Chapter 347 Manifestations of Liver Disease
Pathologic Manifestations
Alterations in hepatic structure and function can be acute or chronic, with varying patterns of reaction of the liver to cell injury. Hepatocyte injury can result in inflammatory cell infiltration or cell death (necrosis), which may be followed by a healing process of scar formation (fibrosis) and, potentially, nodule formation (regeneration). Cirrhosis is the end result of any progressive liver disease.
Injury to individual hepatocytes can result from viral infection, drugs or toxins, hypoxia, immunologic disorders, or inborn errors of metabolism. The evolving process leads to repair, continuing injury with chronic changes, or, in rare cases, to massive hepatic damage.
Cholestasis is an alternative or concomitant response to injury caused by extrahepatic or intrahepatic obstruction to bile flow. Substances that are normally excreted in bile, such as conjugated bilirubin, cholesterol, bile acids, and trace elements, accumulate in serum. Bile pigment accumulation in liver parenchyma can be seen in liver biopsy. In extrahepatic obstruction, bile pigment may be visible in the intralobular bile ducts or throughout the parenchyma as bile lakes or infarcts. In intrahepatic cholestasis, an injury to hepatocytes or an alteration in hepatic physiology leads to a reduction in the rate of secretion of solute and water. Likely causes include alterations in enzymatic or canalicular transporter activity, permeability of the bile canalicular apparatus, organelles responsible for bile secretion, or ultrastructure of the cytoskeleton of the hepatocyte. The end result can be clinically indistinguishable from obstructive cholestasis.
Cirrhosis, defined histologically by the presence of bands of fibrous tissue that link central and portal areas and form parenchymal nodules, is a potential end stage of any acute or chronic liver disease. Cirrhosis can be posthepatitic (after acute or chronic hepatitis) or postnecrotic (after toxic injury), or it can follow chronic biliary obstruction (biliary cirrhosis). Cirrhosis can be macronodular, with nodules of various sizes (up to 5 cm) separated by broad septa, or micronodular, with nodules of uniform size (<1 cm) separated by fine septa; mixed forms occur. The progressive scarring of cirrhosis results in altered hepatic blood flow, with further impairment of liver cell function. Increased intrahepatic resistance to portal blood flow leads to portal hypertension.
The liver can be secondarily involved in neoplastic (metastatic) and non-neoplastic (storage diseases, fat infiltration) processes as well as a number of systemic conditions and infectious processes. The liver can also be affected by chronic passive congestion or acute hypoxia, with hepatocellular damage.
Clinical Manifestations
Hepatomegaly
Enlargement of the liver can be due to several mechanisms (Table 347-1). Normal liver size estimations are based on age-related clinical indices, such as the degree of extension of the liver edge below the costal margin, the span of dullness to percussion, or the length of the vertical axis of the liver, as estimated from imaging techniques. In children, the normal liver edge can be felt up to 2 cm below the right costal margin. In a newborn infant, extension of the liver edge >3.5 cm below the costal margin in the right midclavicular line suggests hepatic enlargement. Measurement of liver span is carried out by percussing the upper margin of dullness and by palpating the lower edge in the right midclavicular line. This may be more reliable than an extension of the liver edge alone. The 2 measurements can correlate poorly.
Table 347-1 MECHANISMS OF HEPATOMEGALY
INCREASE IN THE NUMBER OR SIZE OF THE CELLS INTRINSIC TO THE LIVER
Storage
Inflammation
INFILTRATION OF CELLS
Primary Liver Tumors: Benign
Primary Liver Tumors: Malignant
INCREASED SIZE OF VASCULAR SPACE
INCREASED SIZE OF BILIARY SPACE
IDIOPATHIC
The liver span increases linearly with body weight and age in both sexes, ranging from ∼4.5-5.0 cm at 1 wk of age to ∼7-8 cm in boys and 6.0-6.5 cm in girls by 12 yr of age. The lower edge of the right lobe of the liver extends downward (Riedel lobe) and can be palpated as a broad mass normally in some people. An enlarged left lobe of the liver is palpable in the epigastrium of some patients with cirrhosis. Downward displacement of the liver by the diaphragm (hyperinflation) or thoracic organs can create an erroneous impression of hepatomegaly.
