Chapter 457 Enzymatic Defects
457.1 Pyruvate Kinase Deficiency
Congenital hemolytic anemia occurs in persons homozygous or compound heterozygous for autosomal recessive genes that cause either a marked reduction in red blood cell (RBC) pyruvate kinase (PK) or production of an abnormal enzyme with decreased activity. Generation of adenosine triphosphate (ATP) within RBCs is impaired, and low levels of ATP, pyruvate, and the oxidized form of nicotinamide adenine dinucleotide (NAD+) are found (Fig. 457-1). The concentration of 2,3-diphosphoglycerate is increased; this isomer is beneficial in facilitating oxygen release from hemoglobin but detrimental in inhibiting hexokinase and enzymes of the hexose monophosphate shunt. In addition, an unexplained decrease occurs in the sum of the adenine (ATP, adenosine diphosphate, and adenosine monophosphate) and pyridine (NAD+ and the reduced form of NAD) nucleotides, further impairing glycolysis. As a consequence of decreased ATP, RBCs cannot maintain their potassium and water content; the cells become rigid, and their life span is considerably reduced.
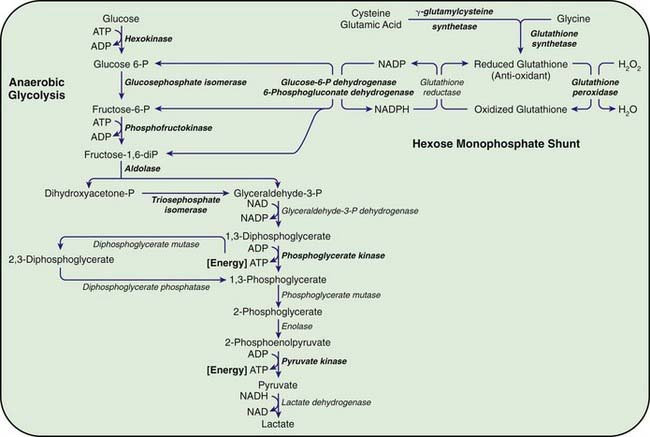
Figure 457-1 Red blood cell metabolism. Glycolysis and the hexose monophosphate pathway. The enzyme deficiencies clearly associated with hemolysis are shown in bold type. ATP, adenosine triphosphate; ADP, adenosine diphosphate; NADP, nicotinamide-adenine dinucleotide phosphate; NADPH, reduced form of NADP.
Etiology
There are 2 mammalian PK genes, but only the PKLR gene is expressed in red cells. The human PKLR gene is located on chromosome 1q21. More than 180 mutations are reported in this structural gene, which codes for a 574–amino acid protein that forms a functional tetramer. These mutations include missense, splice site, and insertion-deletion alterations. Most affected patients are compound heterozygotes for 2 different PK gene defects. The many possible combinations likely account for the variability in clinical severity. The mutations 1456 C to T and 1529 G to A are the most common mutations in the white population.
Clinical Manifestations and Laboratory Findings
The clinical manifestations of PK deficiency vary from severe neonatal hemolytic anemia to mild, well-compensated hemolysis first noted in adulthood. Severe jaundice and anemia may occur in the neonatal period, and kernicterus has been reported. The hemolysis in older children and adults varies in severity, with hemoglobin values ranging from 8 to 12 g/dL associated with some pallor, jaundice, and splenomegaly. Patients with these findings usually do not require transfusion. A severe form of the disease has a relatively high incidence among the Amish of the Midwestern United States. PK deficiency may provide protection against falciparum malaria; there is no demographic support for this observation, however.
Polychromatophilia and mild macrocytosis reflect the elevated reticulocyte count. Spherocytes are uncommon, but a few spiculated pyknocytes may be found. The non-incubated osmotic fragility is normal. Diagnosis relies on demonstration of a marked reduction of RBC PK activity or an increase in the Michaelis-Menten dissociation constant (Km) for its substrate, phosphoenolpyruvate (high Km variant). Other RBC enzyme activity is normal or elevated, reflecting the reticulocytosis. No abnormalities of hemoglobin are noted. The white cells have normal PK activity and must be rigorously excluded from the red cell hemolysates used to measure PK activity. Heterozygous carriers usually have moderately reduced levels of PK activity.
