Chapter 424 Cyanotic Congenital Heart Lesions
Lesions Associated with Decreased Pulmonary Blood Flow
424.1 Tetralogy of Fallot
Tetralogy of Fallot is one of the conotruncal family of heart lesions in which the primary defect is an anterior deviation of the infundibular septum (the muscular septum that separates the aortic and pulmonary outflows). The consequences of this deviation are the 4 components: (1) obstruction to right ventricular outflow (pulmonary stenosis), (2) a malalignment type of ventricular septal defect (VSD), (3) dextroposition of the aorta so that it overrides the ventricular septum, and (4) right ventricular hypertrophy (Fig. 424-1). Obstruction to pulmonary arterial blood flow is usually at both the right ventricular infundibulum (subpulmonic area) and the pulmonary valve. The main pulmonary artery may be small, and various degrees of branch pulmonary artery stenosis may be present. Complete obstruction of right ventricular outflow (pulmonary atresia with VSD) is classified as an extreme form of tetralogy of Fallot (Chapter 424.2). The degree of pulmonary outflow obstruction determines the degree of the patient’s cyanosis and the age of first presentation.
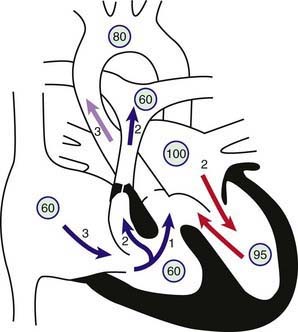
Figure 424-1 Physiology of the tetralogy of Fallot. Circled numbers represent oxygen saturation values. The numbers next to the arrows represent volumes of blood flow (in L/min/m2). Atrial (mixed venous) oxygen saturation is decreased because of the systemic hypoxemia. A volume of 3 L/min/m2 of desaturated blood enters the right atrium and traverses the tricuspid valve. Two liters flows through the right ventricular outflow tract into the lungs, whereas 1 L shunts right to left through the ventricular septal defect (VSD) into the ascending aorta. Thus, pulmonary blood flow is two thirds normal (Qp:Qs [pulmonary-to-systemic blood flow ratio] of 0.7 : 1). Blood returning to the left atrium is fully saturated. Only 2 L of blood flows across the mitral valve. Oxygen saturation in the left ventricle may be slightly decreased because of right-to-left shunting across the VSD. Two liters of saturated left ventricular blood mixing with 1 L of desaturated right ventricular blood is ejected into the ascending aorta. Aortic saturation is decreased, and cardiac output is normal.
Pathophysiology
The pulmonary valve annulus may range from being nearly normal in size to being severely hypoplastic. The valve itself is often bicuspid or unicuspid and, occasionally, is the only site of stenosis. More commonly, the subpulmonic or infundibular muscle, known as the crista supraventricularis, is hypertrophic, which contributes to the subvalvar stenosis and results in an infundibular chamber of variable size and contour. When the right ventricular outflow tract is completely obstructed (pulmonary atresia), the anatomy of the branch pulmonary arteries is extremely variable. A main pulmonary artery segment may be in continuity with right ventricular outflow, separated by a fibrous but imperforate pulmonary valve; the main pulmonary artery may be moderately or severely hypoplastic but still supply part or all of the pulmonary bed; or the entire main pulmonary artery segment may be absent. Occasionally, the branch pulmonary arteries may be discontinuous. Pulmonary blood flow may be supplied by a patent ductus arteriosus (PDA) or by multiple major aortopulmonary collateral arteries (MAPCAs) arising from the ascending and descending aorta and supplying various lung segments.
The VSD is usually nonrestrictive and large, is located just below the aortic valve, and is related to the posterior and right aortic cusps. Rarely, the VSD may be in the inlet portion of the ventricular septum (atrioventricular septal defect). The normal fibrous continuity of the mitral and aortic valves is usually maintained, and if not (due to the presence of a subaortic muscular conus) the classification is usually that of double outlet right ventricle (Chapter 424.5). The aortic arch is right sided in 20% of cases, and the aortic root is usually large and overrides the VSD to varying degrees. When the aorta overrides the VSD by more than 50% and if there is a subaortic conus, this defect is classified as a form of double-outlet right ventricle; however, the circulatory dynamics are the same as that of tetralogy of Fallot.
Systemic venous return to the right atrium and right ventricle is normal. When the right ventricle contracts in the presence of marked pulmonary stenosis, blood is shunted across the VSD into the aorta. Persistent arterial desaturation and cyanosis result, the degree dependent on the severity of the pulmonary obstruction. Pulmonary blood flow, when severely restricted by the obstruction to right ventricular outflow, may be supplemented by a PDA. Peak systolic and diastolic pressures in each ventricle are similar and at systemic level. A large pressure gradient occurs across the obstructed right ventricular outflow tract, and pulmonary arterial pressure is either normal or lower than normal. The degree of right ventricular outflow obstruction determines the timing of the onset of symptoms, the severity of cyanosis, and the degree of right ventricular hypertrophy. When obstruction to right ventricular outflow is mild to moderate and a balanced shunt is present across the VSD, the patient may not be visibly cyanotic (acyanotic or “pink” tetralogy of Fallot). When obstruction is severe, cyanosis will be present from birth and worsen when the ductus begins to close.
Clinical Manifestations
Infants with mild degrees of right ventricular outflow obstruction may initially be seen with heart failure caused by a ventricular-level left-to-right shunt. Often, cyanosis is not present at birth; but with increasing hypertrophy of the right ventricular infundibulum as the patient grows, cyanosis occurs later in the 1st yr of life. In infants with severe degrees of right ventricular outflow obstruction, neonatal cyanosis is noted immediately. In these infants, pulmonary blood flow may be partially or nearly totally dependent on flow through the ductus arteriosus. When the ductus begins to close in the 1st few hours or days of life, severe cyanosis and circulatory collapse may occur. Older children with long-standing cyanosis who have not undergone surgery may have dusky blue skin, gray sclerae with engorged blood vessels, and marked clubbing of the fingers and toes. Extracardiac manifestations of long-standing cyanotic congenital heart disease are described in Chapter 428.
In older children with unrepaired tetralogy, dyspnea occurs on exertion. They may play actively for a short time and then sit or lie down. Older children may be able to walk a block or so before stopping to rest. Characteristically, children assume a squatting position for the relief of dyspnea caused by physical effort; the child is usually able to resume physical activity after a few minutes of squatting. These findings occur most often in patients with significant cyanosis at rest.
Paroxysmal hypercyanotic attacks (hypoxic, “blue,” or “tet” spells) are a particular problem during the 1st 2 yr of life. The infant becomes hyperpneic and restless, cyanosis increases, gasping respirations ensue, and syncope may follow. The spells occur most frequently in the morning on initially awakening or after episodes of vigorous crying. Temporary disappearance or a decrease in intensity of the systolic murmur is usual as flow across the right ventricular outflow tract diminishes. The spells may last from a few minutes to a few hours. Short episodes are followed by generalized weakness and sleep. Severe spells may progress to unconsciousness and, occasionally, to convulsions or hemiparesis. The onset is usually spontaneous and unpredictable. Spells are associated with reduction of an already compromised pulmonary blood flow, which, when prolonged, results in severe systemic hypoxia and metabolic acidosis. Infants who are only mildly cyanotic at rest are often more prone to the development of hypoxic spells because they have not acquired the homeostatic mechanisms to tolerate rapid lowering of arterial oxygen saturation, such as polycythemia.
Depending on the frequency and severity of hypercyanotic attacks, one or more of the following procedures should be instituted in sequence: (1) placement of the infant on the abdomen in the knee-chest position while making certain that the infant’s clothing is not constrictive, (2) administration of oxygen (although increasing inspired oxygen will not reverse cyanosis caused by intracardiac shunting), and (3) injection of morphine subcutaneously in a dose not in excess of 0.2 mg/kg. Calming and holding the infant in a knee-chest position may abort progression of an early spell. Premature attempts to obtain blood samples may cause further agitation and be counterproductive.
Because metabolic acidosis develops when arterial PO2 is <40 mm Hg, rapid correction (within several minutes) with intravenous administration of sodium bicarbonate is necessary if the spell is unusually severe and the child shows a lack of response to the foregoing therapy. Recovery from the spell is usually rapid once the pH has returned to normal. Repeated blood pH measurements may be necessary because rapid recurrence of acidosis may ensue. For spells that are resistant to this therapy, intubation and sedation are often sufficient to break the spell. Drugs that increase systemic vascular resistance, such as intravenous phenylephrine, can improve right ventricular outflow, decrease the right-to-left shunt, and improve the symptoms. β-Adrenergic blockade by the intravenous administration of propranolol (0.1 mg/kg given slowly to a maximum of 0.2 mg/kg) has also been used. Growth and development may be delayed in patients with severe untreated tetralogy of Fallot, particularly when their oxygen saturation is chronically <70%. Puberty may also be delayed in patients who have not undergone surgery.