Examination of the liver should note the consistency, contour, tenderness, and presence of any masses or bruits, as well as assessment of spleen size. Documentation of the presence of ascites and any stigmata of chronic liver disease is important.
Ultrasonography (US) is useful in assessment of liver size and consistency, as well as gallbladder size. Hyperechogenic hepatic parenchyma can be seen with metabolic disease (glycogen storage disease) or fatty liver (obesity, malnutrition, hyperalimentation, corticosteroids).
Gallbladder length normally varies from 1.5-5.5 cm (average, 3.0 cm) in infants to 4-8 cm in adolescents; width ranges from 0.5 to 2.5 cm for all ages. Gallbladder distention may be seen in infants with sepsis. The gallbladder is often absent in infants with biliary atresia.
Jaundice (Icterus)
Yellow discoloration of the sclera, skin, and mucous membranes is a sign of hyperbilirubinemia (Chapter 96.3). Clinically apparent jaundice in children and adults occurs when the serum concentration of bilirubin reaches 2-3 mg/dL (34-51 µmol/L); the neonate might not appear icteric until the bilirubin level is >5 mg/dL (>85 µmol/L). Jaundice may be the earliest and only sign of hepatic dysfunction. Liver disease must be suspected in the infant who appears only mildly jaundiced but has dark urine or acholic (light-colored) stools. Immediate evaluation to establish the cause is required.
Measurement of the total serum bilirubin concentration allows quantitation of jaundice. Bilirubin occurs in plasma in 4 forms: unconjugated bilirubin tightly bound to albumin; free or unbound bilirubin (the form responsible for kernicterus, because it can cross cell membranes); conjugated bilirubin (the only fraction to appear in urine); and δ fraction (bilirubin covalently bound to albumin), which appears in serum when hepatic excretion of conjugated bilirubin is impaired in patients with hepatobiliary disease. The δ fraction permits conjugated bilirubin to persist in the circulation and delays resolution of jaundice. Although the terms direct and indirect bilirubin are used equivalently with conjugated and unconjugated bilirubin, this is not quantitatively correct, because the direct fraction includes both conjugated bilirubin and δ bilirubin. An elevation of the serum bile acid level is often seen in the presence of any form of cholestasis.
Investigation of jaundice in an infant or older child must include determination of the accumulation of both unconjugated and conjugated bilirubin. Unconjugated hyperbilirubinemia might indicate increased production, hemolysis, reduced hepatic removal, or altered metabolism of bilirubin (Table 347-2). Conjugated hyperbilirubinemia reflects decreased excretion by damaged hepatic parenchymal cells or disease of the biliary tract, which may be due to obstruction, sepsis, toxins, inflammation, and genetic or metabolic disease (Table 347-3).
Table 347-2 DIFFERENTIAL DIAGNOSIS OF UNCONJUGATED HYPERBILIRUBINEMIA
INCREASED PRODUCTION OF UNCONJUGATED BILIRUBIN FROM HEME
Hemolytic Disease (Hereditary or Acquired)
DECREASED DELIVERY OF UNCONJUGATED BILIRUBIN (IN PLASMA) TO HEPATOCYTE
DECREASED BILIRUBIN UPTAKE ACROSS HEPATOCYTE MEMBRANE
DECREASED STORAGE OF UNCONJUGATED BILIRUBIN IN CYTOSOL (DECREASED Y AND Z PROTEINS)
DECREASED BIOTRANSFORMATION (CONJUGATION)
ENTEROHEPATIC RECIRCULATION
Pruritus
Intense generalized itching, often with skin excoriation, can occur in patients with cholestasis (conjugated hyperbilirubinemia). Pruritus is unrelated to the degree of hyperbilirubinemia; deeply jaundiced patients can be asymptomatic. Although retained components of bile are likely important, the cause is probably multifactorial, as evidenced by the symptomatic relief of pruritus after administration of various therapeutic agents including bile acid-binding agents (cholestyramine), choleretic agents (ursodeoxycholic acid), opiate antagonists, antihistamines, and antibiotics (rifampin). Surgical diversion of bile (partial external biliary diversion) can also provide relief for medically refractory pruritus.
