Chapter 118 Primary Defects of Antibody Production
Of all of the primary immunodeficiency diseases, those affecting antibody production are most frequent. Selective absence of serum and secretory IgA is the most common defect, with rates ranging from 1/333 to 1/18,000 persons among different races. By contrast, agammaglobulinemia is estimated to occur with a frequency of only 1/10,000 to 1/50,000 persons. Patients with antibody deficiency are usually recognized because they have recurrent infections with encapsulated bacteria predominantly in the upper and lower respiratory tracts; some individuals with selective IgA deficiency or infants with transient hypogammaglobulinemia may have few or no infections (see Table 116-4). The defective gene products for many primary antibody deficiency disorders have been identified (Table 118-1) and localized (Fig. 118-1). Sometimes the defect is not in the B cell itself but in T cells, which are required for complete B-cell function.
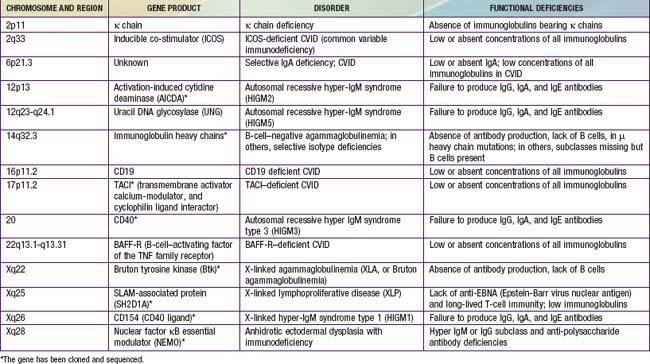
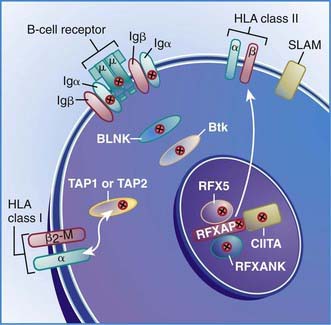
Figure 118-1 Locations of mutant proteins (X) in B cells identified in primary immunodeficiency diseases. β2m, β2 microglobulin; BLNK, B-cell linker adaptor protein; Btk, Bruton tyrosine kinase; HLA, human leukocyte antigen; Ig, immunoglobulin; RFX, RFXAP and CIITA transcription factors; SLAM, signaling lymphocyte activation molecule; TAP1 and TAP2, transporters of processed antigen.
(From Buckley RH: Primary immunodeficiency diseases due to defects in lymphocytes, N Engl J Med 343:1313–1324, 2000.)
X-Linked Agammaglobulinemia (XLA)
Patients with X-linked agammaglobulinemia (XLA), or Bruton agammaglobulinemia, have a profound defect in B-lymphocyte development resulting in severe hypogammaglobulinemia, an absence of circulating B cells, small to absent tonsils, and no palpable lymph nodes.
Genetics and Pathogenesis
The abnormal gene in XLA maps to q22 on the long arm of the X chromosome and encodes the B-cell protein tyrosine kinase Btk (Bruton tyrosine kinase). Btk is a member of the Tec family of cytoplasmic protein tyrosine kinases and is expressed at high levels in all B-lineage cells, including pre–B cells. It appears to be necessary for pre–B-cell expansion and maturation into surface Ig-expressing B cells, but probably has a role at all stages of B-cell development; it has also been found in cells of the myeloid series. More than 500 different mutations in the human Btk gene are recognized; they encompass most parts of the coding portions of the gene. There is not a clear correlation between the location of the mutation and the clinical phenotype (Fig. 118-2). Carriers are detected by mutation analysis, and prenatal diagnosis of affected male fetuses is possible if the mutation is known in the family.
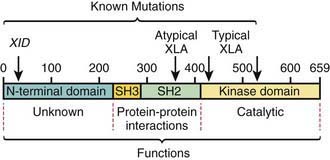
Figure 118-2 Location of mutations in the functional domains of the Bruton tyrosine kinase (Btk) protein. Deletion and point mutations in Btk identified to date in many boys with classic X-linked agammaglobulinemia (XLA) are in the kinase domain, whereas CBA/N XID mice with a less severe B-cell defect have a point mutation at position 28 in the N-terminal domain. More recently, however, boys with classic XLA are also reported to have mutations at the XID mutation site and in the SH2 domain.
