Chapter 470 Hereditary Clotting Factor Deficiencies (Bleeding Disorders)
Hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) are the most common and serious congenital coagulation factor deficiencies. The clinical findings in hemophilia A and hemophilia B are virtually identical. Hemophilia C is the bleeding disorder associated with reduced levels of factor XI (Chapter 470.2). Reduced levels of the contact factors (factor XII, high molecular weight kininogen, and prekallikrein) are associated with significant prolongation of activated partial thromboplastin time (APTT; also referred to as PTT), but are not associated with hemorrhage, as discussed in Chapter 470.3. Other coagulation factor deficiencies that are less common are briefly discussed in subsequent subchapters.
470.1 Factor VIII or Factor IX Deficiency (Hemophilia A or B)
Deficiencies of factors VIII and IX are the most common severe inherited bleeding disorders. Recombinant factor VIII and factor IX concentrates are available to treat patients with hemophilia and thereby avoid the infectious risk of plasma-derived transfusion-transmitted diseases.
Pathophysiology
Factors VIII and IX participate in a complex required for the activation of factor X. Together with phospholipid and calcium, they form the “tenase,” or factor X–activating, complex. Figure 469-1 shows the clotting process as it occurs in the test tube, with factor X being activated by either the complex of factors VIII and IX or the complex of tissue factor and factor VII. In vivo, the complex of factor VIIa and tissue factor activates factor IX to initiate clotting. In the laboratory, prothrombin time (PT) measures the activation of factor X by factor VII and is therefore normal in patients with factor VIII or factor IX deficiency.
After injury, the initial hemostatic event is formation of the platelet plug, together with the generation of the fibrin clot that prevents further hemorrhage. In hemophilia A or B, clot formation is delayed and is not robust. Inadequate thrombin generation leads to failure to form a tightly cross-linked fibrin clot to support the platelet plug. Patients with hemophilia slowly form a soft, friable clot. When untreated bleeding occurs in a closed space, such as a joint, cessation of bleeding may be the result of tamponade. With open wounds, in which tamponade cannot occur, profuse bleeding may result in significant blood loss. The clot that is formed may be friable, and rebleeding occurs during the physiologic lysis of clots or with minimal new trauma.
Clinical Manifestations
Neither factor VIII nor factor IX crosses the placenta; bleeding symptoms may be present from birth or may occur in the fetus. Only 2% of neonates with hemophilia sustain intracranial hemorrhages, and 30% of male infants with hemophilia bleed with circumcision. Thus, in the absence of a positive family history (hemophilia has a high rate of spontaneous mutation), hemophilia may go undiagnosed in the newborn. Obvious symptoms such as easy bruising, intramuscular hematomas, and hemarthroses begin when the child begins to cruise. Bleeding from minor traumatic lacerations of the mouth (a torn frenulum) may persist for hours or days and may cause the parents to seek medical evaluation. Even in patients with severe hemophilia, only 90% have evidence of increased bleeding by 1 yr of age. Although bleeding may occur in any area of the body, the hallmark of hemophilic bleeding is hemarthrosis. Bleeding into the joints may be induced by minor trauma; many hemarthroses are spontaneous. The earliest joint hemorrhages appear most commonly in the ankle. In the older child and adolescent, hemarthroses of the knees and elbows are also common. Whereas the child’s early joint hemorrhages are recognized only after major swelling and fluid accumulation in the joint space, older children are frequently able to recognize bleeding before the physician does. They complain of a warm, tingling sensation in the joint as the first sign of an early joint hemorrhage. Repeated bleeding episodes into the same joint in a patient with severe hemophilia may become a “target” joint. Recurrent bleeding may then become spontaneous because of the underlying pathologic changes in the joint.
