Chapter 650 Disorders of Keratinization
Disorders of Cornification
Disorders of cornification (ichthyoses) are a primary group of inherited conditions characterized clinically by patterns of scaling and histopathologically by hyperkeratosis. They are usually distinguishable on the basis of inheritance patterns, clinical features, associated defects, and histopathologic changes (Table 650-1).
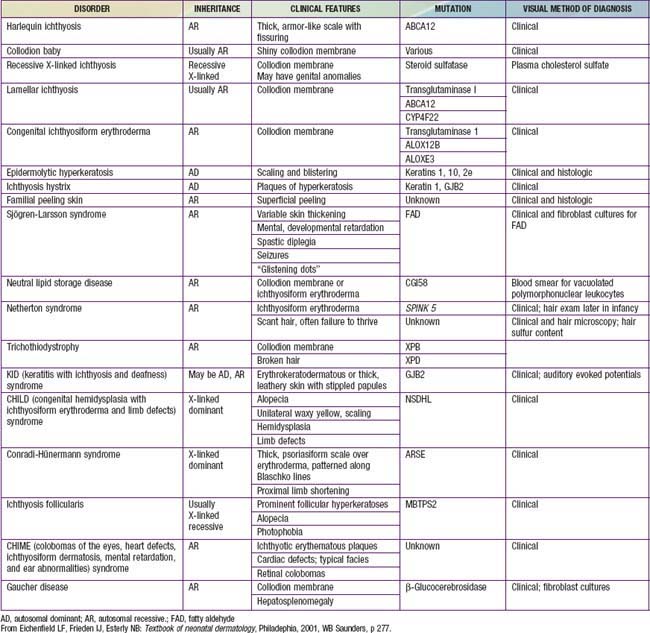
Harlequin Ichthyosis
Etiology/Pathogenesis
Harlequin ichthyosis is caused by mutations in the ABCA12 gene. Mutation in the gene leads to defective lipid transport and ABCA12 activity is required for the generation of long-chain ceramides that are essential for the development of the normal skin barrier.
Clinical Manifestations
At birth, markedly thickened, ridged, and cracked skin forms horny plates over the entire body, disfiguring the facial features and constricting the digits. Severe ectropion and chemosis obscure the orbits, the nose and ears are flattened, and the lips are everted and gaping. Nails and hair may be absent. Joint mobility is restricted, and the hands and feet appear fixed and ischemic. Affected neonates have respiratory difficulty, suck poorly, and are subject to severe cutaneous infection. Most die within the first days to weeks of life, but patients occasionally survive beyond infancy and have severe ichthyosis usually resembling lamellar ichthyosis or congenital ichthyosiform erythroderma.
Histology
Common morphologic abnormalities include hyperkeratosis, accumulation of lipid droplets within corneocytes, and absence of normal lamellar granules.
Treatment
Initial treatment includes high fluid intake to avoid dehydration from transepidermal water loss and use of a humidified heated incubator, emulsifying ointments, careful attention to hygiene, and oral retinoids (1 mg/kg/day). Prenatal diagnosis has been accomplished by fetoscopy, fetal skin biopsy, and microscopic examination of cells from amniotic fluid.
Collodion Baby
Etiology
Collodion baby is not a single entity but a newborn phenotype that is most often seen in babies who will eventually demonstrate lamellar ichthyosis or congenital ichthyosiform erythroderma. Less commonly, collodion babies evolve into babies with other forms of ichthyosis or Gaucher disease. A small subset become otherwise healthy babies without chronic skin disease.
Clinical Manifestations
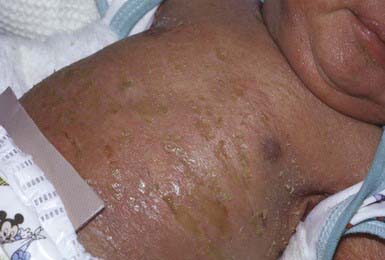
Collodion babies are covered at birth by a thick, taut membrane resembling oiled parchment or collodion (Fig. 650-1), which is subsequently shed. Affected neonates have ectropion, flattening of the ears and nose, and fixation of the lips in an O-shaped configuration. Hair may be absent or may perforate the abnormal covering. The membrane cracks with initial respiratory efforts and, shortly after birth, begins to desquamate in large sheets. Complete shedding may take several weeks, and a new membrane may occasionally form in localized areas.
