chapter 49 Malignant Renal Tumors
Historical Considerations
The introduction of nephrectomy and other subsequent surgical interventions for renal diseases provided the clinical information and histopathologic insight that form the basis of current concepts regarding renal tumors. The first documented nephrectomy was accomplished in 1861 by Wolcott, who operated with the mistaken assumption that the tumor mass was a hepatoma. In 1867, Spiegelberg removed a kidney incidentally in the course of excising an echinococcus cyst. The first planned nephrectomy was performed by Simon in 1869 for persistent ureteral fistula, and this patient survived with cure of the fistula. One year later (1870), the first planned nephrectomy in the United States was successfully accomplished by Gilmore in Mobile, Alabama, for treatment of atrophic pyelonephritis and persistent urinary tract infection (Glenn, 1980; Herr, 2008). Harris (1882) subsequently reported on 100 surgical extirpations of the kidney, a sufficient number to permit analysis of clinical, surgical, and pathologic features of renal disorders that require surgery.
With surgical intervention, tissue became available to pathologists for histologic interpretation. Unfortunately, such interpretation was not always accurate, and there were often serious professional differences of opinion. According to Carson (1928), the first accurate gross description of kidney tumors dates to 1826, with Konig’s observations. In 1855, Robin examined solid tumors apparently arising in the kidney and concluded that renal carcinoma arose from renal tubular epithelium. This interpretation was confirmed by Waldeyer in 1867. Unfortunately, theoretical and practical considerations of renal tumors were confused by Grawitz (1883), who contended that such apparent renal tumors arose from adrenal rests within the kidney. He introduced the terminology struma lipomatodes aberrata renis as descriptive nomenclature for the tumors of clear cells that he believed were derived from the adrenal glands. He based his conclusions not only on the fatty content of the tumors, analogous to that seen in the adrenal glands, but also on the location of the tumors beneath the renal capsule, the approximation to the adrenal glands, the lack of similarity of the cells to uriniferous tubules, and the demonstration of amyloid similar to that seen with adrenal degeneration.
This histogenetic concept was adopted by subsequent investigators, and pathologists of the era readily embraced the idea that renal tumors truly arose from the adrenal glands. In 1894, Lubarch endorsed the idea of a suprarenal origin of renal tumors, and the term hypernephroid tumors, indicating origin above the kidneys, was advocated by Birch-Hirschfeld (Birch-Hirschfeld and Doederlein, 1894). This semantic and conceptual mistake led to the introduction of the term hypernephroma, which predominated in the literature describing parenchymal tumors of primary renal origin. Some clarification of the histopathology of renal tumors was derived from the work of Albarran and Imbert (1903), and the four-volume contribution of Wolff (1883), written between 1883 and 1928, added further historical significance to the understanding of renal tumors (Glenn, 1980).
The modern era has brought an appreciation that renal cell carcinoma (RCC) includes a number of distinct subtypes derived from the various parts of the nephron, each with a unique genetic basis and tumor biology (Linehan et al, 2003, Cohen and McGovern, 2005; Novick, 2007; Rini et al, 2009). Other major advances in the past several decades have included the introduction of radical nephrectomy followed by a trend toward nephron-sparing surgery and, more recently, a variety of minimally invasive approaches (Robson, 1963; Uzzo and Novick, 2001; Abreu and Gill, 2003; Herr, 2005, 2008; Novick, 2007). One common theme that has persisted is that RCC remains primarily a surgical disease—it is still considered the paradigm of the chemorefractory tumor; and although immune-based and targeted molecular approaches have shown promise, overall response rates remain low (Négrier et al, 2002; Milowsky and Nanus, 2003; Escudier et al, 2007; Motzer et al, 2007; Rini et al, 2009). Unfortunately, the incidence of RCC is gradually increasing; and despite a trend toward earlier detection, mortality rates remain high.
Classification
Renal masses can be malignant, benign, or inflammatory as classified by Barbaric (1994) (Table 49–1), or they can be classified based on radiographic appearance (simple cystic, complex cystic, fatty tumors, and others) (Table 49–2). These classification schemes have been updated taking into account current knowledge about the distinct subtypes of RCC and recent advances in our understanding of the various benign and malignant tumors of the kidney (Eble et al, 2004; Cohen and McGovern, 2005; Rini et al, 2009). This approach is practical and should assist in the differential diagnosis of renal masses. Malignant renal tumors include RCC, urothelial-based lesions (addressed elsewhere), sarcomas, embryonic or pediatric tumors, and lymphomas and metastases. Benign renal tumors are diverse and present unique diagnostic challenges (see Chapter 51). Inflammatory and vascular lesions must also be considered in the differential diagnosis.
Table 49–1 Renal Masses Classified by Pathologic Features
| MALIGNANT | BENIGN | INFLAMMATORY |
|---|---|---|
Radiographic Evaluation of Renal Masses
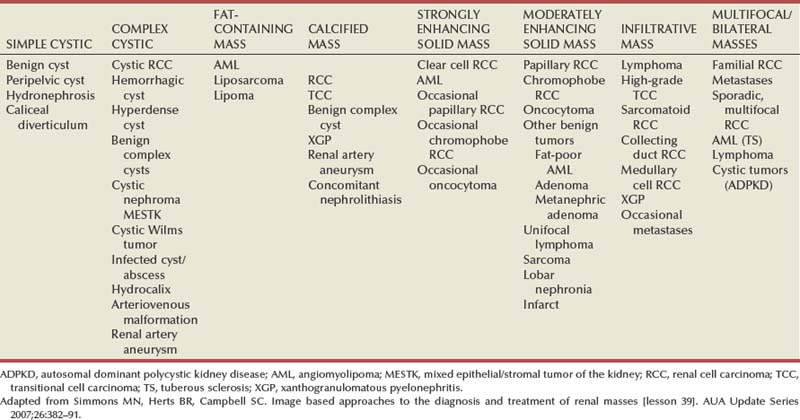
Several radiographic modalities are currently available for detection and evaluation of renal masses, each with relative strengths and limitations (Davidson et al, 1997; Zagoria, 2000; Israel and Bosniak, 2003a; Zhang et al, 2007a; Herts, 2009). A systematic approach is necessary to ensure diligent evaluation of suspected renal masses, given the large differential diagnosis and considerable overlap between benign and malignant renal lesions (Fig. 49–1; see Table 49–2) (Ananthakrishnan et al, 2007; Simmons et al, 2007).
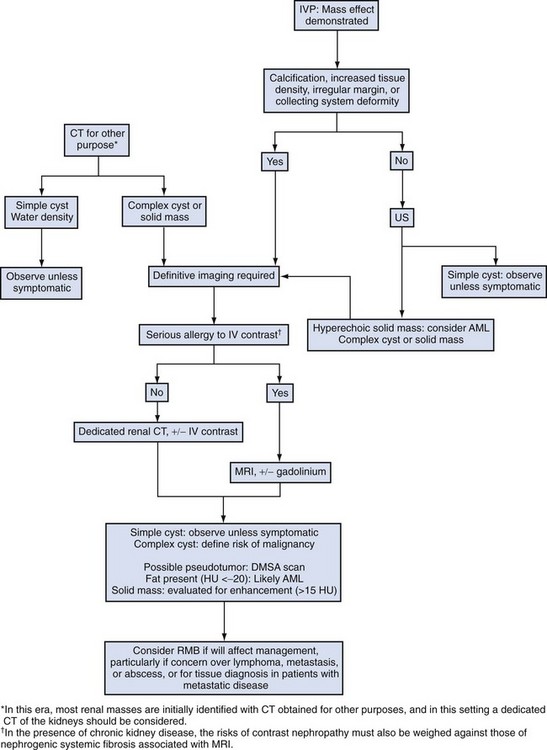
Figure 49–1 Algorithm for radiographic evaluation of renal masses. IVP, intravenous pyelography; US, ultrasonography; AML, angiomyolipoma; CT, computed tomography; DMSA, dimercaptosuccinic acid; MRI, magnetic resonance imaging; RMB, renal mass biopsy.
Although intravenous pyelography was often the first test that indicated a renal mass in the past, it is now only occasionally used for the evaluation of hematuria. The lack of sensitivity and specificity of intravenous pyelography for the detection of parenchyma tumors is well documented. In particular, intravenous pyelography may miss small anterior or posterior lesions that do not distort the collecting system or the contour of the kidney. Features suggestive of malignancy on intravenous pyelography include calcification within the mass, increased tissue density, irregularity of the margin, and distortion of the collecting system (Zagoria, 2000).
When a renal mass is identified by intravenous pyelography, unless the mass has features suggestive of malignancy, ultrasonography should be the next study performed because it is noninvasive, accurate, and relatively inexpensive (Davidson et al, 1997; Paspulati and Bhatt, 2006). Ultrasonography is reliable for differentiation of solid tissue from fluid and can establish the diagnosis of a simple renal cyst. Strict ultrasonographic criteria for simple cysts have been defined and include a smooth cyst wall, a round or oval shape without internal echoes, and through-transmission with strong acoustic shadows posteriorly. If these criteria are met, observation is sufficient in an asymptomatic patient.
In evaluating complicated renal cysts, important ultrasonographic features include thickness and contour of the cyst wall, number and thickness of any septa, presence of any calcifications, density of the renal cyst fluid, and presence of solid components. ultrasonography is helpful in suggesting the fat content of an angiomyolipoma (AML) by its characteristic increased echogenicity (Nelson and Sanda, 2002). A renal mass that is not clearly a simple cyst by strict ultrasound criteria should be evaluated further with computed tomography (CT).
A dedicated (thin-slice) renal CT scan remains the single most important radiographic test for delineating the nature of a renal mass. CT, with and without the administration of contrast material, is necessary to take full advantage of the contrast enhancement characteristics of highly vascular renal parenchymal tumors (Davidson et al, 1997; Zagoria, 2000, Prasad et al, 2008; Ng et al, 2008; Zhang et al, 2007a). In general, any renal mass that enhances with intravenous administration of contrast material on CT by more than 15 Hounsfield units (HU) should be considered an RCC until proved otherwise (Fig. 49–2) (Hartman et al, 2004). Solid masses that also have substantial areas of negative CT attenuation numbers (below −20 HU) indicative of fat are diagnostic of AMLs (Nelson and Sanda, 2002). In 10% to 20% of solid renal masses CT findings are indeterminate, and additional testing or surgical exploration is needed to establish a definitive diagnosis. On occasion, CT demonstrates an enhancing renal segment that is isodense with the remainder of the kidney, suggestive of a renal pseudotumor. Renal pseudotumors may be due to a hypertrophied column of Bertin, renal dysmorphism, or an unusually shaped kidney (Bhatt et al, 2007). In this situation, the diagnosis of a pseudotumor can be confirmed by isotope renography with technetium-labeled dimercaptosuccinic acid or glucoheptonate (Fig. 49–3). These isotope studies demonstrate an area of increased density if the mass is a pseudotumor and an area of decreased density if the mass is a cyst or solid tumor (Israel and Bosniak, 2003a).
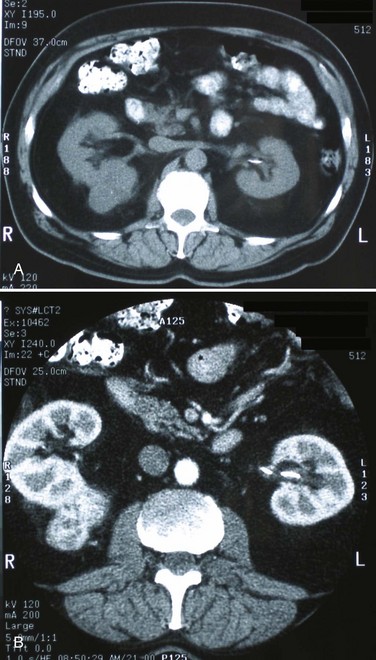
Figure 49–2 A, Unenhanced CT scan shows solid, right posterior renal mass. B, After administration of the contrast agent, CT scan shows that the mass enhances more than 20 HU and is thus highly suggestive of RCC. This mass was excised and confirmed to be a clear cell renal cell carcinoma.
(Courtesy of Dr. Terrence Demos, Maywood, IL.)
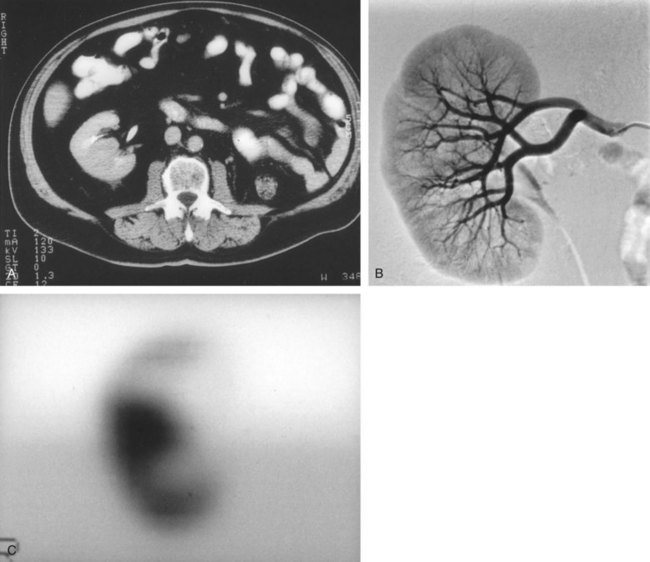
Figure 49–3 A, CT scan after administration of contrast material shows isodense hilar mass in solitary right kidney. B, Right renal arteriography shows no neovascularity. C, Glucoheptonate isotope renal scan shows increased density of mass indicative of hypertrophied column of Bertin.
Magnetic resonance imaging (MRI) is the alternate standard imaging modality for the characterization of a renal mass (Pretorius et al, 2000; Zhang et al, 2004; Bassignani, 2006). A basic consideration in the evaluation of a renal mass is that for such a mass to be considered malignant it must enhance with the intravenous administration of contrast material. Such enhancement can now be determined equally well by magnetic resonance angiography with intravenous gadolinium-labeled diethylenetriaminepentaacetic acid, although the assessment is qualitative rather than quantitative. On T1-weighted scans before and after administration of gadolinium, enhancement (vascularity) of the mass is detected (Fig. 49–4). This technique is most helpful in patients for whom iodinated contrast medium is contraindicated because of severe allergy. One concern with MRI with gadolinium is the uncommon but potentially serious complication of nephrogenic systemic fibrosis (NSF), which is more common in patients with renal insufficiency (Bach and Zhang, 2008). Current recommendations are to avoid MRI, particularly serial studies, in this population whenever possible, and to dialyze patients after the study if severe chronic kidney disease (CKD) is present. MRI was previously the imaging procedure of choice in patients with CKD and a renal mass. Now many radiologists prefer CT with intravenous contrast and careful periprocedural hydration, but decision making must be individualized. Contrast-enhanced ultrasonography using microbubbles has also shown promise for the safe characterization and assessment of enhancement of renal masses and may play an important role in patients with CKD in the future (Simmons et al, 2007).
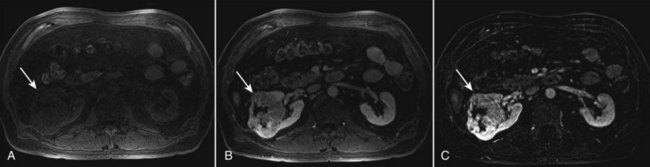
Figure 49–4 Unenhanced and contrast-enhanced MR images of renal cell carcinoma (arrows). A, Unenhanced fat-saturated T1-weighted volume-interpolated breath-hold examination (VIBE) image. B, Contrast-enhanced VIBE image. C, Subtraction image generated by electronically subtracting image A from image B shows a large enhancing mass in the lateral anterior right kidney that is partially exophytic and extends to the central sinus. Although a subjective assessment of enhancement is often possible after contrast administration (B), any signal intensity remaining on the subtraction image (C) represents enhancement after contrast administration.
(Courtesy of Dr. Brian Herts, Cleveland, OH.)
Renal arteriography has a limited role in the diagnostic evaluation of renal masses and is primarily reserved for patients with concomitant renal artery disease. In equivocal cases, the presence or absence of neovascularity may help establish the diagnosis of RCC. However, 20% to 25% of RCCs are angiographically indistinct, even though most of these tumors are not truly avascular and demonstrate contrast enhancement by 10 to 25 HU on CT.
Renal mass biopsy is now being revisited for the evaluation of renal masses (Wunderlich et al, 2005; Lane et al, 2007b; Maturen et al, 2007; Volpe et al, 2007; Lebret et al, 2007; Strope and Wolf, 2008; Wang et al, 2009). Historically the false-negative rate of renal mass biopsy was thought to be 18%, too high to justify routine use. However, most of these “false negatives” were in reality instances in which the mass could not be adequately targeted or the material obtained was insufficient for the pathologist to make a definitive determination. Review of this literature has shown that, although about 15% of renal mass biopsy specimens are nondiagnostic, the real false-negative rate was only 4% in studies prior to 2001 and less than 1% since then (Lane et al, 2007b). Overall accuracy is greater than 80%. Assessment of tumor grade and histologic type, which reflects tumor aggressiveness, is also accurate in the majority of cases (Barocas et al, 2006; Schmidbauer et al, 2008). The risks of clinically significant perinephric bleeding and pneumothorax also appear to be low (<1%), and needle tract seeding is exceedingly rare when centrally located, infiltrative renal masses are excluded. Poorly differentiated transitional cell carcinoma is much higher risk for needle tract seeding than RCC. Given the great heterogeneity in the tumor biology of enhancing clinical T1 renal masses, with 20% benign, 60% indolent RCC, and 20% potentially aggressive RCC, renal mass biopsy is now being considered more frequently, particularly in patients who are potential candidates for a wide variety of treatment options ranging from observation to surgical excision. Younger, healthy patients who are unwilling to accept the uncertainty associated with renal mass biopsy are still typically managed primarily based on radiographic and clinical considerations. More traditional indications for renal mass biopsy include suspicion of renal abscess or when RCC must be differentiated from metastatic malignant disease or renal lymphoma (Herts and Remer, 2000; Anderson et al, 2006; Vasudevan et al, 2006; Beland et al, 2007; Kummerlin et al, 2008a; Somani et al, 2007; Shannon et al, 2008; Volpe et al, 2008).
The differentiation between a benign renal cyst and a cystic RCC remains one of the more common and difficult problems in renal imaging (Balci et al, 1999; Harada et al, 2002; Kausik et al, 2002; Israel and Bosniak, 2005; Warren and McFarlane, 2005). When a complex renal cyst is identified, determination of its benign or malignant nature is based on evaluation of the wall of the lesion; its thickness and contour; the number, contour, and thickness of any septa; the amount, character, and location of any calcifications; the density of fluid in the lesion; the margination of the lesion; and the presence of solid components. Bosniak developed a useful classification scheme primarily based on CT imaging criteria that divides renal cystic lesions into categories that are distinct from one another in terms of the likelihood of malignancy (Bosniak, 1997; Israel and Bosniak, 2005). Category I lesions are uncomplicated, simple, benign cysts of the kidney that are straightforward to diagnose on ultrasonography, CT, or MRI. These are by far the most common renal cystic lesions, and in the absence of associated symptoms, no treatment is necessary.
Category II lesions are minimally complex cysts that are generally benign but have some radiologic findings that cause concern (Fig. 49–5). These lesions include septated cysts, cysts with calcium in the wall or septum, infected cysts, and hyperdense (high-density) cysts (Bosniak, 1997; Israel and Bosniak, 2005). Hyperdense cysts are benign lesions that contain old, degenerated, or clotted blood; therefore, the CT attenuation of their contents is increased (>20 HU). Classic hyperdense renal cysts are small (<3 cm), round, and sharply marginated and do not enhance after the administration of contrast material (Fig. 49–6). This category has now been subdivided to differentiate category II lesions that do not require surveillance from category IIF lesions that mandate surveillance. The nuances involved in this classification are highlighted in Table 49–3. High-quality imaging, preferably CT, and considerable radiologic expertise are required to optimize the characterization of complex renal cystic lesions. The risk of malignancy for category IIF renal cysts is 5% to 10%, and these lesions should be observed with periodic renal imaging (Kausik, 2002; Israel and Bosniak, 2003b, 2005).
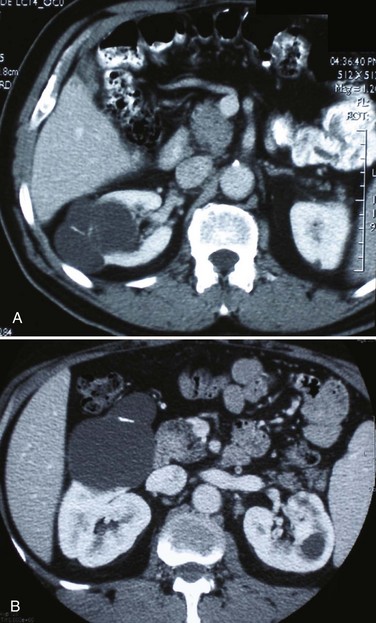
Figure 49–5 Bosniak class II renal cysts. A, CT scan shows right renal cyst with thin internal septation. B, CT scan in another patient shows relatively thin, curvilinear calcification in the septa of the wall of right renal cyst.
(Courtesy of Dr. Terrence Demos, Maywood, IL.)