The pulse is usually normal, as are venous and arterial pressures. In older infants and children, the left anterior hemithorax may bulge anteriorly because of long-standing right ventricular hypertrophy. A substernal right ventricular impulse can usually be detected. A systolic thrill may be felt along the left sternal border in the 3rd and 4th parasternal spaces. The systolic murmur is usually loud and harsh; it may be transmitted widely, especially to the lungs, but is most intense at the left sternal border. The murmur is generally ejection in quality at the upper sternal border, but it may sound more holosystolic toward the lower sternal border. It may be preceded by a click. The murmur is caused by turbulence through the right ventricular outflow tract. It tends to become louder, longer, and harsher as the severity of pulmonary stenosis increases from mild to moderate; however, it can actually become less prominent with severe obstruction, especially during a hypercyanotic spell due to shunting of blood away from the right ventricular outflow through the aortic valve. Either the 2nd heart sound is single, or the pulmonic component is soft. Infrequently, a continuous murmur may be audible, especially if prominent collaterals are present.
Diagnosis
Roentgenographically, the typical configuration as seen in the anteroposterior view consists of a narrow base, concavity of the left heart border in the area usually occupied by the pulmonary artery, and normal overall heart size. The hypertrophied right ventricle causes the rounded apical shadow to be uptilted so that it is situated higher above the diaphragm than normal and pointing horizontally to the left chest wall. The cardiac silhouette has been likened to that of a boot or wooden shoe (“coeur en sabot”) (Fig. 424-2). The hilar areas and lung fields are relatively clear because of diminished pulmonary blood flow or the small size of the pulmonary arteries, or both. The aorta is usually large, and in about 20% of patients it arches to the right, which results in an indentation of the leftward-positioned air-filled tracheobronchial shadow in the anteroposterior view.
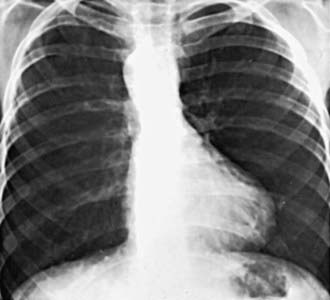
Figure 424-2 Chest x-ray of an 8 yr old boy with the tetralogy of Fallot. Note the normal heart size, some elevation of the cardiac apex, concavity in the region of the main pulmonary artery, right-sided aortic arch, and diminished pulmonary vascularity.
The electrocardiogram demonstrates right axis deviation and evidence of right ventricular hypertrophy. A dominant R wave appears in the right precordial chest leads (Rs, R, qR, qRs) or an RSR′ pattern. In some cases, the only sign of right ventricular hypertrophy may initially be a positive T wave in leads V3R and V1. The P wave is tall and peaked suggesting right atrial enlargement (see Fig. 417-6).
Two-dimensional echocardiography establishes the diagnosis (Fig. 424-3) and provides information about the extent of aortic override of the septum, the location and degree of the right ventricular outflow tract obstruction, the size of the pulmonary valve annulus and main and proximal branch pulmonary arteries, and the side of the aortic arch. The echocardiogram is also useful in determining whether a PDA is supplying a portion of the pulmonary blood flow. In a patient without pulmonary atresia, echocardiography usually obviates the need for catheterization before surgical repair.
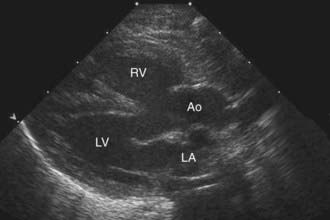
Figure 424-3 Echocardiogram in a patient with the tetralogy of Fallot. This parasternal long-axis two-dimensional view demonstrates anterior displacement of the outflow ventricular septum that resulted in stenosis of the subpulmonic right ventricular outflow tract, overriding of the aorta, and an associated ventricular septal defect. Ao, overriding aorta; LA, left atrium; LV, left ventricle; RV, right ventricle.
Cardiac catheterization demonstrates a systolic pressure in the right ventricle equal to the systemic pressure, since the right ventricle is connected directly to the overriding aorta. If the pulmonary artery is entered, the pressure is markedly decreased, although crossing the right ventricular outflow tract, especially in severe cases, may precipitate a tet spell. Pulmonary arterial pressure is usually lower than normal, in the range of 5-10 mm Hg. The level of arterial oxygen saturation depends on the magnitude of the right-to-left shunt; in “pink tets,” the systemic oxygen saturation may be normal, whereas in a moderately cyanotic patient at rest, it is usually 75-85%.
Selective right ventriculography will demonstrate all of the anatomical features. Contrast medium outlines the heavily trabeculated right ventricle. The infundibular stenosis varies in length, width, contour, and distensibility (Fig. 424-4). The pulmonary valve is usually thickened, and the annulus may be small. In patients with pulmonary atresia and VSD, echocardiography alone is not adequate to assess the anatomy of the pulmonary arteries and MAPCAs. Cardiac CT is extremely helpful, and cardiac catheterization with injection into each arterial collateral is indicated. Complete and accurate information regarding the size and peripheral distribution of the main pulmonary arteries and any collateral vessels (MAPCAs) is important when evaluating these children as surgical candidates.
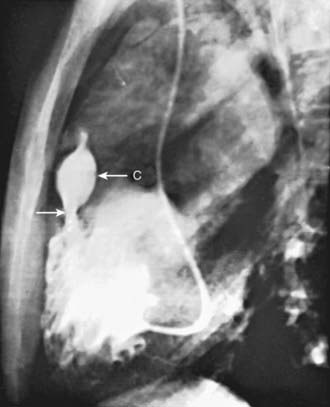
Figure 424-4 Lateral view of a selective right ventriculogram in a patient with the tetralogy of Fallot. The arrow points to an infundibular stenosis that is below the infundibular chamber (C). The narrowed pulmonary valve orifice is seen at the distal end of the infundibular chamber.
Aortography or coronary arteriography outlines the course of the coronary arteries. In 5-10% of patients with the tetralogy of Fallot, coronary artery abnormalities may be present, most commonly an aberrant coronary artery crossing over the right ventricular outflow tract; this artery must not be cut during surgical repair. Verification of normal coronary arteries is important when considering surgery in young infants who may need a patch across the pulmonary valve annulus. Echocardiography can usually delineate the coronary artery anatomy; angiography is reserved for cases in which questions remain.
Complications
Before the age of corrective surgery, patients with tetralogy of Fallot were susceptible to several serious complications. For this reason, most children undergo complete repair (or in some cases palliation) in infancy, and therefore these days these complications are rare. Cerebral thromboses, usually occurring in the cerebral veins or dural sinuses and occasionally in the cerebral arteries, are a sequelae of extreme polycythemia and dehydration. Thromboses occur most often in patients younger than 2 yr. These patients may have iron-deficiency anemia, frequently with hemoglobin and hematocrit levels in the normal range (but too low for cyanotic heart disease). Therapy consists of adequate hydration and supportive measures. Phlebotomy and volume replacement with albumin or saline are indicated in extremely polycythemic patients who are symptomatic.
Brain abscess is less common than cerebral vascular events and extremely rare today. Patients with a brain abscess are usually older than 2 yr. The onset of the illness is often insidious and consists of low-grade fever or a gradual change in behavior, or both. Some patients have an acute onset of symptoms that may develop after a recent history of headache, nausea, and vomiting. Seizures may occur; localized neurologic signs depend on the site and size of the abscess and the presence of increased intracranial pressure. CT or MRI confirms the diagnosis. Antibiotic therapy may help keep the infection localized, but surgical drainage of the abscess is usually necessary (Chapter 596).
Bacterial endocarditis may occur in the right ventricular infundibulum or on the pulmonic, aortic, or, rarely, tricuspid valves. Endocarditis may complicate palliative shunts or, in patients with corrective surgery, any residual pulmonic stenosis or VSD. Heart failure is not a usual feature in patients with tetralogy of Fallot, with the exception of some young infants with “pink” or acyanotic tetralogy of Fallot. As the degree of pulmonary obstruction worsens with age, the symptoms of heart failure resolve and eventually the patient experiences cyanosis, often by 6-12 mo of age. These patients are at increased risk for hypercyanotic spells at this time.
Associated Anomalies
A PDA may be present, and defects in the atrial septum are occasionally seen. A right aortic arch occurs in ≈20% of patients, and other anomalies of the pulmonary arteries and aortic arch may also be seen. Persistence of a left superior vena cava draining into the coronary sinus is common but not a concern. Multiple VSDs are occasionally present and must be diagnosed before corrective surgery. Coronary artery anomalies are present in 5-10% and can complicate surgical repair. Tetralogy of Fallot may also occur with an atrioventricular septal defect, often associated with Down syndrome.