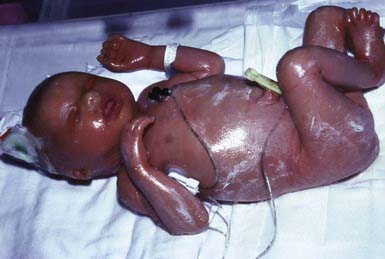
Neonatal morbidity and mortality may be due to cutaneous infection, aspiration pneumonia (squamous material), hypothermia, or hypernatremic dehydration from excessive transcutaneous fluid losses as a result of increased skin permeability. The outcome is uncertain, and accurate prognosis is impossible with respect to the subsequent development of ichthyosis.
Lamellar Ichthyosis and Congenital Ichthyosiform Erythroderma (Nonbullous Congenital Ichthyosiform Erythroderma)
Lamellar ichthyosis and congenital ichthyosiform erythroderma (nonbullous congenital ichthyosiform erythroderma) are the most common types of autosomal recessively inherited ichthyosis. Both forms are present soon or shortly after birth. Most infants with these forms of ichthyosis present with erythroderma and scaling; but among collodion babies, most turn out to have one of these ichthyosis variants.
Etiology/Pathogenesis
Three genes have been identified that cause lamellar ichthyosis (LI). These are LI1 (TGM1), LI2 (ABCA12), and LI3 (CYP4F22). Other genes have also been linked to LI, but they have not yet been identified. Transglutaminase mutations lead to abnormalities in the cornified envelope, whereas defects in ABCA12 cause abnormal lipid transport and those in CYP4F22 produce abnormal lamellar granules.
Three mutations have also been identified as the causes of congenital ichthyosiform erythroderma. These are TGM1, ALOX12B, and ALOX3. The ALOX genes encode for lipoxygenases the function of which is not definitively known. These lipoxygenases are likely to play a role in epidermal barrier formation by affecting lipid metabolism.
Mutations in the gene encoding ichthyn may cause either lamellar ichthyosis or congenital ichthyosiform erythroderma. The function of icthyn is unknown.
Clinical Manifestations
After shedding of the collodion membrane, if present, lamellar ichthyosis evolves into large, quadrilateral, dark scales that are free at the edges and adherent at the center. Scaling is often pronounced and involves the entire body surface, including flexural surfaces (Fig. 650-2). The face is often markedly involved, including ectropion and small, crumpled ears. The palms and soles are generally hyperkeratotic. The hair may be sparse and fine, but the teeth and mucosal surfaces are normal. Unlike in congenital ichthyosiform erythroderma, there is little erythema.
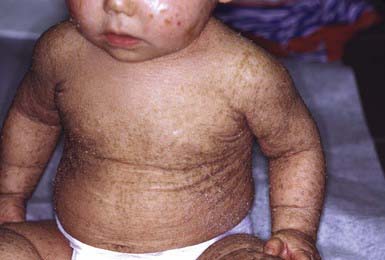
In congenital ichthyosiform erythroderma, erythroderma tends to be persistent, and scales, although they are generalized, are finer and whiter than in lamellar ichthyosis (Fig. 650-3). Hyperkeratosis is particularly noticeable around the knees, elbows, and ankles. Palms and soles are uniformly hyperkeratotic. Patients have sparse hair, cicatricial alopecia, and nail dystrophy. Neither form includes blistering.
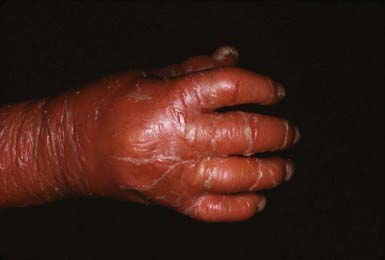
Histology
A markedly thickened stratum corneum and mild, irregular epidermal thickening characterize lamellar ichthyosis. Congenital ichthyosiform erythroderma involves more epidermal thickening with parakeratosis but less hyperkeratosis and hypergranulosis than in lamellar ichthyosis.