Treatment
Phototherapy and exchange transfusions may be indicated for hyperbilirubinemia in newborns. Transfusions of packed RBCs are necessary for severe anemia or for aplastic crises. If the anemia is consistently severe or if frequent transfusions are required, splenectomy should be performed after the child is 5-6 yr of age. Although it is not curative, splenectomy may be followed by higher hemoglobin levels and by strikingly high (30-60%) reticulocyte counts. Death resulting from overwhelming pneumococcal sepsis has followed splenectomy; thus, immunization with vaccines for encapsulated organisms should be given before splenectomy, and prophylactic penicillin should be administered after the procedure.
457.2 Other Glycolytic Enzyme Deficiencies
Chronic nonspherocytic hemolytic anemias of varying severity have been associated with deficiencies of other enzymes in the glycolytic pathway, including hexokinase, glucose phosphate isomerase, and aldolase, which are inherited as autosomal recessive disorders. Phosphofructokinase deficiency, which occurs primarily in Ashkenazi Jews in the USA, results in hemolysis associated with a myopathy classified as glycogen storage disease type VII (Chapter 81.1). Clinically, hemolytic anemia is complicated by muscle weakness, exercise intolerance, cramps, and possibly myoglobinuria. Enzyme assays for phosphofructokinase yield low values for RBCs and muscle.
Triose phosphate isomerase (TPI) deficiency is an autosomal recessive disorder affecting many systems. Affected patients have hemolytic anemia, cardiac abnormalities, and lower motor neuron and pyramidal tract impairment, with or without evidence of cerebral impairment. They usually die in early childhood. The gene for TPI has been cloned and sequenced and is located on chromosome 12.
Phosphoglycerate kinase (PGK) is the first ATP-generating step in glycolysis. At least 23 kindreds with PGK deficiency have been described. PGK is the only glycolytic enzyme inherited on the X chromosome. Affected males may have progressive extrapyramidal disease, myopathy, seizures, and variable mental retardation in conjunction with hemolytic anemia. Nine Japanese patients had neural or myopathic symptoms with hemolysis; 6 had hemolysis alone; 7 had neural or myopathic symptoms alone; and 1 had no symptoms. The gene for PGK is particularly large, spanning 23 kb, and various genetic abnormalities, including nucleotide substitutions, gene deletions, missense, and splicing mutations, result in PGK deficiency.
Deficiencies of Enzymes of the Hexose Monophosphate Pathway
The most important function of the hexose monophosphate pathway is to maintain glutathione in its reduced state (GSH) as protection against the oxidation of RBCs (see Fig. 457-1). Approximately 10% of the glucose taken up by RBCs passes through this pathway to provide the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) necessary for the conversion of oxidized glutathione to GSH. Maintenance of GSH is essential for the physiologic inactivation of oxidant compounds, such as hydrogen peroxide, that are generated within RBCs. If glutathione, or any compound or enzyme necessary for maintaining it in the reduced state, is decreased, the SH groups of the RBC membrane are oxidized and the hemoglobin becomes denatured and may precipitate into RBC inclusions called Heinz bodies. Once Heinz bodies have formed, an acute hemolytic process results from damage to the RBC membrane by the precipitated hemoglobin, the oxidant agent, and the action of the spleen. The damaged RBCs then are rapidly removed from the circulation.
457.3 Glucose-6-Phosphate Dehydrogenase Deficiency and Related Deficiencies
George B. Segel and Lisa R. Hackney
Glucose-6-phosphate dehydrogenase (G6PD) deficiency, the most frequent disease involving enzymes of the hexose monophosphate pathway, is responsible for 2 clinical syndromes, episodic hemolytic anemia, and chronic nonspherocytic hemolytic anemia. The most common manifestations of this disorder are neonatal jaundice and episodic acute hemolytic anemia, which is induced by infections, certain drugs, and, rarely, fava beans. This X-linked deficiency affects more than 400 million people worldwide, representing an overall 4.9% global prevalence. The global distribution of this disorder parallels that of malaria, representing an example of “balanced polymorphism,” in which there is an evolutionary advantage of resistance to falciparum malaria in heterozygous females that outweighs the small negative effect of affected hemizygous males.
