chapter 92 Evaluation and Nonsurgical Management of Benign Prostatic Hyperplasia
The term benign prostatic hyperplasia (BPH) has very different connotations to the pathologist, radiologist, urodynamicist, practicing urologist, and patient. BPH to the pathologist is a microscopic diagnosis characterized by cellular proliferation of the stromal and epithelial elements of the prostate. The radiologist confirms the diagnosis of BPH on the basis of an enlarged prostate either with ultrasound or three-dimensional diagnostic imaging studies of the male pelvis (Haas and Resnick, 2000). The hallmark of BPH to the urodynamicist is the synchronous observation of elevated voiding pressure and a low urinary flow rate in the absence of other disease processes that cause bladder outlet obstruction (BOO) (Nitti, 2000). BPH to the practicing urologist represents a constellation of signs and symptoms that develop in the male population in association with aging and prostatic enlargement presumably caused by BOO (Shapiro and Lepor, 1995), together with ultrasound imaging. The patient is typically concerned about the impact of BPH on quality of life rather than about the presence of cellular proliferation, prostatic enlargement, or elevated voiding pressures.
Because of the diverse connotations of the term, it seems sensible to define BPH as microscopic BPH, macroscopic BPH, or clinical BPH. Microscopic BPH represents histologic evidence of cellular proliferation of the prostate. Macroscopic BPH refers to enlargement of the prostate resulting from microscopic BPH. Clinical BPH represents the lower urinary tract symptoms (LUTS), bladder dysfunction, hematuria, and urinary tract infection (UTI) resulting from macroscopic BPH. Abrams (1994) has suggested using the more clinically descriptive terms benign prostatic enlargement (BPE), bladder outlet obstruction (BOO), and lower urinary tract symptoms to replace BPH, but the time-honored, although limited, original terminology persists.
Microscopic BPH describes a proliferative process of the stromal and epithelial elements of the prostate (Bartsch et al, 1979). The proliferative process originates in the transition zone and the periurethral glands (McNeal, 1978). It is rarely identified in men younger than 40 years of age (Berry et al, 1984). The autopsy incidence of BPH is age dependent, the proliferative process being present in approximately 70% and 90% of men in their seventh and ninth decades of life, respectively. The development of microscopic BPH requires aging and the testes as the source of androgens (Walsh, 1984). Androgens play a passive role in the proliferative process. The specific molecular events that initiate and promote microscopic BPH have yet to be identified and characterized. Growth factors, such as epidermal growth factor (EGF), are involved through autocrine and paracrine stromal-epithelial interactions (Steiner, 2000).
Macroscopic BPH describes an “enlarged” prostate. Digital rectal examination (DRE) provides a relatively crude estimate of prostate size when compared with measurements using transrectal ultrasonography or magnetic resonance imaging (Roehrborn et al, 1997). Knowledge of prostate size may be clinically relevant in terms of selecting appropriate medical or surgical therapy. A strong correlation exists between serum prostate-specific antigen (PSA) levels and prostate volume (Roehrborn et al, 1999), and, as a consequence, in the absence of adenocarcinoma, the PSA value may be used as a surrogate for prostate volume (Roehrborn et al, 2001). The transition zone (inner gland) accounts for the majority of BPH tissue. The transition zone volume can be quantified using transrectal ultrasonography (Lepor et al, 1994) or magnetic resonance imaging (Tempany et al, 1993). There is no consensus regarding the extent of enlargement required to establish the diagnosis of macroscopic BPH; however, prostate volume of approximately 20 mL may be regarded as normal (Garraway et al, 1991) before BPH develops, and volumes of 100 mL or greater are encountered clinically.
The clinical manifestations of BPH include LUTS, poor bladder emptying, urinary retention, an overactive bladder, UTI, hematuria, and renal insufficiency (Jepsen and Bruskewitz, 2000). Historically, the pathophysiology of clinical BPH was attributed to BOO secondary to macroscopic enlargement of the prostate gland (Lepor, 2000). This hypothesis was supported by epidemiologic data suggesting that the prevalence of microscopic BPH, macroscopic BPH, and clinical BPH is age dependent and therefore causally related (Isaacs and Coffey, 1989). This rather oversimplistic concept of the pathophysiology of BPH has been challenged by more recent reports demonstrating only weak relationships between prostate size, severity of BOO, and severity of symptoms (Barry et al, 1993; Bosch et al, 1995; Girman et al, 1995; Yalla et al, 1995). However, there are numerous epidemiologic data to confirm that BPH is a slowly progressive disease and that men with a larger prostate (or higher PSA) are at significantly greater risk of LUTS, impaired quality of life, and complications such as acute urinary retention (AUR) (Roehrborn et al, 2001) (see Chapter 91).
Diagnosis
The complex of symptoms now commonly referred to as “LUTS” is not specific for BPH. Aging men with a variety of lower urinary tract pathologic processes may exhibit similar, if not identical, symptoms. The initial diagnostic challenge in these patients is to establish that the symptoms are, in fact, a result of BPH. This is the primary focus of initial evaluation and diagnostic testing. Fortunately, nonprostatic causes of symptoms can be excluded in a significant majority of patients on the basis of history, physical examination, and urinalysis. Additional diagnostic testing is necessary in patients in whom the diagnosis is still unclear after initial evaluation. These tests may also have a modest (but still unproven) value in predicting the response to treatment. The following recommendations concerning the initial evaluation of men presenting with LUTS reflect the consensus opinion for several independent groups. The American Urological Association (AUA) BPH guidelines were presented in 1994 and 2003, were updated in 2010 (McVary et al, 2010), and are available at www.auanet.org/content/guidelines-and-quality-care/clinical-guidelines.cfm?sub=bph. The European Association of Urology (EAU) has published guidelines on “Assessment, Therapy and Follow-Up of Men with Lower Urinary Tract Symptoms Suggestive of Benign Prostatic Obstruction (BPH Guidelines)” (Madersbacher et al, 2004), and in 2009 a Sixth International Consensus document was published (Abrams et al, 2009). Algorithms for the management of LUTS/BPH are shown in Figures 92-1 to 92-3.
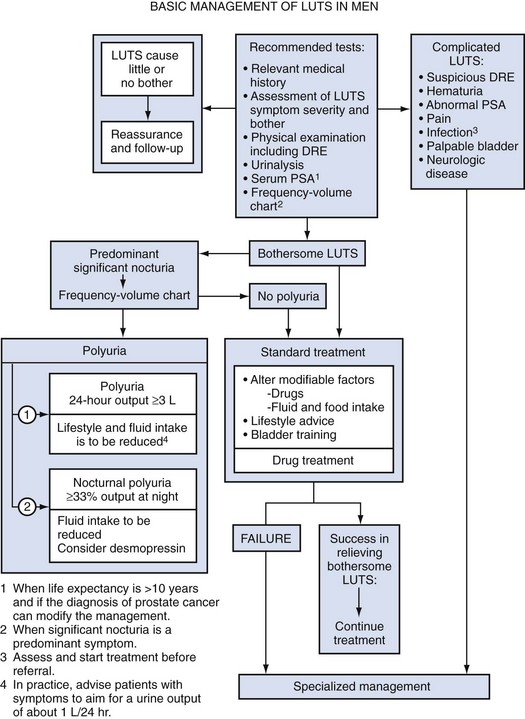
Figure 92–1 International Consensus guideline algorithm for management of lower urinary tract symptoms (2009): Basic management. DRE, digital rectal examination; PSA, prostate-specific antigen.
(From Abrams P, Chapple C, Khoury S, et al. Evaluation and treatment of lower urinary tract symptoms in older men. J Urol 2009;181[4]:1779–87.)
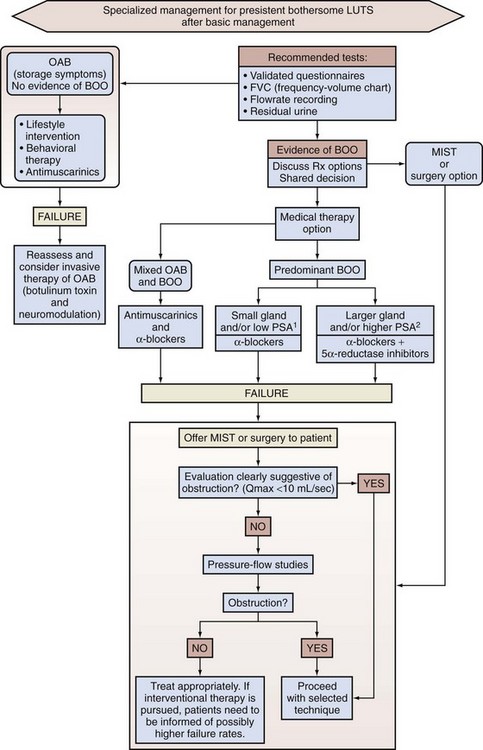
Figure 92–2 International Consensus guideline algorithm for management of lower urinary tract symptoms (LUTS) (2009): Specialized management. 1PSA <1.5 ng; 2PSA >1.5 ng; BOO, bladder outlet obstruction; MIST, minimally invasive surgical treatment; OAB, overactive bladder; PSA, prostate-specific antigen.
(From Abrams P, Chapple C, Khoury S, et al. Evaluation and treatment of lower urinary tract symptoms in older men. J Urol 2009;181[4]:1779–87.)