(From Buckley RH: Breakthroughs in the understanding and therapy of primary immunodeficiency, Pediatr Clin North Am 41:665–690, 1994.)
The expression of Btk in cells of myeloid lineage is of interest because boys with XLA often have neutropenia at the height of an acute infection. It is conceivable that Btk is only one of the signaling molecules participating in myeloid maturation and that neutropenia is observed in XLA only when rapid production of such cells is needed. Some pre–B cells are found in the bone marrow; the percentage of peripheral blood B lymphocytes is <1%. The percentage of T cells is increased, ratios of T-cell subsets are normal, and T-cell function is intact. The thymus is normal.
Six autosomal recessive defects have also been shown to result in agammaglobulinemia with an absence of circulating B cells (see Fig. 118-2), including mutations in the genes encoding: (1) the µ heavy chain gene; (2) the Igα and Igβ signaling molecules; (3) B-cell linker adaptor protein (BLNK); (4) the surrogate light chain, λ5/14.1; and (5) leucine-rich repeat-containing 8 (LRRC8).
Clinical Manifestations
Most boys afflicted with XLA remain well during the 1st 6-9 mo of life by virtue of maternally transmitted IgG antibodies. Thereafter, they acquire infections with extracellular pyogenic organisms, such as Streptococcus pneumoniae and Haemophilus influenzae, unless they are given prophylactic antibiotics or immunoglobulin therapy. Infections include sinusitis, otitis media, pneumonia, or, less often, sepsis or meningitis. Infections with Mycoplasma are also particularly problematic. Chronic fungal infections are seen; Pneumocystis jiroveci pneumonia rarely occurs. Viral infections are usually handled normally with the exceptions of hepatitis viruses and enteroviruses. There were several examples of paralysis when live polio vaccine was administered to these patients and chronic, eventually fatal, central nervous system infections with various echoviruses and coxsackieviruses have occurred in a significant number of them. Echovirus-associated myositis resembling dermatomyositis has also been observed. These observations suggest a primary role for antibody, particularly secretory IgA, in host defense against enteroviruses. Growth hormone deficiency has also been reported in association with XLA.
Diagnosis
The diagnosis of XLA should be suspected if lymphoid hypoplasia is found on physical examination (minimal or no tonsillar tissue and no palpable lymph nodes), and serum concentrations of IgG, IgA, IgM, and IgE are far below the 95% confidence limits for appropriate age- and race-matched controls (Chapter 708), usually with total immunoglobulins <100 mg/dL. Levels of natural antibodies to type A and B red blood cell polysaccharide antigens (isohemagglutinins) and antibodies to antigens given during routine immunizations are abnormally low in this disorder, whereas they are normal in transient hypogammaglobulinemia of infancy. Flow cytometry is an important test to demonstrate the absence of circulating B cells, which will distinguish this disorder from common variable immunodeficiency, the hyper-IgM syndrome and transient hypogammaglobulinemia of infancy.
Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) is a syndrome characterized by hypogammaglobulinemia with phenotypically normal B cells. It has also been called “acquired hypogammaglobulinemia” because of a generally later age of onset of infections. CVID patients may appear similar clinically to those with XLA in the types of infections experienced and bacterial etiologic agents involved, except that echovirus meningoencephalitis is rare in patients with CVID (Table 118-2). In contrast to XLA, the sex distribution in CVID is almost equal, the age of onset is later (although it may be present in infancy), and infections may be less severe.
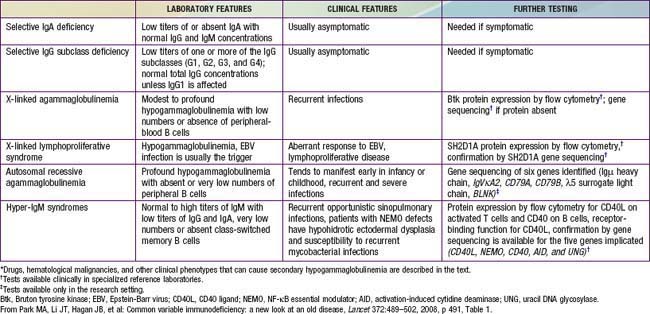
Table 118-2 DIFFERENTIAL DIAGNOSIS FOR PATIENTS WITH SUSPECTED CVID (HYPOGAMMAGLOBULINEMIA WITH RECURRENT INFECTIONS)*
Genetics and Pathogenesis
Most patients have no identified molecular diagnosis. CVID is a category of primary immunodeficiency disorders that likely consists of several different genetic defects with autosomal recessive or dominant inheritance. Genes known to produce the CVID phenotype when mutated include ICOS (inducible co-stimulator) deficiency, SH2DIA (responsible for X-linked lymphoproliferative disease [XLP]), CD19, BAFF-R (B-cell–activating factor of the TNF [tumor necrosis factor] family receptors), and TACI (transmembrane activator, calcium-modulator, and cyclophilin ligand interactor). These mutations in aggregate accounts for less than 10% of all cases of CVID.
