Chapter 561 Goiter
A goiter is an enlargement of the thyroid gland. Persons with enlarged thyroids can have normal function of the gland (euthyroidism), thyroid deficiency (hypothyroidism), or overproduction of the hormones (hyperthyroidism). Goiter may be congenital or acquired, endemic, or sporadic.
The goiter often results from increased pituitary secretion of thyroid-stimulating hormone (TSH) in response to decreased circulating levels of thyroid hormones. Thyroid enlargement can also result from infiltrative processes that may be inflammatory or neoplastic. Goiter in patients with Graves disease and thyrotoxicosis is caused by thyrotropin receptor–stimulating antibodies (TRSAbs).
561.1 Congenital Goiter
Congenital goiter is usually sporadic and can result from a fetal thyroxine (T4) synthetic defect or from administration of antithyroid drugs or iodides during pregnancy for the treatment of maternal thyrotoxicosis. Goitrogenic drugs and iodides cross the placenta and at high doses can interfere with synthesis of thyroid hormone, resulting in goiter and hypothyroidism in the fetus. The concomitant administration of thyroid hormone with the goitrogen does not prevent this effect, because insufficient amounts of T4 cross the placenta. Iodides are included in many proprietary cough preparations used to treat asthma; these preparations should be avoided during pregnancy because they have often been reported to cause congenital goiter. Amiodarone, an antiarrhythmic drug with 37% iodine content, has also caused congenital goiter with hypothyroidism. Even when the infant is clinically euthyroid, there may be retardation of osseous maturation, low levels of T4, and elevated levels of TSH. In women with Graves disease receiving antithyroid drugs, these effects can occur when the mother takes propylthiouracil at only 100-200 mg/24 hr; all such infants should undergo thyroid studies at birth. Administration of thyroid hormone to affected infants may be indicated to treat clinical hypothyroidism, to hasten the disappearance of the goiter, and to prevent brain damage. Because the condition is rarely permanent, thyroid hormone may be safely discontinued after the antithyroid drug has been excreted by the neonate, usually after 1-2 wk.
Enlargement of the thyroid at birth may occasionally be sufficient to cause respiratory distress that interferes with nursing and can even cause death. The head may be maintained in extreme hyperextension. When respiratory obstruction is severe, partial thyroidectomy rather than tracheostomy is indicated (Fig. 561-1).
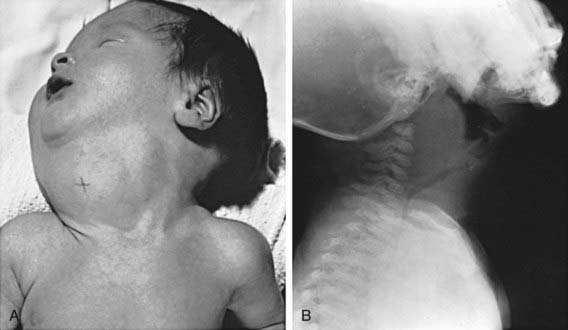
Figure 561-1 Congenital goiter in infancy. A, Large congenital goiter in an infant born to a mother with thyrotoxicosis who had been treated with iodides and methimazole during pregnancy. B, A different infant, 6 wk old, with increasing respiratory distress and cervical mass since birth. The operation revealed a large goiter that almost completely encircled the trachea. Notice the anterior deviation and posterior compression of the trachea. Partial thyroidectomy completely relieved the symptoms. It is apparent why a tracheostomy is not adequate treatment for these infants. The cause for the goiter was not found.
Goiter is almost always present in the infant with neonatal Graves’ disease. These goiters usually are not large; the infant manifests clinical symptoms of hyperthyroidism. The mother often has a history of Graves disease, although discovery of neonatal hyperthyroidism can lead to the diagnosis of maternal Graves’ disease. Thyroid enlargement results from transplacental passage of maternal thyroid-stimulating immunoglobulin (Chapter 562.1). TSH receptor-activating mutations are also a recognized cause of congenital goiter and hyperthyroidism.
