Chapter 63 Congenital Anomalies and Malformations of the Vasculature
Anomalous Venous Connections
An anomalous venous connection abnormally connects a systemic or pulmonary vein to another venous structure or directly to the left or right atrium. The connections arise from failed development of normal embryological venous communications or persistence, lack of regression of normal embryological venous communications, or both.
Anomalous Pulmonary Venous Connections
Anomalous pulmonary venous connection refers to the absence of one or more pulmonary venous connections to the left atrium, without reference to the subsequent drainage of the anomalously disconnected pulmonary vein(s). Total anomalous pulmonary venous connection (TAPVC) refers to the bilateral absence of a connection of both pulmonary veins of each lung to the left atrium. The pulmonary veins connect directly into the right atrium or into one of its tributaries. TAPVC is almost always associated with some type of atrial septal defect (ASD) for life to be sustained beyond the newborn period.1Partial anomalous pulmonary venous connection (PAPVC) is the presence of connection of one or more—but not all—pulmonary veins to the right atrium or one of its tributaries. TAPVC is found in about 2% of autopsied cases with congenital heart disease and has a male predominance. PAPVCs constitute about 0.6% of autopsied cases of congenital heart disease.1 TAPVC is an isolated anomaly in two thirds of patients, and is associated with complex congenital heart disease in the remaining third, especially the heterotaxy syndromes (so-called isomeric TAPVC).
Embryology
In the human embryo, the lung buds and their pulmonary veins connect to the veins of the foregut, the splanchnic plexus. With growth, a new pulmonary venous channel, the primary pulmonary vein, grows as a bulging of the left atrium. At the same time, the intrapulmonary veins lose their connections with the splanchnic plexus and fuse with the primary pulmonary vein. The intrapulmonary veins eventually form the four pulmonary veins and are absorbed into the left atrium (Fig. 63-1). Failure of severance of the connection between the intrapulmonary and splanchnic veins results in anomalous pulmonary venous connections that may be total, partial, bilateral, or unilateral.
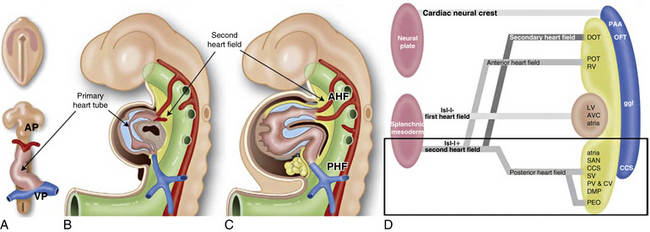
Figure 63-1 Schematic representation of sequential stages of cardiac development and the contribution from first and second heart field.
The primary heart tube is depicted in brown and the myocardium derived from the second heart field (and incorporated later in the heart) in yellow. A, Primary heart tube is formed after fusion of bilateral plates of splanchnic mesoderm in the primitive plate. This tube already has a venous pole (VP), and an arterial pole (AP). B, Lateral view of embryo (≈︀23 days in human), showing primary heart tube surrounded by cardiac jelly (blue), and second heart field situated dorsally to heart. C, Human embryo at 25 days. Primary heart tube has expanded at both VP and AP, with myocardium derived from second heart field (yellow). D, Scheme of nomenclature primary and second heart field. At VP of heart, myocardium is derived from posterior heart field, which contributes to posterior wall of atria, sinoatrial node (SAN), myocardium of sinus venosus (SV), pulmonary veins (PV), and cardinal veins (CV) including the coronary sinus, part of the central conduction system (CCS) and the dorsal mesenchymal protrusion (DMP). The second heart field contribution to VP is discussed in more detail later on in this review.
(Reproduced with permission from Crawford MH, DiMarco JP, Paulus WJ: Cardiology: expert consult, ed 3, St. Louis, 2009, Mosby.)
The drainage site of pulmonary veins may be supradiaphragmatic or infradiaphragmatic (Fig. 63-2). When drainage is supradiaphragmatic, both lungs may drain into a confluence, which may then drain into the left innominate vein and the superior vena cava (SVC), or the supradiaphragmatic drainage may be directly into the left SVC, or indirectly to the SVC via azygos or hemiazygos veins1 (see Fig. 63-2 ).
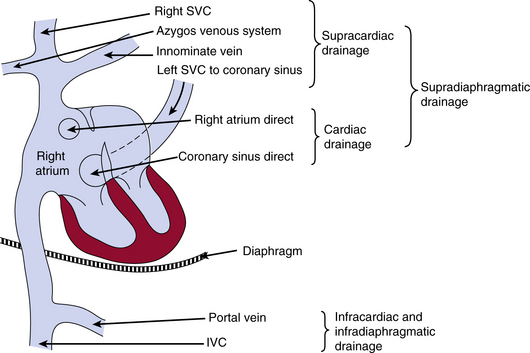
Figure 63-2 Possible sites of drainage for anomalous pulmonary venous connections into the venous system.
IVC, inferior vena cava; SVC, superior vena cava.
(Reproduced with permission from Becker AE, Anderson RH: Pathology of congenital heart disease, London, 1981, Butterworths, pp 47–66, 333.)
Maternal exposure to environmental teratogens such as lead paint or pesticides has been described to cause a familial susceptibility for TAPVC, largely in the presence of a positive family history of cardiac and noncardiac anomalies.2,3 Familial total anomalous pulmonary venous return is most likely an X-linked inheritance and autosomal dominant with variable expression and incomplete penetrance. Until now, two candidate genes have been proposed. The TAPVR1 gene, playing a role in vasculogenesis, maps to 4q12, in which the centromeric regions contain receptor tyrosine kinase genes as a kinase domain receptor (KDR). Recently it has been shown that dysregulation of the PDGFRA (platelet-derived growth factor receptor-α) gene causes PV inflow tract anomalies, including TAPVR.4
Total anomalous pulmonary venous connection
The level of the anomaly relative to the heart or diaphragm classifies TAPVC. Type I denotes anomalous connection at the supracardiac level; type II, anomalous connection at the cardiac level; type III, anomalous connection at the infracardiac level; and type IV, anomalous connection at two or more of those levels.5 The common forms of TAPVC are illustrated in Figure 63-3. In a recent multicenter study6 from the United Kingdom, Ireland, and Sweden with a cohort of 422 liveborn cases, the frequency of supracardiac TAPVC (type I) was 48.6%, infracardiac 26.1%, cardiac level 15.9%, and mixed connections 8.8%. Two cases (0.5%) had common pulmonary vein atresia, and 60 (14.2%) had associated cardiac anomalies. Of the supracardiac TAPVC cases, 73.2% showed connection to the innominate vein, 21% connected to various portions of the SVC, 2.9% connected to the azygos vein, and in 2.9% the connection was unknown (undocumented or unable to determine). The majority of cardiac-type TAPVC cases demonstrated connection to the coronary sinus (86.6%), followed by connections to the right atrium at 11.9%, and no identified connection site in 1.5% of cases. In the case of infracardiac TAPVC, the majority of cases demonstrated pulmonary venous connection to the portal system.6
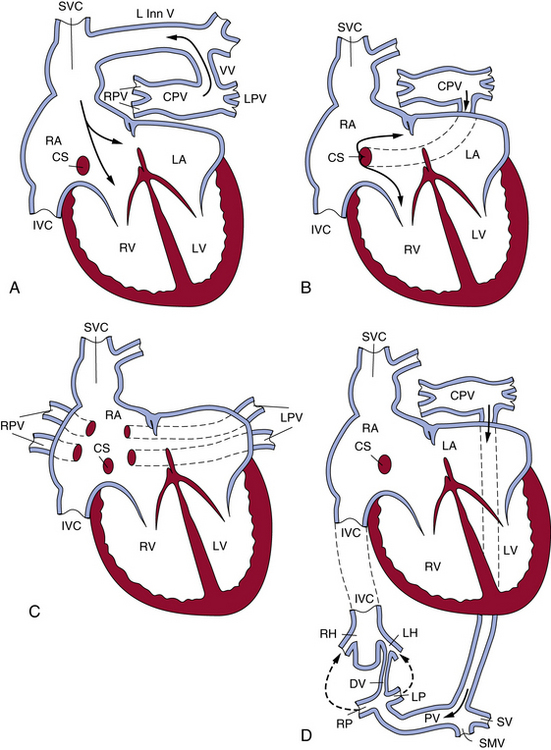
Figure 63-3 The most common patterns of circulation in total anomalous pulmonary connection (TAPVC).
A, TAPVC to left innominate vein by way of a vertical vein. B, TAPVC to coronary sinus. C, TAPVC to right atrium. Right and left pulmonary veins usually enter right atrium separately. D, TAPVC to portal vein. CPV, common pulmonary vein; CS, coronary sinus; DV, ductus venosus; IVC, inferior vena cava; LA, left atrium; LH, left hepatic vein; L Inn V, left innominate vein; LP, left portal vein; LPV, left pulmonary vein; LV, left ventricle; PV, portal vein; RA, right atrium; RH, right hepatic vein; RP, right portal vein; RPV, right pulmonary vein; RV, right ventricle; SMV, superior mesenteric vein; SV, splenic veins; SVC, superior vena cava; VV, vertical vein.
(Reproduced with permission from Lucas RV, Krabill RA: Anomalous venous connections, pulmonary and systemic. In Adams FH, Emmanouilides GC, editors: Moss’ heart disease in infants, children and adolescents, ed 4, Baltimore, 1989, Williams & Wilkins, p 580.)
Size of the ASD has been shown to relate to longevity in TAPVC; the larger the ASD, the longer the survival. Individual pulmonary vein size at diagnosis also is a strong independent predictor of survival in patients with TAPVC.7
Associated cardiac anomalies include transposition of the great arteries, small ventricular septal defects (VSDs),6 pulmonary atresia, coarctation of the aorta (COA), and anomalies of systemic veins. There is a high frequency of TAPVC in patients with congenital heart disease and asplenia (heterotaxy syndromes). In a recent autopsy series of TAPVC associated with asplenia, anomalous pulmonary venous connection to a systemic vein was total in 42 (58%) of 72 and partial in 2 (3%) of 72, with obstruction in 24 (55%) of 44.8
In patients with a widely patent foramen ovale or ASD, which allows free communication between the two atria and mixing of the venous blood, the flow of blood depends on the resistance in the pulmonary and systemic arterial circuits. In cases without pulmonary venous obstruction, the resistance at birth is equal; therefore, the distribution of blood is equal between the pulmonary and systemic circuits. However, within a few weeks of birth, the pulmonary resistance decreases, and a larger proportion of the mixed venous blood returns to the pulmonary circuit, resulting in a nearly three to five times greater pulmonary-to-systemic flow ratio of 3:1 to 5:1, and an equalization of oxygen saturation between the right and left heart. In the presence of pulmonary venous obstruction, there is elevated pulmonary venous pressure that results in pulmonary edema, a decrease in pulmonary flow, pulmonary hypertension (PH), right ventricular (RV) hypertrophy, and ultimately right heart failure.
The signs and symptoms, therefore, depend on the underlying hemodynamics—that is, presence or absence of pulmonary venous obstruction and the extent of mixing of blood between the right and left atrium. If interatrial mixing is inadequate, symptoms occur at birth or shortly thereafter. In TAPVC without pulmonary venous obstruction, patients are usually asymptomatic at birth, but at least 50% become symptomatic within 1 month of life (usually not in the first 12 hours of life). Once symptoms appear, however, they are rapidly progressive. Diagnosis may be established by angiography, echocardiography, or T1-weighted spin-echo magnetic resonance imaging (MRI).9
Partial anomalous pulmonary venous connection
PAPVC is defined when one or more, but not all, pulmonary veins are connected to the right atrium or to a systemic vein. Usually only one lung or part of a lung is involved. The right pulmonary veins are six times more commonly involved than the left pulmonary veins, and the upper lobes of the lung are more commonly involved than the lower. The left-sided pulmonary veins usually connect to the derivatives of the left cardinal system (i.e., coronary sinus and left innominate vein). The right pulmonary veins connect to the derivatives of the right cardinal system (i.e., SVC and inferior vena cava [IVC]) or right atrium. The most common connections are right pulmonary veins to SVC, right pulmonary veins to right atrium, veins of the right lung to the IVC, and left pulmonary veins to the left innominate vein.
The heart usually exhibits mild dilation and hypertrophy of the right atrium and ventricle, with dilation of the pulmonary artery. The left-sided chambers are normal. Usually, ASD accompanies PAPVC; conversely, 9% of cases of ASD have PAPVC. In the case of the right pulmonary vein connecting to the SVC, the right upper and middle lobe veins connect to the SVC. The vein of the right lower lobe usually enters the left atrium but may connect to the right atrium. The lower part of the SVC, below the azygos vein and above the right atrium, is dilated approximately twice the normal size. Most cases have associated ASD of the sinus venosus type, but occasionally a secundum or, rarely, a primum ASD may occur (Fig. 63-4).
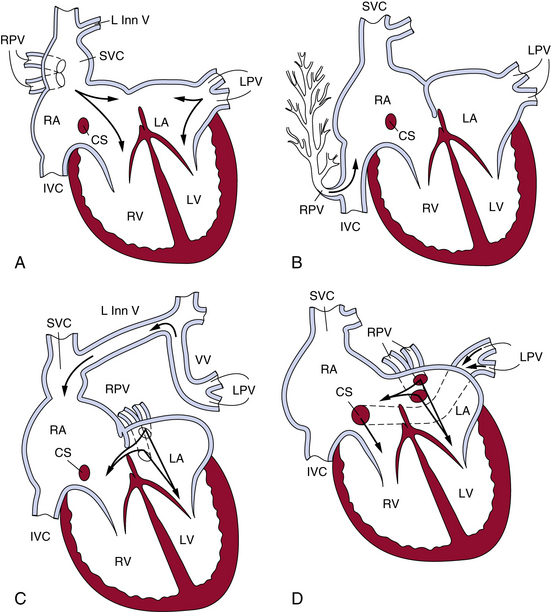
Figure 63-4 The most common patterns of circulation in partial anomalous pulmonary connection (PAPVC).
A, Anomalous connection of right pulmonary veins to superior vena cava. B, Anomalous connections of right pulmonary veins to inferior vena cava in the presence of an intact atrial septum. C, Anomalous connection of left pulmonary veins to left innominate by way of a vertical vein. D, Anomalous connection of left pulmonary veins to coronary sinus. CS, coronary sinus; IVC, inferior vena cava; LA, left atrium; L Inn V, left innominate vein; LPV, left pulmonary vein; LV, left ventricle; RA, right atrium; RPV, right pulmonary vein; RV, right ventricle; SVC, superior vena cava; VV, vertical vein.
(Reproduced with permission from Lucas RV, Krabill RA: Anomalous venous connections, pulmonary and systemic. In Adams FH, Emmanouilides GC, editors: Moss’ heart disease in infants, children and adolescents, ed 4, Baltimore, 1989, Williams & Wilkins, p 580.)
