chapter 51 Benign Renal Tumors
Benign renal neoplasms constitute a rather large and heterogeneous group of renal lesions that can be found in the kidney. These include the simple renal cyst, selected complex renal cysts, cortical and metanephric adenomas, angiomyolipoma, oncocytoma, the rarer cystic nephroma, mixed epithelial/stromal tumor, and leiomyoma, as well as other, even more esoteric tumor types. Management approaches to these lesions can vary widely, from no management for the simple renal cyst to selective embolization for larger angiomyolipomas and surgical extirpation for solid renal masses when the differential diagnosis includes renal cell carcinoma (RCC). With the increased use of cross-sectional abdominal imaging for renal-specific as well as other nonspecific complaints, it is expected that the identification of both benign and malignant renal tumors will continue to increase in the coming years (Patard, 2009). Moreover, with the increase use of, and refinements to, renal mass biopsy, the management of both benign and malignant renal neoplasms is still evolving in a way that the indications for intervention, and the type of intervention, may change significantly in the coming years (Lane et al, 2008; Campbell et al, 2009). Now, with advanced multidimensional imaging techniques, as well as minimally invasive, nephron-sparing surgical approaches, percutaneous ablative approaches, and the concept of active surveillance in the armamentarium of the practicing urologist, management of all renal lesions, including those that are presumptively benign in etiology, continues to evolve (Raj et al, 2007; Benway and Bhayani, 2009; Murphy et al, 2009).
At present, however, the urologist is left primarily with imaging studies such as ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI) to assess whether a renal lesion is benign or malignant before therapeutic decisions are made. And unless the mass has clear radiographic features that suggest a benign etiology, such as clear evidence of fat seen with most angiomyolipomas or the smooth walls and lack of enhancement of a simple or minimally complex cyst, the vast majority of benign renal lesions are diagnosed only after definitive therapy such as surgical intervention has been implemented. There are several clinical features that have been linked to an increased likelihood of a renal mass having a benign etiology, including smaller size, female sex, and older age, but none of these factors can be relied on to avoid intervention if a specific pretherapy diagnosis cannot be made (Kutikov et al, 2006; Snyder et al, 2006; Glassman et al, 2007; Lane et al, 2007; Beisland et al, 2009; Murphy et al, 2009).
In this chapter the most common benign renal neoplasms that occur in the kidney are identified. The discussion is then focused on the etiology and natural history, clinical presentation, imaging characteristics, and treatment options, when treatment is indicated.
Renal Cysts
In much the same way as the molecular biology of RCC was elucidated through the study of familial genetic syndromes such as von Hippel-Lindau disease, the molecular basis for cyst formation has been further elucidated through the genetic analysis of familial renal cystic syndromes, such as autosomal dominant polycystic kidney disease (ADPKD). Through these studies researchers have identified that loss of specific genes, such as PKD1 and/or PKD2 (polycystins) lead to cyst formation in patients with ADPKD. Polycystin-1 and polycystin-2 form a complex that represents a critical ion channel in the kidney, the loss of which results in cyst formation through the loss of intracellular calcium dysregulation and dysfunction of the primary renal cilia, a poorly understood organelle that is in part responsible for normal renal development (Pei, 2003; Weimbs, 2007; Ibraghimov-Beskrovnaya and Bukanov, 2008). Whether these genetic changes are common to sporadic forming “benign” renal cystic disease remains to be proven, but many of the phenotypic and genetic changes noted in the kidneys of patients with familial cystic disease syndromes have been identified in sporadic cystic disease (Qian et al, 1996; Pei, 2001).
Perhaps the best description of the natural history of sporadic renal cysts can be found in the recently updated study by Terada and associates (2008). In this study of 61 patients with simple renal cysts and a mean of 10 years of follow-up, the authors noted that the cysts increased in both size and number over time. The average increase in size of the cysts was 1.9 mm per year, but the authors also noted that the rate of size increase decreased with age. Interestingly, two cysts developed renal neoplasms during the course of the study and no differences were noted in the clinical characteristics of the cysts that developed neoplasms from those that did not. Regarding the risk factors for the development of cysts, increasing age, male gender, presence of hypertension, and the presence of renal insufficiency were all associated with the development of sporadic renal cysts (Terada et al, 2004). Renal cysts remain the most common benign renal lesions, representing more than 70% of asymptomatic renal masses. They can be solitary or multiple and unilateral or bilateral (Terada et al, 2002).
In addition to sporadic cystic disease of the kidney, and the cysts that occur with familial syndromes such as ADPKD and autosomal recessive polycystic disease, cysts are also known to occur in association with end-stage renal disease in patients on dialysis (Bisceglia et al, 2006). There appears to be a higher incidence of RCC associated with the development of acquired renal cystic disease, much like that seen with von Hippel-Lindau disease and tuberous sclerosis, such that the pathogenesis of these cystic lesions may be quite different from that seen with sporadic simple or minimally complex cysts (Truong et al, 2003).
Renal cystic lesions can be imaged through a variety of radiographic techniques, including ultrasonography, CT, as well as MRI. On ultrasonography, simple renal cysts have a smooth wall, are fluid filled with no internal echoes, and have evidence of posterior wall enhancement. Evidence of internal echoes, calcifications or nodularity in the wall, or internal septa on ultrasonography suggest a more complex cyst that might be worthy of further imaging that includes intravenous administration of a contrast agent (Quaia et al, 2008; Eknoyan, 2009).
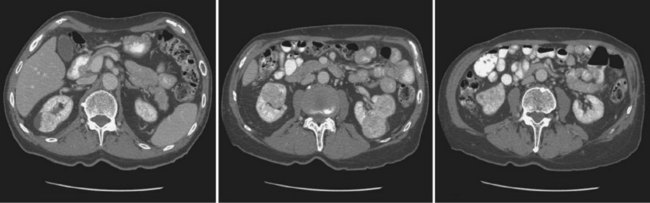
The Bosniak classification for renal cystic lesions, as reviewed in Table 51–1, is perhaps the most useful and widely employed method for characterizing renal cystic lesions and for assessing the likelihood of the presence of a concomitant malignancy within the cyst (Israel and Bosniak, 2005; Warren and McFarlane, 2005). In general, Bosniak class I, II, and IIF cysts are likely to represent benign lesions, thus requiring either no therapy or just continued radiographic follow-up, in the case of class IIF lesions (Fig. 51–1). These recommendations are based on a number of series published in the literature by Bosniak and others that include both radiographic as well as pathologic follow-up (Warren and McFarlane, 2005).

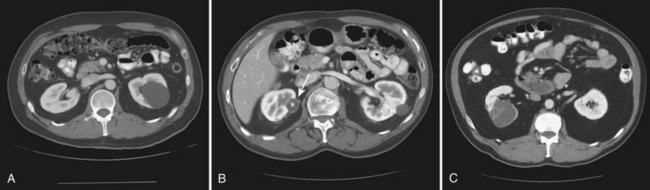
Figure 51–1 A, CT scan of a Bosniak I renal cyst. B, CT scan of a Bosniak II renal cyst. Note internal calcification. C, CT scan of a Bosniak IIF renal cyst. Several thin irregular septations are present within the cyst.
Because of the higher risk of malignancy associated with Bosniak class III and IV lesions, therapy is recommended (Fig. 51–2). The definitive therapy would be surgical excision, although ablative therapies such as cryotherapy or radiofrequency ablation of cystic masses have been reported (Raman et al, 2009) (see Chapter 49).
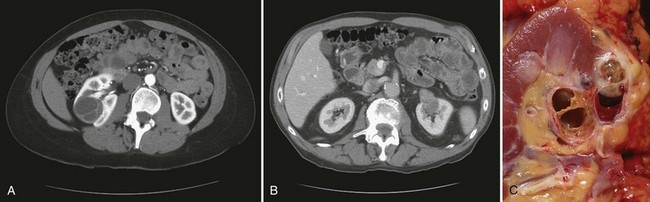
Figure 51–2 A, CT scan of a Bosniak III renal cyst. Thick, irregular septations are present within the cyst. B, CT scan of a Bosniak IV renal cyst, with a solid enhancing nodule. C, Bivalved Bosniak IV renal cyst demonstrating a solid component that proved to be conventional renal cell carcinoma.
The overwhelming majority of simple or minimally complex cysts require no further follow-up or therapy once diagnosed (Eknoyan, 2009). Rarely, benign cystic lesions of the kidney can grow to such a large size that they can cause pain or other symptomatology, including hypertension (Porpiglia et al, 2009; Zerem et al, 2009). Symptoms can also occur as a consequence of hemorrhage within the cyst or spontaneous/traumatic cyst rupture (Hughes et al, 1995; Rainio et al, 2006; Ishikawa et al, 2008; Vaidyanathan et al, 2008).
A variety of therapeutic interventions have been described for benign symptomatic cystic lesions of the kidney. These include aspiration, surgical resection, cyst decortications, as well as sclerotherapy with a variety of different agents (Cho et al, 2008; Ham et al, 2008; Baysal and Soylu, 2009; Canguven et al, 2009; Choi et al, 2009; Porpiglia et al, 2009). Although none of these approaches seems to be better than the others described, it is noted that with aspiration and sclerotherapy there is a higher incidence of cyst recurrence and multiple treatments may be needed to satisfactorily ablate the cystic lesion. A word of caution is warranted regarding the treatment of peripelvic cysts: given their proximity to the vital structures of the kidney, including the renal vessels and collecting system, laparoscopic, rather than percutaneous, approaches may be associated with a higher safety margin and better efficacy (Okumura et al, 2003; Camargo et al, 2005).
Renal Cortical Adenoma
The designation and management of purportedly benign renal cortical adenomas remains a subject of controversy in the urologic literature. These lesions are small, solid cortical renal lesions that are thought to have a benign course (Renshaw, 2002). Histologically, renal adenomas appear as small, well-circumscribed lesions characterized by uniform basophilic or eosinophilic cells with benign-appearing nuclear and cellular features, arranged as tubulopapillary or purely papillary growth. The incidence of renal adenomas increases with age and male sex, and these tumors also have been associated with acquired renal cystic disease that results in end-stage renal failure (Leroy et al, 2001; Denton et al, 2002; Snyder et al, 2006; Ferda et al, 2007). The incidence of renal adenomas in autopsy series ranges from 7% to 23%, although antemortem pathologic diagnosis of renal adenoma is much less common, in part owing to the pathologist’s concern that there are no reliable histopathologic, ultrastructural, or immunohistochemical criteria to distinguish benign from malignant lesions of the kidney (Licht, 1995). In fact, in a more recent study in which immunohistochemical analyses were used to further characterize renal adenomas, it was suggested that these lesions may be linked to the development of papillary RCC and represent a biologic link and continuum as a premalignant precursor (Wang et al, 2007). In this study, researchers examined 542 nephrectomy specimens obtained over 8 years. Seven percent of these demonstrated evidence of renal adenoma; and of these, 47% were associated with a concomitant papillary RCC. Adenomas that arose in the setting of papillary RCC tended to be multiple (61%). Eighty-two percent of adenomas found in the study had a similar immunohistochemistry staining profile as papillary RCC, staining positive for α-methyl-coenzyme A racemase. In other studies, renal adenomas shared similar cytogenetic profiles to papillary RCC, such as trisomy of chromosomes 7 and 17, thus suggesting a biologic link between the two neoplasms (Brunelli et al, 2003a, 2003b).
The overwhelming majority of renal adenomas remain asymptomatic, are undetectable radiographically due to their small size (<1 cm), and require no further therapy. Size of the neoplasm has historically been utilized to differentiate renal adenoma from more malignant neoplasms of the kidney. Thoenes and colleagues (1986) provided a reassessment of the histologic classification of renal tumors and defined renal adenoma as a tumor with nuclear grade I and a diameter of at least 1 cm. For many years, a “3-cm rule” was pervasive in the urologic literature, dating back to the original autopsy studies by Bell (1938) when he noted that only 1 of 38 renal cortical tumors had associated metastases whereas 70 of 106 tumors larger than 3 cm were associated with metastases.
The diagnosis of renal adenoma remains controversial; many believe that all solid renal epithelium-derived masses are potentially malignant and therefore should undergo treatment (Renshaw, 2002).