Spider Angiomas
Vascular spiders (telangiectasias), characterized by central pulsating arterioles from which small, wiry venules radiate, may be seen in patients with chronic liver disease; these are usually most prominent on the face and chest. They presumably reflect altered estrogen metabolism in the presence of hepatic dysfunction.
Palmar Erythema
Blotchy erythema, most noticeable over the thenar and hypothenar eminences and on the tips of the fingers, is also noted in patients with chronic liver disease. This may be due to vasodilation and increased blood flow.
Xanthomas
The marked elevation of serum cholesterol levels (to >500 mg/dL) associated with some forms of chronic cholestasis can cause the deposition of lipid in the dermis and subcutaneous tissue. Brown nodules can develop, 1st over the extensor surfaces of the extremities; rarely, xanthelasma of the eyelids develops.
Portal Hypertension
The portal vein drains the splanchnic area (abdominal portion of the gastrointestinal tract, pancreas, and spleen) into the hepatic sinusoids. Normal portal pressure gradient, the pressure difference between the portal vein and the systemic veins (hepatic veins or inferior vena cava), is 3-6 mm Hg. Clinically significant portal hypertension exists when pressure exceeds a threshold of 10 mm Hg. Portal hypertension is the main complication of cirrhosis, directly responsible for 2 of its most common and potentially lethal complications: ascites and variceal hemorrhage.
Ascites
The onset of ascites in the child with chronic liver disease means that the 2 prerequisite conditions for ascites are present: portal hypertension and hepatic insufficiency. Ascites can also be associated with nephrotic syndrome and other urinary tract abnormalities, metabolic diseases (such as lysosomal storage diseases), congenital or acquired heart disease, and hydrops fetalis. Factors favoring the intra-abdominal accumulation of fluid include decreased plasma colloid osmotic pressure, increased capillary hydrostatic pressure, increased ascitic colloid osmotic fluid pressure, and decreased ascitic fluid hydrostatic pressure. Abnormal renal sodium retention must be considered (Chapter 362).
Variceal Hemorrhage
Gastroesophageal varices are the more clinically significant portosystemic collaterals because of their propensity to rupture and cause life-threatening hemorrhage. Variceal hemorrhage results from increased pressure within the varix, which leads to changes in the diameter of the varix and increased wall tension. When the variceal wall strength is exceeded, physical rupture of the varix results. Given the high blood flow and pressure in the portosystemic collateral system, coupled with the lack of a natural mechanism to tamponade variceal bleeding, the rate of hemorrhage can be striking.
Encephalopathy
Hepatic encephalopathy can involve any neurologic function, and it can be prominent or present in subtle forms such as deterioration of school performance, depression, or emotional outbursts. It can be recurrent and precipitated by intercurrent illness, drugs, bleeding, or electrolyte and acid-base disturbances. The appearance of hepatic encephalopathy depends on the presence of portosystemic shunting, alterations in the blood-brain barrier, and the interactions of toxic metabolites with the central nervous system. Postulated causes include altered ammonia metabolism, synergistic neurotoxins, or false neurotransmitters with plasma amino acid imbalance.
Endocrine Abnormalities
Endocrine abnormalities are more common in adults with hepatic disease than in children. They reflect alterations in hepatic synthetic, storage, and metabolic functions, including those concerned with hormonal metabolism in the liver. Proteins that bind hormones in plasma are synthesized in the liver, and steroid hormones are conjugated in the liver and excreted in the urine; failure of such functions can have clinical consequences. Endocrine abnormalities can also result from malnutrition or specific deficiencies.