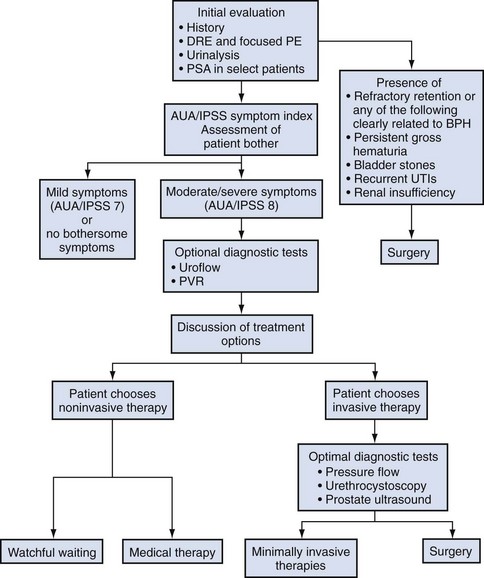
Figure 92–3 American Urological Association (AUA)guideline algorithm for management of benign prostatic hyperplasia (updated 2006). DRE, digital rectal examination; IPSS, International Prostate Symptom Score; PE, physical examination; PSA, prostate-specific antigen; PVR, postvoid residual.
(From Kaplan SA. Update on the American Urological Association guidelines for the treatment of benign prostatic hyperplasia. Rev Urol 2006;8[Suppl. 4]:S10–7.)
Initial Evaluation
Medical History
A detailed medical history should be obtained to identify other causes of voiding dysfunction or cormorbidities that may complicate treatment. Specific additional areas to discuss when taking the history of a man with BPH symptoms include a history of hematuria, UTI, diabetes, nervous system disease (e.g., Parkinson disease or stroke), urethral stricture disease, urinary retention, and aggravation of symptoms by cold or sinus medication. Current use of prescription and over-the-counter medications should be discussed to determine whether the patient is taking drugs that impair bladder contractility (anticholinergic agents) or that increase outflow resistance (α-sympathomimetic agents). A history of prior lower urinary tract surgery raises the possibility of urethral or bladder neck stricture. Use of a voiding diary (recording times and volume) may help identify patients with polyuria or other nonprostatic disorders.
Physical Examination
A DRE and a focused neurologic examination should usually be performed. In addition, examination of the external genitalia is indicated to exclude meatal stenosis or a palpable urethral mass and an abdominal examination is necessary to exclude an overdistended, palpable bladder. The DRE and focused neurologic examination are performed to detect prostate or rectal malignancy, to evaluate anal sphincter tone, and to rule out any neurologic problems that may cause the presenting symptoms. The presence of induration is as important a finding as the presence of a nodule and should be correlated with a serum PSA value so that the need for prostatic biopsy can be assessed and acted upon.
DRE establishes the approximate size of the prostate gland. Estimation of prostate size is important to select the most appropriate pharmacologic or technical approach. DRE provides a sufficiently accurate measurement in most cases. The size of the prostate is not critical in deciding whether active treatment is required. Prostate size does not correlate precisely with symptom severity, degree of urodynamic obstruction, or treatment outcomes (Roehrborn et al, 1986; Simonsen et al, 1987). If a more accurate measurement of prostate volume is needed to determine whether to perform open prostatectomy rather than transurethral resection of the prostate (TURP), or some other procedure such as laser vaporization, ultrasonography (transabdominal or transrectal) is more accurate than cystourethroscopy. It is now established that a larger gland, and consequently a higher PSA, is associated with a greater risk of BPH progression (Roehrborn et al, 2001).
Urinalysis
A urinalysis should be done either by using a dipstick test or microscopic examination of the spun sediment to rule out UTI and hematuria, either of which strongly suggest a non-BPH pathologic process as a cause of symptoms. Because serious urinary tract disorders are relatively uncommon, the positive predictive value of screening for them is low and the effectiveness of early detection and intervention is unproven. However, in older men with BPH and a higher prevalence of these disorders the benefits of an innocuous test such as urinalysis clearly outweigh the harms involved. The test permits the selective use of renal imaging and endoscopy for patients with the greatest chance of benefiting from them. More importantly, urinalysis assists in distinguishing UTIs and bladder cancer from BPH. These conditions may produce urinary tract symptoms (e.g., frequency and urgency) that mimic BPH. If a dipstick approach is used, a test that includes leukocyte esterase and nitrite tests for the detection of pyuria and bacteriuria should be used.
The positive predictive value of urinalysis for cancer or other urologic diseases is around 4% to 26%, depending on the patients screened and the rigor of follow-up studies (Mohr et al, 1986, 1987; Messing et al, 1987). Urine cytology should always be requested in men with severe irritable symptoms and dysuria, especially if they have a smoking history. Carcinoma in-situ of the bladder is a diagnosis that may have serious consequences if overlooked.
Serum Creatinine Measurement
Although the measurement of the serum creatinine concentration was recommended in the initial evaluation of all patients with symptoms of LUTS to exclude renal insufficiency caused by the presence of obstructive uropathy (McConnell et al, 1994; Denis et al, 1998), at the Fifth International Consultation on BPH it was suggested that serum creatinine determination should be optional or secondary. The AUA guidelines on BPH and the Sixth International Consensus report no longer recommend routine creatinine measurement in the standard patient. However, it is well established that BPH patients with renal insufficiency have increased risk for postoperative complications. The risk is 25% for patients with renal insufficiency, compared with 17% for patients without the condition (Mebust et al, 1989). Moreover, the mortality increases up to sixfold for BPH patients treated surgically if they have renal insufficiency (Holtgrewe and Valk, 1962; Melchior et al, 1974). Of 6102 patients evaluated in 25 studies by intravenous urography (IVU) before prostate surgery, 7.6% had evidence of hydronephrosis (McConnell et al, 1994). Of these patients, 33.6% had associated renal insufficiency. Elevated serum creatinine levels in a patient with BPH is an indication for imaging studies (usually ultrasonography) to evaluate the upper urinary tract. In a retrospective analysis of 345 patients who had undergone prostatectomy, 1.7% (n = 6) had occult and progressive renal damage (Mukamel et al, 1979). These patients had minimal or no urinary symptoms and presumably fit the category of patients with “silent prostatism.” Measurement of the serum creatinine concentration is one means to identify such “at risk” patients.
Serum Prostate-Specific Antigen
Prostate cancer can lead to LUTS by producing bladder outflow obstruction similar to BPH. Moreover, localized prostate cancer commonly coexists with BPH. In most men with a 10-year or longer life span the knowledge of concomitant prostate cancer may well alter management of the BPH component. The detection of a large nodular prostate cancer on DRE would no doubt alter therapy; however, the “early detection” of small volume prostate cancer in an 80-year-old man is unlikely to increase life expectancy. A PSA test and DRE increase the detection rate of prostate cancer over DRE alone. Therefore measurement of the serum PSA value should be performed in patients in whom the identification of cancer would clearly alter BPH management (Madersbacher et al, 2004; Kaplan et al, 2006; Abrams et al, 2009). There is significant overlap between the serum PSA values of men with BPH and men with clinically localized prostate cancer. Twenty-eight percent of men with histologically proven BPH have a serum PSA value greater than 4.0 ng/mL (McConnell et al, 1994). Serum PSA trends over time (PSA velocity), measurement of free versus complexed PSA, and PSA density may help to improve the specificity of PSA in men with BPH. Newer markers such as the PCA-3 test can also help to differentiate BPH from prostate cancer.
In the absence of prostate cancer the PSA value provides both a guide to prostate volume and also an indication of the likelihood of response to therapy with 5α-reductase inhibitors. However, in men with BPH already treated with a 5α-reductase inhibitor (e.g., finasteride [Proscar] or dutasteride [Avodart]) serum PSA is reduced 40% to 50% after 6 months of treatment. Failure to establish a baseline (pretreatment) PSA level may therefore complicate interpretation of future PSA values. Men who are taking these agents should have their PSA value doubled to correctly assess their risk of harboring a prostatic adenocarcinoma. Provided that this is done, recent evidence suggests that 5α-reductase therapy actually increases the sensitivity of PSA as a detector of cancer.
Symptom Assessment
The International Prostate Symptom Score (IPSS) is recommended as the symptom scoring instrument to be used for the baseline assessment of symptom severity in men presenting with LUTS (Kaplan et al, 2006; Abrams et al, 2009). When the IPSS system is used, symptoms can be classified as mild (0 to 7), moderate (8 to 19), or severe (20 to 35). The IPSS should also be the primary determinant of treatment response or disease progression in the follow-up period. Although other symptom score questionnaires are used, the IPSS is now the U.S. and international standard.
However, the IPSS cannot be used to establish the diagnosis of BPH. Men (and women) with a variety of lower urinary tract disorders (e.g., infection, tumor, neurogenic bladder disease) will have a high IPSS. Nonetheless, the IPSS is the ideal instrument to grade baseline symptom severity, assess the response to therapy, and detect symptom progression in those men managed by watchful waiting. Optimal treatment decisions in individual patients will also need to take into account how a given level of symptoms affects each man’s quality of life (degree of bothersomeness).
The IPSS was developed from the AUA Symptom Index (AUASI) developed by the Measurement Committee of the AUA (Barry et al, 1992a, 1992b). Each question on the IPSS can yield 0 to 5 points, producing a total symptom score that can range from 0 to 35. This seven-question set is internally consistent (Cronbach alpha, 0.85) and reliable (test-retest correlation, 0.93). The index correlates strongly with patients’ global ratings of their urinary difficulties (r = 0.78) and is sensitive to treatment response.