Key Points: Renal Cortical Adenoma
Metanephric Adenoma
In 1995 Davis and colleagues reported 50 cases of an unusual and novel renal mass lesion with distinctive histologic features and a benign clinical course despite occasional symptomatic presentation and large tumor size. In this series, mean tumor size was 5.5 cm (up to 15 cm) and nearly half of the patients presented with flank pain, gross hematuria, or a palpable mass. Six additional patients presented with polycythemia, and hypercalcemia has also been reported in association with this tumor type, which was designated metanephric adenoma (Davis et al, 1995; Mahoney et al, 1997; Kuroda et al, 2003b). Metanephric adenoma was officially accepted as a primary benign renal tumor based on consensus from the Heidelberg classification (Kovacs et al, 1997). A higher male-to-female ratio was initially reported in the series of Davis and colleagues (1995), but it is likely that there is actually a female predominance as has been reported in other series (Jones et al, 1995; Snyder et al, 2006; Bastide et al, 2009). Incidental presentation is most common, and peak incidence is in the fifth decade of life (Renshaw, 2002). Polycythemia can be seen in 10% of patients and appears to be due to the production of erythropoietin and other cytokines by the tumor (Yoshioka et al, 2007; Bastide et al, 2009). Radiographically these tumors can have peripheral or central calcifications and may be hypovascular on contrast-enhanced CT and hyperechoic on ultrasonography (Bastide et al, 2009).
On microscopic examination these tumors consist of very small, often highly basophilic epithelial cells that form small acini and occasionally tubular or papillary structures within a predominantly acellular stroma (Fig. 51–3). Davis and colleagues (Renshaw, 2002) argued that metanephric adenoma might be histologically related to epithelial Wilms tumor because they believed that it exhibited histologic similarities to the metanephric, hamartomatous elements of nephroblastomatosis. Along these lines, it is interesting to note that many of these tumors exhibit evidence of regression in the form of scarring or calcification. In addition, Muir and colleagues (2001) have shown positive staining for the Wilms tumor protein WT1 and an immunohistochemical staining profile that suggests a histogenetic relationship to Wilms tumor. An alternative theory for the origin of metanephric adenoma was proposed by Brown and associates (1997), who found gain of chromosomes 7 and 17 by fluorescent in-situ hybridization in 8 of 11 of these tumors. These findings suggest a clonal neoplastic disorder potentially related to papillary RCC, but others have argued that this series may have been contaminated by inclusion of some cases of papillary RCC, which can be difficult to differentiate from metanephric adenoma (Brunelli et al, 2003a). Arroyo and colleagues (2001) showed that metanephric adenomas are part of a spectrum of related tumors, bridging metanephric stromal tumors, metanephric adenofibromas, and Wilms tumor. Pesti and colleagues (2001) have described a putative tumor suppressor gene for metanephric adenoma at chromosome 2p13. Various histologic stains have been evaluated to help distinguish metanephric adenoma from other renal neoplasms. The Wilms tumor marker WT1 is frequently expressed in metanephric adenoma (Muir et al, 2001; Bosco et al, 2007). α-Methylacyl-CoA racemase (AMACR) is poorly expressed in metanephric adenoma but highly expressed in papillary RCC (Olgac et al, 2006), whereas S-100 protein expression is very high in metanephric adenoma, weak in Wilms tumor, and absent in papillary RCC (Azabdaftari et al, 2008). Skinnider and colleagues (2005) demonstrated the potential utility of an expression panel to help differentiate papillary RCC and metanephric adenoma using a panel of cytokeratins 7, 8, 18, and 19 and vimentin. Using these various markers appears to have improved the diagnostic yield of percutaneous biopsy and fine-needle aspiration (Bosco et al, 2007; Patel et al, 2009), but the index of initial suspicion needs to be high for biopsy to be considered.
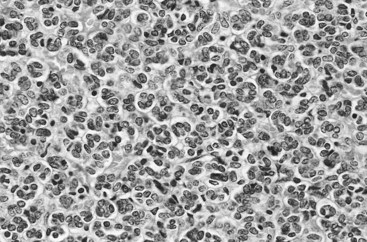
Figure 51–3 Classic metanephric adenoma with small, intensely basophilic cells arranged in an acinar pattern.
Only one case of metastasis has been described in association with classic metanephric adenoma into a regional lymph node, and death related to this entity has not been reported (Renshaw, 2002). However, Picken and colleagues (2001) have described malignant stromal elements associated with a metanephric neoplasm of the kidney in a 21-year-old woman who died of progressive cancer, and they have proposed that there may be a spectrum of metanephric tumors that includes rare, aggressive variants. Atypical histologic features and multifocality in childhood have also been reported (Jain et al, 2007; Kohashi et al, 2009). Given the rarity of this tumor and the lack of highly predictive clinical or radiographic criteria, metanephric adenoma remains primarily a pathologic diagnosis. If radiographic findings raise the index of suspicion, then percutaneous core biopsy with fine-needle aspiration may prove helpful in establishing a diagnosis for nephron-sparing treatment or observation, but most patients will require surgical excision because of concern for malignancy (Fig. 51–4).
Oncocytoma
Renal oncocytoma is the most common benign tumor that appears as an enhancing renal mass on cross-sectional imaging and is presumed to be RCC until surgical excision, representing one of the ultimate challenges in preoperative diagnosis for the urologist. It accounts for 3% to 7% of kidney tumors (Morra and Das, 1993). Oncocytoma initially became accepted as a distinct entity after a report of 13 cases in 1976 by Klein and Valensi. Multiple additional reports since that time, including more recent genotyping studies, confirm it to be a benign histology with a distinct cell of origin and genetic abnormalities (Lieber et al, 1987; Davis et al, 1991; Licht et al, 1993; Perez-Ordonez et al, 1997; Amin et al, 1997; Dechet et al, 1999; Chao et al, 2002; Kuroda et al, 2003a). Cases of metastatic disease have been reported, yet these are considered exceptionally rare and may represent cases of malignant degeneration or pseudometastases (Paner et al, 2005; Oxley et al, 2007).
Grossly these tumors are mahogany or tan, homogeneous, and well circumscribed with a pseudocapsule and typically a central stellate scar (Fig. 51–5). Microscopically the cells are round or polygonal and arranged in a nested growth pattern. The cells are uniform and highly eosinophilic, owing to an abundance of mitochondria (Renshaw, 2002). In up to one third of cases, hemorrhage, extension into perinephric fat, cellular atypia, prominent nucleoli, and pleomorphism may be seen, yet the clinical behavior in these cases is within what is expected with a benign course (Amin et al, 1997; Perez-Ordonez et al, 1997). The most common genetic abnormality is loss of chromosome 1p (Lindgren et al, 2004; Paner et al, 2007). Other common cytogenetic findings include loss of the Y chromosome and chromosome 14q and rearrangements of 11q13 (Schwerdtle et al, 1997; Chao et al, 2002; Polascik et al, 2002; Lindgren et al, 2004). The chromosomal abnormalities typically seen in RCC are not seen in renal oncocytomas, further reinforcing the concept that these tumors are genotypically distinct from RCC (Minor et al, 2003). However, histologically, the greatest dilemma arises from distinguishing chromophobe and clear cell RCC with eosinophilic characteristics from oncocytoma. Hale colloidal iron staining is the classic differentiating marker for oncocytoma, but it can have nonspecific staining and be difficult to interpret (Leroy et al, 2000). Cytokeratin profiles are helpful in distinguishing these histologic findings (Skinnider et al, 2005; Adley et al, 2006). Expression of cytokeratin-7 is seen in 66% of chromophobe RCC and only 5% of oncocytomas, and parvalbumin is expressed in 100% of chromophobe RCC and 47% of oncocytomas (Leroy et al, 2000; Adley et al, 2006). A variety of other markers have been recently described to differentiate oncocytoma from RCC, particularly chromophobe RCC, but the clinical utility for most of these has not yet been fully developed. These include Pax-2, expressed in metanephric tissues and vital for renal tubule development; pattern of claudin-7 and 8 expression, tight junction proteins expressed in distal nephron epithelium; a vimentin expression pattern; c-KIT expression; and S-100 (Pan et al, 2004; Lin et al, 2006; Hes et al, 2007; Rocca et al, 2007; Lechpammer et al, 2008; Gupta et al, 2009; Osunkoya et al, 2009). An “optimal” panel for distinguishing between chromophobe and clear cell RCC and oncocytoma was recommended by Liu and colleagues in 2007, consisting of a combination of three markers (vimentin, glutathione-S-transferase-α, and epithelial cell adhesion molecule). These investigators achieved 100% sensitivity and 100% specificity for the differential diagnosis of chromophobe carcinoma, oncocytoma, and clear cell carcinoma (Liu et al, 2007). The feasibility of molecular signatures and gene profiling is being evaluated as potentially useful tests for the accurate diagnosis of tumors such as oncocytoma (Schuetz et al, 2005; Yang et al, 2006), but such studies are not yet a clinical reality.
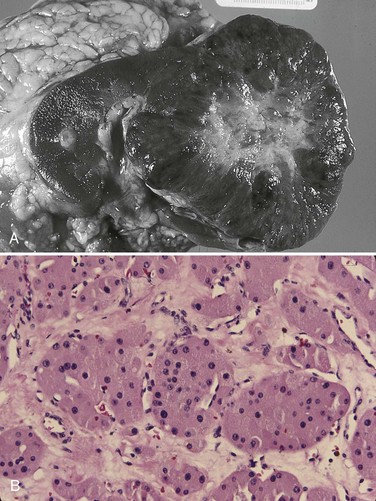
Figure 51–5 A, Bivalved renal oncocytoma demonstrating central scar. B, Oncocytoma with large eosinophilic cells arranged in distinct nests.
As noted previously there are several distinct similarities to chromophobe RCC, which also is derived from the distal renal tubules. Chromophobe RCC, particularly the eosinophilic variant, and oncocytoma are histologically similar and their distinction often requires additional pathologic testing (Weiss et al, 1995; Renshaw, 2002). The commonality of these two tumors is further evidenced in patients with Birt-Hogg-Dubé syndrome, in whom both oncocytomas and chromophobe RCC develop, in addition to cutaneous fibrofolliculomas and spontaneous pneumothoraces (Pavlovich et al, 2005; Toro et al, 2008). Some of the renal tumors display a histology between these two tumors, prompting some to speculate that chromophobe RCC and oncocytoma represent points in a spectrum of neoplasia (Chao et al, 2002; Linehan, 2003; Pavlovich et al, 2005; Toro et al, 2008).
The greatest clinical dilemma remains the inability to confidently differentiate between renal oncocytoma and RCC on clinical or radiographic testing. Both have a similar age at presentation, have a male-to-female predominance, and are similarly sized at presentation. Although oncocytomas were more likely to be asymptomatic at presentation, most RCC are also diagnosed incidentally in the current era, eliminating this clinical scenario as a differentiator (Davis et al, 1991; Licht et al, 1993; Lieber, 1993; Amin et al, 1997; Perez-Ordonez et al, 1997; Dechet et al, 1999). The typical spoke-wheel pattern seen on angiography or the stellate scar on cross-sectional imaging may bring up the question of a renal oncocytoma, but these findings have a poor predictive value by themselves (Davidson et al, 1993; Licht et al, 1993; Licht, 1995). On MRI, oncocytomas may have distinct T1 and T2 signal patterns that can be suggestive but these are not definitive findings (Harmon et al, 1996). T2-weighted images on MRI do not differentiate oncocytoma from RCC (Dann et al, 2006). For lesions undergoing surveillance, the growth rates of RCC and oncocytoma are similar, so growth kinetics also do not help differentiate these tumors either (Chawla et al, 2006; Crispen and Uzzo, 2007; Siu et al, 2007). In 4% to 13% of cases, tumors are multicentric, are bilateral, or have a metachronous presentation (Lieber et al, 1987; Davis et al, 1991; Licht et al, 1993; Amin et al, 1997; Perez-Ordonez et al, 1997; Dechet et al, 1999; Tickoo et al, 1999; Minor et al, 2003). Familial renal oncocytomatosis was initially described (Weirich et al, 1998) in five families in which it presented at a young age as multicentric, bilateral and recurrent oncocytomas. Nonfamilial forms of bilateral multifocal oncocytomas resembling oncocytomatosis can also occur (Fig. 51–6). A recent cytogenetic evaluation of a patient with apparently sporadic oncocytomatosis and hybrid tumors showed different chromosomal losses than the Birt-Hogg-Dubé syndrome (Al-Saleem et al, 2004).
Recent clinical studies highlight the differing incidence of oncocytoma based on age and gender. Cao and colleagues (2005) and Skolarus and colleagues (2008) showed an increasing incidence of oncocytoma in older patients presenting with a small incidentally discovered renal mass. Two different reports highlight that younger females are nearly twice as likely as their male counterparts to have a benign tumor, which includes oncocytoma and angiomyolipoma but is probably largely driven by the higher rates of angiomyolipoma in women (Cao et al, 2005; Snyder et al, 2006).
