Chapter 693 Marfan Syndrome
Marfan syndrome (MFS) is an autosomal dominant disorder caused by mutations in the gene encoding the extracellular matrix (ECM) protein fibrillin-1. It is primarily associated with skeletal, cardiovascular, and ocular pathology, displaying near-complete penetrance but variable expression. Diagnosis is based on clinical findings, some of which are age dependent.
Epidemiology
The incidence of this disorder is about 1/5,000-10,000 births. About 30% of cases are sporadic, due to de novo mutations; new mutations often associate with advanced paternal age.
Pathogenesis
MFS is associated with abnormal biosynthesis of fibrillin-1, a 350-kd ECM protein that is the major constituent of microfibrils. The fibrillin-1 (FBN1) locus resides on the long arm of chromosome 15 (15q21), and the gene is composed of 65 exons. More than 1,000 mutations distributed throughout FBN1 have been identified, many being unique to a given family. With the exception of an early-onset and severe presentation of MFS associated with some mutations in exons 26-27 and 31-32, no clear phenotype-genotype correlation has been identified. There is considerable intrafamily variability, suggesting that epigenetic, modifier gene, environmental, or other unidentified factors might influence expression of the disease.
MFS was traditionally considered to result from a structural deficiency of connective tissues. Reduced fibrillin-1 was thought to lead to a primary derangement of elastic fiber deposition, because both skin and aorta from affected patients show decreased elastin, along with elastic fiber fragmentation. In response to stress (such as hemodynamic forces in the proximal aorta), affected organs were thought to manifest this structural insufficiency with accelerated degeneration. However, it was difficult to reconcile certain manifestations of the disease with this structural deficiency model (i.e., bony overgrowth is more suggestive of excess cell proliferation than structural insufficiency).
Additional research has identified a cytokine-regulatory role for fibrillin-1 that appears to have important implications for MFS. Fibrillin-1 shares significant homology with the latent transforming growth factor β (TGF-β) binding proteins (LTBPs). TGF-β is secreted from cells as part of a large latent complex (LLC) that includes the mature cytokine (TGF-β), a dimer of its processed amino-terminal pro-peptide called latency associated peptide (LAP), and one of three LTBPs. Mice heterozygous for a mutation in the fibrillin-1 gene typical of those that cause MFS in humans (C1039G) display many of the classic features of MFS, including progressive aortic root dilatation. TGF-β signaling has been shown to be increased in the aortas of these mice, as well as the aortic wall from patients with MFS, suggesting that failed ECM sequestration of the LLC by fibrillin-1 leads to increased TGF-β signaling in MFS. Neutralizing antibodies to TGF-β have been shown to reduce the aortic size and improve the aortic wall architecture in these mice.
Aberrant TGF-β signaling might also play a role in the wider spectrum of manifestations of MFS. Increased TGF-β signaling has been observed in many tissues in MFS mice, including the developing lung, skeletal muscle, mitral valve, and dura. Treatment of these mice with agents that antagonize TGF-β signaling attenuates or prevents pulmonary emphysema, skeletal muscle myopathy, and myxomatous degeneration of the mitral valve. Patients with heterozygous mutations in TGF-β receptors 1 and 2 (TGFβR1 and TGFβR2) have MFS-like manifestations yet normal fibrillin-1. Paradoxically, these mutations also appear to result in increased TGF-β signaling in tissues of these patients. The resulting Loeys-Dietz syndrome (LDS) has much phenotypic overlap with MFS, but it also has many discriminating features (see Differential Diagnosis).
Clinical Manifestations
MFS is a multisystem disorder, with cardinal manifestations in the skeletal, cardiovascular, and ocular systems.