Johnson and associates (2009) showed that patients with a low education status are more likely to misunderstand the IPSS whether they are managed in public hospital or university practice. They tend to misrepresent their symptoms and therefore may receive inappropriate treatment. Eight percent of university hospital and almost 25% of public hospital patients under-reported their moderate symptoms as mild, and 33% of university hospital and 16% of public hospital patients over-reported their mild symptoms as moderate. When administered by a medical professional, many of the inaccuracies disappear even though the questionnaire is recommended as “self-administered.” The use of the additional bother score or quality of life question may be useful in guiding appropriate treatment.
Although the IPSS correlates well with quality of life measures (Sagnier et al, 1995), there is still a need for sensitive BPH-specific quality of life instruments. Furthermore, because storage symptoms often predominate there is a need for better methods of quantifying urgency, frequency, and recording any incontinence. The standardization subcommittee of the International Continence Society has usefully categorized the range of likely symptoms into three groups: storage, voiding, and postmicturition (Abrams et al, 2003). Subdividing the IPSS into the four obstructive and three storage questions may also be useful. Other systems such as the Kings Score may be more sensitive to changes in storage symptoms. A voiding diary with frequency and volume recordings also may be helpful.
Clearly, symptom scores alone do not capture the complete picture of a prostate problem as perceived by the individual patient. Symptom impact on a patient’s lifestyle must be considered as well. An intervention may make more sense for a moderately symptomatic patient who finds his symptoms very bothersome than for a severely symptomatic patient who finds his symptoms tolerable.
Additional Diagnostic Tests
Additional testing should be considered after the initial evaluation if there is a significant chance the patient’s LUTS may not be due to BPH. Patients with a normal initial evaluation and only mild symptomatology on the IPSS (scores 0 to 7), or even more moderate symptoms but minimal bother, do not need additional diagnostic evaluation and can be considered for an active surveillance program and observed (Kaplan, 2006). Men who have developed serious complications of BPH should be treated surgically in most cases. Urinary flow rate, postvoid residual (PVR) urine volume, and pressure-flow urodynamic studies are appropriate tests to consider in the evaluation of men with moderate to severe symptoms (IPSS 8 to 35). The value of pressure-flow studies is debated, especially in men who elect watchful waiting or medical therapy as their management option. Cystoscopy should not be done routinely but is optional during later evaluation if invasive treatment is strongly considered. Urinary flow rate and PVR are generally recommended tests (Madersbacher et al, 2004; Kaplan, 2006; Abrams et al, 2009), and frequency-volume chart recordings are recommended by most clinicians (Madersbacher et al, 2004; Abrams et al, 2009).
It may be appropriate for the physician to offer treatment alternatives to the patient without performing any further diagnostic tests. Especially if the patient chooses watchful waiting or noninvasive therapy, invasive diagnostic tests may not be necessary. Conversely, if the patient elects an invasive treatment option, it may be appropriate for the physician to consider further evaluation.
Diagnostic Tests in Men Who Require Surgery for Benign Prostatic Hyperplasia
Surgery is generally recommended if the patient has refractory urinary retention (failing at least one attempt of catheter removal) or any of the following conditions clearly secondary to BPH: recurrent UTI, recurrent gross hematuria (resistant to 5α-reductase inhibitor therapy), bladder stones, renal insufficiency, or large bladder diverticula (McConnell et al, 1994; Denis et al, 1998; Kaplan et al, 2006; Abrams et al 2009). If there is reason to suspect that the patient’s urinary retention may be due to detrusor hypocontractility, then urodynamic studies (e.g., filling cystometry) may be helpful. Pressure-flow urodynamic studies are not informative if the patient cannot urinate. Cystoscopy is appropriate to consider before the operative procedure to help plan the most prudent approach. The presence of infection and hematuria in patients should prompt appropriate evaluation and therapy for these conditions before treatment of BPH.
Uroflowmetry
Uroflowmetry is either generally recommended (by the AUA [Kaplan, 2006]), recommended for specialist investigation (Sixth International Consultation [Abrams et al, 2009]), or recommended generally and required before invasive treatment (EAU [Madersbacher et al, 2004]). Uroflowmetry involves the electronic recording of the urinary flow rate throughout the course of micturition. It is a common, noninvasive urodynamic test used in the diagnostic evaluation of patients presenting with symptoms of BOO. The results of uroflowmetry are nonspecific for causes of the symptoms. For example, an abnormally low flow rate may be caused by an obstruction (e.g., hyperplastic prostate, urethral stricture, meatal stenosis) or by detrusor hypocontractility. The AHCPR Guideline Panel reached the following conclusions regarding uroflowmetry (McConnell et al, 1994), which still apply:
The Fourth International Consultation on BPH concluded that flow rate measurement represents a reproducible way to quantify the strength of the urinary stream and, when used in combination with symptom scores for a small subset of patients (20%), has a high probability of correctly characterizing whether there is BOO (Denis et al, 1998).
Despite its limitations, flow rate recording has demonstrated some sensitivity in identifying BOO due to BPH. Scott and coworkers (1967) and Shoukry and associates (1975) found that PFR correlated better than symptoms with the presence or absence of obstruction as determined by pressure-flow studies. Siroky and coworkers (1979) concluded that uroflowmetry was able to separate physiologically unobstructed and obstructed patients. Gleason and colleagues (1982) found that PFR distinguished between normal men and patients with BPH, urethral stricture, or prostatitis. However, they also noted that a subgroup of patients with a decompensated detrusor muscle could not be separated from the obstructed men on the basis of PFR alone.
Chancellor and colleagues (1991) found that flow rate recording cannot distinguish between BOO and impaired detrusor contractility as the cause for a low PFR. None of eight measured, noninvasive urodynamic parameters was significantly different for 31 patients with outlet obstruction than for 14 patients with impaired detrusor contractility. Abrams and associates (1977, 1979) studied the value of uroflowmetry before prostatectomy. Failure rates for surgery were found to decrease with the addition of flow rate measurement to symptom assessment in preoperative evaluation.
PFR appears to predict surgical outcome in some studies. In one study reported by Jensen and coworkers (1984), 53 patients underwent prostatectomy based on clinical indication alone. All three groups according to level of PFR experienced improvements in their symptom score after surgery, but the group with a PFR less than 10 mL/sec before treatment had a better overall subjective outcome as assessed by global subjective judgment.
In another study, which included men studied with flow rates before and 6 months after prostatectomy (Jensen et al, 1988a), subjective evaluation revealed an overall symptomatic improvement rate of 80% after surgery. The difference in success rates for men falling above or below the cutoff value of PFR = 10 mL/sec was not significant (P = .2). When a PFR cutoff of 15 mL/sec was used, success rates for men above or below the cutoff value differed significantly.
McLoughlin and coworkers (1990), using urodynamic testing and a cutoff value of 12 mL/sec, evaluated 108 men with clinical BPH before and 1 year after surgery and determined that if using a PFR less than 12 mL/sec as an indicator for obstruction then only 3% of patients would have been subjected to an unnecessary TURP. These authors believed that the routine pressure-flow studies or cystometrograms were not indicated but that the screening of flow rates followed by further urodynamic testing in patients with a PFR of greater than 12 mL/sec should be considered. Very low rates do not appear to portend poor treatment outcome. In one study of 84 patients undergoing surgery for symptomatic BPH (Donkervoort et al, 1975), patients with a preoperative PFR less than 7 mL/sec improved symptomatically as much as patients with a PFR greater than 7 mL/sec.
Neither subjectively assessed symptoms nor quantified symptom score analysis correlates strongly with uroflowmetry measurements but each is an independent assessment. Patients with a PFR greater than 15 mL/sec may have somewhat poorer outcomes after surgery than those with a PFR less than 15 mL/sec (although the majority of patients still improve). Other investigators report similar findings for different PFR cutoff values (e.g., 12 mL/sec). Patients with very bothersome symptoms suggestive of clinical BPH but having a PFR greater than 15 mL/sec may benefit from further urodynamic testing (i.e., pressure-flow studies) to reduce the number of surgical treatment failures. A PFR less than 15 mL/sec does not differentiate between outflow obstruction and detrusor impairment. No minimal threshold of PFR reliably diagnoses detrusor failure or predicts a poor surgical outcome.
Postvoid Residual Urine Volume
Postvoid residual urine volume is the amount of fluid remaining in the bladder immediately after the completion of micturition. Studies indicate that PVR urine volume normally ranges from 0.09 to 2.24 mL, with the mean being 0.53 mL (Hinman and Cox, 1967). Seventy-eight percent of normal men have PVR volumes of less than 5 mL, and 100% have volumes of less than 12 mL (DiMare et al, 1963). The Agency for Health Care and Policy Research BPH Guideline Panel reached the following conclusions regarding PVR urine volume (McConnell et al, 1994):
The Fourth International Consultation initially recommended PVR volume determination in the initial assessment and during monitoring of patients under watchful waiting or other conservative treatment regimens (Denis et al, 1998).