Because CVID occurs in 1st-degree relatives of patients with selective IgA deficiency, and some patients with IgA deficiency later become panhypogammaglobulinemic, a large subtype of CVID may have a common genetic basis with IgA deficiency. The high incidence of abnormal immunoglobulin concentrations, autoantibodies, autoimmune disease, and malignancy in both CVID and IgA deficiency and in other members of their families also suggests a shared hereditary influence. This concept is supported by the discovery of a high incidence of C4-A gene deletions and C2 rare gene alleles in the class III major histocompatibility complex (MHC) region in individuals with either IgA deficiency or CVID, suggesting that a common susceptibility gene is on chromosome 6. Only a few human leukocyte antigen (HLA) haplotypes are shared by individuals affected with IgA deficiency and CVID, with at least 1 of 2 particular haplotypes being present in 77% of those affected. In 1 large family with 13 members, 2 had IgA deficiency and 3 had CVID. All of the immunodeficient patients in the family had at least 1 copy of an MHC haplotype that is abnormally frequent in IgA deficiency and CVID: HLA-DQB1 *0201, HLA-DR3, C4B-Sf, C4A-deleted, G11-15, Bf-0.4, C2a, HSP70-7.5, TNFa-5, HLA-B8, and HLA-A1. In a study of 83 multiply affected families with IgA deficiency and CVID, increased allele sharing at chromosome 6p21 in the proximal part of the MHC was observed in a susceptibility locus now designated as IGAD1. More sensitive genetic analysis in 101 multiple-case and 110 single-case families further localized the defect to the HLADQ/DR locus. Environmental factors, particularly drugs such as phenytoin, D-penicillamine, gold, and sulfasalazine are suspected to be triggers for disease expression in individuals with the permissive genetic background.
Most cases of CVID are sporadic or follow an autosomal dominant pattern of inheritance. Patients who lack inducible co-stimulator (ICOS), a surface protein on activated T cells, have an autosomal recessive pattern of inheritance. Nine such patients from 6 families in the Black Forest of Germany have been found to have identical homozygous large genomic deletions of the ICOS gene, suggesting a founder effect. Those who have XLP have an X-linked pattern of inheritance and those with autosomally inherited TACI defects may have heterozygous or homozygous mutations.
Despite normal numbers of circulating immunoglobulin-bearing B lymphocytes and the presence of lymphoid cortical follicles, blood B lymphocytes from CVID patients do not differentiate normally into immunoglobulin-producing cells when stimulated with pokeweed mitogen (PWM) in vitro, even when co-cultured with normal T cells. CVID B cells from some patients can be stimulated both to switch isotype and to synthesize and secrete some immunoglobulin when stimulated with anti-CD40 and IL-4 or IL-10. T cells and T-cell subsets are usually present in normal percentages, although T-cell function is depressed in some patients.
Clinical Manifestations
The serum immunoglobulin and antibody deficiencies in CVID may be as profound as in XLA. Patients with CVID often have autoantibody formation and normal-sized or enlarged tonsils and lymph nodes, and ≈25% of patients have splenomegaly. CVID has also been associated with a sprue-like syndrome with or without nodular follicular lymphoid hyperplasia of the intestine, thymoma, alopecia areata, hemolytic anemia, gastric atrophy, achlorhydria, thrombocytopenia, and pernicious anemia. Lymphoid interstitial pneumonia, pseudolymphoma, B-cell lymphomas, amyloidosis, and noncaseating sarcoid-like granulomas of the lungs, spleen, skin, and liver also occur. There is a 438-fold increase in lymphomas among affected women in the 5th and 6th decades of life. CVID has been reported to resolve transiently or permanently in patients who acquire human immunodeficiency virus (HIV) infection.