Renal Dysfunction
Systemic disease or toxins can affect the liver and kidneys simultaneously, or parenchymal liver disease can produce secondary impairment of renal function. In hepatobiliary disorders, there may be renal alterations in sodium and water economy, impaired renal concentrating ability, and alterations in potassium metabolism. Ascites in patients with cirrhosis may be related to inappropriate retention of sodium by the kidneys and expansion of plasma volume, or it may be related to sodium retention mediated by diminished effective plasma volume. Hepatorenal syndrome (HRS) is defined as functional renal failure in patients with end-stage liver disease. The pathophysiology of HRS is poorly defined, but the hallmark is intense renal vasoconstriction (mediated by hemodynamic, humoral, or neurogenic mechanisms) with coexistent systemic vasodilation. The diagnosis is supported by the findings of oliguria (<1 mL/kg/day), a characteristic pattern of urine electrolyte abnormalities (urine sodium <10 mEq/L, fractional excretion of sodium of <1%, urine : plasma creatinine ratio <10, and normal urinary sediment), absence of hypovolemia, and exclusion of other kidney pathology. The best treatment of HRS is timely liver transplantation, because complete renal recovery can be expected.
Pulmonary Involvement
Hepatopulmonary syndrome is characterized by the typical triad of hypoxemia, intrapulmonary vascular dilations, and liver disease. There is intrapulmonic right-to-left shunting of blood, which results in systemic desaturation. It should be suspected and investigated in the child with chronic liver disease with history of shortness of breath or exercise intolerance and clinical examination findings of cyanosis (particularly of the lips and fingers), digital clubbing, and oxygen saturations <96%, particularly in the upright position. Treatment is timely liver transplantation; successful pulmonary resolution follows.
Recurrent Cholangitis
Ascending infection of the biliary system is often seen in pediatric cholestatic disorders, due most commonly to gram-negative enteric organisms, such as Escherichia coli, Klebsiella, Pseudomonas, and Enterococcus. Liver transplantation is the definitive treatment for recurrent cholangitis, especially when medical therapy is not effective.
Miscellaneous Manifestations of Liver Dysfunction
Nonspecific signs of acute and chronic liver disease include anorexia, which often affects patients with anicteric hepatitis and with cirrhosis associated with chronic cholestasis; abdominal pain or distention resulting from ascites, spontaneous peritonitis, or visceromegaly; malnutrition and growth failure; and bleeding, which may be due to altered synthesis of coagulation factors (biliary obstruction with vitamin K deficiency or excessive hepatic damage) or to portal hypertension with hypersplenism. In the presence of hypersplenism, there can be decreased synthesis of specific clotting factors, production of qualitatively abnormal proteins, or alterations in platelet number and function. Altered drug metabolism can prolong the biologic half-life of commonly administered medications.
347.1 Evaluation of Patients with Possible Liver Dysfunction
Adequate evaluation of an infant, child, or adolescent with suspected liver disease involves an appropriate and accurate history, a carefully performed physical examination, and skillful interpretation of signs and symptoms. Further evaluation is aided by judicious selection of diagnostic tests, followed by the use of imaging modalities or a liver biopsy. Most of the so-called liver function tests do not measure specific hepatic functions: a rise in serum aminotransferase levels reflects liver cell injury, an increase in immunoglobulin levels reflects an immunologic response to injury, or an elevation in serum bilirubin levels can reflect any of several disturbances of bilirubin metabolism (see Tables 347-2 and 347-3). Any single biochemical assay provides limited information, which must be placed in the context of the entire clinical picture. The most cost-efficient approach is to become familiar with the rationale, implications, and limitations of a selected group of tests so that specific questions can be answered. Young infants with cholestatic jaundice should be evaluated promptly to identify patients needing surgical intervention.
For a patient with suspected liver disease, evaluation addresses the following issues in sequence: Is liver disease present? If so, what is its nature? What is its severity? Is specific treatment available? How can we monitor the response to treatment? What is the prognosis?
Biochemical Tests
Laboratory tests commonly used to screen for or to confirm a suspicion of liver disease include measurements of serum aminotransferase, bilirubin (total and fractionated), and alkaline phosphatase (AP) levels, as well as determinations of prothrombin time (PT) or international normalized ratio (INR) and albumin level. These tests are complementary, provide an estimation of synthetic and excretory functions, and might suggest the nature of the disturbance (inflammation or cholestasis).