PVR urine volume measurement can be performed by noninvasive (ultrasound) and invasive (catheterization) methods. Invasive techniques are accurate if performed correctly but carry a small, but clinically significant, risk of discomfort, urethral injury, UTI, and transient bacteremia. Small portable and less expensive devices can be used to measure the PVR urine volume with reported accuracy and are comparable to more expensive ultrasound units and catheterization. Birch and coworkers (1988) reported that of 30 men with BPH, 66% had wide variations in PVR urine volume when three measurements were done on the same day. In 34% of patients there was no difference among the three measurements. In 58%, at least two volumes were significantly different. In 8% of patients, all three were different. In most patients, two measurements were statistically similar whereas the third one yielded quite different results. Bruskewitz and colleagues (1982) found similarly wide variations of the measured amount when they performed repetitive measurements of PVR urine volume (repeated two to five times) by in-and-out catheterization on 47 men before prostatectomy. They also found no correlation between the amount of residual urine and any cystoscopic or urodynamic findings, symptoms, or the presence or absence of a history of UTIs. Most clinical studies demonstrate minimal correlation between PVR urine volume and baseline measurements of symptoms, flow rate, or urodynamic measures of obstruction (Griffiths and Castro, 1970; Shoukry et al, 1975; Abrams and Griffiths, 1979). However, Neal and associates (1987) found a significant association in 253 men between PVR urine volume, age, “below normal” PFR, and high urethral resistance. Low voiding pressure, however, did not correlate well with the PVR amount. The authors concluded that outflow obstruction is related to the development of increasing amounts of PVR urine. In the AUA Outcome Study, Barry and colleagues (1993) found a significant correlation between high PVR urine volumes and low flow rates but no correlation with IPSS.
Traditionally, urologists have assumed that increasing amounts of PVR urine denote BPH progression and are thus an “indication” for surgery. This concept underlies the common inclusion of PVR urine volume in each individual government’s appropriateness criteria. Unfortunately, data are lacking to support the predictive value of PVR urine volume. Andersen (1982) studied 104 men with BPH and reported two patterns of BPH progression. The slow course was characterized by the development of high levels of PVR urine that resulted in decompensation of the detrusor muscle and eventually led to urinary retention. The fast course was associated with uninhibited detrusor contractions. The amount of PVR urine, the presence of uninhibited detrusor contractions, and symptoms correlated poorly in the study. Nevertheless, Andersen recommended the PVR urine volume as a safety parameter when measured longitudinally throughout the clinical course of a patient with prostatism.
Data from the Veterans Affairs (VA) cooperative study group randomized trial comparing TURP with watchful waiting demonstrated that PVR urine volume does not predict the outcome of surgery, and there was little evidence to support criteria that require a certain amount of PVR urine before surgery is justified. Additionally, a high PVR urine volume did predict a slightly higher failure rate for watchful waiting. However, the majority of men with large PVR urine volume did not require surgery during the 3-year duration of the trial. In summary, PVR urine volume is best viewed as a “safety parameter.” Men with significant PVR amounts should certainly be monitored more closely if they elect nonsurgical therapy, particularly if antimuscarinic therapy is chosen.
Pressure-Flow Studies
If the initial evaluation, flow rate, and PVR urine volume are not sufficiently suggestive of BOO, further urodynamic assessment by pressure-flow studies should be considered, especially if an invasive treatment is considered (i.e., surgery) or if surgical treatment has failed (McConnell et al, 1994; Denis et al, 1998; Kaplan et al, 2006; Abrams et al, 2009). Pressure-flow studies differentiate between patients with a low PFR secondary to obstruction and those whose low PFR is caused by impaired detrusor contractility. These studies should be performed when the distinction between the two will affect therapeutic decisions. Patients with a history of neurologic diseases known to affect bladder or sphincteric functions, as well as patients with normal flow rates (PFR > 15 mL/sec) but bothersome symptoms, may also benefit from urodynamic evaluation and especially if surgical therapy is contemplated.
The value of pressure-flow measurement in predicting treatment outcome is uncertain. In a study by Abrams and colleagues (1979), the inclusion of pressure-flow data in the preoperative evaluation and indication for surgery reduced the subjective failure rate to 12%, down from 28% when patients were certified as candidates for surgery without the urodynamic data. However, a 28% failure rate is significantly higher than that reported in other TURP series (McConnell et al, 1994). Jensen and Andersen (1990) recommended invasive urodynamic testing for patients with a PFR greater than 15 mL/sec. For the population in their study this would have resulted in an additional 9% of patients being excluded from surgery and a decrease in failure rate to 8.3%. The support for this recommendation has to be questioned, however, in light of an earlier study by Jensen and coworkers (1988b, 1988c) that found most unsatisfied patients are incorrectly classified preoperatively even with urodynamic testing.
Pressure-flow studies do permit more accurate categorization of patients. Abrams and associates (1979) used pressure-flow plots in addition to flow rate measurement. The study found that in about half the cases the patients with LUTS could be correctly classified as obstructed or nonobstructed by PFR alone but that the addition of the detrusor pressure (Pdet) at PFR allowed correct classification in two thirds of the group. The remaining one third of the patients were assessed by pressure-flow plot. In many of these patients, both Pdet and PFR were low, indicating a decompensating detrusor muscle as the source for the low PFR.
Pressure-flow studies provide much more specific insight into detrusor function and the etiology of voiding dysfunction than do flow rate measurements. However, a number of outcome-based investigations demonstrate a modest additional value of pressure-flow studies over symptom and flow rate evaluation. Discussion of treatment options and the nature of the investigation with the patient is recommended before pressure-flow testing is organized.
Filling Cystometry (Cystometrography)
Filling cystometry adds limited information to the evaluation of most men with LUTS and is not recommended in routine cases. The test may have value in the evaluation of patients with known or suspected neurologic lesions and LUTS, but pressure-flow studies provide more specific information.
Urethrocystoscopy
Urethrocystoscopy is not recommended to determine the need for treatment. Although the linkage between the endoscopic appearance of the lower urinary tract and the treatment outcome is poorly documented, available information suggests that the relationship is minimal (McConnell et al, 1994). The test is recommended for men with LUTS who have a history of microscopic or gross hematuria, urethral stricture disease (or risk factors such as history of urethritis or urethral injury), bladder cancer or suspicion of carcinoma in-situ, or prior lower urinary tract surgery (especially prior TURP). Urethrocystoscopy may be considered in men with moderate to severe symptoms who have chosen (or require) surgical or other invasive therapy to help the surgeon determine the most appropriate technical approach.
For example, if urethrocystoscopy reveals a large middle lobe, transurethral incision of the prostate (TUIP) is unlikely to be successful. The decision to perform an open prostatectomy or laser vaporization may be appropriately influenced by the shape of the gland, as well as its size. Urethroscopy is therefore performed to select (or rule out) specific techniques, not to determine the need for treatment.
Imaging of the Upper Urinary Tract
Upper urinary tract imaging is not recommended in the routine evaluation of men with LUTS unless they also have one or more of the following: hematuria, UTI, renal insufficiency (ultrasonography recommended), history of urolithiasis, or history of urinary tract surgery (McConnell et al, 1994; Denis et al, 1998; Kaplan et al, 2006; Abrams et al, 2009). Intravenous urography before BPH treatment was performed by 73.4% of urologists in the United States in the late 1980s (Holtgrewe et al, 1989) and is associated with a 0.1% incidence of significant adverse events. Generally, ultrasound imaging is now preferred but is unnecessary in the uncomplicated case.
The presence or history of hematuria, renal insufficiency, UTI, and/or history of stones or prior urinary tract surgery increases the likelihood that imaging will demonstrate clinically significant findings (Juul et al, 1989; Andrews et al, 2002). Although there are no conclusive data on the combined incidence of the important clinical predictors just listed, approximately one third of all men with BPH have one or another indication for urinary tract imaging.
Assessing the Effectiveness and Safety of Medical Therapy
The role of treatment for any disease process depends on the magnitude of the clinical effect and the incidence and severity of treatment-related morbidity. Assessing the effectiveness of medical therapies for BPH requires defining clinically relevant end points, identifying quantitative and reliable clinical outcome measures, eliminating investigator and patient bias, accounting for the placebo response, and enrolling the proper number of subjects so that only clinically significant changes are statistically significant. Assessing the safety of medical therapies requires a rigorous effort to identify all treatment-related clinical, biochemical, teratogenic, and mutagenic adverse effects associated with drug treatment.
Clinical End Points
The clinical consequences of BPH includes LUTS and associated reduction of quality of life, detrusor dysfunction characterized by detrusor acontractility, detrusor instability, and detrusor fibrosis; incomplete bladder emptying; acute and chronic urinary retention; UTI; renal insufficiency; and hematuria (Shapiro and Lepor, 1995). The goals of treatment for BPH include relieving LUTS, decreasing BOO, improving bladder emptying, ameliorating detrusor instability, reversing renal insufficiency, and preventing disease progression, which may include a deterioration of symptoms, future episodes of gross hematuria, UTI, AUR, or the need for surgical intervention.
Quantitative Outcome Measures
Symptoms
The primary objective of the AUASI (now IPSS) was to provide a universally accepted instrument to quantify the impact of BPH therapies on LUTS (Barry et al, 1992a, 1992b). There is no standardized format for reporting changes in the AUASI or other quantitative indices of symptom severity after treatment. Symptom response has been reported as a percentage of patients achieving a threshold response or as group mean changes in a symptom score. The literature typically reports the percentage of men achieving between a 30% and a 50% reduction in the symptom score. Expressing the symptom response as a single threshold response does not discriminate the overall magnitude of the clinical effect. When the baseline symptom scores are mild to moderate, small and clinically insignificant changes correspond to large percentage changes. When baseline symptom scores are severe, relatively large absolute changes may not be clinically significant. Symptom outcome should be expressed both as a percentage of patients achieving a threshold reduction response and as group mean changes in the symptom score.