Although most muscular hemorrhages are clinically evident owing to localized pain or swelling, bleeding into the iliopsoas muscle requires specific mention. A patient may lose large volumes of blood into the iliopsoas muscle, verging on hypovolemic shock, with only a vague area of referred pain in the groin. The hip is held in a flexed, internally rotated position owing to irritation of the iliopsoas. The diagnosis is made clinically from the inability to extend the hip but must be confirmed with ultrasonography or CT (Fig. 470-1). Life-threatening bleeding in the patient with hemophilia is caused by bleeding into vital structures (central nervous system, upper airway) or by exsanguination (external trauma, gastrointestinal or iliopsoas hemorrhage). Prompt treatment with clotting factor concentrate for these life-threatening hemorrhages is imperative. If head trauma is of sufficient concern to suggest radiologic evaluation, factor replacement should precede radiologic evaluation. Simply put: “Treat first, image second!” Life-threatening hemorrhages require replacement therapy to achieve a level equal to that of normal plasma (100 IU/dL, or 100%).
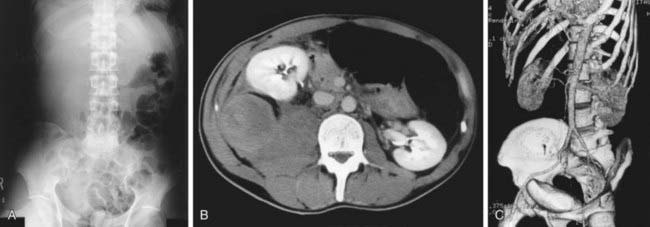
Figure 470-1 Massive hematoma into the iliopsoas muscle in a patient with hemophilia B. A 38-yr-old man with severe deficiency of factor IX (hemophilia B) was admitted for right lower abdominal pain of progressively increasing severity and tenderness. He had had a common cold with severe cough and loss of appetite for approximately 1 wk. A, Abdominal radiograph shows presence of the psoas sign on the right side and left-shifted colon gas. B, CT scan shows massive hematoma in the right iliopsoas muscle, resulting in anterior translocation of the right kidney. C, Reconstructed 3-dimensional image shows more clearly the kidney translocation and the extended, but intact large vessels. These are useful findings for the diagnostic procedures, because progressive right lower abdominal pain may closely simulate acute appendicitis. The hemorrhage was successfully managed by replacement of factor IX for 1 wk without any recurrence. The patient did not have any inhibitors to factor IX.
(From Miyazaki K, Higashihara M: Massive hemorrhage into the iliopsoas muscle, Intern Med 44:158, 2005.)
Patients with mild hemophilia who have factor VIII or factor IX levels > 5 IU/dL usually do not have spontaneous hemorrhages. These individuals may experience prolonged bleeding after dental work, surgery, or injuries from moderate trauma.
Laboratory Findings and Diagnosis
The laboratory screening test that is affected by a reduced level of factor VIII or factor IX is PTT. In severe hemophilia, the PTT value is usually 2-3 times the upper limit of normal. Results of the other screening tests of the hemostatic mechanism (platelet count, bleeding time, prothrombin time, and thrombin time) are normal. Unless the patient has an inhibitor to factor VIII or IX, the mixing of normal plasma with patient plasma results in correction of PTT value. The specific assay for factors VIII and IX will confirm the diagnosis of hemophilia. If correction does not occur on mixing, an inhibitor may be present. In 25-35% of patients with hemophilia who receive infusions of factor VIII or factor IX, a factor-specific antibody may develop. These antibodies are directed against the active clotting site and are termed inhibitors. In such patients, the quantitative Bethesda assay for inhibitors should be performed to measure the antibody titer.
Differential Diagnosis
In young infants with severe bleeding manifestations, the differential diagnosis includes severe thrombocytopenia; severe platelet function disorders, such as Bernard-Soulier syndrome and Glanzmann Thrombasthenia; type 3 (severe) von Willebrand disease; and vitamin K deficiency. Hemostatic screening tests should differentiate these entities from hemophilia.