Treatment
Pruritus may be severe and responds minimally to antipruritic therapy. The unattractive appearance of the child and the bad odor from bacterial colonization of macerated scales may create serious psychological problems. A high-humidity environment in winter and air conditioning in summer reduce discomfort. Generous and frequent applications of emollients and keratolytic agents such as lactic or glycolic acid (5-12%), urea (10-40%), tazarotene (0.1% gel), and retinoic acid (0.1% cream) may lessen the scaling to some extent, although these agents produce stinging if applied to fissured skin. Oral retinoids (1 mg/kg/day) have a beneficial effect in these conditions but do not alter the underlying defect and, therefore, must be administered indefinitely. The long-term risks of these compounds (teratogenic effects and toxicity to bone) may limit their usefulness. Ectropion requires ophthalmologic care and, at times, plastic surgical procedures.
Ichthyosis Vulgaris
Etiology/Pathogenesis
Autosomal dominant or recessive mutations in the filaggrin gene cause ichthyosis vulgaris. Filaggrin is a filament-aggregating protein that assembles the keratin filament cytoskeleton, causing collapse of the granular cells into classic flattened squamous cell shape. Mutations in filaggrin lead to absence or marked reductions in keratohyalin granules.
Clinical Manifestations
Ichthyosis vulgaris is the most common of the disorders of keratinization, with an incidence of 1/250 live births. Onset generally occurs in the 1st yr of life. In most cases, it is trivial, consisting only of slight roughening of the skin surface. Scaling is most prominent on the extensor aspects of the extremities, particularly the legs (Fig. 650-4). Flexural surfaces are spared, and the abdomen, neck, and face are relatively uninvolved. Keratosis pilaris, particularly on the upper arms and thighs, accentuated markings, and hyperkeratosis on the palms and soles, and atopy are relatively common. Scaling is most pronounced during the winter months and may abate completely during warm weather. There is no accompanying disorder of hair, teeth, mucosal surfaces, or other organ systems.
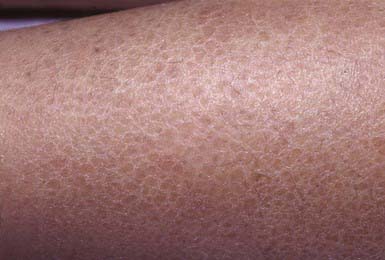
Histology
The histopathologic changes in ichthyosis vulgaris differ from those of other types of ichthyosis in that the hyperkeratosis is associated with a decrease or absence of the granular layer. Abnormally small and crumbly keratohyalin granules are found in epidermal cells on electron microscopy.
X-Linked Ichthyosis
Etiology/Pathogenesis
X-linked ichthyosis involves a deficiency of steroid sulfatase, which hydrolyzes cholesterol sulfate and other sulfated steroids to cholesterol. Cholesterol sulfate accumulates in the stratum corneum and plasma. In the epidermis this accumulation causes malformation of intercellular lipid layers, leading to barrier defects and delay of corneodesmosome degradation, resulting in corneocyte retention.
Clinical Manifestations
Skin peeling may be present at birth but typically begins at 3-6 mo of life. Scaling is most pronounced on the sides of the neck, lower face, preauricular areas, anterior trunk, and the limbs, particularly the legs. The elbow (Fig. 650-5) and knee flexures are generally spared but may be mildly involved. The palms and soles may be slightly thickened but are also usually spared. The condition gradually worsens in severity and extent. Keratosis pilaris is not present, and there is no increased incidence of atopy. Deep corneal opacities that do not interfere with vision develop in late childhood or adolescence and are a useful marker for the disease because they may also be present in carrier females. Some patients have larger deletions on the X chromosome that encompass neighboring genes, generating contiguous gene deletion syndromes. These include Kallmann syndrome (KAL1 gene), which consists of hypogonadotrophic hypogonadism and anosmia, X-linked chondroplasia punctata (ARSE gene), short stature, and ocular albinism. The rate of testicular cancer may be increased in patients with coexistent Kallmann syndrome. There is also an increased risk of attention deficit hyperactivity disorder and autism owing to a contiguous gene defect in neuroglin 4.
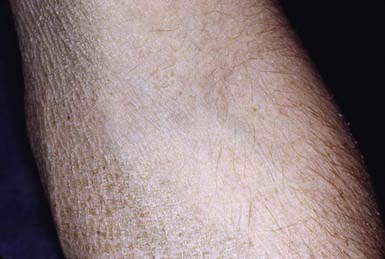
Reduced steroid sulfatase enzyme activity can be detected in fibroblasts, keratinocytes, and leukocytes and, prenatally, in amniocytes or chorionic villus cells. In affected families, an affected male can be detected by restriction enzyme analysis of cultured chorionic villus cell DNA or amniocytes or by in situ hybridization, which identifies steroid sulfatase gene deletions prenatally in chorionic villus cells. A placental steroid sulfatase deficiency in carrier mothers may result in low urinary and serum estriol values, prolonged labor, and insensitivity of the uterus to oxytocin and prostaglandins.
