrogenic etiology of PH is secondary to pulmonary vein stenosis as a complication of pulmonary vein isolation radiofrequency ablation for treatment of atrial fibrillation. In the previous decade, pulmonary vein stenosis was reported in up to 20% of patients undergoing this procedure. However, improved technology and enhanced awareness among operators appears to have resulted in a substantial downward trend in the frequency of this potentially devastating procedural complication, with contemporary case series reporting event rates of 1% to 3%.83
1 McLaughlin V.V., Archer S.L., Badesch D.B., et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573–1619.
2 Farber H.W., Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655–1665.
3 Benza R.L., Miller D.P., Gomberg-Maitland M., et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–172.
4 Proceedings of the 4th World Symposium on Pulmonary Hypertension, February 2008, Dana Point, California, USA. J Am Coll Cardiol. 2009;54(1 Suppl):S1–S117.
5 Simonneau G., Robbins I.M., Beghetti M., et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S43–S54.
6 Leung C.C., Moondra V., Catherwood E., et al. Prevalence and risk factors of pulmonary hypertension in patients with elevated pulmonary venous pressure and preserved ejection fraction. Am J Cardiol. 2010;106(2):284–286.
7 Hoeper M.M., Barbera J.A., Channick R.N., et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S85–S96.
8 Scharf S.M., Iqbal M., Keller C., et al. Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med. 2002;166(3):314–322.
9 Handa T., Nagai S., Miki S., et al. Incidence of pulmonary hypertension and its clinical relevance in patients with sarcoidosis. Chest. 2006;129(5):1246–1252.
10 Baughman R.P., Engel P.J., Taylor L., et al. Survival in sarcoidosis associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest. 2010.
11 Damy T., Goode K.M., Kallvikbacka-Bennett A., et al. Determinants and prognostic value of pulmonary arterial pressure in patients with chronic heart failure. Eur Heart J. 2010;31(18):2280–2290.
12 Maggiorini M., Melot C., Pierre S., et al. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation. 2001;103(16):2078–2083.
13 Lewis G.D., Shah R., Shahzad K., et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116(14):1555–1562.
14 Pengo V., Lensing A.W., Prins M.H., et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350(22):2257–2264.
15 Ha J.W., Chung N., Chang B.C., et al. Acute mitral regurgitation due to leaflet tear after balloon valvotomy. Circulation. 1998;98(19):2095–2097.
16 Zafrir N., Zingerman B., Solodky A., et al. Use of noninvasive tools in primary pulmonary hypertension to assess the correlation of right ventricular function with functional capacity and to predict outcome. Int J Cardiovasc Imaging. 2007;23(2):209–215.
17 Hirakawa S., Suzuki T., Gotoh K., et al. Human pulmonary vascular and venous compliances are reduced before and during left-sided heart failure. J Appl Physiol. 1995;78(1):323–333.
18 Moraes D.L., Colucci W.S., Givertz M.M. Secondary pulmonary hypertension in chronic heart failure: the role of the endothelium in pathophysiology and management. Circulation. 2000;102(14):1718–1723.
19 Furutani H., Zhang X.F., Iwamuro Y., et al. Ca2 + entry channels involved in contractions of rat aorta induced by endothelin-1, noradrenaline, and vasopressin. J Cardiovasc Pharmacol. 2002;40(2):265–276.
20 Kaddoura S., Firth J.D., Boheler K.R., et al. Endothelin-1 is involved in norepinephrine-induced ventricular hypertrophy in vivo. Acute effects of bosentan, an orally active, mixed endothelin ETA and ETB receptor antagonist. Circulation. 1996;93(11):2068–2079.
21 Puwanant S., Park M., Popovic Z.B., et al. Ventricular geometry, strain, and rotational mechanics in pulmonary hypertension. Circulation. 2010;121(2):259–266.
22 Ghio S., Gavazzi A., Campana C., et al. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol. 2001;37(1):183–188.
23 Califf R.M., Adams K.F., McKenna W.J., et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: The Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134(1):44–54.
24 Malouf J.F., Enriquez-Sarano M., Pellikka P.A., et al. Severe pulmonary hypertension in patients with severe aortic valve stenosis: clinical profile and prognostic implications. J Am Coll Cardiol. 2002;40(4):789–795.
25 Bonow R.O., Carabello B.A., Chatterjee K., et al. 2008 focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1998 guidelines for the management of patients with valvular heart disease). Endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52(13):e1–e142.
26 Magne J., Lancellotti P., Pierard L.A. Exercise pulmonary hypertension in asymptomatic degenerative mitral regurgitation. Circulation. 2010;122(1):33–41.
27 Murali S., Kormos R.L., Uretsky B.F., et al. Preoperative pulmonary hemodynamics and early mortality after orthotopic cardiac transplantation: the Pittsburgh experience. Am Heart J. 1993;126(4):896–904.