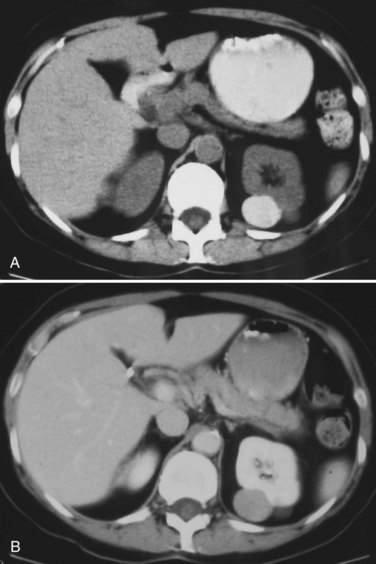
Figure 49–6 Bosniak class II hyperdense cyst. A, Unenhanced CT scan shows small, smooth-walled, high-density left renal cyst. B, CT scan after administration of contrast material shows no enhancement of the cyst. This is an extreme example of a hyperdense cyst.
(Courtesy of Dr. Terrence Demos, Maywood, IL.)

Category III lesions are more complex renal cysts that cannot be confidently distinguished from malignant neoplasms (Kausik, 2002; Israel and Bosniak, 2005). The radiographic features include thickened irregular or smooth walls or septa in which measurable enhancement can be observed (Fig. 49–7). In the absence of a mitigating factor such as renal trauma or infection, surgical exploration is usually indicated in healthy patients. About 50% of these lesions are malignant; the remainder prove to be benign multiloculated, hemorrhagic, or densely calcified cysts (see Table 49–2). Fine-needle aspiration of complex cysts is rarely performed because of concern about sampling error and tumor cell spillage.
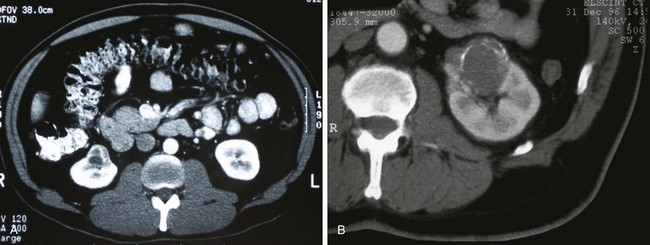
Figure 49–7 Bosniak class III cysts. A, CT scan shows complex right renal cyst with thick and irregular septa and inhomogeneous character. B, CT scan shows somewhat thick-walled, complex left renal cyst also exhibiting irregular calcification and moderate heterogeneity.
(Courtesy of Dr. Terrence Demos, Maywood, IL.)
Category IV lesions have large cystic components; irregular, shaggy margins; and, most important, solid enhancing portions that provide a definitive diagnosis of malignancy (Fig. 49–8) (Bosniak, 1997; Israel and Bosniak, 2003c, 2005). Category IV lesions are almost invariably cystic RCCs that, if localized, require surgical treatment.
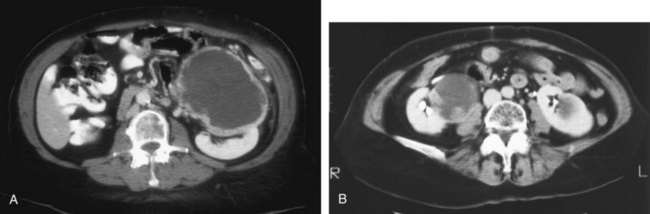
Figure 49–8 Bosniak class IV cysts. A, CT scan shows complex left renal cystic lesion with thick, enhancing walls. B, CT scan shows complex right cystic lesion with enhancing nodular areas and inhomogeneity. Both lesions proved to be renal cell carcinoma.
For radiographically detected solid renal masses, the differential diagnosis is extensive and includes conditions such as RCC, oncocytoma, AML, transitional cell carcinoma, metastatic tumor, abscess, infarct, vascular malformation, and renal pseudotumor (see Table 49–2). The diagnosis of most of these lesions can be established on the basis of the clinical presentation and the characteristic radiographic features (Table 49–4), occasionally combined with endourologic studies or needle biopsy of the mass (Simmons et al, 2007; Dyer et al, 2008). However, it is not possible to reliably distinguish RCC from benign renal neoplasms, including oncocytoma and fat-poor AML, with current diagnostic techniques. Ten to 20 percent of small, solid, CT-enhancing renal masses with features suggestive of RCC prove to be benign after surgical excision (Silver et al, 1997). Although oncocytoma is a benign tumor (see Chapter 51), it can be multifocal and is occasionally associated with RCC in the same or the opposite kidney (Licht, 1995; Dechet, 1999).
Renal Cell Carcinoma
Incidence
RCC, which accounts for 2% to 3% of all adult malignant neoplasms, is the most lethal of the common urologic cancers. Traditionally, 30% to 40% of patients with RCC have died of their cancer, in contrast to the 20% mortality rates associated with prostate and bladder carcinomas (Landis et al, 1999; Pantuck et al, 2001b). Approximately 54,000 new diagnoses of RCC are made each year in the United States, and 13,000 patients die of disease (Russo et al, 2008, Carrizosa and Godley, 2009; Jemal et al, 2009). Overall, approximately 12 new cases are diagnosed per 100,000 population per year, with a male-to-female predominance of 3 : 2 (Landis et al, 1999; Wallen et al, 2007; DeCastro and McKiernon, 2008; Woldrich et al, 2008; Carrizosa and Godley, 2009). This is primarily a disease of the elderly patient, with typical presentation in the sixth and seventh decades of life (Pantuck et al, 2001b; Wallen et al, 2007). Incidence rates are 10% to 20% higher in African Americans for unknown reasons (Chow et al, 1999, Lipworth et al, 2006; Stafford et al, 2008). The majority of cases of RCC are believed to be sporadic; only 2% to 3% are familial (Lipworth et al, 2006).
The incidence of RCC has increased since the 1970s by an average of 3% per year for whites and 4% per year for African-Americans, largely related to the more prevalent use of ultrasonography and CT for the evaluation of a variety of abdominal complaints (Chow et al, 1999; DeCastro and McKiernon, 2008; Kummerlin et al, 2008b). This trend has correlated with an increased proportion of incidentally discovered and localized tumors and with improved 5-year survival rates for patients with this stage of disease (Konnak and Grossman, 1985; Thompson and Peek, 1988; Kessler et al, 1994; Pantuck et al, 2001b; Parsons et al, 2001, Kane et al, 2008). However, other factors must also be at play because Chow and colleagues (1999) have documented a steadily increasing mortality rate from RCC per unit population since the 1980s, and this was observed in all ethnic and both sex groups. They reported that the incidence of advanced tumors per unit population has also increased; and although the proportion of advanced tumors has decreased, the mortality rate per unit population has still been negatively affected (Chow et al, 1999; Hock et al, 2002; Wallen et al, 2007; DeCastro and McKiernon, 2008). This suggests that a deleterious change in tumor biology may have occurred during the past several decades, perhaps related to tobacco use, dietary factors, or exposure to other carcinogens (Chow et al, 1999; Pantuck et al, 2001b; Hock et al, 2002; Parsons et al, 2002; Kane et al, 2008).
RCC in childhood is uncommon, representing only 2.3% to 6.6% of all renal tumors in children (Castellanos et al, 1974; Chan et al, 1983; Freedman et al, 1996; Asanuma et al, 1999; Broecker, 2000). Mean age at presentation in children is 8 to 9 years, and the incidence is similar in boys and in girls. Although Wilms tumor is much more common in younger children, RCC is as common as Wilms tumor during the second decade of life. RCC in children and young adults is more likely to be symptomatic and to exhibit papillary histology, and a predilection for locally advanced, high-grade disease, and unfavorable histologic subtypes has also been reported (Freedman et al, 1996; Renshaw et al, 1999; Sánchez-Ortiz et al, 2004b; Cook et al, 2006; Estrada et al, 2005). TFE3 protein overexpression, which correlates with the presence of ASPL-TFE3 and PRCC-TFE3 gene translocation events involving the X and first chromosomes, is relatively common in children and young adults with RCC and is unique to this population (Heimann et al, 2001; Geller et al, 2008). The clinical significance of TFE3 protein overexpression is not well defined, although preliminary data suggest that these tumors may show differential sensitivity to certain chemotherapeutic agents (Argani et al, 2002; Heimann et al, 2001; Perot et al, 2003; Bruder et al, 2004). Most studies suggest that stage for stage, children and young adults with RCC may respond better to surgical therapy, and a number of long-term survivors have been reported after radical nephrectomy and lymphadenectomy for lymph node–positive disease (Freedman et al, 1996; Asanuma et al, 1999; Abou El Fettouh et al, 2002; Geller et al, 2008; Sánchez-Ortiz et al, 2004b; Geller et al, 2008). An aggressive surgical approach with formal lymphadenectomy has thus been recommended at the time of radical nephrectomy when RCC is suspected in children or young adults (Freedman et al, 1996; Asanuma et al, 1999; Geller et al, 2008; Selle et al, 2006; Bosquet et al, 2008).
Etiology
RCCs were traditionally thought to arise primarily from the proximal convoluted tubules, and this is probably true for the clear cell and papillary variants. However, it is now established that other histologic subtypes of RCC, such as chromophobe and collecting duct RCC, are derived from the more distal components of the nephron (Störkel, 1996; Oyasu, 1998; Pantuck et al, 2001a).
The most generally accepted environmental risk factor for RCC is tobacco exposure, although the relative associated risks have been modest, ranging from 1.4 to 2.5 compared with controls (Table 49–5). All forms of tobacco use have been implicated, and risk increases with cumulative dose or pack-years (Kantor, 1977; La Vecchia et al, 1990; McLaughlin et al, 1995; McLaughlin and Lipworth, 2000; Moyad, 2001; Dhote et al, 2004; Lindblad, 2004; Lipworth et al, 2006; Carrizosa and Godley, 2009). Relative risk is directly related to duration of smoking and begins to fall after cessation, further supporting a cause-and-effect relationship (La Vecchia et al, 1990; McLaughlin et al, 1995; Parker et al, 2003b). Tobacco use accounts for 20% to 30% of cases of RCC in men and 10% to 20% in women (McLaughlin et al, 1995; McLaughlin and Lipworth, 2000).
Table 49–5 Etiology and Environmental Factors of Malignant Renal Tumors
| Established |
| Putative |
Obesity is now accepted as another major risk factor for RCC with an increased relative risk of 1.07 for each unit of rising body mass index (Chow et al, 2000; Bergstrom et al, 2001; Bjorge et al, 2004; Calle and Kaaks, 2004; Reeves et al, 2007). The increased prevalence of obesity likely contributes to the increased incidence of RCC in Western countries, and it has been estimated that more than 40% of cases of RCC in the United States may be causally linked to obesity (Calle and Kaaks, 2004). Potential mechanisms linking obesity to RCC include lipid peroxidation leading to DNA adducts, increased insulin-like growth factor-1 expression, increased circulating estrogen levels, and increased arterionephrosclerosis and local inflammation (Kasiske et al, 1992; Huang et al, 1998; Gago-Dominguez et al, 2002; Calle and Kaaks, 2004).
Hypertension appears to be the third major etiologic factor for RCC. Diuretics and other antihypertensive medications have also been implicated, but the weight of the epidemiologic evidence suggests that it is the underlying disorder, hypertension, rather than the treatment, that increases the risk of RCC (McLaughlin et al, 1995; Yuan et al, 1998; McLaughlin and Lipworth, 2000; Lipworth et al, 2006). The proposed mechanisms are hypertension-induced renal injury and inflammation or metabolic or functional changes in the renal tubules that may increase susceptibility to carcinogens (Lipworth et al, 2006).
Although a number of other potential etiologic factors have been identified in animal models, including viruses, lead compounds, and more than 100 chemicals such as aromatic hydrocarbons, no specific agent has been definitively established as causative in human RCC (Bennington and Beckwith, 1947; Kantor, 1977). The potential role of trichloroethylene exposure has been actively investigated; some studies showed relative risks ranging from twofold to sixfold, but others have argued that inherent biases likely account for these results (Vamvakas et al, 2000; Mandel, 2001; Moyad, 2001; Bruning et al, 2003, Lipworth et al, 2006; Lock and Reed, 2006). However, Brauch and colleagues (1999) reported an increased incidence and unique pattern of von Hippel-Lindau gene (VHL) mutations in this population, which would argue in favor of a potential causative role for this compound. Slightly increased relative risks for RCC have been reported for workers in the metal, chemical, rubber, and printing industries and those exposed to asbestos or cadmium, but the data are not particularly convincing (Kolonel, 1976; Pesch et al, 2000; Moyad, 2001; Hu et al, 2002; Dhote et al, 2004; Lindblad, 2004; Lipworth et al, 2006; Carrizosa and Godley, 2009).
Case-control studies have shown that RCC is more common among individuals with low socioeconomic status and urban background, although the causative factors have not been defined (Kantor, 1977; Goodman et al, 1986; Muscat et al, 1995; Yuan et al, 1998). The typical modern Western diet (high in fat and protein and low in fruits and vegetables), increased intake of dairy products, and increased consumption of coffee or tea have been associated with RCC, but the relative risks have been modest, and conflicting data are available in most instances (Yu et al, 1986; Lindblad et al, 1997; Moyad, 2001; Handa and Kreiger, 2002; Dhote et al, 2004; Lindblad, 2004; Murai and Oya, 2004, Lipworth et al, 2006; Wolk et al, 2006; Carrizosa and Godley, 2009). A family history of RCC may also be a factor; one study showed a relative risk of 2.9 for individuals with a first- or second-degree relative with RCC (Gago-Dominguez et al, 2001).
Other potential iatrogenic causes include Thorotrast (which was used as a contrast agent in the past), and radiation therapy, but, again, the relative risks are low (Wenz, 1967; Romanenko et al, 2000). Vogelzang and colleagues (1998) reported four cases of RCC developing in a previously irradiated field, and a slightly increased incidence of RCC has been reported in men who received retroperitoneal irradiation for the treatment of testicular cancer. Survivors of childhood Wilms tumor also appear to be at increased risk for RCC, possibly related to prior radiation therapy or chemotherapy (Cherullo et al, 2001). An increased incidence of RCC is also observed in patients with end-stage renal failure and certain familial syndromes such as tuberous sclerosis, as discussed later (Ishikawa et al, 1990; Bjornsson et al, 1996; Neumann and Zbar, 1997).
Familial Renal Cell Carcinoma and Molecular Genetics
Since the early 1990s, significant advances have been made in our understanding of the molecular genetics of RCC. Novel familial syndromes of RCC have been identified, and the tumor suppressor genes and oncogenes contributing to the development of both sporadic and familial forms of this malignancy have been characterized (Table 49–6) (Linehan et al, 1995; Zbar et al, 1995; Schmidt et al, 1997; Weirich et al, 1998; Choyke et al, 2003; Linehan et al, 2003; Pavlovich et al, 2003; Zimmer and Iliopoulos, 2003; Pavlovich and Schmidt, 2004; Klatte and Pantuck, 2008; Nathanson and Stephenson, 2009). The impact of this new information should not be underestimated because it has fundamentally changed our perceptions about RCC. We now, more than ever, recognize the distinct nature of the various histologic subtypes of RCC, and advances in molecular genetics have contributed to a major revision in the histologic classification of this malignant neoplasm (Oyasu, 1998; Linehan et al, 2003; Young et al, 2008; Roma and Zhou, 2007; Pfaffenroth and Linehan, 2008; Zhou, 2009). A direct and beneficial impact on management of patients has also been achieved, with molecular targeted agents now extending survival for patients with advanced RCC (Linehan, 2002; Vira et al, 2007; Hudes et al, 2007, Motzer et al, 2007; Lane et al, 2007c; Kroog and Motzer, 2008; Clark and Cookson, 2008).
Table 49–6 Familial Renal Cell Carcinoma (RCC) Syndromes
| SYNDROME | GENETIC ELEMENT | MAJOR CLINICAL MANIFESTATIONS |
|---|---|---|
| von Hippel-Lindau | VHL gene (chromosome 3p25-26) | |
| Hereditary papillary RCC | c-MET proto-oncogene (chromosome 7q31) | |
| Familial leiomyomatosis and RCC | Fumarate hydratase (chromosome 1q42) | |
| Birt-Hogg-Dubé | BHD1 gene (chromosome 17p12q11) |
Transitional tumors*
|
* Also known as hybrid oncocytic tumors and containing features of both chromophobe RCC and oncocytoma.
Data from Linehan, 2002; Choyke et al, 2003; Linehan et al, 2003; Maranchie and Linehan, 2003; Pavlovich et al, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Coleman, 2008; Pfaffenroth and Linehan, 2008; Hansel and Rini, 2008; and Nathanson and Stephenson, 2009.
Knudson and Strong recognized that familial forms of cancer might hold the key to the identification of important regulatory elements known as tumor suppressor genes (Knudson, 1971; Knudson and Strong, 1972). Their observations about the childhood tumor retinoblastoma, in which familial cases tend to be multifocal and early onset, led them to propose a two-hit theory of carcinogenesis. They hypothesized that a gene product that could suppress tumor development must be involved and that both alleles of this “tumor suppressor gene” must be mutated or inactivated for tumorigenesis to occur. Furthermore, Knudson postulated that patients with the familial form of the cancer are born with one mutant allele and that all cells in that organ or tissue are at risk, accounting for the early onset and multifocal nature of the disease. In contrast, sporadic tumors develop only if a mutation occurs in both alleles within the same cell; and because each event occurs with low frequency, most tumors develop late in life and in a unifocal manner (Knudson, 1971; Knudson and Strong, 1972). Knudson’s hypothesis has proved true for retinoblastoma and a number of other tumor types, including RCC (Choyke et al, 2003; Linehan et al, 2003; Pavlovich et al, 2003; Zimmer and Iliopoulos, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006). Identification of familial cases of RCC was particularly important because it allowed linkage analysis between affected family members.
von Hippel-Lindau Disease, VHL Gene, and Genetics of Clear Cell Renal Cell Carcinoma
The familial form of clear cell RCC is von Hippel-Lindau disease. This is a relatively rare autosomal dominant disorder that occurs with a frequency of 1 per 36,000 population. Major manifestations include the development of RCC, pheochromocytoma, retinal angiomas, and hemangioblastomas of the brainstem, cerebellum, or spinal cord (Table 49–7) (Horton et al, 1976; Go et al, 1984; Green, 1986; Jennings et al, 1988; Lamiell et al, 1989; Maher et al, 1990; Neumann and Zbar, 1997; Hansel and Rini, 2008; Nathanson and Stephenson, 2009). All of these tumor types are highly vascular and can lead to substantial morbidity, much of which can be avoided with prompt recognition and careful, skilled management. In particular, central nervous system lesions can lead to paralysis or death and the retinal lesions to blindness if they are not identified and managed in an expedient manner. Other common or important manifestations of von Hippel-Lindau disease include renal and pancreatic cysts, inner ear tumors, and papillary cystadenomas of the epididymis (Neumann and Zbar, 1997). An increased incidence of neuroendocrine tumors of the pancreas has also been reported in von Hippel-Lindau disease (Zbar et al, 1999; Coleman, 2008). Penetrance for all of these traits is far from complete, and some, such as pheochromocytomas, tend to be clustered only in certain families (Table 49–8) (Neumann and Zbar, 1997, Coleman, 2008; Nathanson and Stephenson, 2009). RCC develops in about 50% of patients with von Hippel-Lindau disease and is distinctive for its early age at onset (often in the third, fourth, or fifth decade of life) and for its bilateral and multifocal involvement (Horton et al, 1976; Go et al, 1984; Green, 1986; Jennings et al, 1988; Lamiell et al, 1989; Maher et al, 1990; Neumann and Zbar, 1997; Vira et al, 2007; Nathanson and Stephenson, 2009). With improved management of the central nervous system manifestations of the disease, RCC has now become the most common cause of mortality in patients with von Hippel-Lindau disease (Maher et al, 1990; Neumann and Zbar, 1997). Screening for von Hippel-Lindau disease and important considerations for the management of RCC in von Hippel-Lindau disease are reviewed later in this chapter.
Table 49–7 Manifestations of the von Hippel-Lindau Syndrome
| ORGAN SYSTEM | LESION | INCIDENCE (%) |
|---|---|---|
| Eye | Benign retinal angiomas | 49-59 |
| Central nervous system | Benign hemangioblastomas | 42-72 |
| Kidney | Clear cell renal cell carcinoma | 24-70 |
| Renal cysts | 22-59 | |
| Adrenal gland | Pheochromocytoma | 18 |
| Pancreas | Islet cell tumors | 12 |
| Malignant islet cell tumor | 2 | |
| Pancreatic cysts | 21-72 | |
| Epididymis | Cystadenoma | 10-26 |
| Ear | Endolymphatic sac tumor | 10 |
Data from Horton et al, 1976; Green, 1986; Lamiell et al, 1989; Maher et al, 1990; Neumann and Zbar, 1997; Friedrich, 1999; Choyke et al, 2003; Linehan et al, 2003; Maranchie and Linehan, 2003; Pavlovich et al, 2003; Sudarashan and Linehan, 2006; Coleman, 2008; Hansel and Rini, 2008; and Nathanson and Stephenson, 2009.