The deficiency is caused by inheritance of any of a large number of abnormal alleles of the gene responsible for the synthesis of the G6PD protein. About 140 mutations have been described in the gene responsible for the synthesis of the G6PD protein. Many of these mutations are single base changes leading to amino acid substitutions and destabilization of the G6PD enzyme. The gene for G6PD has been cloned and sequenced. A web-accessible database catalogs G6PD mutations (www.bioinf.org.uk/g6pd). Some of the mutations that cause episodic vs chronic hemolysis are shown in Figure 457-2. Milder disease is associated with mutations near the amino terminus of the G6PD molecule, and chronic nonspherocytic hemolytic anemia is associated with mutations clustered near the carboxyl terminus. The normal enzyme found in most populations is designated G6PD B+. A normal variant, designated G6PD A+, is common in Americans of African descent.
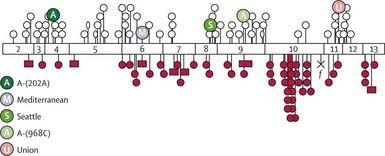
Figure 457-2 Most common mutations along coding sequence of G6PD gene. Exons are shown as open numbered boxes. Open circles are mutations causing classes II and III variants. Filled circles represent sporadic mutations giving rise to severe variants (class I). Open ellipses are mutations causing class IV variants. Cross, a nonsense mutation; f, a splice site mutation; filled square, small deletion. 202A and 968C are the two sites of base substitution in G6PD-A.
(From Cappellini MD, Fiorelli G: Glucose-6-phosphate dehydrogenase deficiency, Lancet 371:64–74, 2008.)
Episodic or Induced Hemolytic Anemia
Etiology
G6PD catalyzes the conversion of glucose 6-phosphate to 6-phosphogluconic acid. This reaction produces NADPH, which maintains glutathione in the reduced, functional state (see Fig. 457-1). Reduced glutathione provides protection against oxidant threats from certain drugs and infections that would otherwise cause precipitation of hemoglobin (Heinz bodies) or damage the RBC membrane.
Synthesis of RBC G6PD is determined by a gene on the X chromosome. Thus, heterozygous females have intermediate enzymatic activity and have 2 populations of RBCs: one is normal, and the other is deficient in G6PD activity. Because they have fewer susceptible cells, most heterozygous females do not have evident clinical hemolysis after exposure to oxidant drugs. Rarely, the majority of RBCs is G6PD deficient in heterozygous females because the inactivation of the normal X chromosome is random and sometimes exaggerated (Lyon-Beutler hypothesis).
Disease involving this enzyme therefore occurs more frequently in males than in females. Approximately 13% of male Americans of African descent have a mutant enzyme (G6PD A−) that results in a deficiency of RBC G6PD activity (5-15% of normal). Italians, Greeks, and other Mediterranean, Middle Eastern, African, and Asian ethnic groups also have a high incidence, ranging from 5% to 40%, of a variant designated G6PD B− (G6PD Mediterranean). In these variants, the G6PD activity of homozygous females or hemizygous males is <5% of normal. Therefore, the defect in Americans of African descent is less severe than that in Americans of European descent. A third mutant enzyme with markedly reduced activity (G6PD Canton) occurs in approximately 5% of the Chinese population.