Genetics and Classification
Hemophilia occurs in approximately 1 : 5,000 males, with 85% having factor VIII deficiency and 10-15% having factor IX deficiency. Hemophilia shows no apparent racial predilection, appearing in all ethnic groups. The severity of hemophilia is classified on the basis of the patient’s baseline level of factor VIII or factor IX, because factor levels usually correlate with the severity of bleeding symptoms. By definition, 1 IU of each factor is defined as that amount in 1 mL of normal plasma referenced against a standard established by the World Health Organization (WHO); thus, 100 mL of normal plasma has 100 IU/dL (100% activity) of each factor. For ease of discussion, henceforth in this chapter, we use the term % activity to refer to the percentage found in normal plasma (100% activity). Factor concentrates are also referenced against an international WHO standard, so treatment doses are usually referred to in IU. Severe hemophilia is characterized as having <1% activity of the specific clotting factor, and bleeding is often spontaneous. Patients with moderate hemophilia have factor levels of 1-5% and usually require mild trauma to induce bleeding. Individuals with mild hemophilia have levels >5%, may go many years before the condition is diagnosed, and frequently require significant trauma to cause bleeding. The hemostatic level for factor VIII is >30-40%, and for factor IX, it is >25-30%. The lower limit of levels for factors VIII and IX in normal individuals is approximately 50%.
The genes for factors VIII and IX are carried near the terminus of the long arm of the X chromosome and are therefore X-linked traits. The majority of patients with hemophilia have reduced clotting factor protein; 5-10% of those with hemophilia A and 40-50% of those with hemophilia B make a dysfunctional protein. Approximately 45-50% of patients with severe hemophilia A have the same mutation, in which there is an internal inversion within the factor VIII gene that results in production of no protein. This mutation can be detected in the blood of patients or carriers and in the amniotic fluid by molecular techniques. African Americans often have a different FVIII haplotype, and this difference may be the reason that African Americans have higher inhibitor formation (see later). Because of the multiple genetic causes of either factor VIII or factor IX deficiency, most cases of hemophilia are classified according to the amount of factor VIII or factor IX clotting activity. In the newborn, factor VIII values may be artificially elevated because of the acute-phase response elicited by the birth process. This artificial elevation may cause a mildly affected patient to have normal or near-normal levels of factor VIII. Patients with severe hemophilia do not have detectable levels of factor VIII. In contrast, factor IX levels are physiologically low in the newborn. If severe hemophilia is present in the family, an undetectable level of factor IX is diagnostic of severe hemophilia B. In some patients with mild factor IX deficiency, the presence of hemophilia can be confirmed only after several weeks of life.
Through lyonization of the X chromosome, some female carriers of hemophilia A or B have sufficient reduction of factor VIII or factor IX to produce mild bleeding disorders. Levels of these factors should be determined in all known or potential carriers to assess the need for treatment in the event of surgery or clinical bleeding.
Because factor VIII is carried in plasma by von Willebrand factor, the ratio of factor VIII to von Willebrand factor is sometimes used to diagnose carriers of hemophilia. When possible, specific genetic mutations should be identified in the propositus and used to test other family members who are at risk of either having hemophilia or being carriers.
Treatment
Early, appropriate therapy is the hallmark of excellent hemophilia care. When mild to moderate bleeding occurs, values of factor VIII or factor IX must be raised to hemostatic levels, in the 35-50% range. For life-threatening or major hemorrhages, the dose should aim to achieve levels of 100% activity.
Calculation of the dose of recombinant factor VIII (FVIII) or recombinant factor IX (FIX) is as follows:
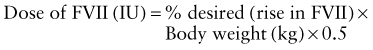
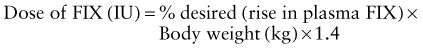
For factor VIII, the correction factor is based on the volume of distribution of factor VIII. For factor IX, the correction factor is based on the volume of distribution and the observed rise in plasma level after infusion of recombinant factor IX.
Table 470-1 summarizes the treatment of some common types of hemorrhage in a patient with hemophilia.