Skeletal System
Disproportionate overgrowth of the long bones is often the most striking and immediately evident manifestation of MFS. Anterior chest deformity is caused by overgrowth of the ribs, pushing the sternum anteriorly (pectus carinatum) or posteriorly (pectus excavatum). Overgrowth of arms and legs can lead to an arm span >1.05 times the height or a reduced upper to lower segment ratio (in the absence of severe scoliosis). Arachnodactyly (overgrowth of the fingers) is generally a subjective finding. The combination of long fingers and loose joints leads to the characteristic Walker-Murdoch or wrist sign: full overlap of the distal phalanges of the thumb and fifth finger when wrapped around the contralateral wrist (Fig. 693-1). The Steinberg or thumb sign is present when the distal phalanx of the thumb fully extends beyond the ulnar border of the hand when folded across the palm (Fig. 693-2), with or without active assistance by the patient or examiner.

Figure 693-1 Wrist sign. When the wrist is grasped by the contralateral hand, the thumb overlaps the terminal phalanx of the 5th digit.
(From McBride ART, Gargan M: Marfan syndrome, Curr Orthop 20:418–423, 2006.)
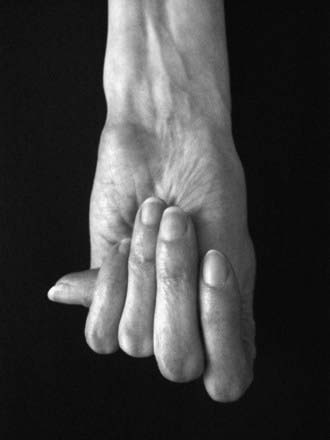
Figure 693-2 Thumb sign. When the hand is clenched without assistance, the entire thumbnail projects beyond the border of the hand.
(From McBride ART, Gargan M: Marfan syndrome, Curr Orthop 20:418–423, 2006.)
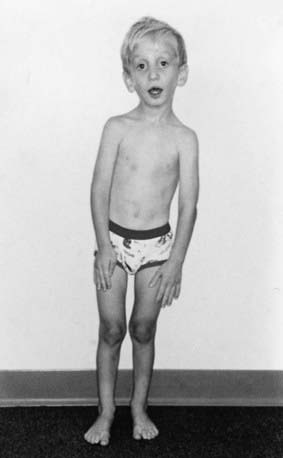
Thoracolumbar scoliosis is commonly present and can contribute to the systemic score of the diagnosis (Table 693-1). Protrusio acetabuli (inward bulging of the acetabulum into the pelvic cavity), which is generally asymptomatic in young adults, is best identified with radiographic imaging. Pes planus (flat feet) is commonly present and varies from mild and asymptomatic to severe deformity, wherein medial displacement of the medial malleolus results in collapse of the arch and often reactive hip and knee disturbances. Curiously, a subset of patients with the disorder present with an exaggerated arch (pes cavus). Although joint laxity or hypermobility is often identified, joints can be normal or even develop contractures. Reduced extension of the elbows is common and can contribute to the systemic score of the diagnosis (see Table 693-1). Contracture of the fingers (camptodactyly) is commonly observed, especially in children with severe and rapidly progressive MFS. Several craniofacial manifestations are often present and can contribute to the diagnosis (Fig. 693-3). These include a long narrow skull (dolicocephaly), recession of the eyeball within the socket (enophthalmos), recessed lower mandible (retrognathia) or small chin (micrognathia), malar hypoplasia (malar flattening), and downward-slanting palpebral fissures.
Table 693-1 Diagnostic Criteria for Marfan Syndrome (MFS)
In the absence of a family history of MFS, a diagnosis can be reached in 1 of 4 scenarios:
In the absence of a family history of MFS, alternative diagnoses to MFS include:
In the presence of a family history of MFS, a diagnosis can be reached in 1 of 3 scenarios:
SCORING OF SYSTEMIC FEATURES (IN POINTS)†
CRITERIA FOR CAUSAL FBN1 MUTATION
US/LS, upper segment/lower segment ratio.
* Without discriminating features of SGS, LDS, or vEDS (as defined in Table 693-2) and after TGFBR1/2, collagen biochemistry, COL3A1 testing if indicated. Other conditions/genes will emerge with time.
† Maximum total: 20 points; score ≥7 indicates systemic involvement.
From Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan syndrome, J Med Genet 47:476–485, 2010.
Cardiovascular System
Manifestations of MFS in the cardiovascular system are conveniently divided into those affecting the heart and those affecting the vasculature. Within the heart, the atrioventricular (AV) valves are most often affected. Thickening of the AV valves is common and often associated with prolapse of the mitral and/or tricuspid valves. Variable degrees of regurgitation may be present. In children with early onset and severe MFS, insufficiency of the mitral valve can lead to congestive heart failure, pulmonary hypertension, and death in infancy; this manifestation represents the leading cause of morbidity and mortality in young children with the disorder (Chapter 422.3). Aortic valve dysfunction is generally a late occurrence, attributed to stretching of the aortic annulus by an expanding root aneurysm. Both the aortic and AV valves seem to be more prone to calcification in persons with MFS.
Ventricular dysrhythmia has been described in children with MFS (Chapter 429). In association with mitral valve dysfunction, supraventricular arrhythmia (e.g., atrial fibrillation or supraventricular tachycardia) may be seen. There is also an increased prevalence of prolonged QT interval on electrocardiographic surveys of patients with MFS. Dilated cardiomyopathy, beyond that explained by aortic or mitral valve regurgitation, seems to occur with increased prevalence in patients with MFS, perhaps implicating a role for fibrillin-1 in the cardiac ventricles. However, the incidence seems to be low, and the occurrence of mild to moderate ventricular systolic dysfunction is often attributed to mitral or aortic insufficiency or to the use of β-adrenergic receptor blockade.
Aortic aneurysm and dissection remain the most life-threatening manifestations of MFS. This finding is age dependent, prompting life-long monitoring by echocardiography or other imaging modalities. Dilatation at the sinuses of Valsalva can begin in utero in severe cases, although some unequivocally affected persons never reach an aortic size that requires surgical intervention. In contrast to atherosclerotic aneurysms and some other forms of ascending aortic aneurysms, dilatation in MFS is generally greatest at (and often restricted to) the aortic root. Normal aortic dimensions vary with both age and body size, hence proper interpretation of aortic dimensions mandates comparison to age-dependent nomograms. The two most important determinants of risk of dissection of the aorta are the maximal dimension and a family history of dissection.
Surgical repair of the aorta is recommended when its greatest diameter reaches about 50 mm in adults. Early intervention is considered in those with a family history of early dissection. There are no definitive methods to guide the timing of surgery in childhood. The observation that dissection is extremely rare in this age group, irrespective of aortic size, has prompted many centers to adopt the adult criterion of 50 mm. Early surgery is often undertaken given a rapid rate of growth (>10 mm in a year) or the emergence of significant aortic regurgitation. Most patients with acute aortic dissection have classic symptoms, including severe chest pain, often radiating along the path of dissection. This path almost invariably begins at the aortic root (type A), and dissection can remain isolated (type II) or can propagate along the length of the descending aorta (type I). Acute-onset congestive heart failure typically indicates severe aortic valve insufficiency, complicating the aortic dissection. Depending on the involvement of the carotid arteries, some patients can have neurologic sequelae, due to cerebrovascular injury. Involvement of the coronary arteries can lead to myocardial infarction or sudden cardiac death. The mechanism of death usually includes rupture into the pericardial sac with subsequent pericardial tamponade. Chronic aortic dissection and intimal tears usually occur more insidiously, often without chest pain. Dilatation of the main pulmonary artery or descending thoracic or abdominal aorta can also occur, although relatively rarely.
Ocular System
Ectopia lentis (dislocation of the ocular lens) of any degree constitutes a major part of the diagnostic criteria for Marfan syndrome (see Table 693-1), although it is not unique to the disorder. Ectopia lentis occurs in around 60-70% of patients with the disorder. When identified, it should prompt further assessment for MFS, although homocystinuria, Weill-Marchesani, and familial ectopia lentis are also associated with this condition. Other manifestations include early and severe myopia, flat cornea, increased axial length of the globe, hypoplastic iris, and ciliary muscle hypoplasia, causing decreased miosis. Patients with MFS can also have retinal detachment and a predisposition for early cataracts or glaucoma.