Congenital absence of the pulmonary valve produces a distinct syndrome that is usually marked by signs of upper airway obstruction (Chapter 422.1). Cyanosis may be absent, mild, or moderate; the heart is large and hyperdynamic; and a loud to-and-fro murmur is present. Marked aneurysmal dilatation of the main and branch pulmonary arteries results in compression of the bronchi and produces stridulous or wheezing respirations and recurrent pneumonia. If the airway obstruction is severe, reconstruction of the trachea at the time of corrective cardiac surgery may be required to alleviate the symptoms.
Absence of a branch pulmonary artery, most often the left, should be suspected if the roentgenographic appearance of the pulmonary vasculature differs on the two sides; absence of a pulmonary artery is often associated with hypoplasia of the affected lung. It is important to recognize the absence of a pulmonary artery because occlusion of the remaining pulmonary artery during surgery seriously compromises the already reduced pulmonary blood flow.
As one of the conotruncal malformations, tetralogy of Fallot can be associated with DiGeorge syndrome or Shprintzen velocardiofacial syndrome, also known by the acronym CATCH 22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia). Cytogenetic analysis using fluorescence in situ hybridization demonstrates deletions of a large segment of chromosome 22q11 known as the DiGeorge critical region. Deletion or mutation of the gene encoding the transcription factor Tbx1 has been implicated as a possible cause of DiGeorge syndrome, although several other genes have been identified as possible candidates or modifier genes.
Treatment
Treatment of tetralogy of Fallot depends on the severity of the right ventricular outflow tract obstruction. Infants with severe tetralogy require urgent medical treatment and surgical intervention in the neonatal period. Therapy is aimed at providing an immediate increase in pulmonary blood flow to prevent the sequelae of severe hypoxia. The infant should be transported to a medical center adequately equipped to evaluate and treat neonates with congenital heart disease under optimal conditions. Prolonged, severe hypoxia may lead to shock, respiratory failure, and intractable acidosis and will significantly reduce the chance of survival, even when surgically amenable lesions are present. It is critical that normal body temperature be maintained during the transfer since cold increases oxygen consumption, which places additional stress on a cyanotic infant, whose oxygen delivery is already limited. Blood glucose levels should be monitored because hypoglycemia is more likely to develop in infants with cyanotic heart disease.
Neonates with marked right ventricular outflow tract obstruction may deteriorate rapidly because, as the ductus arteriosus begins to close, pulmonary blood flow is further compromised. The intravenous administration of prostaglandin E1 (0.01-0.20 µg/kg/min), a potent and specific relaxant of ductal smooth muscle, causes dilatation of the ductus arteriosus and usually provides adequate pulmonary blood flow until a surgical procedure can be performed. This agent should be administered intravenously as soon as cyanotic congenital heart disease is clinically suspected and continued through the preoperative period and during cardiac catheterization. Because prostaglandin can cause apnea, an individual skilled in neonatal intubation should be readily available.
Infants with less severe right ventricular outflow tract obstruction who are stable and awaiting surgical intervention require careful observation. Acyanotic patients can fairly quickly progress to having cyanotic episodes. Prevention or prompt treatment of dehydration is important to avoid hemoconcentration and possible thrombotic episodes. Oral propranolol (0.5-1 mg/kg every 6 hr) had been used in the past to decrease the frequency and severity of hypercyanotic spells, but with the excellent surgical results available today, surgical treatment is now indicated as soon as spells begin.
Infants with symptoms and severe cyanosis in the 1st mo of life usually have marked obstruction of the right ventricular outflow tract. Two options are available in these infants. The first is corrective open heart surgery performed in early infancy and even in the newborn period in critically ill infants. This approach has widespread acceptance today with excellent short- and long-term results and has supplanted palliative shunts (see later) for most cases. Early total repair carries the theoretical advantage that early physiologic correction allows for improved growth of the branch pulmonary arteries. In infants with less severe cyanosis who can be maintained with good growth and absence of hypercyanotic spells, primary repair is performed electively at between 4 and 6 mo of age.
Corrective surgical therapy consists of relief of the right ventricular outflow tract obstruction by resecting obstructive muscle bundles and by patch closure of the VSD. If the pulmonary valve is stenotic, as it usually is, a valvotomy is performed. If the pulmonary valve annulus is too small or the valve is extremely thickened, a valvectomy may be performed, the pulmonary valve annulus split open, and a transannular patch placed across the pulmonary valve ring. The surgical risk of total correction in major centers is <5%. A right ventriculotomy was once the standard approach; a transatrial-transpulmonary approach is routinely performed to reduce the long-term risks of a large right ventriculotomy. In patients in whom repair has been delayed to childhood, increased bleeding in the immediate postoperative period may be a complicating factor due to their extreme polycythemia.
The second option, more common in previous years, is a palliative systemic-to-pulmonary artery shunt (Blalock-Taussig shunt) performed to augment pulmonary artery blood flow. The rationale for this surgery, previously the only option for these patients, is to augment pulmonary blood flow to decrease the amount of hypoxia and improve linear growth, as well as augment growth of the branch pulmonary arteries. The modified Blalock-Taussig shunt is currently the most common aortopulmonary shunt procedure and consists of a Gore-Tex conduit anastomosed side to side from the subclavian artery to the homolateral branch of the pulmonary artery (Fig. 424-5). Sometimes the shunt is brought directly from the ascending aorta to the main pulmonary artery and in this case is called a central shunt. The Blalock-Taussig operation can be successfully performed in the newborn period with shunts 3-4 mm in diameter and has also been used successfully in premature infants.
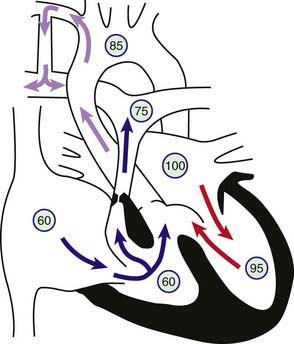
Figure 424-5 Physiology of a Blalock-Taussig shunt in a patient with the tetralogy of Fallot. Circled numbers represent oxygen saturation values. The intracardiac shunting pattern is as described for Figure 424-1. Blood shunting left to right across the shunt from the right subclavian artery to the right pulmonary artery increases total pulmonary blood flow and results in a higher oxygen saturation than would exist without the shunt (see Fig. 424-1).
Postoperative complications after a Blalock-Taussig shunt include chylothorax, diaphragmatic paralysis, and Horner syndrome. Postoperative pulmonary overcirculation leading to symptoms of cardiac failure may be caused by too large a shunt; its treatment is described in Chapter 436. Vascular problems other than a diminished radial pulse and occasional long-term arm length discrepancy are rarely seen in the upper extremity supplied by the subclavian artery used for the anastomosis.
After a successful shunt procedure, cyanosis diminishes. The development of a continuous murmur over the lung fields after the operation indicates a functioning anastomosis. A good continuous shunt murmur may not be heard until several days after surgery. The duration of symptomatic relief is variable. As the child grows, more pulmonary blood flow is needed and the shunt eventually becomes inadequate. When increasing cyanosis develops rapidly, thrombosis of the shunt should be suspected, often requiring emergent surgery.
Prognosis
After successful total correction, patients are generally asymptomatic and are able to lead unrestricted lives. Uncommon immediate postoperative problems include right ventricular failure, transient heart block, residual VSD with left-to-right shunting, and myocardial infarction from interruption of an aberrant coronary artery. Postoperative heart failure (particularly in patients with a large transannular outflow patch) may require anticongestive therapy. The long-term effects of isolated, surgically induced pulmonary valvular insufficiency are still being defined as more patients with repaired tetralogy of Fallot reach middle age, but insufficiency is generally well tolerated through adolescence. Many patients after tetralogy repair and all of those with transannular patch repairs have a to-and-fro murmur at the left sternal border, usually indicative of mild outflow obstruction and mild to moderate pulmonary insufficiency. Patients with more marked pulmonary valve insufficiency also have moderate to marked heart enlargement and may develop tricuspid regurgitation as the tricuspid valve annulus dilates. These patients will develop a holosystolic murmur at the lower left sternal border. Patients with a severe residual gradient across the right ventricular outflow tract may require reoperation, but mild to moderate obstruction is usually present and does not require reintervention.
Follow-up of patients 5-20 yr after surgery indicates that the marked improvement in symptoms is generally maintained. Asymptomatic patients nonetheless have lower than normal exercise capacity, maximal heart rate, and cardiac output. These abnormal findings are more common in patients who underwent placement of a transannular outflow tract patch and may be less frequent when surgery is performed at an early age. As these children move into adolescence and adulthood, some (more commonly those with transannular patches) will develop right ventricular dilation due to severe pulmonary regurgitation. After reaching adulthood, careful lifelong follow-up by a specialist in adult congenital heart disease is important. Serial echocardiography and magnetic resonance angiography (MRA) are valuable tools for assessing the degree of right ventricular dilation, the presence of right ventricular dysfunction, and for quantifying the regurgitant fraction. Valve replacement is indicated for those patients with increasing right ventricular dilation and tricuspid regurgitation.