28 Gorlitzer M., Ankersmit J., Fiegl N., et al. Is the transpulmonary pressure gradient a predictor for mortality after orthotopic cardiac transplantation? Transpl Int. 2005;18(4):390–395.
29 Chen J.M., Levin H.R., Michler R.E., et al. Reevaluating the significance of pulmonary hypertension before cardiac transplantation: determination of optimal thresholds and quantification of the effect of reversibility on perioperative mortality. J Thorac Cardiovasc Surg. 1997;114(4):627–634.
30 Zimpfer D., Zrunek P., Sandner S., et al. Post-transplant survival after lowering fixed pulmonary hypertension using left ventricular assist devices. Eur J Cardiothorac Surg. 2007;31(4):698–702.
31 Hopkins N., McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat. 2002;201(4):335–348.
32 Harris P., Heath D. Influence of respiratory gases. In: Harris P., Heath D. The human pulmonary circulation. ed 2. London: Churchill Livingston; 1986:456–483.
33 Dumas J.P., Bardou M., Goirand F., et al. Hypoxic pulmonary vasoconstriction. Gen Pharmacol. 1999;33(4):289–297.
34 Diebold I., Djordjevic T., Hess J., et al. Rac-1 promotes pulmonary artery smooth muscle cell proliferation by upregulation of plasminogen activator inhibitor-1: role of NFkappaB-dependent hypoxia-inducible factor-1alpha transcription. Thromb Haemost. 2008;100(6):1021–1028.
35 Smith T.G., Talbot N.P., Privat C., et al. Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: two randomized controlled trials. JAMA. 2009;302(13):1444–1450.
36 Hyduk A., Croft J.B., Ayala C., et al. Pulmonary hypertension surveillance–United States, 1980–2002. MMWR Surveill Summ. 2005;54(5):1–28.
37 Weitzenblum E. Chronic cor pulmonale. Heart. 2003;89(2):225–230.
38 Oswald-Mammosser M., Weitzenblum E., Quoix E., et al. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure. Chest. 1995;107(5):1193–1198.
39 Incalzi R.A., Fuso L., De Rosa M., et al. Electrocardiographic signs of chronic cor pulmonale: a negative prognostic finding in chronic obstructive pulmonary disease. Circulation. 1999;99(12):1600–1605.
40 Ryu J.H., Daniels C.E., Hartman T.E., et al. Diagnosis of interstitial lung diseases. Mayo Clin Proc. 2007;82(8):976–986.
41 Patel N.M., Lederer D.J., Borczuk A.C., et al. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007;132(3):998–1006.
42 Tan R.T., Kuzo R., Goodman L.R., et al. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Medical College of Wisconsin Transplant Group. Chest. 1998;113(5):1250–1256.
43 McQuillan B.M., Picard M.H., Leavitt M., et al. Clinical correlates and reference intervals for pulmonary artery systolic pressure among echocardiographically normal subjects. Circulation. 2001;104(23):2797–2802.
44 Devaraj A., Wells A.U., Meister M.G., et al. Detection of pulmonary hypertension with multidetector CT and echocardiography alone and in combination. Radiology. 2010;254(2):609–616.
45 Shahar E., Whitney C.W., Redline S., et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163(1):19–25.
46 Penaloza D., Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation. 2007;115(9):1132–1146.
47 Penaloza D., Sime F. Chronic cor pulmonale due to loss of altitude acclimatization (chronic mountain sickness). Am J Med. 1971;50(6):728–743.
48 Ribeiro A., Lindmarker P., Johnsson H., et al. Pulmonary embolism: one-year follow-up with echocardiography Doppler and five-year survival analysis. Circulation. 1999;99(10):1325–1330.
49 Hoeper M.M., Mayer E., Simonneau G., et al. Chronic thromboembolic pulmonary hypertension. Circulation. 2006;113(16):2011–2020.
50 Klepetko W., Mayer E., Sandoval J., et al. Interventional and surgical modalities of treatment for pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):73S–80S.
51 Jais X., Ioos V., Jardim C., et al. Splenectomy and chronic thromboembolic pulmonary hypertension. Thorax. 2005;60(12):1031–1034.
52 Bunn H.F., Nathan D.G., Dover G.J., et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;116(5):687–692.
53 Eldor A., Rachmilewitz E.A. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36–43.
54 Bonderman D., Jakowitsch J., Adlbrecht C., et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2005;93(3):512–516.
55 Fedullo P.F., Auger W.R., Kerr K.M., et al. Chronic thromboembolic pulmonary hypertension. N Engl J Med. 2001;345(20):1465–1472.
56 Tardivon A.A., Musset D., Maitre S., et al. Role of CT in chronic pulmonary embolism: comparison with pulmonary angiography. J Comput Assist Tomogr. 1993;17(3):345–351.