The clinical significance of changes in the AUASI score were reported by Barry and colleagues (1995). There were 1165 subjects who participated in a randomized double-blind placebo-controlled study of medical therapy and completed the AUASI at baseline and after 3 months of treatment. The absolute and percentage changes in AUASI and BPH Impact Index scores were correlated with five global ratings of symptom improvement. The group mean changes in AUASI for subjects rating their improvement as markedly, moderately, or slightly improved, unchanged, or worse were −8.8, −5.1, −3.0, −0.7, and +2.7, respectively. The relationship between the patients’ global ratings of improvement and the AUASI and BPH Impact Index changes were dependent on the baseline AUASI score. This important study provides the data required to determine sample sizes and interpret the clinical significance of symptom improvement in BPH clinical trials. A 3-point change appears perceptible to symptomatic men.
Bladder Outlet Obstruction
Experimental animal models of BOO have demonstrated profound changes in bladder ultrastructure, cellular composition, metabolism, and function resulting from BOO (Levin et al, 2000). These experimental observations must be cautiously extrapolated to man, because the response to BOO depends on the species and the severity and duration of obstruction. Animal studies demonstrate that under experimental conditions BOO causes alterations likely to adversely affect bladder function. The justification for measuring and treating BOO in men with BPH is to reverse or prevent these deleterious consequences of BOO.
Because synchronous pressure-flow urodynamic measurements do not correlate with severity of bladder dysfunction, severity of symptoms, or response to therapy, it is difficult to require these studies when evaluating the effectiveness of medical therapy for BPH. Long-term studies are needed to determine whether urodynamic measurements predict disease progression. At the present time the primary use of urodynamic testing is to discriminate the differential diagnosis of men presenting with multiple potential causes for LUTS.
Uroflowmetry represents a noninvasive and inexpensive but indirect indicator of urinary performances measure of BOO (Siroky, 1990). The reporting of PFR has been standardized (Abrams et al, 2003). At the lower spectrum of PFR a relatively small absolute change (i.e., 4 to 6 mL/sec) corresponds to a relatively high percentage change, whereas at the higher end of PFR a relatively large absolute change (i.e., 12 to 17 mL/sec) corresponds to a relatively modest percent change. The clinical significance of the changes in PFR cannot be defined, owing to the lack of correlations with relevant clinical, physiologic, or biochemical outcomes.
Bladder Emptying
The clinical significance of PVR urine volume measurement is controversial. Barry and colleagues (1993) reported no correlation between AUASI score and PVR amount. It has been suggested that the PVR urine volume may predispose to UTI and irreversible bladder dysfunction secondary to stasis and overdistention. There are no data clearly documenting that the incidence of UTI is related to the PVR urine volume. Another limitation of PVR urine measurements is variability over short intervals of time (Bruskewitz et al, 1982). It is imperative to measure the PVR volume on several occasions if this parameter will influence treatment decisions.
There is no standardization for reporting changes in PVR urine volume. Typically, the data are presented as absolute group mean changes. The majority of BPH clinical trials exclude subjects with high baseline PVR amount (>300 mL) because of the potential risks of randomization to a placebo or ineffective treatment group. Therefore the majority of subjects enrolled in clinical trials have clinically insignificant baseline PVR urine volumes, potentially undermining the relevance of most trials to “real world” practice.
Bladder Overactivity
The definition of bladder overactivity (detrusor instability) is the development of a detrusor contraction exceeding 15 cm H2O at a bladder volume less than 300 mL (Jepsen and Bruskewitz, 2000). The clinical significance of an overactive bladder (OAB) in men with BPH is unresolved. There is no evidence that men with detrusor instability electing watchful waiting are predisposed to develop disease progression. The presence of an OAB does not reliably predict response to medical or surgical treatment. Therefore improvement of an OAB is not a standard outcome measure in clinical trials.
Urinary Tract Infection, Renal Insufficiency, and Hematuria
Unlike other manifestations of BPH, the diagnosis of UTI, renal insufficiency, and hematuria is not controversial and the requirements for measurement are noninvasive and inexpensive. Because these events are not disease or gender specific, one must be cautious in assuming a causal relationship to BPH. There is no convincing evidence that UTI in the aging male population is associated with either PVR urine or BOO. It is reasonable to assume that renal insufficiency occurs secondary to urinary retention if renal failure is reversed after catheter drainage. Hematuria may be associated with prostatic vascularity and may sometimes respond to medical therapy with a 5α-reductase inhibitor.
Because the incidences of UTI, renal insufficiency, and hematuria are relatively uncommon and non–disease-specific events in the aging male population, it would be extremely difficult to design a prospective study to determine whether any BPH treatment prevents these events in an unselected cohort of men.
Eliminating Bias
Bias may be defined as a systematic error or difference between the true value and that actually attained from all causes other than sampling ability. The only mechanism to ensure that the potential bias of the subject and the investigator does not influence the outcome is a randomized, placebo-controlled, double-blind design. Because subjects are typically randomized to receive a drug or its matching placebo, any effect of the investigators’ bias would occur equally in the intervention and control groups.
The importance of eliminating bias cannot be overemphasized in BPH clinical trials. Some patients are very enthusiastic about receiving the “new” treatment. In the absence of a blinded randomization, these patients may be disproportionately directed into the active treatment groups. A subject receiving a known placebo would be reluctant to report any adverse events or clinical response. An investigator may be inclined to censor various outcomes if treatment group assignment is known. Although subjective outcome measures such as symptoms are more likely to be influenced by the placebo response, quantitative outcome measurements such as the PVR urine volume and PFR are also subject to a placebo response.
The placebo effect can be substantial in trials of drug treatment of BPH. Trials should therefore include a placebo run-in period before recording baseline values. Therefore these baseline values have already incorporated the placebo effect before any comparison is made. Ideally a 4-week placebo run-in period before initiation of treatment should be included in any trial design.
Similarly, the statistical concept of “regression toward mean” should also be taken into account in trial design. If measuring urinary symptom scores or PFRs, for instance, then in any population there will be some individuals whose values will be recorded at the extremes of the range for that population. These individuals, when followed up with sequential measurements of the same parameters, will tend to produce values that are less extreme and closer to the mean for the population being studied. Again, incorporating a placebo run-in period will allow this process to occur, at least to a degree, so that subsequent measurements from the baseline values determined after the placebo run-in period are more likely to be in response to a true treatment effect rather than to these two potentially confounding and misleading processes.
Sample Size
It is a general misconception that the validity of a clinical trial is directly proportional to the number of subjects enrolled. One of the objectives of a clinical trial is to determine whether the difference observed between two different treatment groups is clinically relevant. If the sample size of the clinical trial is properly determined in the early planning phase, a statistically significant outcome represents a clinically significant outcome. Calculation of sample size with provisions for adequate levels of significance and power is an essential part of planning a trial. Enrolling an excessive number of subjects may result in an overpowered study; that is, a small and clinically insignificant difference may be statistically significant. Conversely, enrolling insufficient numbers of subjects may result in an underpowered study; that is, a large and clinically significant difference may not be statistically significant. The larger the number of subjects enrolled in a study, the smaller is the change that is required to achieve statistical significance. Therefore the reader must examine the magnitude of the between-group difference and make a judgment about clinical significance.
Adverse Events
For a drug to enter into clinical investigation in humans it must be shown to elicit no significant chemical, behavioral, physiologic, teratogenic, mutagenic, or carcinogenic effects in at least two animal models. The typical untoward or adverse events captured in a clinical trial include physical findings, laboratory results, and complaints. A comprehensive physical examination and a battery of general laboratory tests are obtained at baseline and at the completion of the trial to capture any untoward events. The subjects’ complaints are typically captured at each study visit. The adverse clinical effects may be expected or unexpected.
Complaints may be elicited by means of a checklist or by the subjects’ unsolicited recall of an event. Adverse clinical events are typically greater with a checklist. The assessment of adverse events should determine the frequency and severity of the events and whether an event was severe enough to terminate participation in the study. The majority of clinical trials are powered based on outcome measures and not adverse events. There is a tendency for studies to be underpowered to detect serious adverse events that may therefore only show up later in postmarketing surveillance studies.
Nonsurgical Therapy
Watchful Waiting or “Self-Help”
A significant proportion of men with LUTS will not choose medical or surgical intervention because the symptoms are not bothersome, the complications of treatment are perceived to be greater than the inconvenience of the symptoms, and there is a reluctance to take a daily pill owing to side effects and/or the cost of treatment. Reassured that the symptoms are not caused by cancer or other serious genitourinary pathology, or that the delay in treatment will not have irreversible consequences, watchful waiting is often the patient-driven treatment of choice in the absence of absolute indications for intervention. Of 670 consecutive men with BPH referred to 39 urologists in the Netherlands, 41% elected watchful waiting (Stoevelaar et al, 1999). It is unreasonable to discourage an informed patient with severe symptoms and no other consequences of BPH from pursuing watchful waiting despite the safety and effectiveness of medical therapy. Watchful waiting does not imply the total absence of intervention. The severity and bother due to symptoms may be improved by simple measures such as decreasing total fluid intake especially before bedtime, moderating the intake of alcohol- and caffeine-containing products, and maintaining timed voiding schedules.
