Chapter 426 Other Congenital Heart and Vascular Malformations
426.1 Anomalies of the Aortic Arch
Right Aortic Arch
In this abnormality, the aorta curves to the right and, if it descends on the right side of the vertebral column, is usually associated with other cardiac malformations. It is found in 20% of cases of tetralogy of Fallot and is also common in truncus arteriosus. A right aortic arch without other cardiac anomalies is not associated with symptoms. It can often be visualized on the chest roentgenogram. The trachea is deviated slightly to the left of the midline rather than to the right, as in the presence of a normal left arch. On a barium esophagogram, the esophagus is indented on its right border at the level of the aortic arch.
Vascular Rings
Congenital abnormalities of the aortic arch and its major branches result in the formation of vascular rings around the trachea and esophagus with varying degrees of compression (Table 426-1). The origin of these lesions can best be appreciated by reviewing the embryology of the aortic arch (Fig. 414-1). The most common anomalies include (1) double aortic arch (Fig. 426-1A), (2) right aortic arch with a left ligamentum arteriosum, (3) anomalous innominate artery arising farther to the left on the arch than usual, (4) anomalous left carotid artery arising farther to the right than usual and passing anterior to the trachea, and (5) anomalous left pulmonary artery (vascular sling). In the latter anomaly, the abnormal vessel arises from an elongated main pulmonary artery or from the right pulmonary artery. It courses between and compresses the trachea and the esophagus. Associated congenital heart disease may be present in 5-50% of patients, depending on the vascular anomaly.
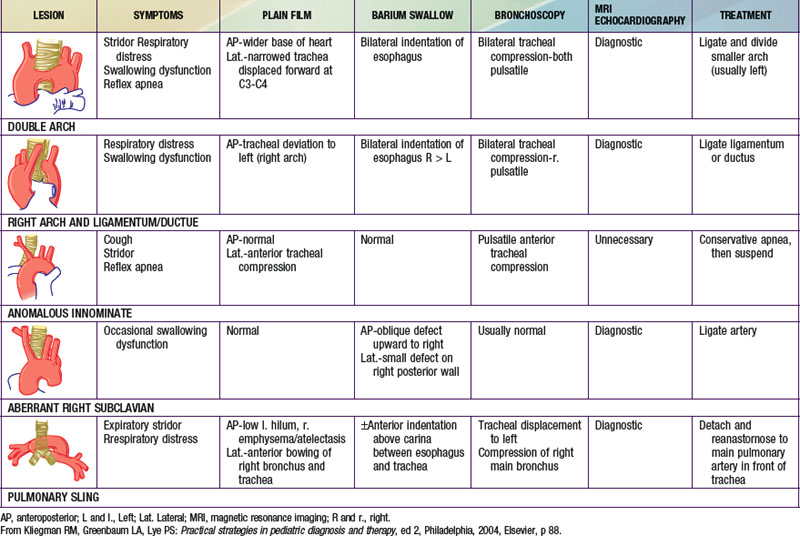
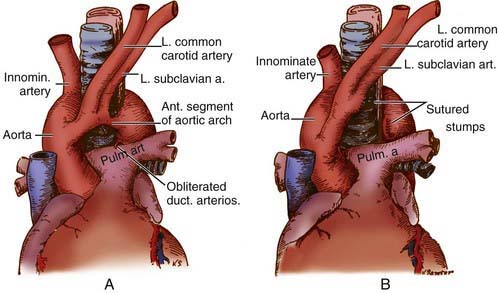
Figure 426-1 Double aortic arch. A, Small anterior segment of the double aortic arch (most common type). B, Operative procedure for release of the vascular ring.
Clinical Manifestations
If the vascular ring produces compression of the trachea and esophagus, symptoms are frequently present during infancy. Chronic wheezing is exacerbated by crying, feeding, and flexion of the neck. Extension of the neck tends to relieve the noisy respiration. Vomiting may also be a component. Affected infants may have a brassy cough, pneumonia, or rarely, sudden death from aspiration.
Diagnosis
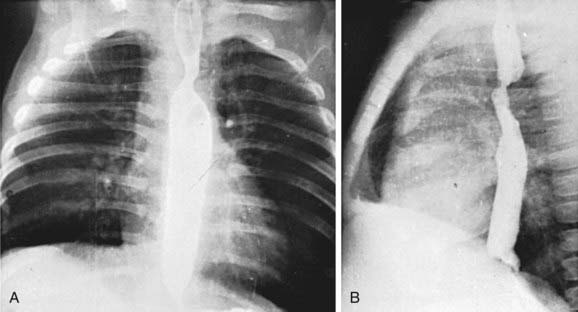
Standard roentgenographic examination is not usually helpful, however, in the past, performing a barium esophagogram was the standard method of diagnosis (Fig. 426-2). Echocardiography in combination with either MRI or CT will usually define the lesion. Cardiac catheterization is reserved for cases with associated anomalies or in rare cases where these other modalities are not diagnostic. Bronchoscopy may be helpful in more severe cases to determine the extent of airway narrowing.