Pulmonary System
Several factors can result in pulmonary disease in patients with MFS. Pectus excavatum or progressive scoliosis can contribute to a restrictive pattern of lung disease. Widening of the distal airspaces with or without discrete bullae or (often apical) blebs can predispose to spontaneous pneumothorax, which occurs in up to 15% of patients. During assessment of pulmonary volumes and function, one should recognize that long-bone overgrowth affecting the lower extremities can lead to a reduction in the normalized forced vital capacity and total lung capacity. However, if normalized to thoracic size or sitting height, pulmonary function testing is often normal in patients with the disorder.
Skin and Integument
In contrast with other connective tissue disorders (e.g., Ehlers-Danlos syndrome), patients with MFS typically have normal skin texture and elasticity. The most common manifestation in the skin is striae atrophicae, which occurs in about two thirds of patients. In contrast to striae in those without a connective tissue disorder, stretch marks tend to occur in patients with MFS in the absence of obesity, rapid gain in muscle mass, or pregnancy, and at sites not associated with increased skin distention (e.g., the anterior shoulder or lower back). Another common manifestation is inguinal hernia, either occurring at birth or acquired in adolescence. There is an increased risk of surgical and recurrent hernias in MFS.
Dural Ectasia
Widening of the dural sac or root sleeves (dural ectasia) is present in 63-92% of MFS patients. Although dural ectasia can result in lumbar back pain, it is often asymptomatic and should be assessed by lumbosacral imaging with CT or MRI. When present, it contributes to the systemic score of the diagnostic criteria, although the specificity and predictive value of this criterion are unknown. Incidence of dural ectasia in other connective tissue disorders has not been rigorously assessed, although roughly 24% of patients with Ehlers-Danlos syndrome and many patients with LDS also show it.
Family and Genetic History
The current criteria established for the diagnosis of MFS emphasize the need for a first-degree relative to independently meet the criteria before including family history as a major criterion for diagnosis. The criteria that are required to meet a causal mutation in a patient with MFS are described in Table 693-1.
Diagnosis
Given the complexity of the diagnostic evaluation for MFS and the differential diagnosis, the evaluation should be coordinated by an individual or team with extensive experience in the diagnosis of connective tissue disorders. Diagnosis is based on criteria that include both clinical and molecular analyses (see Table 693-1).
In the absence of a conclusive family history of MFS, the diagnosis can be established in 4 distinct scenarios:
In an individual with a positive family history of MFS (where a family member has been independently diagnosed using the above criteria), the diagnosis can be established:
In the case of scenarios 2 and 3 (i.e., a systemic score ≥7 or aortic root dilatation), features suggestive of SGS, LDS, or vEDS must again be excluded and appropriate alternative genetic testing should be performed (as above).
Special consideration should be given to young individuals (<20 yr old) without a positive family history of MFS, who may not fit into one of the 4 proposed scenarios. In the absence of an FBN1 mutation, if insufficient systemic features (<7) and/or borderline aortic root measurements (Z-score <3) are present, the term “nonspecific connective tissue disorder” should be used until follow-up echocardiographic evaluation shows aortic root dilation (Z-score ≥3). If the presence of an FBN1 mutation is identified in a sporadic or familial case, but aortic root measurements are still below a Z-score of 3, the term “potential MFS” should be used until the aorta reaches threshold. Neonatal MFS is not considered as a separate category, but rather represents the severe end of the disease spectrum.
In adults (>20 yr), there are 3 main categories of alternative diagnoses: ectopia lentis syndrome (ELS), MASS phenotype (myopia, mitral valve prolapse, borderline [Z<2] aortic root enlargement, skin and skeletal findings), and mitral valve prolapse syndrome (MVPS).