Conduction disturbances can occur after surgery. The atrioventricular node and the bundle of His and its divisions are in close proximity to the VSD and may be injured during surgery; however, permanent complete heart block after tetralogy repair is rare. When present, it should be treated by placement of a permanently implanted pacemaker. Even transient complete heart block in the immediate postoperative period is rare; it may be associated with an increased incidence of late-onset complete heart block and sudden death. In contrast, right bundle branch block is quite common on the postoperative electrocardiogram. The duration of the QRS interval has been shown to predict both the presence of residual hemodynamic derangement and the long-term risk of arrhythmia and sudden death. Research is ongoing to determine the effectiveness of biventricular pacing (in which a pacemaker is used to resynchronize the activation of the right and left ventricles) in improving hemodynamics in those patients with long ventricular conduction delays.
A number of children have premature ventricular beats after repair of the tetralogy of Fallot. These beats are of concern in patients with residual hemodynamic abnormalities; 24-hr electrocardiographic (Holter) monitoring studies should be performed to be certain that occult short episodes of ventricular tachycardia are not occurring. Exercise studies may be useful in provoking cardiac arrhythmias that are not apparent at rest. In the presence of complex ventricular arrhythmias or severe residual hemodynamic abnormalities, prophylactic antiarrhythmic therapy, catheter ablation, or implantation of an implantable defibrillator is warranted. Re-repair is indicated if significant residual right ventricular outflow obstruction or severe pulmonary insufficiency is present, because arrhythmias may improve after hemodynamics are restored to a more normal level.
Apitz C, Webb GD, Redington AN. Tetralogy of Fallot. Lancet. 2009;374:1462-1470.
Boudjemline Y, Fermont L, Le Bidois J, et al. Prevalence of 22q11 deletion in fetuses with conotruncal cardiac defects: a 6-year prospective study. J Pediatr. 2001;138:520-524.
Dubin AM, Feinstein JA, Reddy VM, et al. Electrical resynchronization: a novel therapy for the failing right ventricle. Circulation. 2003;107:2287-2289.
Greenway SC, Pereira AC, Lin JC, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931-935.
Griffin HR, Hall DH, Topf A, et al. Genetic variation in VEGF does not contribute significantly to the risk of congenital cardiovascular malformation. PLoS ONE. 2009;4:e4978.
Jonas RA. Early primary repair of tetralogy of Fallot. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009;12:39-47.
Malhotra SP, Hanley FL. Surgical management of pulmonary atresia with ventricular septal defect and major aortopulmonary collaterals: a protocol-based approach. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009:145-151.
Momenah TS, El Oakley R, Al Najashi K, et al. Extended application of percutaneous pulmonary valve implantation. J Am Coll Cardiol. 2009;53:1859-1863.
Reddy VM, McElhinney DB, Amin Z, et al. Early and intermediate outcomes after repair of pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries: experience with 85 patients. Circulation. 2000;101:1826-1832.
Schultz AH, Wernovsky G. Late outcomes in patients with surgically treated congenital heart disease. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2005;8:145-156.
Wald RM, Haber I, Wald R, et al. Effects of regional dysfunction and late gadolinium enhancement on global right ventricular function and exercise capacity in patients with repaired tetralogy of Fallot. Circulation. 2009;119:1370-1377.
424.2 Tetralogy of Fallot with Pulmonary Atresia
Pathophysiology
Tetralogy of a Fallot with pulmonary atresia is the most extreme form of the tetralogy of Fallot. The pulmonary valve is atretic (absent), and the pulmonary trunk may be hypoplastic or atretic as well. The entire right ventricular output is ejected into the aorta. Pulmonary blood flow is then dependent on collateral vessels or major aortopulmonary collateral arteries (MAPCAs) or, less commonly, on a PDA. The ultimate prognosis depends on the degree of development of the branch pulmonary arteries, which needs to be assessed by cardiac catheterization. If the pulmonary arteries are severely hypoplastic and fail to grow after palliative shunt procedures, heart-lung transplantation may be the only therapy (Chapter 437.2). Pulmonary atresia with VSD is also associated with the CATCH 22 deletion and DiGeorge syndrome. The association of severe tracheomalacia or bronchomalacia with these severe forms of tetralogy/pulmonary atresia may complicate postoperative recovery.
Clinical Manifestations
Patients with pulmonary atresia and VSD have findings similar to those in patients with severe tetralogy of Fallot. Cyanosis usually appears within the 1st few hr or days after birth; however, the prominent systolic murmur associated with the tetralogy is usually absent. The 1st heart sound may be followed by an ejection click caused by the enlarged aortic root, the 2nd heart sound is single and loud, and continuous murmurs of a PDA or bronchial collateral flow may be heard over the entire precordium, both anteriorly and posteriorly. Most patients are moderately cyanotic and are initially stabilized with a prostaglandin E1 infusion pending cardiac catheterization or CT scan to further delineate the anatomy. Patients with several large MAPCAs may be less cyanotic and, once the diagnosis is confirmed, can be taken off prostaglandin while awaiting palliative surgical intervention. Some patients may even develop symptoms of heart failure caused by increased pulmonary blood flow via these collateral vessels.
Diagnosis
The chest roentgenogram demonstrates a varying heart size, depending on the amount of pulmonary blood flow, a concavity at the position of the pulmonary arterial segment, and often the reticular pattern of bronchial collateral flow. The electrocardiogram shows right ventricular hypertrophy. The echocardiogram identifies aortic override, a thick right ventricular wall, and atresia of the pulmonary valve. Pulsed and color Doppler echocardiographic studies show an absence of forward flow through the pulmonary valve, with pulmonary blood flow being supplied by MAPCAs, which can usually be seen arising from the descending aorta. At cardiac catheterization, right ventriculography reveals a large aorta, opacified immediately by passage of contrast medium through the VSD, but with no dye entering the lungs through the right ventricular outflow tract. Careful delineation of the native pulmonary arteries, if present, to determine whether they are continuous or discontinuous and whether they arborize to all lung segments is important in planning surgical repair. The location and arborization of all MAPCAs is also determined by selective contrast injection.
Treatment
The surgical procedure of choice depends on whether the main pulmonary artery segment is present and, if so, on the size and branching pattern of the branch pulmonary arteries. If these arteries are well developed, a one-stage surgical repair with a homograft conduit between the right ventricle and pulmonary arteries and closure of the VSD is feasible. If the pulmonary arteries are hypoplastic, extensive reconstruction may be required. This usually involves several staged surgical procedures. If the native pulmonary artery is small, a connection made between the aorta and the hypoplastic native pulmonary artery (aortopulmonary window) in the newborn period induces growth. At 3-4 mo of age, the multiple MAPCAs are gathered together (unifocalization procedure) and eventually incorporated into the final repair along with the native pulmonary arteries. This may be accomplished through successive lateral thoracotomies, or through a single midline sternotomy if the anatomy is more favorable.
To be a candidate for full repair, the pulmonary arteries must be of adequate size to accept the full volume of right ventricular output. Complete repair includes closure of the VSD and placement of a homograft conduit from the right ventricle to the pulmonary artery. At the time of reparative surgery, previous shunts are taken down. Because of patient growth as well as homograft narrowing due to proliferation of intimal tissue and calcification, replacement of the homograft conduit replacement is usually required in later life, and multiple replacements may be needed. Patients with obstruction of the very distal branches of the pulmonary arteries may undergo transcatheter balloon dilatation of the multiple branch pulmonary arterial stenoses, as these distal branches may be difficult to reach surgically.
424.3 Pulmonary Atresia with Intact Ventricular Septum
Pathophysiology
In pulmonary atresia with an intact ventricular septum, the pulmonary valve leaflets are completely fused to form a membrane and the right ventricular outflow tract is atretic. Because no VSD is present, no egress of blood from the right ventricle can occur. Any blood that enters the right ventricle will regurgitate back across the tricuspid valve into the right atrium. Right atrial pressure increases, and blood shunts via the foramen ovale into the left atrium, where it mixes with pulmonary venous blood and enters the left ventricle (Fig. 424-6). The combined left and right ventricular output is pumped solely by the left ventricle into the aorta. In a newborn with pulmonary atresia, the only source of pulmonary blood flow occurs via a PDA. The right ventricle and tricuspid valve are usually hypoplastic, although the degree of hypoplasia varies considerably. Patients who have a small right ventricular cavity also tend to be those with the smallest tricuspid valve annulus, which limits right ventricular inflow. Patients with pulmonary atresia and intact ventricular septum may have coronary sinusoidal channels within the right ventricular wall that communicate directly with the coronary arterial circulation. The high right ventricular pressure results in desaturated blood flowing retrograde via these channels into the coronary arteries. Sometimes there are also stenoses of the coronary arteries proximal to where the sinusoids enter, so that distal coronary artery flow is dependent on flow from the right ventricle (known as RV dependent coronary circulation). The prognosis in patients with these sinusoids and proximal stenosis of the coronary arteries is more guarded than in those patients without sinusoids or with sinusoids but no coronary stenoses. Rarely, the proximal coronary artery may be totally absent.