Associated cardiovascular defects are common, occurring in up to 80% of patients.10 Major cardiac anomalies are found in about 20% of cases and include tetralogy of Fallot, VSD, single ventricle, COA, transposition of the great arteries, and hypoplastic left heart syndrome. Indications for surgery for isolated PAPVC include pulmonary-to-systemic flow ratio of over 2.0.10
When all right pulmonary veins drain the right middle and lower lobes and enter the IVC, either above or below the diaphragm, the pattern of the pulmonary veins is altered, giving a “fir tree” appearance (see Fig. 63-4). This malformation is called scimitar syndrome, as initially described by Chassinat in 1836,11,12 and derives its name from the shape of the anomalous vein on chest radiographs.13 Classically, this syndrome is associated with right lung anomalies, dextrocardia of the heart, hypoplasia of the right pulmonary artery, and anomalous connection between aorta and right lung. In the European Congenital Heart Surgeons Association (ECHSA) multicenter study, 65% of patients also presented with secundum-type ASDs, and 16% had a concomitant VSD.14
In connection of left pulmonary veins to the left innominate vein, the veins of the left upper lobe or the whole left lung connect to the left innominate vein via the vertical vein. Atrial septal defect of the secundum type usually is present, and the septum is rarely intact (see Fig. 63-4). Other sites of PAPVC are the left pulmonary veins draining into the coronary sinus, IVC, right SVC, right atrium, or left subclavian vein. In rare cases, veins of both lungs drain anomalously, but a small segment of the pulmonary venous system drains normally. That condition has been termed by Edwards subtotal anomalous pulmonary venous connection.
Cor Triatriatum
Cor triatriatum is a relatively rare cardiac anomaly (0.4% of autopsied cases with congenital heart disease, ratio of males to females 1.5:1).15 In this condition, the pulmonary veins enter an accessory chamber lying posterior to the left atrium and joining the left atrium through a narrow opening. The following broad classification has been suggested by a number of authors16,17:
1. Accessory atrial chamber that receives all pulmonary veins and communicates with the left atrium; no other connections (classic cor triatriatum).
2. Accessory atrial chamber that receives part of pulmonary veins and does not communicate with left atrium.
Physiologically, when blood from the accessory atrial chamber drains into the right atrium, the hemodynamic features are those of TAPVC. A stenotic opening between the accessory atrium and the left atrium, however, results in features of severe pulmonary obstruction. In subtotal cor triatriatum when obstruction affects only one lung, the result is reflex pulmonary arterial obstruction with decreased flow through that portion of the lung. The rest of the unobstructed lung receives increased flow, but there is no elevation in pulmonary arterial pressure (PAP).17
The classic cor triatriatum patients have onset of symptoms in the first few years of life. Patients usually have history of breathlessness and frequent respiratory infections. Right heart failure is usually present along with signs of pulmonary hypertension. Surgery has been advocated as early as the neonatal period,18 and balloon dilation may be successful in older children.19
Some patients remain asymptomatic until adulthood20 or may present with sick sinus syndrome in advanced years.21 In adults, clinical features on presentation can mimic those of mitral stenosis because of the obstructive properties of the membrane22; they include dyspnea with increased pulmonary capillary wedge pressure (PCWP) on exertion.23 As with other anomalies of the pulmonary venous system, associated anomalies may occur, including atrioventricular septal defect.24
Pulmonary vascular changes in cor triatriatum are progressive medial thickening and intimal fibrosis in pulmonary arteries and veins, accompanied by lymphangiectasia. In contrast to persistent left-to-right shunts, no plexiform lesions or more advanced stages of pulmonary vascular disease occur, which may explain the reversibility of PH due to congenital pulmonary venous obstruction.25
Congenital Stenosis of Pulmonary Veins
Congenital pulmonary vein stenosis is often associated with other anomalies, such as anomalous connections, cor triatriatum, and VSDs.26 The isolated form generally comes to clinical attention because of symptoms related to pulmonary hypertension.27 In its most severe form, congenital pulmonary vein stenosis is a progressive disease with rapid PH and rare survival beyond the first year of life.28
There are two types of stenosis of pulmonary veins. One is localized stenosis of the pulmonary veins at the junction of the left atrium, which may involve one or more pulmonary veins; the other is characterized by narrowing of the lumen of the pulmonary vein, either intra- or extrapulmonary in location, and is called hypoplasia of the pulmonary veins. The latter type is occasionally present in patients with pulmonary artery atresia or hypoplastic left heart syndrome. Clinically, patients have a history of respiratory symptoms, which may progress to right heart failure. Because of poor surgical results, balloon angioplasty and stenting currently are being evaluated as noninvasive treatments for pulmonary artery stenosis. In the most severe forms, lung transplantation appears to be an important option.28
Anomalous Systemic Venous Connections
Anomalous systemic venous connections, especially persistent left SVC, are usually asymptomatic and of little functional significance because systemic venous flow to the right atrium is not impaired. However, if they are not recognized, systemic venous anomalies may be accidentally severed during cardiac surgery. A classification based on embryological principles includes anomalies of the cardinal venous system, anomalies of the IVC, and anomalies of the valves of the sinus venosus.16
Anomalies of the cardinal venous system include persistent left SVC; persistent left SVC connecting to the right or left atrium, with and without failure of development of coronary sinus; right SVC connected to the left atrium; and anomalies of the coronary sinus. Anomalies of the IVC include double IVC, left IVC, and many lesser anomalies that are usually of no clinical significance, except that they may present problems to the surgeon.29,30 Infrahepatic interruption of the IVC with drainage via the enlarged azygous vein to the SVC has been found to occur in 2.9% of congenital heart defects.16 Anomalies of the valves of the sinus venosus involve the eustachian and thebesian valves and crista terminalis. Minor abnormal persistence results in larger valves and a Chiari network. Large outgrowth of the valve of the sinus venosus may result in complete or partial subdivision of the right atrium.
As with anomalous pulmonary veins, anomalous systemic venous return is a hallmark of the heterotaxy syndromes, especially asplenia. In a recent autopsy review of 72 cases of asplenia, the SVC was bilateral in 51 cases (71%) and unilateral in 21 (29%). In nine cases of bilateral SVC, one of the SVCs was partly or totally atretic. Although the IVC was never interrupted, a prominent azygos vein was found in six cases (8%). An intact coronary sinus was rare.8
Cor Triatriatum Dexter
Cor triatriatum dexter is an unusual cardiac abnormality with division between the sinus and primitive atrial portions of the right atrium. Symptoms in adults with isolated cor triatriatum dexter include lifelong exertional cyanosis and dyspnea.31
Congenital Coronary Artery Anomalies
The incidence of coronary artery anomalies is between 0.46% and 1.55% in angiographic series and 0.3% in autopsy series. The major anomalies will be discussed briefly.
Anomalous Left Main Coronary Artery
The most common coronary anomaly resulting in clinical symptoms is an aberrant left main coronary artery arising in the right coronary sinus of Valsalva. There is a male/female ratio of 4:1 to 9:1. Sudden death occurs in up to two thirds of individuals with this anomaly, 75% of which occur during exercise.32 Most patients are adolescents or young adults, although death may occur as young as 1 month of age. There are often premonitory symptoms of syncope or chest pain, but stress electrocardiograms (ECGs) and stress echocardiograms are often negative.
Pathologically, there are several variants to this anomaly. The common feature is the presence of the left main ostium within the right sinus. This ostium is typically near the commissure, and in some cases actually lies above the commissure between the right and left sinuses. Often the ostium is somewhat malformed and slitlike, and an ostial ridge is present. The proximal artery lies within the aortic media and traverses between the aorta and pulmonary trunk.
The pathophysiology of sudden death in patients with aberrant left main coronary artery may be related to compression of the left main by the pulmonary trunk and aorta, diastolic compression of the vessel lying within the aortic media, and poor filling during diastole because of ostial ridges or slitlike ostia.
Anomalous Right Coronary Artery
In contrast to anomalous left main coronary artery, anomalous right coronary artery from the left sinus is usually an incidental finding, although up to one third of patients may die suddenly. In the majority of cases (67%), the anomalous vessel courses between the aorta and pulmonary trunk, with the remainder usually coursing posterior to the aorta.32 Almost 50% of sudden deaths are exercise related, and most deaths occur in young and middle-aged adults (<35 years).
Grossly, there are two ostia located in the left sinus of Valsalva. The ostium supplying the right coronary artery may have features similar to anomalous left ostia located in the right sinus. Namely, there may be upward displacement, location near the commissure, and slitlike ostia with ostial ridges. The proximal anomalous right coronary generally also courses between the aorta and pulmonary trunk. The pathophysiology of sudden death is similar to that of anomalous left coronary artery, and like that anomaly, evidence of acute or remote ischemia in the ventricular myocardium is not often found.
Anomalous Left Circumflex Artery
Anomalous left circumflex artery is the most common anomaly of the origin of a coronary artery, accounting for 28% of anomalies identified by cardiac catheterization and with an incidence of 1 in 300.33 This anomaly is considered benign. Awareness of this anomaly is important during cardiac surgery to avoid problems with myocardial protection or during prosthetic valve replacement.
Origin from Pulmonary Trunk
The left main coronary artery arises from the pulmonary trunk in 1/50,000 to 1/300,000 autopsies. Most cases are identified in the first year of life, and sudden death occurs in approximately 40% of cases. In a minority of cases (≈︀20%), sufficient myocardial collaterals develop from the normally structured right coronary artery, enabling potential survival into adulthood. In these cases, a continuous murmur may be present with other symptoms including angina pectoris, myocardial infarction (MI), dyspnea (MI), and syncope. Sudden death usually occurs at rest, but may occur after strenuous activity in older children. Pathologically, the aberrant artery arises in the left pulmonary sinus in 95% of cases. Typically, the artery appears thin-walled and veinlike, and the right coronary artery, although normal in location, is tortuous.
Angiographic and Other Imaging in Diagnosis of Anomalous Coronary Arteries
Angiographically identifying an anomalous right or left main coronary artery from the contralateral coronary sinus depends on the vessel’s course between the aorta and pulmonary trunk. This particular course is angiographically distinct in the right anterior oblique projection when the left main forms a cranial-posterior loop (Fig. 63-5). An alternative method is to perform simultaneous pulmonary and coronary arteriography, or more practically, insert a pulmonary artery catheter as an angiographic marker for the location of the pulmonary vessels, and then perform an angiogram of the aberrant coronary artery in the steep anteroposterior caudal projection.
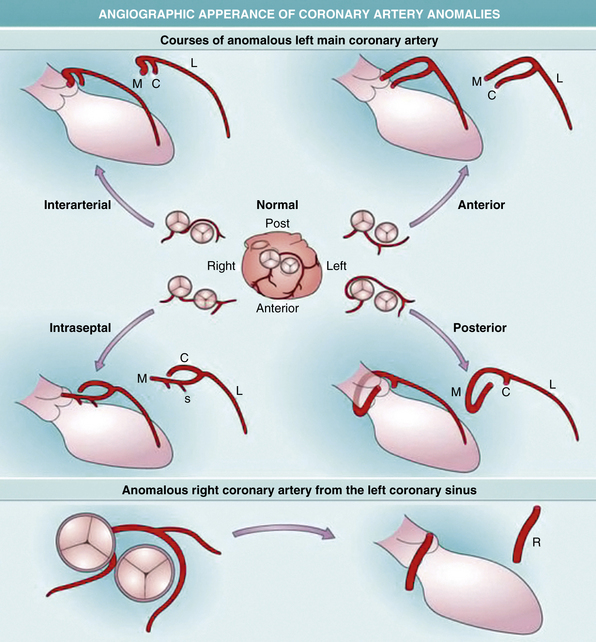
Figure 63-5 Development of coronary arteries.
Top, Movement of the proepicardial organ (PEO) to and over the heart. Bottom, Mesenchymal migration and differentiation. The PEO (blue) is seen as an outgrowth from the dorsal body wall that moves to the looping heart (red). Next, migrating epithelium is seen spreading over the heart. In cross-section, the epithelium is seen as a single cell layer. Epithelial/mesenchymal transition provides cells that migrate into the myocardium. Vasculogenic cell differentiate and link to form plexi that induce other mesenchymal cells to become smooth muscle. These plexi are remodeled into definitive arteries, and the most proximal points of the major coronaries finally link up with the aorta.
(Reproduced with permission from Reese DE, Mikawa T, Bader DM: Development of the coronary vessel system. Circ Res 91:761–768, 2002.)
Standard two-dimensional (2D) echocardiography has been used as a screening procedure for coronary anomalies in athletes. An echocardiographic screening study of 3650 athletes found 3 cases of anomalous right (n = 2) and left (n = 1) coronary artery from the contralateral aortic sinus.34 However, the specificity of transthoracic echocardiography (TTE) is insufficient for this test to serve as an accurate screening tool.35 Alternative noninvasive procedures in the case of inadequate screening echocardiograms include transesophageal echocardiography (TEE) and the recent use of three-dimensional coronary magnetic resonance angiography (3D-CRMA). In one study, 3D-CRMA confirmed the anomalous origin of coronary arteries in 8 out of 15 (53%) pediatric cases, without the use of contrast medium or β-blockers, which could prove advantageous in the evaluation of younger patients.36
Malformations Affecting the Great Vessels
Coarctation of the Aorta
Coarctation of the aorta occurs as a congenital narrowing of the aortic arch, either as a discrete narrowing or one of some length that is usually located adjacent to the junction of the ductus arteriosus.37 The obstruction may be in the form of uniform tubular narrowing of some part of the aortic arch system, usually the isthmus (which lies between the left subclavian artery and the ductus arteriosus) or as a shelflike coarctation within the arch (Box 63-1). The latter is the more common of the two lesions. The narrowing may vary in severity but becomes significant only when there is a pressure gradient across the area of narrowing. That usually occurs when there is greater than 50% cross-sectional area reduction, but there may be lesser narrowing for tubular coarctation.
 Box 63-1 Classification of Coarctation of Aorta
Box 63-1 Classification of Coarctation of Aorta
Discrete Coarctation of Aorta (COA)
Modified from JE Edwards: Congenital malformations of the heart and great vessels. In Gould SE, editor: Pathology of the heart and blood vessels, ed 3, Springfield, Ill., 1968, Thomas, pp 391–454.