Recurrent or chronic infections include pneumonia, sinusitis, otitis media, and diarrhea (bacterial, giardiasis). Repeated pulmonary infections may produce bronchiectasis, while live polio virus vaccines may produce paralysis. Sepsis and meningitis with encapsulated bacteria occur more frequently than the general population. There is often a delay in the diagnosis of 5-10 yr between the 1st infections and a definitive diagnosis.
Selective IgA Deficiency
An isolated absence or near absence (<10 mg/dL) of serum and secretory IgA is the most common well-defined immunodeficiency disorder, with a disease frequency as high as 0.33% in some populations. This condition is also occasionally associated with ill health.
The basic defect resulting in IgA deficiency is unknown. Phenotypically normal blood B cells are present. IgA deficiency occasionally remits spontaneously or after discontinuation of phenytoin (Dilantin) therapy. The occurrence of IgA deficiency in both males and females and in members of successive generations within families suggests autosomal dominant inheritance with variable expressivity. This defect also occurs commonly in pedigrees containing individuals with CVID. Indeed, IgA deficiency may evolve into CVID, and the finding of rare alleles and deletions of MHC class III genes in both conditions suggests that the susceptibility gene common to these 2 conditions may reside in the MHC class III region on chromosome 6. IgA deficiency is noted in patients treated with the same drugs associated with producing CVID (phenytoin, D-penicillamine, gold, and sulfasalazine), suggesting that environmental factors may trigger this disease in a genetically susceptible person.
Clinical Manifestations
Infections occur predominantly in the respiratory, gastrointestinal, and urogenital tracts. Bacterial agents responsible are the same as in other antibody deficiency syndromes. Intestinal giardiasis is common. Children with IgA deficiency vaccinated intranasally with killed poliovirus produce local IgM and IgG antibodies. Serum concentrations of other immunoglobulins are usually normal in patients with selective IgA deficiency, although IgG2 (and other) subclass deficiency has been reported, and IgM (usually elevated) may be monomeric.
Patients with IgA deficiency often have IgG antibodies against cow’s milk and ruminant serum proteins. These antiruminant antibodies may cause false-positive results in immunoassays for IgA that use goat (but not rabbit) antisera. IgA deficiency is associated with a sprue-like syndrome, which may or may not respond to a gluten-free diet. The incidence of autoantibodies, autoimmune diseases, and malignancy is increased. Serum antibodies to IgA are reported in as many as 44% of patients with selective IgA deficiency. If these antibodies are of the IgE isotype, they can cause severe or fatal anaphylactic reactions after intravenous administration of blood products containing IgA. Only 5-times washed (in 200-mL volumes) normal donor erythrocytes (frozen blood would have this done routinely), or blood products from other IgA-deficient individuals, should be administered to patients with IgA deficiency. Administration of intravenous immunoglobulin (IVIG), which is >99% IgG, is not indicated because most IgA-deficient patients make IgG antibodies normally. Many IVIG preparations contain sufficient IgA to cause anaphylactic reactions.
IgG Subclass Deficiencies
Some patients have deficiencies of 1 or more of the 4 subclasses of IgG despite normal or elevated total IgG serum concentration. Some patients with absent or very low concentrations of IgG2 also have IgA deficiency. Other patients with IgG2 deficiency have gone on to develop CVID, suggesting that the presence of IgG subclass deficiency may be a marker for more generalized immune dysfunction. The biologic significance of the numerous moderate deficiencies of IgG subclasses that have been reported is difficult to assess, particularly because commercial laboratory measurement of IgG subclasses is problematic. IgG subclass measurement is not cost-effective in evaluating immune function in the child with recurrent infection. The more relevant issue is a patient’s capacity to make specific antibodies to protein and polysaccharide antigens, because profound deficiencies of antipolysaccharide antibodies have been noted even in the presence of normal concentrations of IgG2. IVIG should not be administered to patients with IgG subclass deficiency unless they are shown to have a deficiency of antibodies to a broad array of antigens.