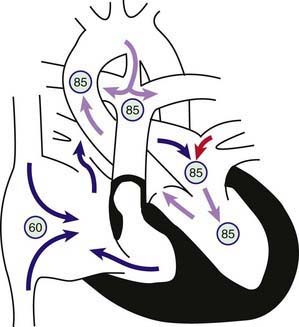
Figure 424-6 Physiology of pulmonary atresia with an intact ventricular septum. Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. A small amount of the blood entering the right atrium may cross the tricuspid valve, which is often stenotic as well. The right ventricular cavity is hypertrophied and may be hypoplastic. No outlet from the right ventricle exists because of the atretic pulmonary valve; thus, any blood entering the right ventricle returns to the right atrium via tricuspid regurgitation. Most of the desaturated blood shunts right to left via the foramen ovale into the left atrium, where it mixes with fully saturated blood returning from the lungs. The only source of pulmonary blood flow is via the patent ductus arteriosus. Aortic and pulmonary arterial oxygen saturation will be identical (definition of a total mixing lesion).
Clinical Manifestations
As the ductus arteriosus closes in the 1st hours/days of life, infants with pulmonary atresia and an intact ventricular septum become markedly cyanotic since their only source of pulmonary blood flow is removed. Untreated, most patients die within the 1st wk of life. Physical examination reveals severe cyanosis and respiratory distress. The 2nd heart sound, representing only aortic closure, is single and loud. Often, no murmurs are audible; sometimes a systolic or continuous murmur can be heard secondary to ductal blood flow. A harsh holosytolic murmur may be heard at the lower left sternal border if there is significant tricuspid regurgitation.
Diagnosis
The electrocardiogram shows a frontal QRS axis between 0 and +90 degrees, the amount of leftward shift reflecting the degree of hypoplasia of the right ventricle. Tall, spiked P waves indicate right atrial enlargement. QRS voltages are consistent with left ventricular dominance or hypertrophy; right ventricular forces are decreased in proportion to the decreased size of the right ventricular cavity. Most patients with small right ventricles have decreased right ventricular forces, but, occasionally, patients with larger, thickened right ventricular cavities may have evidence of right ventricular hypertrophy. The chest roentgenogram shows decreased pulmonary vascularity, the degree depending on the size of the branch pulmonary arteries and the patency of the ductus. Unlike in patients with pulmonary atresia and tetralogy of Fallot, the presence of major collateral vessels (MAPCAs) is rare.
The two-dimensional echocardiogram is useful in estimating right ventricular dimensions and the size of the tricuspid valve annulus, which have been shown to be of prognostic value. Echocardiography can often suggest the presence of sinusoidal channels but cannot be used to evaluate coronary stenoses. Thus, cardiac catheterization is necessary for complete evaluation. Pressure measurements reveal right atrial and right ventricular hypertension. Ventriculography demonstrates the size of the right ventricular cavity, the atretic right ventricular outflow tract, the degree of tricuspid regurgitation, and the presence or absence of intramyocardial sinusoids filling the coronary vessels. Aortography shows filling of the pulmonary arteries via the PDA and is helpful in determining the size and branching patterns of the pulmonary arterial bed. An aortogram, or if necessary, selective coronary angiography is performed to evaluate for the presence of proximal coronary artery stenosis (right ventricular dependent coronary circulation).
Treatment
Infusion of prostaglandin E1 (0.01-0.20 µg/kg/min) is usually effective in keeping the ductus arteriosus open before intervention, thus reducing hypoxemia and acidemia before surgery. The choice of surgical procedure depends on whether there is an RV dependent coronary circulation and on the size of the right ventricular cavity. In patients with only mild to moderate right ventricular hypoplasia without sinusoids, or in patients with sinusoids but no evidence of coronary stenoses, a surgical pulmonary valvotomy is carried out to relieve outflow obstruction. Often, the right ventricular outflow tract is widened with a patch. To preserve adequate pulmonary blood flow, an aortopulmonary shunt may also be performed during the same procedure. An alternative approach utilizes interventional catheterization, in which the imperforate pulmonary valve is first punctured either with a wire or a radiofrequency ablation catheter, followed by a balloon valvuloplasty. If this course is taken, it may take days to weeks before the right ventricular muscle regresses enough for the patient to be weaned from prostaglandin, and many of these patients will still require surgical intervention. The aim of surgery or interventional catheterization is to encourage growth of the right ventricular chamber by allowing some forward flow through the pulmonary valve while using the shunt to ensure adequate pulmonary blood flow. Later, if the tricuspid valve annulus and right ventricular chamber grow to adequate size, the shunt is taken down and any remaining atrial level shunt can be closed. If the right ventricular chamber remains too small for use as a pulmonary ventricle, then the patient is treated as a single ventricle circulation, with a Glenn procedure followed by a modified Fontan procedure (Chapter 424.4) allowing blood to bypass the hypoplastic right ventricle by flowing to the pulmonary arteries directly from the venae cavae. When coronary artery stenoses are present and retrograde coronary perfusion occurs from the right ventricle via myocardial sinusoids, the prognosis is more guarded because of a higher risk of arrhythmias, coronary ischemia, and sudden death. It is important for these patients not to try to open the right ventricular outflow tract, as dropping the right ventricular pressure will reduce coronary perfusion, leading to ischemia. These patients are usually treated with an aortopulmonary shunt, followed by the Glenn and Fontan procedure. Although at higher risk than those without coronary stenoses, recent reports suggest that these palliative procedures can still be successful. Some of these infants, especially those with total atresia of a proximal coronary artery, are referred instead for heart transplantation.
Ekman-Joelsson BM, Berggren H, Boll AB, et al. Abnormalities in myocardial perfusion after surgical correction of pulmonary atresia with intact ventricular septum. Cardiol Young. 2008;18:89-95.
Guleserian KJ, Armsby LB, Thiagarajan RR, et al. Natural history of pulmonary atresia with intact ventricular septum and right-ventricle-dependent coronary circulation managed by the single-ventricle approach. Ann Thorac Surg. 2006;81:2250-2257.
Hirata Y, Chen JM, Quaegebeur JM, et al. Pulmonary atresia with intact ventricular septum: limitations of catheter-based intervention. Ann Thorac Surg. 2007;84:574-579.
L’Ecuyer TJ, Poulik JM, Vincent JA. Myocardial infarction due to coronary abnormalities in pulmonary atresia with intact ventricular septum. Pediatr Cardiol. 2001;22:68-70.
424.4 Tricuspid Atresia
Pathophysiology
In tricuspid atresia, no outlet from the right atrium to the right ventricle is present; the entire systemic venous return leaves the right atrium and enters the left side of the heart by means of the foramen ovale or, most often, through an atrial septal defect (Fig. 424-7). The physiology of the circulation and the clinical presentation will depend on the presence of other congenital heart defects, most notably on whether the great vessels are normally related or are transposed (aorta arising from the right ventricle, pulmonary artery from the left ventricle). In patients with normally related great vessels, left ventricular blood supplies the systemic circulation via the aorta. Blood also usually flows into the right ventricle via a VSD (if the ventricular septum is intact, the right ventricle will be completely hypoplastic and pulmonary atresia will be present [Chapter 424.3]). Pulmonary blood flow (and thus the degree of cyanosis) depends on the size of the VSD and the presence and severity of any associated pulmonic stenosis. Pulmonary blood flow may be augmented by or be totally dependent on a PDA. The inflow portion of the right ventricle is always missing in these patients, but the outflow portion is of variable size. The clinical presentation of patients with tricuspid atresia and normally related great vessels will depend on the degree of pulmonary obstruction. Patients with at least moderate degrees of pulmonary stenosis are recognized in the early days or weeks of life by decreased pulmonary blood flow and cyanosis. Alternatively, in those with a large VSD and minimal or no right ventricular outflow obstruction, pulmonary blood flow may be high; these patients have only mild cyanosis and present with signs of pulmonary overcirculation and heart failure.