Finally, some patients will remain difficult to classify due to overlap of phenotypes from different entities, the evolving nature of these connective tissue diseases, the absence of a mutation after screening of the appropriate genes, or divergence between the phenotype and the genotype. However, these patients should be uncommon and will hopefully benefit from better definition of still unrecognized conditions in the future.
Differential Diagnosis
Several disorders are included in the differential diagnosis of MFS on the basis of similar skeletal, cardiac, or ophthalmologic manifestations (Table 693-2). Many patients referred for possible MFS are found to have evidence of a systemic connective tissue disorder, including long limbs, deformity of the thoracic cage, striae atrophicae, mitral valve prolapse, and mild and nonprogressive dilatation of the aortic root, but do not meet diagnostic criteria for MFS. This constellation of features not fulfilling the diagnostic requirements of MFS is referred to by the acronym MASS phenotype, emphasizing the mitral, aortic, skin, and skeletal manifestations. The MASS phenotype can segregate in large pedigrees and remain stable over time. This diagnosis is most challenging in the context of an isolated and young patient. In this setting, careful follow-up is needed to distinguish MASS phenotype from emerging MFS, especially in children.
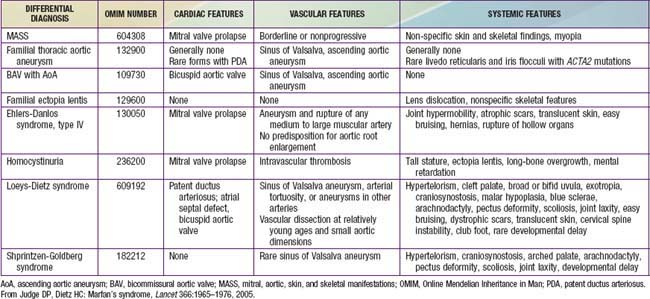
Other fibrillinopathies, such as familial mitral valve prolapse (MVP) syndrome and familial ectopia lentis, also include subdiagnostic manifestations and can be due to mutations in the gene encoding fibrillin-1. Also included in the differential diagnosis of the disorder is homocystinuria, caused by a deficiency of cystathionine β-synthase. Patients with homocystinuria often have tall stature, long-bone overgrowth, and ectopia lentis, but they do not typically have aortic enlargement or dissection. By contrast with MFS, the inheritance of homocystinuria (Chapter 79.3) is autosomal recessive, and affected persons often have mental retardation, a predisposition to thromboembolism, and a high incidence of coronary artery atherosclerosis. Observation of severely elevated concentrations of plasma homocystine is an efficient mechanism to distinguish homocystinuria from MFS.
Familial thoracic aortic aneurysm (FTAA) syndrome segregates as a dominant trait and can show vascular disease identical to MFS, including aortic root aneurysm and dissection. However, these persons generally do not show any of the systemic manifestations of MFS. Other families show an association between bicommissural aortic valve and ascending aortic aneurysm, which can also segregate as a dominant trait. Here, maximum dilatation often occurs further up in the ascending aorta, beyond the sinotubular junction. There is emerging evidence that bicommissural aortic valve and aneurysm both represent primary manifestations of a single gene defect, and that family members of a proband can have aneurysm without accompanying valve disease. These patients typically do not show any systemic features of a connective tissue disorder.
Unlike with MFS, many families with both FTAA syndrome and with bicommissural aortic valve and ascending aortic aneurysm show reduced penetrance. Although a number of genetic loci and genes have been described for thoracic aortic aneurysm syndrome, they do not account for a significant percentage of cases to allow efficient molecular testing, mandating ongoing clinical follow-up of at-risk family members. In most cases, the management principles that have been generated for MFS have proved effective for these other forms of familial aortic aneurysm.