The impact of watchful waiting was examined in a study of 556 subjects with moderate symptoms of BPH randomized to TURP versus watchful waiting (Wasson et al, 1995). The changes in all outcome measures were significantly greater in the TURP group. A relevant outcome for patients selecting watchful waiting is disease progression. During 3 years of follow-up, treatment failure was observed in 23 (8.2%) and 47 (17%) of subjects randomized to TURP and watchful waiting, respectively. Treatment failure in the watchful waiting group was most often the result of increasing PVR volume or symptom score. Significant renal impairment was not seen.
Brown and associates (2007) evaluated the effectiveness of self-management as a first-line intervention for men with LUTS in a randomized controlled trial set in a teaching hospital and a district general hospital in London. One hundred forty men were randomized between standard care and “self-management,” which comprised three small group sessions of relevant urinary education and lifestyle advice. Self-management significantly reduced the frequency of treatment failure and reduced urinary symptoms. Because of the large observed benefit of self-management, these investigators suggested a large multicenter trial to confirm whether self-management could be considered as first-line treatment for men with LUTS.
Medical Therapy
Medical therapies extensively investigated for BPH include α-adrenergic blockers, 5α-reductase inhibitors, aromatase inhibitors, and numerous plant extracts. Newer therapies include antimuscarinic drugs and phosphodiesterase inhibitors (PDEIs) and several combinations of these agents. α-Adrenergic blockers and 5α-reductase inhibitors, and the combination of both of these, are emphasized in this chapter because the safety and efficacy of drugs in these classes have been critically examined and these drugs are widely prescribed for the treatment of BPH. Aromatase inhibitors are briefly reviewed for historical interest. Plant extracts are also reviewed, because these agents are widely used in some parts of the world, despite the lack of properly designed clinical trials. Because plant extracts are not classified as drugs, the marketing and claims are not critically scrutinized by regulatory agencies.
The Impact of Medical Therapy
Prior to the 1980s, prostatectomy was the only widely accepted intervention for BPH. The enthusiasm for medical therapy has been supported in part by the limitations of prostatectomy, which include the morbidity of the surgical procedure, failure to invariably achieve a successful outcome, and a small but significant re-treatment rate (Lepor, 1993). Although medical therapies do not achieve the same level of efficacy as prostatectomy, the attractive features of medical therapy relative to prostatectomy are that clinically significant outcomes are obtained with fewer, less serious, and reversible side effects (Lepor, 1993). Because the indication for intervention in the overwhelming majority of patients with BPH is to improve quality of life by relieving symptoms (Emberton et al, 2008), the lower morbidity of medical therapy is of paramount importance in patient-driven treatment decisions.
In 1990, TURP was second only to cataract surgery in terms of expenditures paid by the U.S. Medicare program. Medical therapy is currently considered the preferred treatment alternative for those individuals who lack absolute indications for surgery. Because the overwhelming majority of men undergoing TURP lack absolute indications for intervention (Mebust et al, 1989) and prefer nonsurgical options (Emberton et al, 2008), the number of prostatectomies performed throughout the world has decreased significantly. A survey of the U.S. Medicare database also revealed that the absolute number of prostatectomies decreased from 250,000 in 1987 to 116,000 in 1996 to 88,000 in 2000 (Wasson, 2000) and stabilized since then. This 55% reduction in TURP has occurred despite the progressively increasing number of men enrolled in the Medicare program and overall increase in U.S. spending on BPH (Wei et al, 2005). Similar reductions in TURPs have been reported from France, Canada, Denmark, and Germany.
Approximately 30% of American men older than 50 years have moderate to severe symptoms (Chute et al, 1993; Lepor and Machi, 1993). Based on the demographics of the U.S. population, 6.5 to 8.7 million men are eligible to discuss BPH treatment options (Wei et al, 2005; Jacobsen et al, 1995). The overwhelming majority of these men would not elect prostatectomy, owing to the risks associated with surgical intervention. These individuals are potential candidates for medical therapy.
Selecting Candidates for Medical Therapy
The ideal candidate for medical therapy should have symptoms that are bothersome and negatively affect quality of life so that the patient is willing to make a lifetime commitment to medical therapy, providing the drug is effective and adverse experiences are either nonexistent or minimal. There are currently no scientific data to support offering medical therapy to individuals presenting with absolute indications for intervention. Individuals presenting with recurrent urinary retention, recurrent UTIs, renal insufficiency, bladder calculi, and recurrent gross hematuria may develop life-threatening consequences from their BPH if it is not managed surgically. Until properly controlled clinical studies unequivocally demonstrate favorable outcomes, patients presenting with absolute indications for intervention should be discouraged from selecting medical therapy. If informed patients are willing to accept potential risks, medical therapy may be offered with a proviso for careful follow-up and future prostatectomy if medical therapy proves ineffective.
Preventing Benign Prostatic Hyperplasia with Medical Therapy
A potential role of medical therapy is to prevent the development of BPH or its progression. There are several factors limiting the enthusiasm for preventing the development of BPH. The clinical manifestations of BPH are rarely life threatening. Preventative intervention would have to be initiated before the fifth decade of life coinciding with the development of BPH (Partin, 2000). The long-term exposure to drug-induced adverse events and the prohibitive costs are the primary limitations of prevention therapy. In addition, effective medical and surgical therapy exists when BPH ultimately does becomes clinically evident. Because there are no clinical, biochemical, or genetic predictors of BPH development or progression, every man is potentially at risk. The ability to identify those individuals who are predisposed to develop clinical BPH refractory to medical therapy would provide a more compelling rationale for prophylaxis. There is good evidence that men with very large prostates (and usually higher PSA values) are at greater risk for developing urinary retention (Jacobsen et al, 1997) and that medical therapy (finasteride or dutasteride) can significantly decrease this risk of developing urinary retention (McConnell et al, 1998; Roehrborn et al, 2004). The decision to offer preventative therapy for urinary retention depends on the risk of the events, cost associated with treatment, and patient preferences for intervention.
Therapy with α-Adrenergic Blockers
Rationale
The rationale for α-adrenergic blockers in the treatment of BPH is based on the hypothesis that the pathophysiology of clinical BPH is in part caused by BOO, which is mediated by α1-adrenergic receptors associated with prostatic smooth muscle (Caine, 1986) (Fig. 92–4). The importance of this dynamic obstruction was supported by morphometric studies demonstrating that smooth muscle is one of the dominant cellular constituents of BPH, accounting for 40% of the area density of the hyperplastic prostate (Shapiro et al, 1992). Caine and coworkers (1975) reported that the human prostate contracts in the presence of the α-adrenergic agonist norepinephrine. Several investigators subsequently demonstrated that the tension of prostate smooth muscle is mediated by the α1 receptor (Hieble et al, 1985; Lepor et al, 1988; Gup et al, 1989). Lepor and colleagues (1988) were the first investigators to characterize the α1 receptor in the human prostate using radioligand binding studies. These investigators subsequently reported that 98% of the α1 receptors are localized to the prostatic stroma (Kobayashi et al, 1994). The importance of the adrenergic innervation of the prostate was further supported by the observation of high levels of norepinephrine in the human prostate (Lepor et al, 1990). Although the finding of high levels of smooth muscle α1 receptors and norepinephrine in the human prostate suggests an important role of the adrenergic innervation in prostatic function, it cannot be assumed that these factors are directly responsible for clinical BPH. Lepor and associates (1990) reported no significant differences between norepinephrine levels, α1 receptor density, or isometric contractile responses to phenylephrine (Gup et al, 1989) in BPH tissues obtained from men with symptomatic and asymptomatic BPH. Other investigators have shown α1 receptor levels are higher in prostatic adenoma relative to prostatic capsule (Yamada et al, 1987; Kawabe et al, 1990). These observations simply show regional differences of α1 receptors in the prostate and do not prove that clinical BPH is caused by upregulation of the α1 receptor.
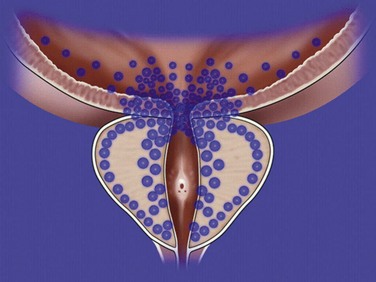
The most definitive evidence that blockade of prostate α1 receptors relieves BOO was the observed direct relationship between the area density of prostate smooth muscle and the change in the PFR in 26 subjects undergoing prostatic biopsy before initiating α-adrenergic blocker therapy with terazosin (Hytrin) (Shapiro et al, 1992). Although the prostates of those subjects achieving symptom improvement had a significantly greater group mean area density of smooth muscle compared with those of nonresponders, a direct relationship between prostate smooth muscle area density and change in symptom scores was not observed. These observations suggest that nonprostate smooth muscle–mediated α1 receptor events may also be responsible for the effectiveness of α-adrenergic blockade and that α1 receptor–mediated symptom improvement and decreases in BOO are mediated by different mechanisms.