Immunoglobulin Heavy- and Light-Chain Deletions
Some completely asymptomatic individuals have been documented to have a total absence of IgG1, IgG2, IgG4, and/or IgA1 due to gene deletions. These abnormalities were discovered fortuitously in 16 individuals, 15 of whom had no history of undue susceptibility to infection, and all of whom produced antibodies of all other isotypes in normal quantities. These patients illustrate the importance of assessing specific antibody formation before deciding to initiate IVIG therapy in IgG subclass-deficient patients.
Hyper-IgM Syndrome
The hyper-IgM syndrome is genetically heterogeneous and characterized by normal or elevated serum IgM levels associated with low or absent IgG, IgA, and IgE serum levels, indicating a defect in the class-switch recombination (CSR) process. Causative mutations have been identified in 2 genes on the X chromosome, the CD40 ligand (hyper-IgM syndrome type 1, HIGM1) and NEMO (nuclear factor κB [NF-κB] essential modulator, XHM-ED) genes; and 3 genes on autosomal chromosomes, the AICDA gene (hyper-IgM type 2, HIGM2) on chromosome 12, the uracil DNA glycosylase gene (UNG, hyper-IgM type 5, HIGM5), on chromosome 12, and the CD40 gene (hyper-IgM type 3, HIGM3) on chromosome 20. Distinctive clinical features permit presumptive recognition of the type of mutation in these patients, thereby aiding proper choice of therapy. All such patients should undergo molecular analysis to ascertain the affected gene for purposes of genetic counseling, carrier detection, and decisions regarding definitive therapy.
X-Linked Hyper-IgM Caused by Mutations in the CD40 Ligand: Hyper-IgM Type 1 (HIGM1)
HIGM1 is caused by mutations in the gene that encodes the CD40 ligand (CD154, CD40L), which is expressed on activated T helper cells. Boys with this syndrome have very low serum concentrations of IgG and IgA, with a usually normal or sometimes elevated concentration of polyclonal IgM, may or may not have small tonsils, usually have no palpable lymph nodes, and often profound neutropenia.
Genetics and Pathogenesis
B cells from boys with the CD40 ligand defect are capable of synthesizing not only IgM but also IgA and IgG when co-cultured with normal activated T helper cells, indicating that the B cells are actually normal in this condition and that the defect is in the T cells. The abnormal gene is localized to Xq26, and the gene product, CD154 (CD40L), is the ligand for CD40, which is present on B cells and monocytes. CD154 is upregulated on activated T cells. Mutations in CD154 result in an inability to signal B cells to undergo isotype switching, and thus the B cells produce only IgM. The failure of T cells to interact with B cells through this receptor-ligand pair also causes a failure of upregulation of the B cell and monocyte surface molecules CD80 and CD86 that interact with CD28/CTLA4 on T cells, resulting in failure of “cross talk” between immune system cells. The failure of interaction of the molecules of those pathways results in a propensity for tolerogenic T-cell signaling and defective recognition of tumor cells. More than 73 distinct point mutations or deletions in the gene encoding CD154 have been identified in 87 unrelated families, giving rise to frame shifts, premature stop codons, and single amino acid substitutions, most of which are clustered in the domain with homology to TNF, located in the carboxy-terminal region.
Clinical Manifestations
Similar to patients with XLA, boys with the CD40 ligand defect become symptomatic during the 1st or 2nd yr of life with recurrent pyogenic infections, including otitis media, sinusitis, pneumonia, and tonsillitis. Lymph node histology shows only abortive germinal center formation with severe depletion and phenotypic abnormalities of follicular dendritic cells. These patients have normal numbers of circulating B lymphocytes, marked susceptibility to P. jiroveci pneumonia, and are frequently profoundly neutropenic. Circulating T cells are also present in normal number and in vitro responses to mitogens are normal, but there is decreased antigen-specific T-cell function. In a study of patients with the CD40 ligand defect, 23.3% had died at a mean age at death of 11.7 yr. In addition to opportunistic infections such as P. jiroveci pneumonia, there is an increased incidence of extensive verruca vulgaris lesions, Cryptosporidium enteritis, subsequent liver disease, and increased risk of malignancy. Because of the poor prognosis, the treatment of choice is an HLA-identical stem cell transplant at an early age. Alternative treatment for this condition is monthly infusion of IVIG. In patients with severe neutropenia, the use of G-CSF (granulocyte colony stimulating factor) has been beneficial.