Table 470-1 TREATMENT OF HEMOPHILIA

With the availability of recombinant replacement products, prophylaxis is the standard of care for most children with severe hemophilia, to prevent spontaneous bleeding and early joint deformities. A study comparing prophylaxis with aggressive episodic treatment provides evidence for the superiority of prophylaxis in preventing debilitating joint disease. If target joints develop, “secondary” prophylaxis is often initiated.
With mild factor VIII hemophilia, the patient’s endogenously produced factor VIII can be released by the administration of desmopressin acetate (DDAVP). In patients with moderate or severe factor VIII deficiency, the stored levels of factor VIII in the body are inadequate, and desmopressin treatment is ineffective. The risk of exposing the patient with mild hemophilia to transfusion-transmitted diseases and the cost of recombinant products warrant the use of desmopressin, if it is effective. A concentrated intranasal form of desmopressin acetate, not the enuresis or pituitary replacement dose, can also be used to treat patients with mild hemophilia A. The dose is 150 µg (1 puff) for children weighing <50 kg and 300 µg (2 puffs) for children and young adults weighing >50 kg. Most centers administer a trial of desmopressin to determine the level of factor VIII achieved after its infusion. Desmopressin is not effective in the treatment of factor IX–deficient hemophilia.
Prophylaxis
Many patients are now given lifelong prophylaxis to prevent spontaneous joint bleeding. The National Hemophilia Foundation recommends that prophylaxis be considered optimal therapy for children with severe hemophilia. Usually, such programs are initiated with the first joint hemorrhage. Young children often require the insertion of a central catheter to ensure venous access. Such programs are expensive but are highly effective in preventing or greatly limiting the degree of joint pathology. Treatment is usually provided every 2-3 days to maintain a measurable plasma level of clotting factor (1-2%) when assayed just before the next infusion (trough level). Whether prophylaxis should be continued into adulthood has not yet been adequately studied. If moderate arthropathy develops, prevention of future bleeding will require higher plasma levels of clotting factors. In the older child who is not given primary prophylaxis, secondary prophylaxis is frequently initiated if a target joint develops.
Supportive Care
Although it is easy to tell parents that their child should avoid trauma, this advice is practically useless. Toddlers are active, are curious about everything, and injure themselves easily. Effective measures include anticipatory guidance, including the use of car seats, seatbelts, and bike helmets, and the importance of avoiding high-risk behaviors. Older boys should be counseled to avoid violent contact sports, but this issue is a challenge. Boys with severe hemophilia often sustain hemorrhages in the absence of known trauma. Early psychosocial intervention helps the family achieve a balance between overprotection and permissiveness. Patients with hemophilia should avoid aspirin and other nonsteroidal anti-inflammatory drugs that affect platelet function. The child with a bleeding disorder should receive the appropriate vaccinations against hepatitis B, even though recombinant products may avoid exposure to transfusion-transmitted diseases. Patients exposed to plasma-derived products should be screened periodically for hepatitis B and C, HIV, and abnormalities in liver function.
Chronic Complications
Long-term complications of hemophilia A and B include chronic arthropathy, the development of an inhibitor to either factor VIII or factor IX, and the risk of transfusion-transmitted infectious diseases. Although an aggressive, or prophylactic, approach to treatment has reduced the problems of chronic arthropathy, these problems have not been eliminated.
Historically, chronic arthropathy has been the major long-term disability associated with hemophilia. The natural history of untreated hemophilia is one of cyclic recurrent hemorrhages into specific joints, including hemorrhages into the same (target) joint. In young children, the joint distends easily and a large volume of blood may fill the joint until tamponade ensues or therapy intervenes. After joint hemorrhage, proteolytic enzymes are released by white blood cells into the joint space, and heme iron induces macrophage proliferation, leading to inflammation of the synovium. The synovium thickens and develops frondlike projections into the joint that are susceptible to being pinched and may induce further hemorrhage. The cartilaginous surface becomes eroded and ultimately may even expose raw bone, leaving the joint susceptible to articular fusion. In the older patient with advanced arthropathy, bleeding into the target joint, with its thickened synovium, causes severe pain, because the joint may have little space to accommodate blood. Once a target joint is seen to be developing, the patient is usually given short-or long-term prophylaxis to prevent progression of the arthropathy and reduce inflammation.