When no causative factor is identifiable, a defect in synthesis of thyroid hormone should be suspected. Neonatal screening programs find congenital hypothyroidism caused by such a defect in 1/30,000-50,000 live births. It is advisable to treat immediately with thyroid hormone and to postpone more-detailed studies for later in life. If a specific defect is suspected, genetic tests to identify a mutation may be undertaken (Chapter 559). Because these defects are transmitted by recessive genes, a precise diagnosis is helpful for genetic counseling. Monitoring subsequent pregnancies with ultrasonography can be useful in detecting fetal goiters (Chapter 90).
Pendred’s syndrome, characterized by familial goiter and neurosensory deafness, is caused by a mutation in the pendrin gene, which codes for a chloride-iodide transport protein present in the thyroid gland and cochlea. This defect results in abnormal iodide organification in the thyroid and can cause a goiter at birth. The more common presentation is a euthyroid goiter and deafness later in life.
Iodine deficiency as a cause of congenital goiter is rare in developed countries but persists in isolated endemic areas (see later). More important is the recognition that severe iodine deficiency early in pregnancy can cause neurologic damage during fetal development, even in the absence of goiter. The iodine deficiency can result in maternal and fetal hypothyroidism, preventing the partially protective transfer of maternal thyroid hormones.
When the “goiter” is lobulated, asymmetric, firm, or large to an unusual degree, a teratoma within or in the vicinity of the thyroid must be considered in the differential diagnosis (Chapter 563).
Bizhanova A, Kopp P. Minireview: the sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinol. 2009;150:1084-1090.
Caron P, Moya CM, Malet D, et al. Compound heterozygous mutations in the thyroglobulin gene (1143delC and 6725G → A [R2223H]) resulting in fetal goitrous hypothyroidism. J Clin Endocrinol Metab. 2003;88:3546-3553.
Hashimoto H, Hashimoto K, Suehara N. Successful in utero treatment of fetal goitrous hypothyroidism: case report and review of the literature. Fetal Diagn Ther. 2006;21:360-365.
Koksal N, Akturk B, Saglan H, et al. Reference values for neonatal thyroid volumes in a moderately iodine-deficient area. J Endocrinol Invest. 2008;31:642-646.
Moreno JC, Klootwijk W, van Toor H, et al. Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med. 2008;358:1811-1818.
Remuzzi G, Garattni S. Elimination of iodine-deficiency disorders in Tibet. Lancet. 2008;371:1980-1981.
Teng W, Shan Z, Teng X, et al. Effect of iodine intake on thyroid diseases in China. N Engl J Med. 2006;354:2782-2893.
Zimmernamm MB, Jooste PL, Pandav CS. Iodine-deficiency disorders. Lancet. 2008;372:1251-1262.
561.2 Intratracheal Goiter
One of the many ectopic locations of thyroid tissue is within the trachea. The intraluminal thyroid lies beneath the tracheal mucosa and is often continuous with the normally situated extratracheal thyroid. The thyroid tissue is susceptible to goitrous enlargement, which involves the normally situated and the ectopic thyroid. When there is obstruction of the airway associated with a goiter, it must be ascertained whether the obstruction is extratracheal or endotracheal. If obstructive manifestations are mild, administration of sodium L-thyroxine usually causes the goiter to decrease in size. When symptoms are severe, surgical removal of the endotracheal goiter is indicated.
561.3 Endemic Goiter and Cretinism
Etiology
The association between dietary deficiency of iodine and the prevalence of goiter or cretinism is well established. A moderate deficiency of iodine can be overcome by increased efficiency in the synthesis of thyroid hormone. Iodine liberated in the tissues is returned rapidly to the gland, which resynthesizes triiodothyronine (T3) preferentially at a higher rate than normal. This increased activity is achieved by compensatory hypertrophy and hyperplasia (goiter), which satisfy the demands of the tissues for thyroid hormone. In geographic areas where deficiency of iodine is severe, decompensation and hypothyroidism can result. It is estimated that 1 billion persons in developing countries live in areas of iodine deficiency.