X-Linked Hyper-IgM Caused by Mutations in the Gene Encoding Nuclear Factor κB (NF-κB) Essential Modulator (NEMO, OR IKKγ); XHM-ED
This syndrome in males is characterized most often clinically as anhydrotic ectodermal dysplasia with associated immunodeficiency (EDA-ID). The condition results from missense mutations in the IKBKG gene at position 28q on the X chromosome that encodes nuclear factor κB (NF-κB) essential modulator (NEMO), a regulatory protein required for the activation of the transcription factor NF-κB. Germ line loss-of-function mutations cause the X-linked dominant condition incontinentia pigmenti in females and are lethal in male fetuses. Mutations in the coding region of IKBKG are associated with EDA-ID. The immunodeficiency is variable, with most patients showing impaired antibody responses to polysaccharide antigens. Some patients with EDA-ID have hyper-IgM. Pharmacologic inhibitors of NF-κB activation have been shown to downregulate CD154 mRNA and protein levels, suggesting the mechanism of hyper-IgM in this condition. The hyper-IgM patients with this defect should be easily recognizable because of the presence of ectodermal dysplasia, although there is a report of this condition without ectodermal dysplasia.
Autosomal Recessive Hyper-IgM Caused by Mutations in the Gene for Activation-Induced Cytidine Deaminase (AICDA): Hyper-IgM Type 2 (HIGM2)
An autosomal recessive form of hyper-IgM syndrome is caused by mutations in the gene for activation-induced cytidine deaminase (AICDA).
Genetics and Pathogenesis
Patients with autosomal hyper-IgM usually have normal numbers of circulating B lymphocytes, but, in contrast to patients with the CD40 ligand defect, B cells from these patients are not able to switch from IgM-secreting to IgG-, IgA-, or IgE-secreting cells, even when co-cultured with monoclonal antibodies to CD40 and a variety of cytokines. When their B cells are cultured in vitro, they spontaneously secrete large amounts of IgM, but this is not further augmented by the addition of IL-4 or anti-CD40 with IL-4 or other cytokines. Thus, in these patients, there is truly an intrinsic B-cell abnormality. The defect in many such patients has been identified as due to mutations in a gene on chromosome 12p13 that encodes AICDA. AICDA is a single-stranded (SS) DNA deaminase required for somatic hypermutation (SHM) and CSR of immunoglobulin genes. Histologic examination of the enlarged lymph nodes reveals the presence of giant germinal centers (5-10 times larger than normal) filled with highly proliferating B cells. Proliferating B cells co-express IgM, IgD, and CD38, a phenotype previously described for a small B-cell subset corresponding to germinal center (GC) founder cells. These cells are thought to correspond to a transitional stage between follicular mantle and GC B cells, at the onset of somatic mutation of the Ig variable region gene and antigen-driven selection. Deficiency of AICDA results in impaired terminal differentiation of B cells, a failure of CSR, and lack of immunoglobulin gene SHM.
Clinical Manifestations
Concentrations of serum IgG, IgA, and IgE are very low in AICDA deficiency. In contrast to the CD40 ligand defect, however, the serum IgM concentration in patients with AICDA deficiency is usually markedly elevated and polyclonal. Patients with this form of hyper-IgM have lymphoid hyperplasia, are generally older at age of onset, do not have susceptibility to P. jiroveci pneumonia, often do have isohemagglutinins, and are much less likely to have neutropenia unless it occurs on an autoimmune basis. They have a tendency, however, to develop autoimmune and inflammatory disorders including diabetes mellitus, polyarthritis, autoimmune hepatitis, hemolytic anemia, immune thrombocytopenia, Crohn disease, and chronic uveitis. With early diagnosis and monthly infusions of IVIG, as well as good management of infections with antibiotics, patients with AICDA mutations generally have a more benign course than do boys with the CD40 ligand defect.
Autosomal Recessive Hyper-IgM Caused by Mutations in the Gene for Uracil DNA Glycosylase (UNG); Hyper-IgM Type 5 (HIGM5)
Another cause of the hyper-IgM syndrome is a deficiency of uracil DNA glycosylase.
Genetics and Pathogenesis
AICDA deaminates cytosine into uracil in targeted DNA, which is followed by uracil removal by UNG. Profoundly impaired class-switch recombination was found in 3 hyper-IgM patients reported to have UNG deficiency. Their clinical characteristics were similar to those with AICDA deficiency, with increased susceptibility to bacterial infections and lymphoid hyperplasia. The patients had a markedly elevated serum IgM and profoundly decreased serum IgG and IgA concentrations. Their B cells had an intrinsic defect in CSR when stimulated with anti-CD40 and IL-4 and constitutively produced high quantities of IgM. They had only a partial defect in SHM, however.