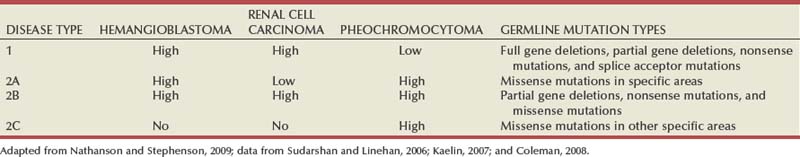
Early clues to the genetic elements involved in the development of RCC came from cytogenetics. These studies demonstrated a common loss of chromosome 3 in kidney cancer, particularly the clear cell variant, and led to intensive efforts to find a tumor suppressor gene in this region (Zbar et al, 1987; Seizinger et al, 1988; Hosoe et al, 1990; Lerman et al, 1991). Reports by Kovacs and colleagues (1989) and Cohen and associates (1979) of translocations involving chromosome 3 further implicated this chromosome as an important regulatory element. Southern blot testing and analysis for restriction fragment length polymorphisms with a wide variety of genetic markers subsequently demonstrated loss of heterozygosity in distinct regions on the short arm of chromosome 3 (reviewed by Jennings et al, 1995). Sophisticated molecular genetic linkage studies in patients with von Hippel-Lindau disease eventually led to the identification of the VHL tumor suppressor gene (Latif et al, 1993). This gene, which is located at chromosome 3p25-26, has now been completely sequenced, and its role as a tumor suppressor gene for both the sporadic and the familial forms of clear cell RCC has been confirmed (Gnarra et al, 1994; Linehan et al, 1995; Zbar, 1995; Furge and Teh, 2009; Nathanson and Stephenson, 2009). The VHL gene consists of three exons, and it encodes a protein of 213 amino acids. A large number of common mutations or “hot spots” in the gene have been identified, and a direct correlation between genotype and phenotype has been established in some cases (Gnarra et al, 1994; Linehan et al, 1995; Zbar, 1995; Brauch et al, 2000; Pavlovich and Schmidt, 2004; Kaelin, 2007). For instance, missense mutations (type 2 mutations) that result in a full-length but nonfunctional protein are commonly found in families with von Hippel-Lindau disease that develop pheochromocytomas, whereas deletions leading to a truncated protein (type 1 mutations) are typically found in families that do not develop pheochromocytomas (see Table 49–8) (Crossey et al, 1994; Linehan et al, 1995; Maher and Kaelin, 1997; Neumann and Zbar, 1997; Walther et al, 1999c; Hes et al, 2000; Friedrich, 2001; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Coleman, 2008). The identification of this tumor suppressor gene represented a major advance in the field and required close collaboration between clinical urologic-oncologists and molecular geneticists. The important historical steps in solving this challenging puzzle were reviewed by Linehan and colleagues (1995) and Zbar (1995), who spearheaded this important effort.
As with most tumor suppressor genes, both alleles of the VHL gene must be mutated or inactivated for development of the disease; the observed inheritance patterns have conformed to Knudson’s hypotheses. As expected, almost all patients with von Hippel-Lindau disease were found to have germline mutations of one allele of the VHL tumor suppressor gene, and autosomal dominant inheritance from the affected parent was confirmed (Gnarra et al, 1994; Linehan et al, 1995, 2003). The second allele is commonly lost by gene or chromosome deletion (Zbar, 1995). Also, as predicted, most sporadic clear cell RCCs were found to harbor mutations or other genetic mechanisms, such as hypermethylation, that inactivated both alleles of the VHL gene (Zbar, 1995; Linehan et al, 2003; Nathanson and Stephenson, 2009). However, they differ in that both mutations must be acquired after birth, accounting for the late onset and the unifocal nature of the sporadic form of the disease.
Subsequent work has focused on the function of the VHL protein and its potential mechanisms of action. The VHL protein is known to bind to elongins B and C, CUL-2, and RBX1 to form an E3 ubiquitin ligase complex and thereby modulates the degradation of important regulatory proteins (Gorospe et al, 1999; Lisztwan et al, 1999; Zbar et al, 1999; Wiesener et al, 2001; George and Kaelin, 2003; Linehan et al, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Vira et al, 2007). A critically important function of the VHL protein complex is to target the hypoxia-inducible factors 1 and 2 (HIF-1 and HIF-2) for ubiquitin-mediated degradation, keeping the levels of HIFs low under normal conditions. The HIFs are intracellular proteins that play an important role in regulating cellular responses to hypoxia, starvation, and other stresses. Inactivation or mutation of the VHL gene leads to dysregulated expression of the HIFs (Maxwell et al, 1999; Yu et al, 2001), and they begin to accumulate in the cell. This, in turn, leads to a severalfold upregulation of the expression of vascular endothelial growth factor (VEGF), the primary angiogenic growth factor in RCC, contributing to the pronounced neovascularity associated with clear cell RCC (Gnarra et al, 1996; Iliopoulos et al, 1996; Gunningham et al, 2001; Igarashi et al, 2002; Linehan et al, 2003; Sudarshan and Linehan, 2006; Vira et al, 2007). HIFs also upregulate the expression of tumor growth factor-α, platelet-derived growth factor (PDGF), glucose transporter (Glut 1), erythropoietin, and carbonic anhydrase IX (CA-IX), a tumor-associated antigen with specificity for clear cell RCC (Fig. 49–9) (Zbar et al, 1999; Wykoff et al, 2000; Turner et al, 2002; Wiesener et al, 2002; Linehan et al, 2003; Grabmaier et al, 2004; Sudarshan and Linehan, 2006; Vira et al, 2007). In addition, the VHL protein appears to influence the cell cycle, cellular differentiation, and intracellular processing of important matrix molecules such as fibronectin, and it may impact the metastatic process by upregulating the chemokine receptor CXCR4. All of these functions may contribute to the pathogenesis and distinctive character of this disease (Lieubeau-Teillet et al, 1998; Kamada et al, 2001; Bindra et al, 2002; Hergovich et al, 2003; Linehan et al, 2003; Na et al, 2003; Pavlovich and Schmidt, 2004; Klatte and Pantuck, 2008).
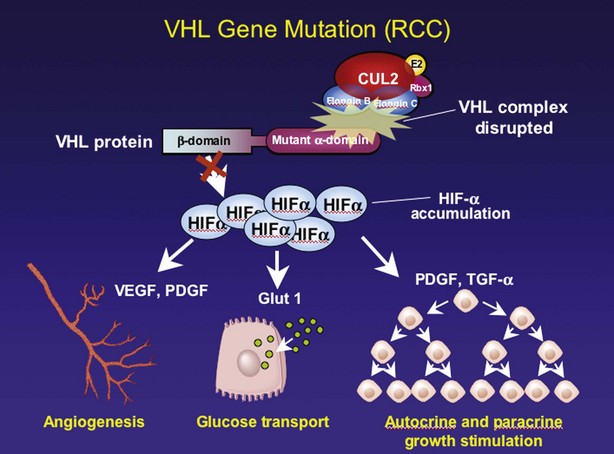
Figure 49–9 Biologic functions of the von Hippel-Lindau (VHL) protein. The wild-type VHL protein targets hypoxia-inducible factor-α (HIF-α) for degradation. Mutation of the VHL gene allows HIF-α to accumulate, leading to increased expression of vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), Glut 1, and transforming growth factor-α (TGF-α). This, in turn, has important implications with respect to tumor angiogenesis, metabolic activity, and autocrine and paracrine stimulation. RCC, renal cell carcinoma.
(From Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol 2003;170:2163–72.)
Other genetic elements potentially involved in the development of sporadic clear cell RCC include additional loci on the short arm of chromosome 3 and the TP53 and PTEN tumor suppressor genes. Sophisticated molecular analyses, including complete gene sequencing and assessment for inactivation of the promoter by hypermethylation, has failed to reveal VHL gene abnormalities in a small proportion of sporadic clear cell RCCs, and the search for additional regulatory elements has continued (Clifford et al, 1998b; Hamano et al, 2002; Banks et al, 2006). Loss of heterozygosity has also been observed at 3p12-p14 and 3p21.2-p21.3 and is particularly common in tumors with wild-type VHL status (Shridhar et al, 1997; van den Berg and Buys, 1997; Clifford et al, 1998a; Lott et al, 1998; Velickovic et al, 2001; Bodmer et al, 2002). The functional importance of these loci is suggested by experiments showing that the transfer of fragments of chromosome 3 containing only these genetic elements can suppress tumorigenesis in RCC cell lines (Van den Berg and Buys, 1997; Lovell et al, 1999; Bodmer et al, 2002). A candidate tumor suppressor gene at 3p12 has been described and may contribute to a VHL-independent pathway to RCC (Lovell et al, 1999). Increased immunostaining for TP53 has been reported in 6% to 40% of RCCs, with some studies suggesting a correlation with tumor grade and stage (Reiter et al, 1993; Uhlman et al, 1994; Haitel et al, 1999). However, the data regarding TP53 in RCC have been controversial, and no clear consensus is available at this time. Of more relevance is the PTEN tumor suppressor gene, which is downregulated in a subset of RCC tumors (Alimov et al, 1999; Kondo et al, 2001; Brenner et al, 2002; Velickovic et al, 2002; Horiguchi et al, 2003; Shin et al, 2003; Hara et al, 2004; Robb et al, 2007; Hager et al, 2007; Pantuck et al, 2007; Klatte and Pantuck, 2008). The PTEN protein inhibits phosphatidylinositol-3-kinase–dependent activation of protein kinase B (Akt), a key intermediary in the mammalian target of rapamycin (mTOR) pathway (Kim et al, 2009). Loss of PTEN leads to constitutive activation of mTOR, which promotes tumorigenesis, and this pathway has proven to be fertile ground for pharmacologic intervention (see Tumor Biology and Clinical Implications and Chapter 50) (Hudes et al, 2007; Klatte and Pantuck, 2008; Kim et al, 2009; Hudes, 2009b).
Oncogenes, such as c-MYC, c-ERBB1, c-Ha-RAS, c-FOS, and RAF-1, have also been studied in clear cell RCC, but the available data suggest limited involvement (Slamon et al, 1984). Downregulation of DNA mismatch repair genes may contribute to genetic instability in RCC and allow accumulation of multiple genetic defects (Deguchi et al, 2003).
Familial Papillary Renal Cell Carcinoma and Genetics of Papillary Renal Cell Carcinoma
Several studies have documented distinct cytogenetic findings in non–clear cell histiotypes of RCC; chromosome 3 and VHL gene abnormalities are uncommon in these variants (Störkel et al, 1997; Oyasu, 1998; Sudarshan and Linehan, 2006; Vira et al, 2007). These observations suggested a distinct genetic basis for non–clear cell RCC. Papillary RCC, the second most common histologic subtype of RCC, is characterized by trisomy for chromosomes 7 and 17 as well as abnormalities on chromosomes 1, 12, 16, 20, and Y (Störkel et al, 1997; Oyasu, 1998; Pavlovich et al, 2003). In 1995, Zbar and colleagues at the National Cancer Institute reported a second familial syndrome of RCC—hereditary papillary RCC (HPRCC). This followed a number of isolated case reports that suggested clustering of papillary RCCs within certain families (Zbar et al, 1994). In Zbar and colleagues’ series (1995) there were 10 families with 41 affected members (29 men and 12 women). Median age at diagnosis was 45 years, and most patients developed multifocal and bilateral papillary RCC. Type 1 papillary RCC is typically found in this syndrome rather than type 2, which is commonly seen in the hereditary leiomyomatosis and RCC syndrome. Unlike von Hippel-Lindau disease, most patients with HPRCC do not develop tumors in other organ systems (Czene and Hemminki, 2003; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009). Mean survival in affected individuals was only 52 years in Zbar and colleagues’ series, although the number of patients dying of RCC was not defined. The development of CKD due to a combination of malignant replacement of the renal mass and loss of functioning nephrons secondary to various interventions is a potential contributor to morbidity and mortality in this syndrome (Ornstein et al, 2000). CT is the preferred imaging modality for patients with HPRCC because it has the greatest sensitivity for detecting the small, hypovascular lesions that are common in this syndrome (Sudarshan and Linehan, 2006; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009).
Studies of families with HPRCC demonstrate an autosomal dominant mode of transmission, similar to all of the familial RCC syndromes, and provide insight into the molecular genetics of HPRCC as well as a subset of patients with sporadic papillary RCC (Zbar et al, 1995; Schmidt et al, 1997; Linehan et al, 2003, Vira et al, 2007). Again, molecular linkage analysis in affected families played a key role in the discovery of this gene, which was localized to chromosome 7q31. However, in this case, the inciting event is activation of a proto-oncogene, rather than inactivation of a tumor suppressor gene. Missense mutations of the c-MET proto-oncogene at 7q31 were found to segregate with the disease, implicating it as the relevant genetic locus (Schmidt et al, 1997; Pavlovich et al, 2003). The protein product of this gene is the receptor tyrosine kinase for the hepatocyte growth factor (also known as scatter factor), and its activation leads to cellular proliferation and other potentially tumorigenic effects (Vira et al, 2007). Most of the mutations in HPRCC have been found in the tyrosine kinase domain of c-MET and apparently lead to constitutive activation (Schmidt et al, 1997; Pavlovich et al, 2003; Sudarshan and Linehan, 2006). The mutated MET protein can transform NIH 3T3 murine fibroblasts and is tumorigenic in immunodeficient murine models. Trisomy for chromosome 7, which is commonly found in HPRCC, develops primarily through duplication of the chromosome harboring the mutant allele of the c-MET proto-oncogene and effectively increases the dosage of the activated receptor (Zhuang et al, 1998; Sudarshan and Linehan, 2006; Hansel and Rini, 2008). Relatively early onset and multifocality in HPRCC are due to inheritance of the mutated c-MET gene, which places all the cells in the kidney at risk from birth, but the incomplete penetrance and variable clinical courses associated with this syndrome suggest that additional genetic loci or epigenetic phenomena may modulate the phenotype (Choyke et al, 2003; Linehan et al, 2003; Pavlovich et al, 2003; Pavlovich and Schmidt, 2004; Schmidt at al, 2004; Vira et al, 2007). Schmidt and colleagues (2004) have described three more families with HPRCC and have shown age-dependent penetrance and a variable clinical course according to the site of mutation and family involved. Whereas tumors in HPRCC tend to be less aggressive than their sporadic counterparts, it is clear that some can metastasize and become lethal (Schmidt et al, 2004; Vira et al, 2007). Schmidt and colleagues report c-MET mutations in 13% of patients with sporadic papillary RCC, suggesting that this molecular defect also contributes to a subset of this disease population (Schmidt et al, 1999; Sudarshan and Linehan, 2006; Vira et al, 2007). Small molecule inhibitors of the c-MET receptor are currently in development and may prove useful for the management of HPRCC and the subset of patients with sporadic RCC who harbor this mutation (Bellon et al, 2008; Hansel and Rini, 2008; Pfaffenroth et al, 2008).
Hereditary Leiomyomatosis and Renal Cell Carcinoma
In 2001, Launonen and colleagues described a new familial renal cancer syndrome in which patients commonly develop cutaneous and uterine leiomyomas and type 2 papillary RCC (Choyke et al, 2003; Kiuru and Launonen, 2004; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Sudarshan et al, 2007). Mean age at diagnosis is in the early 40s (Hansel and Rini, 2008; Nathanson and Stephenson, 2009). Renal tumors in this syndrome are unusual for familial RCC in that they are often solitary and unilateral, and they are more likely to be aggressive than other forms of familial RCC (Linehan et al, 2003; Maranchie and Linehan, 2003; Pavlovich and Schmidt, 2004; Sudarshan and Linehan, 2006; Grubb et al, 2007; Hansel and Rini, 2008). Collecting duct RCC, another highly malignant variant of RCC, has also been observed in this syndrome, which was named hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome (Linehan et al, 2003; Grubb et al, 2007; Nathanson and Stephenson, 2009). The histologic hallmark of these tumors is large, prominent eosinophilic nuclei and nucleoli with perinucleolar clearing (Grubb et al, 2007; Merino et al, 2007).
The HLRCC locus was mapped to a region on 1q42-44, and this was later shown to be the site of the fumarate hydratase gene (Tomlinson et al, 2002; Linehan et al, 2003; Toro et al, 2003; Pavlovich and Schmidt, 2004; Alam et al, 2005). Fumarate hydratase is an essential enzyme in the Krebs cycle of oxidative metabolism. Again, autosomal dominant inheritance was observed, and this appears to be a tumor suppressor gene rather than an oncogene (Linehan et al, 2003; Pavlovich and Schmidt, 2004). The mechanistic link between a metabolic enzyme located in the mitochondria and tumorigenesis is still an enigma and remains an active area of investigation (Pollard et al, 2003; Pavlovich and Schmidt, 2004). One hypothesis is that fumarate accumulation may stabilize HIF-1 by preventing its hydroxylation, which targets it for degradation (Isaacs et al, 2005; Sudarshan and Linehan, 2006; Ratcliffe, 2007; Coleman, 2008; Hansel and Rini, 2008; Pfaffenroth and Linehan, 2008).
Penetrance for RCC in HLRCC is lower than for the cutaneous and uterine manifestations, with only a minority (20%) of patients developing RCC (Choyke et al, 2003; Sudarshan and Linehan, 2006; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009). In contrast, almost all individuals with this syndrome will develop cutaneous leiomyomas and uterine fibroids (if female), usually manifesting at the age of 20 to 35 years (Sudarshan and Linehan, 2006; Vira et al, 2007). A high proportion of women have had a hysterectomy for fibroids before formal diagnosis of HLRCC (Coleman, 2008). Leiomyosarcomas of the uterus have been reported in HLRCC, although they appear to be uncommon (Sudarshan and Linehan, 2006; Pfaffenroth and Linehan, 2008; Nathanson and Stephenson, 2009). Prompt surgical management of the renal tumors is recommended in this syndrome, given their tendency toward invasive and aggressive behavior (Linehan et al, 2003; Grubb et al, 2005b, 2007; Sudarshan and Linehan, 2006; Vira et al, 2007; Coleman, 2008). This is in contrast to other familial syndromes of RCC, for which management tends to be more conservative, as discussed later (Grubb et al, 2007; Vira et al, 2007; Pfaffenroth and Linehan, 2008).
Birt-Hogg-Dubé Syndrome
Birt-Hogg-Dubé syndrome, in which patients develop cutaneous fibrofolliculomas, lung cysts, spontaneous pneumothoraces, and a variety of renal tumors primarily derived from the distal nephron, is named after three Canadian physicians who first described the cutaneous lesions in 1977 (Toro et al, 1999; Linehan et al, 2003; Pavlovich et al, 2003, 2005; Pavlovich and Schmidt, 2004; Adley et al, 2006; Hansel and Rini, 2008). The renal tumors typically include chromophobe RCC, oncocytomas, and hybrid or transitional tumors that exhibit features of both of these entities. However, other forms of RCC, including a substantial proportion of clear cell RCC, have been observed in this syndrome (Pavlovich et al, 2002; Adley et al, 2006). Overall penetrance for renal tumors is 20% to 40%, but when they occur they are often bilateral and multifocal (Choyke et al, 2003; Pavlovich et al, 2002; Linehan et al, 2003; Pavlovich and Schmidt, 2004: Pfaffenroth and Linehan, 2008; Toro et al, 2008). Average age at renal tumor diagnosis is approximately 50 years (Pavlovich et al, 2005). Most renal tumors in Birt-Hogg-Dubé syndrome have limited biologic aggressiveness, although metastatic behavior and lethality have been reported (Pavlovich et al, 2005)
The BHD gene responsible for this syndrome has been mapped to chromosome 17p12q11.2 and is now fully sequenced (Khoo et al, 2001). Recent studies have shown that the gene product is folliculin, which appears to be a tumor suppressor gene, although its function with respect to this syndrome is still under investigation (Adley et al, 2006; Vira et al, 2007; Toro et al, 2008). One hypothesis is that folliculin may interface with the mTOR pathway (Toro et al, 2008; Nathanson and Stephenson, 2009). Germline mutations in this gene have been found in 88% of kindreds (Toro et al, 2008). As with all of the other well-characterized familial RCC syndromes, an autosomal dominant pattern of inheritance is observed and genetic testing is now available (see Table 49–6) (Choyke et al, 2003; Maranchie and Linehan, 2003; Pavlovich et al, 2005; Adley et al, 2006; Toro et al, 2008).
Tumor Biology and Clinical Implications
Resistance to Cytotoxic Therapy
RCC is a prototype of the chemorefractory tumor because it has only demonstrated limited or modest responses to traditional chemotherapeutics (Motzer and Russo, 2000; Rini et al, 2009). Study of the tumor biology of RCC provides insight into its refractory nature and, through elucidation of the VEGF and mTOR pathways, is beginning to yield agents with clinical benefit for advanced disease (Table 49–9) (Rini et al, 2009).
Table 49–9 Tumor Biology and Clinical Implications
| BIOLOGIC CHARACTERISTIC | CLINICAL IMPLICATIONS |
|---|---|
| Expression of multidrug resistance | |
| Immunogenic | |
| Angiogenic | |
| Dependence on mTOR pathway |
IL-2, interleukin-2; RCC, renal cell carcinoma; VEGF, vascular endothelial growth factor.
Expression of multidrug resistance (MDR) proteins, such as MDR-1 (also known as P-glycoprotein) and MDR-related proteins, which act as energy-dependent efflux pumps for a wide variety of hydrophobic compounds, contribute to the chemorefractory nature of advanced RCC (Mickisch, 1994; Chapman and Goldstein, 1995). However, the resistance of RCC to cisplatin and other agents that are not handled by MDR proteins, and the downregulation of MDR-1 in high-grade tumors and metastases (Tobe et al, 1995), suggests redundancy in resistance mechanisms. The ancillary benefit of this refractoriness has been an impetus for clinical investigations of immunomodulators and targeted-molecular therapies, which have markedly altered our paradigms for the management of patients with advanced RCC (see Chapter 50).