Inhibitor Formation
Infusion of the deficient clotting factor may initiate an immune response in patients with either factor VIII or factor IX deficiency. Inhibitors are antibodies directed against factor VIII or factor IX that block the clotting activity. Failure of a bleeding episode to respond to appropriate replacement therapy is usually the first sign of an inhibitor. Less often, inhibitors are identified during routine follow-up testing. Inhibitors develop in approximately 25-35% of patients with hemophilia A; the percentage is somewhat lower in patients with hemophilia B, many of whom make an inactive dysfunctional protein that renders them less susceptible to an immune response. Highly purified factor IX or recombinant factor IX seems to increase the frequency of inhibitor development, and some anti–factor IX inhibitors induce anaphylaxis. Many patients who have an inhibitor lose it with continued regular infusions. Others have a higher titer of antibody with subsequent infusions and may need to go through desensitization programs, in which high doses of factor VIII for hemophilia A or factor IX for hemophilia B are infused in an attempt to saturate the antibody and permit the body to develop tolerance. Factor IX immune tolerance programs have resulted in nephrotic syndrome in some patients. Rituximab has been used, off label (i.e., in a use not approved by the U.S. Food and Drug Administration [FDA]), as an alternate therapy for patients with high inhibitor titers in whom immune tolerance programs have failed. If desensitization fails, bleeding episodes are treated with either recombinant factor VIIa or activated prothrombin complex concentrates. The use of these products bypasses the inhibitor in many instances but may increase the risk of thrombosis. Patients with inhibitors require referral to a center that cares for many such patients and has a comprehensive hemophilia program.
In the past, plasma-derived treatment products transmitted hepatitis B and C as well as HIV to large numbers of patients with hemophilia. In the era of recombinant products, the risk of acquiring such infections should be minimal, but patients should receive appropriate immunizations against hepatitis B. Those who are exposed to blood products should be monitored for transfusion-related infections. Reports have also identified the transmission of variant Creutzfeldt-Jakob disease to patients receiving therapeutic plasma and may warrant study of patients with hemophilia for prion transmission from plasma-derived factor concentrates.
Comprehensive Care
Today, patients with hemophilia are best managed through comprehensive hemophilia care centers. Such centers are dedicated to patient and family education as well as to the prevention and/or treatment of the complications of hemophilia, including chronic joint disease and inhibitor development as well as infection, such as hepatitis B and C or HIV. Such centers involve a team of physicians, nurses, orthopedists, physical therapists, and psychosocial workers, among others. Education remains crucial in hemophilia care, because patients who are receiving prophylaxis may be less “experienced” in recognizing bleeding episodes than affected children from previous eras.
Berntorp E, Abshire T. The von Willebrand disease prophylaxis network: exploring a treatment concept. J Thromb Haemost. 2006;4:2511-2512.
Berntorp E, Shapiro A, Astermark J, et al. Inhibitor treatment in haemophilias A and B: summary statement for the 2006 international consensus conference. Haemophilia. 2006;12(Suppl 6):1-7.
DiMichele DM. Immune tolerance: critical issues of factor dose, purity and treatment complications. Haemophilia. 2006;12(Suppl 6):81-85.
Greninger DA, Saint-Remy JM, Jacquemin M, et al. The use of factor VIII/von Willebrand factor concentrate for immune tolerance induction in haemophilia A patients with high-titre inhibitors: association of clinical outcome with inhibitor epitope profile. Haemophilia. 2008;14:295-302.
Hermans C, Altisent C, Batorova A, et al. Replacement therapy for invasive procedures in patients with haemophilia: literature review, European survey and recommendations. Haemophilia. 2009;15:639-658.
Key NS, Negrier C. Coagulation factor concentrates: past, present, and future. Lancet. 2007;370:439-448.