Important to the differential is Loeys-Dietz syndrome (LDS). As in MFS, patients with LDS show malar hypoplasia, arched palate, retrognathia, pectus deformity, scoliosis, joint laxity, dural ectasia, and aortic root aneurysms and dissection. Although their fingers tend to be long, overgrowth of the long bones is subtle or absent, and they do not show ectopia lentis. Unique features of LDS include a high incidence of hypertelorism, broad or bifid uvula, arterial tortuosity in large and medium-sized vessels (predominantly neck vessels) and a high risk of aneurysm and dissection throughout the arterial tree. Aortic root aneurysms tend to dissect at younger ages and smaller dimensions than in MFS, often leading to death in early childhood. In view of the severe nature of vascular disease in LDS, correct diagnosis and aggressive management is essential in these patients.
Less-consistent features of LDS include blue sclerae, translucent skin, easy bruising, craniosynostosis, cleft palate, Chiari type I malformation of the brain, learning disability, congenital heart disease (patent ductus arteriosus, atrial septal defect, bicommissural aortic valve), and clubfoot deformity. There is significant overlap between LDS and the Shprintzen-Goldberg syndrome that includes craniosynostosis, hypertelorism, arched palate, learning disability, bone overgrowth, pectus deformity, and scoliosis. In contrast to LDS, vascular disease is a rare manifestation of Shprintzen-Goldberg syndrome.
Laboratory Findings
Laboratory studies should document a negative urinary cyanide nitroprusside test or specific amino acid studies to exclude cystathionine synthase deficiency (homocystinuria). Although it is estimated that most, if not all, people with classic MFS have an FBN1 mutation, the large size of this gene and the extreme allelic heterogeneity in MFS have frustrated efficient molecular diagnosis. The yield of mutation screening varies based on technique and clinical presentation. Recent experience suggests that >95% of people with typical MFS will have an identifiable FBN1 mutation. It remains unclear whether the “missing” mutations are simply atypical in character or location within FBN1 or occur in another gene. Some reports suggest that mutations in TGFβR2 are occasionally seen in classic MFS; whether or not these patients have discriminating features of LDS remains to be determined.
Other diseases in the differential such as MVP syndrome, MASS phenotype, familial ectopia lentis, Weill-Marchesani syndrome, and Shprintzen-Goldberg syndrome have all been associated with mutations in the FBN1 gene. It is often difficult or impossible to predict the phenotype from the nature or location of a FBN1 mutation in MFS. Hence, molecular genetic techniques can contribute to the diagnosis, but they do not substitute for comprehensive clinical evaluation and follow-up. The absence or presence of an FBN1 mutation is not sufficient to exclude or establish the diagnosis, respectively.
Treatment
Management focuses on preventing complications and genetic counseling. In view of the potential complexity of management required by some patients, referral to a multidisciplinary center with experience in MFS is advisable. The pediatrician should work in concert with pediatric subspecialists to coordinate a rational approach to monitoring and treatment of potential complications. Yearly evaluations for cardiovascular disease, scoliosis, or ophthalmologic problems are imperative.
Current and Future Therapies
Most therapies currently available or under investigation aim to diminish aortic complications. They can be categorized as activity restrictions, surgeries, endocarditis prophylaxis, current pharmacologic approaches and emerging or experimental strategies.
Activity Restrictions
Physical therapy can improve neuromuscular tone in infancy, and aerobic exertion in moderation is recommended during childhood and adulthood. Strenuous physical exertion, competitive athletics, and particularly isometric activities such as weight lifting are associated with an increased risk of aortic complications or ocular problems such as retinal detachment and should be discouraged. Consensus guidelines have been developed for activity limitations.
Surgery for Aortic Aneurysm
Successful surgical repair of aortic root aneurysms may be the single most important cause of improvement in life expectancy for patients with MFS to date. Surgical outcome is more favorable if undertaken on an elective rather than an urgent or emergent basis. Therefore, aortic root surgery in MFS should be recommended for patients with an aortic root diameter ≥50 mm and considered for those with a rapid rate of enlargement (>5-10 mm/yr) or a family history of early aortic dissection. Preserving the native aortic valve at the time of repair is desirable to avoid the need for lifelong anticoagulation with warfarin, although the long-term durability of the native aortic valve in MFS remains to be fully elucidated. Replacing the aortic root with a pulmonary root autograft (Ross procedure) is not recommended, due to the observations of progressive enlargement of the pulmonary root when exposed to systemic pressure and eventual autograft failure.