Historically fine-needle aspiration or core biopsy was compromised by high false-negative or nondiagnostic specimens, given the difficulty in differentiating oncocytoma with RCC subtypes (Weiss et al, 1995; Campbell et al, 1997). However, the diagnostic accuracy of percutaneous biopsy has markedly improved, particularly when a core biopsy is done in addition to a fine-needle aspiration and bolstered with the use of immunostains (Liu and Fanning, 2001; Barocas et al, 2006; Lebret et al, 2007; Volpe et al, 2007; Kummerlin et al, 2008; Schmidbauer et al, 2008), prompting some investigators to revisit the role of the biopsy in the management of some patients with an incidental renal tumor (Shah et al, 2005; Lebret et al, 2007; Volpe et al, 2007). When multiple tumors are present, one must consider the possibility of RCC coexisting with oncocytoma, which in some series has been shown to be as high as 32% (Davis et al, 1991; Licht et al, 1993; Licht, 1995; Gudbjartsson et al, 2005). Consideration of biopsy in these cases must be thoughtfully considered to provide a sampling of all sites of disease that are of concern.
Treatment options for a known oncocytoma range from observation to thermal ablation, laparoscopic or open partial nephrectomy, and even radical nephrectomy depending on the clinical scenario and uncertainty regarding the diagnosis (Licht, 1995; Romis et al, 2004; Gudbjartsson et al, 2005; Crispen and Uzzo, 2007). If oncocytoma is highly suspected and surgery is indicated, a nephron-sparing approach is preferred given the benign nature of these lesions and the very low probability of recurrence (Licht, 1995; Romis et al, 2004; Gudbjartsson et al, 2005). Frozen section analysis is usually not sensitive enough to differentiate the eosinophilic appearance of oncocytomas with eosinophilic RCCs and should not be used to guide surgical strategy. Thermal ablation, although sometimes reported as a treatment option, commits the patient to long-term radiographic surveillance, given the lower success rates of these procedures and the unknown long-term outcomes, tests that would be considered unnecessary should surgical excision be performed. In most cases the treatment options are isolated to observation, particularly for the older or sicker patient, and surgical resection, particularly for the younger healthier patient.
Key Points: Oncocytoma
Angiomyolipoma
Angiomyolipoma accounts for less than 10% of renal tumors, with autopsy series and ultrasound-screened populations showing a 0.3% and 0.13% incidence in the general population (Eble, 1998). It is a benign neoplasm consisting of thick-walled aneurysmal vessels, smooth muscle, and varying levels of mature adipose tissue (Tamboli et al, 2000; Nelson et al, 2002; Bissler et al, 2004). Initially considered to be a form of hamartoma or choristoma, recent evidence suggests a neoplastic origin with evidence of a monoclonal, rather than polyclonal, source (Green et al, 1996; Sepp et al, 1996; Kattar et al, 1999). Angiomyolipoma is now considered to be of neural crest origin, possibly derived from perivascular epithelioid cells (PEC). Originally described in 1911 by Fischer, its current name was given by Morgan in 1951 (Eble, 1998). The tumor strongly expresses estrogen receptor-β as well as androgen receptor, is predominantly found in females, and is rare before puberty, suggesting a potential hormonal influence (Boorjian et al, 2008).
The typical sporadic presentation is of a middle-aged woman with a single asymptomatic tumor. Sporadic angiomyolipomas appear to have a slow growth rate and are usually detected incidentally (Seyam et al, 2008). Angiomyolipoma is the most common renal neoplasm associated with spontaneous perirenal hemorrhage, closely followed by RCC (Zhang et al, 2003). Skolarus and colleagues (2008) suggested that there may be a decreased incidence of sporadic angiomyolipoma with increasing age. These tumors are most often sporadic but can also be associated with the autosomal dominant tuberous sclerosis complex (TSC). Twenty to 30 percent of angiomyolipomas are in patients with TSC, and approximately 50% of patients with TSC develop angiomyolipomas (Eble, 1998; Neumann et al, 1998; Tamboli et al, 2000; Lendvay et al, 2003; Minor et al, 2003). TSC-associated angiomyolipomas typically present at a younger age (mean age 30 years); there is less female-to-male predominance (2 : 1) with multiple, bilateral, and symptomatic tumors (Eble, 1998; Neumann et al, 1998; Lendvay et al, 2003). Because of the variable penetrance of the TSC mutation, the classic triad of seizures, adenoma sebaceum, and mental retardation may not be present because this mutation is seen in a minority of patients (Steiner et al, 1993). Patients with TSC also develop renal cysts and may be at higher risk of developing RCC. Lymphangioleiomyomatosis is also significantly associated with renal involvement in TSC (Rakowski et al, 2006).
Similar to RCC, most angiomyolipomas are now diagnosed incidentally during workup of unrelated complaints (Lemaitre et al, 1997; Seyam et al, 2008). The literature correlating tumor size and symptoms, however, is derived prior to this era, when most angiomyolipomas were diagnosed after the development of symptoms. The Wunderlich syndrome, or massive retroperitoneal hemorrhage, representing the most significant complication of renal angiomyolipoma, was reported in up to 10% of patients and could be associated with significant morbidity and potential mortality if not promptly treated (Oesterling et al, 1986; Steiner et al, 1993; Eble, 1998). The most common cause of spontaneous retroperitoneal hemorrhage is angiomyolipoma, followed by RCC (Zhang et al, 2003). Pregnancy appears to increase the risk of hemorrhage from angiomyolipoma, a factor that can influence clinical decision making (Eble, 1998).
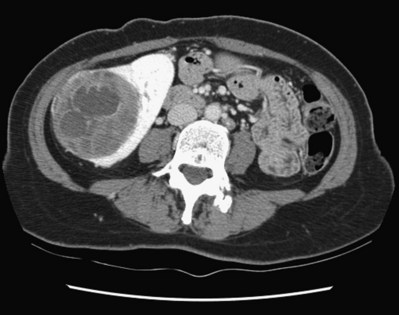
Angiomyolipoma is the only benign renal tumor that is confidently diagnosed on cross-sectional imaging (Fig. 51–7). The presence of fat (confirmed on nonenhanced thin-cut CT by a value of −20 Hounsfield Units [HU] or less) within a renal lesion is considered the diagnostic hallmark (Jinzaki et al, 1997; Lemaitre et al, 1997; Bosniak et al, 1998; Simpfendorfer et al, 2009). Findings of more than 20 pixels with attenuation less than −20 HU and of more than 5 pixels with attenuation less than −30 HU have been shown to have a positive predictive value of 100% (Simpfendorfer et al, 2009). Ultrasonography shows a well-circumscribed, highly echogenic lesion with shadowing (Siegel et al, 1996; Lemaitre et al, 1997). On angiography (or CT-angiography) aneurysmal dilation is found in 50% of angiomyolipomas (Lemaitre et al, 1997). The size of the aneurysms has been reported to correlate with the risk of rupture (Yamakado et al, 2002). MRI can be used in difficult cases or in lieu of CT, with findings on fat-suppressed images being highly suggestive of the diagnosis.
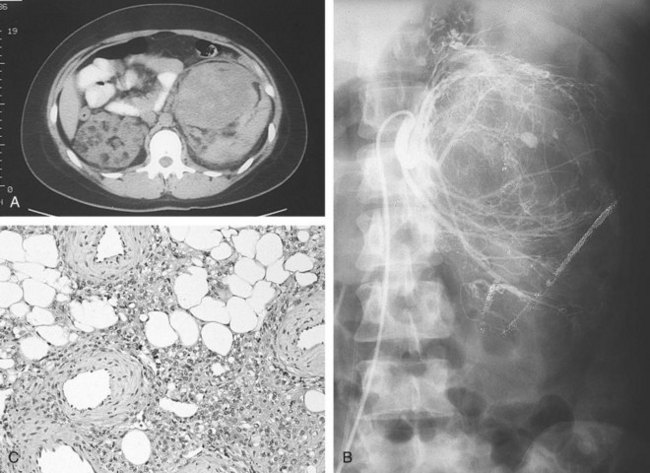
Figure 51–7 A, CT scan demonstrating large bilateral renal angiomyolipomas in a patient with tuberous sclerosis. B, Renal angiogram shows increased vascularity and aneurysmal dilation characteristic of angiomyolipoma. C, Typical microscopic appearance of angiomyolipoma with admixture of mature adipose tissue, smooth muscle, and thick-walled blood vessels.
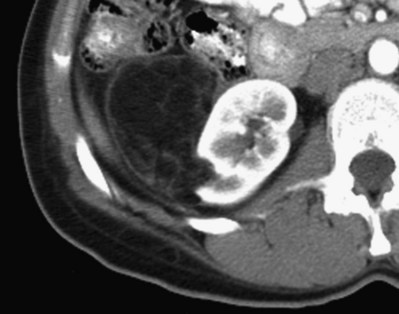
Despite the radiographic hallmarks of angiomyolipoma the diagnosis can be confused in the three following situations: confusion with liposarcoma, possibility of fat-containing RCC, and possibility of a fat-poor angiomyolipoma resembling an RCC. Large angiomyolipomas can be confused with retroperitoneal liposarcomas, which are very rare. A good quality, high-resolution CT however will invariably show several features that are characteristic of angiomyolipoma, the primary one being a small indentation of the renal parenchyma even when the tumor envelops the kidney (Fig. 51–8), whereas liposarcomas will compress or only extrinsically push the renal parenchyma (Clark and Novick, 2001; Wang et al, 2002). Although RCC containing fat density has also been reported, these extremely rare cases also contained calcifications, a finding usually almost never seen with angiomyolipoma (Henderson et al, 1997; Lemaitre et al, 1997; Roy et al, 1998). Most recently a report of two cases of RCC with very small amounts of intratumoral fat but without calcification was reported, adding to two other reported cases in the literature (Hayn et al, 2009). Fat-poor angiomyolipoma, seen in 14% of cases or more, is much more difficult to diagnose, owing to the paucity of mature adipose tissue (Lemaitre et al, 1997; Kim et al, 2004; Milner et al, 2006). Lane and colleagues (2008) showed that fat-poor angiomyolipoma was more commonly single, smaller, and found in older patients. Various imaging modalities have attempted to accurately identify these lesions preoperatively and differentiate them from RCC, with mixed success. On CT many appear to have hyperattenuation on noncontrast imaging and have a homogeneous and prolonged enhancement pattern (Jinzaki et al, 1997; Kim et al, 2004; Hafron et al, 2005; Milner et al, 2006; Silverman et al, 2007). Detailed pixel distribution analysis cannot differentiate between fat-poor angiomyolipoma and RCC (Catalano et al, 2008). Ultrasonography can show a hyperechoic or isoechoic lesion, suggesting a fat-poor angiomyolipoma (Jinzaki et al, 1997). Because of the nonspecific nature of these findings most patients are often treated as having a presumed RCC. However, these radiographic findings may prompt the attentive urologist to consider a percutaneous biopsy if the suspicion is raised by imaging. Percutaneous biopsy can play an important role in diagnosis in these cases, because a core biopsy should be eminently accurate in the diagnosis of angiomyolipoma (Lebret et al, 2007; Silverman et al, 2007).
Although an invariably benign nature of angiomyolipoma is well accepted, extrarenal occurrences are occasionally seen and have been reported in the hilar lymph nodes, retroperitoneum, and liver, with direct extension into the venous system (Eble, 1998; Türker Köksal et al, 2000; Gögüs et al, 2001; Nelson and Sanda, 2002; Lin et al, 2003; Bissler and Kingswood, 2004; Akcali et al, 2006; Haritharan et al, 2006; Blick et al, 2008; Schade et al, 2008). Even in these cases a benign clinical course follows, indicating multicentric origin rather than malignancy with metastasis. Many angiomyolipomas exhibit regions of cellular atypia, and the pathologic differential diagnosis can include a number of subtypes of sarcoma, including fibrosarcoma, leiomyosarcoma, and liposarcoma, depending on the relative amounts of adipose, vascular, or smooth muscle tissue present (Wang et al, 2002). Positive immunoreactivity for HMB-45, a monoclonal antibody raised against a melanoma-associated antigen, is characteristic for angiomyolipoma and can be used to differentiate this tumor from sarcoma and other tumors (Eble, 1998). There have been two reports of high-grade and eventually lethal leiomyosarcoma arising within an angiomyolipoma. Christiano and colleagues (1999) have described a highly pleomorphic angiomyolipoma that was associated with the development of multiple pulmonary nodules, the majority of which stained positive for HMB-45 (Ferry et al, 1991). They believed that this case represented a malignant transformation of angiomyolipoma, which, if it does occur, must be exceedingly rare. However, these cases may also have represented the more recently described entity of epithelioid angiomyolipoma. In 1997, Eble and colleagues published their experience of five patients who had angiomyolipoma with a predominant epithelioid component; and more recently this malignant epithelioid variant of angiomyolipoma that can metastasize has been further described in patients with and without TSC, many of whom succumbed to the disease (Pea et al, 1998; L’Hostis et al, 1999; Martignoni et al, 2000; Cibas et al, 2001; Menè et al, 2001; Nelson and Sanda, 2002; Saito et al, 2002; Bissler and Kingswood, 2004; Huang et al, 2007; Limaiem et al, 2008; Matsuyama et al, 2008; Moudouni et al, 2008; Zanelli et al, 2008; Kato et al, 2009). This phenotype is characterized by epithelioid cells that are cytokeratin negative and HMB-45 positive. Whether this extremely rare variant represents malignant degeneration of a preexistent angiomyolipoma or a de-novo tumor without a benign precursor remains unknown.