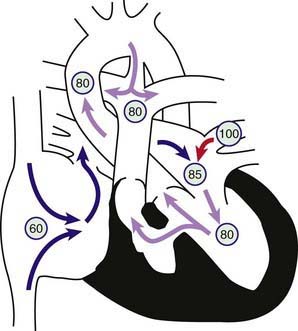
Figure 424-7 Physiology of tricuspid atresia with normally related great vessels. Circled numbers represent oxygen saturation values. Right atrial (mixed venous) oxygen saturation is decreased secondary to systemic hypoxemia. The tricuspid valve is nonpatent, and the right ventricle may manifest varying degrees of hypoplasia. The only outlet from the right atrium involves shunting right to left across an atrial septal defect or patent foramen ovale to the left atrium. There, desaturated blood mixes with saturated pulmonary venous return. Blood enters the left ventricle and is ejected either through the aorta or via a ventricular septal defect (VSD) into the right ventricle. In this example, some pulmonary blood flow is derived from the right ventricle, the rest from a patent ductus arteriosus (PDA). In patients with tricuspid atresia, the PDA may close or the VSD may grow smaller and result in a marked decrease in systemic oxygen saturation.
In patients with tricuspid atresia and transposition of the great arteries, left ventricular blood flows directly into the pulmonary artery, whereas systemic blood must traverse the VSD and right ventricle to reach the aorta. In these patients, pulmonary blood flow is usually massively increased and heart failure develops early. If the VSD is restrictive, aortic blood flow may be compromised. Coarctation of the aorta is not uncommon in this setting.
Clinical Manifestations
Some degree of cyanosis is usually evident at birth, with the extent depending on the degree of limitation to pulmonary blood flow (as noted above). An increased left ventricular impulse may be noted, in contrast to most other causes of cyanotic heart disease, in which an increased right ventricular impulse is usually present. The majority of patients have holosystolic murmurs audible along the left sternal border; the 2nd heart sound is usually single. Pulses in the lower extremities may be weak or absent in the presence of transposition with coarctation of the aorta. Patients with tricuspid atresia are at risk for spontaneous narrowing or even closure of the VSD, which can occasionally occur rapidly and lead to a marked increase in cyanosis.
Diagnosis
Roentgenographic studies show either pulmonary under circulation (usually in patients with normally related great vessels) or over circulation (usually in patients with transposed great vessels). Left axis deviation and left ventricular hypertrophy are generally noted on the electrocardiogram (except in those patients with transposition of the great arteries), and these features distinguish tricuspid atresia from most other cyanotic heart lesions. Thus the combination of cyanosis and left axis deviation on the electrocardiogram is highly suggestive of tricuspid atresia. In the right precordial leads, the normally prominent R wave is replaced by an rS complex. The left precordial leads show a qR complex, followed by a normal, flat, biphasic, or inverted T wave. RV6 is normal or tall, and SV1 is generally deep. The P waves are usually biphasic, with the initial component tall and spiked in lead II. Two-dimensional echocardiography reveals the presence of a fibromuscular membrane in place of a tricuspid valve, a variably small right ventricle, VSD, and the large left ventricle (Fig. 424-8). The relationship of the great vessels (normal or transposed) can be determined. The degree of obstruction at the level of the VSD or at the right ventricular outflow tract can be determined by Doppler examination. Blood flow through a patent ductus can be evaluated by color flow and pulsed Doppler.
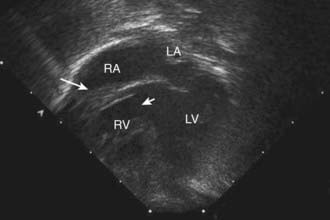
Figure 424-8 Echocardiogram demonstrating tricuspid atresia. The floor of the right atrium consists of a fibromuscular membrane (large arrow) instead of the normal tricuspid valve apparatus. The large secundum atrial septal defect can be seen between the right and left atria. The small arrow shows the ventricular septal defect. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
Cardiac catheterization, indicated usually only if questions remain after echocardiography, shows normal or slightly elevated right atrial pressure with a prominent a wave. If the right ventricle is entered through the VSD, the pressure may be lower than on the left if the VSD is restrictive in size. Right atrial angiography shows immediate opacification of the left atrium from the right atrium followed by left ventricular filling and visualization of the aorta. Absence of direct flow to the right ventricle results in an angiographic filling defect between the right atrium and the left ventricle.
Treatment
Management of patients with tricuspid atresia depends on the adequacy of pulmonary blood flow. Severely cyanotic neonates should be maintained on an intravenous infusion of prostaglandin E1 (0.01-0.20 µg/kg/min) until a surgical aortopulmonary shunt procedure can be performed to increase pulmonary blood flow. The Blalock-Taussig procedure (Chapter 424.1) or a variation is the preferred anastomosis. Rare patients with restrictive atrial-level communications also benefit from a Rashkind balloon atrial septostomy (Chapter 425.2) or surgical septectomy.
Infants with increased pulmonary blood flow because of an unobstructed pulmonary outflow tract (more often patients with aortopulmonary transposition) may require pulmonary arterial banding to decrease the symptoms of heart failure and protect the pulmonary bed from the development of pulmonary vascular disease. Infants with just adequate pulmonary blood flow who are well balanced between cyanosis and pulmonary overcirculation can be watched closely for the development of increasing cyanosis, which may occur as the VSD begins to get smaller or the pulmonary outflow becomes narrower and is an indication for surgery.
The next stage of palliation for patients with tricuspid atresia involves the creation of an anastomosis between the superior vena cava and the pulmonary arteries (bidirectional Glenn shunt; Fig. 424-9A). This procedure is performed at usually between 3 and 6 mo of age. The benefit of the Glenn shunt is that it reduces the volume load on the left ventricle and may lessen the chance of left ventricular dysfunction developing later in life.
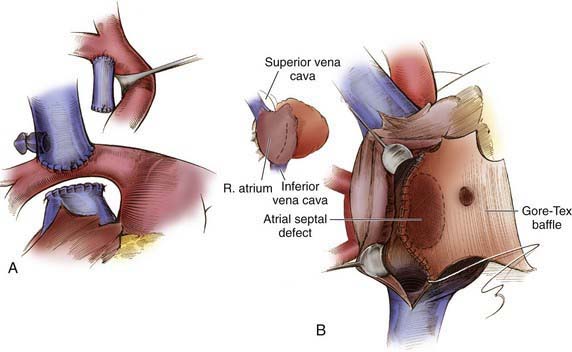
Figure 424-9 A, Bidirectional Glenn shunt showing the superior vena cava–right pulmonary anastomosis. B, A modified Fontan procedure (cavopulmonary isolation) is completed with placement of a baffle to convey inferior vena cava blood along the lateral wall of the right atrium to the superior vena cava orifice. A 4 mm fenestration is sometimes made on the medial aspect of the polytetrafluoroethylene baffle.
(From Castañeda AR, Jonas RA, Mayer JE Jr, et al: Single-ventricle tricuspid atresia. In Cardiac surgery of the neonate and infant, Philadelphia, 1994, WB Saunders.)
The modified Fontan operation is the preferred approach to later surgical management. It is usually performed between 1.5 and 3 yr of age, usually after the patient is ambulatory. Initially, this procedure was performed by anastomosing the right atrium or atrial appendage directly to the pulmonary artery. A modification of the Fontan procedure, known as a cavopulmonary isolation procedure, involves anastomosing the inferior vena cava to the pulmonary arteries, either via a baffle that runs along the lateral wall of the right atrium (lateral tunnel Fontan; see Fig. 424-9B) or via a homograft or Gore-Tex tube running outside the heart (external conduit Fontan). The advantage of these later approaches is that blood flows by a more direct route into the pulmonary arteries, thereby decreasing the possibility of right atrial dilatation and markedly reducing the incidence of postoperative pleural effusions, which were common with the earlier method. In a completed Fontan repair, desaturated blood flows from both venae cavae directly into the pulmonary arteries. Oxygenated blood returns to the left atrium, enters the left ventricle, and is ejected into the systemic circulation. The volume load is completely removed from the left ventricle, and the right-to-left shunt is abolished. Because of the reliance on passive filling of the pulmonary circulation, the Fontan procedure is contraindicated in patients with elevated pulmonary vascular resistance, in those with pulmonary artery hypoplasia, and in patients with left ventricular dysfunction. The patient must also not have significant mitral insufficiency. Patients who are not in normal sinus rhythm are at increased risk and if a pacemaker is required in these patients, dual chamber pacing is the preferred approach.
Postoperative problems after the Fontan procedure include marked elevation of systemic venous pressure, fluid retention, and pleural or pericardial effusions. In the past, pleural effusions were a problem in 30-40% of patients using the standard Fontan procedure, but the cavopulmonary isolation procedure now in use reduces this risk to about 5%. Some centers use a fenestration at the time of the Fontan, consisting of a small communication between the inferior vena cava and the pulmonary artery conduit and the left atrium. This serves as a “pop-off” during early postoperative recovery and may hasten hospital discharge. The fenestration will result in some amount of right-to-left shunting, and is therefore usually closed with a catheter closure device after the immediate postoperative period.