Seawater is rich in iodine; the iodine content of fish and shellfish is also high. Endemic goiter is therefore rare in populations living along the coast. Iodine is deficient in the water and native foods in the Pacific West and the Great Lakes areas of the United States. Deficiency of dietary iodine is even greater in certain Alpine valleys, the Himalayas, the Andes, the Congo, and the highlands of Papua New Guinea. In areas such as the United States, where iodine is provided in foods from other areas and in iodized salt, endemic goiter has disappeared. Iodized salt in the United States contains potassium iodide (100 µg/g), which provides excellent prophylaxis. Further iodine intake in the United States is contributed by iodates used in baking, iodine-containing coloring agents, and iodine-containing disinfectants used in the dairy industry. The recommended daily allowance of iodine is as follows:
The intake of iodine in adults in the United States decreased by approximately 50% from the 1970s to the 1990s, as reflected by a drop in median urinary iodine excretion from 320 µg/L to 145 µg/L. This decrease appears to have stabilized; the most recent NHANES (National Health and Nutrition Examination Survey) from 2001-2002 reports median urinary iodine excretion of 167.8 µg/L. However, approximately 15% of women of reproductive age have iodine excretion <100 µg/L.
Clinical Manifestations
If the deficiency of iodine is mild, thyroid enlargement does not become noticeable except when there is increased demand for the hormone during periods of rapid growth, as in adolescence and during pregnancy. In regions of moderate iodine deficiency, goiter observed in school children can disappear with maturity and reappear during pregnancy or lactation. Iodine-deficient goiters are more common in girls than in boys. In areas where iodine deficiency is severe, as in the hyperendemic highlands of Papua New Guinea, nearly half the population has large goiters, and endemic cretinism is common (Fig. 561-2).
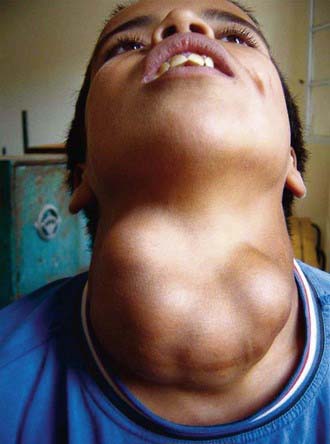
Figure 561-2 A 14 yr old boy with a large nodular goiter was seen in 2004, in an area of severe iodine-deficiency disorders in northern Morocco. He had tracheal and esophageal compression and hoarseness, probably due to damage to the recurrent laryngeal nerves.
(From Zimmernamm MB, Jooste PL, Pandav CS: Iodine-deficiency disorders, Lancet 372:1251–1262, 2008.)
Serum T4 levels are often low in persons with endemic goiter, although clinical hypothyroidism is rare. This is true in New Guinea, the Congo, the Himalayas, and South America. Despite low serum T4 levels, serum TSH concentrations are often normal or only moderately increased. In such patients, circulating levels of T3 are elevated. Moreover, T3 levels are also elevated in patients with normal T4 levels, indicating a preferential secretion of T3 by the thyroid in this disease.
Endemic cretinism is the most serious consequence of iodine deficiency; it occurs only in geographic association with endemic goiter. The term endemic cretinism includes 2 different but overlapping syndromes—a neurologic type and a myxedematous type. The incidence of the 2 types varies among different populations. In Papua New Guinea, the neurologic type occurs almost exclusively, whereas in the Congo, the myxedematous type predominates. Both types are found in all endemic areas, and some persons have intermediate or mixed features.
The neurologic syndrome is characterized by mental retardation, deaf-mutism, disturbances in standing and gait, and pyramidal signs such as clonus of the foot, the Babinski sign, and patellar hyperreflexia. Affected persons are goitrous but euthyroid, have normal pubertal development and adult stature, and have little or no impaired thyroid function. Persons with the myxedematous syndrome also are mentally retarded and deaf and have neurologic symptoms, but in contrast to the neurologic type they have delayed growth and sexual development, myxedema, and absence of goiter. Serum T4 levels are low and TSH levels are markedly elevated. Delayed skeletal maturation may extend into the 3rd decade or later. Ultrasonographic examination shows thyroid atrophy.