Epidermolytic Hyperkeratosis (Bullous Congenital Ichthyosiform Erythroderma)
Etiology/Pathogenesis
Epidermolytic hyperkeratosis is an autosomal dominant trait that has been shown to be due to defects in either keratin 1 or keratin 10. These keratins are required to form the keratin-intermediate filaments in cells of the suprabasilar layers of the epidermis.
Clinical Manifestations
The clinical manifestations are initially characterized by the onset at birth of widespread blisters and erosions on a background of generalized erythroderma (Fig. 650-6). Recurrent blistering may be widespread in neonates and may cause diagnostic confusion with other blistering disorders. With time, the blister formation ceases, erythema decreases, and generalized hyperkeratosis develops. The scales are small, hard, and verrucous. Distinctive, parallel hyperkeratotic ridges develop over the joint flexures, including the axillary, popliteal, and antecubital fossae, and on the neck and hips. Palmoplantar keratoderma is associated with keratin 1 defects. The hair, nails, mucosa, and sweat glands are normal. Malodorous secondary bacterial infection is common and requires appropriate antibiotic therapy.
Pathology
The histopathology is diagnostic of epidermolytic hyperkeratosis, consisting of hyperkeratosis, degeneration of the epidermal granular layer with an increased number of keratohyalin granules, clear spaces around nuclei, and indistinct cellular boundaries of cells in the upper epidermis. On electron microscopic examination, keratin-intermediate filaments are clumped, and many desmosomes are attached to only one keratinocyte instead of connecting neighboring keratinocytes. Localized forms of the disease may resemble epidermal nevi or keratoderma of the palms and soles but share the distinctive histopathologic changes of epidermolytic hyperkeratosis.
Treatment
Treatment of epidermolytic hyperkeratosis is difficult. Morbidity is increased in the neonatal period as a result of prematurity, sepsis, and fluid and electrolyte imbalance. Bacterial colonization of macerated scales produces a distinctive bad odor that can be controlled somewhat by use of an antibacterial cleanser. Intermittent oral antibiotics are generally necessary. Keratolytic agents are often poorly tolerated. Oral retinoids (1 mg/kg/day) may produce significant improvement. Prenatal diagnosis for affected families is possible by examination of DNA extracts from chorionic villus cells or amniocytes, provided that the specific mutation in the affected parent is known.
Erythrokeratoderma Variabilis
Etiology/Pathogenesis
An autosomal dominant disorder, erythrokeratoderma variabilis (EKV) is caused by mutations in connexins 31 and 30.3. Connexins are proteins that form gap junctions between cells that allow for transport and signaling between neighboring epidermal cells.
Clinical Manifestations
EKV usually manifests in the early months of life, progresses in childhood, and stabilizes in adolescence. It is characterized by two distinctive manifestations: sharply demarcated, hyperkeratotic plaques (Fig. 650-7A) and transient figurate erythema (Fig. 650-7B). The distribution is generalized but sparse; sites of predilection are the face, buttocks, axillae, and extensor surfaces of the limbs. The palms and soles may be thickened, but hair, teeth, and nails are normal.
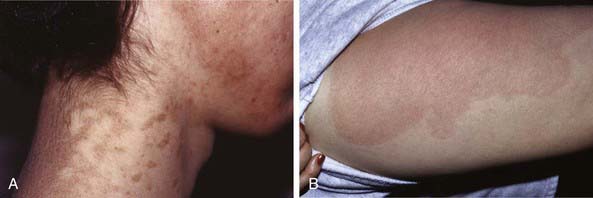
Symmetric Progressive Erythrokeratoderma
Etiology/Pathogenesis
Symmetric progressive erythrokeratoderma is an autosomal dominant disorder caused by mutations in the gene encoding loricrin. Loricrin is a major component of the epidermal cornified cell envelope.