Immunobiology and Immune Tolerance
Several lines of evidence demonstrate that RCC is immunogenic, and this knowledge has stimulated intensive efforts to harness the immune system to improve outcomes for patients with advanced disease. Tumor-infiltrating immune cells can be readily isolated from RCC, including (1) cytotoxic T cells with specificity for antigens on tumor cells and (2) dendritic cells and helper T cells, which express interleukin (IL)-1 and IL-2 and function as antigen-presenting cells (Finke et al, 1992; Gaudin et al, 1995; Elsasser-Beile et al, 1999; Schwaab et al, 1999). Of the tumor-associated antigens for RCC, carbonic anhydrase IX (CA-IX or MN-9) has demonstrated the most specificity (Liao et al, 1997; McKiernan et al, 1997; Shuch et al, 2008). This antigen, which is recognized by the G250 monoclonal antibody, is expressed almost ubiquitously by clear cell RCC and only rarely by other RCC subtypes. Immunohistochemical analysis of CA-IX expression has been investigated as a diagnostic and a prognostic marker for clear cell RCC (Bui et al, 2003; Divgi et al, 2007; Leibovich et al, 2007). In normal tissues, the expression of CA-IX is restricted to the gastric mucosa, large bile ducts, and pancreas, and its expression in normal renal epithelial cells is suppressed by wild-type VHL protein (McKiernan et al, 1997; Ivanov et al, 1998; Vissers et al, 1999). CA-IX has also been investigated for reverse transcriptase-polymerase chain reaction detection of circulating RCC cells in the peripheral blood (McKiernan et al, 1999; De la Taille et al, 2000; Ohlmann et al, 2006), and CA-IX based tumor vaccine protocols have been developed (Hernandez et al, 2002; Lamers et al, 2006; Kim et al, 2007). Radioactively labeled G250 has shown promise for the detection of RCC metastases by radionuclide scanning (Brouwers et al, 2003), and more recently by positron emission tomography (Divgi et al, 2007). All these potential applications of CA-IX are, at present, promising but experimental.
A second class of factors that may modulate immunotherapeutic responses in kidney cancer is the B7 family of cell surface glycoproteins, which are expressed on various immune and nonimmune cells (Thompson et al, 2007b). B7-H1 is a T-cell coregulatory molecule that is normally expressed by macrophage lineage cells, can be induced on activated T lymphocytes, and is aberrantly expressed by RCC (Thompson et al, 2007b). Tumor-associated B7-H1 impairs antigen-specific T-cell function, and blockade of B7-H1 has been shown to potentiate antitumoral responses in preclinical models (Thompson et al, 2004). Consequently, blockade of tumor-associated B7-H1 has garnered much attention in the recent literature as a potential means to boost antitumoral immune responses. Thompson and associates (2006) have shown that B7-H1 expression by clear cell RCC tumors correlates with aggressive pathologic features and is associated with an increased risk of disease progression, even after multivariate adjustment. Consequently, abrogation of B7-H1 signaling is being investigated as a means to facilitate the response of RCC to various immunotherapeutics (Thompson et al, 2007b).
Clinical observations such as validated responses to immunotherapy, prolonged disease stabilization, and occasional spontaneous tumor regression also support the immunogenicity of RCC. The response of RCC to immunomodulators, such as interleukin (IL)-2, interferon-alfa, and tumor-infiltrating lymphocytes, argues in favor of an important role for the immune system in the tumor biology of RCC (Rosenberg et al, 1998; Motzer and Russo, 2000; Coppin et al, 2005). Indeed, high-dose IL-2 remains the only treatment with curative potential for patients with metastatic RCC, with durable and complete regression of disease accomplished in a finite proportion (3%-5%) of patients (Coppin et al, 2008). The estimated incidence of spontaneous regression of RCC has ranged from 0.3% to as high as 7%, although most experienced clinicians believe that the lower figure is more accurate (Oliver et al, 1989; Vogelzang et al, 1992). Most spontaneous regressions have been noted in patients with pulmonary metastases and have occurred after cytoreductive nephrectomy, but regression of primary RCC has also been reported in the absence of any form of treatment (Vogelzang et al, 1992). Remission can be durable, and this phenomenon, although rare, is thought to be real and has been assumed to be due to immune surveillance, although other possibilities cannot be excluded (Papac, 1998; Young, 1998; Coppin et al, 2008).
Unfortunately, response rates of immunotherapy for RCC have been disappointing, typically ranging from 15% to 20%, despite a variety of creative treatment strategies, suggesting immune tolerance (Motzer and Russo, 2000; Ng et al, 2002; Coppin et al, 2008). A number of observations support impaired immune surveillance in RCC, and a variety of mechanisms affecting virtually all levels of regulation of the immune system have been proposed. Defects in transcriptional regulation via nuclear factor-kappaB (NF-κB) are present in the tumor-infiltrating lymphocytes and dendritic cells of 60% of RCCs (Finke et al, 2001; Thornton et al, 2004). Defective NF-κB signaling impairs lymphocyte function, predisposes lymphocytes to apoptosis, and leads to deficient recruitment and activation of dendritic cells (Alexander et al, 1993; Kolenko et al, 1997; Troy et al, 1998; Ling et al, 1998; Uzzo et al, 1999a; Kallfelz et al, 1999). Improved understanding of the mechanisms contributing to immunotolerance in RCC should suggest novel and rational strategies for improving outcomes for patients with advanced disease. For example, in addition to its antiangiogenic activity, sunitinib also appears to stimulate antitumor immunity by reversing myeloid-derived suppressor cell-mediated immunosuppression (Ko et al, 2009). Although the current targeted therapies appear to have significant impact on other aspects of RCC pathogenesis, such as angiogenesis, it remains to be seen whether augmentation of the antitumoral immune responses seen with some agents contributes to their clinical activity.
Angiogenesis and Targeted Pathways
RCC has long been recognized as one of the most vascular of cancers as reflected by the distinctive neovascular pattern exhibited on renal angiography (Fig. 49–10). Dependence on angiogenesis is also suggested by preclinical studies demonstrating blockade of the growth and metastasis of RCC by several well-established antiangiogenic agents (Fujioka et al, 1996; Tan et al, 1996; Dhanabal et al, 1999; Drevs et al, 2000). The primary angiogenesis inducer in clear cell RCC appears to be VEGF, which is suppressed by the wild-type VHL protein under normal conditions and is dramatically upregulated during tumor development (Gnarra et al, 1996; Iliopoulos et al, 1996; Tomisawa et al, 1999; Gunningham et al, 2001; Igarashi et al, 2002; Linehan et al, 2003; Sudarshan and Linehan, 2006; Vira et al, 2007; Lane et al, 2007d). Increased levels of VEGF have been found in the serum and urine of patients with RCC, and a correlation with stage and grade has been reported (Tsuchiya et al, 1998; Chang et al, 2001; Horstmann et al, 2005). Elevated serum levels of basic fibroblast growth factor and other putative angiogenesis inducers for this malignant neoplasm have also been reported in RCC patients compared with normal control subjects (Campbell, 1997; Slaton et al, 2001; Currie et al, 2002; Yagasaki et al, 2003). Functional relevance of VEGF has been demonstrated by studies showing increased levels of VEGF transcript in most hypervascular tumors, whereas the less common hypovascular counterparts exhibit reduced expression of VEGF (Takahashi et al, 1994).
Figure 49–10 Neovascularity associated with renal cell carcinoma. Renal angiogram shows left renal mass exhibiting markedly increased neovascularity within renal tumor.
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2007-2009. All Rights Reserved.)
VEGF is actually a family consisting of several subtypes, most of which are regulated by HIFs and VHL (also see earlier under Familial Renal Cell Carcinoma and Molecular Genetics) and bind to one or more of the corresponding VEGF receptor family members (Lane et al, 2007d). VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) are receptor tyrosine kinases that are the target of several multi–tyrosine kinase inhibitors with activity against RCC (Carmeliet, 2005; Hicklin and Ellis, 2005). Upon binding of ligand (VEGF), key tyrosine residues along the intracellular portion of the VEGFR are phosphorylated, which leads to the binding of specific intracellular factors and activation of corresponding pathways. Pathways known to be activated by phosphorylation of VEGFRs include the Raf-MEK-Erk and phosphatidylinositol-3-kinase/Akt/mTOR pathways that promote endothelial cell survival and proliferation (Carmeliet, 2005; Hicklin and Ellis, 2005). However, the promiscuity of the interactions between the various ligands, receptors, and downstream effectors leads to a host of effects that may be difficult to predict in the absence of analyses investigating the complete microenvironment of the cancer or endothelial cell. This promiscuity is likely a major reason that two therapeutic agents that have the “same” mode of action are found to have disparate clinical or off-target effects. In contrast, bevacizumab is a monoclonal antibody that binds to VEGF and sequesters the ligand so that it cannot interact with VEGFR; its clinical activity is therefore almost certainly directly related to this activity.
Given the dependence of RCC on angiogenesis and the absence of generally effective forms of systemic therapies, it is not surprising that RCC has been targeted for antiangiogenic approaches. Initial clinical trials identified several antiangiogenic compounds such as TNP-470, roquinimex, and thalidomide with limited activity in patients with advanced RCC (de Wit et al, 1997; Stadler et al, 1998). Thalidomide, for example, has shown only rare responses despite its potent antiangiogenic effects, and toxicity can be substantial, including thrombotic events and neurologic morbidity (Daliani et al, 2002; Escudier et al, 2002b; Nathan et al, 2002; Fanelli et al, 2003; Matthews and McCoy, 2003). More promising initial results were reported for bevacizumab, a humanized anti-VEGF antibody, which was associated with a significant delay in time to progression for patients with metastatic RCC compared with placebo (Yang et al, 2003a). Bevacizumab therapy commonly leads to shrinkage in the total tumor burden, although in this initial experience objective partial responses were uncommon and there were no complete responses, consistent with a tumoristatic rather than a tumoricidal mechanism of action. A number of other multiple kinase inhibitors that target the VEGF pathway were subsequently tested in clinical trials and found to have substantial activity in patients with advanced RCC, culminating in approval of two agents, sunitinib (Sutent) and sorafenib (Nexavar), for this indication by the U.S. Food and Drug Administration (FDA) in December 2005 and January 2006 (Motzer et al, 2005, 2006). These agents are now commonly used for front-line and treatment-refractory metastatic RCC (see further details in Chapter 50). Several other antiangiogenic compounds are currently in clinical trials investigating these and other potential applications for the treatment of advanced RCC.
In general, antiangiogenic agents offer a number of potential advantages in that they are not genotoxic and target stable endothelial cells rather than genetically unstable tumor cells. They are thus unlikely to induce secondary cancers or tumor resistance. In addition, many antiangiogenic agents have proved to be synergistic with one another and with cytotoxic therapies, and their side effects should not overlap those of conventional forms of therapy (Campbell, 1997; Lane et al, 2007d).
Signal Transduction, Cell Cycle Regulation, and Other Targeted Molecular Pathways
Aberrant activation of additional signal transduction pathways in RCC may also contribute to altered cell cycle kinetics and represent excellent targets for therapeutic intervention. One such regulatory pathway in RCC is the mammalian target of rapamycin (mTOR) pathway, which interfaces with Akt (protein kinase B) and the PTEN tumor suppressor gene (Brenner et al, 2002; Velickovic et al, 2002; Horiguchi et al, 2003; Shin et al, 2003; Hara et al, 2004; Robb et al, 2007; Pantuck et al, 2007; Klatte and Pantuck, 2008). Expression of mTOR is upregulated by various growth factors or by mutation or loss of PTEN (Kim et al, 2009). Through complex pathways involving a variety of intermediaries, the mTOR pathway leads to increased expression of HIF-1 and other growth-promoting and potentially tumorigenic sequelae (Klatte and Pantuck, 2008; Kim et al, 2009). Inhibition of mTOR with temsirolimus (Torisel), now also FDA approved, has yielded prolonged survival in patients with poor-risk, metastatic RCC, confirming the clinical relevance of this pathway (Hudes et al, 2007; Hudes, 2009b).
Other important growth regulatory elements in RCC include the insulin-like growth factor axis (Takahashi et al, 2005), telomerase, and BCL2. Insulin-like growth factor receptor expression has been documented in RCC, and Parker and colleagues (2003a) have correlated this with decreased survival. Increased telomerase activity, which has been found in 56% to 93% of RCCs, may also affect the cell cycle by maintaining telomere length (Mehle et al, 1996; Yoshida et al, 1998; Orlando et al, 2001). Progressive telomere loss occurs each time a normal cell divides and eventually leads to growth inhibition and cellular senescence. Increased expression of BCL2, which protects against programmed cell death, has also been reported in RCC and may contribute to tumor viability and treatment failure (Huang et al, 1999; Gobe et al, 2002).
Proliferative index, as defined by proliferating cell nuclear antigen (PCNA) or Ki-67 staining, has correlated with pathologic parameters and clinical outcomes in RCC, suggesting that regulation of the cell cycle plays an important role in the tumor biology of RCC (deRiese et al, 1993; Delahunt et al, 1995; Tannapfel et al, 1996; Bui et al, 2004; Tollefson et al, 2007). Increased expression of tumor growth factor-α and its receptor tyrosine kinase, the epidermal growth factor receptor (EGFR), have been reported in RCC and may contribute to tumorigenesis by promoting cell proliferation or transformation through an autocrine mechanism (Gomella et al, 1989; Mydlo et al, 1989; Sargent et al, 1989; Lager et al, 1994; Moch et al, 1997; Ramp et al, 1997). The functional relevance of EGFR in the development of RCC has also been suggested by preclinical studies testing the efficacy of the C225 monoclonal antibody, which neutralizes EGFR and blocks tumor growth and metastasis (Prewett et al, 1998; Brouwers et al, 2004). Unfortunately, phase II clinical trials using agents that target EGFR, including erlotinib (Tarceva), gefitinib (Iressa), and ABX-EGF, demonstrated a lack of substantial activity in patients with advanced RCC (Rini et al, 2008). Based on these disappointing results, agents targeting the EGFR pathway have fallen out of favor, although there may be a role for their use in patients with cancers that express high levels of EGFR (Ravaud et al, 2008).
The hepatocyte growth factor and its receptor, the c-MET proto-oncogene, may also contribute to the pathogenesis of RCC (Sweeney et al, 2002a). The role of activating mutations of the c-MET proto-oncogene in the etiology of hereditary papillary RCC has already been discussed, but data suggest that upregulated expression of this ligand may occur in most of the histologic subtypes of RCC (Natali et al, 1996; Pisters et al, 1997; Schmidt et al, 1997; Clifford et al, 1998a; Horie et al, 1999). Hepatocyte growth factor is expressed by proximal tubular cells in the normal kidney, where it is involved in the branching tubulogenesis of the developing kidney and regeneration after renal injury. In vitro, hepatocyte growth factor has mitogenic and morphogenic effects on renal epithelial cells (Clifford et al, 1998a). Increased serum levels of hepatocyte growth factor have also been reported in most patients with RCC, independent of histologic subtype, and activation of this factor by phosphorylation at two sites is associated with cancer progression (Petri et al, 1997; Miyata et al, 2006). Taken together, these data suggest that hepatocyte growth factor and its receptor may play an important role in the tumor biology of RCC, although constitutive activation of the receptor, which may be the most potent mechanism, appears to be primarily limited to familial papillary RCC.
Proteases, Adhesion, and the Extracellular Matrix
Interactions among cancer cells, adjacent cells, and the surrounding matrix can strongly influence their pathogenic potential. Altered intracellular processing and secretion of fibronectin and other matrix proteins is found in RCC, representing one consequence of VHL gene mutation (Lieubeau-Teillet et al, 1998; Ohh et al, 1998). This fundamental defect most likely has important effects on tumor biology, given the important role of the matrix in regulating cellular differentiation and tumor invasiveness and metastasis. Increased expression of proteases, such as plasmin and the matrix metalloproteinases, has correlated with reduced survival in RCC and may also contribute to the aggressive behavior of RCC (Walther et al, 1997a; Kugler et al, 1998). Downregulation of E-cadherin and cadherin-6, which mediate adhesion between cancer cells, is well documented in RCC and has correlated with poor outcomes in most studies (Morton et al, 1995; Paul et al, 2004). Aberrant regulation of α-catenins, the cytoplasmic proteins that bind cadherins and mediate their effects on the cytoskeleton, has also been observed in RCC, and a correlation with compromised survival has been reported (Shimazui et al, 1996; Paul et al, 1997).
Other studies have defined the adhesion molecules that facilitate interactions between tumor cells and endothelial cells in RCC. Steinbach and colleagues (1996) have shown that sialyl-Lewisx/endothelial leukocyte adhesion molecule-1 and VLA-4/vascular cell adhesion molecule-1 interactions regulate this process, which presumably influences the ability of tumor cells to move into or out of the vascular system during the metastatic cascade.
Pathology
Most RCCs are round to ovoid and circumscribed by a pseudocapsule of compressed parenchyma and fibrous tissue rather than a true histologic capsule. Unlike upper tract transitional cell carcinomas, most RCCs are not grossly infiltrative, with the notable exception of collecting duct RCC and some sarcomatoid variants (Farrow, 1997). Tumor size has averaged between 4 and 8 cm in most series but can vary from a few millimeters to large enough to fill the entire abdomen. Tumors smaller than 3 cm were previously classified as benign adenomas, but some small tumors have been associated with metastases (Nguyen and Gill, 2009), and most pathologists agree that with the exception of oncocytomas and some small (<1 cm) low-grade papillary adenomas there are no reliable histologic or ultrastructural criteria to differentiate benign from malignant renal epithelial tumors (Farrow, 1997; Eble et al, 2004) (see Chapter 51). When they are bivalved, RCCs consist of yellow, tan, or brown tumor interspersed with fibrotic, necrotic, or hemorrhagic areas; few are uniform in gross appearance. Cystic degeneration is found in 10% to 25% of RCCs and appears to be associated with a better prognosis compared with purely solid RCC (Corica et al, 1999; Koga et al, 2000; Onishi et al, 2001; Nassir et al, 2002; Imura et al, 2004; Webster et al, 2007). Calcification can be stippled or plaquelike and is found in 10% to 20% of RCCs.
Nuclear features can be highly variable. Grading has been based primarily on nuclear size and shape and the presence or absence of prominent nucleoli. Fuhrman’s system (Table 49–10) has been most generally adopted and is now recognized as an independent prognostic factor for RCC generally and for clear cell RCC in particular (Fuhrman et al, 1982; Pantuck et al, 2001a; Lang et al, 2005; Lohse et al, 2005; Zhou, 2009).
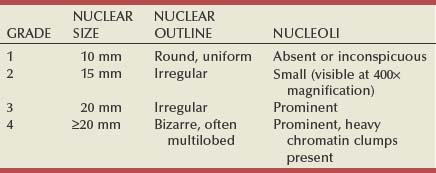
Aggressive local behavior is not uncommon with RCC and can be expressed in a variety of ways. Frank invasion and perforation of the renal capsule, renal sinus, or collecting system are found in approximately 20% of cases, although displacement of these structures is a more common finding. Further spread to involve adjacent organs or the abdominal wall is often precluded by the Gerota fascia, although some high-grade RCCs are able to overcome this natural barrier. One unique feature of RCC is its predilection for involvement of the venous system, which is found in 10% of RCCs, more often than in any other tumor type (Skinner et al, 1972; Schefft et al, 1978). This is most commonly manifested in the form of a contiguous tumor thrombus that can extend into the inferior vena cava (IVC) as high as the right atrium (Blute et al, 2007). Many such tumor thrombi are highly vascularized by arterial blood flow (Novick et al, 1990), and some directly invade the wall of the renal vein or vena cava, which correlates with compromised prognosis (Zini et al, 2008).
Most sporadic RCCs are unilateral and unifocal. Bilateral involvement can be synchronous or asynchronous and is found in 2% to 4% of sporadic RCCs, although it is considerably more common in patients with familial forms of RCC, such as von Hippel-Lindau disease (Farrow, 1997; Linehan et al, 2003). Multicentricity, which is found in 10% to 20% of cases, is more common in association with papillary histology and familial RCC (Mukamel et al, 1988; Cheng et al, 1991; Whang et al, 1995; Campbell et al, 1997a; Richstone et al, 2004; Krambeck et al, 2008). Satellite lesions are often small and difficult to identify by preoperative imaging, intraoperative ultrasonography, or visual inspection; they appear to be the main factor contributing to local recurrence after partial nephrectomy (Campbell et al, 1996a; Richstone et al, 2004). Microsatellite analysis suggests a clonal origin for most multifocal RCC within the same kidney (Junker et al, 2002), but tumor in the contralateral kidney is likely to be an independent growth if it is synchronous or a metastasis if it is asynchronous (Kito et al, 2002). Molecular analyses, such as gene expression profiling, may help to determine whether an asynchronous tumor is a second primary tumor or a metastasis (Lane et al, 2009).