The severity of the liver disease may be reflected in clinical signs or biochemical alterations. Clinical signs include encephalopathy, variceal hemorrhage, worsening jaundice, apparent shrinkage of liver mass owing to massive necrosis, or onset of ascites. Biochemical alterations include hypoglycemia, acidosis, hyperammonemia, electrolyte imbalance, continued hyperbilirubinemia, marked hypoalbuminemia, or a prolonged PT or INR that is unresponsive to parenteral administration of vitamin K.
Acute liver cell injury (parenchymal disease) due to viral hepatitis, drug- or toxin-induced liver disease, shock, hypoxemia, or metabolic disease is best reflected by a marked increase in serum aminotransferase levels. Cholestasis (obstructive disease) involves regurgitation of bile components into serum; the serum levels of total and conjugated bilirubin and serum bile acids are elevated. Elevations in serum AP, 5′ nucleotidase (5′NT), and γ-glutamyl transpeptidase (GGT) levels are also sensitive indicators of obstruction or inflammation of the biliary tract. Fractionation of the total serum bilirubin level into conjugated and unconjugated bilirubin fractions helps to distinguish between elevations caused by processes such as hemolysis and those caused by hepatic dysfunction. A predominant elevation in the conjugated bilirubin level provides a relatively sensitive index of hepatocellular disease or hepatic excretory dysfunction.
Alanine aminotransferase (ALT, serum glutamate pyruvate transaminase) is liver specific, whereas aspartate aminotransferase (AST, serum glutamic-oxaloacetic transaminase) is derived from other organs in addition to the liver. The most marked rises of AST and ALT levels can occur with acute hepatocellular injury; a several thousand–fold elevation can result from acute viral hepatitis, toxic injury, hypoxia, or hypoperfusion. After blunt abdominal trauma, parallel elevations in aminotransferase levels can provide an early clue to hepatic injury. A differential rise or fall in AST and ALT levels sometimes provides useful information. In acute hepatitis, the rise in ALT may be greater than the rise in AST. In alcohol-induced liver injury, fulminant echovirus infection, and various metabolic diseases, more predominant rises in the AST level are reported. In chronic liver disease or in intrahepatic and extrahepatic biliary obstruction, AST and ALT elevations may be less marked. Elevated serum aminotransferase levels are seen in nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH), chronic liver disorders seen in obese children; the notable characteristic is histology similar to alcoholic-induced liver injury in the absence of alcohol abuse.
Hepatic synthetic function is reflected in serum albumin and protein levels and in the PT or INR. Examination of serum globulin concentration and of the relative amounts of the globulin fractions may be helpful. Patients with autoimmune hepatitis often have high gamma-globulin levels and increased titers of anti–smooth muscle, antinuclear, and anti–liver-kidney-microsome antibodies. Antimitochondrial antibodies may also be found in patients with autoimmune hepatitis. A resurgence in α-fetoprotein levels can suggest hepatoma, hepatoblastoma, or hereditary tyrosinemia. Hypoalbuminemia caused by depressed synthesis can complicate severe liver disease and serve as a prognostic factor. Deficiencies of factor V and of the vitamin K–dependent factors (II, VII, IX, and X) can occur in patients with severe liver disease or fulminant hepatic failure. If the PT or INR is prolonged as a result of intestinal malabsorption of vitamin K (resulting from cholestasis) or decreased nutritional intake of vitamin K, parenteral administration of vitamin K should correct the coagulopathy, leading to normalization within 12-24 hr. Unresponsiveness to vitamin K suggests severe hepatic disease. Persistently low levels of factor VII are evidence of a poor prognosis in fulminant liver disease.