Clinical Manifestations
The disorder manifests in childhood as large, fixed, geographic and symmetric, fine, scaling, hyperkeratotic, erythematous plaques primarily on the extremities, buttocks, face, ankles, and wrists. The primary feature distinguishing this disorder from EKV is the lack of variable erythema seen in the latter condition.
Ichthyosiform Dermatoses
Several rare and distinct syndromes include ichthyosis as a constant feature.
Sjögren-Larsson Syndrome
Etiology/Pathogenesis
The autosomal recessive inborn error of metabolism known as Sjögren-Larsson syndrome is an abnormality of fatty alcohol oxidation that results from a deficiency of fatty aldehyde dehydrogenase (ALDH3A2), a component of the fatty alcohol–nicotinamide adenine dinucleotide oxidoreductase enzyme complex.
Clinical Manifestations
The clinical picture of Sjögren-Larsson syndrome consists of ichthyosis, mental retardation, and spasticity. The ichthyosis is generalized but is accentuated on the flexures and the lower abdomen and consists of erythroderma, fine scaling, larger platelike scales, and dark hyperkeratosis. The degree of scale varies markedly from patient to patient. Most individuals have palmoplantar hyperkeratosis. The skin changes may be identical to the other forms of ichthyosis, and diagnosis is often delayed until the onset of neurologic symptoms. Pruritus is severe and hypohidrosis is common. Glistening dots in the foveal area are a cardinal ophthalmologic sign. About half the patients have primary retinal degeneration. Motor and speech developmental delays are usually noted before 1yr of age, and spastic diplegia or tetraplegia, epilepsy, and mental retardation generally become evident in the first 3 yr of life. Some patients may walk with the aid of braces, but most are confined to wheelchairs. This deficiency can be demonstrated in cultured skin fibroblasts of affected patients and carriers and, prenatally, in cultured chorionic villus cells and amniocytes from affected fetuses. Elevation of urinary leukotriene B4 (LTB4) may provide an easier approach to diagnosis.
Netherton Syndrome
Etiology/Pathogenesis
A rare autosomal recessive disorder, Netherton syndrome is caused by mutations in the SPINK 5 gene, which encodes a serine protease inhibitor (LEKT1).
Clinical Manifestations
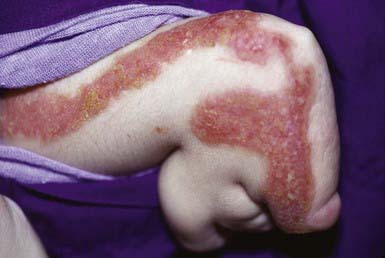
Netherton syndrome is characterized by ichthyosis (usually ichthyosis linearis circumflexa but occasionally the lamellar or congenital types of ichthyosiform erythroderma), trichorrhexis invaginata and other hair shaft anomalies, and atopic diathesis. The disorder manifests at birth or in the first few months of life as generalized erythema and scaling. The trunk and limbs have diffuse erythema and superimposed migratory, polycyclic, and serpiginous hyperkeratotic lesions (Fig. 650-8), some with a distinctive double-edged margin of scale. Lichenification or hyperkeratosis tends to persist in the antecubital and popliteal fossae. The face and scalp may remain erythematous and scaling. Many hair shaft deformities, most notably, trichorrhexis invaginata, have been described in most patients with Netherton syndrome.

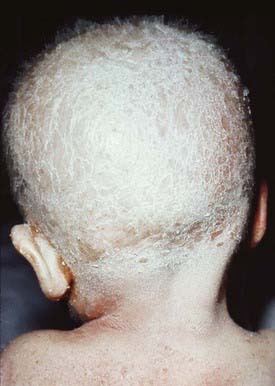
The ichthyosis is present in the first 10 days of life and may be especially marked around the eyes, mouth, and perineal area. The erythroderma is often intensified after infection. Infants may suffer from failure to thrive, recurrent bacterial and candidal infections, elevated serum immunoglobulin (Ig) E values, and marked hypernatremic dehydration. The most frequent allergic manifestations are urticaria, angioedema, atopic dermatitis, and asthma. Scalp hair is sparse and short and fractures easily (Fig. 650-9); eyebrows, eyelashes, and body hair are also abnormal. The characteristic hair abnormality can be identified with light microscopy; in the newborn, it may best be identified in eyebrow hair.