Autosomal Recessive Hyper-IgM Caused by Mutations in CD40: Hyper-IgM Type 3 (HIGM3)
Patients with autosomal recessive hyper-IgM who failed to express CD40 on their B-cell surfaces, resulting from mutations in the CD40 gene, were identified.
Genetics and Pathogenesis
CD40 is a type I integral membrane glycoprotein encoded by a gene on chromosome 20 and belonging to the TNF and nerve growth factor receptor superfamily. It is expressed on B cells, macrophages, dendritic cells, and a few other types of cells. Mutations in the CD40 gene cause an autosomal recessive form of hyper-IgM syndrome that is clinically indistinguishable from HIGM1, resulting from the X-linked CD40 ligand (CD154) defect. In contrast to the CD40 ligand defect, however, the B cells in the autosomal recessive condition are intrinsically abnormal and cannot isotype switch. The T cells are normal except to the extent that they cannot cause upregulation of CD80 and CD86 on B cells and macrophages to interact with CD28/CTLA4 on T cells.
X-Linked Lymphoproliferative Disease
X-linked lymphoproliferative (XLP) disease, also referred to as Duncan disease after the original kindred in which it was described, is an X-linked recessive trait characterized by an inadequate immune response to infection with Epstein-Barr virus (EBV).
Genetics and Pathogenesis
The defective gene in XLP was localized to Xq25, cloned, and the gene product was initially named SAP (for SLAM-associated protein), but is now known officially as SH2D1A. SLAM (signaling lymphocyte activation molecule) is an adhesion molecule that is upregulated on both T and B cells with infection and other stimulation. SH2D1A is highly expressed in thymocytes and peripheral blood T and NK cells, with a prevalent expression on Th1 cells. Its presence on B lymphocytes is unclear. Thus, although antibody deficiency is frequently present, this is really a T- and NK-cell defect. SH2D1A competes with SHP-2 for binding to SLAM and, as such, is a regulatory molecule (see Fig. 119-1). In XLP patients, the absence of SH2D1A can lead to an uncontrolled cytotoxic T-cell immune response to EBV. The SH2D1A protein associates permissively with 2B4 on NK cells; thus, selective impairment of 2B4-mediated NK-cell activation also contributes to the immunopathology of XLP.
XLP type 2 is less common and is due to a mutation in XIAP (X-linked inhibitor of apoptosis protein); disease manifestations are similar to XLP.
Clinical Manifestations
Affected males are usually healthy until they acquire EBV infection. The mean age of presentation is <5 yr. There are 3 major clinical phenotypes: (1) fulminant, often fatal, infectious mononucleosis (50% of cases); (2) lymphomas, predominantly involving B-lineage cells (25%); or (3) acquired hypogammaglobulinemia (25%). There is a marked impairment in production of antibodies to the EBV nuclear antigen (EBNA), whereas titers of antibodies to the viral capsid antigen (VCA) have ranged from absent to markedly elevated. XLP has an unfavorable prognosis; 70% of affected boys die by age 10 yr. Only 2 XLP patients are known to have survived beyond 40 yr of age. Unless there is a family history of XLP, diagnosis prior to the onset of complications is difficult because affected individuals are asymptomatic initially. Using mutation analysis, it is possible to identify affected males within identified kindreds before they develop primary EBV infection. Approximately half of the few patients with XLP given HLA-identical related or unrelated stem cell transplants are currently surviving without signs of the disease.
Two pedigrees have been reported in which boys in one arm of each pedigree were diagnosed with CVID, whereas those in the other arms had fulminant infectious mononucleosis. The family members with CVID never gave a history of infectious mononucleosis. All affected members of each pedigree had the same distinct SH2D1A mutation, however, despite the different clinical phenotypes. Because the SH2D1A mutation was the same but the phenotype varied in these families, XLP should be considered in all males with a diagnosis of CVID, particularly if there is more than one male family member with this phenotype.