Pathogenesis
The pathogenesis of the neurologic syndrome has been attributed to iodine deficiency and hypothyroxinemia during pregnancy, leading to fetal and postnatal hypothyroidism. Although some investigators have attributed brain damage to a direct effect of elemental iodine deficiency in the fetus, most believe the neurologic symptoms are caused by fetal and maternal hypothyroxinemia. There is evidence for the presence of thyroid hormone receptors in the fetal brain as early as 7 wk of gestation. Although the normal fetal thyroid gland does not begin to produce significant amounts of thyroid hormone until mid-gestation, there is measurable T4 in the coelomic fluid as early as 6 weeks, almost certainly of maternal origin. These lines of evidence support a role for maternal thyroid hormone in fetal brain development in the first trimester. In addition, there is evidence of transplacental passage of maternal thyroid hormone into the fetus, which normally might ameliorate the effects of fetal hypothyroidism on the developing nervous system in the second half of pregnancy. Thus, iodine deficiency in the mother affects fetal brain development both in the first trimester and throughout pregnancy. Intake of iodine after birth is often sufficient for normal or only minimally impaired thyroid function.
The pathogenesis of the myxedematous syndrome leading to thyroid atrophy is more bewildering. Searches for additional environmental factors that might provoke continuing postnatal hypothyroidism have led to incrimination of selenium deficiency, goitrogenic foods, thiocyanates, and Yersinia (Table 561-1). Studies from western China suggest that thyroid autoimmunity might play a role. Children with myxedematous cretinism with thyroid atrophy, but not children with euthyroid cretinism, were found to have thyroid growth-blocking immunoglobulins of the kind found in infants with sporadic congenital hypothyroidism. Others are skeptical about any role of thyroid growth-blocking immunoglobulins to explain these findings.
Table 561-1 GOITROGENS AND THEIR MECHANISM
| GOITROGEN | MECHANISM |
|---|---|
| FOODS | |
| Cassava, lima beans, linseed, sorghum, sweet potato | Contain cyanogenic glucosides that are metabolized to thiocyanates that compete with iodine for uptake by the thyroid |
| Cruciferous vegetables such as cabbage, kale, cauliflower, broccoli, turnips, rapeseed | Contain glucosinolates; metabolites compete with iodine for uptake by the thyroid |
| Soy, millet | Flavonoids impair thyroid peroxidase activity |
| INDUSTRIAL POLLUTANTS | |
| Perchlorate | Competitive inhibitor of the sodium-iodine symporter, decreasing iodine transport into the thyroid |
| Others (e.g., disulfides from coal processes) | Reduce thyroidal iodine uptake |
| Smoking | An important goitrogen; smoking during breast-feeding is associated with reduced iodine concentrations in breast milk; high serum concentration of thiocyanate due to smoking might compete with iodine for active transport into the secretory epithelium of the lactating breast |
| NUTRIENTS | |
| Selenium deficiency | Accumulated peroxides can damage the thyroid, and deiodinase deficiency impairs thyroid hormone synthesis |
| Iron deficiency | Reduces heme-dependent thyroperoxidase activity in the thyroid and might blunt the efficacy of iodine prophylaxis |
| Vitamin A deficiency | Increases TSH stimulation and goiter through decreased vitamin A–mediated suppression of the pituitary TSH-β gene |
TSH, thyroid-stimulating hormone.
From Zimmernamm MB, Jooste PL, Pandav CS: Iodine-deficiency disorders, Lancet 372:1251–1262, 2008.