Knight C, Dano AM, Kennedy-Martin T. A systematic review of the cost-effectiveness of rFVIIa and APCC in the treatment of minor/moderate bleeding episodes for haemophilia patients with inhibitors. Haemophilia. 2009;15:405-419.
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535-544.
Mannucci PM. Back to the future: a recent history of haemophilia treatment. Haemophilia. 2008;14(Suppl 3):10-18.
Montgomery RR, Cox Gill J, Di Paola J. Hemophilia and von Willebrand disease. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1487-1524.
Pipe SW. Recombinant clotting factors. ThrombHaemost. 2008;99:840-850.
Ross C, Goldenberg NA, Hund D, et al. Athletic participation in severe hemophilia: bleeding and joint outcomes in children on prophylaxis. Pediatrics. 2009;124:1267-1272.
Street AM, Ljung R, Lavery SA. Management of carriers and babies with haemophilia. Haemophilia. 2008;14(Suppl 3):181-187.
Tagariello G, Iorio A, Santagostino E, et al. Comparison of the rates of joint arthroplasty in patients with severe factor VIII and IX deficiency: an index of different clinical severity of the two coagulation disorders. Blood. 2009;114:779-784.
Viel KR, Ameri A, Abshire TC, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. 2009;360:1618-1626.
470.2 Factor XI Deficiency (Hemophilia C)
Factor XI deficiency is an autosomal deficiency associated with mild to moderate bleeding symptoms. It is frequently encountered in Ashkenazi Jews but has been found in many other ethnic groups. In Israel, 1-3/1,000 individuals are homozygous for this deficiency.
The bleeding tendency is not as severe as in factor VIII or factor IX deficiency. The bleeding associated with factor XI deficiency is not correlated with the amount of factor XI. Some patients with severe deficiency may have minimal or no symptoms at the time of major surgery. Because factor XI augments thrombin generation and leads to activation of the fibrinolytic inhibitor thrombin-activatable fibrinolysis inhibitor (TAFI), surgical bleeding is more prominent in sites of high fibrinolytic activity like the oral cavity. Unless the patient previously had surgery without bleeding, replacement therapy should be considered and given preoperatively, depending on the nature of the surgical procedure. No approved concentrate of factor XI is available in the USA; therefore, the physician must use fresh frozen plasma (FFP).
Bleeding during minor surgery can be controlled with local pressure. Patients undergoing dental extractions can be monitored closely and may benefit from treatment with fibrinolytic inhibitors like aminocaproic acid, with plasma replacement therapy used only if hemorrhage occurs. In a patient with homozygous deficiency of factor XI, PTT is often longer than it is in patients with either severe factor VIII or factor IX deficiency. The paradox of fewer clinical symptoms in combination with longer PTT is surprising, but it occurs because factor VIIa can activate factor IX in vivo. The deficiency of factor XI can be confirmed by specific factor XI assays. Plasma infusions of 1 IU/kg usually increase the plasma concentration by 2%. Thus, infusion of plasma at 10-15 mL/kg will result in a plasma level of 20-30%, which is usually sufficient to control moderate hemorrhage. Frequent infusions of plasma would be necessary to achieve higher levels of factor XI. Because the half-life of factor XI is usually ≥48 hr, maintaining adequate levels of factor XI commonly is not difficult.
Chronic joint bleeding is rarely a problem in factor XI deficiency, and for most patients, the deficiency is a concern only at the time of major surgery unless there is a second underlying hemostatic defect (e.g., von Willebrand disease).
470.3 Deficiencies of the Contact Factors (Nonbleeding Disorders)
J. Paul Scott and Robert R. Montgomery
Deficiency of the “contact factors” (factor XII, prekallikrein, and high molecular weight kininogen) causes prolonged PTT but no bleeding symptoms. Because these contact factors function at the step of initiation of the intrinsic clotting system by the reagent used to determine PTT, the PTT is markedly prolonged when these factors are absent. Thus, there is the paradoxical situation in which PTT is extremely prolonged with no evidence of clinical bleeding. It is important that individuals with these findings be well informed about the meaning of their clotting factor deficiency because they do not need treatment, even for major surgery.