All RCCs are, by definition, adenocarcinomas, derived from renal tubular epithelial cells (Zambrano et al, 1999; Pantuck et al, 2001a; Renshaw, 2002; Zhou, 2009). Most RCCs share ultrastructural features, such as surface microvilli and complex intracellular junctions, with normal proximal tubular cells, and are believed to be derived from this region of the nephron (reviewed in Farrow, 1997). Similarly, immunohistochemistry for lectins and other cell surface antigens has supported derivation from the proximal convoluted tubule (Kim and Kim, 2002). However, more recent data suggest that this information applies primarily to the more common clear cell and papillary variants of RCC (Table 49–11), whereas most other histologic subtypes of RCC appear to be derived from more distal elements of the nephron (Störkel et al, 1997; Zambrano et al, 1999; Pantuck et al, 2001a; Renshaw, 2002; Linehan et al, 2003; Pavlovich and Schmidt, 2004).
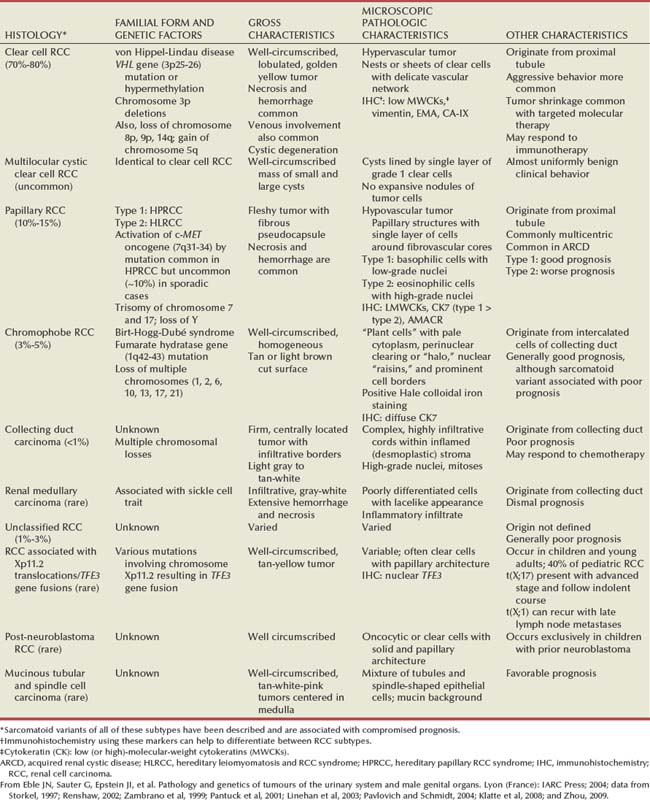
Since the early 1990s the histologic classification of RCC has undergone several major revisions (Zambrano et al, 1999; Renshaw, 2002; Linehan et al, 2003; Eble et al, 2004; Zhou, 2009). Traditionally, RCC was divided into four histologic subtypes: clear cell, granular cell, tubulopapillary, and sarcomatoid. On the basis of advances in the molecular genetics of RCC and a more discerning interpretation of histologic and ultrastructural features, a newer classification scheme was proposed by Kovacs (1993). This classification system was approved by an international consensus workshop of clinicians and researchers in the field (Weiss et al, 1995; Störkel et al, 1997; Zambrano et al, 1999; Pantuck et al, 2001a; Renshaw, 2002; Linehan et al, 2003; Pavlovich and Schmidt, 2004). In this system, granular cell tumors were reclassified into other categories based on distinct histopathologic features, chromophobe RCC was recognized as a new RCC subtype, and sarcomatoid features were categorized as variants of other histologic subtypes rather than a distinct tumor type (Weiss et al, 1995; Störkel et al, 1997; Oyasu, 1998; Zambrano et al, 1999; Renshaw, 2002; Linehan et al, 2003; Pavlovich and Schmidt, 2004). Current practice is to identify the primary histologic subtype and comment on the presence and extent of sarcomatoid differentiation rather than to separate these tumors into a distinct category, although the prognostic implications have not changed (Mian et al, 2002; Cheville et al, 2004; Eble et al, 2004; Zhou, 2009). Depending on well-defined histologic and ultrastructural criteria, granular cell tumors were reclassified as papillary RCC, eosinophilic variants of chromophobe RCC, or combined with clear cell RCC (Weiss et al, 1995; Kovacs et al, 1997; Störkel et al, 1997; Oyasu, 1998; Zambrano et al, 1999; Renshaw, 2002; Zhou, 2009). Another important development was the identification of renal medullary carcinoma that is common in young African-Americans with sickle cell trait (Davis et al, 1995b; Störkel et al, 1997; Hakimi et al, 2007). With additional advances in ancillary pathologic studies, including electron microscopy, immunohistochemistry, molecular genetics and cytogenetics, several additional unique subtypes of RCC have been identified since the implementation of the 1993 classification system. Based on these findings, an updated classification of malignant epithelial tumors of the kidney was presented by the World Health Organization in 2004 (see Table 49–11; Eble et al, 2004).
The 2004 World Health Organization classification reflects current understanding of RCC not as a single malignant neoplasm but rather a group comprising several different tumor subtypes, each with a distinct genetic basis and unique clinical features (see Table 49–11). Important changes include the addition of several RCC subtypes with distinct pathologic and clinical features that were previously grouped within “conventional” or unclassified RCC, such as RCC associated with XP11.2 translocations/TFE3 gene fusions, which has microscopic features of both clear cell and papillary RCC and occurs primarily in children and young adults (Argani et al, 2001; Camparo et al, 2008; Geller et al, 2008), and the indolent mucinous tubular and spindle cell carcinoma (Hes et al, 2002; Ferlicot et al 2005; Fine et al, 2006). Sophisticated gene expression profiling and proteomic analyses support the individuality of each of these tumor subtypes and hold great promise for differentiating additional subtypes in the future (Takahashi et al, 2003, 2004; Amy-Bazille et al, 2004; Furge et al, 2004; Sugimura et al, 2004; Yang et al, 2004, 2006; Young et al, 2008). This is clearly a field in evolution, with changes stimulated by basic science advances and astute clinical observation.
Clear Cell Renal Cell Carcinoma
Clear cell RCC accounts for 70% to 80% of all RCCs, representing the garden variety of RCC formerly known as “conventional” RCC (Störkel et al, 1997; Eble et al, 2004; Rini et al, 2009). These tumors are typically yellow when they are bivalved and are highly vascular, containing a network of delicate vascular sinusoids interspersed between sheets or acini of tumor cells (Fig. 49–11). On microscopic examination, clear cell RCC can include clear cell, granular cell, or mixed types. Clear cells are typically round or polygonal with abundant cytoplasm containing glycogen, cholesterol, cholesterol esters, and phospholipids, all of which are readily extracted by the solvents used in routine histologic preparations, contributing to the clear appearance of the tumor cells (Farrow, 1997). However, granular cells, which have eosinophilic cytoplasm and abundant mitochondria, can predominate. Two to 5 percent of clear cell RCC demonstrate sarcomatoid features, and clear cell RCC is more likely to exhibit venous tumor extension than any other subtype of RCC (Rabbani et al, 2004). In general, patients with clear cell RCC have a worse prognosis compared with papillary or chromophobe RCC, even after stratification for stage and grade (Lau et al, 2002; Cheville et al, 2003; Beck et al, 2004). However, most responders in immunotherapy protocols have had clear cell RCC, and these protocols are now being reserved primarily for this population (Childs et al, 2000; Drachenberg and Childs, 2003). Chromosome 3 alterations and VHL mutations are common in clear cell RCC, and mutation or inactivation of this gene has been found in a majority of sporadic cases (Clifford et al, 1998b; Zambrano et al, 1999; Renshaw, 2002; Linehan et al, 2003). The familial form of clear cell RCC, the von Hippel-Lindau syndrome, has already been reviewed.
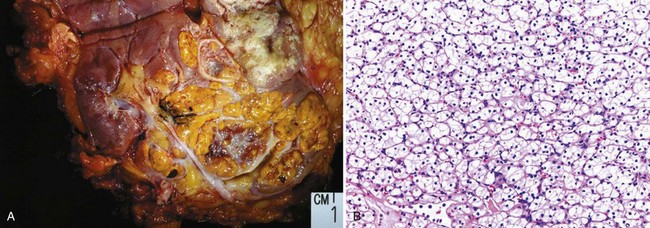
Figure 49–11 A, Clear cell renal cell carcinoma (RCC) with typical golden yellow color. B, Low-power view of typical microscopic appearance of a low-grade clear cell RCC demonstrating a delicate vascular network interspersed within homogeneous nests of cells with clear cytoplasm.
(Courtesy of Dr. Ming Zhou, Cleveland, OH.)
Papillary Renal Cell Carcinoma
Papillary RCC, which has also been designated chromophilic RCC in previous classification schemes, is the second most common histologic subtype (Störkel et al, 1997; Eble et al, 2004; Pignot et al, 2007; Gontero et al, 2008). It represents 10% to 15% of all RCCs, although it is more commonly found in patients with end-stage renal failure and acquired renal cystic disease (Ishikawa and Kovacs, 1993; Störkel et al, 1997). On microscopic examination, most tumors in this category consist of basophilic or eosinophilic cells arranged in papillary or tubular configuration; previously, more than 50% or 75% of the tumor had to exhibit such architectural features to qualify as a papillary RCC (Fig. 49–12). One unique feature of papillary RCC is its tendency toward multicentricity, which approaches 40% in many series (Renshaw and Corless, 1995; Amin et al, 1997a; Campbell et al, 1997b; Chow et al, 2001; Zhou, 2009).
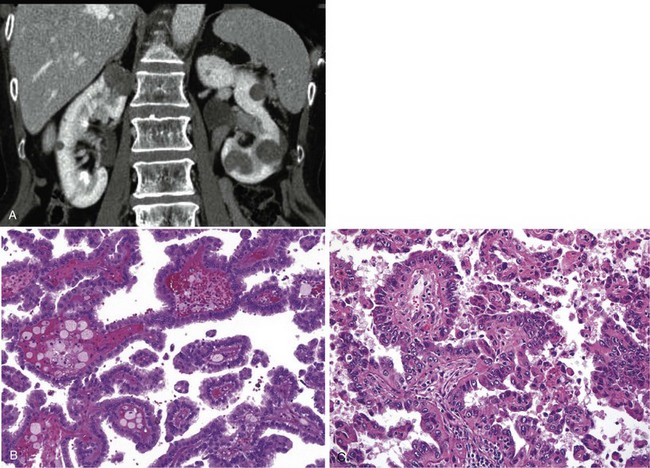
Figure 49–12 A, Papillary renal cell carcinoma (RCC) often presents as multiple, small mildly enhancing renal tumors, as demonstrated on this CT image. B, Microscopic appearance of type 1 papillary RCC demonstrating basophilic cells with scant cytoplasm and low-grade nuclei. C, In contrast, type 2 papillary RCC consists of eosinophilic cells with abundant granular cytoplasm and high-grade nuclei.
(Courtesy of Dr. Ming Zhou, Cleveland, OH.)
Two distinct variants of papillary RCC have been described by characteristic cytogenetics, immunostaining profiles and gene expression profiling (see Fig. 49–12) (Renshaw et al, 1997; Störkel et al, 1997; Oyasu, 1998; Delahunt et al, 2001; Eble et al, 2004; Yang et al, 2005). Type 1 papillary RCC, the more common form, consists of basophilic cells with scant cytoplasm; type 2 papillary RCC include potentially more aggressive variants with eosinophilic cells and abundant granular cytoplasm (Delahunt et al, 2001; Amsellem-Ouazana et al, 2002; Leroy et al, 2002; Renshaw, 2002; Choyke et al, 2003; Gunawan et al, 2003; Pignot et al, 2007). The two subtypes of papillary RCC correspond with two familial RCC syndromes: hereditary papillary RCC syndrome (type 1) and hereditary leiomyomatosis and RCC syndrome (type 2) (Rosner et al, 2009). Although mounting molecular and genetic evidence indicate that these 2 subtypes appear to represent distinct entities, subclassification of papillary RCC into type 1 and type 2 is not routinely practiced within the community of genitourinary pathologists at present (Eble et al, 2004; Yang et al, 2005; Pignot et al, 2007). First, although patients with hereditary leiomyomatosis and RCC syndrome have compromised survival relative to those with hereditary papillary RCC syndrome, oncologic outcomes in patients with sporadic forms of these entities are not well defined (Pignot et al, 2007; Gontero et al, 2008). Moreover, evolving definitions of these entities and corresponding differences in interpretation and classification may contribute to the current lack of consensus about this issue (Pignot et al, 2007; Gontero et al, 2008).
The cytogenetic abnormalities associated with papillary RCC are characteristic and include trisomy of chromosomes 7 and 17 and loss of the Y chromosome (Kovacs et al, 1989). Other common findings include gain of chromosomes 12, 16, and 20 and loss of heterozygosity on chromosome 14 (Oyasu, 1998; Brunelli et al, 2003b; Kuroda et al, 2003a; Pavlovich and Schmidt, 2004; Zhou, 2009). VHL mutations are rare in papillary RCC, confirming distinct genetic pathways to tumorigenesis (Kenck et al, 1996; Zambrano et al, 1999; Linehan et al, 2003; Rosner et al, 2009). Papillary RCC is more likely to be hypovascular, perhaps owing to the lack of VHL mutations that regulate VEGF, the primary proangiogenic molecule in RCC (Blath et al, 1976; Herts et al, 2002; Kim JK et al, 2002). As discussed earlier, activating mutations of the c-MET proto-oncogene located on chromosome 7, which encodes the receptor for hepatocyte growth factor, appear to be common and pathogenic in inherited papillary RCC (Schmidt et al, 1997; Pavlovich and Schmidt, 2004). Indeed, this genetic defect is now being targeted for novel treatment approaches with use of small molecule inhibitors (Schmidt et al, 1997; Linehan et al, 2003; Rosner et al, 2009). Interestingly, these mutations have been detected in only about 10% of sporadic papillary RCCs, suggesting divergent molecular pathways (Schmidt et al, 1999).
The prognosis associated with papillary RCC remains controversial. Many older studies suggested a tendency toward low-grade disease, and one literature review indicated that 80% of papillary RCCs are confined to the kidney, which has obvious prognostic implications (Blath et al, 1976; Mancilla-Jimenez et al, 1976; Boczko et al, 1979; Mydlo and Bard, 1987; Farrow, 1997; Chow et al, 2001). On the other hand, more recent studies contain a somewhat increased proportion of high-grade and advanced tumors, often leading to the patient’s demise due in part to the ineffectiveness of current systemic therapies against papillary RCC (Lager et al, 1995; Amin et al, 1997a; Renshaw, 2002; Margulis et al, 2008). One recent study indicates that although venous tumor thrombus was less common with papillary RCC than clear cell RCC, outcomes in this subgroup of patients were compromised (Margulis et al, 2008). Conversely, although lymph node involvement was more common with papillary RCC than clear cell RCC in the same study, patients with lymphatic involvement with papillary RCC had higher cancer-specific survival at 5 years (65% vs. 19%, P = .03). At present, most authors believe that papillary RCC, and type 1 papillary RCC in particular, carry a better prognosis than clear cell RCC when compared grade for grade and stage for stage (Moch et al, 2000; Lau et al, 2002; Amin et al, 2002; Beck et al, 2004; Zhou, 2009).
Papillary adenomas are small (≤5 mm) tumors that resemble papillary RCC under the microscope, are often well encapsulated and low grade, and are commonly found at autopsy (Farrow, 1997; Renshaw, 2002). These lesions, which possess many of the same genetic alterations found in larger papillary RCCs, are benign neoplasms (see Chapter 51).
Chromophobe Renal Cell Carcinoma
Chromophobe RCC, first described by Theones and colleagues in 1985, is a distinctive histologic subtype of RCC that appears to be derived from the cortical portion of the collecting duct (Störkel et al, 1997). It represents 3% to 5% of all RCCs (Oyasu, 1998; Eble et al, 2004; Klatte et al, 2008a). The tumor cells typically exhibit a relatively transparent cytoplasm with a fine reticular pattern that has been described as a “plant cell” appearance (Fig. 49–13). The chromophobic nature of this classic variant is responsible for the name of this histologic subtype (Nagashima, 2000; Kuroda et al, 2003a). However, eosinophilic variants of chromophobic RCC have also been described and constitute about 30% of cases (Theones et al, 1988; Störkel et al, 1997; Latham et al, 1999; Kuroda et al, 2003a). In either case, a perinuclear clearing or “halo” is typically found and electron microscopic findings consist of numerous 150- to 300-nm microvesicles, which are the single most distinctive and defining feature of chromophobe cell carcinoma (Nagashima, 2000; Krishnan and Truong, 2002). These microvesicles characteristically stain positive for Hale colloidal iron, indicating the presence of a mucopolysaccharide unique to chromophobe RCC (see Fig. 49–13) (Theones et al, 1988). Most chromophobe RCCs also stain positive for various cytokeratins and most are negative for vimentin (Cochand-Priollet et al, 1997). Genetic analysis has revealed multiple chromosomal losses, most frequently the whole chromosomes 1, 2, 6, 10, 13, 17, and 21, and flow cytometric analysis has demonstrated hypodiploid DNA content in most cases (Schwerdtle et al, 1996; Bugert et al, 1997; Iqbal et al, 2000; Polascik et al, 2002). Some studies have reported an increased incidence of TP53 mutations in this histologic subtype and upregulated expression of the c-KIT oncogene has also been reported (Contractor et al, 1997; Yamazaki et al, 2003; Pan et al, 2004; Petit et al, 2004; Huo et al, 2005). Chromophobe RCC is commonly seen in the Birt-Hogg-Dubé syndrome, but most cases are sporadic (Pavlovich et al, 2002).
Figure 49–13 A, Chromophobe renal cell carcinoma (RCC) typically appears as a well-circumscribed, homogeneous, tan tumor. B, Chromophobe RCC with admixture of classic (chromophobic) and eosinophilic cells. Characteristic features include distinct cytoplasmic borders, perinuclear halos, and nuclear “raisins.” The classic variant is notable for its “plant cell” appearance. C, Chromophobe RCC stains positive for Hale colloidal iron and demonstrates multiple microvesicles on analysis by electron microscopy.
(Courtesy of Dr. Ming Zhou, Cleveland, OH.)
Most studies of the clinical behavior of chromophobe RCC suggest a better prognosis for localized chromophobe RCC than for clear cell RCC but a poor outcome in the subset of patients with sarcomatoid features or metastatic disease (Polascik et al, 2002; Lau et al, 2002; Cindolo et al, 2005a; Cheville et al, 2003; Peyromaure et al, 2003; Beck et al, 2004; Klatte et al, 2008a). Most early reports suggested a tendency to remain localized despite growth to large size, as well as a predominance of low-grade disease (Theones et al, 1988). Subsequent reports have verified that chromophobe RCC generally presents at an earlier stage, with more than 90% of patients remaining cancer free for 5 or more years after treatment (Crotty et al, 1995; Campbell et al, 1996b; Peyromaure et al, 2003; Cindolo et al, 2005a; Klatte et al, 2008a). However, a more adverse prognosis has been reported for high-grade tumors, including those with sarcomatoid differentiation (Campbell et al, 1996b; Renshaw et al, 1999; Kuroda et al, 2003a; Klatte et al, 2008a). Lymph node and distant metastases are commonly seen with such variants, and systemic disease is poorly responsive to IL-2 (Renshaw et al, 1999; Motzer et al, 2002; Klatte et al, 2008a). Initial data with the mTOR inhibitor temsirolimus indicate that this agent may have some activity for metastatic chromophobe RCC; among 18 patients receiving treatment, 16 had stable disease and 1 had a partial response (Stadler et al, 2007). Clearly, further clinical evaluation will be required to identify the most effective therapeutic agents for patients with metastatic, non–clear cell RCC.
Collecting Duct Carcinoma
Carcinoma of the collecting ducts of Bellini is a relatively rare subtype of RCC, accounting for less than 1% of all RCCs (Kennedy et al, 1990; Rumpelt et al, 1991; Carter et al, 1992; Störkel et al, 1997; Srigley and Eble, 1998; Swartz et al, 2002). Many reported cases have occurred in younger patients, often in the third, fourth, or fifth decades of life (Carter et al, 1992). Most patients are symptomatic at presentation, and up to 50% have metastatic disease at the time of detection (Tokuda et al, 2006). Ulex europaeus agglutinin 1 reactivity and positivity for E-cadherin and c-KIT help to distinguish this entity from aggressive papillary RCC, but this staining profile can also be present in urothelial carcinoma and differential diagnosis often requires careful examination of multiple sections (Rumpelt et al, 1991; Oyasu, 1998; Polascik et al, 2002; Kobayashi et al, 2008). Expression of high-molecular-weight cytokeratin was initially believed to support a collecting duct origin, but more recent studies suggest that this is not a reliable marker for collecting duct carcinoma (Rumpelt et al, 1991; Oyasu, 1998; Polascik et al, 2002; Kobayashi et al, 2008). Small collecting duct carcinomas can arise in a medullary pyramid, but most are large, infiltrative masses and extension into the cortex is common (Pickhardt et al, 2001; Kobayashi et al, 2008). On microscopic examination, these tumors consist of an admixture of dilated tubules and papillary structures typically lined by a single layer of cuboidal cells, often creating a cobblestone appearance. Deletions on chromosome 1q and monosomy of chromosomes 6, 8, 11, 18, 21, and Y have been reported, but the number of tumors analyzed thus far has been limited (Fuzesi et al, 1992; Steiner et al, 1996; Polascik et al, 2002). Most reported cases of collecting duct carcinoma have been high grade, advanced stage, and unresponsive to conventional therapies (Carter et al, 1992; Chao et al, 2002b; Polascik et al, 2002; Mejean et al, 2003; Tokuda et al, 2006; Kobayashi et al, 2008). Reflecting the fact that collecting duct RCC may share features in common with transitional cell carcinoma, some patients with advanced collecting duct RCC have responded to cisplatin- or gemcitabine-based chemotherapy (Milowsky et al, 2002; Peyromaure et al, 2003; Kobayashi et al, 2008).