Treatment
In many developing countries, administration of a single intramuscular injection of iodinated poppy seed oil to women prevents iodine deficiency during future pregnancies for about 5 yr. This form of therapy given to children younger than 4 yr of age with myxedematous cretinism results in a euthyroid state in 5 mo. Older children respond poorly and adults not at all to iodized oil injections, indicating an inability of the thyroid gland to synthesize hormone; these patients require treatment with T4. Through the efforts of the World Health Organization and its program of universal salt iodization, endemic iodine deficiency worldwide has been reduced by approximately 50%. In the Xinjiang province of China, where the usual methods of iodine supplementation had failed, iodination of irrigation water has increased iodine levels in soil, animals, and human beings. In other countries, iodinated salt in school meal programs gives children the dietary iodine they need. Still, political, economic, and practical obstacles have limited penetration of iodized food into regular diets around the world.
Benmiloud M, Chaouki ML, Gutekunst R, et al. Oral iodized oil for correcting iodine deficiency: optimal dosing and outcome indicator selection. J Clin Endocrinol Metab. 1994;79:20-24.
Boyages SC, Halpern JP, Maberly GF, et al. A comparative study of neurological and myxedematous endemic cretinism in western China. J Clin Endocrinol Metab. 1988;67:1262-1271.
Caldwell KL, Jones R, Hollowell JG. Urinary iodine concentration: United States National Health and Nutrition Examination Survey 2001-2002. Thyroid. 2005;15:692-699.
DeLange F. Iodine deficiency as a cause of brain damage. Postgrad Med J. 2001;77:217-220.
Delange F, de Benoist B, Pretell E, et al. Iodine deficiency in the world: where do we stand at the turn of the century? Thyroid. 2001;11:437-447.
Fuse Y, Saito N, Tsuchiya T, et al. Smaller thyroid gland volume with high urinary iodine excretion in Japanese schoolchildren: normative reference values in an iodine-sufficient area and comparison with the WHO/ICCIDD reference. Thyroid. 2007;17:145-155.
Maberly GF, Haxton DP, van der Haar F. Iodine deficiency: consequences and progress toward elimination. Food Nutr Bull. 2003;24(Suppl):S91-S98.
Remuzzi G, Garattini S. Elimination of iodine-deficiency disorders in Tibet. Lancet. 2008;371:1980-1981.
World Health Organization, United Nations Children’s Fund, and International Council for the Control of Iodine Deficiency Disorders. Assessment of the iodine deficiency disorders and monitoring their elimination. Geneva: World Health Organization; 2001.
561.4 Acquired Goiter
Most acquired goiters are sporadic and develop from a variety of causes; patients are usually euthyroid but may be hypothyroid or hyperthyroid. The most common cause of acquired goiter is lymphocytic thyroiditis (Chapter 560). A rare cause in children is subacute thyroiditis (De Quervain disease) (Chapter 560). Other causes include excess iodide ingestion and certain drugs, including amiodarone and lithium. Intrinsic biochemical defects in the synthesis of thyroid hormone are almost always associated with goiter; milder defects occur later in childhood. The occurrence of the disorder in siblings, onset in early life, and possible association with hypothyroidism (goitrous hypothyroidism) are important clues to the diagnosis.
Iodide Goiter
A small percentage of patients treated with iodide preparations for prolonged periods acquire goiters. Iodides are commonly included for their expectorant effect in cough medicines and in proprietary mixtures for asthma. Goiters resulting from iodide administration are firm and diffusely enlarged, and in some instances hypothyroidism develops. In normal persons, acute administration of large doses of iodide inhibits the organification of iodine and the synthesis of thyroid hormone (Wolff-Chaikoff effect). This effect is short-lived and does not lead to permanent hypothyroidism. When iodide administration continues, an autoregulatory mechanism in normal persons limits iodide trapping and permits the level of iodide in the thyroid to decrease and organification to proceed normally. In patients with iodide-induced goiter, this escape does not occur because of an underlying abnormality of biosynthesis of thyroid hormone. The persons most susceptible to the development of iodide goiter are those with lymphocytic thyroiditis or with a subclinical inborn error in thyroid hormone synthesis and those who have had a partial thyroidectomy.