470.4 Factor VII Deficiency
J. Paul Scott and Robert R. Montgomery
Factor VII deficiency is a rare autosomal bleeding disorder usually detected only in the homozygous state. Severity of bleeding varies from mild to severe with hemarthroses, spontaneous intracranial hemorrhage, and mucocutaneous bleeding, especially nosebleeds and menorrhagia. Patients with this deficiency have markedly prolonged PT but normal PTT. Factor VII assays show a marked reduction in factor VII. Because the plasma half-life of factor VII is 2-4 hr, therapy with FFP is difficult and is often complicated by fluid overload. A commercial concentrate of recombinant factor VIIa has been shown in case reports to be effective in treating some patients with factor VII deficiency, but this concentrate has not been approved by the FDA for this indication.
470.5 Factor X Deficiency
J. Paul Scott and Robert R. Montgomery
Factor X deficiency is a rare (estimated 1/1,000,000) autosomal disorder with variable severity. Mild deficiency results in mucocutaneous and post-traumatic bleeding, whereas severe deficiency results in spontaneous hemarthroses and intracranial hemorrhages. Factor X deficiency is the result of either a quantitative deficiency or a dysfunctional molecule. A reduced factor X level is associated with prolongation of both PT and PTT. In patients with hereditary factor X deficiency, factor X levels can be increased with use of either FFP or prothrombin complex concentrate. The half-life of factor X is approximately 30 hr, and its volume of distribution is similar to that of factor IX. Thus, 1 U/kg will increase the plasma level of factor X by 1%.
Although it is rarely a problem in pediatric patients, systemic amyloidosis may be associated with factor X deficiency, owing to the adsorption of factor X on the amyloid protein. In the setting of amyloidosis, transfusion therapy often is not successful because of the rapid clearance of factor X.
470.6 Prothrombin (Factor II) Deficiency
Prothrombin deficiency is caused either by a markedly reduced prothrombin level (hypoprothrombinemia) or by functionally abnormal prothrombin (dysprothrombinemia). Laboratory testing in homozygous patients shows prolonged PT and PTT. Factor II, or prothrombin, assays show a markedly reduced prothrombin level. Mucocutaneous bleeding in infancy and post-traumatic bleeding later are common. Patients are treated with either FFP or, rarely, prothrombin complex concentrates. In prothrombin deficiency, FFP is useful, because the half-life of prothrombin is 3.5 days. Administration of 1 IU/kg of prothrombin will increase the plasma activity by 1%.
470.7 Factor V Deficiency
Deficiency of factor V is an autosomal recessive, mild to moderate bleeding disorder that has also been termed parahemophilia. Hemarthroses occur rarely; mucocutaneous bleeding and hematomas are the most common symptoms. Severe menorrhagia is a frequent symptom in women. Laboratory evaluation shows prolonged PTT and PT. Specific assays for factor V show a reduction in factor V levels. FFP is the only currently available therapeutic product that contains factor V. Factor V is lost rapidly from stored FFP. Patients with severe factor V deficiency are treated with infusions of FFP at 10 mL/kg every 12 hr. Rarely, a patient with a negative family history of bleeding has an acquired antibody to factor V. Often, such a patient does not bleed because the factor V in platelets prevents excessive bleeding.
470.8 Combined Deficiency of Factors V and VIII
J. Paul Scott and Robert R. Montgomery
Combined deficiency of factors V and VIII occurs secondary to the absence of an intracellular transport protein that is responsible for transporting factors V and VIII from the endoplasmic reticulum to the Golgi compartments. This explains the paradoxical deficiency of 2 factors, one encoded on chromosome 1 and the other on the X chromosome. Bleeding symptoms are often milder than for hemophilia A and are treated with FFP to replace both factors V and VIII.