Treatment
Surgery is advised for symptomatic patients who have evidence of tracheal compression. The anterior vessel is usually divided in patients with a double aortic arch (see Fig. 426-1B). Compression produced by a right aortic arch and left ligamentum arteriosum is relieved by division of the latter. Anomalous innominate or carotid arteries cannot be divided; attaching the adventitia of these vessels to the sternum usually relieves the tracheal compression. An anomalous left pulmonary artery is corrected by division at its origin and re-anastomosis to the main pulmonary artery after it has been brought in front of the trachea. Severe tracheomalacia, if present, may require reconstruction of the trachea as well.
Azarow KS, Pearl RH, Hoffman MA, et al. Vascular ring: does magnetic resonance imaging replace angiography? Ann Thorac Surg. 1992;53:882-885.
Bertrand J-M, Chartrand C, Lamarre A, et al. Vascular ring: clinical and physiological assessment of pulmonary function following surgical correction. Pediatr Pulmonol. 1986;2:378-383.
Kussman BD, Geva T, McGowan FX. Cardiovascular causes of airway compression. Paediatr Anaesth. 2004;14:60-74.
Murdison KA, Andrews BA, Chin AJ. Ultrasonographic display of complex vascular rings. J Am Coll Cardiol. 1990;15:1645-1653.
van Son JA, Julsrud PR, Hagler DJ, et al. Imaging strategies for vascular rings. Ann Thorac Surg. 1994;57:604-610.
426.2 Anomalous Origin of the Coronary Arteries
Anomalous Origin of the Left Coronary Artery from the Pulmonary Artery (Alcapa)
In anomalous origin of the left coronary artery from the pulmonary artery, the blood supply to the left ventricular myocardium is severely compromised. Soon after birth, as pulmonary arterial pressure falls, perfusion pressure to the left coronary artery becomes inadequate; myocardial ischemia, infarction, and fibrosis result. In some cases, interarterial collateral anastomoses develop between the right and left coronary arteries. Blood flow in the left coronary artery is then reversed, and it empties into the pulmonary artery, a condition known as the “myocardial steal” syndrome. The left ventricle becomes dilated, and its performance is decreased. Mitral insufficiency is a frequent complication secondary to a dilated valve ring or infarction of a papillary muscle. Localized aneurysms may also develop in the left ventricular free wall. Occasional patients have adequate myocardial blood flow during childhood and, later in life, a continuous murmur and a small left-to-right shunt via the dilated coronary system (aorta to right coronary to left coronary to pulmonary artery).
Clinical Manifestations
Evidence of heart failure becomes apparent within the 1st few months of life, and may be exacerbated by respiratory infection. Recurrent attacks of discomfort, restlessness, irritability, sweating, dyspnea, and pallor occur and probably represent angina pectoris. Cardiac enlargement is moderate to massive. A gallop rhythm is common. Murmurs may be of the nonspecific ejection type or may be holosystolic due to mitral insufficiency. Older patients with abundant intercoronary anastomoses may have continuous murmurs and minimal left ventricular dysfunction. During adolescence, they may experience angina during exercise. Rare patients with an anomalous right coronary artery may also have such clinical findings.
Diagnosis
Roentgenographic examination confirms the cardiomegaly. The electrocardiogram resembles the pattern described in lateral wall myocardial infarction in adults. A QR pattern followed by inverted T waves is seen in leads I and aVL. The left ventricular surface leads (V5 and V6) may also show deep Q waves and exhibit elevated ST segments and inverted T waves (Fig. 426-3). Two-dimensional echocardiography can usually suggest the diagnosis; however, echocardiography is not always reliable in diagnosing this condition. On two-dimensional imaging alone, the left coronary artery may appear as though it is arising from the aorta. Color Doppler ultrasound examination has improved the accuracy of diagnosis of this lesion, demonstrating the presence of retrograde flow in the left coronary artery. CT or MRI may be helpful in confirming the origin of the coronary arteries. Cardiac catheterization is diagnostic; aortography shows immediate opacification of the right coronary artery only. This vessel is large and tortuous. After filling of the intercoronary anastomoses, the left coronary artery is opacified, and contrast can be seen to enter the pulmonary artery. Pulmonary arteriography may also opacify the origin of the anomalous left coronary artery. Selective left ventriculography usually demonstrates a dilated left ventricle that empties poorly and mitral regurgitation.