Endocarditis Prophylaxis
The American Heart Association (AHA) no longer routinely recommends the use of antibiotic prophylaxis for persons with structural or valvular heart disease, but exceptions are made for select groups at the greatest risk for bad outcomes from infectious endocarditis. The Professional Advisory Board of the National Marfan Foundation believes that patients with MFS (as well as other patients with certain connective tissue disorders who are at high risk for severe and rapidly progressive degeneration and dysfunction of multiple heart valves and for early and recurrent cardiovascular surgeries) should continue to receive prophylaxis for subacute bacterial endocarditis (SBE). SBE prophylaxis is especially important because it remains unknown but possible that the myxomatous valves typical of MFS represent a preferred substrate for endocarditis.
Current Pharmacologic Approaches
Based on the putative role of hemodynamic stress in aortic dilatation in MFS, β-blockers have traditionally been considered the standard of care, due to their negative inotropic and chronotropic effects. While multiple small observational studies have supported a protective effect of β-blockers in MFS, and a reduced rate of aortic growth was seen in MFS mice treated with propanolol, other analyses have concluded that there is insufficient evidence to support their routine use. Calcium channel antagonists (e.g., verapamil) and angiotensin converting enzyme (ACE) inhibitors (e.g., enalapril) offer alternative therapeutic options. However, there is again no conclusive evidence supporting the use of either of these classes of agent in the treatment of MFS.
Emerging and Experimental Strategies
There is extensive evidence linking angiotensin II signaling to TGF-β activation and signaling. In addition, direct activation of TGF-β-responsive signaling cascades by angiotensin II (independent of TGF-β ligands) has been reported. In a mouse model of MFS, the angiotensin II receptor type 1 blocker (ARB) losartan has been shown to completely prevent pathologic aortic root growth and to normalize both aortic wall thickness and architecture, findings that were absent in placebo-treated and propanolol-treated mice. These data suggest the potential for productive aortic wall remodeling in MFS after TGF-β inhibition. Remarkably, improvements in pulmonary and skeletal muscle pathology in these mice also occurred with losartan, further supporting the conclusion that this treatment works by decreasing TGF-β signaling rather than by simply reducing hemodynamic stress on structurally predisposed tissues.
In support of its relevance to humans, a study assessing the effect of ARBs in a small cohort of pediatric patients with MFS who had severe aortic root enlargement despite previous alternate medical therapy, showed that ARBs significantly slowed the rate of aortic root and sinotubular junction dilatation (both of which occur in MFS), whereas the distal ascending aorta (which does not normally become dilated in MFS) remained unaffected. In light of this growing body of evidence, a multicenter prospective clinical trial to assess the potential therapeutic benefit of ARBs in patients with MFS is under way.
TGF-β Neutralizing Antibodies
Several studies have shown the ability of antibodies that antagonize TGF-β to modulate the manifestations of MFS in mice, including defective pulmonary alveolar septation, myxomatous atrioventricular valves, skeletal muscle myopathy, and aortic root aneurysms. Currently, therapeutic delivery of TGF-β neutralizing antibody is not available for humans. However, a humanized anti–TGF-β1 monoclonal antibody (CAT-192) is currently under investigation for treatment of other TGF-β-related disorders, and its use remains a potential strategy for treating MFS.
Other FDA-Approved Agents
It has been proposed that ACE inhibitors might prove as effective as, or more effective than, ARBs in treating MFS. By limiting the production of angiotensin II, ACE inhibitors effectively limit signaling through the type 1 and type 2 angiotensin receptors (AT1R and AT2R, respectively). By contrast, ARBs selectively block the AT1R and might actually increase signaling through the AT2R. Given that AT2R signaling can promote cell death but also can limit TGF-β signaling, the relative benefit of the two approaches remains controversial. Several small studies suggest there is some protection afforded by ACE inhibitors in MFS, but this issue will only be definitively resolved with additional experimentation and clinical experience.