For optimal management of angiomyolipoma, treatment must be individualized. The management should take into account the size of the tumor, presence of symptoms, and patient factors. In particular, the risk of hemorrhage must be weighed during the evaluation. In general, most symptomatic angiomyolipomas have been relatively large and most studies in the literature have focused on a 4-cm cut point (Steiner et al, 1993; Nelson and Sanda, 2002). On the basis of an extensive literature review, Oesterling and coworkers (1986) reported that 82% of patients with angiomyolipomas larger than 4 cm in diameter were symptomatic, with 9% in hemorrhagic shock at the time of presentation; in contrast, patients with smaller tumors were symptomatic 23% of the time. Echoing these findings, Dickinson and colleagues (1998) reported that all 18 patients with angiomyolipomas smaller than 4 cm in their series were asymptomatic, whereas 7 of 13 patients with angiomyolipomas of 4 to 8 cm and 5 of 6 patients with tumors larger than 8 cm required intervention, primarily related to pain or bleeding. These observations have been confirmed and extended by a number of investigators (Blute et al, 1988; Steiner et al, 1993; Lemaitre et al, 1995; De Luca et al, 1999; Seyam et al, 2008). Steiner and colleagues (1993) reported that patients with angiomyolipomas larger than 4 cm were symptomatic 52% of the time, with 30% requiring surgical intervention, whereas patients with smaller tumors never required surgery and were asymptomatic 76% of the time. Although it was primarily retrospective, limited follow-up with a mean of 4 years was available for 24 patients with 28 tumors in this series. Interval growth was documented in 6 of 13 tumors with diameter of more than 4 cm and in 4 of 15 tumors smaller than 4 cm. A slower growth rate and a low risk of hemorrhage for smaller tumors was also supported by Kennelly and colleagues (1994), who observed 17 angiomyolipomas with tumor size of less than 4 cm for a mean of 3.8 years. Similarly, De Luca and colleagues (1999) studied 32 incidentally discovered angiomyolipomas smaller than 5 cm in diameter and found that 92% remained asymptomatic and unchanged in size. Even so, occasional larger angiomyolipomas undergoing observation, some for up to 18 years, can remain asymptomatic (Kennelly et al, 1994; Hadley et al, 2006; Danforth et al, 2007), reinforcing the concept that size represents a continuum of risk, not an absolute phenomenon, and highlighting the need for individualized tailoring of treatment recommendations. Multicentric angiomyolipomas and those in patients with tuberous sclerosis represent a special group that has demonstrated increased growth rates of approximately 20% per year, in contrast to a mean growth rate of 5% per year for solitary, sporadic angiomyolipomas (Steiner et al, 1993; Nelson and Sanda, 2002; Harabayashi et al, 2004; Seyam et al, 2008).
Although no large prospective studies evaluating the long-term outcomes of angiomyolipomas have been performed, the information reviewed here allows one to propose general guidelines for management. Asymptomatic, smaller tumors, which by convention have been those with a diameter less than 4 cm, can be observed expectantly, with repeat initial imaging at 6 to 12 months to define the growth rate and clinical significance. Repeat imaging can be lengthened once stability is established, with follow-up performed only annually or biannually for smaller tumors (Oesterling et al, 1986; De Luca et al, 1999; Matin et al, 2008). Intervention should be considered for larger tumors, particularly if the patient is symptomatic, taking into account the patient’s age, comorbidities, and other related factors. Women of childbearing age and patients with limited access to surveillance or to emergency care should also consider a proactive approach (Nelson and Sanda, 2002). A nephron-sparing approach, by either selective embolization or open or laparoscopic/robotic partial nephrectomy, is clearly preferred in patients with angiomyolipomas requiring intervention. Preservation of renal tissue remains a priority in those with TSC or multicentric angiomyolipoma and particularly in patients with underlying renal insufficiency. The feasibility and efficacy of partial nephrectomy by either wedge resection or enucleation in patients with angiomyolipoma is well established, with preservation of renal function achieved even in patients with large lesions in a solitary kidney (Fazeli-Matin and Novick, 1998; Boorjian et al, 2007; Minervini et al, 2007). Selective embolization is reported by some to be the preferred modality, and data from 76 patients in six series have documented long-term success in most cases (Nelson and Sanda, 2002; Harabayashi et al, 2004). However, a substantial proportion of patients experienced persistent or recurrence of symptoms or hemorrhage and most of these required repeated procedures, including embolization or surgery (Hamlin et al, 1997; Han et al, 1997; Kehagias et al, 1998; Mourikis et al, 1999; Nelson and Sanda, 2002; Lenton et al, 2008). The overall complication rate with embolization in these series was 10%, similar to rates of partial nephrectomy (Boorjian et al, 2007), and included hemorrhage, abscess formation, or sterile liquefaction of the tumor requiring percutaneous drainage or surgical intervention. These data highlight the need for extended follow-up after selective embolization, which would not be required after partial nephrectomy (Nelson and Sanda, 2002). Selective embolization should be considered as first-line therapy in patients with acute or potentially life-threatening hemorrhage, because surgical exploration in this setting is often associated with total nephrectomy (Pappas et al, 2006; Chang et al, 2007). Ablative therapies such as radiofrequency ablation (Prevoo et al, 2008) and cryoablation (Bachmann et al, 2005; Byrd et al, 2006; Littrup et al, 2007; Caviezel et al, 2008) have also been utilized for the treatment of angiomyolipoma, but follow-up remains short, the evaluation of success remains poorly defined, and the duration for continued radiographic surveillance is unknown, thus committing the patient to multiple, long-term imaging. Ablative therapies may have their best role for the treatment of patients with TSC who have multicentric angiomyolipomas or older patients with comorbidities who require treatment and are not candidates for embolization.
Key Points: Angiomyolipoma
Cystic Nephroma and Mixed Epithelial/Stromal Tumor
Although considered separate entities by the 2004 World Health Organization classification of renal neoplasms, cystic nephroma and mixed epithelial/stromal tumors of the kidney (MEST) are rare benign renal neoplasms that have overlapping clinical, morphologic, and immunohistochemical characteristics (Lopez-Beltran et al, 2006; Turbiner et al, 2007; Lane et al, 2008; Montironi et al, 2008). Recent demonstration of striking similarities between global gene expression profiles in cystic nephroma and MEST further strengthens the notion that these two entities represent opposite ends of the spectrum of the same biologic process (Zhou et al, 2009). Female predilection and history hormonal ablation therapy in male patients, combined with the frequent expression of estrogen and progesterone receptors, suggest that the sex-steroid hormones might play a role in the pathogenesis of these rare lesions (Turbiner et al, 2007; Lane et al, 2008; Montironi et al, 2008; Stamatiou et al. 2008).
Cystic Nephroma
Cystic nephroma is a characteristic renal lesion with a bimodal age distribution and a benign clinical course (Tamboli et al, 2000). Diagnostic peaks occur primarily in the first 2 to 3 years of life, predominantly in males, and again in the fourth and fifth decades with a significant (8 : 1) female prevalence (Madewell et al, 1983; Upadhyay and Neely, 1989; Castillo et al, 1991; Stamatiou et al, 2008; Kuzgunbay et al, 2009). As with other renal lesions, presenting signs can include abdominal mass, pain, and hematuria, but the majority of cystic nephromas are incidental findings (Madewell et al, 1983; Kuzgunbay et al, 2009). Several familial cases have been reported in the literature, and there have been anecdotal reports of sarcoma and clear cell carcinoma arising from cystic nephroma (Bal et al, 2005; Omar et al, 2006; Raj et al, 2006; Ashley and Reinberg, 2007).
Radiologically, most cystic nephromas are solitary, centrally located, and widely variable in size (mean 9 cm) and commonly demonstrate curvilinear calcifications, herniation into the renal collecting system, and septal enhancement (Fig. 51–9A, B) (Madewell et al, 1983; Turbiner et al, 2007). Consequently, reliable radiologic differentiation between cystic nephroma and cystic RCC in adults or Wilms tumor in children is not possible (Vujanic et al, 2000).
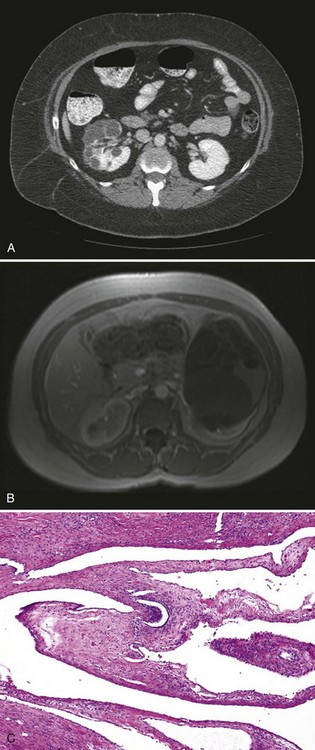
Figure 51–9 Cystic nephroma. CT (A) and MR (B) images do not allow reliable distinction from cystic renal cell carcinoma or cystic Wilms tumor. C, The variably sized cystic spaces are lined by flattened epithelium (low magnification).
Histologically, cystic nephromas are well encapsulated by a thick fibrous pseudocapsule and are composed of cysts lined by flattened, cuboidal, or hobnail epithelium (see Fig. 51–9C). The stromal component can range from dense paucicellular collagen to markedly cellular fascicles of spindle cells, closely resembling ovarian stroma (Tamboli et al, 2000). Immunohistochemical studies reveal affinity of the epithelial component for cytokeratins, whereas stromal components frequently stain positive for CD10, calretinin, inhibin, estrogen, and progesterone receptors (Turbiner et al, 2007; Montironi et al, 2008).
Because of concern for cystic Wilms tumor, most children with cystic nephromas continue to be managed by radical nephrectomy, whereas a nephron-sparing approach with partial nephrectomy, if feasible, is an attractive option in adults.
Mixed Epithelial/Stromal Tumor
MEST is a rare benign adult renal neoplasm with a variable admixture of epithelial and mesenchymal components (Adsay et al, 2000). Previously these tumors have been described as congenital mesoblastic nephroma, leiomyomatous renal hamartoma, solid and cystic biphasic tumor, cystic hamartoma, solitary multilocular cyst, and adult metanephric stromal tumor (Adsay et al, 2000; Pierson et al, 2001; Mai et al, 2007). Similar to cystic nephromas, MEST exhibits striking female predominance with a diagnostic peak in the fourth decade. The majority of women diagnosed with MEST had a history of receiving estrogen replacement therapy, and the only male patient in the largest MEST series had a long history of androgen deprivation therapy for prostate cancer (Adsay et al, 2000). Presenting signs and symptoms of MEST are similar to those of cystic nephroma with the majority detected incidentally (Adsay et al, 2000; Turbiner et al, 2007; Montironi et al, 2008).
Radiologic appearance of MEST is of a complex cystic renal mass, typically classifying these tumors as Bosniak class III to IV lesions, indistinguishable from cystic RCC (Fig. 51–10A) (Adsay et al, 2000). A typical MEST has a benign clinical course, but recently a case of malignant transformation to a sarcomatoid carcinoma and several cases of local recurrence of a malignant stromal component with a dismal clinical course have been described (Adsay et al, 2000; Nakagawa et al, 2004).