118.1 Treatment of B-Cell Defects
Except for the CD40 ligand defect and XLP, for which stem cell transplantation is recommended, judicious use of antibiotics to treat documented infections and regular administration of intravenous immunoglobulins are the only effective treatments for primary B-cell disorders. The most common forms of replacement therapy are either intravenous or subcutaneous immunoglobulin (IVIG or SCIG). Broad antibody deficiency should be carefully documented before such therapy is initiated. The rationale for the use of IVIG or SCIG is to provide missing antibodies, not to raise the serum IgG or IgG subclass level. The development of safe and effective immunoglobulin preparations is a major advance in the treatment of patients with severe antibody deficiencies, although it is expensive and there have been national shortages. Almost all commercial preparations are isolated from normal plasma by the Cohn alcohol fractionation method or a modification of this method. Cohn fraction II is then further treated to remove aggregated IgG. Additional stabilizing agents such as sugars, glycine, and albumin are added to prevent reaggregation and protect the IgG molecule during lyophilization. The ethanol used in preparation of immunoglobulin inactivates HIV; and an organic solvent/detergent step inactivates hepatitis B and C viruses. Some preparations are also nanofiltered to remove infectious agents. Most commercial lots are produced from plasma pooled from more than 60,000 donors and therefore contain a broad spectrum of antibodies. Each pool must contain adequate levels of antibody to antigens in various vaccines, such as tetanus and measles. However, there is no standardization based on titers of antibodies to more clinically relevant organisms, such as Streptococcus pneumoniae and Haemophilus influenzae type b.
The IVIG and SCIG preparations available in the USA have similar efficacy and safety. Rare transmission of hepatitis C virus has occurred in the past, but the potential transmission of hepatitis C virus has been resolved by additional treatment with an organic solvent/detergent mixture. There has been no documented transmission of HIV by any of these preparations. IVIG or SCIG at a dose of 400 mg/kg/mo achieves trough IgG levels close to the normal range. Systemic reactions may occur, but rarely are these true anaphylactic reactions. Anaphylactic reactions caused by a patient’s IgE antibodies to IgA in the IVIG or SCIG preparation may occur in patients with CVID or IgA deficiency. Newly diagnosed patients with CVID should be screened through the American Red Cross for anti-IgA antibodies. If anti-IgA antibodies are detected, IVIG therapy should consist of the one available immunoglobulin preparations containing almost no IgA (Gammagard S/D, Baxter).
Fasth A, Nystrom J. Quality of life and health-care resource utilization among children with primary immunodeficiency receiving home treatment with subcutaneous human immunoglobulin. J Clin Immunol. 2008;28:370-378.
The Medical Letter: Subcutaneous immune globulin (SGIC). Med Lett. 2007;49:31-32.
Moore ML, Quinn JM. Subcutaneous immunoglobulin replacement therapy for primary antibody deficiency: advancements into the 21st century. Ann Allerg Asthma Immunol. 2008;101:114-121.
Aghamohammadi A, Mohammadi J, Parvaneh N, et al. Progression of selective IgA deficiency to common variable immunodeficiency. Int Arch Allergy Immunol. 2008;147:87-92.
Bonilla FA, Geha RS. Common variable immunodeficiency. Pediatr Res. 2009;65:13R-19R.
Durandy A, Taubenheim N, Peron S, et al. Pathophysiology of B-cell intrinsic immunoglobulin class switch recombination deficiencies. Adv Immunol. 2007;94:275-306.
Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev. 2009;22:396-414.
Geha RS, Notarangelo LD, Casanova JL, et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol. 2007;120:776-794.
Lougaris V, Badolato R, Ferrari S, et al. Hyper immunoglobulin M syndrome due to CD40 deficiency: clinical, molecular, and immunological features. Immunol Rev. 2005;203:48-66.
Oksenhendler E, Gérard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 46, 2008. 1547–1154
Park MA, Li JT, Hagan JB, et al. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489-502.
Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308-316.
Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967-1976.
Urschel S, Kayikci L, Wintergerst U, et al. Common variable immunodeficiency disorders in children: delayed diagnosis despite typical clinical presentation. J Pediatr. 2009;154:888-894.
van Zelm MC, Reisli I, van der BM, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901-1912.
Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85:193-202.
Wood P, Stanworth S, Burton J, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol. 2007;149:410-423.
Yong PF, Salzer U, Grimbacher B. The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunol Rev. 2009;229:101-113.