Late complications of the Fontan procedure include baffle obstruction causing superior or inferior vena cava syndrome, vena cava or pulmonary artery thromboembolism, protein-losing enteropathy, supraventricular arrhythmias (atrial flutter, paroxysmal atrial tachycardia), and hepatic cirrhosis due to persistently elevated central venous pressures. Left ventricular dysfunction may be a late occurrence, usually not until adolescence or young adulthood. Heart transplantation is a successful treatment option for pediatric patients with “failed” Fontan circuits but is a somewhat riskier procedure in adults.
Anderson RH, Cook AC. Morphology of the functionally univentricular heart. Cardiol Young. 2004;14(Suppl 1):3-12.
Bernstein D, Naftel D, Chin C, et al. Pediatric Heart Transplant Study. Outcome of listing for cardiac transplantation for failed Fontan: a multi-institutional study. Circulation. 2006;114:273-280.
Blaufox AD, Sleeper LA, Bradley DJ, et al. Functional status, heart rate, and rhythm abnormalities in 521 Fontan patients 6 to 18 years of age. J Thorac Cardiovasc Surg. 2008;136:100-107.
Mair DD, Puga FJ, Danielson GK. The Fontan procedure for tricuspid atresia: early and late results of a 25-year experience with 216 patients. J Am Coll Cardiol. 2001;37:933-939.
Marsden AL, Bernstein AJ, Reddy VM, et al. Evaluation of a novel Y-shaped extracardiac Fontan baffle using computational fluid dynamics. J Thorac Cardiovasc Surg. 2009;137:394-403.
Shirai LK, Rosenthal DN, Reitz BA, et al. Arrhythmias and thromboembolic complications after the extracardiac Fontan operation. J Thorac Cardiovasc Surg. 1998;115:499-505.
Shiraishi S, Yagihara T, Kagisaki K, et al. Impact of age at Fontan completion on postoperative hemodynamics and long-term aerobic exercise capacity in patients with dominant left ventricle. Ann Thorac Surg. 2009;87:555-560.
van den Bosch AE, Roos-Hesselink JW, Van Domburg R, et al. Long-term outcome and quality of life in adult patients after the Fontan operation. Am J Cardiol. 2004;93:1141-1145.
424.5 Double-Outlet Right Ventricle
Double-outlet right ventricle (DORV) is characterized when both the aorta and pulmonary artery arise from the right ventricle. The outlet from the left ventricle is through VSD into the right ventricle. Normally, the aortic and mitral valves are in fibrous continuity; however, in DORV, the aortic and mitral valves are separated by a smooth muscular conus, similar to that seen under the normal pulmonary valve. In DORV, the great arteries may be normally related, with the aorta closer to the VSD, or malposed, with the pulmonary artery closer to the VSD. The great artery closest to the VSD may override the defect by a variable amount but is at least 50% committed to the right ventricle. When the VSD is subaortic, the defect may be viewed as part of a continuum with the tetralogy of Fallot, and the physiology as well as the history, physical examination, electrocardiogram, and roentgenograms are depending on the degree of pulmonary stenosis, similar to the situation in tetralogy of Fallot (Chapter 424.1). If the VSD is subpulmonic, there may be subvalvar, valvar, or supravalvar aortic stenosis, and coarctation is a possibility as well. This is known as the Taussig-Bing malformation. The clinical presentation of these patients will be dependent on the degree of aortic obstruction, but since the pulmonary artery is usually wide open, will usually include some degree of pulmonary overcirculation and heart failure. If the aortic obstruction is severe or there is a coarctation, poor pulses, hypoperfusion, and cardiovascular collapse are possible presenting signs.
The two-dimensional echocardiogram demonstrates both great vessels arising from the right ventricle and mitral-aortic valve discontinuity. The relationships between the aorta and pulmonary artery to the VSD can be delineated, and the presence of either pulmonary obstruction or aortic obstruction can be evaluated. Cardiac catheterization is not necessarily required if the echocardiogram is straightforward. Angiography will show that the aortic and pulmonary valves lie in the same horizontal plane and that both arise predominantly or exclusively from the right ventricle.
Surgical correction depends on the relationship of the great vessels to the VSD. If the VSD is subaortic, the repair may be similar to that used for tetralogy of Fallot, or consist of creating an intraventricular tunnel so that the left ventricle ejects blood through the VSD, into the tunnel, and into the aorta. The pulmonary obstruction is relieved either with an outflow patch or with a right ventricular to pulmonary artery homograft conduit (Rastelli operation). If the VSD is subpulmonic, the great vessels can be switched (Chapter 424.6) and the Rastelli operation performed. However, if there is substantial aortic obstruction, or if one of the ventricles is hypoplastic, then a Norwood-style single ventricle repair may be necessary (Chapter 425.10). In small infants, palliation with an aortopulmonary shunt provides symptomatic improvement and allows for adequate growth before corrective surgery is performed.
424.6 Transposition of the Great Arteries with Ventricular Septal Defect and Pulmonary Stenosis
This combination of anomalies may mimic tetralogy of Fallot in its clinical features (Chapter 424.1). However, because of the transposed great vessels, the site of obstruction is in the left as opposed to the right ventricle. The obstruction can be either valvular or subvalvular; the latter type may be dynamic, related to the interventricular septum or atrioventricular valve tissue, or acquired, as in patients with transposition and VSD after pulmonary arterial banding.
The age at which clinical manifestations initially appear varies from soon after birth to later infancy, depending on the degree of pulmonic stenosis. Clinical findings include cyanosis, decreased exercise tolerance, and poor physical development, similar to those described for tetralogy of Fallot; the heart is usually more enlarged. The pulmonary vasculature as seen on the roentgenogram is dependent on the degree of pulmonary obstruction. The electrocardiogram usually shows right axis deviation, right and left ventricular hypertrophy, and sometimes tall, spiked P waves. Echocardiography confirms the diagnosis and is useful in sequential evaluation of the degree and progression of the left ventricular outflow tract obstruction. Cardiac catheterization, if necessary, shows that pulmonary arterial pressure is low and that oxygen saturation in the pulmonary artery exceeds that in the aorta. Selective right and left ventriculography demonstrates the origin of the aorta from the right ventricle, the origin of the pulmonary artery from the left ventricle, the VSD, and the site and severity of the pulmonary stenosis.
An infusion of prostaglandin E1 (0.01-0.20 µg/kg/min) should be started in neonates who present with cyanosis. When necessary, balloon atrial septostomy is performed to improve atrial-level mixing and to decompress the left atrium (Chapter 425.2). Cyanotic infants may be palliated with an aortopulmonary shunt (Chapter 424.1) followed by a Rastelli operation when older as the preferred corrective procedure. The Rastelli procedure achieves physiologic and anatomic correction by (1) closure of the VSD using an interventricular tunnel so that left ventricular blood flow is directed to the aorta, and (2) connection of the right ventricle to the pulmonary artery via an extracardiac homograft conduit between the right ventricle and the distal pulmonary artery (Fig. 424-10). These conduits will eventually become stenotic or functionally restrictive with growth of the patient and require replacement. Patients with milder degrees of pulmonary stenosis amenable to simple valvotomy may be able to undergo complete correction with an arterial switch procedure (Chapter 425.2) and closure of the VSD. Surgical correction by the Mustard operation (Chapter 425.2) with simultaneous closure of the VSD and relief of left ventricular outflow obstruction may be an alternative when the position of the VSD is not suitable for a Rastelli operation; however, this procedure leaves the right ventricle as the systemic pumping chamber and has fallen out of favor.
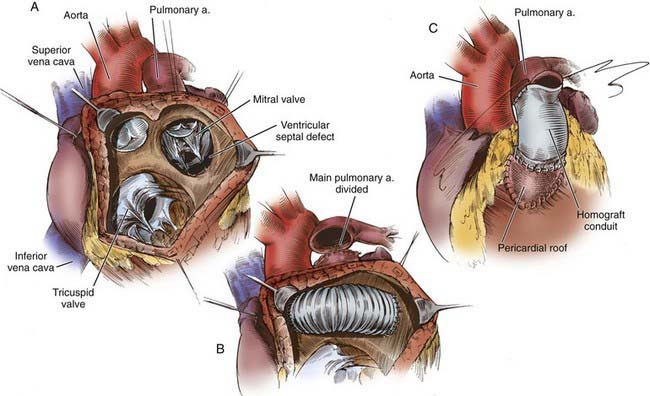
Figure 424-10 A, Taussig-Bing type of double-outlet right ventricle with subpulmonary stenosis necessitating repair by the Rastelli technique. B, The main pulmonary artery is divided and oversewn proximally. The pulmonary valve lies within the baffle pathway. C, Completion of the Rastelli repair with a right ventricle-pulmonary artery allograft conduit.
(From Castañeda AR, Jonas RA, Mayer JE Jr, et al: Single-ventricle tricuspid atresia. In Cardiac surgery of the neonate and infant, Philadelphia, 1994, WB Saunders.)