Interpretation of results of biochemical tests of hepatic structure and function must be made in the context of age-related changes. The activity of AP varies considerably with age. Normal growing children have significant elevations of serum AP activity originating from influx into serum of the isoenzyme that originates in bone, particularly in rapidly growing adolescents. An isolated increase in AP does not indicate hepatic or biliary disease if other liver function test results are normal. Other enzymes such as 5′NT and GGT are increased in cholestatic conditions, and may be more specific for hepatobiliary disease. 5′NT is not found in bone. GGT exhibits high enzyme activity in early life that declines rapidly with age. Cholesterol concentrations increase throughout life. Cholesterol levels may be markedly elevated in patients with intra- or extrahepatic cholestasis and decreased in severe acute liver disease such as hepatitis.
Interpretation of serum ammonia values must be carried out with caution because of variability in their physiologic determinants and the inherent difficulty in laboratory measurement.
Liver Biopsy
Liver biopsy combined with clinical data can suggest a cause for hepatocellular injury or cholestatic disease in most cases. Specimens of liver tissue can be used to determine a precise histologic diagnosis in patients with neonatal cholestasis, chronic hepatitis, NAFLD or NASH, metabolic liver disease, intrahepatic cholestasis, congenital hepatic fibrosis, or undefined portal hypertension; for enzyme analysis to detect inborn errors of metabolism; and for analysis of stored material such as iron, copper, or specific metabolites. Liver biopsies can monitor responses to therapy or detect complications of treatment with potentially hepatotoxic agents, such as aspirin, anti-infectives (erythromycin, minocycline, ketoconazole, isoniazid), antimetabolites, antineoplastics, or anticonvulsant agents.
In infants and children, needle biopsy of the liver is easily accomplished percutaneously. The amount of tissue obtained, even in small infants, is usually sufficient for histologic interpretation and for biochemical analyses, if the latter are deemed necessary. Percutaneous liver biopsy can be performed safely in infants as young as 1 wk of age. Patients usually require only conscious sedation and local anesthesia. Contraindications to the percutaneous approach include prolonged PT or INR; thrombocytopenia; suspicion of a vascular, cystic, or infectious lesion in the path of the needle; and severe ascites. If administration of fresh frozen plasma or of platelet transfusions fails to correct a prolonged PT, INR, or thrombocytopenia, a tissue specimen can be obtained via alternative techniques. Considerations include either the open laparotomy (wedge) approach by a general surgeon or the transjugular approach under US and fluoroscopic guidance by an experienced pediatric interventional radiologist in an appropriately equipped fluoroscopy suite. The risk of development of a complication such as hemorrhage, hematoma, creation of an arteriovenous fistula, pneumothorax, or bile peritonitis is small.
Hepatic Imaging Procedures
Various techniques help define the size, shape, and architecture of the liver and the anatomy of the intrahepatic and extrahepatic biliary trees. Although imaging might not provide a precise histologic and biochemical diagnosis, specific questions can be answered, such as whether hepatomegaly is related to accumulation of fat or glycogen or is due to a tumor or cyst. These studies can direct further evaluation such as percutaneous biopsy and make possible prompt referral of patients with biliary obstruction to a surgeon. Choice of imaging procedure should be part of a carefully formulated diagnostic approach, with avoidance of redundant demonstrations by several techniques.
A plain x-ray study can suggest hepatomegaly, but a carefully performed physical examination gives a more reliable assessment of liver size. The liver might appear less dense than normal in patients with fatty infiltration or more dense with deposition of heavy metals such as iron. A hepatic or biliary tract mass can displace an air-filled loop of bowel. Calcifications may be evident in the liver (parasitic or neoplastic disease), in the vasculature (portal vein thrombosis), or in the gallbladder or biliary tree (gallstones). Collections of gas may be seen within the liver (abscess), biliary tract, or portal circulation (necrotizing enterocolitis).
US provides information about the size, composition, and blood flow of the liver. Increased echogenicity is observed with fatty infiltration; mass lesions as small as 1-2 cm may be shown. US has replaced cholangiography in detecting stones in the gallbladder or biliary tree. Even in neonates, US can assess gallbladder size, detect dilatation of the biliary tract, and define a choledochal cyst. In infants with biliary atresia, US findings might include small or absent gallbladder; nonvisualization of the common duct; and presence of the triangular cord sign, a triangular or tubular-shaped echogenic density in the bifurcation of the portal vein, representing fibrous remnants at the porta hepatis. In patients with portal hypertension, Doppler US can evaluate patency of the portal vein, demonstrate collateral circulation, and assess size of spleen and amount of ascites. Relatively small amounts of ascitic fluid can also be detected. The use of Doppler US has been helpful in determining vascular patency after liver transplantation.