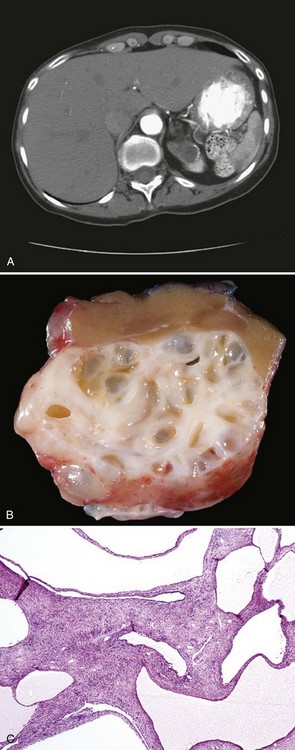
Figure 51–10 Mixed epithelial and stromal tumor. A, CT scan characteristics are not distinguishable from renal cell carcinoma. B, Gross photograph of a partial nephrectomy specimen demonstrating a well-circumscribed mass composed of variably sized cysts separated by thick white septa. C, Medium-power magnification shows cysts lined by hobnailed cells and spindle cell stroma.
Grossly, MEST appears encapsulated and ranges from 2 to 24 cm (mean 6 cm) (Adsay et al, 2000; Mai et al, 2007). Involvement of renal hilum and compression of the pelvicalyceal system is common, but gross infiltration of adjacent renal parenchyma is not seen (see Fig. 51–10B). The mesenchymal component is characterized by spindle cells showing variable degrees of smooth muscle, fibroblastic, or myofibroblastic differentiation with interspersed collagen bundles. The epithelial components vary from regular tubules to complex tubulopapillary structures with or without cystic dilatation, lined by cuboidal to flattened epithelium that may show clear cell changes and have a characteristic hobnail appearance (see Fig. 51–10C) (Adsay et al, 2000; Antic et al, 2006; Turbiner et al, 2007; Montironi et al, 2008). As in cystic nephroma, the epithelial components stain positive for cytokeratins whereas estrogen and progesterone receptor staining has been observed in the majority of the mesenchymal elements of MEST (Adsay et al, 2000).
A preoperative diagnosis of MEST should be considered in perimenopausal women receiving hormone therapy; however, because radiologic differentiation from RCC is not reliable, surgical intervention, preferably with a nephron-sparing approach should be offered to appropriately selected patients.
Key Points: Cystic Nephroma and Mixed Epithelial/Stromal Tumor
Leiomyoma
Leiomyomas are rare, benign tumors that may arise from smooth muscle cells anywhere along the genitourinary tract (Tamboli et al, 2000). In the kidney these tumors most commonly arise from the renal capsule, but renal pelvis and renal vein sites of origin have been reported (Wells et al, 1981; Steiner et al, 1990; O’Brien et al, 1992; Rao et al, 2001). Leiomyomas are found at autopsy with a frequency of 4.2% to 5.2%, but only a minority are discovered clinically, representing approximately 1.5% of all benign renal tumors treated surgically (Romero et al, 2005). As with other renal lesions, in the age of widespread utilization of computerized tomographic abdominal imaging, the vast majority of leiomyomas are found incidentally (Romero et al, 2005; Derchi et al, 2008).
Renal leiomyomas have a characteristic appearance of a small exophytic renal mass with or without enhancement arising from renal capsule, but conclusive radiologic differentiation from RCC is not possible (Fig. 51–11A) (Steiner et al, 1990; Derchi et al, 2008).
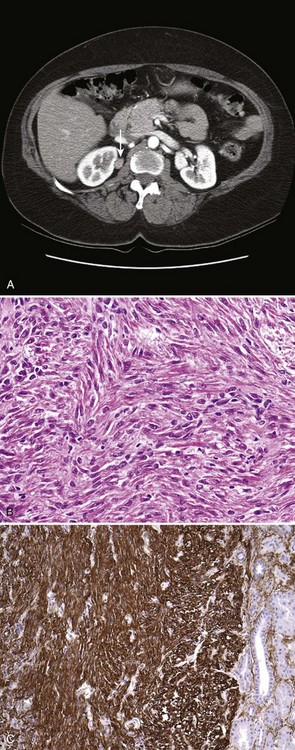
Figure 51–11 Renal leiomyoma. A, CT scan with characteristic appearance of a small renal mass arising from the renal capsule. B, Medium-power magnification shows uniform spindle cells with thin cigar-shaped nuclei, without any pleomorphism. C, Strong positive immunohistochemical staining with smooth muscle actin in the leiomyoma. Note lack of smooth-muscle actin staining in the normal renal tubules on the right.
Grossly, leiomyomas are well encapsulated and range from purely cystic to mixed solid/cystic or solid in appearance. Histologic examination reveals intersecting fascicles of smooth muscle with no evidence of hypercellularity, pleomorphism, mitotic activity, or necrosis (see Fig. 51–11B) (Steiner et al, 1990; Tamboli et al, 2000). Immunohistochemical stains confirm the smooth muscle nature of the tumor with strong diffuse positive staining for smooth muscle markers desmin and caldesmon (see Fig. 51–11C) (Romero et al, 2005). Large lesions have traditionally been managed with radical nephrectomy, but nephron-sparing approaches should be considered for peripherally located small lesions.
Other Benign Renal Tumors
A multitude of rare benign tumors derived from the various mesenchymal components of the kidney have been described and include multiple histiotypes, such as fibromas, lipomas, lymphangiomas, and hemangiomas (Ligato et al, 1999; Tamboli et al, 2000). Current radiologic methods do not allow for conclusive differentiation of these benign tumors from malignant renal lesions, and surgical excision is often needed for pathologic confirmation.
Among these, noteworthy is reninoma, which is a benign tumor of the renal juxtaglomerular cell apparatus (Wong et al, 2008). With fewer than 100 cases reported in the literature, females in the third and fourth decade are most commonly affected (Martin et al, 2001; Rubenstein et al, 2002; Wong et al, 2008). Clinical presentation is dominated by hypersecretion of renin and includes hypertension and hypokalemia and associated symptoms such as polydipsia, polyuria, myalgia, and headaches (Schonfeld et al, 1991; Rubenstein et al, 2002). The radiologic appearance is that of a small (<3 cm) solid hypovascular renal mass; and surgical excision, preferably sparing the remaining renal parenchyma, results in rapid decline of plasma rennin levels, normalization of blood pressure, and resolution of the associated symptoms (Dunnick et al, 1983; Schonfeld et al, 1991; Tanabe et al, 2001). Histologic examination reveals sheets of polygonal to spindle-shaped cells with indistinct cell borders, abundant eosinophilic cytoplasm, and minimal atypia (Martin et al, 2001). Strong immunostaining for factor VIII and factor VIII–related antigens is characteristic and confirms derivation from endothelial cell lineage (Sanfilippo et al, 1982). Although a benign clinical course is expected, one case of malignant reninoma has been documented in the literature (Duan et al, 2004).
Antic T, Perry KT, et al. Mixed epithelial and stromal tumor of the kidney and cystic nephroma share overlapping features: reappraisal of 15 lesions. Arch Pathol Lab Med. 2006;130(1):80-85.
Barocas DA, Rohan SM, et al. Diagnosis of renal tumors on needle biopsy specimens by histological and molecular analysis. J Urol. 2006;176(5):1957-1962.
Boorjian SA, Frank I, et al. The role of partial nephrectomy for the management of sporadic renal angiomyolipoma. Urology. 2007;70(6):1064-1068.
Castillo OA, Boyle ETJr, et al. Multilocular cysts of kidney: a study of 29 patients and review of literature. Urology. 1991;37(2):156-162.
Davis CJJr, Barton JH, Sesterhenn IA, Mostofi FK. Metanephric adenoma. Clinicopathological study of fifty patients. Am J Surg Pathol. 1995;10(10):1101-1114.
Dechet CB, Bostwick DG, Blute ML, et al. Renal oncocytoma: multifocality, bilateralism, metachronous tumor development and coexistent renal cell carcinoma. J Urol. 1999;162(1):40-42.
Eble J, Amin M, et al. Epithelioid angiomyolipoma of the kidney: a report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol. 1997;21(10):1123-1130.
Eknoyan G. A clinical view of simple and complex renal cysts. J Am Soc Nephrol. 2009;20(9):1874-1876.
Hafron J, Fogarty JD, et al. Imaging characteristics of minimal fat renal angiomyolipoma with histologic correlations. Urology. 2005;66(6):1155-1159.
Harabayashi T, Shinohara N, et al. Management of renal angiomyolipomas associated with tuberous sclerosis complex. J Urol. 2004;171(1):102-105.
Ibraghimov-Beskrovnaya O, Bukanov N. Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci. 2008;65(4):605-619.
Israel GM, Bosniak MA. An update of the Bosniak renal cyst classification system. Urology. 2005;66(3):484-488.
Lane BR, Aydin H, et al. Clinical correlates of renal angiomyolipoma subtypes in 209 patients: classic, fat poor, tuberous sclerosis associated and epithelioid. J Urol. 2008;180(3):836-843.
Lopez-Beltran A, Scarpelli M, et al. 2004 WHO classification of the renal tumors of the adults. Eur Urol. 2006;49(5):798-805.
Qian F, Watnick TJ, et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979-987.
Seyam RM, Bissada NK, et al. Changing trends in presentation, diagnosis and management of renal angiomyolipoma: comparison of sporadic and tuberous sclerosis complex-associated forms. Urology. 2008;72(5):1077-1082.
Siu W, Hafez KS, et al. Growth rates of renal cell carcinoma and oncocytoma under surveillance are similar. Urol Oncol. 2007;25(2):115-119.
Terada N, Arai Y, et al. The 10-year natural history of simple renal cysts. Urology. 2008;71(1):7-11. discussion 11–2
Warren KS, McFarlane J. The Bosniak classification of renal cystic masses. BJU Int. 2005;95(7):939-942.
Zhou M, Kort E, et al. Adult cystic nephroma and mixed epithelial and stromal tumor of the kidney are the same disease entity: molecular and histologic evidence. Am J Surg Pathol. 2009;33(1):72-80.
Adley BP, Papavero V, et al. Diagnostic value of cytokeratin 7 and parvalbumin in differentiating chromophobe renal cell carcinoma from renal oncocytoma. Anal Quant Cytol Histol. 2006;28(4):228-236.
Adsay NV, Eble JN, et al. Mixed epithelial and stromal tumor of the kidney. Am J Surg Pathol. 2000;24(7):958-970.
Akcali Y, Karahan OI, et al. Angiomyolipoma with cavoatrial extension. Eur Urol. 2006;50(3):605-606.
Al-Saleem T, Cairns P, et al. The genetics of renal oncocytosis: a possible model for neoplastic progression. Cancer Genet Cytogenet. 2004;152(1):23-28.
Amin MB, Crotty TB, Tickoo SK, Farrow GM. Renal oncocytoma: a reappraisal of morphologic features with clinicopathologic findings in 80 cases. Am J Surg Pathol. 1997;21(1):1-12.
Antic T, Perry KT, et al. Mixed epithelial and stromal tumor of the kidney and cystic nephroma share overlapping features: reappraisal of 15 lesions. Arch Pathol Lab Med. 2006;130(1):80-85.
Arroyo MR, Green DM, et al. The spectrum of metanephric adenofibroma and related lesions: clinicopathologic study of 25 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol. 2001;25(4):433-444.
Ashley RA, Reinberg YE. Familial multilocular cystic nephroma: a variant of a unique renal neoplasm. Urology. 2007;70(1):179. e9–10
Azabdaftari G, Alroy J, et al. S100 protein expression distinguishes metanephric adenomas from other renal neoplasms. Pathol Res Practice. 2008;204(10):719-723.
Bachmann A, Sulser T, et al. Retroperitoneoscopy-assisted cryoablation of renal tumors using multiple 1.5 mm ultrathin cryoprobes: a preliminary report. Eur Urol. 2005;47(4):474-479.
Bal N, Kayaselcuk F, et al. Familial cystic nephroma in two siblings with pleuropulmonary blastoma. Pathol Oncol Res. 2005;11(1):53-56.
Barocas DA, Rohan SM, et al. Diagnosis of renal tumors on needle biopsy specimens by histological and molecular analysis. J Urol. 2006;176(5):1957-1962.
Bastide C, Rambeaud JJ, et al. Metanephric adenoma of the kidney: clinical and radiological study of nine cases. BJU Int. 2009;103(11):1544-1548.
Baysal T, Soylu A. Percutaneous treatment of simple renal cysts with n-butyl cyanoacrylate and iodized oil. Diagn Interv Radiol. 2009;15(2):148-152.
Beisland C, Hjelle KM, et al. Observation should be considered as an alternative in management of renal masses in older and comorbid patients. Eur Urol. 2009;55(6):1419-1427.
Bell E. A classification of renal tumors with observations on the frequency of the various types. J Urol. 1938;39(5):238-243.
Benway BM, Bhayani SB. Approach to the small renal mass: weighing treatment options. Curr Urol Rep. 2009;10(1):11-16.