Other work has suggested that doxycycline might improve aortic root pathology in MFS. Matrix metalloproteinases (MMPs) 2 and 9 are extracellular TGF-β-responsive proteinases that cause elastic fiber damage. Doxycycline, through its broad inhibition of MMPs, has been shown to reduce aortic root growth and rupture in mouse models of MFS.
Prognosis
Although prognosis is highly variable, the average life expectancy for people with MFS approaches normal, particularly in those who underwent aortic root surgery after 1980. However, there is still morbidity and early mortality associated with the disorder, placing both physical and mental stresses on patients and their families. Awareness of these issues and referral for support services can facilitate a positive perspective toward the condition.
Genetic Counseling
The heritable nature of MFS makes recurrence risk (genetic) counseling mandatory. Approximately 30% of cases are the first affected individuals in their families. Fathers of these sporadic cases are, on average, 7 to 10 years older than fathers in the general population. This paternal age effect suggests that these cases represent new dominant mutations with minimal recurrence risk to the future offspring of the normal parents. Due to rare reports of gonadal mosaicism in a phenotypically normal parent, the recurrence risk for parents of a sporadic case can be reported as low but not zero. Each child of an affected parent, however, has a 50% risk of inheriting a number 15 chromosome with the MFS mutation and thus being affected. Recurrence risk counseling is best accomplished by professionals with expertise in the issues surrounding the disorder.
Ahimastos AA, Aggarwal A, D’Orsa KM, et al. Effect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome. JAMA. 2007;298:1539-1546.
Brooke BS, Habashi JP, Judge DP, et al. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787-2795.
Davies RR, Goldstein LJ, Coady MA, et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. 2002;73:17-27.
de Oliveira NC, David TE, Ivanov J, et al. Results of surgery for aortic root aneurysm in patients with Marfan syndrome. JThorac Cardiovasc Surg. 2003;125:789-796.
DePaepe A, Devereux RB, Dietz HC, et al. Revised criteria for Marfan syndrome. Am J Med Genet. 1996;62:417-426.
Faivre L, Masurel-Paulet A, Collard-Béroud G, et al. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009;123:391-398.
Gelb BD. Marfan’s syndrome and related disorders—more tightly connected than we thought. N Engl J Med. 2006;355:841-844.
Glesby MJ, Pyeritz RE. Association of mitral valve prolapse and systemic abnormalities of connective tissue: a phenotypic continuum. JAMA. 1989;262:523-528.
Habashi JP, Judge D, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117-121.
Johnson JN, Hartman TK, Pianosi PT, et al. Cardiorespiratory function after operation for pectus excavatum. J Pediatr. 2008;153:359-364.
Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366:1965-1976.
Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117:2802-2813.
Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275-281.
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476-485.
Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sport participation for young patients with genetic cardiovascular diseases. Circulation. 2004;109:2807-2816.
Matt P, Schoenhoff F, Habashi J, et al. Circulating transforming growth factor-β in Marfan syndrome. Circulation. 2009;120:526-532.
Meijboom LJ, Vos FE, Timmermans J, et al. Pregnancy and aortic root growth in the Marfan syndrome: a prospective study. Eur Heart J. 2005;26:914-920.
Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407-411.
Rhee D, Solowiejczyk D, Altmann K, et al. Incidence of aortic root dilation in pectus excavatum and its association with Marfan syndrome. Arch Pediatr Adolesc Med. 2008;162:882-885.
Tierney ESS, Feingold B, Printz BF, et al. Beta-blocker therapy does not alter the rate of aortic root dilation in pediatric patients with Marfan syndrome. J Pediatr. 2007;150:77-82.
Yetman AT, Bornemeier RA, McCrindle BW. Usefulness of enalapril versus propranolol or atenolol for prevention of aortic dilation in patients with the Marfan syndrome. Am J Cardiol. 2005;95:1125-1127.