Incidence
Isolated COA is the fifth or sixth most common anomaly of all the congenital heart diseases, with estimates of 1 in 3000 to 4000 live births.38–41 In the New England regional study of congenital heart defects, COA accounted for 7.5% of anomalies in infants younger than 1 year of age.42 That may be an underestimation, since COA in newborn infants may not be detected because of similar blood pressure in the upper and lower extremities.43 The male-to-female ratio is 1.74:1.44 In older patients with isolated COA, the incidence is also higher in males. Most cases of COA appear to be sporadic, with no evidence of a mendelian pattern of inheritance.38 However, congenital heart disease has been reported in approximately 4% of the offspring of female COA patients.40 In addition, a recent nonparametric linkage analysis suggests a genetic basis for a subset of COA cases. McBride et al. demonstrated possible susceptibility loci on chromosomes 2p23, 10q21, and 16p12 in a cohort of 289 individuals from 43 separate families.40,45 Coarctation of the aorta is the most common cardiovascular defect found in Turner’s syndrome. Noncardiac abnormalities that have been reported with COA are hypospadias, clubfoot, and ocular defects.44
Pathology
The severity of luminal narrowing in the region of the coarctation at autopsy reveals 42% severe (pinhole) stenosis, 25% atresia, and 33% moderate narrowing in individuals 2 years of age and older.46 In cases of COA containing a contraductal shelf, there is an infolding of the aortic media into the lumen located opposite the ductus arteriosus, marked on the adventitial side by a localized indentation like a waist in the aortic wall.47 The aorta distal to the coarctation shows poststenotic dilation, and the wall of the aorta is thinner, whereas proximally where the pressure is higher, the wall is thicker. The shelf of the coarctation may be pre- or postductal, but most often is juxtaductal.48 Flow patterns in the fetus with various congenital defects determine the development of the aortic isthmus. In a normal fetus, the level of blood flow across the aortic isthmus is lower (25% of the combined total ventricular output) than after birth; therefore, the aortic isthmus at birth is narrower than the descending thoracic aorta.48 In congenital heart disease with reduced pulmonary arterial flow (pulmonary stenosis), the diameter of the isthmus is wider than normal because it carries greater flow in the fetus, and coarctation is virtually unknown. In lesions interfering with left ventricular (LV) outflow (mitral and aortic stenosis), the aortic isthmus is underdeveloped. When localized juxtaductal coarctation is present, aortic obstruction is not present during fetal life, but when the ductus arteriosus begins to close at birth, obstruction will appear after birth.49 The ductus closes from the pulmonary end; obstruction of the aortic end and a gradient across the coarctation may be delayed.
The ductal tissue itself plays an important role in the mechanism of coarctation formation. Normally, ductal tissue, composed mostly of smooth muscle cells (SMCs), extends only partially around the circumference of the aorta. In a patient with left-sided obstruction, there is right-to-left flow through the ductus in utero. This results in the migration of ductal tissue into the adjacent aortic wall, resulting in a circumferential distribution.48 Ho and Anderson, using a serial section technique, have confirmed that ductal tissue completely surrounds the juxtaductal aorta such that “the ductal and descending aorta form a common channel of structural continuity, and the isthmus enters this channel” rather than the ductus entering the isthmus descending aortic region.50,51
Collateral circulation
Olney and Stephens and Bahn et al. have reported that collateral circulation in infants who die with coarctation is poorly developed52,53 (Fig. 63-6). Bahn et al. noted that this depends on the location of the coarctation in relation to the ductus arteriosus; if the blood from the ductus enters the aorta proximal to the coarctation (preductal), then collateral circulation will develop53 (Fig. 63-7). If the ductal blood flow enters below the coarctation (postductal), however, there is little stimulation during fetal life for development of collateral circulation. In that situation, postnatal closure of the ductus results in obstructive hypertension and hypovolemia, which together result in LV failure.
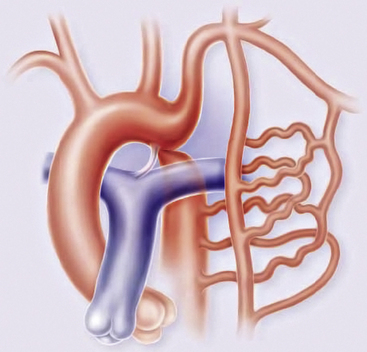
Figure 63-6 Coarctation of the aorta (COA).
Coarctation causes severe obstruction of blood flow in descending thoracic aorta. Descending aorta and its branches are perfused by collateral channels from axillary and internal thoracic arteries through intercostal arteries.
(Reproduced with permission from Brickner ME: Congenital heart disease in adults. N Engl J Med 342:256–263, 2000.)
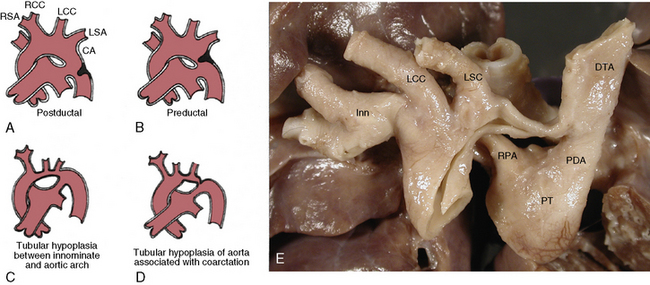
Figure 63-7 Coarctation of aorta (COA).
A, Postductal. B, Preductal with patent ductus arteriosus (PDA). C, Tubular hypoplasia of aortic arch between innominate and subclavian artery with patent ductus arteriosus. D, Tubular hypoplasia (between common carotid and left subclavian) associated with COA (arrow). E, Gross photography of heart and aorta with preductal COA. DTA, descending thoracic aorta; Inn, innominate artery; LCC, left common carotid artery; LSC, left subclavian artery; PT, pulmonary trunk; RPA, right pulmonary artery.
(A-D modified from Moller JH, Amplatz K, Edwards JE: Congenital heart disease, Kalamazoo, Mich., 1974, Upjohn, pp 46–51.)
The collateral circulation in COA between the proximal aorta and the distal aorta usually is present to some extent at birth but develops further as the patient ages. Collateral circulation primarily involves branches of both subclavian arteries, especially the internal mammary, vertebral, costocervical, and thyrocervical trunks, which carry blood to the lower limbs, usually through the third and fourth intercostal arteries and subscapular arteries. The subclavian arterial branches become greatly enlarged and are responsible for the classic signs of coarctation, such as rib notching, which extends from the third to the eighth rib, and parascapular pulsations.37,54,55 Collateral circulation also reaches the lower limbs through the internal mammary to the superior epigastric, which in turn connects with the inferior epigastric and joins the iliac arteries. The anterior spinal artery may provide additional collateral channels through its communication with the vertebral arteries above the coarctation and with intercostals and vertebral arteries below the coarctation.37,54
Development of good collateral circulation and clinical manifestations vary depending on the presence of stenosis of the left subclavian artery, which is an important source of collateral circulation; rib notching is seen only on the right side. If the right subclavian artery arises as a fourth branch and is distal to the coarctation, it is not a source of collateral flow, and rib notching occurs only on the left side.38
Associated conditions
Coarctation of the aorta is commonly associated with bicuspid aortic valve (BAV), but the exact incidence remains speculative (27%-46%).51,56 Among 250 patients with COA studied by Tawes et al.,56 bicuspid valve disease was present in 32 (13%). The most common lesion is stenosis and, unusually, incompetence, which occurs on the basis of bicuspid valve with persistent hypertension.57
There is a high incidence (85%) of major cardiac defects associated with COA in neonates48; and infants aged 1 to 11 months, however, the incidence drops to 52%. After 1 year of age, the incidence drops further to 40% (excluding bicuspid valve), as reported by Kirklin and Barratt-Boyes.57 Infants undergoing operation in the first 3 months of life have a 60% incidence of other congenital cardiac anomalies, compared to 25% of those between 6 and 12 months.57
Patent ductus arteriosus (PDA) is present in virtually all neonates and is considered part of the coarctation complex.57 Ventricular septal defect occurs in 30% to 36% of cases of COA.38,54 Coarctation of the aorta in transposition of the great arteries occurs in 10%, and ASD in 6% to 7%.58 Mitral valve disease, which causes mitral stenosis and regurgitation, is present in fewer than 10%.58
Complications
Aortic rupture usually occurs in the second or third decade and normally involves the ascending aorta, with resultant tamponade. Aortic rupture also may occur distal to the coarctation where the aorta is thin and dilated (poststenotic dilation).57 Some ruptures may be accompanied by aortic dissection.57 Women with coarctation are at high risk of aortic dissections during pregnancy. Stroke, as reported by Liberthson et al., usually occurs in older patients: older than age 40, 21%; age 11 to 39, 8%; and younger than age 11, less than 1%.58 The main cause is usually rupture of a congenital berry aneurysm in the circle of Willis and is secondary to the presence of hypertension.59
Aortic Arch Anomalies
Malformations of the aortic arch are a complex group of lesions first described in the 18th century. Classification of the aortic arch malformations is presented in Box 63-2. Vascular ring is the broad term used to describe an aortic arch malformation in which the trachea and esophagus are compressed. The first clinical description of tracheal and esophageal compression by a double aortic arch is credited to Wolman in 1939.60
Incidence
The true incidence of aortic arch malformations and vascular rings is difficult to determine because so many lesions are asymptomatic. Congenital heart defects involving the outflow tract, aortic arch, ductus arteriosus, and pulmonary arteries account for 20% to 30% of all congenital heart defects.61 The incidence of vascular rings is thought to be approximately 1% of congenital heart disease. Conotruncal malformations, including interrupted aortic arch and isolated anomalies of the aortic arch, are included in the constellation of findings in patients with the 22q11.2 deletion syndrome (del 22q11.2).62
In 1948, Edwards proposed the hypothetical double aortic arch model to conceptualize all the known and possible aortic arch malformations.63 Today there is a better understanding of the embryology, and it has been shown that the final arrangement and morphology of the great arteries requires reciprocal signaling between endothelial cells (ECs) lining the pharyngeal aortic arteries and their surrounding SMCs and mesenchyme, derived from neural crest cells (NCC)64 (Fig. 63-8). Septation of the truncus into the aorta and pulmonary artery is accompanied by migration of the cardiac NCC into the pharyngeal arches and the heart, and this occurs in the mouse embryo at 9 (E9.0) days onwards. The NCC also contribute to the bilateral symmetrical aortic arches that arise from the aortic sac and undergo extensive remodeling, resulting in formation of the aorta, ductus arteriosus, and proximal subclavian, carotid, and pulmonary arteries. The NCC are remodeled to give rise to segments of the mature aortic arch, which is present from E12.5 days onwards.61 The left and right third arch arteries form the common carotid arteries (CCAs), the right fourth arch artery forms the proximal portion of the right subclavian artery, the left sixth arch artery forms the ductus arteriosus, and the right sixth arch artery regresses.65 Recent work with transgenic mice has shown which factors may be important for normal development of the arch vessels. For example, disruption of the forkhead transcription factor Mfh1 causes hypoplasia of the fourth aortic arch artery in mice, resulting in absence of the transverse aortic arch, resembling the interruption of the aortic arch in man.66
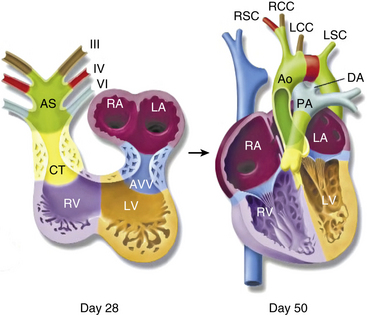
Figure 63-8 Schematic of development of arch vessels and outflow tract of the heart.
Neural crest cells populate the bilaterally symmetrical aortic arch arteries (III, IV, and V) and aortic sac (AS) that together contribute to specific segments of the mature aortic arch, also color coded. Mesenchymal cells form the cardiac valve from the conotruncal (CT) and atrioventricular valve (AVV) segments. Corresponding days of human embryonic development are indicated. Ao, aorta; DA, ductus arteriosus; LA, left atrium; LCC, left common carotid; LSC, left subclavian artery; LV, left ventricle; PA, pulmonary artery; RCC, right common carotid artery; RSC, right subclavian artery; RV, right ventricle.
(Reproduced with permission from Srivastava D, Olson EN: A genetic blueprint for cardiac development. Nature 407:221–226, 2000.)
As already noted, aortic arch vascular ring malformations can encircle and compress the trachea, esophagus, or both.67
Double aortic arch
The double aortic arch is the most frequent type of aortic arch malformation to result in a vascular ring. The ascending aorta arises normally, and as it leaves the pericardium it divides into two branches, a left and a right aortic arch that join posteriorly to form the descending aorta. The left arch passes anteriorly and to the left of the trachea in the usual position and then becomes the descending aorta by the ligamentum arteriosum or the ductus arteriosus. The right aortic arch passes to the right and then posterior to the esophagus to join the left-sided descending aorta, thereby completing the vascular ring.67 From each arch arise a carotid and a subclavian artery (Fig. 63-9). The arches are usually not equal in size, the right arch commonly the larger of the two. One arch may be represented by a single atretic segment; in that case, the right arch usually persists.
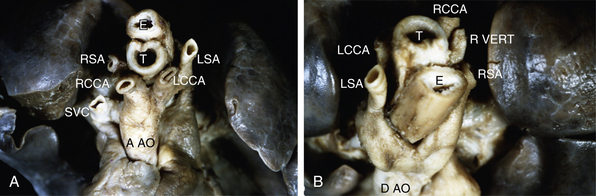
Figure 63-9 A, Double aortic arch, anterior/cranial view. Ascending aorta bifurcates into an anterior left branch, supplying left common carotid artery and left subclavian artery, and a posterior right branch, supplying right common carotid and right subclavian arteries. B, Double aortic arch, posterior/cranial view. Continuation of aorta viewed from behind demonstrates anterior left branch wrapping around trachea and esophagus, as well as right posterior branch emerging from under esophagus. Distal aorta continues as a centrally located structure. Aao, ascending aorta; DAo, descending aorta; E, esophagus; LCCA, left common carotid artery; LSA, left subclavian artery; RCCA, right common carotid artery; RSA, right subclavian artery; Rvert, right vertebral artery; SVC, superior vena cava; T, trachea.
It is theoretically possible, using the double aortic arch model, that the ductus arteriosus could be bilateral or on the right or left side only. No case of functional double arch with bilateral ductus arteriosus has been reported. The descending aorta may be on the right, on the left, or occasionally in the midline.63,67–69 Associated cardiac anomalies in most series are low but were reported to be as high as 45% in the series by Kocis et al. in 1997,70 and these include tetralogy of Fallot and transposition of the great arteries.68
Most patients present at the time of diagnosis with many symptoms, the most common being those arising from the respiratory system. Wheezing is the most common, followed by stridor, pneumonia, upper respiratory tract infection, respiratory distress, cough, and respiratory cyanosis. If such symptoms occur in the newborn or young infant and occur recurrently, it should alert the physician to the possibility of the presence of arch anomalies.71 Surgical repair is the ideal and is usually performed approximately 18 months after the initial diagnosis.