Classification of α-Adrenergic Blockers
α-Adrenergic blockers may be classified according to α receptor selectivity and serum elimination half-life (Table 92–1)
Table 92–1 Classification of α-Adrenergic Blockers and Recommended Doses
| CLASS OF α-ADRENERGIC BLOCKER | DOSE |
|---|---|
| Nonselective | |
| Phenoxybenzamine | 10 mg bid |
| α1 | |
| Prazosin | 2 mg bid |
| IR Alfuzosin | 2.5 mg tid |
| Indoramin | 20 mg bid |
| Long-Acting α1 | |
| Terazosin | 5 or 10 mg qd |
| Doxazosin | 4 or 8 mg qd |
| Alfuzosin SR | 10 mg qd |
| Subtype Selective | |
| Tamsulosin | 0.4 mg qd |
| Silodosin | 8 mg qd |
Phenoxybenzamine, a nonselective α blocker, was shown to be highly effective for BPH (Caine et al, 1976, 1978). The limitation of phenoxybenzamine was the high incidence and severity of adverse clinical events. Berthelsen and Pettinger (1977) described two subtypes of the α receptor (α1 and α2). Prazosin was one of the first α1 receptor antagonists to be investigated for the treatment of BPH (Hedlund et al, 1983). The efficacy of phenoxybenzamine and prazosin are comparable; however, prazosin is better tolerated, implying that efficacy and toxicity are mediated primarily by the α1 and α2 receptors, respectively (Lepor, 1989). Prazosin and other α1 antagonists, including intermediate-release (IR) alfuzosin (Jardin et al, 1991) and indoramin (Ramsay et al, 1985), require at least twice-daily dosing, owing to the relatively short serum elimination half-lives. The next advance in the development of α-adrenergic blockers was the development of advanced drugs with serum elimination half-lives that allowed for once-a-day dosing. Terazosin (Hytrin) (Lepor et al, 1992) and doxazosin (Cardura) (Gillenwater et al, 1995), tamsulosin (Flomax) (Chapple et al, 1997; Narayan and Tewari, 1998; Wilt et al, 2002b), and extended-release alfuzosin (UroXatral) (McNeill et al, 2005; van Kerrebroeck et al, 2000) are long-acting α-adrenergic blockers that have been shown to be safe and effective for the treatment of BPH.
Molecular cloning studies have identified three subtypes of the α1 receptor (Andersson et al, 1997). Price and coworkers (1993) reported that the mRNA encoding the α1A receptor is predominant in the human prostate. The fact that the α1A mRNA is translated does not mean the encoded protein is translated. Lepor and associates reported that using autoradiographic (Kobayashi et al, 1994) and immunohistochemical (Walden et al, 1997) techniques, the α1A and α1B receptors are predominant in the human stroma and epithelium, respectively. Prostate smooth muscle tension has been shown to be mediated by the α1A receptor (Forray et al, 1994).
Tamsulosin is a once-daily administered α1 antagonist that exhibits some modest degree of selectivity for the α1A versus the α1B receptor and no selectivity for the α1A versus the α1D receptor (Foglar et al, 1995). The pharmaceutical industry has developed α1 antagonists that are 1000-fold selective for the α1A receptor versus α1B/α1D (Forray et al, 1994). Recently, silodosin (Rapaflo) has been introduced. This agent shows 162:1 selectivity for α1A versus α1B adrenoceptors and is achieving promising results.
Interpreting the α-Adrenergic Blocker Literature
Meta-analyses derived from the α-adrenergic blocker literature are often misleading because all of the data for a given drug are combined independent of dose and study design.
Study Designs
Four study designs have been used to investigate α-adrenergic blockers for BPH: titration to fixed dose, titration to response, titration to maximal dose, and randomized dose withdrawal.
Subjects enrolled in titration to fixed dose studies receive one of several predetermined final doses independent of clinical response unless significant adverse effects are encountered. An advantage of this study design is that dose-dependent efficacy and safety of different doses are determined. A disadvantage is the requirement for a large sample size to identify statistically significant differences between placebo and all of the treatment groups.
Titration to response design allows the investigators to titrate the dose to a threshold response or maximal dose. An advantage of this design is a smaller sample size because all subjects receiving active treatment are analyzed as a composite group independent of final dose. A disadvantage of this design is that the maximal therapeutic effect may be underestimated if the titration is not to maximal response. The data are also misleading if expressed in terms of group mean changes according to final dose because all nonresponders are titrated to the maximal dose in the absence of toxicity.
A randomized dose withdrawal design begins with an open-label dose titration. All responders are randomized to active drug or placebo. An advantage of this design is the enrichment of responders. A disadvantage is that the results are not generalizable to untreated patients.
A titration to maximal dose design, like titration to response, requires a relatively small sample size because there is only one active treatment group. This study design defines maximal clinical response achievable in practice, providing the maximal dose is also the most efficacious tolerable dose.
Dose Response
Multicenter, randomized, placebo-controlled studies have consistently shown that symptom and flow improvement is dependent on the dose of the α1 blockers. The differences between the effectiveness of different doses were often not statistically significant because these dose-ranging studies were not adequately powered to show significant differences between dose groups. MacDiarmid and coworkers (1999) have provided the most compelling evidence for a positive correlation relationship between dose and effectiveness of α1 blockers in the treatment of BPH. Responders to 4 mg of doxazosin were randomized in a double-blind manner to receive 4 mg or 8 mg of doxazosin. The improvement observed in the 8-mg group was 3.7 symptom units greater than in the 4-mg group (P = .03). In phase 3 trials, the impact of dose observed in the responders is diluted by the lack of effect in the nonresponders. In clinical practice, nonresponders are withdrawn from treatment.
Review of the Literature
Several reviews have summarized the extensive clinical experiences with α-adrenergic blockade in BPH (Chapple, 1998; Djavan and Marberger, 1999; Lowe, 1999; Lepor, 2000; Roehrborn et al, 2004; Kaplan, 2008). Nonselective and short-acting α1 antagonists are used less commonly in clinical practice, owing to tolerance and the requirement for multiple daily dose. Randomized, double-blind, placebo-controlled studies have reported the safety and efficacy of phenoxybenzamine (Caine et al, 1978; Abrams et al, 1982), prazosin (Hedlund et al, 1983; Kirby et al, 1987; LeDuc et al, 1990; Ruutu et al, 1991; Chapple et al, 1992), indoramin (Iacovou and Dunn, 1987; Chow et al, 1990; Stott and Abrams, 1991), and IR alfuzosin (Ramsay et al, 1985; Carbin et al, 1991; Jardin et al, 1991; Hansen et al, 1994). With the exception of alfuzosin, these studies typically enrolled relatively small numbers of subjects into short-term single-dose studies without quantitative assessment of symptom improvement. Multicenter, randomized, double-blind, placebo-controlled studies have examined the safety and efficacy of the long-acting α-adrenergic blockers terazosin, doxazosin, tamsulosin, and slow-release (SR) alfuzosin. Subjects enrolled in these studies generally presented with moderate to severe symptoms, PVR less than 300 mL, and no absolute indications for surgical intervention. Representative studies are reviewed to illustrate the safety, efficacy, and most effective use of α-adrenergic blockers in BPH. The reader is referred to the original articles for more comprehensive outcome assessments.
Terazosin
Randomized, double-blind, multicenter, placebo-controlled studies have consistently demonstrated the efficacy and safety of terazosin for BPH (Di Silverio, 1992; Lepor et al, 1992; Lloyd et al, 1992; Brawer et al, 1993; Elhilali et al, 1996; Lepor et al, 1996; Roehrborn et al, 1996) (Table 92–2). The multicenter, double-blind, parallel-group, randomized, placebo-controlled study of once-a-day administration of terazosin to patients with symptomatic BPH reported by Lepor and associates (1992) is representative of the expectations of terazosin therapy. Two hundred eighty-five patients entered the double-blind treatment receiving either placebo or 2, 5, or 10 mg of terazosin once daily. Statistically significant decreases from baseline obstructive, irritative, and total symptom scores were observed for all terazosin treatment groups. The level of improvements in the symptom scores were dose dependent. The 10-mg terazosin treatment group exhibited significantly greater decreases in mean irritative and total symptom scores relative to the placebo group. The 5- and 10-mg terazosin treatment groups exhibited a significantly greater mean decrease in obstructive scores relative to the placebo group. The percentages of patients experiencing a greater than 30% improvement in the total symptom scores for the placebo, 2-, 5-, and 10-mg treatment groups were 40%, 51%, 57%, and 69%, respectively (Fig. 92–5). The percentage of patients experiencing greater than 30% improvement in total symptom score in the 10-mg treatment groups was significantly greater than that of the placebo group.
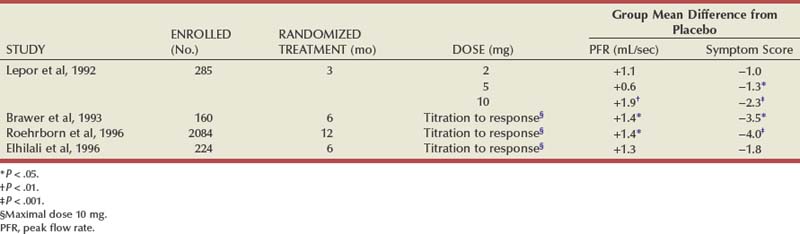
Figure 92–5 Two hundred eighty-five patients were enrolled in a randomized double-blind study comparing placebo (Plb) and 2 mg, 5 mg, and 10 mg terazosin administered once daily. Percentages of patients experiencing more than 30% improvement in total symptom scores and peak urinary flow rates are shown.