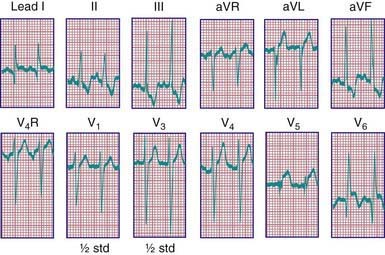
Figure 426-3 Electrocardiogram of a 3 mo old child with anomalous origin of the left coronary artery from the pulmonary artery. Lateral myocardial infarction is present as evidenced by abnormally large and wide Q waves in leads I, V5, and V6; an elevated ST segment in V5 and V6; and inversion of TV6.
Treatment and Prognosis
Untreated, death often occurs from heart failure within the 1st 6 mo of life. Those who survive generally have abundant intercoronary collateral anastomoses. Medical management includes standard therapy for heart failure (diuretics, angiotensin-converting enzyme inhibitors) and for controlling ischemia (nitrates, β-blocking agents).
Surgical treatment consists of detaching the anomalous coronary artery from the pulmonary artery and anastomosing it to the aorta to establish normal myocardial perfusion. A seriously ill infant with a tiny left coronary artery may present a difficult technical problem. In patients who have already sustained a significant myocardial infarction, cardiac transplantation may be the best option (Chapter 437.1).
Anomalous Origin of the Right Coronary Artery from the Pulmonary Artery
Anomalous origin of the right coronary artery from the pulmonary artery is rarely manifested in infancy or early childhood. The left coronary artery is enlarged, whereas the right is thin-walled and mildly enlarged. In early infancy, perfusion of the right coronary artery is from the pulmonary artery, whereas later, perfusion is from collaterals of the left coronary vessels. Angina and sudden death can occur in adolescence or adulthood. When recognized, this anomaly should be repaired by re-anastomosis of the right coronary artery to the aorta.
Ectopic Origin of the Coronary Artery from the Aorta with Aberrant Proximal Course
In ectopic origin of the coronary artery from the aorta with an aberrant proximal course, the aberrant artery may be a left, right, or major branch coronary artery. The site of origin may be the wrong sinus of Valsalva or a proximal coronary artery. The ostium may be hypoplastic, slitlike, or of normal caliber. The aberrant vessel may pass anteriorly, posteriorly, or between the aorta and right ventricular outflow tract; it may tunnel in the conal or interventricular septal tissue. Obstruction resulting from hypoplasia of the ostia, tunneling between the aorta and right ventricular outflow tract or interventricular septum, and acute angulation produces myocardial infarction. Unobstructed vessels produce no symptoms. Patients with this extremely rare abnormality are often initially seen with severe myocardial infarction, ventricular arrhythmias, angina pectoris, or syncope; sudden death may occur, especially in young athletes.
Diagnostic modalities include an electrocardiogram, stress testing, two-dimensional echocardiography, CT or MRI, radionuclide perfusion scan, and cardiac catheterization with selective coronary angiography.
Treatment is indicated for obstructed vessels and consists of aortoplasty with re-anastomosis of the aberrant vessel or, occasionally, coronary artery bypass grafting. The management of asymptomatic infants with these forms of ectopic coronary origin remains controversial.
Chang RR, Allada V. Electrocardiographic and echocardiographic features that distinguish anomalous origin of the left coronary artery from pulmonary artery from idiopathic dilated cardiomyopathy. Pediatr Cardiol. 2001;22:3-10.
Frommelt PC, Berger S, Pelech AN, et al. Prospective identification of anomalous origin of left coronary artery from the right sinus of Valsalva using transthoracic echocardiography: Importance of color Doppler flow mapping. Pediatr Cardiol. 2001;22:327-332.
Johnsrude CL, Perry JC, Cecchin F, et al. Differentiating anomalous left main coronary artery originating from the pulmonary artery in infants from myocarditis and dilated cardiomyopathy by electrocardiogram. Am J Cardiol. 1995;75:71-74.
Schmidt KG, Cooper MJ, Silverman NH, et al. Pulmonary artery origin of the left coronary artery: diagnosis by two-dimensional echocardiography, pulsed Doppler ultrasound and color flow mapping. J Am Coll Cardiol. 1988;11:396-402.
426.3 Pulmonary Arteriovenous Fistula
Fistulous vascular communications in the lungs may be large and localized or multiple, scattered, and small. The most common form of this unusual condition is the Osler-Weber-Rendu syndrome (hereditary hemorrhagic telangiectasia type I), which is also associated with angiomas of the nasal and buccal mucous membranes, gastrointestinal tract, or liver. Mutations in the endoglin gene, a cell surface component of the transforming growth factor-β receptor complex causes this syndrome. The usual communication is between the pulmonary artery and pulmonary vein; direct communication between the pulmonary artery and left atrium is extremely rare. Desaturated blood in the pulmonary artery is shunted through the fistula into the pulmonary vein, thus bypassing the lungs, and then enters the left side of the heart resulting in systemic arterial desaturation and, sometimes, clinically detectable cyanosis. The shunt across the fistula is at low pressure and resistance, so pulmonary arterial pressure is normal; cardiomegaly and heart failure are not present.