Renal Medullary Carcinoma
Renal medullary carcinoma is a subtype of RCC that occurs almost exclusively in association with the sickle cell trait. It is typically diagnosed in young African-Americans, often in the third decade of life, and many cases are both locally-advanced and metastatic at the time of diagnosis (Davis et al, 1995b; Polascik et al, 2002; Swartz et al, 2002; Zhou, 2009). Most patients do not respond to therapy and succumb to their disease in a few to several months (Davis et al, 1995b; Herring et al, 1997; Figenshau et al, 1998; Polascik et al, 2002). Mean survival in Davis and coworkers’ series (1995b), which consisted of 34 patients, was only 15 weeks. This tumor shares many histologic features with collecting duct carcinoma, and some consider it a subtype of collecting duct carcinoma or at least a closely related tumor (Störkel et al, 1997; Polascik et al, 2002; Swartz et al, 2002). Renal medullary carcinoma is thought to arise from the calyceal epithelium near the renal papillae but is often highly infiltrative (Davidson et al, 1995; Davis et al, 1995b). The site of origin (renal papillae) and association with sickle cell trait suggest that a relatively hypoxic environment may contribute to tumorigenesis.
Sarcomatoid Differentiation
Sarcomatoid differentiation is found in 1% to 5% of RCCs, most commonly in association with clear cell RCC or chromophobe RCC, but variants of most other subtypes of RCC have been described (Weiss et al, 1995; Störkel et al, 1997; Oyasu, 1998; Delahunt, 1999; dePeralta-Venturina et al, 2001; Kuroda et al, 2003e; Cheville et al, 2004; Zhou, 2009). Most authors now believe that sarcomatoid lesions represent poorly differentiated regions of other histologic subtypes of RCC rather than independently derived tumors (DeLong et al, 1993; Oyasu, 1998; Delahunt, 1999; Eble et al, 2004). A thorough search for epithelial-derived malignant components is almost always fruitful; it is rare to find a truly pure sarcomatoid renal mass (Delahunt, 1999). For this reason, this entity is no longer recognized as a distinct histologic subtype of RCC (Eble et al, 2004). Sarcomatoid differentiation is characterized by spindle cell histology, positive staining for vimentin, infiltrative growth pattern, aggressive local and metastatic behavior, and poor prognosis (Fig. 49–14) (Ro et al, 1987; DeLong et al, 1993; Cangiano et al, 1999; Eble et al, 2004). Invasion of adjacent organs is common, and median survival has been less than 1 year in most series (Ro et al, 1987; Cangiano et al, 1999; Escudier et al, 2002a; Mian et al, 2002; Nanus et al, 2004; Dall’Oglio et al, 2005; Zhou, 2009). Multimodal approaches should be considered if performance status allows based on the extremely poor prognosis with surgery alone and selected reports demonstrating modestly improved response rates in patients receiving IL-2–based immunotherapy, chemotherapy, or targeted molecular therapy after surgery (Cangiano et al, 1999; Bangalore et al, 2001; Fujiwara et al, 2008; Rini et al, 2009).
Figure 49–14 A, Clear cell renal cell carcinoma (RCC) with sarcomatoid differentiation demonstrating extension into the perinephric fat. B, High-grade RCC with typical spindle-cell appearance on the left indicating a component of sarcomatoid differentiation.
(Courtesy of Dr. Ming Zhou, Cleveland, OH.)
Unclassified Renal Cell Carcinoma
Unclassified RCC represents a small minority of cases (<3%) of presumed RCC with features that remain indeterminate even after careful analysis (Zisman et al, 2002a). Most are poorly differentiated and are associated with a highly aggressive biologic behavior and a particularly poor prognosis (Zisman et al, 2002a; Karakiewicz et al, 2007). Included within this “catch-all” category are RCCs with extensive sarcomatoid differentiation and no discernible epithelial component. Advances in molecular diagnostics, such as gene expression profiling, may enable further classification of unusual tumors that previously would have fallen into this category and identify candidate pathways for targeted molecular therapeutics (Yang et al, 2006; Young et al, 2008).
Clinical Presentation
Because of the sequestered location of the kidney within the retroperitoneum, many renal masses remain asymptomatic and nonpalpable until they are advanced. With the more pervasive use of noninvasive imaging for the evaluation of a variety of nonspecific symptom complexes, more than 50% of RCCs are now detected incidentally (Pantuck et al, 2000). Several studies have shown that such tumors are more likely to be confined to the kidney, and a positive impact on survival has been reported, although the potential contributions of lead and length time biases have not been defined (Konnak and Grossman, 1985; Thompson and Peek, 1988; Kessler et al, 1994; Tsui et al, 2000b; Parsons et al, 2001; Lee et al, 2002; Leslie et al, 2003; Nguyen et al, 2006; DeCastro and McKiernan, 2008; Kane et al, 2008).
Symptoms associated with RCC can be due to local tumor growth, hemorrhage, paraneoplastic syndromes, or metastatic disease (Table 49–12). Flank pain is usually due to hemorrhage and clot obstruction, although it can also occur with locally advanced or invasive disease. The classic triad of flank pain, gross hematuria, and palpable abdominal mass is now rarely found (Jayson and Sanders, 1994). This is fortunate because this constellation of findings almost always denotes advanced disease, and some refer to it as the “too late triad.” Before the advent of ultrasonography and CT, most patients with RCC presented with one or more of these signs or symptoms, and many were incurable. Other indicators of advanced disease include constitutional symptoms, such as weight loss, fever, and night sweats, and physical examination findings such as palpable cervical lymphadenopathy, nonreducing varicocele, and bilateral lower extremity edema due to venous involvement. A minority of patients present with symptoms directly related to metastatic disease, such as bone pain or persistent cough. A less common but important presentation of RCC is that of spontaneous perirenal hemorrhage, although the underlying mass is often obscured by the blood. Zhang and colleagues (2002) have shown that more than 50% of patients with perirenal hematoma of unclear etiology have an occult renal tumor, most often AML or RCC. Repeat CT a few months later will often provide a definitive diagnosis.
Table 49–12 Clinical Presentation of Renal Cell Carcinoma
Paraneoplastic syndromes are found in 20% of patients with RCC, and few tumors are associated with the diversity of such syndromes (Table 49–13). In fact, RCC was previously referred to as the internist’s tumor because of the predominance of systemic rather than local manifestations (Sufrin et al, 1989; Gold et al, 1996; Moein and Dehghani, 2000; DeLuca et al, 2002; Kamra et al, 2002; Hagel et al, 2005; Costanzi et al, 2008). Now, a more appropriate name would be the radiologist’s tumor, given the frequency of incidental detection (Parsons et al, 2001; DeCastro and McKiernan, 2008; Kane et al, 2008). Nevertheless, it is still important to evaluate for paraneoplastic phenomena because they can be a source of major morbidity and can affect clinical decision making. Under normal circumstances, the kidney produces 1,25-dihydroxycholecalciferol, renin, erythropoietin, and various prostaglandins, all of which are tightly regulated to maintain homeostasis. RCC may produce these substances in pathologic amounts, and it may also elaborate a variety of other physiologically important factors, such as parathyroid hormone–like peptides, lupus-type anticoagulant, human chorionic gonadotropin, insulin, and various cytokines and inflammatory mediators (Sufrin et al, 1989; Gold et al, 1996; Ather et al, 2002; Elias, 2005). These substances are believed to be responsible for the development of constitutional symptoms such as weight loss, fever, and anemia as well as some of the distinct paraneoplastic syndromes observed with this malignancy (Altundag et al, 2005; Elias, 2005).
Table 49–13 Incidence of Systemic Syndromes Associated with Renal Cell Carcinoma
| SYNDROME | % |
|---|---|
| Elevated erythrocyte sedimentation rate | 55.6 |
| Hypertension | 37.5 |
| Anemia | 36.3 |
| Cachexia, weight loss | 34.5 |
| Pyrexia | 17.2 |
| Abnormal liver function | 14.4 |
| Hypercalcemia | 4.9 |
| Polycythemia | 3.5 |
| Neuromyopathy | 3.2 |
| Amyloidosis | 2.0 |
From Gold PJ, Fefer A, Thompson JA. Paraneoplastic manifestations of renal cell carcinoma. Semin Urol Oncol 1996;14:216–22.
Hypercalcemia has been reported in up to 13% of patients with RCC and can be due to either paraneoplastic phenomena or osteolytic metastatic involvement of the bone (Sufrin et al, 1989; Gold et al, 1996; Magera et al, 2004; Klatte et al, 2007d; Pepper et al, 2007; Schwarzberg and Michaelson, 2009). The production of parathyroid hormone–like peptides is the most common paraneoplastic etiology, although tumor-derived 1,25-dihydroxycholecalciferol and prostaglandins may contribute in a minority of cases (Goldberg et al, 1964; Mangin et al, 1988; Sufrin et al, 1989; Gold et al, 1996; Walther et al, 1997b; Magera et al, 2004; Massfelder et al, 2004; Klatte et al, 2007d; Pepper et al, 2007). Recent data suggest that the expression of parathyroid hormone–like peptides is suppressed by the wild-type VHL protein, and these peptides may act as potent growth factors for RCC (Massfelder et al, 2004). This may account in part for the observation that patients with RCC who present with hypercalcemia have a compromised prognosis, with a relative risk of death from cancer progression of 1.78 compared with patients with normal serum calcium levels (Magera et al, 2004). The signs and symptoms of hypercalcemia are often nonspecific and include nausea, anorexia, fatigue, and decreased deep tendon reflexes. Medical management predominates and includes vigorous hydration followed by diuresis with furosemide and the selective use of bisphosphonates, corticosteroids, or calcitonin (reviewed in Gold et al, 1996; Coleman, 2004; Pepper et al, 2007; Schwarzberg and Michaelson, 2009). Bisphosphonate therapy is now established as standard of care for patients with hypercalcemia of malignancy, as long as renal function is adequate (Schwarzberg and Michaelson, 2009). Zoledronic acid, 4 mg intravenously every 4 weeks, appears to be particularly effective in patients with RCC but must be withheld in the presence of renal insufficiency (Lipton et al, 2003, 2004; Coleman et al, 2004; Schwarzberg and Michaelson, 2009). Indomethacin has also proved useful in a minority of cases (Gold et al, 1996; Walther et al, 1997b). More definite management includes nephrectomy and occasional metastasectomy, depending on the clinical circumstances. Systemic therapy to reduce the burden of disease is also desirable but difficult to achieve in patients with RCC (Goldberg et al, 1980). Hypercalcemia related to extensive osteolytic metastases is much more difficult to palliate because it is not amenable to surgical approaches, but many such patients may respond to bisphosphonate therapy (Lipton et al, 2003, 2004; Coleman et al, 2004). Some patients with hypercalcemia related to osteolytic metastases may also benefit from focused radiation therapy if limited sites of involvement can be identified (Gold et al, 1996).
Hypertension and polycythemia are other important paraneoplastic syndromes commonly found in patients with RCC (Moein and Dehghani, 2000). Hypertension associated with RCC can be secondary to increased production of renin directly by the tumor; compression or encasement of the renal artery or its branches, effectively leading to renal artery stenosis; or arteriovenous fistula within the tumor (Robertson et al, 1967; Sufrin et al, 1989). Less common causes include polycythemia, hypercalcemia, ureteral obstruction, and increased intracranial pressure associated with cerebral metastases (Sufrin et al, 1989). Polycythemia associated with RCC can be due to increased production of erythropoietin, either directly by the tumor or by the adjacent parenchyma in response to hypoxia induced by tumor growth (Gross et al, 1994; Wiesener et al, 2007).
One of the more fascinating paraneoplastic syndromes associated with RCC is nonmetastatic hepatic dysfunction, or Stauffer syndrome, which has been reported in 3% to 20% of cases (Stauffer, 1961; Rosenblum, 1987; Giannakos et al, 2005; Moria et al, 2006; Young et al, 2008). Almost all patients with Stauffer syndrome have an elevated serum alkaline phosphatase level, 67% have elevated prothrombin time or hypoalbuminemia, and 20% to 30% have elevated serum bilirubin or transaminase levels (Sufrin et al, 1989). Other common findings include thrombocytopenia and neutropenia, and typical symptoms include fever and weight loss, which is not surprising given that many patients are found to harbor discrete regions of hepatic necrosis (Sufrin et al, 1989; Gold et al, 1996). Hepatic metastases must be excluded. Biopsy, when indicated, often demonstrates nonspecific hepatitis associated with a prominent lymphocytic infiltrate (Hanash, 1982). Elevated serum levels of IL-6 have been found in patients with Stauffer syndrome, and it is believed that this and other cytokines may play a pathogenic role (Blay et al, 1997). Hepatic function normalizes after nephrectomy in 60% to 70% of cases. Persistence or recurrence of hepatic dysfunction is almost always indicative of the presence of viable tumor and thus represents a poor prognostic finding (reviewed in Sufrin, 1989).
A variety of other less common but distinct paraneoplastic syndromes associated with RCC have been reviewed by Sufrin and colleagues (1989), including Cushing syndrome, hyperglycemia, galactorrhea, neuromyopathy, clotting disorders, and cerebellar ataxia (Ather et al, 2002; Kamra et al, 2002; Hagel et al, 2005; Mohammed et al, 2006; Ammar et al, 2008).
In general, treatment of paraneoplastic syndromes associated with RCC has required surgical excision or systemic therapy and, except for hypercalcemia, medical therapies have not proved helpful.
Screening and Clinical Associations
A number of factors make screening for RCC appealing (Cohn and Campbell, 2000; Herts, 2005; Carrizosa and Godley, 2009). Most important, RCC remains primarily a surgical disease requiring early diagnosis to optimize the opportunity for cure. Unfortunately, our ability to salvage patients with advanced disease remains limited. Consistent with these observations, several studies have demonstrated an apparent advantage to early or incidental diagnosis of RCC (Konnak and Grossman, 1985; Thompson and Peek, 1988; Kessler et al, 1994; Cohn and Campbell, 2000; Tsui et al, 2000b; Parsons et al, 2001; Lee et al, 2002; Leslie et al, 2003).
The primary factor that limits the widespread implementation of screening for RCC is the relatively low incidence of RCC in the general population (approximately 12 cases per 100,000 population/year) (Wallen et al, 2007; DeCastro and McKiernon, 2008; Woldrich et al, 2008; Carrizosa and Godley, 2009). In this setting a screening test must be almost 100% specific to avoid an unacceptably high false-positive rate, which would lead to unnecessary, expensive, and potentially harmful diagnostic or therapeutic procedures. In addition, even if the test were 100% sensitive and specific, the yield from screening would be so low that it would not be considered cost effective (Cohn and Campbell, 2000; Carrizosa and Godley, 2009). Even when one considers populations with established risk factors for RCC, such as male sex, increased age, and heavy tobacco use, generalized screening would be difficult to justify because the increase in relative risk associated with each of these factors is at best twofold to threefold (Paganini-Hill et al, 1988; Cohn and Campbell, 2000; Carrizosa and Godley, 2009). Another confounding factor is the prevalence of clinically insignificant tumors such as renal adenomas, which are found at autopsy in 10% to 20% of individuals, and other benign or slow-growing tumors (Xipell, 1971; Bosniak et al, 1995; Cohn and Campbell, 2000; Pantuck et al, 2000; Parsons et al, 2001). There is clearly a risk that such clinically insignificant lesions could be detected, leading to unnecessary evaluation and treatment (Pantuck et al, 2000; Parsons et al, 2001). All these factors recommend against generalized screening efforts for the detection of RCC.
Review of the literature describing the use of dipstick analysis for hematuria and ultrasonography or CT for screening for RCC supports these conclusions (Cohn and Campbell, 2000; Herst, 2005; Carrizosa and Godley, 2009). Urinalysis is simple and inexpensive, but the yield of RCC in several screening studies has been exceedingly low (Mohr et al, 1986; Thompson, 1987; Mariani et al, 1989; Murakami et al, 1990). In part, this may be because small RCCs are often not associated with hematuria (gross or microscopic) because this is a parenchymal rather than an urothelium-based malignant neoplasm (Tosaka et al, 1990). The incidence of RCC in ultrasound or CT screening studies has ranged from 23 to 300 per 100,000 population, and an apparent advantage of screening has been debated because an increased proportion of organ-confined tumors has been found in screened populations compared with historical controls (Sohma et al, 1989; Tosaka et al, 1990; Spouge et al, 1996; Tsuboi et al, 2000; Filipas et al, 2003; Fenton and Weiss, 2004; Herts, 2005; Malaeb et al, 2005; Turney et al, 2006; Carrizosa and Godley, 2009). However, although the yield of RCC has been higher than expected, it is still relatively low; and it is unlikely that such efforts would be considered cost effective (Cohn and Campbell, 2000; Herts, 2005). Overall, the yield of RCC in such studies is an order of magnitude lower than the yield from prostate-specific antigen–based screening for prostate cancer, and many of the same controversies about lead and length time biases that have plagued the debate about screening for prostate cancer also apply to RCC (Cohn and Campbell, 2000; Pantuck et al, 2000; Parsons et al, 2001; Fenton and Weiss, 2004; Herts, 2005; Carrizosa and Godley, 2009). Because of these considerations, it is difficult to justify generalized screening efforts for RCC given the currently available technology.
Several investigators are now reporting novel molecular assays to detect RCC-related biomarkers in the urine or serum that may substantially alter our perspective about screening for RCC. These assays can detect microsatellite alterations in the DNA, VHL gene mutations or hypermethylation, expression of RCC-specific proteins such as CA-9, or upregulation of angiogenic factors, including VEGF (Eisenberger et al, 1999; De la Taille et al, 2000; Chang et al, 2001; Ashida et al, 2003; Battagli et al, 2003; Chen and Getzenberg, 2004; Hoque et al, 2004; Uzzo et al, 2004). Proteomic profiling of the urine to detect RCC-specific markers also holds much promise for the future (Rogers et al, 2003).
For now, however, the focus of screening for RCC must be on well-defined target populations, such as patients with end-stage renal disease and acquired renal cystic disease, tuberous sclerosis, and familial RCC (Table 49–14). Eighty percent of patients with end-stage renal disease eventually develop acquired renal cystic disease, and 1% to 2% of this subgroup develops RCC (Ishikawa et al, 1980, 1990; Matson and Cohen, 1990; Levine et al, 1991; Cheuk et al, 2002; Brown, 2004). Overall, the relative risk of RCC in patients with end-stage renal disease has been estimated to be 5- to 20-fold higher than that in the general population (Ishikawa et al, 1990, 1991, 2004; Matson and Cohen, 1990; Levine et al, 1991; Levine, 1992; Cowie, 2002; Denton et al, 2002; Brown, 2004; Farivar-Mohseni et al, 2006). Fifteen percent of patients with RCC in the setting of end-stage renal disease have metastases at the time of presentation, and many such patients die of malignant progression (Levine et al, 1991; Brown, 2004; Ishikawa, 2004). Given these considerations, screening for RCC is recommended in this population, which is substantial, representing almost 300,000 patients in the United States alone. There are, however, a number of problems associated with screening this population of patients. These include concerns about short life expectancy, increased incidence of adenomas (20% to 40% vs. 10% to 20% in the general population), complexity of imaging given the altered architecture associated with acquired renal cystic disease, and inevitable cost-related issues (Mindell, 1989; Levine et al, 1991, 1997; Sarasin et al, 1995; Cowie, 2002). A reasonable compromise for patients with end-stage renal disease is to target those without other major comorbidities, to delay screening until the third year on dialysis, and to take into account the sex and type of renal replacement therapy, although data about the last factors are admittedly controversial (Brown, 2004; Carrizosa and Godley, 2009). Ishizuka and colleagues (2000) have demonstrated elevated serum levels of VEGF in dialysis patients with RCC, suggesting a potential role for biomarkers for screening this population in the future. Interestingly, recent data suggest that renal transplant recipients remain at high risk for RCC in the native kidneys, and Neuzillet and colleagues (2005) have recommended continued periodic radiologic screening even after transplantation (Ianhez et al, 2007).