Clinical Manifestations
Most individuals with G6PD deficiency are asymptomatic, with no clinical manifestations of illness unless triggered by infection, drugs, or ingestion of fava beans. Typically, hemolysis ensues in about 24-48 hr after a patient has ingested a substance with oxidant properties. In severe cases, hemoglobinuria and jaundice result, and the hemoglobin concentration may fall precipitously. Drugs that elicit hemolysis in these individuals include aspirin, sulfonamides, rasburicase, and antimalarials, such as primaquine (Table 457-1). The degree of hemolysis varies with the inciting agent, amount ingested, and severity of the enzyme deficiency. In some individuals, ingestion of fava beans also produces an acute, severe hemolytic syndrome, known as favism. Fava beans contain divicine, isouramil, and convicine, which ultimately lead to production of hydrogen peroxide and other reactive oxygen products. Favism is thought to be more frequently associated with the G6PD B− variant.
Table 457-1 AGENTS PRECIPITATING HEMOLYSIS IN GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
MEDICATIONS
Antibacterials
Antimalarials
Others
CHEMICALS
ILLNESS
From Asselin BL, Segel GB: In Rakel R, editor: Conn’s current therapy, Philadelphia, 1994, WB Saunders, p 341.
In the G6PD A− variant, the stability of the folded protein dimer is impaired, and this defect is accentuated as the RBCs age. Thus, hemolysis decreases as older red cells are destroyed, even if administration of the drug is continued. This recovery results from the age-labile enzyme, which is abundant and more stable in younger RBCs. The associated reticulocytosis produces a compensated hemolytic process in which the blood hemoglobin may be only slightly decreased, despite continued exposure to the offending agent.
G6PD deficiency can produce hemolysis in the neonatal period. In G6PD A–, spontaneous hemolysis and hyperbilirubinemia have been observed in preterm infants. In newborns with the G6PDB− and G6PD Canton varieties, hyperbilirubinemia and even kernicterus may occur. Neonates with coinheritance of G6PD deficiency and a mutation of the promoter of uridine-diphosphate-glucuronyl transferase (UGT1A1), seen in Gilbert syndrome, have more severe neonatal jaundice. When a pregnant woman ingests oxidant drugs, they may be transmitted to her G6PD-deficient fetus, and hemolytic anemia and jaundice may be apparent at birth.
Laboratory Findings
The onset of acute hemolysis usually results in a precipitous fall in hemoglobin and hematocrit. If the episode is severe, the hemoglobin binding proteins, such as haptoglobin, are saturated, and free hemoglobin may appear in the plasma and subsequently in the urine. Unstained or supravital preparations of RBCs reveal precipitated hemoglobin, known as Heinz bodies. The RBC inclusions are not visible on the Wright-stained blood film. Cells that contain these inclusions are seen only within the first 3-4 days of illness because they are rapidly cleared from the blood. Also, the blood film may contain red cells with what appears to be a bite taken from their periphery and polychromasia (evidence of bluish, larger RBCs), representing reticulocytosis (Fig. 457-3).
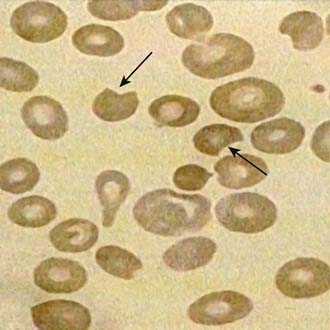
Figure 457-3 Morphologic erythrocyte changes (anisopoikilocytosis, bite cells) during acute hemolysis in a G6PD-deficient patient. Arrows show bite cells. Anisopoikilocytosis is abnormality in the shape or size of erythrocytes.
(From Cappellini MD, Fiorelli G: Glucose-6-phosphate dehydrogenase deficiency, Lancet 371:64–74, 2008.)
Diagnosis
The diagnosis depends on direct or indirect demonstration of reduced G6PD activity in RBCs. By direct measurement, enzyme activity in affected persons is ≤10% of normal, and the reduction of enzyme activity is more extreme in Americans of European descent and in Asians than in Americans of African descent. Satisfactory screening tests are based on decoloration of methylene blue, reduction of methemoglobin, or fluorescence of NADPH. Immediately after a hemolytic episode, reticulocytes and young RBCs predominate. These young cells have significantly higher enzyme activity than do older cells in the A− variety. Testing may therefore have to be deferred for a few weeks before a diagnostically low level of enzyme can be shown. The diagnosis can be suspected when G6PD activity is within the low-normal range in the presence of a high reticulocyte count. G6PD variants also can be detected by electrophoretic and molecular analysis.