Right aortic arch
A right aortic arch is present when the ascending aorta and arch pass anterior to the right mainstem bronchus. There are two main types of right aortic arch: without a retroesophageal component (mirror-image branching) and with a retroesophageal component. Right aortic arch exists in 0.1% of the population.72
Right Aortic Arch without Retroesophageal Component
In the usual right aortic arch, there is mirror-image branching of the arch vessels compared to the normal left arch. The first vessel is a left innominate artery, the second and third are the right common carotid and right subclavian arteries (Fig. 63-10). The majority of patients have a left ductus arteriosus arising from the left pulmonary artery and inserting into the left subclavian artery. Bilateral patent ductus is associated with congenital cardiac anomalies, usually tetralogy of Fallot or truncus arteriosus.69 This abnormality does not produce symptoms but may be picked up radiographically.
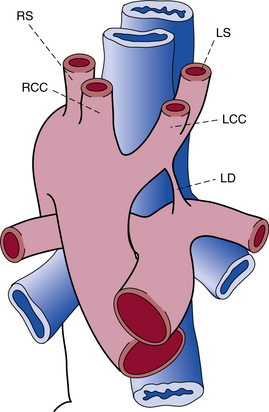
Figure 63-10 Right aortic arch with mirror-image branching and left ductus arteriosus.
LCC, left common carotid artery; LD, left ductus arteriosus; LS, left subclavian artery; RCC, right common carotid artery; RS, right subclavian artery.
(Reproduced with permission from Bankl H: Congenital malformations of the heart and great vessels. Synopsis of pathology, embryology, and natural history, Baltimore, 1977, Urban & Schwarzenberg, pp 159–166.)
Right Aortic Arch with Retroesophageal Component
A vascular ring is usually present in the right aortic arch with retroesophageal component. The right aortic arch extends to the left, behind the esophagus, in the form of a diverticulum. The vascular ring is formed from the right arch and a left-sided ductus arteriosus arising from the left pulmonary artery, with extension into the upper descending thoracic aorta. This group of abnormalities also includes anomalous retroesophageal left subclavian artery. Right aortic arch with mirror-image pattern is associated with congenital heart anomalies, and the most common are tetralogy of Fallot and truncus arteriosus.73
Left aortic arch
A vascular ring is frequently associated with left aortic arch.
Aberrant Right Subclavian Artery
The most common left arch abnormality is aberrant right subclavian artery. The right subclavian artery arises as the fourth branch of the aortic arch, distal to the left subclavian artery. Aberrant right subclavian artery does not result in a vascular ring unless there is a right ductus arteriosus extending from the right pulmonary artery to the right subclavian artery (Fig. 63-11). This anomaly is of importance because of its frequent association with tetralogy of Fallot and the difficulty of using this vessel for the Blalock-Taussig anastomosis.74 Aberrant right subclavian artery is also associated with COA.68
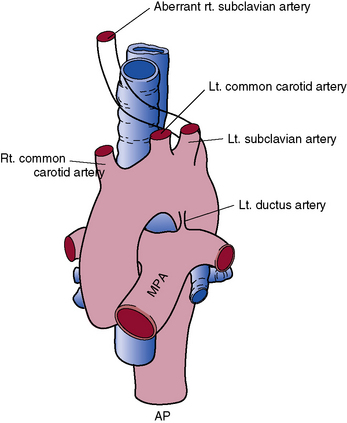
Ductus arteriosus sling
An aberrant ductus arteriosus extending from the right pulmonary artery between the trachea and esophagus to the aorta near the origin of an aberrant right subclavian artery has been reported.75 The patient may have dyspnea and wheezing, which are relieved by surgical division of the vessel.
Cervical aortic arch/interrupted aortic arch
Complete interruption of the aortic arch is different from coarctation of the aortic arch in that there is no continuity of the aorta. Interrupted aortic arch is a rare congenital malformation occurring in 3 per million live births. It is defined as a loss of continuity between the ascending and descending thoracic aorta and has poor prognosis without surgical treatment. Almost all cases have a PDA and associated intracardiac anomalies such as VSD, subaortic stenosis, and truncus arteriosus. The symptoms of complete interruption of the aortic arch are severe right-to-left shunting. In infants, the clinical presentation is severe congestive heart failure (CHF). If untreated, 90% of infants will die at a median age of 4 days. Only rare cases in adults have been reported.76
Clinical presentation and diagnosis
Patients with vascular rings present within the first 6 months of life, and close to 50% have symptoms at birth. Those with associated cardiac defects may present earlier. The signs and symptoms of tracheal and esophageal compression vary with the severity of compression. Infants may present with stridor, wheeze, and recurrent respiratory infections. They often demonstrate poor feeding and dysphagia for solid foods.56 Surgery is indicated when esophageal and tracheal compression symptoms are severe.
Anomalies of the Pulmonary Trunk and Arteries
Isolated pulmonary artery abnormalities are rare and can be divided into (1) those with anomalous arterial supply to one lung in the presence of separate aortic and pulmonary valves (and without inherent interposition of ductal tissue) and (2) those with lungs receiving normally connected pulmonary arteries.
Origin of Right or Left Pulmonary Artery from Ascending Aorta
When the right pulmonary artery arises from the aorta, it usually arises from the right or posterior aspect of the ascending aorta.77 The origin is usually within 1 to 3 cm of the aortic valve, and the right pulmonary artery is larger than the left.77 The pulmonary vascular bed of both lungs may be similar, especially in patients dying within the first 6 months of life.78 In older patients, however, hypertensive vascular disease is usually present and similar in extent in both lungs.77 This lesion is seen as an isolated defect in 20% of cases, but most often it is associated with PDA (50%).79,80 Occasionally, the lesion coexists with left pulmonary vein stenosis.77 The pulmonary stenosis may be tubular or membraneous.77
The origin of the left pulmonary artery from the ascending aorta is more rare than the right pulmonary artery from the aorta, and is usually seen in association with right aortic arch.77 This anomaly is isolated in 40% of cases, and the most commonly associated lesion is tetralogy of Fallot. Part of one lung may receive anomalous vascular supply, called sequestration of the lung. A distinct form of sequestration, scimitar syndrome (see earlier under “Partial Anomalous Pulmonary Venous Connection”), involves abnormal arterial supply as well as venous drainage into the IVC.81 The more severe type of sequestration occurs when one lung is supplied completely from a systemic source.
Pulmonary arteries may arise from the pulmonary trunk, but the left artery connects to the right lung and vice versa.81 This anomaly has been described in truncus arteriosus, as well as when the pulmonary trunk is normally connected to the right ventricle.81
Aortopulmonary Communication
Aortopulmonary communication or window is a distinct anatomical lesion with communication between the ascending aorta and the pulmonary trunk, and the presence of separate aortic and pulmonary valves.82 The defect is a true window, with no length to the communication between the aorta and pulmonary trunk. Usually the defect is single, large, and oval; infrequently (<10% of cases) the defect is small.83 The communication is usually situated in the left lateral wall of the ascending aorta (close to the origin of the left coronary artery) in communication with the right wall of the pulmonary trunk (inferior to the origin of the right pulmonary artery).83,84 It is not surprising, therefore, that the right or, more rarely, the left coronary arteries may be seen arising from the pulmonary trunk. Rarely, the communication may be more downstream.83–85 Because the defect is often large, it is not surprising that pulmonary vascular disease may develop early, similar to that seen in VSDs and patent ductus arteriosus.82,83 Aortopulmonary window is associated with other cardiovascular malformations, such as VSDs, tetralogy of Fallot, subaortic stenosis, and infrequently, right aortic arch, ASD, or patent ductus arteriosus.83–86
Vascular Anomalies
Before the advent of the system proposed by Mulliken and Glowacki in 1982, proper classification of vascular anomalies was replete with inaccurate and misleading terminology. In 1996, building on the Mulliken and Glowacki system, the International Society for the Study of Vascular Anomalies (ISSVA) established a more appropriate classification system, dividing benign vascular anomalies into two major categories: (1) vascular tumors and (2) vascular malformations (VM)87 (also see Chapter 64). Although there are several types of vascular tumors, hemangiomas represent the overwhelming number of vascular tumors encountered in pediatrics, are considered benign vascular neoplasms characterized by endothelial proliferation (initially rapidly progressing), and are not considered to cause significant arteriovenous shunting. Vascular malformations, on the other hand, are considered slow-growing congenital anomalies associated with arteriovenous shunting, and histologically are characterized by a proliferation of heterogeneous and often dysplastic vascular elements, including arteries, dysplastic arteries, veins, and arterialized veins. Vascular malformations are further subdivided according to the Modified Hamburg Classification, including a (1) primary classification based on the predominant vascular defect, and (2) an embryological subclassification based on anatomy and the stage of developmental arrest.88–91 Vascular malformations can be further divided into groups based on vessel type and flow characteristics. Capillary, venous, and lymphatic malformations are slow-flow lesions, and arteriovenous malformations (AVMs) and fistulas (AVFs) are fast-flow lesions; further combined lesions may also occur. Either proliferation (i.e., VMs, hemangiomas) may be associated with syndromes, especially when multiple.
Vascular Malformations
Slow-flow malformations
Capillary Malformations
Clinically, the most well known of the capillary malformations are known as fading capillary stains, port-wine stains, or venocapillary malformations, which represent some of the most common vascular malformations of the skin.87 As a group, capillary malformations are a generic term to describe a heterogenous group of vascular stains of the skin, also including lymphaticovenous malformations and the malformations observed in Klippel-Trénaunay’s and Parkes-Weber’s syndromes. In the case of the facial port-wine stain or the facial nevus flammeus, histological sections may show only rare dilated capillary-like vessels in young children, or collections of haphazardly arranged dilated vessels in the papillary and reticular dermis in older patients92 (Fig. 63-12).
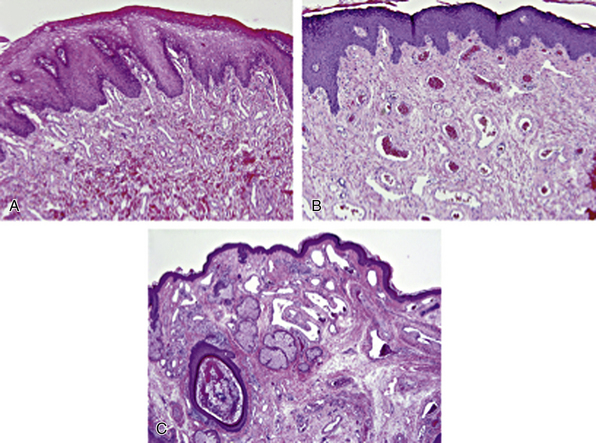
Figure 63-12 Capillary malformations (CM).
A, Facial CM with lip hypertrophy in a 4-year-old boy, showing an excessive number of thin-walled venule-like channels with narrow lumens (hematoxylin-eosin [H&E] stain × 100). B, Facial CM with lip hypertrophy in a 17-year-old boy has an increased number of enlarged vein-like channels with both thin and thick, mostly fibrous walls. Intervascular fibrous tissue is increased (H&E stain × 100). C, Facial CM with thickening and nodules in a 35-year-old man with Sturge-Weber’s syndrome. Nasal skin shows a nodular cluster of large, abnormal veinlike channels. Fibrosis and follicular dilation and keratin plugging are present (H&E stain × 40).
(Reproduced with permission from Gupta A, Kozakewich H: Histopathology of vascular anomalies. Clin Plast Surg 38:31–44, 2011.)
Telangiectasias
According to the updated ISSVA classification, telangiectasias are considered a slow-flow type of capillary malformation manifesting as localized dilations of capillaries and venules that are presumed congenital and may form part of an inherited syndrome. Unlike hemangiomas, there is no proliferation of vessels, but rather an idiopathic dilation of preexisting capillaries and venules. However, the term hemangioma has often been used, especially clinically, for these lesions. Most forms of telangiectasias are present in the skin, but internal organs including the brain can be affected. There are generally few or no symptoms attributable to the telangiectasia itself, other than cosmetic problems when it involves the skin, or hemorrhagic complications of gastrointestinal hemangiomas. Incidental telangiectasias of the brain are found predominantly in the pons, and have only rarely been reported to cause symptoms by bleeding.
Cutaneous telangiectasias
The most common congenital cutaneous telangiectasia is the nevus flammeus, or ordinary birthmark. Nevus flammeus appears as mottled macular lesion on the head and neck, and usually regresses. The nevus vinosus, or port-wine stain, is a specialized form of nevus flammeus that demonstrates no tendency to fade and often becomes elevated, reminiscent of a true hemangioma. Unlike true hemangiomas, telangiectasias appear histologically as congested normal vessels that are separated by intervening tissue. Indeed, in most cases there is no detectable histopathological abnormality after processing the tissue for examination.
Telangiectasia syndromes
Ataxia telangiectasia is an autosomal dominant inherited disease that is characterized by cerebellar degeneration, immunodeficiency, oculocutaneous telangiectasia, cancer risk, and radiosensitivity. The vascular manifestations are heralded by appearance in childhood of telangiectasias of the bulbar conjunctivae and skin of the face and extremities.93 Ocular telangiectasias often do not appear until several years after the ataxia. The patients generally succumb to an underlying immunological abnormality that results in recurrent infections and the development of lymphoproliferative disorders. The most common type of malignancy is lymphoma, usually of the B-cell type. Leukemias also occur.94 Histological examination of the telangiectasias demonstrates the presence of dilated subpapillary venous channels.