424.7 Ebstein Anomaly of the Tricuspid Valve
Pathophysiology
Ebstein anomaly consists of downward displacement of an abnormal tricuspid valve into the right ventricle. The defect arises from failure of the normal process by which the tricuspid valve is separated from the right ventricular myocardium (Chapter 414). The anterior cusp of the valve retains some attachment to the valve ring, but the other leaflets are adherent to the wall of the right ventricle. The right ventricle is thus divided into 2 parts by the abnormal tricuspid valve: the 1st, a thin-walled “atrialized” portion, is continuous with the cavity of the right atrium; the 2nd, often smaller portion consists of normal ventricular myocardium. The right atrium is enlarged as a result of tricuspid valve regurgitation, although the degree is extremely variable. In more severe forms of Ebstein anomaly, the effective output from the right side of the heart is decreased due to a combination of the poorly functioning small right ventricle, tricuspid valve regurgitation, and obstruction of the right ventricular outflow tract produced by the large, sail-like, anterior tricuspid valve leaflet. In newborns, right ventricular function may be so compromised that it is unable to generate enough force to open the pulmonary valve in systole, thus producing “functional” pulmonary atresia. Some infants have true anatomic pulmonary atresia. The increased volume of right atrial blood shunts through the foramen ovale (or through an associated atrial septal defect) to the left atrium and produces cyanosis (Fig. 424-11).
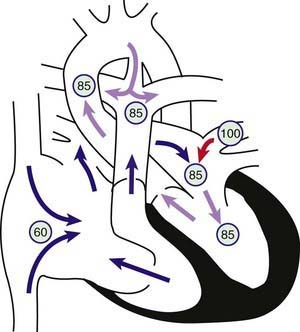
Figure 424-11 Physiology of Ebstein anomaly of the tricuspid valve. Circled numbers represent oxygen saturation values. Inferior displacement of the tricuspid valve leaflets into the right ventricle has resulted in a thin-walled, low-pressure “atrialized” segment of right ventricle. The tricuspid valve is grossly insufficient (clear arrow). Right atrial blood flow is shunted right to left across an atrial septal defect or patent foramen ovale into the left atrium. Some blood may cross the right ventricular outflow tract and enter the pulmonary artery; however, in severe cases, the right ventricle may generate insufficient force to open the pulmonary valve, and “functional pulmonary atresia” results. In the left atrium, desaturated blood mixes with saturated pulmonary venous return. Blood enters the left ventricle and is ejected via the aorta. In this example, some pulmonary blood flow is derived from the right ventricle, the rest from a patent ductus arteriosus (PDA). Severe cyanosis will develop in neonates with a severe Ebstein anomaly when the PDA closes.
Clinical Manifestations
The severity of symptoms and the degree of cyanosis are highly variable and depend on the extent of displacement of the tricuspid valve and the severity of right ventricular outflow tract obstruction. In many patients, symptoms are mild and may be delayed until the teenage years or young adult life; the patient may initially have fatigue or palpitations as a result of cardiac dysrhythmias. The atrial right-to-left shunt is responsible for cyanosis and polycythemia. Jugular venous pulsations, a index of central venous pressure, may be normal or increased in those with tricuspid insufficiency. On palpation, the precordium is quiet. A holosystolic murmur caused by tricuspid regurgitation is audible over most of the anterior left side of the chest. A gallop rhythm is common and often associated with multiple clicks at the lower left sternal border. A scratchy diastolic murmur may also be heard at the left sternal border. This murmur may mimic a pericardial friction rub.
Newborns with severe forms of Ebstein anomaly have marked cyanosis, massive cardiomegaly, and long holosystolic murmurs. Death may result from cardiac failure, hypoxemia, and pulmonary hypoplasia. Spontaneous improvement may occur in some neonates as pulmonary vascular resistance falls and improves the ability of the right ventricle to provide pulmonary blood flow. The majority are dependent on a PDA, and thus on a prostaglandin infusion, for pulmonary blood flow.
Diagnosis
The electrocardiogram usually shows a right bundle branch block without increased right precordial voltage, normal or tall and broad P waves, and a normal or prolonged P-R interval. Wolff-Parkinson-White syndrome (Chapter 429) may be present and these patients may have episodes of supraventricular tachycardia. On roentgenographic examination, heart size varies from slightly enlarged to massive box-shaped cardiomegaly caused by enlargement of the right atrium. In newborns with severe Ebstein anomaly, the heart may totally obscure the pulmonary fields. Echocardiography is diagnostic and shows the degree of displacement of the tricuspid valve leaflets, a dilated right atrium, and any right ventricular outflow tract obstruction (Fig. 424-12). Pulsed and color Doppler examination demonstrates the degree of tricuspid regurgitation. In severe cases, the pulmonary valve may appear immobile and pulmonary blood flow may come solely from the ductus arteriosus. It may be difficult to distinguish true from functional pulmonary valve atresia. Cardiac catheterization, which is not usually necessary, confirms the presence of a large right atrium, an abnormal tricuspid valve, and any right-to-left shunt at the atrial level. The risk of arrhythmia is significant during catheterization and angiographic studies.
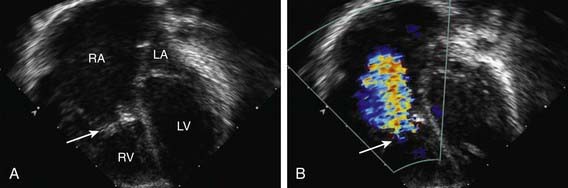
Figure 424-12 Echocardiographic demonstration of Ebstein anomaly of the tricuspid valve. A, Subcostal, four-chamber, two-dimensional view showing severe displacement of the tricuspid valve leaflets (large arrow) inferiorly into the right ventricle. The location of the tricuspid valve annulus is outlined by the two arrowheads. The portion of the right ventricle between the valve annulus and the valve leaflets is the “atrialized” component. B, Color Doppler examination showing severe regurgitation of the dysplastic tricuspid valve. Note that the regurgitant turbulent flow (arrow) begins halfway into the right ventricular chamber, at the location of the displaced valve leaflets. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
Prognosis and Complications
The prognosis in Ebstein anomaly is extremely variable and depends on the severity of the defect. The prognosis is more guarded for neonates or infants with intractable symptoms and cyanosis. Patients with milder degrees of Ebstein anomaly usually survive well into adult life. There is an association of a form of left ventricular cardiomyopathy, isolated left ventricular non-compaction, in 18% of patients with Ebstein anomaly, and the severity of the left ventricular dysfunction directly impacts the prognosis.
Treatment
Neonates with severe hypoxia who are prostaglandin dependent have been treated with an aortopulmonary shunt alone, by repair of the tricuspid valve, or by surgical patch closure of the tricuspid valve, atrial septectomy, and placement of an aortopulmonary shunt (with eventual single ventricle repair using the Fontan procedure [Chapter 424.4]). Many infants with Ebstein anomaly who have undergone valve repair will still have enough regurgitation that a Glenn shunt is performed to reduce the volume load on the right ventricle (Chapter 424.4). In older children with mild or moderate disease, control of supraventricular dysrhythmias is of primary importance; surgical treatment may not be necessary until adolescence or young adulthood. In patients with severe tricuspid regurgitation, repair or replacement of the abnormal tricuspid valve along with closure of the atrial septal defect is carried out. In some older patients, a bidirectional Glenn shunt is also performed, with the superior vena cava anastomosed to the pulmonary arteries. This procedure reduces the volume of blood that the dysfunctional right side of the heart has to pump, thus creating a “one-and-one-half ventricle repair.”
Attenhofer Jost CH, Connolly HM, O’Leary PW, et al. Left heart lesions in patients with Ebstein anomaly. Mayo Clin Proc. 2005;80:361-368.
Brown ML, Dearani JA, Danielson GK, et al. The outcomes of operations for 539 patients with Ebstein anomaly. J Thorac Cardiovasc Surg. 2008;135:1120-1136.
Knott-Craig CJ, Goldberg SP, Overholt ED, et al. Repair of neonates and young infants with Ebstein’s anomaly and related disorders. Ann Thorac Surg. 2007;84:587-592.
Knott-Craig CJ, Goldberg SP. Management of neonatal Ebstein’s anomaly. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2007:112-116.
Quinonez LG, Dearani JA, Puga FJ, et al. Results of the 1.5-ventricle repair for Ebstein anomaly and the failing right ventricle. J Thorac Cardiovasc Surg. 2007;133:1303-1310.
Schreiber C, Cook A, Ho SY, et al. Morphologic spectrum of Ebstein’s malformation: revisitation relative to surgical repair. J Thorac Cardiovasc Surg. 1999;117:148-155.
Starnes VA, Pitlick PT, Bernstein D, et al. Ebstein’s anomaly appearing in the neonate. J Thorac Cardiovasc Surg. 1991;101:1082-1087.