Chapter 525 Bartter and Gitelman Syndromes and Other Inherited Tubular Transport Abnormalities
525.1 Bartter Syndrome
Rajasree Sreedharan and Ellis D. Avner
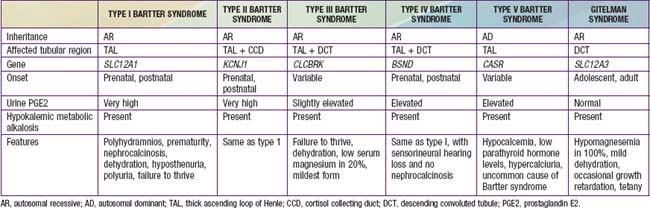
Bartter syndrome is a group of disorders characterized by hypokalemic metabolic alkalosis with hypercalciuria and salt wasting (Chapter 52) (Table 525-1). Antenatal Bartter syndrome (types I, II, IV) (also called hyperprostaglandin E syndrome) typically manifests in infancy and has a more-severe phenotype than classic Bartter syndrome (type III), including maternal polyhydramnios, neonatal salt wasting, and severe episodes of recurrent dehydration. The milder phenotype, classic Bartter syndrome, manifests in childhood with failure to thrive and a history of recurrent episodes of dehydration. A phenotypically related disease, Gitelman syndrome has a distinct genetic defect and is discussed in Chapter 525.2 (see Table 525-1). One distinct variant of antenatal Bartter syndrome is associated with sensorineural deafness (type IV).
Pathogenesis
The biochemical features of Bartter syndrome, including hypokalemic metabolic alkalosis with hypercalciuria, resemble those seen with chronic use of loop diuretics and reflect a defect in sodium, chloride, and potassium transport in the ascending loop of Henle. The loss of sodium and chloride, with resultant volume contraction, stimulates the renin-angiotensin II-aldosterone (RAA) axis. Aldosterone promotes sodium uptake and potassium secretion, exacerbating the hypokalemia. It also stimulates hydrogen ion secretion distally, worsening the metabolic alkalosis. Hypokalemia stimulates prostaglandin synthesis, which further activates the RAA axis. Bartter syndrome has been associated with 5 distinct genetic defects in loop of Henle transporters (see Table 525-1). Each contributes, in some manner, to sodium and chloride transport. Mutations in the genes that encode the Na+/K+/2Cl− transporter (NKCC2, the site of action of furosemide), the luminal potassium channel (ROMK), combined chloride channel (CLC-Ka, CLC-Kb), or subunit of chloride channels (barttin) cause neonatal Bartter syndrome. Isolated defects in the genes that produce the basolateral chloride channel ClC-Kb cause classic Bartter syndrome.
Clinical Manifestations
A history of maternal polyhydramnios with or without prematurity may be elicited. Dysmorphic features, including triangular facies, protruding ears, large eyes with strabismus, and drooping mouth may be present on physical examination. Consanguinity suggests the presence of an autosomal recessive disorder. Older children can have a history of recurrent episodes of polyuria with dehydration, failure to thrive, and the classic biochemical abnormalities of a hypokalemic metabolic alkalosis. Urinary calcium levels are typically elevated, as are urinary potassium and sodium levels. Serum renin, aldosterone, and prostaglandin E levels are often markedly elevated, particularly in the more-severe antenatal form. Blood pressure is usually normal, although patients with the antenatal form can have severe salt wasting, resulting in dehydration and hypotension. Renal function is typically normal. Nephrocalcinosis, resulting from hypercalciuria, may be seen on ultrasound examination (types I, II, III, V).
Diagnosis
The diagnosis is usually made based on clinical presentation and laboratory findings. The diagnosis in the neonate or infant is suggested by severe hypokalemia, usually <2.5 mmol/L, with metabolic alkalosis. Hypercalciuria is typical; hypomagnesemia is seen in a minority of patients but is more common in Gitelman syndrome. Because features of Bartter syndrome resemble chronic use of loop diuretics, diuretic abuse should be considered in the differential diagnosis, even in young children. Chronic vomiting can also give a similar clinical picture but can be distinguished by measurement of urinary chloride, which is elevated in Bartter syndrome and low in patients with chronic vomiting. Histologically, kidneys demonstrate hyperplasia of the juxtaglomerular apparatus. Renal biopsy is rarely performed to diagnose this condition.
Treatment and Prognosis
Treatment of Bartter syndrome is directed at preventing dehydration, maintaining nutritional status, and correcting hypokalemia. Potassium supplementation, often at very high doses, is required; potassium-sparing (aldosterone antagonists) diuretics may be of value. Even with appropriate therapy, serum potassium values might not normalize, particularly in patients with the neonatal form. Infants and young children require a high sodium diet and at times sodium supplementation. Indomethacin, a prostaglandin inhibitor, can also be effective. If hypomagnesemia is present, magnesium supplementation is required. With close attention to electrolyte balance, volume status, and growth, the long-term prognosis is generally good. In a minority of patients, chronic hypokalemia, nephrocalcinosis, and chronic indomethacin therapy can lead to chronic interstitial nephritis and chronic renal failure.