Currently, the genetic basis is believed related to mutations in the ataxia-telangiectasia gene (ATM). Since the cloning of ATM in 1995,95 more that 100 ATM mutations occurring in ataxia-telangiectasia patients have been documented. The mutations are broadly distributed throughout the ATM gene.96 The product of the ATM gene is a 350-kD protein. ATM is involved in DNA damage recognition and cell cycle control in response to ionizing radiation damage. There is evidence that ATM may also have a more general signaling role.97 Affected homozygotes are at increased risk for a wide range of malignancies; however, the elevated risk of malignancy is not observed among A-T heterozygotes, with the possible exception of breast carcinoma.98
Generalized essential telangiectasia is another localized form of telangiectasia associated with recurrent cutaneous telangiectasis.99 Another form of telangiectasia that can result in gastrointestinal bleeding is watermelon stomach, or gastric antral vascular ectasia. Watermelon stomach has been increasingly recognized as an important cause of occult gastrointestinal blood loss and anemia. The histological hallmark of this entity is superficial capillary ectasia of gastric antral mucosa and microvascular thrombosis in the lamina propria. Endoscopic findings of the longitudinal antral folds containing visible columns of tortuous red ectatic vessels (watermelon stripes) are pathognomonic.100 Watermelon stomach is usually isolated, but has been reported in patients with autoimmune diseases, especially scleroderma.101–103 The precise nature of the vascular defects is unclear, but underlying fibromuscular dysplasia (FMD) of the gastric arteries has been suggested as a cause for the superficial lesions.100
Venous Malformations
Unlike AVMs, vascular malformations are slow-flow lesions composed entirely of veins, which do not show significant enhancement by radiographic procedures.104 The imaging characteristics, especially those of magnetic resonance (MR), are otherwise similar to those of hemangiomas; hence the past use of the terms cavernous hemangioma, venous hemangioma, and cavernous angioma still present in textbooks.105 Typically, there is a focus of attachment to muscle in those occurring in deep sites. Venous hemangiomas have been reported in a variety of sites, including the mediastinum, mesentery, skeletal muscle, and retroperitoneum.106–111
Venous malformations histologically are composed of nodules of irregular venous-type vascular channels that may vary in size. This lobular or grouped arrangement of vessels is helpful for distinguishing these benign from malignant vascular proliferations. Mast cells and factor XIII–positive interstitial cells are a consistent feature. The spaces are lined by a mitotically inactive flat endothelium highlighted by CD31, surrounded by smooth muscle that appears attenuated or absent relative to the size of the vessel.105
The blue rubber bleb nevus syndrome (BRBNS) is a developmental disorder originally identified by the presence of distinctive cutaneous blue nevi, especially of the tongue, lips, neck, and gastrointestinal system, with a predilection for the small bowel.112 The cutaneous blue nevi are in fact nodular venous malformations involving the dermis and subcutaneous tissue.105 More recently, central nervous system (CNS) vascular malformations, including venous anomalies, have been associated with BRBNS.113,114 Colonoscopy with laser photocoagulation is a nonoperative method of controlling bleeding from colonic hemangiomas, although surgical resection may be necessary to control chronic gastrointestinal blood loss.115 Virtually all internal organs, including the orbit,116 have been reported as involved in BRBNS, as well as multiple cutaneous sites. There is often a diffuse, mild, consumptive coagulopathy.117
BRBNS can be considered a particular manifestation of venous malformation that has been shown to be either sporadic or familial. In the latter, it is inherited in an autosomal dominant fashion. A genetic basis for BRBNS remains inconclusive. Analyses from differing groups have identified a locus chromosome, 9p, responsible for venous malformation in a single large kindred,118 an activating mutation in the gene that encodes for the kinase domain of the EC receptor TIE2 in another familial genetic analysis,119,120 and sporadic somatic mutations in TIE2.121
Fast-flow vascular malformations
Arteriovenous Malformations
Arteriovenous malformations are rare fast-flow lesions consisting of abnormal vascular communications between arteries and veins that occur without an intervening capillary bed and that result in the formation of a mass. These malformations are also referred to as arteriovenous fistulas, arteriovenous hemangiomas, arteriovenous aneurysms, and racemose or cirsoid aneurysms.
Arteriovenous Malformations of the Central Nervous system, Head, and Neck
Although AVMs are described in almost every organ of the body, approximately 50% are located in the head and neck region, including the CNS.122–129 Congenital AVMs are often multiple and have a female predominance. Occasionally, there may be familial AVMs in the absence of a defined syndrome.130 Arteriovenous malformations of the CNS may result in seizures or subarachnoid hemorrhage (SAH) and are treated with a combination of surgery, radiosurgery, embolization, and radiation.130,131 When the malformation results in vein of Galen steal, treatment consists of coil embolization.132 There can be an associated venous malformation within the brain.133 Presenting symptoms of oral and maxillary AVMs vary, including soft-tissue swelling, pain, changes in skin and mucosal color, erythematous and bleeding gingiva, bruit, and paresthesias. The radiological appearance is not pathognomonic. Arteriovenous malformations of the cranial bones can cause bleeding after dental surgery and are also treated with embolization.134 Some are thought to be hamartomas134 and can be associated with other vascular anomalies, such as persistent trigeminal artery.135 Treatment of osseous AVMs of the head and neck include direct transosseous injection of cyanoacrylate.
Arteriovenous Malformations of the Lung
Pulmonary AVMs were first described at autopsy in the 19th century, but the first clinical diagnosis based on the triad of cyanosis, clubbing, and polycythemia was made in 1939.136 Pulmonary AVM causes a shunt of venous blood from the pulmonary arteries to the pulmonary veins, thus decreasing arterial oxygen saturation. Although most patients are asymptomatic, pulmonary AVMs can cause dyspnea from right-to-left shunts. Because of paradoxical emboli, various CNS complications have been described, including stroke and brain abscesses. There is a strong association between pulmonary AVMs and hereditary hemorrhagic telangiectasia (HHT). Up to 36% of patients with solitary pulmonary AVM and 60% of patients with multiple pulmonary AVMs have associated HHT.137,138
Chest radiography and contrast-enhanced computed tomography (CT) are essential initial diagnostic tools, but pulmonary angiography is the gold standard to establish the presence of shunting. Contrast echocardiography is useful for diagnosis and monitoring after treatment. Most patients should be treated. Therapeutic options include angiographic embolization with metal coil or balloon occlusion and surgical excision.139
Occasionally a pulmonary AVM receives blood from a systemic artery in addition to a pulmonary artery.140 Pulmonary AVMs may be single or multiple, small or large enough to involve an entire lung. These can be associated with cerebral AVMs in patients with HHT.141 They may present during pregnancy with hemoptysis, often heralding HHT, and treatment with embolization is indicated if the patient is symptomatic.133
Cyanosis is present since childhood and occasionally since birth. Neurological symptoms may be due to brain abscesses resulting from a loss of the filtering function of the lung.142 A decrease in arterial oxygen saturation occurs with pulmonary AVM, and pulmonary angiography demonstrates the malformation with early filling of the left atrium.
The frequency of fatal complications is significant in pulmonary AVM.137 These include rupture, hemorrhage, endocarditis, and brain abscess. Surgery is indicated with segmental resection whenever possible to preserve the maximum amount of lung tissue, but lobectomy may be necessary.142
Arteriovenous Malformations of Other Sites
Other locations for AVM include the gastrointestinal tract, heart, liver, and kidney. Those of the gastrointestinal tract typically present with bleeding or mucosal ulceration.143 They may occur anywhere in the gastrointestinal tract, and endoscopically are raised lesions that may show nonspecific histological alterations.144 Those of the kidney and pelvis may present with a variety of symptoms and are initially treated with embolization.145 Multiplicity and early age at onset are signs that the lesion may be part of a syndrome, especially Klippel-Trénaunay’s syndrome or HHT.
The physical findings in AVM are closely related to the location and size of the lesion. Patients may present with swelling, pain, or hemorrhage. If located near the skin, the lesion is often a pulsatile mass with a thrill or bruit. There may be erythema or cyanosis distal to the lesion. Cutaneous or mucosal AVMs may ulcerate and bleed, or there may be thrombosis or cellulitis or both superficially. Hypertrophy of an extremity can occur if a large shunt is present. Cardiomegaly in infancy is occasionally due to an unsuspected AVM, and large aneurysms, particularly in the brain or liver, may cause neonatal heart failure. Central nervous system AVMs may present as repeated episodes of intracranial hemorrhage or as seizure disorders secondary to gliosis or atrophy of the adjacent cortex.
Angiographically, the lesions have multiple anomalous arterial branches and anastomoses with early filling of the venous system. Vessels are dilated, elongated, and tortuous. Magnetic resonance imaging is capable of differentiating AVMs from other types of hemangiomas, both in the CNS and the skin and subcutaneous tissue.122 Arteriovenous malformations may be present at birth, become apparent soon after birth, or remain asymptomatic until adulthood.125 Unlike capillary hemangiomas in infants, AVMs do not regress but grow with the growth of the child.
A specialized form of AVM that occurs in the gastric mucosa is termed Dieulafoy’s disease and is prone to massive upper gastrointestinal hemorrhage. Indications for the treatment of AVMs include congestive heart failure, cosmetic deformity, hemorrhage, or ulceration. Coil embolization of hepatic AVMs may improve heart failure, although there is risk of hepatic damage.104 Other complications of embolization therapy include stroke,128 skin slough, and blindness.123 Treatment of AVMs is tailored to the particular location and size of the lesion. A variety of therapeutic modalities have been used to treat AVMs, including radiation, surgery, and embolization techniques. Surgical treatment of AVMs involves resection. Isolated ligation of the vessel is not curative; collateral circulation can reestablish flow to the lesion.123,128
Arteriovenous malformations can recur if not completely excised.127 The morbidity of surgery relates to hemorrhage, disseminated intravascular coagulation (DIC), and cosmetic deformity.127 A variety of materials have been used for embolization, including autologous materials such as fat or muscle, hemostatic agents such as gelatin sponges or polyvinyl alcohol, and methyl methacrylate or silicon spheres. More recently, coils and balloons have been used. The amount of shunting and the diameter of shunts determine the size of the embolization particles.
Pathology
The pathological diagnosis of AVM rests on the presence of communicating arteries and veins in a vascular lesion. In the larger AVM, the gross appearance of the specimen is one of multiple blood-filled spaces, often aneurysmally dilated (Fig. 63-13). A smaller AVM removed for pathological examination may require extensive sectioning to demonstrate the lesion, and ultimately the diagnosis may rest on radiological correlation. Microscopically, AVM is marked by the presence of arteries and veins with little intervening tissue. The direct communication between an artery and a vein may be difficult to locate. Vessels are often elongated and dilated, and the structure of the vessel walls in AVM is usually abnormal. Artery walls are thinned or may be hypertrophied, with disruption and loss of elastic lamina and medial smooth muscle (Fig. 63-14). The medial smooth muscle may also form nodules projecting into the vessel lumen. Vein walls become thickened or arterialized, with the acquisition of internal elastic lamina. Rarely, the malformation may be composed entirely of veins, representing the so-called venous hemangioma. Elastic stains can be helpful, but it is not always possible in AVM to classify a vessel as an artery or a vein.
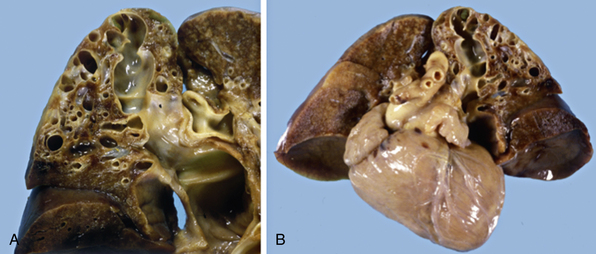
Figure 63-13 Pulmonary arteriovenous malformations (AVM).
A, Gross image of a large pulmonary AVM demonstrating aneurysmally dilated spaces. B, Pulmonary AVM in left upper lobe of lung from a 4-day-old male infant born with low Apgar scores and cyanosis. The infant died in congestive heart failure (CHF).
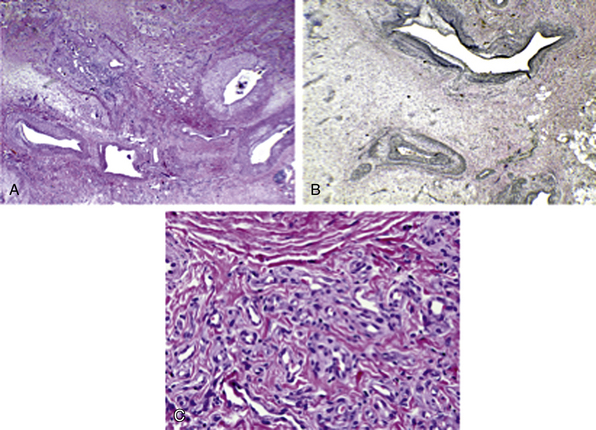
Figure 63-14 Arteriovenous malformations (AVM).
A, AVM shows malformed arteries and veins (hematoxylin-eosin [H&E] stain × 20). B, AVM with Verhoeff-van Gieson (VVG) stain highlights the disrupted internal elastic lamina in arteries, with transition to indeterminate type elastic pattern (top vessel, VVG special stain ×4). C, Small vessel (proliferative component) in AVM has foci of small channels with plump endothelium and pericytes (H&E stain × 400).
(Reproduced with permission from Gupta A, Kozakewich H: Histopathology of vascular anomalies. Clin Plast Surg 38:31–44, 2011.)
Combined Vascular Malformations: Capillary-Venous-Lymphatic Malformations
This category encompasses lesions with a complex combination of vessel types, thus the designation of capillary-lymphatic-venous malformations (CLVMs).87 CLVMs are the most common vascular malformation associated with Klippel-Trénaunay’s syndrome (also see Chapter 64).
Cardiac hemangiomas/arteriovenous malformations
Vascular malformations within the heart include coronary-cameral fistula and cardiac hemangiomas. Hemangiomas involving the myocardium are a diverse group of lesions that represent either hamartomatous malformations or, less likely, benign neoplasms, despite the designation of a “hemangioma.” The histological classification includes those composed of multiple dilated thin-walled vessels (cavernous type), smaller vessels resembling capillaries (capillary type), and dysplastic malformed arteries and veins (arteriovenous hemangioma, cirsoid aneurysm). Cardiac hemangiomas often have combined features of cavernous, capillary, and arteriovenous hemangiomas, and many contain fibrous tissue and fat. These features are reminiscent of intramuscular hemangiomas of skeletal muscle.
An intramuscular cardiac hemangioma has a superficial resemblance to an AVM, with the presence of heterogeneous vessel types, including muscularized arteries, veins, and capillaries. In contrast to capillary hemangiomas, they are infiltrative lesions and occur within the myocardium. They are histologically identical to intramuscular hemangiomas within skeletal muscle and may possess, in addition to the vessels, fat and fibrous tissue. Because of the latter features, some intramuscular cardiac hemangiomas are misclassified as lipomas or fibrolipomas.
Most cardiac hemangiomas are discovered incidentally, but patients may present with dyspnea on exertion, arrhythmias, right-sided heart failure, pericarditis, pericardial effusion, and failure to thrive.146 Patients may have associated vascular syndromes (e.g., Kasabach-Merritt; see Chapter 64).
Whereas chest roentgenograms are abnormal in the majority of cases, the diagnosis of a cardiac tumor is rarely made on the basis of plain radiographs alone. A characteristic tumor blush on coronary arteriography suggests the diagnosis of a cardiac hemangioma.147 Echocardiography usually directs the diagnosis toward a cardiac mass. An enhanced-contrast CT scan or MRI can establish the diagnosis of hypervascularized cardiac tumor.
The most frequent locations are the lateral wall of the left ventricle (21%), the anterior wall of the right ventricle (21%), the interventricular septum (17%), and occasionally the RV outflow tract.148
Cardiac hemangiomas are often large, and gross appearance depends on the size of the vascular spaces in the tumor. The capillary type is frequently slightly raised from the endocardial surface and appears red to purple. Intramuscular types will appear infiltrative. Cavernous hemangiomas are usually large and are also poorly circumscribed.