CT scanning provides information similar to that obtained by US but is less suitable for use in patients <2 yr of age because of the small size of structures, the paucity of intra-abdominal fat for contrast, and the need for heavy sedation or general anesthesia. MRI is a useful alternative. Magnetic resonance cholangiography can be of value in differentiating biliary tract lesions. CT scan or MRI may be more accurate than US in detecting focal lesions such as tumors, cysts, and abscesses. When enhanced by contrast medium, CT scanning can reveal a neoplastic mass density only slightly different from that of a normal liver. When a hepatic tumor is suspected, CT scanning is the best method to define anatomic extent, solid or cystic nature, and vascularity. CT scanning can also reveal subtle differences in density of liver parenchyma, the average liver attenuation coefficient being reduced with fatty infiltration. Increases in density can occur with diffuse iron deposition or with glycogen storage. In differentiating obstructive from nonobstructive cholestasis, CT scanning or MRI identifies the precise level of obstruction more often than US. Either CT scanning or US may be used to guide percutaneously placed fine needles for biopsies, aspiration of specific lesions, or cholangiography.
Radionuclide scanning relies on selective uptake of a radiopharmaceutical agent. Commonly used agents include technetium 99m-labeled sulfur colloid, which undergoes phagocytosis by Kupffer cells; 99mTc-iminodiacetic acid agents, which are taken up by hepatocytes and excreted into bile in a fashion similar to bilirubin; and gallium-67, which is concentrated in inflammatory and neoplastic cells. The anatomic resolution possible with hepatic scintiscans is generally less than that obtained with CT scanning, MRI, or US.
The 99mTc-sulfur colloid scan can detect focal lesions (tumors, cysts, abscesses) >2-3 cm in diameter. This modality can help to evaluate patients with possible cirrhosis and with patchy hepatic uptake and a shift of colloid uptake from liver to bone marrow.
The 99mTc-substituted iminodiacetic acid dyes can differentiate intrahepatic cholestasis from extrahepatic obstruction in neonates. Imaging results are best when scanning is preceded by a 5-7 day period of treatment with phenobarbital to stimulate bile flow. After intravenous injection, the isotope is normally detected in the bowel within 1-2 hr. In the presence of extrahepatic obstruction, excretion of the isotope is delayed; accordingly, serial scans should be made for up to 24 hr after injection. Early in the course of biliary atresia, hepatocyte function is usually good; uptake (clearance) occurs rapidly, but excretion into the intestine is absent. In contrast, uptake is poor in parenchymal liver disease, such as neonatal hepatitis, but excretion into the bile and intestine eventually ensues.
Cholangiography, direct visualization of the intrahepatic and extrahepatic biliary tree after injection of opaque material, may be required in some patients to evaluate the cause, location, or extent of biliary obstruction. Percutaneous transhepatic cholangiography with a fine needle is the technique of choice in infants and young children. The likelihood of opacifying the biliary tract is excellent in patients in whom CT scanning, MRI, or ultrasonography demonstrates dilated ducts. Percutaneous transhepatic cholangiography has been used to outline the biliary ductal system.
Endoscopic retrograde cholangiopancreatography (ERCP) is an alternative method of examining the bile ducts in older children. The papilla of Vater is cannulated under direct vision through a fiberoptic endoscope, and contrast material is injected into the biliary and pancreatic ducts to outline the anatomy.
Selective angiography of the celiac, superior mesenteric, or hepatic artery can be used to visualize the hepatic or portal circulation. Both arterial and venous circulatory systems of the liver can be examined. Angiography is often required to define the blood supply of tumors before surgery and is useful in the study of patients with known or presumed portal hypertension. The patency of the portal system, the extent of collateral circulation, and the caliber of vessels under consideration for a shunting procedure can be evaluated. MRI can provide similar information.