Refsum Syndrome (Chapters 80.2 and 605.5)
Etiology/Pathogenesis
There are 2 types of Refsum syndrome. The classic form is autosomal recessive and caused by mutations in the PAHX gene that result in an increase in phytanic acid. A novel Refsum-like disorder has been mapped to chromosome 20 (20p11.21-q12), but the gene remains unidentified. The infantile forms of Refsum syndrome are also autosomal recessive and caused by mutations in the PEX1, PEX2, or PEX26 genes. These are peroxisomal abnormalities that lead to an increase in very long chain fatty acids, di- and tri-hydroxycholestanoic acid, and pipecolic acid as well as phytanic acid.
Clinical Manifestations
Refsum syndrome is a multisystem disorder that becomes symptomatic in the 2nd or 3rd decade of life. The ichthyosis may be generalized, is relatively mild, and resembles ichthyosis vulgaris. The ichthyosis may also be localized to the palms and soles. Chronic polyneuritis with progressive paralysis and ataxia, retinitis pigmentosa, anosmia, deafness, bony abnormalities, and electrocardiographic changes are the most characteristic features. The condition is diagnosed through lipid analysis of the blood or skin, which shows elevated phytanic acid values.
The infantile form begins, as suggested by the name, early in life, and in addition to the changes seen in the classic form, affected patients have hepatomegaly, abnormal bile acid profiles, developmental delay, and mental retardation.
Chondrodysplasia Punctata (Chapter 80.2)
Etiology/Pathogenesis
Chondrodysplasia punctata (CPD) is a clinically and genetically heterogeneous condition. X-linked dominant CPD, also known as Conradi-Hünermann syndrome, is the best-characterized form. There is also an X-linked recessive form caused by mutation in the ARSE gene. Rhizomelic chondrodysplasia punctata type 1 is an autosomal recessive disorder caused by mutations in the PEX7 gene, which encodes the peroxisomal type 2 targeting signal (PTS2) receptor. CPD can also be caused by maternal vitamin K deficiency or warfarin teratogenicity.
Clinical Manifestations
These heterogeneous disorders are marked by ichthyosis and bone changes. Nearly all patients with the X-linked dominant form and approximately 25% of those with the recessive type have cutaneous lesions, ranging from severe, generalized erythema and scaling to mild hyperkeratosis. Rhizomelic chondrodysplasia punctata is associated with cataracts, hypertelorism, optic nerve atrophy, disproportionate shortening of the proximal extremities, psychomotor retardation, failure to thrive, and spasticity; most affected patients die in infancy. Patients with the X-linked dominant form have asymmetric, variable shortening of the limbs and a distinctive ichthyosiform eruption at birth. Thick, yellow, tightly adherent, keratinized plaques are distributed in a whorled pattern over the entire body. The eruption typically resolves in infancy and may be superseded by a follicular atrophoderma and patchy alopecia.
Additional features in all variants include cataracts and abnormal facies with saddle nose and frontal bossing. The pathognomonic defect, termed chondrodysplasia punctata, is stippled epiphyses in the cartilaginous skeleton. This defect, which is seen in various settings and inherited disorders, often in association with peroxisomal deficiency and disturbance of cholesterol biosynthesis, disappears by 3-4 yr of age.
Other Syndromes with Ichthyosis
A number of other rare syndromes with ichthyosis as a consistent feature include the following: keratitis with ichthyosis and deafness (KID syndrome, connexin 26 gene), ichthyosis with defective hair having a banded pattern under polarized light and a low sulfur content (trichothiodystrophy), multiple sulfatase deficiency, neutral lipid storage disease with ichthyosis (Chanarin-Dorfman syndrome/CGI58 gene), and CHILD syndrome (Fig. 650-10; congenital hemidysplasia with ichthyosiform erythroderma and limb defects; NSDHL gene).
Palmoplantar Keratodermas
Excessive hyperkeratosis of the palms and soles may occur as a manifestation of a focal or generalized congenital hereditary skin disorder or may result from such chronic skin diseases as psoriasis, eczema, pityriasis rubra pilaris, lupus erythematosus, or postinfectious arthritis syndrome.