The clinical manifestations depend on the magnitude of the shunt. Large fistulas are associated with dyspnea, cyanosis, clubbing, a continuous murmur, and polycythemia. Hemoptysis is rare, but when it occurs, it may be massive. Features of the Osler-Weber-Rendu syndrome are seen in ≈50% of patients (or other family members) and include recurrent epistaxis and gastrointestinal tract bleeding. Transitory dizziness, diplopia, aphasia, motor weakness, or convulsions may result from cerebral thrombosis, abscess, or paradoxical emboli. Soft systolic or continuous murmurs may be audible over the site of the fistula. The electrocardiogram is normal. Roentgenographic examination of the chest may show opacities produced by large fistulas; multiple small fistulas may be visualized by fluoroscopy (as abnormal pulsations), MRI, or CT. Selective pulmonary arteriography demonstrates the site, extent, and distribution of the fistulas.
Treatment consisting of excision of solitary or localized lesions by lobectomy or wedge resection results in complete disappearance of symptoms. In most instances, fistulas are so widespread that surgery is not possible. Any direct communication between the pulmonary artery and the left atrium can be obliterated.
Patients who have undergone a Glenn cavopulmonary anastomosis for cyanotic congenital heart disease (Chapter 424.4) are also at risk for the development of pulmonary arteriovenous malformations. In these patients, the arteriovenous malformations are usually multiple and the risk increases over time after the Glenn procedure. These malformations rarely occur after the heart disease is fully palliated by completion of the Fontan operation. This finding suggests that the pulmonary circulation requires an as yet undetermined hepatic factor to suppress the development of arteriovenous malformations. The hallmark of the development of these malformations is a decrease in the patient’s oxygen saturation. The diagnosis can often be made with contrast echocardiography; cardiac catheterization is the definitive test. Completion of the Fontan circuit, so that inferior vena cava blood flow (containing hepatic venous drainage) is routed through the lungs, usually results in improvement or resolution of the malformations.
Feinstein JA, Moore P, Rosenthal DN, et al. Comparison of contrast echocardiography versus cardiac catheterization for detection of pulmonary arteriovenous malformations. Am J Cardiol. 2002;89:281-285.
Freedom RM, Yoo SJ, Perrin D. The biological “scrabble” of pulmonary arteriovenous malformations: considerations in the setting of cavopulmonary surgery. Cardiol Young. 2004;14:417-437.
Marchuk DA. Genetic abnormalities in hereditary hemorrhagic telangiectasia. Curr Opin Hematol. 1998;5:332-338.
Paterson A. Imaging evaluation of congenital lung abnormalities in infants and children. Radiol Clin North Am. 2005;43:303-323.
Srivastava D, Preminger T, Lock JE, et al. Hepatic venous blood and the development of pulmonary arteriovenous malformations in congenital heart disease. Circulation. 1995;92:1217-1222.
426.4 Ectopia Cordis
In the most common thoracic form of ectopia cordis, the sternum is split and the heart protrudes outside the chest. In other forms, the heart protrudes through the diaphragm into the abdominal cavity or may be situated in the neck. Associated intracardiac anomalies are common. Pentalogy of Cantrell consists of ectopia cordis, midline supraumbilical abdominal defect, deficiency of the anterior diaphragm, defect of the lower sternum, and an intracardiac defect (either a ventricular septal defect, tetralogy of Fallot or diverticulum of the left ventricle). Death may occur early in life, usually from infection, cardiac failure, or hypoxemia. Surgical therapy for neonates without overwhelmingly severe cardiac anomalies consists of covering the heart with skin without compromising venous return or ventricular ejection. Repair or palliation of associated defects is also necessary.
426.5 Diverticulum of the Left Ventricle
Left ventricular diverticulum is a rare anomaly, where the diverticulum protrudes into the epigastrium. The lesion may be isolated or associated with complex cardiovascular anomalies. A pulsating mass is usually visible and palpable in the epigastrium. Systolic or systolic-diastolic murmurs produced by blood flow into and out of the diverticulum may be audible over the lower part of the sternum and the mass. The electrocardiogram shows a pattern of complete or incomplete left bundle branch block. Roentgenograms of the chest may or may not show the mass. Associated abnormalities include defects of the sternum, abdominal wall, diaphragm, and pericardium (see earlier). Surgical treatment of the diverticulum and associated cardiac defects can be performed in selected cases. Occasionally, a diverticulum may be small and not associated with clinical signs or symptoms. These small diverticula are diagnosed at the time of echocardiographic examination for other indications.