Diagnostic Approach to Infants with Jaundice
The North American Society for Pediatric Gastroenterology, Hepatology and Nutrition has published an algorithm for the evaluation of cholestatic jaundice in neonates and young infants. Well-appearing infants can have cholestatic jaundice. Biliary atresia and neonatal hepatitis are the most common causes of cholestasis in early infancy. Biliary atresia portends a poor prognosis unless it is identified early. The best outcome for this disorder is with early surgical reconstruction (45-60 days of age). History, physical examination, and the detection of a conjugated hyperbilirubinemia via examination of total and direct bilirubin are the 1st steps in evaluating the jaundiced infant (Fig. 347-1). Consultation with a pediatric gastroenterologist should be sought early in the course of the evaluation.
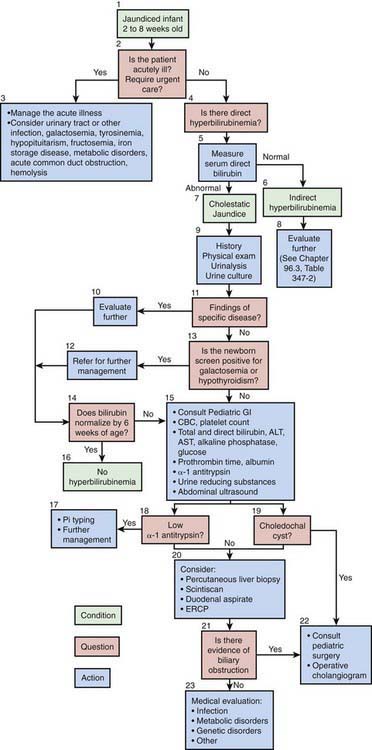
Figure 347-1 Cholestasis clinical practice guideline. Algorithm for a 2-8 wk old. ALT, alanine aminotransferase; AST, aspartate aminotransferase; ERCP, endoscopic retrograde cholangiopancreatography.
(Moyer V, Freese DK, Whitington PF, et al; North American Society for Pediatric Gastroenterology, Hepatology and Nutrition: Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition, J Pediatr Gastroenterol Nutr 39:115–128, 2004.)
Batres LA, Maller ES. Laboratory assessment of liver function and injury in children. In: Suchy FS, Sokol RJ, Balistreri WF, editors. Liver disease in children. ed 2. Philadelphia: Lippincott Williams & Wilkins; 2001:155-170.
Moyer V, Freese DK, Whitington PF, et al. North American Society for Pediatric Gastroenterology, Hepatology and Nutrition: Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2004;39:115-128.
Balistreri WF. Pediatric hepatology. A half-century of progress. Clin Liver Dis. 2000;4:191-210.
Balistreri WF. Bile acid therapy in pediatric hepatobiliary disease: the role of ursodeoxycholic acid. J Pediatr Gastroenterol Nutr. 1997;24:573-589.
Bezerra JA, Balistreri WF. Cholestatic syndromes of infancy and childhood. Semin Gastrointest Dis. 2001;12:54-65.
Feranchak AP, Ramirez RO, Sokol RJ. Medical and nutritional management of cholestasis. In: Suchy FS, Sokol RJ, Balistreri WF, editors. Liver disease in children. ed 2. Philadelphia: Lippincott Williams & Wilkins; 2001:195-238.
Garcia-Tsao G. Current management of the complications of cirrhosis and portal hypertension: variceal hemorrhage, ascites and spontaneous bacterial peritonitis. Gastroenterology. 2001;120:726-748.
Ryckman FC, Alonso MH. Causes and management of portal hypertension in the pediatric population. Clin Liver Dis. 2001;5:789-818.
Ryckman FC, Alonso MH, Bucuvalas JC, Balistreri WF. Liver transplantation in children. In: Suchy FS, Sokol RJ, Balistreri WF, editors. Liver disease in children. ed 2. Philadelphia: Lippincott Williams & Wilkins; 2001:949-974.