Vascular Tumors
According to the modified ISSVA classification, vascular tumors include infantile hemangiomas, congenital hemangiomas, tufted angiomas, pyogenic granulomas, kaposiform hemangioendotheliomas, and the capillary hemangioblastoma/hemangioma associated with von Hippel-Lindau’s syndrome (see Chapter 64). Readers are also referred to a recent histological review.92
Fibromuscular Dysplasia
Fibromuscular dysplasia is a generic term for a group of structural abnormalities of one or more layers of medium-sized and large arteries that result in luminal narrowing by fibrous, smooth muscle, or fibromuscular tissue, with or without associated aneurysms and dissections of the media. Fibromuscular Dysplasia is an uncommon noninflammatory and nonatherosclerotic angiopathy with a predilection for the renal and carotid arteries. Young to middle-aged predominantly Caucasian females are most commonly affected. Renal involvement is the most common (60%-75%), followed by cervicocranial arteries (25%-30%), visceral arteries (9%), and the arteries of the extremities (5%). The disease consists of a heterogeneous group of histological changes that ultimately lead to arterial narrowing. Clinical manifestations reflect the arterial bed involved, most commonly hypertension (renal), stroke (carotid), and abdominal pain. Classification is based on the histopathological localization and pattern of structural abnormalities. The latter determines the angiographic appearance.
Fibromuscular dysplasia is a pathological diagnosis and has been classified into intimal, medial, and periarterial subtypes. The characteristic angiographic changes described as “string of beads,” focal, and tubular can be used to make the diagnosis in the appropriate clinical setting. The most common lesions become symptomatic as high-grade stenosis producing renovascular hypertension, or as an embolic source in the cerebral circulation. Treatment is reserved for symptomatic lesions. Most simple lesions are effectively treated by catheter-based intervention. Surgical therapy is warranted for more complex lesions. Both produce durable long-term results.149
The cause(s) of FMD remains obscure and may include estrogen exposure, mechanical factors, ischemic factors, environmental toxins such as cigarette smoke, and autoantibodies. There is an underlying genetic predisposition to the development of FMD. Caucasians are much more likely to develop FMD than blacks, and many cases are familial, with an autosomal dominant transmission and variable penetrance. However, no specific genetic abnormalities have been reported up to now.
Histological Subtypes
The traditional histological classification of FMD has been established based on the layer of vessel involved150 (Fig. 63-15). Classically, FMD has been divided into medial (Fig. 63-16), intimal (Fig. 63-17), and adventitial types (Fig. 63-18). Medial FMD accounts for up to 95% of cases and has been further subdivided into medial fibroplasia, perimedial fibroplasia, and medial hyperplasia.151 Medial fibroplasia is shown to correlate with either a string-of-beads appearance or multiple stenoses on angiography in 62% of cases. The tubular type with long, concentric stenosis is seen in 14%, and the focal type with solitary stenosis less than 1 cm in length occurs in 7%.
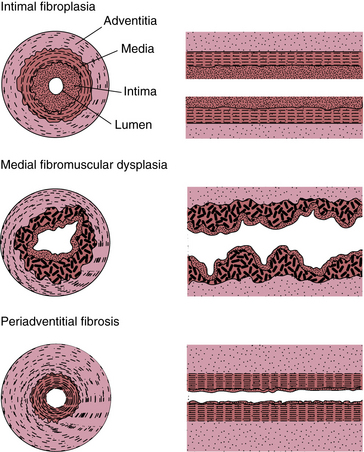
Figure 63-15 Schematic showing transverse and longitudinal patterns of involvement of the arteries by the major types of fibromuscular dysplasia (FMD).
(Modified from Harrison EG Jr, McCormack LJ: Pathologic classification of renal arterial disease in renovascular hypertension. Mayo Clin Proc 46:161–167, 1971; and Luschner TF, Lie JT, Stanson AW, et al: Arterial fibromuscular dysplasia. Mayo Clin Proc 69:931–952, 1987.)
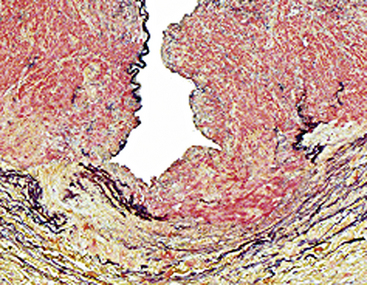
Figure 63-16 Arterial dysplasia, medial type.
High magnification demonstrates a gap in the arterial media. A succession of such defects results in the string-of-beads appearance on angiogram.
(Reproduced with permission from Virmani R, Burke AP, Farb A: Arterial dysplasia, aneurysms, and dissections. In Virmani R, Burke AP, Farb A, editors: Atlas of cardiovascular pathology, Philadelphia, 1996, Saunders, pp 184–193.)
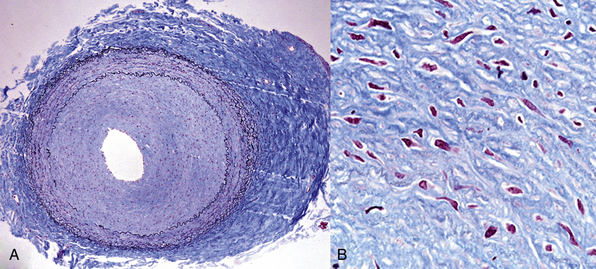
Figure 63-17 Fibromuscular dysplasia (FMD), primarily intimal.
A 55-year-old woman underwent coronary artery bypass graft (CABG) surgery. Her internal mammary artery (demonstrated histologically) was grossly cordlike, as were other arteries in the mediastinum. A, Low magnification shows marked concentric thickening of the intima, with a relatively normal media. There is also a degree of adventitial scarring. B, Higher magnification of the intima shows smooth muscle cells (SMCs) within a proteoglycan matrix.
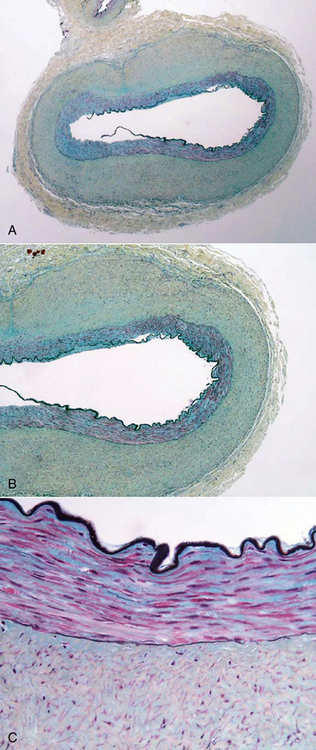
Figure 63-18 Fibromuscular dysplasia (FMD), primarily adventitial.
A, Low magnification of an artery with normal media and intima but markedly thickened adventitia. B, Adventitial scarring is seen at intermediate magnification. C, Normal internal elastic lamina, relatively normal media and cellular adventitia.
Medial fibroplasia is characterized by fibromuscular ridges of hyperplastic SMCs and fibrous tissue, which project into the lumen of the affected artery. Perimedial hyperplasia implies a predominance of fibrous tissue, and the proliferation is primarily in the outer layer of the media. Pure medial hypertrophy is quite rare, and represents a concentric hypertrophy of the medial wall. The wall is relatively smooth, lacks fibrosis, and causes severe stenosis. Angiographically and grossly, the stenosis is usually subtotal, tubular, and smooth.
Intimal hyperplasia, which represents less than 5% of lesions, is characterized by proliferation of fibrous tissue, including smooth muscle cells, within the intima. There is no lipid deposition or inflammatory cell infiltrate, and the internal elastic lamina is preserved. Endarteritis secondary to inflammatory conditions or trauma may be associated with intimal fibroplasia in muscular arteries that may mimic the histological picture of intimal fibroplasias. The morphological appearance may be virtually indistinguishable from that of secondary or reactive intimal fibroplasia seen after endarterectomy or in the initial proliferative stage of atherosclerosis. Intimal fibroplasia may also occur concomitantly with medial FMD. In adventitial or periarterial fibroplasia, the primary lesion is a dense collagenous replacement of the loose fibrous tissue of the arterial adventitia. This fibrosis may extend into the surrounding adipose and connective tissues. In periarterial fibroplasia, the media and intima are normal.
The histological classification of FMD is generally based on the layer of vessel involved. Although this classification is well established in the literature, we, as well as others,152 have not always found this classification useful and reproducible. A simplified classification based on the presence of smooth muscle hyperplasia has recently been proposed.152
Fibromuscular Dysplasia of Renal Arteries
In children and young adults, FMD of the renal arteries is the most common cause of renovascular hypertension.153 The renal arteries are the most common site of symptomatic FMD, accounting for about 75% of cases. Multivessel FMD, which involves renal as well as other arteries, occurs in approximately 25% of patients with renal FMD.
The potential of FMD involving transplant kidneys and affecting the remaining donor kidney has led to the routine use of digital subtraction (DSA) and magnetic resonance angiography (MRA) in potential kidney donors.154–158
Although the cause of FMD of the renal arteries is unknown, there is an association with the HLA-DRw6 antigen. In one series of patients, this HLA antigen was more common in the 33 FMD patients than in the 61 renal transplant donor control subjects (odds ratio (OR), 2.5; P = 0.03).159
Patients with renal FMD are often young women. The diagnosis of FMD is usually made upon investigation for unexplained hypertension. The right renal artery is more often involved than the left, and cases of bilateral involvement are often associated with extrarenal FMD. In a large recent series of 104 unrelated hypertensive patients (94 women) with renal artery FMD, 81 patients had multifocal, 16 had unifocal, and 7 patients both types of stenosis. Fifty-four patients had bilateral FMD. The documented prevalence of familial cases was 11% in this series; familial cases all exhibited the multifocal type and were more commonly bilateral.160 The distal two thirds of the artery is typically involved, with frequent extension into the branch arteries. Helical CT angiography (CTA), especially the combination of transverse sections and maximum-intensity projection (MIP) reconstructions, can reliably reveal renal artery FMD.161 Complications of arterial occlusion and renal infarction may result from arterial dissection, embolization originating in the aneurysm, and aneurysmal thrombosis.
Fibromuscular Dysplasia of Aortic Arch Vessels
Fibromuscular dysplasia has been described involving the carotid, subclavian, and vertebral arteries.162–164 The internal carotid artery (ICA) is typically involved at the level of C1-C2 and is usually “typical” medial FMD, resulting in a classic string-of-beads angiographic appearance. Heterozygous α1-antitrypsin deficiency may be a genetic risk factor for the development of FMD of the internal carotid artery.165 Fibromuscular dysplasia is a rare cause of cerebral ischemia; however, FMD of the ICA is typically diagnosed after symptoms such as transient ischemic attacks (TIAs), amaurosis fugax, minor stroke, or nonfocalized ischemic cerebral symptoms. Fibromuscular dysplasia of the arch vessels is increasingly being found incidentally. Surgical intervention in symptomatic cases includes thromboendarterectomy of the bifurcation, venous interposition graft, and venous patching.166 Recently, endovascular therapy has been applied with increasing frequency. Stent placement has been used for FMD-related aneurysms, and balloon angioplasty for patients with TIA or stroke.167
Patients with cerebrovascular FMD have a high incidence of berry aneurysms of the circle of Willis, especially the intracranial ICA and the middle cerebral artery (MCA). Aneurysms in these locations are likely to be found in females with other vascular lesions caused by FMD or other aneurysms. In contrast, aneurysms of the anterior cerebral artery (ACA) are more likely sporadic and found in men. The prevalence of intracranial aneurysms in patients with cervical ICA and/or vertebral artery FMD is approximately 7%, which is not as high as the 21% to 51% prevalence previously reported.168
Subclavian and vertebral FMD is less likely to have a characteristic angiographic appearance and has been termed atypical FMD on the basis of radiological findings. Fibromuscular dysplasia involving more distal branches of the aortic arch, such as the brachial artery, is unusual.169
Cerebrovascular FMD and FMD of the subclavian arteries are often asymptomatic and may be an incidental angiographic finding. The overall incidence of this disease, and therefore the proportion of cases that are asymptomatic, are unknown. Symptoms occur if there is significant stenosis of major cephalic or subclavian arteries or embolization from thrombosed areas of FMD. Symptoms include transient ischemic attacks, strokes, and SAH from associated berry aneurysms of the circle of Willis, subclavian steal syndrome, and weakness or claudication of the arms. Complications of cerebrovascular FMD include arterial dissections and carotid cavernous fistulas. Fibromuscular dysplasia of the cerebrovascular circulation and subclavian arteries is more often multifocal and bilateral than isolated renal artery FMD. Patients with subclavian FMD often have systemic FMD, including FMD of the renal arteries.170
Fibrous Dysplasia of Miscellaneous Sites
Fibromuscular dysplasia has been described in the visceral arteries,171–173 iliac,174 axillary, brachial and coronary arteries, and aorta.175 Typical arteries involved in FMD of the visceral arteries are the celiac artery, mesenteric arteries, hepatic artery,176 and splenic artery. These are often found in association with renal artery FMD.173,177
Segmental arterial mediolysis, a rare form of mesenteric arteriopathy that typically results in spontaneous dissections and hemoperitoneum in hypertensive patients, may be a form of mesenteric arterial dysplasia. Symptoms related to stenotic lesions of the visceral arteries are rare owing to well-developed collateral vessels. Fibromuscular dysplasia of miscellaneous arteries are prone to similar complications of renal FMD and FMD of the aortic arch, including dissection and embolization.
1 Burroughs J.T., Edwards J.E. Total anomalous pulmonary venous connection. Am Heart J. 1960;59:913–931.
2 Jackson L.W., Correa-Villasenor A., Lees P.S., et al. Parental lead exposure and total anomalous pulmonary venous return. Birth Defects Res A Clin Mol Teratol. 2004;70:185–193.
3 Ruggieri M., Abbate M., Parano E., et al. Scimitar vein anomaly with multiple cardiac malformations, craniofacial, and central nervous system abnormalities in a brother and sister: familial scimitar anomaly or new syndrome? Am J Med Genet. 2003;116A:170–175.
4 Bleyl SB, Saijoh Y, Bax NA, et al: Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: integrating evidence from human genetics and model organisms, Hum Mol Genet 19:1286–1301.
5 Darling R.C., Rothney W.B., Criaig J.M. Total pulmonary venous drainage into the right side of the heart. Report of 17 autopsied cases not associated with other major cardiovascular anomalies. Lab Invest. 1957;6:44–64.
6 Seale A.N., Uemura H., Webber S.A., et al. Total anomalous pulmonary venous connection: morphology and outcome from an international population-based study. Circulation. 2010;122:2718–2726.
7 Jenkins K.J., Sanders S.P., Orav E.J., et al. Individual pulmonary vein size and survival in infants with totally anomalous pulmonary venous connection. J Am Coll Cardiol. 1993;22:201–206.
8 Rubino M., Van Praagh S., Kadoba K., et al. Systemic and pulmonary venous connections in visceral heterotaxy with asplenia. Diagnostic and surgical considerations based on seventy-two autopsied cases. J Thorac Cardiovasc Surg. 1995;110:641–650.