57 Riedel M., Stanek V., Widimsky J., et al. Long-term follow-up of patients with pulmonary thromboembolism. Late prognosis and evolution of hemodynamic and respiratory data. Chest. 1982;81(2):151–158.
58 Saouti N., de Man F., Westerhof N., et al. Predictors of mortality in inoperable chronic thromboembolic pulmonary hypertension. Respir Med. 2009;103(7):1013–1019.
59 Jais X., D’Armini A.M., Jansa P., et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol. 2008;52(25):2127–2134.
60 Suntharalingam J., Hughes R.J., Goldsmith K., et al. Acute haemodynamic responses to inhaled nitric oxide and intravenous sildenafil in distal chronic thromboembolic pulmonary hypertension (CTEPH). Vasc Pharmacol. 2007;46(6):449–455.
61 Jamieson S.W. Pulmonary thromboendarterectomy. Heart. 1998;79(2):118–120.
62 Thistlethwaite P.A., Madani M., Jamieson S.W. Pulmonary thromboendarterectomy surgery. Cardiol Clin. 2004;22(3):467–478. vii
63 Jamieson S.W., Kapelanski D.P., Sakakibara N., et al. Pulmonary endarterectomy: experience and lessons learned in 1,500 cases. Ann Thorac Surg. 2003;76(5):1457–1462. discussion 1462–1454
64 Skoro-Sajer N., Hack N., Sadushi-Kolici R., et al. Pulmonary vascular reactivity and prognosis in patients with chronic thromboembolic pulmonary hypertension: a pilot study. Circulation. 2009;119(2):298–305.
65 Hamilton-Craig C., Kermeen F. Dunning JJ, et al: Cardiovascular magnetic resonance prior to surgical treatment of chronic thrombo-embolic pulmonary hypertension. Eur Heart J. 2010;31(9):1040.
66 Bonderman D., Skoro-Sajer N., Jakowitsch J., et al. Predictors of outcome in chronic thromboembolic pulmonary hypertension. Circulation. 2007;115(16):2153–2158.
67 Gladwin M.T., Sachdev V., Jison M.L., et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886–895.
68 Haque A.K., Gokhale S., Rampy B.A., et al. Pulmonary hypertension in sickle cell hemoglobinopathy: a clinicopathologic study of 20 cases. Hum Pathol. 2002;33(10):1037–1043.
69 Gibson W.H., Roughton F.J. The kinetics and equilibria of the reactions of nitric oxide with sheep haemoglobin. J Physiol. 1957;136(3):507–524.
70 Aslan M., Freeman B.A. Redox-dependent impairment of vascular function in sickle cell disease. Free Radic Biol Med. 2007;43(11):1469–1483.
71 Hammerman S.I., Klings E.S., Hendra K.P., et al. Endothelial cell nitric oxide production in acute chest syndrome. Am J Physiol. 1999;277(4 Pt 2):H1579–H1592.
72 Maron B.A., Zhang Y.Y., Handy D.E., et al. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009;284(12):7665–7672.
73 Iyamu E.W., Cecil R., Parkin L., et al. Modulation of erythrocyte arginase activity in sickle cell disease patients during hydroxyurea therapy. Br J Haematol. 2005;131(3):389–394.
74 Morris C.R., Morris S.M.Jr. Hagar W, et al: Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168(1):63–69.
75 Brodsky R.A. Advances in the diagnosis and therapy of paroxysmal nocturnal hemoglobinuria. Blood Rev. 2008;22(2):65–74.
76 Hill A., Rother R.P., Wang X., et al. Effect of eculizumab on haemolysis-associated nitric oxide depletion, dyspnoea, and measures of pulmonary hypertension in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2010;149(3):414–425.
77 Kato G.J., Wang Z., Machado R.F., et al. Endogenous nitric oxide synthase inhibitors in sickle cell disease: abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br J Haematol. 2009;145(4):506–513.
78 Manodori A.B., Barabino G.A., Lubin B.H., et al. Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood. 2000;95(4):1293–1300.
79 Werdehoff S.G., Moore R.B., Hoff C.J., et al. Elevated plasma endothelin-1 levels in sickle cell anemia: relationships to oxygen saturation and left ventricular hypertrophy. Am J Hematol. 1998;58(3):195–199.
80 Phelan M., Perrine S.P., Brauer M., et al. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. 1995;96(2):1145–1151.
81 Minniti C.P., Machado R.F., Coles W.A., et al. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br J Haematol. 2009;147(5):737–743.
82 Barst R.J., Mubarak K.K., Machado R.F., et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. Br J Haematol. 2010;149(3):426–435.
83 Holmes D.R.Jr, Monahan K.H., Packer D. Pulmonary vein stenosis complicating ablation for atrial fibrillation: clinical spectrum and interventional considerations. JACC Cardiovasc Interv. 2009;2(4):267–276.