Table 49–14 Screening for Renal Cell Carcinoma: Target Populations
| Patients with End-Stage Renal Disease |
| Patients with Known von Hippel-Lindau Syndrome |
| Relatives of Patients with von Hippel-Lindau Syndrome |
| Relatives of Patients with Other Familial Forms of Renal Cell Carcinoma |
| Patients with Tuberous Sclerosis |
| Patients with Autosomal-Dominant Polycystic Kidney Disease |
An increased incidence of RCC has also been debated in tuberous sclerosis, an autosomal dominant disorder in which patients can develop adenoma sebaceum (a distinctive skin lesion), epilepsy, mental retardation, and renal cysts and AMLs (Shapiro et al, 1984; Bernstein et al, 1986; Washecka and Hanna, 1991; Aoyama et al, 1996; Bjornsson et al, 1996; Robertson et al, 1996; Sampson, 1996; Tello et al, 1998; Choyke et al, 2003; Lendvay and Marshall, 2003; Narayanan, 2003; Cohen and Zhou, 2005; Radkowski et al, 2006). Many cases of RCC in this syndrome have been characterized by early onset and multifocality, suggesting a genetic predisposition (Washecka and Hanna, 1991; Lendvay and Marshall, 2003). In addition, the Eker rat, which is mutant for the rodent homologue of the TSC2 gene responsible for the development of tuberous sclerosis in humans, develops RCC at high frequency, as do TSC2-deficient knockout mice (Yeung et al, 1994; Kobayashi et al, 1997, 1999; Hino et al, 1999; Lendvay and Marshall, 2003; McDorman and Wolf, 2002). Furthermore, an increased incidence of TSC2 mutations has been found in human RCC (Duffy et al, 2002; Lendvay and Marshall, 2003), and Liu and colleagues (2003) have shown that loss of this tumor suppressor gene leads to upregulated expression of VEGF through an mTOR and HIF-2α–mediated mechanism, analogous to the role of the VHL protein. Such biologic and clinical observations argue in favor of an increased predisposition for RCC in this syndrome, which is consistent with most, although admittedly not all, relevant demographic data (Shapiro et al, 1984; Bernstein et al, 1986; Washecka and Hanna, 1991; Aoyama et al, 1996; Bjornsson et al, 1996; Robertson et al, 1996; Sampson, 1996; Tello et al, 1998; Lendvay and Marshall, 2003; Martignoni et al, 2003; Cohen and Zhou, 2005; Rakowski et al, 2006; Nathanson and Stephenson, 2009). A reasonable conclusion is that periodic renal imaging should be pursued in patients with tuberous sclerosis; such a policy will also facilitate follow-up for the development and progression of AML.
Screening for RCC in autosomal dominant polycystic kidney disease (ADPKD) was previously recommended, but several factors have prompted a reassessment of this policy. More recent studies suggest no significantly increased risk of RCC in ADPKD, and imaging is extremely difficult in this population related to the altered intrarenal architecture (Torres et al, 1985; Gregoire et al, 1987; Keith et al, 1994; Sessa et al, 1997; Soderdahl et al, 1997; Cohn and Campbell, 2000; Gupta et al, 2000; Mosetti et al, 2003). The increased incidence of adenomas in ADPKD would also mitigate against a potential benefit of screening (Torres et al, 1985; Gregoire et al, 1987). Taken together, these considerations suggest that screening for RCC in patients with ADPKD should not be pursued.
Special consideration should also be given to von Hippel-Lindau disease in any discussion of the value of screening for RCC. This syndrome should be considered in any patient with early-onset or multifocal RCC or RCC in combination with any of the following: a history of visual or neurologic disorders; a family history of blindness, central nervous system tumors, or renal cancer; or coexistent pancreatic cysts, epididymal lesions, or inner ear tumors (Neumann and Zbar, 1997; Choyke et al, 2003; Linehan et al, 2003; Pavlovich and Schmidt, 2004; Vira et al, 2007; Pfaffenroth and Linehan, 2008; Coleman, 2008; Hansel and Rini, 2008; Nathanson and Stephenson, 2009). Patients suspected of having von Hippel-Lindau disease, or the appropriate relatives of those with documented disease, should strongly consider genetic evaluation. The entire VHL gene has now been sequenced and sophisticated molecular analysis is readily available and offers a number of important advantages (Zbar et al, 1999; Linehan et al, 2003; Vira et al, 2007; Pfaffenroth and Linehan, 2008). Patients with germline mutations can be identified and offered clinical and radiographic screening that can identify the major manifestations of von Hippel-Lindau disease at a presymptomatic phase, allowing potential amelioration of the considerable morbidity associated with this syndrome (Glenn et al, 1992; Maranchie and Linehan, 2003; Vira et al, 2007; Pfaffenroth and Linehan, 2008). Investigators at the National Institutes of Health have recommended that such patients be evaluated with (1) annual physical examination and ophthalmologic evaluation beginning in infancy; (2) estimation of urinary catecholamines at the age of 2 years and every 1 to 2 years thereafter; (3) MRI of the central nervous system biannually beginning at the age of 11 years; (4) ultrasound examination of the abdomen and pelvis annually beginning at the age of 11 years, followed by CT every 6 months if cysts or tumors develop; and (5) periodic auditory examinations (Choyke et al, 1995; Friedrich, 1999; Maranchie and Linehan, 2003). Less intensive protocols have also been advocated, although all relevant organ systems should be addressed (Fraser et al, 2007; Foulkes and Hodgson, 1998). Individuals who are found to be wild type for both alleles of VHL also benefit because they can be spared much of the expense and anxiety associated with such intensive surveillance protocols.
Molecular screening is also available for patients suspected of having hereditary papillary RCC and other familial forms of RCC and should be discussed with appropriate family members (Schmidt et al, 1997; Linehan et al, 2003; Zbar et al, 2007; Vira et al, 2007; Pfaffenroth and Linehan, 2008; Coleman, 2008; Hansel and Rini, 2009, Nathanson and Stephenson, 2009). Again, individuals at risk, as defined by the presence of mutations of the c-MET proto-oncogene or other relevant genetic alterations, and those with suggestive clinical or family histories should be evaluated with abdominal ultrasonography or CT at periodic intervals. Further testing may be indicated according to the syndrome involved.
Staging
Until the 1990s the most commonly used staging system for RCC was Robson’s modification of the system of Flocks and Kadesky, and this schema is still embedded in the mindset of many urologists (Fig. 49–15) (Robson, 1963; Robson et al, 1969). In retrospect, the limitations of this classification scheme are readily evident. The primary problem can be found in stage III, where tumors with lymphatic metastases, a very poor prognostic finding, were combined with those with venous involvement, many of which can be treated and potentially cured with an aggressive surgical approach (Gettman and Blute, 2002; Leibovich et al, 2003c; Nguyen and Campbell, 2006). Further imprecision resulted from the fact that the extent of venous involvement was not delineated in this system, and tumor size, an important prognostic parameter, was not incorporated. The net effect was that the prognostic significance of the various stages was blunted, with some studies reporting equivalent survival for patients with stage II and stage III tumors (Skinner et al, 1971).
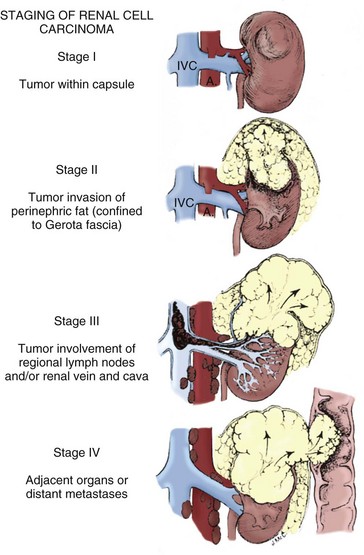
Figure 49–15 Staging of renal cell carcinoma as proposed by Holland, in accordance with classification systems developed by Robson, Murphy, and Flocks and Kadesky. A, aorta; IVC, inferior vena cava.
(From Holland JM. Cancer of the kidney: natural history and staging. Cancer 1973;32:1030.
The tumor, nodes, and metastasis (TNM) system proposed by the Union International Contre le Cancer (UICC) represented a major improvement because it separated tumors with venous involvement from those with lymphatic invasion and defined the anatomic extent of disease more explicitly (Beahrs et al, 1988; Leibovich et al, 2003b; Nguyen and Campbell, 2006; DeCastro and McKiernon, 2008). Another advantage of the TNM system is that it has facilitated comparison of clinical and pathologic data from various centers across the globe (Leung and Ghavamian, 2002; Leibovich et al, 2003b; Nguyen and Campbell, 2006).
In 2009 the American Joint Committee on Cancer (AJCC) proposed a revision of the TNM system that is now the recommended staging system for RCC (Table 49–15) (Edge et al, 2010). The TNM classification for RCC has undergone several modifications in the past 3 decades in an effort to more accurately reflect tumor biology and prognosis and to guide clinical management. It is important to be cognizant of these changes when comparing studies from different eras (Nguyen and Campbell, 2006). In 1997 the previous division of stages T1 and T2 at tumor size of 2.5 cm was abandoned because several studies showed no prognostic significance at this level. Analysis of the Surveillance, Epidemiology, and End Results (SEER) program database demonstrated survival differences associated with 5-, 7.5-, and 10-cm cut points, and the 7-cm cut point between stages T1 and T2 was adopted because it reflected the mean tumor size in the database (Guinan et al, 1995a; Gettman et al, 2001b; Ficcarra et al, 2006; Salama et al, 2005). In the 2002 version stage T1 was subdivided: T1a represents tumor size of 4 cm or less, and T1b represents tumor size between 4 and 7 cm, reflecting data in the literature demonstrating excellent outcomes for patients with small (≤4 cm), unilateral, confined tumors managed by either partial or radical nephrectomy (Butler et al, 1994; Lerner et al, 1996; Hafez et al, 1999; Igarashi et al, 2001; Frank et al, 2004a, 2005b, Nguyen and Campbell, 2006). The most recent change for organ-confined tumors is a subdivision of T2 tumors: T2a tumor represents tumors between 7 and 10 cm, and T2b represents tumors greater than 10 cm (see Table 49–15), supported by a number of studies demonstrating prognostic relevance at this breakpoint (Frank et al, 2005b; Klatte et al, 2007c).
Other major revisions in 2009 included a reclassification of tumors with adrenal metastasis, venous thrombi, and lymphatic involvement, representing a substantial departure from previous staging paradigms for RCC (Table 49–16) (Edge et al, 2010). Contiguous extension of tumor into the ipsilateral adrenal gland is now classified as T4 and metastatic involvement of either adrenal as M1, reflecting likely patterns of dissemination. The poor prognosis of adrenal involvement from RCC is well documented and supported this important change (Sagalowsky et al, 1994; Sandock et al, 1997; Paul et al, 2001a; Von Knobloch et al, 2004; Lam et al, 2005a; Shvarts et al, 2005a; Siemer et al, 2005; Thompson et al, 2005a; Nguyen and Campbell, 2006; Kirkali et al, 2007). The favorable prognosis of isolated renal venous thrombi prompted a downgrading from stage T3b to stage T3a in the 2009 version (Moinzadeh and Libertino, 2004; DeKernion, 2005; Leibovich et al, 2005; Shvarts et al, 2005a; Thompson et al, 2005a; Ficarra and Artibani, 2006; Gofrit et al, 2007; Ficarra et al, 2007b; Margulis et al, 2007c). Finally, lymphatic extension, which previously was subdivided based on the number of involved nodes, has now been compressed to simplify this aspect of the staging process, because prognostic relevance of the previous version was not observed (Edge et al, 2010).
Table 49–16 2010 Revision of AJCC Staging for Kidney Cancer (7th Edition): Summary of Substantial Changes
Data from Edge SB, Byrd DR, Compton CC, et al, editors. AJCC cancer staging manual. 7th ed. New York: Springer; 2010. p. 479–89.
TNM staging classically is defined by the most advanced feature demonstrated by the tumor, yet important prognostic information can be lost in the process. Many tumors exhibit multiple adverse findings, such as high-level tumor thrombus along with ipsilateral adrenal involvement. Ideally all of the relevant anatomic staging information would be captured, at least parenthetically (e.g., pT4 [ipsilateral adrenal involvement; also exhibiting IVC thrombus above the diaphragm]). Future staging systems will ideally capture all of this information, because a number of investigators have demonstrated strong prognostic relevance to such combinations of parameters (Liebovich et al, 2005; Shvarts et al, 2005a; Thompson et al, 2005a; Fujita et al, 2007; Ficarra et al, 2007a, 2007b; Klatte et al, 2007c; Terrone et al, 2008).
The clinical staging of renal malignant disease begins with a thorough history, physical examination, and judicious use of laboratory tests (Nguyen and Campbell, 2006; DeCastro and McKiernon, 2008). Presentation with systemic symptoms such as significant weight loss (>10% of body weight), cachexia, or poor performance status all suggest advanced disease, as do physical examination findings of a palpable mass or lymphadenopathy. A nonreducing varicocele and lower extremity edema suggest venous involvement. Abnormal liver function test results, elevated serum alkaline phosphatase or lactate dehydrogenase level or sedimentation rate, hypercalcemia, and significant anemia point to the probability of advanced disease (Gelb, 1997; Srigley et al, 1997; Nguyen and Campbell, 2006; Lane and Kattan, 2008).
The radiographic staging of RCC can be accomplished in most cases with a high-quality abdominal CT scan and a routine chest radiograph, with selective use of MRI and other studies as indicated (reviewed in Bechtold and Zagoria, 1997; Choyke et al, 2001; Griffin et al, 2007, Zhang et al, 2007a; Ng et al, 2008; Herts, 2009). MRI can be reserved primarily for patients with locally advanced malignant disease, equivocal venous involvement, or allergy to intravenous contrast material (Choyke, 1997; Pretorius et al, 2000; Choyke et al, 2001; Zhang et al, 2007a; Herts, 2009). CT findings suggestive of extension into the perinephric fat include perinephric stranding (Fig. 49–16), which is a nonspecific finding, or a distinct soft tissue density within the perinephric space, which is a more definitive but uncommon finding (Bechtold and Zagoria, 1997; Herts, 2009). Overall, the accuracy of CT or MRI for detection of involvement of the perinephric fat is low, reflecting the fact that extracapsular spread often occurs microscopically (Pretorius et al, 2000; Choyke et al, 2001; Leung and Ghavamian, 2002; Kamel et al, 2004; Zhang et al, 2007a). Many of these potentially locally advanced cases are managed with radical nephrectomy, so the clinical relevance of this imprecision in staging is blunted. Ipsilateral adrenal involvement can be assessed with reasonable accuracy through a combination of preoperative CT and intraoperative inspection. Patients with an enlarged or indistinct adrenal gland on CT, extensive malignant replacement of the kidney, or palpably abnormal adrenal gland are at risk for ipsilateral adrenal involvement and should be managed accordingly (Sagalowsky et al, 1994; Sandock et al, 1997; Paul et al, 2001a; Sawai et al, 2002; Kobayashi et al, 2003; Zhang et al, 2007a; Ng et al, 2008; Lane et al, 2009). Enlarged hilar or retroperitoneal lymph nodes 2 cm or more in diameter on CT almost always harbor malignant change, but this should be confirmed by surgical exploration or percutaneous biopsy if the patient is not a surgical candidate. Many smaller nodes prove to be inflammatory rather than neoplastic and should not preclude surgical therapy (Studer et al, 1990; Choyke et al, 2001; Israel and Bosniak, 2003a; Zhang et al, 2007a; Bach and Zhang, 2008; Ng et al, 2008; Herts, 2009). MRI can add specificity to the evaluation of retroperitoneal nodes by distinguishing vascular structures from lymphatic ones (Bechtold and Zagoria, 1997; Bassignani, 2006). MRI is still the premier study for evaluation of invasion of tumor into adjacent structures and for surgical planning in these challenging cases (Bechtold and Zagoria, 1997; Choyke, 1997; Pretorius et al, 2000; Choyke et al, 2001; Bassignani, 2006; Bach and Zhang, 2008; Herts, 2009). Obliteration of the fat plane between the tumor and adjacent organs (e.g., the liver) can be a misleading finding on CT and should prompt further imaging with MRI. In reality, surgical exploration is often required to make an absolute differentiation.
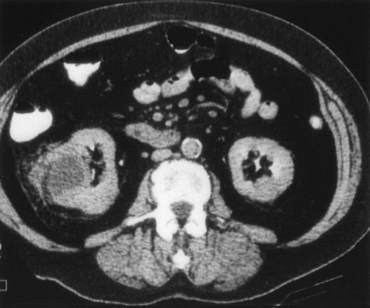
Figure 49–16 Contrast-enhanced CT scan shows right renal tumor with perinephric stranding suggesting invasion of the perinephric fat.
The sensitivities of CT for detection of renal venous tumor thrombus and IVC involvement are 78% and 96%, respectively (Bechtold and Zagoria, 1997; Zhang et al, 2007a; Ng et al, 2008; Herts, 2009). CT findings suggestive of venous involvement include venous enlargement, abrupt change in the caliber of the vein, and intraluminal areas of decreased density or filling defects. The diagnosis is strengthened by the demonstration of collateral vessels. Most false-negative findings occur in patients with right-sided tumors in whom the short length of the vein and the mass effect from the tumor combine to make detection of the tumor thrombus difficult (Bechtold and Zagoria, 1997; Herts, 2009). Fortunately, most such cases are readily identified and dealt with intraoperatively. MRI is well established as the premier study for the evaluation and staging of IVC tumor thrombus, although recent data suggest that multiplanar CT is likely equivalent (Choyke et al, 1987; Goldfarb et al, 1990; Kallman et al, 1992; Bechtold and Zagoria, 1997; Choyke, 1997; Oto et al, 1998; Sun et al, 1999; Pretorius et al, 2000; Sohaib et al, 2002; Israel and Bosniak, 2003a; Ergen et al, 2004; Hallscheidt et al, 2005; Zhang et al, 2007a; Ng et al, 2008). MRI and multiplanar CT are noninvasive methods that provide reliable information about both the cephalad and caudal extent of the thrombus and can often distinguish bland from tumor thrombus (Pretorius et al, 2000; Choyke et al, 2001, Bassignani, 2006; Bach and Zhang, 2008; Herts, 2009). Venacavography is now best reserved for patients with equivocal MRI or CT findings or for patients who cannot tolerate or have other contraindications to cross-sectional imaging. Transesophageal echocardiography also appears to be accurate for establishing the cephalad extent of the tumor thrombus, but it is invasive and provides no distinct advantages over MRI or CT in the preoperative setting (Glazer and Novick, 1997). Doppler ultrasonography is operator dependent and does not provide the anatomic resolution available with MRI or multiplanar CT (Habboub et al, 1997).
Metastatic evaluation in all cases should include a routine chest radiograph, careful and systematic review of the abdominal and pelvic CT or MRI findings, and liver function tests (Choyke et al, 1987; Griffin et al, 2007; Zhang et al, 2007a; Ng et al, 2008; Herts, 2009). Most investigators agree that a bone scintiscan can be reserved for patients with elevated serum alkaline phosphatase or bone pain and that a chest CT scan can be reserved for patients with pulmonary symptoms or an abnormal chest radiograph (Lim and Carter, 1993; Seaman et al, 1996; Choyke et al, 2001). However, patients with locally advanced disease, enlarged retroperitoneal lymph nodes, or significant comorbid disease may mandate more thorough imaging to rule out metastatic disease and to aid in treatment planning (Choyke et al, 2001; Griffin et al, 2007). As always, evaluation and management must be individualized on the basis of the clinical circumstances. Shvarts and colleagues (2004) have shown that performance status is a powerful predictor of bone metastasis. In their analysis, patients with good performance status (Eastern Cooperative Oncology Group performance status score of 0), no evidence of extraosseous metastases, and no bone pain were at extremely low risk and did not benefit from bone scintigraphy. They recommended a bone scintiscan for all other patients, and the incidence of bone metastasis in this group was above 15%.
Positron emission tomography (PET) has also been investigated for patients with high risk of metastatic RCC, with most studies showing good specificity but suboptimal sensitivity. At present its best role is for patients with equivocal findings on conventional imaging. In this setting an abnormal PET scan may indicate metastatic disease and could strongly influence further evaluation and management (Hoh et al, 1998; Ramdave et al, 2001; Nimeh et al, 2002; Jadvar et al, 2003; Kang et al, 2004; Lawrentschuk et al, 2006; Griffin et al, 2007; Powles et al, 2007; Bouchelouche et al, 2008; Nieh, 2009). PET/CT combined with radiolabeled monoclonal antibody to CA-9 is also being explored in this population for molecular imaging of clear cell RCC (Brouwers et al, 2002; Divgi et al, 2007; Zhang, 2008). Biopsy of the primary tumor and/or potential metastatic sites is also selectively required as part of the staging process.
Prognosis
Important prognostic factors for cancer-specific survival in patients with localized RCC include specific clinical signs or symptoms, tumor-related factors, and various laboratory findings (Table 49–17) (Lane and Kattan, 2008). Overall, tumor-related factors such as pathologic stage, tumor size, nuclear grade, and histologic subtype have the greatest utility on an independent basis. However, an integrative approach, combining a variety of factors that have proved to have independent value on multivariate analysis, appears to be most powerful (Kattan et al, 2001; Zisman et al, 2001b; Frank et al, 2002; Sorbellini et al, 2005; Lane and Kattan, 2008; Parker et al, 2009; Isbarn and Karakiewicz, 2009). Patient-related factors, such as age, CKD, and co-morbidity, have a significant impact on overall survival in some patients and should be a primary consideration during treatment planning for patients with localized RCC (Hollingsworth et al, 2006; Berger et al, 2009; Pettus et al, 2008).