Lithium carbonate, which is used to treat bipolar disorder, also causes goiters and mild hypothyroidism. Lithium competes with iodide, resulting in decreased T4 and T3 synthesis and release; the mechanism producing the goiter or hypothyroidism is similar to that described for iodide goiter. Lithium and iodide also act synergistically to produce goiter; their combined use should be avoided.
Amiodarone, a drug used to treat cardiac arrhythmias, can cause thyroid dysfunction with goiter because it is rich in iodine. It is also a potent inhibitor of 5′-deiodinase, preventing conversion of T4 to T3. It can cause hypothyroidism, particularly in patients with underlying autoimmune disease; in other patients, it can cause hyperthyroidism.
Simple Goiter (Colloid Goiter)
A few children with euthyroid goiters have simple goiters, a condition of unknown cause not associated with hypothyroidism or hyperthyroidism and not caused by inflammation or neoplasia. The condition predominates in girls and has a peak incidence before and during the pubertal years. Histologic examination of the thyroid either is normal or reveals variable follicular size, dense colloid, and flattened epithelium. The goiter may be small or large. It is firm in half the patients and occasionally is asymmetric or nodular. Levels of TSH are normal or low, scintiscans are normal, and thyroid antibodies are absent. Differentiation from lymphocytic thyroiditis might not be possible without a biopsy, but biopsy is usually not indicated. Therapy with thyroid hormone can help prevent progression to a large multinodular goiter, although it is difficult to separate any treatment effects from the natural history, which is for the goiter to decrease in size. Patients should be reevaluated periodically, because some have antibody-negative lymphocytic thyroiditis and therefore are at risk for changes in thyroid function (Chapter 560).
Multinodular Goiter
Rarely, a firm goiter with a lobulated surface and single or multiple palpable nodules is encountered. Areas of cystic change, hemorrhage, and fibrosis may be present. The incidence of this condition has decreased markedly with the use of iodine-enriched salt. A mild goitrogenic stimulus, acting over a long time, is thought to be the cause. Ultrasonographic examination can reveal multiple echo-free and echogenic lesions that are nonfunctioning on scintiscans. Thyroid studies are usually normal. Some children with chronic lymphocytic thyroiditis develop multinodular goiter; TSH may be elevated, and thyroid antibodies may be present.
Children can develop toxic multinodular goiter, characterized by a suppressed TSH and hyperthyroidism. The condition occurs in children with McCune-Albright syndrome (usually resulting in hyperthyroidism), with TSH receptor activating mutations, and it has been described in 3 children (including 2 siblings) with digital anomalies and cystic kidney disease. Dominant nodules within a multinodular goiter, particularly those not suppressed by replacement therapy with T4, may be an indication for evaluation by fine-needle aspiration because malignancy cannot readily be ruled out.
Akcurin S, Turkkahraman D, Tysoe C, et al. A family with a novel TSH receptor activating germline mutation (p. Ala485Val). Eur J Pediatr. 2008;167:1231-1237.
Brix TH, Kyvik KO, Hegedus L. Major role of genes in the etiology of simple goiter in females: A population-based twin study. J Clin Endocrinol Metab. 1999;84:3071-3075.
Daneman D, Davy T, Mancer K, et al. Association of multinodular goiter, cystic renal disease, and digital anomalies. J Pediatr. 1985;107:270-272.
De Vries L, Bulvik S, Phillip M. Chronic autoimmune thyriditis in children and adolescents: at presentation and during long-term follow-up. Arch Dis Child. 2009;94:33-37.
Feuillan PP, Shawker T, Rose SR, et al. Thyroid abnormalities in the McCune-Albright syndrome: Ultrasonography and hormonal studies. J Clin Endocrinol Metab. 1990;71:1596-1601.
Gopalakrishnan S, Chugh PK, Chhillar M, et al. Goitrous autoimmune thyroiditis in a pediatric population: a longitudinal study. Pediatrics. 2008;122:e670-e674.
Jaruratanasirkul S, Leethanaporn K, Suchat K. The natural clinical course of children with an initial diagnosis of simple goiter: A 5-year longitudinal follow-up. J Pediatr Endocrinol Metab. 2000;13:1109-1113.