525.2 Gitelman Syndrome
Rajasree Sreedharan and Ellis D. Avner
Gitelman syndrome (often called a “Bartter syndrome variant”) is a rare autosomal recessive cause of hypokalemic metabolic alkalosis, with distinct features of hypocalciuria and hypomagnesemia. Patients with Gitelman syndrome typically present in late childhood or early adulthood (see Table 525-1).
Pathogenesis
The biochemical features of Gitelman syndrome resemble those of chronic use of thiazide diuretics. Thiazides act on the sodium chloride co-transporter NCCT, present in the distal convoluted tubule. Through linkage analysis and mutational studies, defects in the gene encoding NCCT have been demonstrated in patients with Gitelman syndrome.
Clinical Manifestations
Patients with Gitelman syndrome typically present at a later age than those with Bartter syndrome. Patients often have a history of recurrent muscle cramps and spasms, presumably caused by low serum magnesium levels. They usually do not have a history of recurrent episodes of dehydration. Biochemical abnormalities include hypokalemia, metabolic alkalosis, and hypomagnesemia. The urinary calcium level is usually very low (in contrast to the elevated urinary calcium level often seen in Bartter syndrome), and the urinary magnesium level is elevated. Renin and aldosterone levels are usually normal, and prostaglandin E secretion is not elevated. Growth failure is less prominent in Gitelman syndrome than in Bartter syndrome, but it may be present.
Diagnosis
The diagnosis of Gitelman syndrome is suggested in an adolescent or adult presenting with hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria.
Treatment
Therapy is directed at correcting hypokalemia and hypomagnesemia with supplemental potassium and magnesium. Sodium supplementation or treatment with prostaglandin inhibitors is generally not necessary because patients typically do not have episodes of volume depletion or elevated prostaglandin E excretion.
525.3 Other Inherited Tubular Transport Abnormalities
Rajasree Sreedharan and Ellis D. Avner
Inherited abnormalities in distinct transporters in each segment of the nephron have now been identified and the molecular defects have been characterized. Renal tubular acidosis and nephrogenic diabetes insipidus are discussed in detail in Chapters 523 and 524, respectively. Cystinuria is an autosomal recessive disorder seen primarily in patients of Middle Eastern descent and is characterized by recurrent stone formation. The disease is caused by a defective high-affinity transporter for L-cystine and dibasic amino acids present in the proximal tubule.
Dent disease is an X-linked proximal tubulopathy with characteristic abnormalities that include low-molecular-weight (LMW) proteinuria, hypercalciuria, and other features of Fanconi syndrome, such as glycosuria, aminoaciduria, and phosphaturia. Although some patients develop nephrocalcinosis, nephrolithiasis, progressive renal failure, and hypophosphatemic rickets, patients with Dent disease typically do not have proximal renal tubular acidosis or extrarenal manifestations. Since the turn of the century, loss-of-function mutations of the CLCN5 gene, which is located in Xp11.22 and encodes a renal Cl−/H+ antiporter (ClC-5), have been reported consistently in patients with Dent disease. Genetic heterogeneity of Dent disease in some patients who exhibit mutations in the gene for OCRL1 (responsible for Lowe syndrome) also meets Dent disease criteria:Dent-2 disease. Dent disease includes X-linked recessive nephrolithiasis with renal failure, X-linked recessive hypophosphatemic rickets, and idiopathic LMW proteinuria seen in Japanese children.
Mutations in an extracellular basolateral calcium sensing receptor (CASR), normally present in the loop of Henle can cause a dominant Bartter syndrome–like picture. These patients’ predominant symptoms are hypocalcemic hypercalciuria, which differentiates them from patients with Bartter syndrome.
In the distal convoluted tubule, gain-of-function mutations in WNK1 and loss-of-function mutations in WNK4, both serine threonine kinases, lead to excessive NCCT-mediated salt reabsorption with the clinical picture of pseudohypoaldosteronism type 2 (familial hyperkalemic hypertension [FHH], or Gordon syndrome).
In the collecting duct, gain-of-function mutations of the gene that encodes the epithelial sodium channel causes an inherited form of hypertension, Liddle syndrome. Patients with this disorder have constitutive sodium uptake in the collecting duct, with hypokalemia and suppressed aldosterone. Conversely, loss-of-function mutations cause pseudohypoaldosteronism, characterized by severe sodium wasting and hyperkalemia. A variant of the latter disorder is associated with systemic abnormalities, including defects in sweat chloride, and can resemble cystic fibrosis.
Bökenkamp A, Böckenhauer D, Cheonh HI, et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr. 2009;155:94-99.
Chadha V, Alon US. Hereditary renal tubular disorders. Sem Nephrol. 2009;29:399-411.
Cho HY, Lee BH, Choi HJ, et al. Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol. 2008;23:243-249.
Devuyst O, Konrad M, Jeunemaitre X, et al. Tubular disorders of electrolyte regulation. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:929-978.
Garnier A, Dreux S, Vargas-Poussou R, et al. Bartter syndrome prenatal diagnosis based on amniotic fluid biochemical analysis. Pediatr Res. 2010;67(3):300-303.
Kleta R, Bockenhauer D. Bartter syndrome and other salt-losing tubulopathies. Nephron Physiol. 2006;104:73-80.
Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4:560-570.
Sung CC, Chen YS, Lin SH. Family paralysis. Lancet. 2011;377:352.