470.9 Fibrinogen (Factor I) Deficiency
J. Paul Scott and Robert R. Montgomery
Congenital afibrinogenemia is a rare autosomal recessive disorder in which there is an absence of fibrinogen. Patients with this disorder do not bleed as frequently as patients with hemophilia and rarely have hemarthroses. Affected patients may present in the neonatal period with gastrointestinal hemorrhage or hematomas after vaginal delivery. In addition to marked prolongation of PT and PTT, thrombin time is prolonged. In the absence of consumptive coagulopathy, an unmeasurable fibrinogen level is diagnostic. In addition to the quantitative deficiency of fibrinogen, a number of dysfunctional fibrinogens have been reported (dysfibrinogenemia). Rarely patients with dysfibrinogenemia present with thrombosis. Currently, no fibrinogen concentrates are commercially available. Because the plasma half-life of fibrinogen is 2-4 days, treatment with either FFP or cryoprecipitate is effective. The hemostatic level of fibrinogen is >60 mg/dL. Each bag of cryoprecipitate contains 100-150 mg of fibrinogen. Some clinical assays for fibrinogen are inhibited by high doses of heparin. Thus, a markedly prolonged thrombin time associated with a low fibrinogen level should be evaluated with determination of reptilase time. Prolonged reptilase time confirms that functional levels of fibrinogen are low and that heparin is not present.
470.10 Factor XIII Deficiency (Fibrin-Stabilizing Factor or Transglutaminase Deficiency)
Because factor XIII is responsible for the cross linking of fibrin to stabilize the fibrin clot, symptoms of delayed hemorrhage are secondary to instability of the clot. Typically, patients have trauma 1 day and then have a bruise or hematoma the next day. Clinical symptoms include mild bruising, delayed separation of the umbilical stump beyond 4 wk in neonates, poor wound healing, and recurrent spontaneous abortions in women. Rare kindreds with XIII deficiency with hemarthroses and intracranial hemorrhage have been described. Results of the usual screening tests for hemostasis are normal in patients with factor XIII deficiency. Screening tests for factor XIII deficiency are based on the observation that there is increased solubility of the clot because of the failure of cross linking. The normal clot remains insoluble in the presence of 5M urea, whereas in a patient with XIII deficiency, the clot dissolves. More specific assays for factor XIII are immunologic. Because the half-life of factor XIII is 5-7 days and the hemostatic level is 2-3% activity, infusion of FFP or cryoprecipitate will correct the deficiency in patients with factor XIII deficiency. Plasma contains 1 IU/dL, and cryoprecipitate contains 75 IU/bag. In patients with significant bleeding symptoms, prophylaxis can be achieved with infusion of cryoprecipitate every 3-4 wk.
470.11 Antiplasmin or Plasminogen Activator Inhibitor Deficiency
Deficiency of either antiplasmin or plasminogen activator inhibitor, both of which are antifibrinolytic proteins, results in increased plasmin generation and premature lysis of fibrin clots. Affected patients have a mild bleeding disorder characterized by mucocutaneous bleeding but rarely have joint hemorrhages. Because results of the usual hemostatic tests are normal, further work-up of a patient with a positive bleeding history should include euglobulin clot lysis time (if available), which measures fibrinolytic activity and yields a shortened result in the presence of these deficiencies. Specific assays for α2-antiplasmin and plasminogen activator inhibitor are available. Bleeding episodes are treated with FFP; bleeding in the oral cavity may respond to aminocaproic acid.
Agren A, Wiman B, Schulman S. Laboratory evidence of hyperfibrinolysis in association with low plasminogen activator inhibitor type 1 activity. Blood Coagul Fibrinolysis. 2007;18:657-660.
Bauer KA. Rare coagulation factor abnormalities. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1525-1532.
Carpenter SL, Mathew P. Alpha2-antiplasmin and its deficiency: fibrinolysis out of balance. Haemophilia. 2008;14:1250-1254.