Chapter 398 Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis (PAP) is a rare cause of interstitial lung disease characterized by the intra-alveolar accumulation of pulmonary surfactant. Histopathologic examination shows distal air spaces to be filled with a granular, eosinophilic material that stains positively with periodic acid–Schiff reagent and is diastase resistant. Two clinically distinct forms of PAP have been described in children: a fulminant, usually fatal form manifesting shortly after birth (termed congenital PAP) and a gradually progressive type manifesting in older infants and children that is similar to the PAP observed in adults. PAP in older individuals has historically been classified as either primary or secondary to a number of recognized conditions, although this terminology is evolving as specific etiologies for PAP are identified. In the congenital form, alveolar proteinosis is a histologic feature that is found along with alveolar type II cell hyperplasia, interstitial thickening and inflammation, and alveolar macrophage accumulation in what are now being classified as disorders of surfactant metabolism or surfactant dysfunction (Chapter 399).
Etiology and Pathophysiology
Disordered signaling of granulocyte-macrophage colony-stimulating factor (GM-CSF) is the major underlying cause of primary PAP in children and adults. Most cases of primary or idiopathic PAP in older children and adults are mediated by autoantibodies directed against GM-CSF, which can be detected in serum and bronchoalveolar lavage (BAL) fluid. These autoantibodies block binding of GM-CSF to its receptor, thereby inhibiting alveolar macrophage function and surfactant clearance. Mutations in the gene encoding the α subunit of the GM-CSF receptor (CSF2RA) in children with primary alveolar proteinosis demonstrate a genetic basis for some forms of primary PAP in childhood. Deficiency in expression of the β subunit of the GM-CSF receptor has been observed in infants with alveolar proteinosis, although the underlying molecular mechanism for absence of receptor expression has not been elucidated. Alveolar proteinosis has also been reported in children, including young infants, with lysinuric protein intolerance (LPI), a rare autosomal recessive disorder caused by mutations in the cationic amino acid transporter SLC7A7 (Chapter 79.14). These children generally present with vomiting, hyperammonemia, and failure to thrive, although their pulmonary disease may prove fatal. The relationship between the basic defect and the development of PAP is unclear, although a case of recurrence of the disease after lung transplantation suggests that alveolar macrophage dysfunction is likely important in the pathogenesis of PAP associated with LPI. PAP has also been recognized in association with some subtypes of Niemann-Pick disease.
Secondary alveolar proteinosis also may occur in association with infection, particularly in immunocompromised individuals. However, because the same pathologic process occurs in severely immunodeficient mice raised in a pathogen-free environment, it is not clear whether this phenotype results from a secondary infection or the underlying immunodeficiency. Environmental exposures to dust, silica, and chemicals have also been associated with the development of secondary alveolar proteinosis.
Clinical Manifestations
Infants with the congenital form of PAP present in the immediate newborn period and rapidly demonstrate respiratory failure. There is no gender difference in frequency. Congenital PAP is clinically and radiographically indistinguishable from more common disorders of the newborn that lead to respiratory failure, including pneumonia, generalized bacterial infection, respiratory distress syndrome, and total anomalous pulmonary venous return with obstruction. The differential diagnosis also includes primary persistent pulmonary hypertension (Chapter 95.07), meconium aspiration (Chapter 95.06), and alveolar capillary dysplasia, although the radiologic findings of these disorders usually differ. The incidence of congenital PAP is unknown but thought to be low. Alveolar proteinosis in older infants and children is also rare. Boys are affected three times as often as girls. Usually there is no identifiable etiologic factor (primary), although PAP may occur in association with malignancy or infection, particularly in the setting of systemic immunosuppression or congenital immunodeficiency, or following exposure to several inciting agents, such as dust and chemicals (secondary). Older infants and children with PAP present with dyspnea, fatigue, cough, weight loss, chest pain, or hemoptysis. In the later stages, cyanosis and digital clubbing may be seen. Pulmonary function changes include decreased diffusing capacity of carbon monoxide (DLCO), lung volumes with a restrictive abnormality, and arterial blood gas values indicating marked hypoxemia and/or chronic respiratory acidosis.