9 Masui T., Seelos K.C., Kersting-Sommerhoff B.A., et al. Abnormalities of the pulmonary veins: evaluation with MR imaging and comparison with cardiac angiography and echocardiography. Radiology. 1991;181:645–649.
10 Hijii T., Fukushige J., Hara T. Diagnosis and management of partial anomalous pulmonary venous connection. A review of 28 pediatric cases. Cardiology. 1998;89:148–151.
11 Chassinat R. Observations d’anomalies anatomiques remarquables de l’appareil circulatoire, avec hépatocéle congéniale, symptom particulier. Arch Gen Med. 1836;11:80–91.
12 Sehgal A., Loughran-Fowlds A. Scimitar syndrome. Indian J Pediatr. 2005;72:249–251.
13 Midyat L., Demir E., Askin M., et al. Eponym. Scimitar syndrome. Eur J Pediatr. 2010;169:1171–1177.
14 Vida V.L., Padalino M.A., Boccuzzo G., et al. Scimitar syndrome: a European Congenital Heart Surgeons Association (ECHSA) multicentric study. Circulation. 2010;122:1159–1166.
15 Perry L.W., Scott L.P.3rd. Cor triatriatum: clinical and pathophysiological features. Clin Proc Child Hosp Dist Columbia. 1967;23:294–304.
16 Herlong J.R., Jaggers J.J., Ungerleider R.M. Congenital Heart Surgery Nomenclature and Database Project: pulmonary venous anomalies. Ann Thorac Surg. 2000;69:S56–S69.
17 Buchholz S., Jenni R. Doppler echocardiographic findings in 2 identical variants of a rare cardiac anomaly, “subtotal” cor triatriatum: a critical review of the literature. J Am Soc Echocardiogr. 2001;14:846–849.
18 Tueche S. Cor triatriatum dextrum. Surgical treatment in a neonate. Acta Cardiol. 2003;58:39–40.
19 Huang T.C., Lee C.L., Lin C.C., et al. Use of Inoue balloon dilatation method for treatment of cor triatriatum stenosis in a child. Catheter Cardiovasc Interv. 2002;57:252–256.
20 Chen Q., Guhathakurta S., Vadalapali G., et al. Cor triatriatum in adults: three new cases and a brief review. Tex Heart Inst J. 1999;26:206–210.
21 Jeong J.W., Tei C., Chang K.S., et al. A case of cor triatriatum in an eighty-year-old man: transesophageal echocardiographic observation of multiple defects. J Am Soc Echocardiogr. 1997;10:185–188.
22 Slight R.D., Nzewi O.C., Sivaprakasam R., et al. Cor triatriatum sinister presenting in the adult as mitral stenosis. Heart. 2003;89:e26.
23 Rorie M., Xie G.Y., Miles H., et al. Diagnosis and surgical correction of cor triatriatum in an adult: combined use of transesophageal echocardiography and catheterization. Catheter Cardiovasc Interv. 2000;51:83–86.
24 Goel A.K., Saxena A., Kothari S.S. Atrioventricular septal defect with cor triatriatum: case report and review of the literature. Pediatr Cardiol. 1998;19:243–245.
25 Endo M., Yamaki S., Ohmi M., et al. Pulmonary vascular changes induced by congenital obstruction of pulmonary venous return. Ann Thorac Surg. 2000;69:193–197.
26 Victor S., Nayak V.M. Deringing procedure for congenital pulmonary vein stenosis. Tex Heart Inst J. 1995;22:166–169.
27 Omasa M., Hasegawa S., Bando T., et al. A case of congenital pulmonary vein stenosis in an adult. Respiration. 2004;71:92–94.
28 Spray T.L., Bridges N.D. Surgical management of congenital and acquired pulmonary vein stenosis. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 1999;2:177–188.
29 Yee E.S., Turley K., Hsieh W.R., et al. Infant total anomalous pulmonary venous connection: factors influencing timing of presentation and operative outcome. Circulation. 1987;76:83–87. III
30 Lincoln C.R., Rigby M.L., Mercanti C., et al. Surgical risk factors in total anomalous pulmonary venous connection. Am J Cardiol. 1988;61:608–611.
31 Dobbertin A., Warnes C.A., Seward J.B. Cor triatriatum dexter in an adult diagnosed by transesophageal echocardiography: a case report. J Am Soc Echocardiogr. 1995;8:952–957.
32 Taylor A.J., Rogan K.M., Virmani R. Sudden cardiac death associated with isolated congenital coronary artery anomalies. J Am Coll Cardiol. 1992;20:640–647.
33 Yamanaka O., Hobbs R.E. Coronary artery anomalies in 126,595 patients undergoing coronary arteriography. Cathet Cardiovasc Diagn. 1990;21:28–40.
34 Zeppilli P., dello Russo A., Santini C., et al. In vivo detection of coronary artery anomalies in asymptomatic athletes by echocardiographic screening. Chest. 1998;114:89–93.
35 Frescura C., Basso C., Thiene G., et al. Anomalous origin of coronary arteries and risk of sudden death: a study based on an autopsy population of congenital heart disease. Hum Pathol. 1998;29:689–695.
36 Clemente A., Del Borrello M., Greco P., et al. Anomalous origin of the coronary arteries in children: diagnostic role of three-dimensional coronary MR angiography. Clin Imaging. 2010;34:337–343.
37 Edwards J.E. Anomalous pulmonary venous connections. Gould S.E., ed. Pathology of the heart and blood vessels. 1968.
38 Gersony W.M. Coarctation of the aorta. In: Adams F.H., Emmanouilides G.C. Heart disease in infants, children and adolescents. Baltimore: William & Wilkins; 1989:243.
39 Fixler D.E., Pastor P., Chamberlin M., et al. Trends in congenital heart disease in Dallas County births. 1971–1984. Circulation. 1990;81:137–142.
40 Tanous D., Benson L.N., Horlick E.M. Coarctation of the aorta: evaluation and management. Curr Opin Cardiol. 2009;24:509–515.
41 Grech V. Diagnostic and surgical trends, and epidemiology of coarctation of the aorta in a population-based study. Int J Cardiol. 1999;68:197–202.
42 Fyler D.C., Rothman K.J., Buckley L.P., et al. The determinants of five year survival of infants with critical congenital heart disease. Cardiovasc Clin. 1981;11:393–405.
43 Edwards J.E. Anomalous pulmonary venous connection. In: Gould S.E., ed. Pathology of the heart and blood vessels. ed 3. Springfield: Thomas; 1968:455–478.
44 Campbell M., Polani P.E. The aetiology of coarctation of the aorta. Lancet. 1961;1:463–468.
45 McBride K.L., Zender G.A., Fitzgerald-Butt S.M., et al. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur J Hum Genet. 2009;17:811–819.
46 Reifenstein G.H., Levine S.A., Gross R.E. Coarctation of the aorta: a review of 104 autopsied cases of the “adult type”, 2 years of age or older. Am Heart J. 1947;33:146.
47 Pellegrino A., Deverall P.B., Anderson R.H., et al. Aortic coarctation in the first three months of life. An anatomopathological study with respect to treatment. J Thorac Cardiovasc Surg. 1985;89:121–127.
48 Shinebourne E.A., Elseed A.M. Relation between fetal flow patterns, coarctation of the aorta, and pulmonary blood flow. Br Heart J. 1974;36:492–498.
49 Rudolph A.M., Heymann M.A., Spitznas U. Hemodynamic considerations in the development of narrowing of the aorta. Am J Cardiol. 1972;30:514–525.
50 Ho S.Y., Anderson R.H. Coarctation, tubular hypoplasia, and the ductus arteriosus. Histological study of 35 specimens. Br Heart J. 1979;41:268–274.
51 Becker A.E., Becker M.J., Edwards J.E. Anomalies associated with coarctation of aorta: particular reference to infancy. Circulation. 1970;41:1067–1075.
52 Olney M.B., Stephens H.B. Coarctation of the aorta in children: observations in fourteen cases. J Pediatr. 1950;37:192–203.
53 Bahn R.C., Edwards J.E., DuShane J.W. Coarctation of the aorta in children: observations in fourteen cases. Pediatrics. 1951;8:192–203.
54 Moller J.H., Amplatz K., Edwards J.E. Congenital heart disease. Kalamazoo: Upjohn; 1974.
55 Edwards J.E. Pathologic considerations in coarctation of the aorta. Proc Staff Meet Mayo Clin. 1948:23.
56 Tawes R.L.Jr, Berry C.L., Aberdeen E. Congenital bicuspid aortic valves associated with coarctation of the aorta in children. Br Heart J. 1969;31:127–128.
57 Kirklin J.W., Barratt-Boyes B.G. Coarctation of the aorta and interrupted aortic arch. In: Cardiac surgery. New York: Wiley. 1993.
58 Liberthson R.R., Pennington D.G., Jacobs M.L., et al. Coarctation of the aorta: review of 234 patients and clarification of management problems. Am J Cardiol. 1979;43:835–840.
59 Lucas R.V., Krabill R.A. Anomalous venous connections, pulmonary and systemic. In: Adams F.H., Emmanouilides G.C. Heart disease in infants, children, and adolescents. Baltimore: Williams & Wilkins; 1989:580.
60 Wolman L.J. Syndrome of constricting double aortic arch in infancy report of a case. J Pediatr. 1939;14:527.
61 Creazzo T.L., Godt R.E., Leatherbury L., et al. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–286.
62 McDonald-McGinn D., Emanuel B.S., Zackai E.H. 22q11.2 Deletion syndrome. GeneReviews. Seattle: University of Washington; 2005.
63 Edwards J.E. Anomalies of the derivatives of the aortic arch system. Med Clin North Am. 1948;6:925.
64 High F.A., Jain R., Stoller J.Z., et al. Murine jagged1/notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. J Clin Invest. 2009;119:1986–1996.
65 Conway S.J., Kruzynska-Frejtag A., Kneer P.L., et al. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21.
66 Iida K., Koseki H., Kakinuma H., et al. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development. 1997;124:4627–4638.
67 Kirklin J.W., Baratt-Boyes B.G. Vascular rings and slings. cardiac surgery. New York: Wiley. 1986.
68 Stewart J.R., Kincaid O.W., Edwards J.E. An atlas of vascular rings and related malformations of the aortic arch system. Springfield: Thomas; 1964.
69 Sissman N.J. Anomalies of the aortic arch complex. In: Adams F.H., Emmanouilides G.C. Heart disease in infants, children, and adolescents. Baltimore: Williams & Wilkins; 1983:199–215.
70 Kocis K.C., Midgley F.M., Ruckman R.N. Aortic arch complex anomalies: 20-year experience with symptoms, diagnosis, associated cardiac defects, and surgical repair. Pediatr Cardiol. 1997;18:127–132.
71 Woods R.K., Sharp R.J., Holcomb G.W.3rd, et al. Vascular anomalies and tracheoesophageal compression: a single institution’s 25-year experience. Ann Thorac Surg. 2001;72:434–438. discussion 438–439
72 Moes C.A., Freedom R.M. Rare types of aortic arch anomalies. Pediatr Cardiol. 1993;14:93–101.
73 Gil-Jaurena J.M., Murtra M., Goncalves A., et al. Aortic coarctation, vascular ring, and right aortic arch with aberrant subclavian artery. Ann Thorac Surg. 2002;73:1640–1642.
74 Taussig H.B. Congenital malformations of the heart, ed 2. Cambridge, Mass: Harvard University Press; 1960. vol 2
75 Binet J.P., Conso J.F., Losay J., et al. Ductus arteriosus sling: report of a newly recognised anomaly and its surgical correction. Thorax. 1978;33:72–75.
76 Messner G., Reul G.J., Flamm S.D., et al. Interrupted aortic arch in an adult single-stage extra-anatomic repair. Tex Heart Inst J. 2002;29:118–121.
77 Kirklin J.W., Baratt-Boyes B.G. Origin of the right or left pulmonary artery from the ascending aorta. cardiac surgery. New York: Wiley. 1993.
78 Keane J.F., Maltz D., Bernhard W.F., et al. Anomalous origin of one pulmonary artery from the ascending aorta. Diagnostic, physiological and surgical considerations. Circulation. 1974;50:588–594.
79 Calder A.L., Brandt P.W., Barratt-Boyes B.G., et al. Variant of tetralogy of Fallot with absent pulmonary valve leaflets and origin of one pulmonary artery from the ascending aorta. Am J Cardiol. 1980;46:106–116.
80 Penkoske P.A., Castaneda A.R., Fyler D.C., et al. Origin of pulmonary artery branch from ascending aorta. Primary surgical repair in infancy. J Thorac Cardiovasc Surg. 1983;85:537–545.
81 Becker A.E., Anderson R.H. Malformations of the pulmonary trunk and arteries. pathology of congenital heart disease. London: Butterworths. 1981.
82 Becker A.E., Anderson R.H. Aortopulmonary communications. Pathology of Congenital Heart Disease. London: Butterworths. 1981.
83 Kirklin J.W., Baratt-Boyes B.G. Aortopulmonary window. Cardiac surgery, ed 2. New York: Wiley. 1993.
84 Neufeld H.N., Lester R.G., Adams P.Jr, et al. Aorticopulmonary septal defect. Am J Cardiol. 1962;9:12–25.
85 Luisi S.V., Ashraf M.H., Gula G., et al. Anomalous origin of the right coronary artery with aortopulmonary window: functional and surgical considerations. Thorax. 1980;35:446–448.
86 Blieden L.C., Moller J.H. Aorticopulmonary septal defect. An experience with 17 patients. Br Heart J. 1974;36:630–635.
87 Huang J.T., Liang M.G. Vascular malformations. Pediatr Clin North Am. 2010;57:1091–1110.
88 Gloviczki P., Duncan A., Kalra M., et al. Vascular malformations: an update. Perspect Vasc Surg Endovasc Ther. 2009;21:133–148.
89 Belov S. Anatomopathological classification of congenital vascular defects. Semin Vasc Surg. 1993;6:219–224.
90 Lee B.B., Laredo J., Lee T.S., et al. Terminology and classification of congenital vascular malformations. Phlebologie. 2007;22:249–252.
91 Rutherford R.B. Classification of peripheral congenital vascular malformations. In: Ernst C., Stanley J., eds. Current therapy in vascular surgery, vol 22. St. Louis: Mosby; 1995.
92 Gupta A., Kozakewich H. Histopathology of vascular anomalies. Clin Plast Surg. 2011;38:31–44.
93 Woods C.G., Taylor A.M. Ataxia telangiectasia in the British Isles: the clinical and laboratory features of 70 affected individuals. Q J Med. 1992;82:169–179.
94 Gatti R.A. Ataxia-telangiectasia. Dermatol Clin. 1995;13:1–6.
95 Uhrhammer N., Bay J.O., Bignon Y.J. Seventh International Workshop on Ataxia-Telangiectasia. Cancer Res. 1998;58:3480–3485.
96 Concannon P., Gatti R.A. Diversity of ATM gene mutations detected in patients with ataxia-telangiectasia. Hum Mutat. 1997;10:100–107.