Bisceglia M, Galliani CA, et al. Renal cystic diseases: a review. Adv Anat Pathol. 2006;13(1):26-56.
Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66(3):924-934.
Blick C, Ravindranath N, et al. Bilateral renal angiomyolipomas with invasion of the renal vein: a case report. Sci World J. 2008;8:145-148.
Blute M, Malek R, et al. Angiomyolipoma: clinical metamorphosis and concepts for management. J Urol. 1988;139(1):20-24.
Boorjian SA, Frank I, et al. The role of partial nephrectomy for the management of sporadic renal angiomyolipoma. Urology. 2007;70(6):1064-1068.
Boorjian SA, Sheinin Y, et al. Hormone receptor expression in renal angiomyolipoma: clinicopathologic correlation. Urology. 2008;72(4):927-932.
Bosco M, Galliano D, et al. Cytologic features of metanephric adenoma of the kidney during pregnancy: a case report. Acta Cytol. 2007;51(3):468-472.
Bosniak MA, Megibow AJ, Hulnick DH, et al. CT diagnosis of renal angiomyolipoma: the importance of detecting small amounts of fat. AJR Am J Roentgenol. 1998;151(3):497-501.
Brown JA, Anderl KL, Borell TJ, et al. Simultaneous chromosome 7 and 17 gain and sex chromosome loss provide evidence that renal metanephric adenoma is related to papillary renal cell carcinoma. J Urol. 1997;158(2):370-374.
Brunelli M, Eble JN, et al. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol. 2003;16(10):1053-1059.
Brunelli M, Eble JN, et al. Metanephric adenoma lacks the gains of chromosomes 7 and 17 and loss of Y that are typical of papillary renal cell carcinoma and papillary adenoma. Mod Pathol. 2003;16(10):1060-1063.
Byrd GF, Lawatsch EJ, et al. Laparoscopic cryoablation of renal angiomyolipoma. J Urol. 2006;176(4 pt 1):1512-1516. discussion 1516
Camargo AH, Cooperberg MR, et al. Laparoscopic management of peripelvic renal cysts: University of California, San Francisco, experience and review of literature. Urology. 2005;65(5):882-887.
Campbell SC, Novick AC, et al. Prospective evaluation of fine needle aspiration of small, solid renal masses: accuracy and morbidity. Urology. 1997;50(1):25-29.
Campbell SC, Novick AC, et al. Guideline for management of the clinical T1 renal mass. J Urol. 2009;182(4):1271-1279.
Canguven O, Goktas C, Yencilek F, et al. A new technique for simple renal cyst: cystoretroperitoneal shunt. Adv Urol. 2009:906013.
Cao Y, Paner GP, et al. Renal neoplasms in younger adults: analysis of 112 tumors from a single institution according to the new 2004 World Health Organization classification and 2002 American Joint Committee on Cancer Staging System. Arch Pathol Lab Med. 2005;129(4):487-491.
Castillo OA, Boyle ETJr, et al. Multilocular cysts of kidney: a study of 29 patients and review of literature. Urology. 1991;37(2):156-162.
Catalano OA, Samir AE, et al. Pixel distribution analysis: can it be used to distinguish clear cell carcinomas from angiomyolipomas with minimal fat? Radiology. 2008;247(3):738-746.
Caviezel A, Terraz S, et al. Percutaneous cryoablation of small kidney tumours under magnetic resonance imaging guidance: medium-term follow-up. Scand J Urol Nephrol. 2008;42(5):412-416.
Chang YH, Wang LJ, et al. The efficacy and outcomes of urgent superselective transcatheter arterial embolization of patients with ruptured renal angiomyolipomas [erratum appears in J Trauma 2007;63(5):1190]. J Trauma. 2007;62(6):1487-1490.
Chao DH, Zisman A, Pantuck AJ, et al. Changing concepts in the management of renal oncocytoma. Urology. 2002;59(5):635-642.
Chawla SN, Crispen PL, et al. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol. 2006;175(2):425-431.
Cho DS, Ahn HS, et al. Sclerotherapy of renal cysts using acetic acid: a comparison with ethanol sclerotherapy. Br J Radiol. 2008;81(972):946-949.
Choi YD, Ham WS, et al. Clinical experience of single-session percutaneous aspiration and OK-432 sclerotherapy for treatment of simple renal cysts: 1-year follow-up. J Endourol. 2009;23(6):1001-1006.
Christiano A, Yang X, et al. Malignant transformation of renal angiomyolipoma. J Urol. 1999;161(6):1900-1901.
Cibas ES, Goss GA, Kulke MH, et al. Malignant epithelioid angiomyolipoma (‘sarcoma ex angiomyolipoma’) of the kidney: a case report and review of the literature. Am J Surg Pathol. 2001;25(1):121-126.
Clark PE, Novick AC. Exophytic noninvasive growth pattern of renal angiomyolipomas: implications for nephron sparing surgery. J Urol. 2001;165(2):513-514.
Crispen PL, Uzzo RG. The natural history of untreated renal masses. BJU Int. 2007;99(5 Pt. B):1203-1207.
Danforth TL, Lane BR, et al. Conservative management of giant symptomatic angiomyolipomas in patients with the tuberous sclerosis complex. BJU Int. 2007;100(4):794-797.
Dann P, Thakur R, et al. Are T2-weighted images necessary in renal mass characterization? Eur J Radiol. 2006;59(1):112-116.
Davidson AJ, Hayes WS, Hartman DS, et al. Renal oncocytoma and carcinoma: failure of differentiation with CT. Radiology. 1993;186(3):693-696.
Davis CJJr, Barton JH, Sesterhenn IA, Mostofi FK. Metanephric adenoma: clinicopathological study of fifty patients. Am J Surg Pathol. 1995;10(10):1101-1114.
Davis CJ, Mostofi FK, Sesterhenn IA, Ho CK. Renal oncocytoma: clinicopathological study of 166 patients. J Urogenital Pathol. 1991;1:41-52.
De Luca S, Terrone C, Rossetti SR. Management of renal angiomyolipoma: a report of 53 cases. BJU Int. 1999;83(3):215-218.
Dechet CB, Bostwick DG, Blute ML, et al. Renal oncocytoma: multifocality, bilateralism, metachronous tumor development and coexistent renal cell carcinoma. J Urol. 1999;162(1):40-42.
Denton MD, Magee CC, et al. Prevalence of renal cell carcinoma in patients with ESRD pre-transplantation: a pathologic analysis. Kidney Int. 2002;61(6):2201-2209.
Derchi LE, Grenier N, Heinz-Peer G, et al. Imaging of renal leiomyomas. Acta Radiol. 2008 Sep;49(7):833-838.
Dickinson M, Ruckle H, et al. Renal angiomyolipoma: optimal treatment based on size and symptoms. Clin Nephrol. 1998;49(5):281-286.
Duan X, Bruneval P, et al. Metastatic juxtaglomerular cell tumor in a 52-year-old man. Am J Surg Pathol. 2004;28(8):1098-1102.
Dunnick NR, Hartman DS, et al. The radiology of juxtaglomerular tumors. Radiology. 1983;147(2):321-326.
Eble J. Angiomyolipoma of kidney. Semin Diagn Pathol. 1998;15(1):21-40.
Eble J, Amin M, et al. Epithelioid angiomyolipoma of the kidney: a report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol. 1997;21(10):1123-1130.
Eknoyan G. A clinical view of simple and complex renal cysts. J Am Soc Nephrol. 2009;20(9):1874-1876.
Fazeli-Matin S, Novick A. Nephron-sparing surgery for renal angiomyolipoma. Urology. 1998;52(4):577-583.
Ferda J, Hora M, et al. Computed tomography of renal cell carcinoma in patients with terminal renal impairment. Eur J Radiol. 2007;63(2):295-301.
Ferry JA, Malt RA, Young RH. Renal angiomyolipoma with sarcomatous transformation and pulmonary metastases. Am J Surg Pathol. 1991;15(11):1083-1088.
Glassman D, Chawla SN, et al. Correlation of pathology with tumor size of renal masses. Can J Urol. 2007;14(4):3616-3620.
Gögüs C, Safak M, Erekul S, et al. Angiomyolipoma of the kidney with lymph node involvement in a 17-year old female mimicking renal cell carcinoma: a case report. Int Urol Nephrol. 2001;33(4):617-618.
Green AJ, Sepp T, et al. Clonality of tuberous sclerosis hamatomas shown by non-random X-chromosome inactivation. Hum Genet. 1996;97(2):240-243.
Gudbjartsson T, Hardarson S, et al. Renal oncocytoma: a clinicopathological analysis of 45 consecutive cases. BJU Int. 2005;96(9):1275-1279.
Gupta R, Balzer B, et al. Diagnostic implications of transcription factor Pax 2 protein and transmembrane enzyme complex carbonic anhydrase IX immunoreactivity in adult renal epithelial neoplasms. Am J Surg Pathol. 2009;33(2):241-247.
Hadley DA, Bryant LJ, et al. Conservative treatment of renal angiomyolipomas in patients with tuberous sclerosis. Clin Nephrol. 2006;65(1):22-27.
Hafron J, Fogarty JD, et al. Imaging characteristics of minimal fat renal angiomyolipoma with histologic correlations. Urology. 2005;66(6):1155-1159.
Ham WS, Lee JH, et al. Comparison of multiple session 99% ethanol and single session OK-432 sclerotherapy for the treatment of simple renal cysts. J Urol. 2008;180(6):2552-2556.
Hamlin JA, Smith DC, Taylor FC, et al. Renal angiomyolipomas: long-term follow-up of embolization for acute hemorrhage. Can Assoc Radiol J. 1997;48(3):191-198.
Han YM, Kim JK, Roh BS, et al. Renal angiomyolipoma: selective arterial embolization—effectiveness and changes in angiomyogenic components in long-term follow-up. Radiology. 1997;204(1):65-70.
Harabayashi T, Shinohara N, et al. Management of renal angiomyolipomas associated with tuberous sclerosis complex. J Urol. 2004;171(1):102-105.
Haritharan T, Sritharan S, et al. Renal angiomyolipoma with inferior vena caval involvement. Med J Malaysia. 2006;61(4):493-495.
Harmon WJ, King BF, Lieber MM. Renal oncocytoma: magnetic resonance imaging characteristics. J Urol. 1996;155(3):863-867.
Hayn MH, Cannon GMJr, et al. Renal cell carcinoma containing fat without associated calcifications: two case reports and review of literature. Urology. 2009;73(2):443. e5–7
Henderson RJ, Germany R, Peavy PW, et al. Fat density in renal cell carcinoma: demonstration with computerized tomography. J Urol. 1997;157(4):1347-1348.
Hes O, Michal M, et al. Vimentin reactivity in renal oncocytoma: immunohistochemical study of 234 cases. Arch Pathol Lab Med. 2007;131(12):1782-1788.
Huang KH, Huang CY, et al. Malignant epithelioid angiomyolipoma of the kidney. J Formosan Med Assoc. 2007;106(2 Suppl.):S51-S54.
Hughes CR, Stewart PFJr, et al. Renal cyst rupture following blunt abdominal trauma: case report. J Trauma. 1995;38(1):28-29.
Ibraghimov-Beskrovnaya O, Bukanov N. Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci. 2008;65(4):605-619.
Ishikawa E, Kudo M, et al. Intracystic hemorrhage in a patient of polycystic kidney with renocolic fistula diagnosed by contrast-enhanced ultrasonography. Intern Med. 2008;47(22):1977-1979.
Israel GM, Bosniak MA. An update of the Bosniak renal cyst classification system. Urology. 2005;66(3):484-488.
Jain M, Rastogi A, et al. Atypical metanephric adenoma—a case report and review of literature. Int Urol Nephrol. 2007;39(1):123-127.
Jinzaki M, Tanimoto A, Narimatsu Y, et al. Angiomyolipoma: imaging findings in lesions with minimal fat. Radiology. 1997;205(2):497-502.
Jones EC, Pins M, Dickersin GR, Young RH. Metanephric adenoma of the kidney: a clinicopathological, immunohistochemical, flow cytometric, cytogenetic, and electron microscopic study of seven cases. Am J Surg Pathol. 1995;19(6):615-626.
Kato I, Inayama Y, et al. Epithelioid angiomyolipoma of the kidney. Pathol Int. 2009;59(1):38-43.
Kattar MM, Grignon DJ, et al. Chromosomal analysis of renal angiomyolipoma by comparative genomic hybridization: evidence for clonal origin. Hum Pathol. 1999;30(3):295-299.
Kehagias D, Mourikis D, et al. Management of renal angiomyolipoma by selective arterial embolization. Urol Int. 1998;60(2):113-117.