Diagnosis
Histopathologic examination of lung biopsy specimens remains the gold standard for diagnosis of PAP in children. In patients with alveolar proteinosis caused by mechanisms other than surfactant protein-B (SP-B) deficiency, immunohistochemical staining reveals abundant quantities of alveolar and intracellular surfactant proteins A and B. Latex agglutination tests for the presence of anti–GM-CSF antibodies in BAL fluid or blood are highly sensitive and specific for the acquired forms of alveolar proteinosis. Elevations of GM-CSF in peripheral blood suggest a GM-CSF receptor defect, and molecular analysis of these genes should be pursued. The examination of sputum or BAL fluid for surfactant components has been used diagnostically in adults, but these methods have not been validated in children. Examination of peripheral blood and/or bone marrow for clonogenic stimulation of monocyte-macrophage precursors, GM-CSF receptor and ligand expression, and GM-CSF binding are available through research protocols.
Treatment
The natural history of primary PAP is significantly more variable, making prognostic and therapeutic decisions difficult. Total lung lavage has been associated with prolonged remissions of PAP in adults and remains a therapeutic option for patients with the later-onset form of PAP (Figs. 398-1 and 398-2). It is ineffective in the congenital form of the disease because of inborn errors of surfactant metabolism. The role of repeated BAL in children has not been well studied. It may provide a temporizing measure in some circumstances and may benefit in patients with autoimmune or secondary PAP. In addition, trials have now suggested that subcutaneous or inhaled administration of recombinant GM-CSF may improve pulmonary function in some adults with later-onset PAP. Although experience is limited, both of these interventions have been unsuccessful for the long-term management of PAL in newborn infants. Lung transplantation is a therapeutic option, but its use is limited by concerns about disease recurrence in the absence of a defined primary mechanism of lung injury.
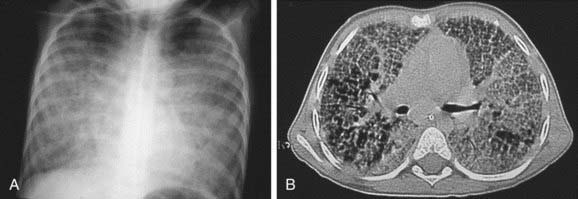
Figure 398-1 Severe pulmonary alveolar proteinosis in a 5 yr old boy before therapeutic lung lavage. A, Chest radiograph shows diffuse alveolo-interstitial infiltrates. B, CT scan demonstrates major air-space opacities and crazy-paving pattern.
(From De Blic J: Pulmonary alveolar proteinosis in children, Paediatr Resp Rev 5:316–322, 2004.)
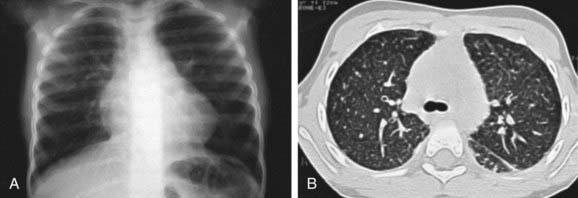
Figure 398-2 Same patient as in Figure 398-1 after 12 therapeutic lung lavages. A, Chest radiograph demonstrates improvement of alveolo-interstitial infiltrates. B, CT scan shows regression of the air-space opacities with a residual micronodular pattern.
(From De Blic J: Pulmonary alveolar proteinosis in children, Paediatr Resp Rev 5:316–322, 2004.)
De Blic J. Pulmonary alveolar proteinosis in children. Paediatr Resp Rev. 2004;5:316-322.
Ikegami M, Whitsett JA, Chroneos ZC, et al. IL-4 increases surfactant and regulates metabolism in vivo. Am J Physiol. 2000;278:175-180.
Kitamura T, Uchida K, Tanaka N, et al. Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2000;162:658-662.
Martinez-Moczygemba M, Doan ML, Elidemir O, et al. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFR gene in the X chromosome pseudoautosomal region 1. J Exp Med. 2008;205:2711-2716.
Seymour JF, Presneill JJ, Schoch OD, et al. Therapeutic efficacy of granulocyte-macrophage colony-stimulating factor in patients with idiopathic acquired alveolar proteinosis. Am J Respir Crit Care Med. 2001;163:524-531.
Suzuki T, Sakagami T, Rubin BK, et al. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med. 2008;205:2703-2710.
Uchida K, Beck DC, Yamamoto T, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007;356:567-578.
Uchida K, Nakata K, Trapnell BC, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. 2004;103:1089-1098.