Clinical findings suggestive of a compromised prognosis in patients with presumed localized RCC include symptomatic presentation, weight loss of more than 10% of body weight, and poor performance status (Gelb, 1997; Srigley et al, 1997; Kattan et al, 2001; Zisman et al, 2001b; Kim HL et al, 2003; Kontak and Campbell, 2003; Schips et al, 2003; Patard et al, 2004a). Anemia, thrombocytosis, hypercalcemia, albuminuria, elevated serum alkaline phosphatase, C-reactive protein, lactate dehydrogenase, or erythrocyte sedimentation rate, and other paraneoplastic signs or symptoms have also correlated with poor outcomes for patients with RCC (Gelb, 1997; Srigley et al, 1997; Symbas et al, 2000; Jacobsen et al, 2000; O’Keefe et al, 2002; Kim HL et al, 2003; Kontak and Campbell, 2003; Vaglio et al, 2003; Patard et al, 2004a; Karakiewicz et al, 2007; Magera et al, 2008). Although abnormal values are more common in patients with advanced RCC, some of these abnormalities, including hypercalcemia, anemia, and elevated erythrocyte sedimentation rate, were independent predictors of cancer-specific mortality in patients with localized clear cell RCC after accounting for other major prognostic factors (Magera et al, 2008).
Pathologic stage has proved to be the single most important prognostic factor for RCC (Thrasher and Paulson, 1993; Delahunt, 1998; Kontak and Campbell, 2003; Leibovich et al, 2005a; Lane and Kattan, 2008; Kanao et al, 2009). The RCC TNM staging system clearly distinguishes between patient groups with different predicted cancer-specific outcomes (Table 49–18), confirming that the extent of locoregional or systemic disease at diagnosis is the primary determinant of outcome for this disease (Bassil et al, 1985; Hermanek and Schrott, 1990; Kontak and Campbell, 2003; Lane and Kattan, 2008). Previous studies confirmed that the 2002 modification of the TNM system provided better prognostic ability than its predecessors (Ficarra et al, 2005; Frank et al, 2005a), and it is anticipated that the 2010 TNM staging system will impart further improvements (Frank et al, 2005a; Leibovich et al, 2005a; Thompson et al, 2005a).
Several studies demonstrate 5-year survival rates of 70% to 90% for organ-confined disease and document a 15% to 20% reduction in survival associated with invasion of the perinephric fat (Kontak and Campbell, 2003; Leibovich et al, 2005a; Lane and Kattan, 2008). Renal sinus involvement is classified along with perinephric fat invasion as T3a, and several studies suggest that these patients may be at even higher risk for metastasis related to increased access to the venous system (Bonsib et al, 2000; Uzzo et al, 2002; Thompson et al, 2005a; Margulis et al, 2007b; Bedke et al, 2009; Bertini et al, 2009). Collecting system invasion has also been shown to confer poorer prognosis in otherwise organ-confined RCC (Uzzo et al, 2002; Klatte et al, 2007a). Several reports have shown that most patients with direct or metastatic ipsilateral adrenal involvement, which is found in 1% to 2% of cases, eventually succumb to systemic disease progression, suggesting a hematogenous route of dissemination or a highly invasive phenotype (Sagalowsky et al, 1994; Sandock et al, 1997; Paul et al, 2001b; Von Knobloch et al, 2004; Siemer et al, 2005; Thompson et al, 2005a). The most recent staging system now reclassifies tumor as T4 if there is direct invasion of the adrenal gland or otherwise as M1, to reflect this poor prognosis (Guinan et al, 1997; Thompson et al, 2005a).
Venous involvement was once thought to be a very poor prognostic finding for RCC, but several reports demonstrate that many patients with tumor thrombi can be salvaged with an aggressive surgical approach. These studies document 45% to 69% 5-year survival rates for patients with venous tumor thrombi as long as the tumor is otherwise confined to the kidney (Novick et al, 1990; Thrasher and Paulson, 1993; Staehler and Brkovic, 2000; Quek et al, 2001; Zisman et al, 2003; Leibovich et al, 2005b; Blute et al, 2007; Haferkamp et al, 2007; Klatte et al, 2007b; Wotkowicz et al, 2008; Subramanian et al, 2009). At one extreme, Golimbu and associates (1986) reported 84% 5-year survival in the best of circumstances—tumor thrombus limited to the main renal vein and tumor otherwise confined to the kidney. Patients with venous tumor thrombi and concomitant lymph node or systemic metastases have markedly decreased survival, and those with tumor extending into the perinephric fat have intermediate survival (Montie et al, 1991; Glazer and Novick, 1996; Gettman et al, 1999; Naitoh et al, 1999; Sweeney et al, 2002b; Bissada et al, 2003; Kim HL et al, 2004c; Moinzadeh and Libertino, 2004; Leibovich et al, 2005b; Klatte et al, 2007b; Wotkowicz et al, 2008). The most recent version of the TNM system advocates capturing all such adverse features during the staging process.
The prognostic significance of the cephalad extent of tumor thrombus has been controversial, and it is difficult to compare various series because of selection biases and related covariables (Leibovich et al, 2005b; Wotkowicz et al, 2008). In several series the incidence of advanced locoregional or systemic disease increased with the cephalad extent of the tumor thrombus, accounting for the reduced survival associated with tumor thrombus extending into or above the level of the hepatic veins (Sosa et al, 1984; Thrasher and Paulson, 1993; Kim HL et al, 2004a; Wotkowicz et al, 2008). However, other data suggest that the cephalad extent of tumor thrombus is not of prognostic significance as long as the tumor is otherwise confined (Libertino et al, 1987; Glazer and Novick, 1996; Staehler and Brkovic, 2000; Blute et al, 2007). Direct invasion of the wall of the vein appears to be a more important prognostic factor than level of tumor thrombus and is now classified as pT3c independent of the level of tumor thrombus (Hatcher et al, 1991; Zini et al, 2008).
The major drop in prognosis comes in patients whose tumor extends beyond the Gerota fascia to involve contiguous organs (stage T4), which is rarely associated with 5-year survival, and in patients with lymph node or systemic metastases (Thrasher and Paulson, 1993; Thompson et al, 2005a). Lymph node involvement has long been recognized as a dire prognostic sign because it is associated with 5- and 10-year survival rates of 5% to 30% and 0% to 5%, respectively (Bassil et al, 1985; Phillips and Taneja, 2004). Systemic metastases also portend a particularly poor prognosis for RCC, traditionally with 1-year survival of less than 50%, 5-year survival of 5% to 30%, and 10-year survival of 0% to 5%, although these numbers have improved modestly in the era of targeted treatments (Motzer et al, 1999; Motzer and Russo, 2000; Négrier et al, 2002; Sella et al, 2003; Rini et al, 2009). Patients presenting with synchronous metastases fare worse, with many patients dying of disease progression within a year (Motzer et al, 1999, 2004; Mekhail et al, 2005; Leibovich et al, 2005b; Rini et al, 2009). For patients with asynchronous metastases the metastasis-free interval has proved to be a useful prognosticator because it reflects the tempo of disease progression (Maldazys and deKernion, 1986; Négrier et al, 2002; Motzer et al, 2004; Mekhail et al, 2005; Rini et al, 2009). Other important prognostic factors for patients with systemic metastases include performance status, number and sites of metastases, anemia, hypercalcemia, elevated alkaline phosphatase or lactate dehydrogenase levels, thrombocytosis, and sarcomatoid histology (Maldazys and deKernion, 1986; Motzer et al, 1999; Motzer and Russo, 2000; Négrier et al, 2002; Leibovich et al, 2003b; Motzer et al, 2004; Mekhail et al, 2005; Escudier et al, 2007; Choueiri et al, 2007; Lane and Kattan, 2008). The presence of bone, brain, and/or liver metastases, and multiple metastatic sites have been associated with further compromise in prognosis (Négrier et al, 2002; Leibovich et al, 2005b; Mekhail et al, 2005; Escudier et al, 2007). These factors have been used to effectively categorize patients with metastatic RCC as low, intermediate, and poor risk, with corresponding differences in median survival (Table 49–19) (Motzer et al, 1999; Boumerhi et al, 2003; Motzer et al, 2004; Mekhail et al, 2005; Choueiri et al, 2007; Escudier et al, 2007). These risk groups provide important information for determining the likelihood of benefit a patient may expect to receive after cytoreductive nephrectomy and/or resection of other metastatic disease.
Another significant prognostic factor for RCC is tumor size, which has proved to be an independent prognostic factor for both organ-confined and invasive RCC (Kattan et al, 2001; Kontak and Campbell, 2003; Lane and Kattan, 2008). Giuliani and colleagues (1990) reported 5-year survival rates of 84% for patients with tumor diameter less than 5 cm, 50% for tumors between 5 and 10 cm, and 0% for tumors more than 10 cm in diameter. To a large extent, this is due to a strong correlation between tumor size and pathologic tumor stage, but several studies have subsequently demonstrated that tumor size can function as an independent prognostic factor (Guinan et al, 1995b; Kattan et al, 2001; Sorbellini et al, 2005; Crispen et al, 2008b; Nguyen and Gill, 2009). Larger tumors are more likely to exhibit clear cell histology and high nuclear grade, and both of these factors correlate with a compromised prognosis (Frank et al, 2003b; Lane et al, 2007a; Thompson et al, 2009). A review of 1771 patients with organ-confined RCC showed 10-year cancer-specific survival rates of 90% to 95%, 80% to 85%, and 75% for patients with pT1a, pT1b, and pT2 tumor, respectively (Patard et al, 2004a). Many other studies have also shown a particularly favorable prognosis for the unilateral pT1a tumors that are now being discovered with increased frequency. In series from the Cleveland Clinic and the Mayo Clinic, such tumors were associated with greater than 95% 5-year cancer-specific survival rates, whether they were managed with nephron-sparing surgery or radical nephrectomy (Butler et al, 1994; Lerner et al, 1996; Cheville et al, 2001; Gill et al, 2007; Lane and Gill, 2007; Crispen et al, 2008b).
Other important prognostic factors for RCC include nuclear grade, histologic subtype and symptomatic presentation. Several grading systems for RCC have been proposed on the basis of nuclear size and morphology and presence or absence of nucleoli. Unfortunately, interobserver variability is common in the assignment of nuclear grade; there is no ideal classification system that can overcome the subjectivity of this exercise. Nevertheless, almost all the proposed grading systems have provided prognostic information for RCC, and nuclear grade has proved in most cases to be an independent prognostic factor when subjected to multivariate analysis (Goldstein, 1997; Ficarra et al, 2001; Kattan et al, 2001; Zisman et al, 2001b; Lohse et al, 2002, 2005; True, 2002; Kontak and Campbell, 2003; Patard et al, 2004c; Lang et al, 2005; Lane and Kattan, 2008; Ficarra et al, 2009).
Fuhrman’s classification system has been the most generally adopted grading system for RCC. In Fuhrman and colleagues’ original report (1982) the 5-year survival rates for grades 1 to 4 were 64%, 34%, 31%, and 10%, respectively, and nuclear grade proved to be the most significant prognostic factor for organ-confined tumors in this series. Subsequent reports have demonstrated correlations between Fuhrman’s nuclear grade and tumor stage, tumor size, venous tumor thrombi, and lymph node and systemic metastases (Bretheau et al, 1995; Lang et al, 2005; Lohse et al, 2005; Ficarra et al, 2009). Significant differences have been consistently observed between low-grade (grades 1/2) and high-grade (grade 3/4) tumors with some difficulty distinguishing the intermediate grades. Based on these studies, some have recommended changing to a three-tiered system, but this remains a matter of investigation (Medeiros et al, 1997; Lang et al, 2005; Lane and Kattan, 2008; Zhou 2009). In addition, although significant differences according to nuclear grade have been reported in series that have included patients with all types of RCC or clear cell RCC alone, the relevance of the Fuhrman classification system to evaluation of other subtypes of RCC is not entirely clear. Papillary RCC may be better subgrouped into type 1 and type 2, and oncocytic neoplasms may be better classified as chromophobe RCC, hybrid oncolytic tumors, and oncocytomas, although whether these provide better prognostic information about cancer recurrence has not been determined (see Pathology).
Histologic subtype also carries prognostic significance, although, again, primarily at the ends of the spectrum. The presence of sarcomatoid differentiation or collecting duct, renal medullary, or unclassified histologic subtype denotes a poor prognosis (Carter et al, 1992; Davis et al, 1995b; Chao et al, 2002b; Escudier et al, 2002a; Mian et al, 2002; Polascik et al, 2002; Mejean et al, 2003; Cheville et al, 2004; Nanus et al, 2004; Tokuda et al, 2006; Kobayashi et al, 2008; Zhou 2009). Several studies now suggest that clear cell RCC may have a worse prognosis on average compared with papillary or chromophobe RCC, although there are clearly poorly differentiated tumors in each of these subcategories that can be lethal (Moch et al, 2000; Amin et al, 2002; Lau et al, 2002; Cheville et al, 2003; Krejci et al, 2003; Beck et al, 2004; Patard et al, 2005; Lane and Kattan, 2008; Crispen et al, 2008b; Dall’Oglio et al, 2008; Lam et al, 2008; Margulis et al, 2008; Klatte et al, 2008b; Rothman et al, 2009). Finally, several subtypes of RCC are predictably indolent, including multiloculated cystic clear cell RCC and mucinous tubular and spindle cell carcinoma.
For patients with clinically localized disease, mode of presentation (incidental vs. symptomatic) can be combined with other predictive elements to better stratify patients after primary surgical management (Kattan et al, 2001; Patard et al, 2004a; Sorbellini et al, 2005; Karakiewicz et al, 2007; Lane and Kattan, 2008). In addition, patients with systemic symptoms suggestive of metastatic spread have significantly poorer outcomes than those with only local symptoms, such as hematuria or flank pain (Kattan et al, 2001; Sorbellini et al, 2005).
Dozens of genes that may have prognostic or therapeutic significance for patients with RCC have been identified using high-throughput technologies (Takahashi et al, 2003; Zhao et al, 2006; Lane et al, 2008b). Gene expression profiling (cDNA microarrays) can quantify the levels of thousands of individual messenger RNA transcripts within an individual tumor sample. Alterations in gene expression can then be correlated with the amount and location of specific gene products (proteins) using immunohistochemical staining of cancer specimens (Kim HL et al, 2004a; Liu et al, 2004; Parker et al, 2009). Construction of tissue microarrays can facilitate the screening of hundreds of tumors, but interpretation of results can be challenging due to tumor heterogeneity and the selection of only a small amount of tissue for analysis (Kim HL et al, 2004a; Liu et al, 2004; Leibovich et al, 2007). Furthermore, when evaluating the potential value of a new marker, it is important to consider its contribution after accounting for other known prognostic factors (George and Bukowski, 2007; Tunuguntia and Jorda, 2008).
Several molecular markers appear to serve as independent prognostic factors for RCC and have provided important insights into tumor biology (see Tumor Biology) (Bui et al, 2001; Han et al, 2003; Crispen et al, 2008b; Nogueira and Kim, 2008; Parker et al, 2009). One such factor is CA-IX, which is regulated by the VHL gene and overexpressed in most clear cell RCCs (Bui et al, 2003, 2004; Leibovich et al, 2007). Although initial studies indicated that decreased expression of CA-IX is independently associated with poorer survival in patients with metastatic RCC (Bui et al, 2003; Kim et al, 2005), this association does not appear to apply for patients with localized disease (Kim et al, 2005; Leibovich et al, 2007). CA-IX also may serve as a marker for response to systemic therapy, making CA-IX immunostaining of particular value for patients with advanced disease (Bui et al, 2004; Atkins et al, 2005; Cho et al, 2007). B7-H1 is a T-cell coregulatory molecule that is a strong independent predictor of disease progression for RCC (Thompson et al, 2006; Parker et al, 2009). This association holds even after accounting for other molecular factors and the major clinical and pathologic predictors (Krambeck et al, 2007; Parker et al, 2009). Increased proliferative index as assessed by Ki-67 has also been correlated with reduced survival in clear cell RCC (Bui et al, 2004; Klatte et al, 2009; Parker et al, 2009). Although initial data indicated that Ki-67 expression was a surrogate for histologic necrosis, more recent studies have found Ki-67 to be an independent predictor and have incorporated it into predictive algorithms (Tollefson et al, 2007; Klatte et al, 2009; Parker et al, 2009). Other factors that appear to be useful include cell cycle regulators, such as the tumor suppressor TP53 (Kim HL et al, 2004a; Shvarts et al, 2005b; Klatte et al, 2009); various growth factors and their receptors, including members of the VEGF family (Jacobsen et al, 2000; Rivet et al, 2008; Phyoc et al, 2008; Klatte et al, 2009); adhesion molecules; and other factors, such as survivin (Parker et al, 2006, 2009; Byun et al, 2007; Krambeck et al, 2007).
Several investigators have now developed tools that combine various prognostic factors, and this has greatly improved our predictive capacity for patients with RCC (see Table 49–19) (Kattan et al, 2001; Zisman et al, 2001b; Frank et al, 2002; Sorbellini et al, 2005; Lane and Kattan, 2008). For instance, Kattan and colleagues (2001) have combined manner of presentation (incidental vs. local or systemic symptoms), tumor histology, tumor size, and pathologic stage to develop a nomogram that predicts cancer-free survival after nephrectomy. Tumor grade was not included in this analysis because its role for non–clear cell RCC has not been clearly defined. A subsequent analysis from this same group focused only on patients with clear cell RCC and incorporated tumor grade, assessment of tumor necrosis, and vascular invasion to further improve prognostication (Fig. 49–17) (Sorbellini et al, 2005). Such nomograms provide an individual assessment of risk that clinicians can use during patient counseling (www.nomograms.org). Although several predictive algorithms incorporate histologic necrosis (Frank et al, 2002; Sorbellini et al, 2005), the utility of this predictor has been called into question in some recent studies because its assessment has not been standardized and it is not routinely reported at many centers (Isbarn and Karakiewicz, 2009).
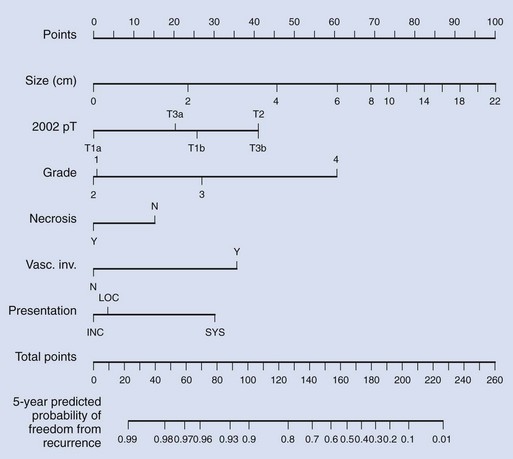
Figure 49–17 Postoperative nomogram predicting recurrence of clear cell renal cell carcinoma after nephrectomy. Locate the tumor size on the size axis. Draw a line upward to the points axis to determine how many points toward recurrence the patient receives for this parameter. Repeat this process for the other axes, each time drawing straight upward to the points axis. Sum the points achieved for each predictor and locate the sum on the total points axis. Draw a straight line down to find the probability that the patient will remain free of recurrence for 5 years, assuming the patient does not die of another cause first.
(Adapted from Sorbellini M, Kattan MW, Snyder ME, et al. A postoperative prognostic nomogram predicting recurrence for patients with conventional clear cell renal cell carcinoma. J Urol 2005;173:48–51.)
Pantuck and colleagues (2001a) have also performed an integrated analysis of prognostic factors for patients with all stages of RCC with encouraging results. A sophisticated multivariate analysis revealed three independent prognostic factors that were most robust for predicting outcomes, namely, TNM stage, performance status, and tumor grade (Zisman et al, 2001b). The UCLA integrated staging system (UISS) was subsequently modified to identify patients with localized or metastatic disease at low, intermediate, and high risk of disease progression and has been validated internally and externally (Zisman et al, 2002b; Patard et al, 2004c; Cindolo et al, 2006, 2008; Parker et al, 2009). Molecular factors such as TP53, Ki-67, VEGF family members, and CA-IX have also been incorporated into UISS-based algorithms to predict outcomes for patients with localized or metastatic RCC (Kim et al, 2005; Klatte et al, 2009).
Another predominant model that provides individualized information for patients with clear cell RCC is the SSIGN score, which incorporates 1997 TNM stage, tumor size, nuclear grade, and presence of tumor necrosis to predict recurrence and survival after radical nephrectomy (Frank et al, 2002). Estimated cancer-specific survival according to the SSIGN score was based on data from more than 1800 patients and has subsequently been validated in several other datasets (Ficarra et al, 2006, 2009; Fujii et al, 2008; Zigeuner et al, 2010). Thompson and colleagues (2007d) have also developed a dynamic outcome prediction model that provides patients with cancer-specific survival rates that improve as the disease-free interval following surgery increases. Most recently, the group at Mayo Clinic has developed a sequential approach in which the predicted outcomes for patients at various risk of recurrence according to clinical and pathologic factors can be further stratified based on molecular data incorporated into a BioScore (Parker et al, 2009). The expression of B7-H1, survivin and Ki-67 each added independent predictive ability after accounting for either the UISS or SSIGN score, especially for patients at intermediate or high risk of recurrence (Parker et al, 2009).
TNM staging systems and prognostic algorithms have different purposes. The TNM staging system is used to provide a universal language for communication between clinicians and patients and is based solely on the anatomic extent of cancer dissemination. A wealth of literature now supports the notion that algorithms that incorporate multiple predictive elements, such as nomograms and artificial neural networks, outperform risk assessment based on expert opinion or simpler models, such as classic staging systems (Dawes et al, 1989; Ross et al, 2002; Isbarn and Karakiewicz, 2009; Shariat et al, 2009). The development and use of these predictive tools can help guide counseling and follow-up of patients with RCC and identify patients more likely to benefit from specific interventions.