Diffuse Hyperkeratosis of Palms and Soles (Unna-Thost, Vorner)
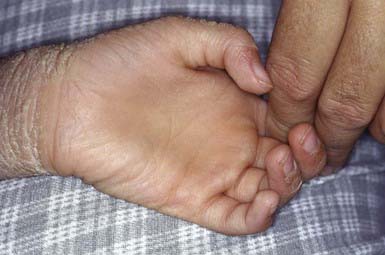
Unna-Thost and Vorner type palmoplantar keratodermas (PPKs), although clinically inseparable, were thought to be separate entities. They were separated histologically by the presence (Vorner) or absence (Unna-Thost) of epidermolytic hyperkeratosis. They represent the clinical spectrum of the same disease caused by mutations in keratin (KRT1 and KRT9 genes). This autosomal dominant disorder manifests in the first few months of life as erythema that gradually progresses to sharply demarcated, hyperkeratotic, scaling plaques over the palms (Fig. 650-11) and soles. The margins of the plaques often remain red; plaques may extend along the lateral aspects of the hands and feet and onto the volar wrists and the heels. Hyperhidrosis is usually present, but hair, teeth, and nails are usually normal. Striate (DSG1, DSP, KRT1 genes) and punctate forms of palmar and plantar hyperkeratosis represent distinct entities.
Mal de Meleda (SLURP-1 Gene)
A rare, progressive autosomal recessive condition, mal de Meleda is characterized by erythema and thick scales on the palms, fingers, soles, and flexor aspects of the wrists, knees, and elbows. Hyperhidrosis, nail thickening or koilonychia, and eczema may also occur.
Vohwinkel Palmoplantar Keratoderma (Mutilating Keratoderma)
Vohwinkel PPK is a progressive autosomal dominant disease consisting of honeycombed hyperkeratosis of palms and soles, sparing the arches; starfish-like and linear keratoses on the dorsum of the hands, fingers, feet, and knees; and ainhum-like constriction of the digits that sometimes leads to autoamputation. Varying degrees of alopecia may be seen. Two forms have been identified. Vohwinkel PPK with ichthyosis is caused by mutations in the loricrin gene, and Vohwinkel PPK with deafness by mutations in connexin 26.
Papillon-Lefèvre Syndrome (Cathepsin C Gene)
An autosomal recessive erythematous hyperkeratosis of the palms and soles, Papillon-Lefèvre syndrome sometimes extends to the dorsal hands and feet, elbows, and knees later in childhood. The PPK may be either diffuse, striate, or punctuate. This syndrome is characterized by periodontal inflammation, leading to loss of teeth by age 4-5 yr if untreated.
Other Syndromes
Keratoderma of palms and soles also occurs as a feature of some forms of ichthyosis and ectodermal dysplasia. Richner-Hanhart syndrome is an autosomal recessive palmoplantar keratoderma with corneal ulcers, progressive mental impairment, and a deficiency of tyrosine aminotransferase, which leads to tyrosinemia. Pachyonychia congenita is transmitted as an autosomal dominant trait with variable expressivity. The classic type I form (Jadassohn-Lewandowski syndrome) is due to mutations in the gene for keratin 16. Major features of the syndrome are onychogryphosis; palmoplantar keratoderma; follicular hyperkeratosis, especially of the elbows and knees; and oral leukokeratosis. The nail dystrophy is the most striking feature and may be present at birth or develop early in life. The nails are thickened and tubular, projecting upward at the free edge to form a conical roof over a mass of subungual keratotic debris. Repeated paronychial inflammation may result in shedding of the nails. The feature seen most consistently among patients with this condition is keratoderma of the palms and soles. Additional associated features include hyperhidrosis of the palms and soles, and bullae and erosions on the palms and soles. Some patients have shown a selective cell-mediated defect in recognition and processing of Candida. Surgical removal of the nails and excision of the nail matrix have been helpful in some patients.
Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129:1319-1321.
Hwang S, Schwartz RA. Keratosis pilaris: a common follicular hyperkeratosis. Cutis. 2008;82:177-180.
Lane EB, McLean WH. Keratins and skin disorders. J Pathol. 2004;204:355-366.
Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol. 2006;16:349-359.
Rajpopat S, Moss C, Mellerio J, et al. Harlequin ichthyosis. Arch Dermatol. Feb 2011. [Epub ahead of print]
Sandilands A, Sutherland C, Irvine AD, et al. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122(Pt 9):1285-1294.
Shwayder T. Disorders of keratinization: diagnosis and management. Am J Clin Dermatol. 2004;5:17-29.
Smith FJD, Irvine AD, Terron-Kwiatkiwski A, et al. Loss -of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38:337-342.