Kennelly M, Grossman H, et al. Outcome analysis of 42 cases of renal angiomyolipoma. J Urol. 1994;152(6 pt 1):1988-1991.
Kim JK, Park SY, et al. Angiomyolipoma with minimal fat: differentiation from renal cell carcinoma at biphasic helical CT [see comment]. Radiology. 2004;230(3):677-684.
Klein M, Valensi Q. Proximal tubular adenomas of kidney with so-called oncocytic features: a clinicopathologic study of 13 cases of a rarely reported neoplasm. Cancer. 1976;38(2):906-914.
Kohashi K, Oda Y, et al. Multifocal metanephric adenoma in childhood. Pathol Int. 2009;59(1):49-52.
Kovacs G, Akhtar M, et al. The Heidelberg classification of renal cell tumours. J Pathol. 1997;183(2):131-133.
Kummerlin I, ten Kate F, et al. Core biopsies of renal tumors: a study on diagnostic accuracy, interobserver, and intraobserver variability. Eur Urol. 2008;53(6):1219-1225.
Kuroda N, Tol M, Hiroi M, et al. Review of renal oncocytoma with focus on clinical and pathobiological aspects. Histol Histopathol. 2003;18(3):935-942.
Kuroda N, Tol M, et al. Review of metanephric adenoma of the kidney with focus on clinical and pathobiological aspects. Histol Histopathol. 2003;18(1):253-257.
Kutikov A, Fossett LK, et al. Incidence of benign pathologic findings at partial nephrectomy for solitary renal mass presumed to be renal cell carcinoma on preoperative imaging. Urology. 2006;68(4):737-740.
Kuzgunbay B, Turunc T, Bolat F, Kilinc F. Adult cystic nephroma: a case report and a review of the literature. Urol Oncol. 2009;27(4):407-409.
L’Hostis H, Deminiere C, et al. Renal angiomyolipoma: a clinicopathologic, immunohistochemical, and follow-up study of 46 cases. Am J Surg Pathol. 1999;23(9):1011-1020.
Lane BR, Aydin H, et al. Clinical correlates of renal angiomyolipoma subtypes in 209 patients: classic, fat poor, tuberous sclerosis associated and epithelioid. J Urol. 2008;180(3):836-843.
Lane BR, Babineau D, et al. A preoperative prognostic nomogram for solid enhancing renal tumors 7 cm or less amenable to partial nephrectomy. J Urol. 2007;178(2):429-434.
Lane BR, Campbell SC, et al. Adult cystic nephroma and mixed epithelial and stromal tumor of the kidney: clinical, radiographic, and pathologic characteristics. Urology. 2008;71(6):1142-1148.
Lane BR, Samplaski MK, et al. Renal mass biopsy—a renaissance? J Urol. 2008;179(1):20-27.
Lebret T, Poulain JE, et al. Percutaneous core biopsy for renal masses: indications, accuracy and results. J Urol. 2007;178(4 Pt. 1):1184-1188. discussion 1188
Lechpammer M, Resnick MB, et al. The diagnostic and prognostic utility of claudin expression in renal cell neoplasms. Mod Pathol. 2008;21(11):1320-1329.
Lemaitre L, Claudon M, Dubrulle F, Mazeman E. Imaging of angiomyolipomas. Semin Ultrasound CT MR. 1997;18(2):100-114.
Lemaitre L, Robert Y, Dubrulle F, et al. Renal angiomyolipoma: growth followed up with CT and/or US. Radiology. 1995;197(3):598-602.
Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169(5):1635-1642.
Lenton J, Kessel D, et al. Embolization of renal angiomyolipoma: immediate complications and long-term outcomes. Clin Radiol. 2008;63(8):864-870.
Leroy X, Lemaitre L, et al. Bilateral renal oncocytosis with renal failure. Arch Pathol Lab Med. 2001;125(5):683-685.
Leroy X, Moukassa D, Copin MC, et al. Utility of cytokeratin 7 for distinguishing chromophobe renal cell carcinoma from renal oncocytoma. Eur Urol. 2000;37(4):484-487.
Licht MR. Renal adenoma and oncocytoma. Semin Urol Oncol. 1995;13(4):262-266.
Licht MR, Novick AC, Tubbs RR, et al. Renal oncocytoma: clinical and biological correlates. J Urol. 1993;150(5):1380-1383.
Lieber MM. Renal oncocytoma. Urol Clin North Am. 1993;20(2):355-359.
Lieber MM, Hosaka Y, Tsukamoto T. Renal oncocytoma. World J Urol. 1987;5(2):71-79.
Ligato S, Ro JY, et al. Benign tumors and tumor-like lesions of the adult kidney. I. Benign renal epithelial neoplasms. Adv Anat Pathol. 1999;6(1):1-11.
Limaiem F, Mekni A, et al. Renal epithelioid angiomyolipoma: a case report and literature review. Pathologica. 2008;100(1):31-35.
Lin F, Yang W, et al. Expression of S-100 protein in renal cell neoplasms. Hum Pathol. 2006;37(4):462-470.
Lin WY, Chuang CK, et al. Renal angiomyolipoma with lymph node involvement: a case report and literature review. Chang Gung Medical J. 2003;26(8):607-610.
Lindgren V, Paner GP, et al. Cytogenetic analysis of a series of 13 renal oncocytomas. J Urol. 2004;171(2 Pt. 1):602-604.
Linehan W. Molecular targeting of VHL gene pathway in clear cell kidney cancer. J Urol. 2003;170(2 Pt. 1):593-594.
Littrup PJ, Ahmed A, et al. CT-guided percutaneous cryotherapy of renal masses. J Vasc Intervent Radiol. 2007;18(3):383-392.
Liu J, Fanning CV. Can renal oncocytomas be distinguished from renal cell carcinoma on fine-needle aspiration specimens? A study of conventional smears in conjunction with ancillary studies. Cancer. 2001;93(6):390-397.
Liu L, Qian J, et al. Immunohistochemical analysis of chromophobe renal cell carcinoma, renal oncocytoma, and clear cell carcinoma: an optimal and practical panel for differential diagnosis. Arch Pathol Lab Med. 2007;131(8):1290-1297.
Lopez-Beltran A, Scarpelli M, et al. 2004 WHO classification of the renal tumors of the adults. Eur Urol. 2006;49(5):798-805.
Madewell JE, Goldman SM, et al. Multilocular cystic nephroma: a radiographic-pathologic correlation of 58 patients. Radiology. 1983;146(2):309-321.
Mahoney CP, Cassady C, Weinberger E, et al. Humoral hypercalcemia due to an occult renal adenoma. Pediatr Nephrol. 1997;11(3):339-342.
Mai KT, Elkeilani A, et al. Mixed epithelial and stromal tumour (MEST) of the kidney: report of 14 cases with male and PEComatous variants and proposed histopathogenesis. Pathology. 2007;39(2):235-240.
Martignoni G, Pea M, et al. Renal angiomyolipoma with epithelioid sarcomatous transformation and metastases: demonstration of the same genetic defects in the primary and metastatic lesions. Am J Surg Pathol. 2000;24(6):889-894.
Martin SA, Mynderse LA, et al. Juxtaglomerular cell tumor: a clinicopathologic study of four cases and review of the literature. Am J Clin Pathol. 2001;116(6):854-863.
Matin S, Tamboli P, et al. Renal angiomyolipoma. In: Bukowski RM, Novick AC, editors. Clinical management of renal tumors. Totowa (NJ): Humana Press, 2008.
Matsuyama A, Hisaoka M, et al. Sclerosing variant of epithelioid angiomyolipoma. Pathol Int. 2008;58(5):306-310.
Menè P, Festuccia F, et al. Malignant epithelioid renal angiomyolipoma in a case of tuberous sclerosis with multiple organ involvement. Contrib Nephrol, 136, 2001, 299-305.
Milner J, McNeil B, et al. Fat poor renal angiomyolipoma: patient, computerized tomography and histological findings. J Urol. 2006;176(3):905-909.
Minervini A, Giubilei G, et al. Simple enucleation for the treatment of renal angiomyolipoma. BJU Int. 2007;99(4):887-891.
Minor LD, Picken MM, Campbell SC. Benign renal tumors. AUA Update. 2003;22:170-175.
Montironi R, Mazzucchelli R, et al. Cystic nephroma and mixed epithelial and stromal tumour of the kidney: opposite ends of the spectrum of the same entity? Eur Urol. 2008;54(6):1237-1246.
Morra MN, Das S. Renal oncocytoma: a review of histogenesis, histopathology, diagnosis and treatment. J Urol. 1993;150(2 Pt. 1):295-302.
Moudouni SM, Tligui M, et al. Malignant epithelioid renal angiomyolipoma involving the inferior vena cava in a patient with tuberous sclerosis. Urol Int. 2008;80(1):102-104. discussion 104
Mourikis D, Chatziioannou A, et al. Selective arterial embolization in the management of symptomatic renal angiomyolipomas. Eur J Radiol. 1999;32(3):153-159.
Muir TE, Cheville J, Lager DJ. Metanephric adenoma, nephrogenic rests, and Wilms’ tumor: a histologic and immunophenotypic comparison. Am J Surg Pathol. 2001;25(10):1290-1296.
Murphy AM, Buck AM, Benson MC, McKiernan JM. Increasing detection rate of benign renal tumors: evaluation of factors predicting for benign tumor histologic features during past two decades. Urology. 2009;73(6):1293-1297.
Nakagawa T, Kanai Y, et al. Malignant mixed epithelial and stromal tumours of the kidney: a report of the first two cases with a fatal clinical outcome. Histopathology. 2004;44(3):302-304.
Nelson C, Sanda M. Contemporary diagnosis and management of renal angiomyolipoma. J Urol. 2002;168(4 Pt. 1):1315-1325.
Neumann HP, Schwarzkopf G, Henske EP. Renal angiomyolipomas, cysts, and cancer in tuberous sclerosis complex. Semin Pediatr Neurol. 1998;5(4):269-275.
O’Brien A, Sinnott B, et al. Leiomyoma of the renal pelvis. Br J Urol. 1992;70(3):331-332.
Oesterling JE, Fishman EK, et al. The management of renal angiomyolipoma. J Urol. 1986;135(6):1121-1124.
Okumura A, Fuse H, et al. Laparoscopic ablation of peripelvic renal cysts. Int Urol Nephrol. 2003;35(3):307-310.
Olgac S, Hutchinson B, et al. Alpha-methylacyl-CoA racemase as a marker in the differential diagnosis of metanephric adenoma. Mod Pathol. 2006;19(2):218-224.
Omar AM, Khattak AQ, et al. Cystic renal cell carcinoma arising from multilocular cystic nephroma of the same kidney. Int Braz J Urol. 2006;32(2):187-189. discussion 189
Osunkoya AO, Cohen C, et al. Claudin-7 and claudin-8: immunohistochemical markers for the differential diagnosis of chromophobe renal cell carcinoma and renal oncocytoma. Hum Pathol. 2009;40(2):206-210.
Oxley JD, Sullivan J, et al. Metastatic renal oncocytoma. J Clin Pathol. 2007;60(6):720-722.
Pan CC, Chen PC, et al. Overexpression of KIT (CD117) in chromophobe renal cell carcinoma and renal oncocytoma. Am J Clin Pathol. 2004;121(6):878-883.
Paner GP, Lindgren V, et al. High incidence of chromosome 1 abnormalities in a series of 27 renal oncocytomas: cytogenetic and fluorescence in situ hybridization studies. Arch Pathol Lab Med. 2007;131(1):81-85.
Paner GP, Turk TM, et al. Passive seeding in metanephric adenoma: a review of pseudometastatic lesions in perinephric lymph nodes. Arch Pathol Lab Med. 2005;129(10):1317-1321.
Pappas P, Leonardou P, et al. Urgent superselective segmental renal artery embolization in the treatment of life-threatening renal hemorrhage. Urol Int. 2006;77(1):34-41.
Patard JJ. Incidental renal tumours. Curr Opin Urol. 2009;19(5):454-458.
Patel NP, Geisinger KR, et al. Fine needle aspiration biopsy of metanephric adenoma: a case report. Acta Cytol. 2009;53(3):327-331.
Pavlovich CP, Grubb RL3rd, Hurley K, et al. Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J Urol. 2005;173(5):1482-1486.
Pea M, Bonetti F, et al. Apparent renal cell carcinomas in tuberous sclerosis are heterogeneous: the identification of malignant epithelioid angiomyolipoma. Am J Surg Pathol. 1998;22(2):180-187.
Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med. 2001;7(4):151-156.