97 Lavin M.F., Khanna K.K. ATM: the protein encoded by the gene mutated in the radiosensitive syndrome ataxia-telangiectasia. Int J Radiat Biol. 1999;75:1201–1214.
98 Yuille M.A., Coignet L.J. The ataxia telangiectasia gene in familial and sporadic cancer. Recent Results Cancer Res. 1998;154:156–173.
99 Checketts S.R., Burton P.S., Bjorkman D.J., et al. Generalized essential telangiectasia in the presence of gastrointestinal bleeding. J Am Acad Dermatol. 1997;37:321–325.
100 Novitsky Y.W., Kercher K.W., Czerniach D.R., et al. Watermelon stomach: pathophysiology, diagnosis, and management. J Gastrointest Surg. 2003;7:652–661.
101 Elkayam O., Oumanski M., Yaron M., et al. Watermelon stomach following and preceding systemic sclerosis. Semin Arthritis Rheum. 2000;30:127–131.
102 Watson M., Hally R.J., McCue P.A., et al. Gastric antral vascular ectasia (watermelon stomach) in patients with systemic sclerosis. Arthritis Rheum. 1996;39:341–346.
103 Goel A., Christian C.L. Gastric antral vascular ectasia (watermelon stomach) in a patient with Sjogren’s syndrome. J Rheumatol. 2003;30:1090–1092.
104 Hisamatsu K., Ueeda M., Ando M., et al. Peripheral arterial coil embolization for hepatic arteriovenous malformation in Osler-Weber-Rendu disease; useful for controlling high output heart failure, but harmful to the liver. Intern Med. 1999;38:962–968.
105 Abbot M.E. Coarctation of the aorta of the adult type II. A statistical study and historical retrospect of 200 recorded cases with autopsy of stenosis or obliteration of the descending aorta in subjects over the age of two years. Am Heart J. 1928;3:574–618.
106 Abe K., Akata S., Ohkubo Y., et al. Venous hemangioma of the mediastinum. Eur Radiol. 2001;11:73–75.
107 Igarashi J., Hanazaki K. Retroperitoneal venous hemangioma. Am J Gastroenterol. 1998;93:2292–2293.
108 Ichikawa M.M., Ishida-Yamamoto A., Hashimoto Y., et al. Venous hemangioma. An immunohistochemical and ultrastructural study. Acta Derm Venereol. 1997;77:382–384.
109 Itosaka H., Tada M., Sawamura Y., et al. Vanishing tumor of the temporalis muscle: repeated hemorrhage in an intramuscular venous hemangioma. AJNR Am J Neuroradiol. 1997;18:983–985.
110 Tada M., Sawamura Y., Abe H., et al. Venous hemangioma of the temporalis muscle. Neurol Med Chir (Tokyo). 1996;36:23–25.
111 Hanatate F., Mizuno Y., Murakami T. Venous hemangioma of the mesoappendix: report of a case and a brief review of the Japanese literature. Surg Today. 1995;25:962–964.
112 Bedocs P.M., Gould J.W. Blue rubber-bleb nevus syndrome: a case report. Cutis. 2003;71:315–318.
113 Gabikian P., Clatterbuck R.E., Gailloud P., et al. Developmental venous anomalies and sinus pericranii in the blue rubber-bleb nevus syndrome. case report, J Neurosurg. 2003;99:409–411.
114 Eiris-Punal J., Picon-Cotos M., Viso-Lorenzo A., et al. Epileptic disorder as the first neurologic manifestation of blue rubber bleb nevus syndrome. J Child Neurol. 2002;17:219–222.
115 Morris L., Lynch P.M., Gleason W.A.Jr, et al. Blue rubber bleb nevus syndrome: laser photocoagulation of colonic hemangiomas in a child with microcytic anemia. Pediatr Dermatol. 1992;9:91–94.
116 McCannel C.A., Hoenig J., Umlas J., et al. Orbital lesions in the blue rubber bleb nevus syndrome. Ophthalmology. 1996;103:933–936.
117 Rodrigues D., Bourroul M.L., Ferrer A.P., et al. Blue rubber bleb nevus syndrome. Rev Hosp Clin Fac Med Sao Paulo. 2000;55:29–34.
118 Gallione C.J., Pasyk K.A., Boon L.M., et al. A gene for familial venous malformations maps to chromosome 9p in a second large kindred. J Med Genet. 1995;32:197–199.
119 Calvert J.T., Riney T.J., Kontos C.D., et al. Allelic and locus heterogeneity in inherited venous malformations. Hum Mol Genet. 1999;8:1279–1289.
120 Vikkula M., Boon L.M., Carraway K.L.3rd, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190.
121 Limaye N., Wouters V., Uebelhoer M., et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41:118–124.
122 Awad I.A., Robinson J.R., Mohanty S., et al. Mixed vascular malformations of the brain: clinical and pathogenetic considerations. Neurosurgery. 1993;33:179–188.
123 Coleman C.C. Diagnosis and treatment of congenital arteriovenous fistulas of the head and neck. Am J Surg. 1973;126:424–428.
124 Finn M.C., Celowack J., Mulliken J.B. Congenital vascular lesions: clinical application of a new classification. J Pediatr Surg. 1983;18:894–900.
125 Garcia-Gonzalez R., Gonzalez-Palacios J., Maganto-Pavon E. Congenital renal arteriovenous fistula (cirsoid aneurysm). Urology. 1984;24:495–498.
126 Jellinger K. Vascular malformations of the central nervous system: a morphologic overview. Neurosurg Rev. 1986;9:177–216.
127 Trout H.H.3rd, McAllister H.A.Jr, Giordano J.M., et al. Vascular malformations. Surgery. 1985;97:36–41.
128 Trout H.H. Management of patients with hemangiomas and arteriovenous malformations. Surg Clin North Am. 1986;66:333–345.
129 Watson W.L., McCarthy W.D. Blood and lymph vessel tumors–a report of 1,056 cases. Surg Gynecol Obstet. 1940;71:569–583.
130 Herzig R., Burval S., Vladyka V., et al. Familial occurrence of cerebral arteriovenous malformation in sisters: case report and review of the literature. Eur J Neurol. 2000;7:95–100.
131 Irie K., Nagao S., Honma Y., et al. Treatment of arteriovenous malformation of the brain–preliminary experience. J Clin Neurosci. 2000;7:24–29.
132 Brunelle F. Arteriovenous malformation of the vein of Galen in children. Pediatr Radiol. 1997;27:501–513.
133 Yanaka K., Hyodo A., Nose T. Venous malformation serving as the draining vein of an adjoining arteriovenous malformation. Case report and review of the literature. Surg Neurol. 2001;56:170–174.
134 Kacker A., Heier L., Jones J. Large intraosseous arteriovenous malformation of the maxilla - a case report with review of literature. Int J Pediatr Otorhinolaryngol. 2000;52:89–92.
135 Nakai Y., Yasuda S., Hyodo A., et al. Infratentorial arteriovenous malformation associated with persistent primitive trigeminal artery–case report. Neurol Med Chir (Tokyo). 2000;40:572–574.
136 Smith H.L., Horton B.T. Arteriovenous fistula of the lung associated with polycythemia vera; report of a case in which the diagnosis was made clinically. Am Heart J. 1939;18:589–594.
137 Burke C.M., Safai C., Nelson D.P., et al. Pulmonary arteriovenous malformations: a critical update. Am Rev Respir Dis. 1986;134:334–339.
138 Hodgson C.EA. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous fistula: survey of a large family. N Engl J Med. 1959;26:625–638.
139 Khurshid I., Downie G.H. Pulmonary arteriovenous malformation. Postgrad Med J. 2002;78:191–197.
140 Bosher L.H., Blake A., Byrd B.R. An analysis of the pathologic anatomy of pulmonary arteriovenous aneurysms with particular reference to the applicability of local excision. Surgery. 1959;45:91–104.
141 Tripathy U., Kaul S., Bhosle K., et al. Pulmonary arteriovenous fistula with cerebral arteriovenous malformation without hereditary hemorrhagic telangiectasia. Unusual case report and literature review. J Cardiovasc Surg (Torino). 1997;38:677–680.
142 Chow L.T., Chow W.H., Ma K.F. Pulmonary arteriovenous malformation. Progressive enlargement with replacement of the entire right middle lobe in a patient with concomitant mitral stenosis. Med J Aust. 1993;158:632–634.
143 Aida K., Nakamura H., Kihara Y., et al. Duodenal ulcer and pancreatitis associated with pancreatic arteriovenous malformation. Eur J Gastroenterol Hepatol. 2002;14:551–554.
144 Hayakawa H., Kusagawa M., Takahashi H., et al. Arteriovenous malformation of the rectum: report of a case. Surg Today. 1998;28:1182–1187.
145 Game X., Berlizot P., Hassan T., et al. Congenital pelvic arteriovenous malformation in male patients: a rare cause of urological symptoms and role of embolization. Eur Urol. 2002;42:407–412.
146 Kojima S., Sumiyoshi M., Suwa S., et al. Cardiac hemangioma: a report of two cases and review of the literature. Heart Vessels. 2003;18:153–156.
147 Grebenc M.L., de Christenson ML R.osado., Burke A.P., et al. Primary cardiac and pericardial neoplasms: radiologic-pathologic correlation. Radiographics. 2000;20:1073–1103. quiz 1110–1071, 1112
148 Burke A., Johns J.P., Virmani R. Hemangiomas of the heart. A clinicopathologic study of ten cases. Am J Cardiovasc Pathol. 1990;3:283–290.
149 Curry T.K., Messina L.M. Fibromuscular dysplasia: when is intervention warranted? Semin Vasc Surg. 2003;16:190–199.
150 Slovut D.P., Olin J.W. Fibromuscular dysplasia. N Engl J Med. 2004;350:1862–1871.
151 Hata J., Hosoda Y. Perimedial fibroplasia of the renal artery. Arch Pathol Lab Med. 1979;103:220–223.
152 Alimi Y., Mercier C., Pellissier J.F., et al. Fibromuscular disease of the renal artery: a new histopathologic classification. Ann Vasc Surg. 1992;6:220–224.
153 Fenves A.Z., Ram C.V. Fibromuscular dysplasia of the renal arteries. Curr Hypertens Rep. 1999;1:546–549.
154 Andreoni K.A., Weeks S.M., Gerber D.A., et al. Incidence of donor renal fibromuscular dysplasia: does it justify routine angiography? Transplantation. 2002;73:1112–1116.
155 Indudhara R., Kenney, Bueschen A.J., et al. Live donor nephrectomy in patients with fibromuscular dysplasia of the renal arteries. J Urol. 1999;162:678–681.
156 Williams M.E., Shaffer D. ACE inhibitor-induced transplant acute renal failure due to donor fibromuscular dysplasia. Nephrol Dial Transplant. 1999;14:760–764.
157 Wolters H.H., Vowinkel T., Schult M., et al. Fibromuscular dysplasia in a living donor: early post-operative allograft artery stenosis with successful venous interposition. Nephrol Dial Transplant. 2002;17:153–155.
158 Blondin D, Lanzman R, Schellhammer F, et al: Fibromuscular dysplasia in living renal donors: still a challenge to computed tomographic angiography, Eur J Radiol 75:67–71.
159 Sang C.N., Whelton P.K., Hamper U.M., et al. Etiologic factors in renovascular fibromuscular dysplasia. A case-control study. Hypertension. 1989;14:472–479.
160 Pannier-Moreau I., Grimbert P., Fiquet-Kempf B. v Possible familial origin of multifocal renal artery fibromuscular dysplasia. J Hypertens. 1997;15:1797–1801.
161 Beregi J.P., Louvegny S., Gautier C., et al. Fibromuscular dysplasia of the renal arteries: comparison of helical CT angiography and arteriography. AJR Am J Roentgenol. 1999;172:27–34.
162 Luschner T.F., Lie J.T., Stanson A.W., et al. Arterial fibromuscular dysplasia. Mayo Clin Proc. 1987;62:931–952.
163 Perry M.O. Fibromuscular dysplasia. Surg Gynecol Obstet. 1974:97–104.
164 Sato S., Hata J. Fibromuscular dysplasia. Its occurrence with a dissecting aneurysm of the internal carotid artery. Arch Pathol Lab Med. 1982;106:332–335.
165 Schievink W.I., Meyer F.B., Parisi J.E., et al. Fibromuscular dysplasia of the internal carotid artery associated with alpha1-antitrypsin deficiency. Neurosurgery. 1998;43:229–233. discussion 233–224
166 Van Damme H., Sakalihasan N., Limet R. Fibromuscular dysplasia of the internal carotid artery. Personal experience with 13 cases and literature review. Acta Chir Belg. 1999;99:163–168.
167 Olin J.W., Sealove B.A. Diagnosis, management, and future developments of fibromuscular dysplasia. J Vasc Surg. 2011;53:826–836. e821
168 Cloft H.J., Kallmes D.F., Kallmes M.H., et al. Prevalence of cerebral aneurysms in patients with fibromuscular dysplasia: a reassessment. J Neurosurg. 1998;88:436–440.
169 Suzuki H., Daida H., Sakurai H., et al. Familial fibromuscular dysplasia of bilateral brachial arteries. Heart. 1999;82:251–252.
170 Leventer R.J., Kornberg A.J., Coleman L.T., et al. Stroke and fibromuscular dysplasia: confirmation by renal magnetic resonance angiography. Pediatr Neurol. 1998;18:172–175.
171 Kojima A., Shindo S., Kubota K., et al. Successful surgical treatment of a patient with multiple visceral artery aneurysms due to fibromuscular dysplasia. Cardiovasc Surg. 2002;10:157–160.
172 Safioleas M., Kakisis J., Manti C. Coexistence of hypertrophic cardiomyopathy and fibromuscular dysplasia of the superior mesenteric artery. N Engl J Med. 2001;344:1333–1334.
173 Sandmann W., Schulte K.M. Multivisceral fibromuscular dysplasia in childhood: case report and review of the literature. Ann Vasc Surg. 2000;14:496–502.
174 Verhelst H., Lauwers G., Schroe H. Fibromuscular dysplasia of the external iliac artery. Acta Chir Belg. 1999;99:171–173.
175 Suarez W.A., Kurczynski T.W., Bove E.L. An unusual type of combined aortic coarctation due to fibromuscular dysplasia. Cardiol Young. 1999;9:323–326.
176 Jones H.J., Staud R., Williams R.C.Jr. Rupture of a hepatic artery aneurysm and renal infarction: 2 complications of fibromuscular dysplasia that mimic vasculitis. J Rheumatol. 1998;25:2015–2018.
177 Ebaugh J.L., Chiou A.C., Morasch M.D., et al. Staged embolization and operative treatment of multiple visceral aneurysms in a patient with fibromuscular dysplasia–a case report. Vasc Surg. 2001;35:145–148.