Prevention and Treatment
Prevention of hemolysis constitutes the most important therapeutic measure. When possible, males belonging to ethnic groups with a significant incidence of G6PD deficiency (e.g., Greeks, southern Italians, Sephardic Jews, Filipinos, southern Chinese, Americans of African descent, and Thais) should be tested for the defect before known oxidant drugs are given. The usual doses of aspirin and trimethoprim-sulfamethoxazole do not cause clinically relevant hemolysis in the A− variety. Aspirin administered in doses used for acute rheumatic fever (60-100 mg/kg/24 hr) may produce a severe hemolytic episode. Infants with severe neonatal jaundice who belong to these ethnic groups also require testing for G6PD deficiency because of their heightened risk for this defect. If severe hemolysis has occurred, supportive therapy may require blood transfusions, although recovery is the rule when the oxidant agent is discontinued.
Chronic Hemolytic Anemias Associated with Deficiency of G6pd or Related Factors
Chronic nonspherocytic hemolytic anemia has been associated with profound deficiency of G6PD caused by enzyme variants, particularly those defective in quantity, activity, or stability. The gene defects leading to chronic hemolysis are located primarily in the region of the NADP binding site near the carboxyl terminus of the protein (see Fig. 457-2). These include the Loma Linda, Tomah, Iowa, Beverly Hills, Nashville, Riverside, Santiago de Cuba, and Andalus variants. Persons with G6PD B− enzyme deficiency occasionally have chronic hemolysis, and the hemolytic process may worsen after ingestion of oxidant drugs. Splenectomy is of little value in these types of chronic hemolysis.
Other enzyme defects may impair the regeneration of GSH as an oxidant “sump” (see Fig. 457-1). Mild, chronic nonspherocytic anemia has been reported in association with decreased RBC GSH, resulting from γ-glutamylcysteine or glutathione synthetase deficiencies. Deficiency of 6-phosphogluconate dehydrogenase (6PDG) has been associated primarily with drug-induced hemolysis, and hemolysis with hyperbilirubinemia has been related to a deficiency of glutathione peroxidase in newborn infants.
Ayi K, Min-Oo G, Serghides L, et al. Pyruvate kinase deficiency and malaria. N Engl J Med. 2008;358:1805-1810.
Beutler E. Glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. 1994;331:169-173.
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64-74.
Fujii H, Miwa S. Other erythrocyte enzyme deficiencies associated with non-haematological symptoms: phosphoglycerate kinase and phosphofructokinase deficiency. Baillieres Best Pract Res Clin Haematol. 2000;13:141-148.
Kaplan M, Beutler E, Vreman HJ, et al. Neonatal hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient heterozygotes. Pediatrics. 1999;104:68-74.
Kaplan M, Muraca M, Hammerman C, et al. Bilirubin conjugation, reflected by conjugated bilirubin fractions, in glucose-6-phosphate dehydrogenase-deficient neonates: a determining factor in the pathogenesis of hyperbilirubinemia. Pediatrics. 1998;102:E37.
Martinov MV, Plotnikov AG, Vitvitsky VM, et al. Deficiencies of glycolytic enzymes as a possible cause of hemolytic anemia. Biochim Biophys Acta. 2000;1474:75-87.
McMullin MF. The molecular basis of disorders of red cell enzymes. J Clin Pathol. 1999;52:241-244.
Min-Oo G, Gros P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol. 2005;7:753-763.
Valentin C, Pissard S, Martin J, et al. Triose phosphate isomerase deficiency in 3 French families: two novel null alleles, a frameshift mutation (TPI Alfortville) and an alteration in the initiation codon (TPI Paris). Blood. 2000;96:1130-1135.
Zanella A, Bianchi P. Red cell pyruvate kinase deficiency: from genetics. Baillieres Best Pract Res Clin Haematol. 2000;13:57-81.
Zanella A, Fermo E, Bianchi P, et al. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130:11-25.