Pei Y. Molecular genetics of autosomal dominant polycystic kidney disease. Clin Invest Med. 2003;26(5):252-258.
Perez-Ordonez B, Hamed G, Campbell S, et al. Renal oncocytoma: a clinicopathologic study of 70 cases. Am J Surg Pathol. 1997;21(8):871-883.
Pesti T, Sükösd F, Jones EC, Kovacs G. Mapping a tumor suppressor gene to chromosome 2p13 in metanephric adenoma by microsatellite allelotyping. Hum Pathol. 2001;32(1):101-104.
Picken MM, Curry JL, Lindgren V, et al. Metanephric adenosarcoma in a young adult: morphologic, immunophenotypic, ultrastructural, and fluorescence in situ hybridization analyses: a case report and review of the literature. Am J Surg Pathol. 2001;25(11):1451-1457.
Pierson CR, Schober MS, et al. Mixed epithelial and stromal tumor of the kidney lacks the genetic alterations of cellular congenital mesoblastic nephroma. Hum Pathol. 2001;32(5):513-520.
Polascik TJ, Bostwick DG, Cairns P. Molecular genetics and histopathologic features of adult distal nephron tumors. Urology. 2002;60(6):941-946.
Porpiglia F, Fiori C, et al. Retroperitoneal decortication of simple renal cysts vs decortication with wadding using perirenal fat tissue: results of a prospective randomized trial. BJU Int. 2009;103(11):1532-1536.
Prevoo W, van den Bosch MA, et al. Radiofrequency ablation for treatment of sporadic angiomyolipoma. Urology. 2008;72(1):188-191.
Qian F, Watnick TJ, et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979-987.
Quaia E, Bertolotto M, et al. Comparison of contrast-enhanced sonography with unenhanced sonography and contrast-enhanced CT in the diagnosis of malignancy in complex cystic renal masses. AJR Am J Roentgenol. 2008;191(4):1239-1249.
Rainio J, De Giorgio F, et al. Death from renal cyst: spontaneous or traumatic rupture? Am J Forensic Med Pathol. 2006;27(2):193-195.
Raj GV, Bach AM, et al. Predicting the histology of renal masses using preoperative Doppler ultrasonography. J Urol. 2007;177(1):53-58.
Raj GV, Yowell C, et al. Malignant cystic nephroma. Can J Urol. 2006;13(6):3348-3350.
Rakowski SK, Winterkorn EB, et al. Renal manifestations of tuberous sclerosis complex: incidence, prognosis, and predictive factors. Kidney Int. 2006;70(10):1777-1782.
Raman JD, Hall DW, et al. Renal ablative therapy: radiofrequency ablation and cryoablation. J Surg Oncol. 2009;100(8):639-644.
Rao VS, Naik R, et al. Leiomyoma of the kidney—report of two cases. Indian J Pathol Microbiol. 2001;44(3):343-344.
Renshaw A. Subclassification of renal cell neoplasms: an update for the practicing pathologist. Histopathology. 2002;41(4):283-300.
Rocca PC, Brunelli M, et al. Diagnostic utility of S100A1 expression in renal cell neoplasms: an immunohistochemical and quantitative RT-PCR study. Mod Pathol. 2007;20(7):722-728.
Romero FR, Kohanim S, et al. Leiomyomas of the kidney: emphasis on conservative diagnosis and treatment. Urology. 2005;66(6):1319.
Romis L, Cindolo L, et al. Frequency, clinical presentation and evolution of renal oncocytomas: multicentric experience from a European database. Eur Urol. 2004;45(1):53-57. discussion 57
Roy C, Tuchmann C, Lindner V, et al. Renal cell carcinoma with a fatty component mimicking angiomyolipoma on CT. Br J Radiol. 1998;71(849):977-979.
Rubenstein JN, Eggener SE, et al. Juxtaglomerular apparatus tumor: a rare, surgically correctable cause of hypertension. Rev Urol. 2002;4(4):192-195.
Saito K, Fujii Y, Kasahara I, et al. Malignant clear cell “sugar” tumor of the kidney: clear cell variant of epithelioid angiomyolipoma. J Urol. 2002;168(6):2533-2534.
Sanfilippo F, Pizzo WV, et al. Immunohistochemical studies of cell differentiation in a juxtaglomerular tumor. Arch Pathol Lab Med. 1982;106(12):604-607.
Schade GR, Gofrit ON, et al. Renal angiomyolipoma with intravascular extension into the inferior vena cava: a case report and review of the literature. Can J Urol. 2008;15(2):4012-4015.
Schmidbauer J, Remzi M, et al. Diagnostic accuracy of computed tomography-guided percutaneous biopsy of renal masses. Eur Urol. 2008;53(5):1003-1011.
Schonfeld AD, Jackson JA, et al. Renin-secreting juxtaglomerular tumor causing severe hypertension: diagnosis by computerized tomography-directed needle biopsy. J Urol. 1991;146(6):1607-1609.
Schuetz AN, Yin-Goen Q, et al. Molecular classification of renal tumors by gene expression profiling. J Mol Diagn. 2005;7(2):206-218.
Schwerdtle RF, Winterpacht A, Störkel S, et al. Loss of heterozygosity studies and deletion mapping identify two putative chromosome 14q tumor suppressor loci in renal oncocytomas. Cancer Res. 1997;57(22):5009-5012.
Sepp T, Yates JR, et al. Loss of heterozygosity in tuberous sclerosis hamartomas. J Med Genet. 1996;33(11):962-964.
Seyam RM, Bissada NK, et al. Changing trends in presentation, diagnosis and management of renal angiomyolipoma: comparison of sporadic and tuberous sclerosis complex-associated forms. Urology. 2008;72(5):1077-1082.
Shah RB, Bakshi N, et al. Image-guided biopsy in the evaluation of renal mass lesions in contemporary urological practice: indications, adequacy, clinical impact, and limitations of the pathological diagnosis. Hum Pathol. 2005;36(12):1309-1315.
Siegel CL, Middleton WD, Teefey SA, McClennan BL. Angiomyolipoma and renal cell carcinoma: US differentiation. Radiology. 1996;198(3):789-793.
Silverman SG, Mortele KJ, et al. Hyperattenuating renal masses: etiologies, pathogenesis, and imaging evaluation. RadioGraphics. 2007;27(4):1131-1143.
Simpfendorfer C, Herts BR, et al. Angiomyolipoma with minimal fat on MDCT: can counts of negative-attenuation pixels aid diagnosis? AJR Am J Roentgenol. 2009;192(2):438-443.
Siu W, Hafez KS, et al. Growth rates of renal cell carcinoma and oncocytoma under surveillance are similar. Urol Oncol. 2007;25(2):115-119.
Skinnider BF, Folpe AL, et al. Distribution of cytokeratins and vimentin in adult renal neoplasms and normal renal tissue: potential utility of a cytokeratin antibody panel in the differential diagnosis of renal tumors [see comment]. Am J Surg Pathol. 2005;29(6):747-754.
Skolarus TA, Serrano MF, et al. The distribution of histological subtypes of renal tumors by decade of life using the 2004 WHO classification. J Urol. 2008;179(2):439-443. discussion 443–4
Snyder ME, Bach A, et al. Incidence of benign lesions for clinically localized renal masses smaller than 7 cm in radiological diameter: influence of sex. J Urol. 2006;176(6 Pt. 1):2391-2395. discussion 2395–6.
Stamatiou K, Polizois K, et al. Cystic nephroma: a case report and review of the literature. Cases J. 2008;1(1):267.
Steiner MS, Goldman SM, Fishman EK, Marshall FF. The natural history of renal angiomyolipoma. Urology. 1993;150(6):1782-1786.
Steiner MS, Quinlan D, et al. Leiomyoma of the kidney: presentation of 4 new cases and the role of computerized tomography. J Urol. 1990;143(5):994-998.
Tamboli P, Ro JY, Amin MB, et al. Benign tumors and tumor-like lesions of the adult kidney. II. Benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
Tanabe A, Naruse M, et al. Dynamic computer tomography is useful in the differential diagnosis of juxtaglomerular cell tumor and renal cell carcinoma. Hypertens Res. 2001;24(4):331-336.
Terada N, Arai Y, et al. Risk factors for renal cysts. BJU Int. 2004;93(9):1300-1302.
Terada N, Arai Y, et al. The 10-year natural history of simple renal cysts. Urology. 2008;71(1):7-11. discussion 11–12
Terada N, Ichioka K, et al. The natural history of simple renal cysts. J Urol. 2002;167(1):21-23.
Thoenes W, Storkel S, et al. Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas): the basic cytological and histopathological elements and their use for diagnostics. Pathol Res Pract. 1986;181(2):125-143.
Tickoo SK, Reuter VE, Amin MB, et al. Renal oncocytosis: a morphologic study of fourteen cases. Am J Surg Pathol. 1999;23(9):1094-1101.
Toro JR, Wei MH, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45(6):321-331.
Truong LD, Choi YJ, et al. Renal cystic neoplasms and renal neoplasms associated with cystic renal diseases: pathogenetic and molecular links. Adv Anat Pathol. 2003;10(3):135-159.
Turbiner J, Amin MB, et al. Cystic nephroma and mixed epithelial and stromal tumor of kidney: a detailed clinicopathologic analysis of 34 cases and proposal for renal epithelial and stromal tumor (REST) as a unifying term. Am J Surg Pathol. 2007;31(4):489-500.
Türker Köksal I, Tunç M, Kiliçaslan I, et al. Lymph nodal involvement by renal angiomyolipoma. Int J Urol. 2000;7(10):386-389.
Upadhyay AK, Neely JA. Cystic nephroma: an emerging entity. Ann R Coll Surg Engl. 1989;71(6):381-383.
Vaidyanathan S, Hughes PL, et al. Spontaneous rupture of an infected renal cyst and external drainage through a lumbar surgical scar in a male patient with cervical spinal cord injury: a case report. J Med Case Reports. 2008;2:154.
Volpe A, Kachura JR, et al. Techniques, safety and accuracy of sampling of renal tumors by fine needle aspiration and core biopsy. J Urol. 2007;178(2):379-386.
Vujanic GM, Jenney ME, et al. Juxtaposed cystic nephroma and Wilms’ tumor. Pediatr Dev Pathol. 2000;3(1):91-94.
Wang KL, Weinrach DM, et al. Renal papillary adenoma—a putative precursor of papillary renal cell carcinoma. Hum Pathol. 2007;38(2):239-246.
Wang LJ, Wong YC, Chen CJ, See LC. Computerized tomography characteristics that differentiate angiomyolipomas from liposarcomas in the perinephric space. J Urol. 2002;167(2 Pt. 1):490-493.
Warren KS, McFarlane J. The Bosniak classification of renal cystic masses. BJU Int. 2005;95(7):939-942.
Weimbs T. Polycystic kidney disease and renal injury repair: common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Renal Physiol. 2007;293(5):F1423-F1432.
Weirich G, Glenn G, Junker K, et al. Familial renal oncocytoma: clinicopathological study of 5 families. J Urol. 1998;160(2):335-340.
Weiss LM, Gelb AB, Medeiros LJ. Adult renal epithelial neoplasms. Am J Clin Pathol. 1995;103(5):624-635.
Wells FC, Naylor CP, et al. Leiomyoma of the renal vein. J R Soc Med. 1981;74(7):542-545.
Wong L, Hsu TH, et al. Reninoma: case report and literature review. J Hypertens. 2008;26(2):368-373.
Yamakado K, Tanaka N, et al. Renal angiomyolipoma: relationships between tumor size, aneurysm formation, and rupture. Radiology. 2002;225(1):78-82.
Yang XJ, Sugimura J, et al. Classification of renal neoplasms based on molecular signatures. J Urol. 2006;175(6):2302-2306.
Yoshioka K, Miyakawa A, et al. Production of erythropoietin and multiple cytokines by metanephric adenoma results in erythrocytosis. Pathol Int. 2007;57(8):529-536.
Zanelli M, Cortecchia S, et al. Epithelioid angiomyolipoma of the kidney: case report. Pathologica. 2008;100(3):202-205.
Zerem E, Imamovic G, et al. Simple renal cysts and arterial hypertension: does their evacuation decrease the blood pressure? J Hypertens. 2009;27(10):2074-2078.
Zhang JQ, Fielding JR, Zou KH. Etiology of spontaneous perirenal hemorrhage: a meta-analysis. J Urol. 2003;167(4):1593-1596.
Zhou M, Kort E, et al. Adult cystic nephroma and mixed epithelial and stromal tumor of the kidney are the same disease entity: molecular and histologic evidence. Am J Surg Pathol. 2009;33(1):72-80.