(From Lepor H. Medical therapy for benign prostatic hyperplasia. Urology 1993;42:483–501.)
A statistically significant improvement from baseline was seen in the peak and mean urinary flow rates for all the treatment groups. The effect of terazosin on PFR was also dose dependent. The 10-mg treatment group exhibited a significantly greater increase from baseline in peak and mean urinary flow rates relative to the placebo group. The percentages of patients experiencing a greater than 30% increase in PFR in the placebo, 2-, 5-, and 10-mg treatment groups were 26%, 40%, 35%, and 52%, respectively. A significantly greater proportion of patients in the 10-mg terazosin treatment group exhibited a greater than 30% improvement in PFR compared with the placebo group. Overall, the adverse events in the four treatment groups were minor and reversible. Although a higher incidence of asthenia, flu syndrome, and dizziness were observed in the terazosin treatment groups, the differences from placebo were not statistically significant. There was a significantly greater incidence of postural hypotension in the 5-mg terazosin group than in the placebo group. The incidence of syncope for all terazosin-treated patients was less than 0.5%.
The relationships between percentage change in total symptom score and PFR versus baseline age, prostate size, PFR, PVR, and total symptom score were examined to identify clinical or urodynamic factors that predicted response to terazosin therapy. No significant association was observed between treatment effect and any of these baseline factors.
There is legitimate concern whether the results of multicenter studies conducted primarily at tertiary medical centers is generalizable to community practice. Roehrborn and coworkers (1996) reported the results of the Hytrin Community Assessment Trial (HYCAT), which enrolled 2084 men into a 1-year randomized double-blind study comparing the safety and effectiveness of terazosin versus placebo. The overwhelming majority of the subjects were enrolled by urologists in community practice. The daily dose of terazosin was titrated up to 10 mg based on the discretion of the principal investigators. The symptom scores in the placebo and terazosin group throughout the study are shown in (Fig. 92–6). The treatment-related improvement (terazosin minus placebo) in the AUASI score and in urinary PFR was 1.4 mL/sec and 3.9 symptom units, respectively. The treatment-related incidences of dizziness, asthenia, and peripheral edema were 5.9%, 4.6%, and 3.1%, respectively.
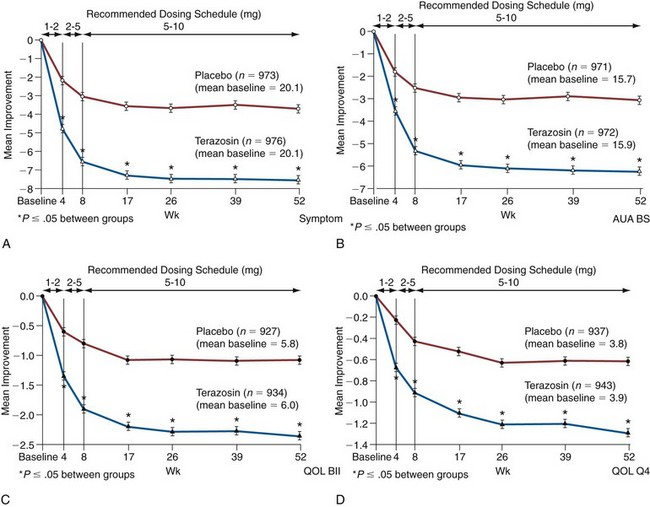
Figure 92–6 Time course of improvements in the American Urological Association (AUA) Symptom Index (A), the AUA Bothersome Score (AUA BS) (B), BPH Impact Index (QOL BII) (C), and Quality of Life question (QOL Q4) (D) for placebo and terazosin treatment groups. A lowering of the score represents clinical improvement. Weeks 1 to 2 and 2 to 5 are the mandatory titration periods for 1 and 2 mg of terazosin (and placebo), respectively.
(A to D, redrawn from Roehrborn CG. Effectiveness and safety of terazosin versus placebo in the treatment of men with symptomatic benign prostatic hyperplasia in the HYCAT study. Urology 1996;47:159–68.)
Lepor (1995) presented an interim report of 494 patients entered into an open-label extension study demonstrating the durable clinical response of terazosin. Durations of follow-up ranged from 3 to 42 months. The percentage of patients on final terazosin doses of 1, 2, 5, 10, and 20 mg were 7%, 12%, 26%, 34%, and 21%, respectively. Of the 494 patients, 213 (43.1%) withdrew prematurely: 55 (11%) because of therapeutic failure, 96 (19%) because of adverse events, and 62 (13%) because of administrative reasons.
At all follow-up visits the group mean PFRs were significantly higher than baseline values (Fig. 92–7). At baseline, PFR was 10.0 mL/sec. From 3 to 42 months, improvement ranged from 2.3 to 4.0 mL/sec. Between months 3 and 42, at least a 30% improvement in PFR from baseline was observed in 40% to 59% of the patients. At all follow-up intervals, the group mean Boyarsky symptom scores were significantly lower than at baseline; this was true of obstructive, irritative, and total scores (Fig. 92–8). From 3 months onward, improvement ranged from 4.0 to 5.4 points. Between months 3 and 42, at least a 30% improvement in total symptom score from baseline was observed in 62.4% to 77.1% of the patients.
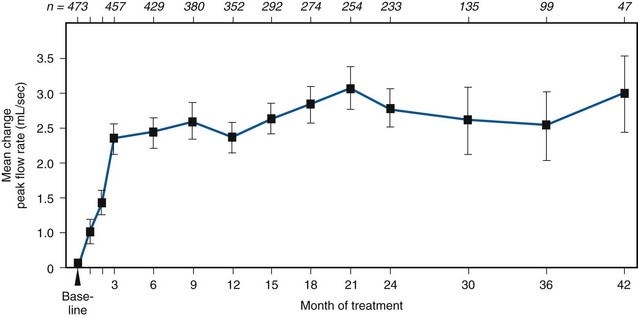
Figure 92–7 Mean change in peak flow rate between baseline and 42 months in terazosin-treated patients. The numbers across the top of the graph indicate the number of patients available at each time interval. All data points were significantly different from baseline at the P ≤ .05 level.
(From Lepor H and the Multicenter Study Group. Long-term efficacy and safety of terazosin in patients with benign prostatic hyperplasia. Urology 1995;45:406–13.)
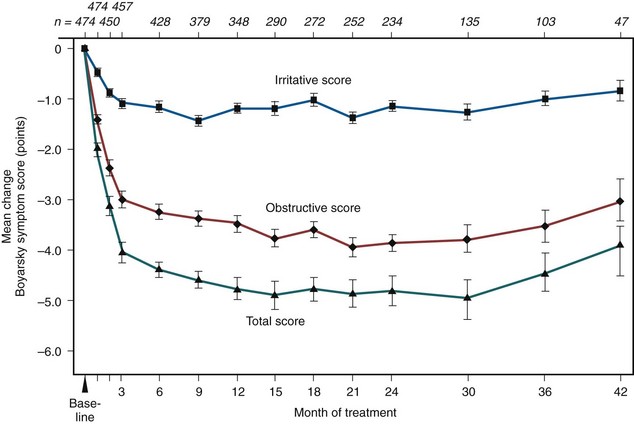
Figure 92–8 Mean change in Boyarsky symptom scores from baseline to 42 months. Baseline scores were 10.5 for total score, 6.2 for obstructive scores, and 4.3 for irritative scores. The numbers across the top of the graph indicate number of patients available at each time interval. All changes were significant at the P ≤ .05 level.
(From Lepor H and the Multicenter Study Group. Long-term efficacy and safety of terazosin in patients with benign prostatic hyperplasia. Urology 1995;45:406–13.)
Kirby (1998b) has examined the mean changes in blood pressure according to whether subjects were normotensive or hypertensive at baseline (Table 92–3). In normotensive patients, small, clinically insignificant decreases in blood pressure were generally noted. Untreated hypertensive men had larger and clinically significant decreases in blood pressure. In men with medically controlled hypertension, terazosin had no clinically significant effect on blood pressure, whereas in men with poorly controlled medically treated hypertension, terazosin significantly lowered blood pressure. In all clinical circumstances, terazosin’s effect on blood pressure was consistently physiologically desirable. The ability to treat two common coexisting conditions (BPH and hypertension) is a potentially desirable feature of the drug.
Table 92–3 Effect of Terazosin on Blood Pressure in Normotensive and Hypertensive Men
| Treatment-Related Changes in Systolic Blood Pressure/Diastolic Blood Pressure (mm Hg) | ||
|---|---|---|
| Normotensives | Hypertensives | |
| No antihypertensive treatment | −3.1/−1.7 | −13.7/−10.7 |
| Antihypertensive treatment | −2.8/−0.2 | −12.6/−11.5 |
Doxazosin
The half-life of doxazosin is longer than that of terazosin (22 vs. 12 hours). The efficacy, safety, and durability of clinical response of doxazosin has been demonstrated in multicenter, randomized, double-blind, placebo-controlled studies (Chapple et al, 1994; Fawzy et al, 1995; Gillenwater et al, 1995) (Table 92–4) and a long-term open-label extension study (Lepor, 1995). Fawzy and associates (1995) reported a 16-week multicenter, randomized, double-blind, placebo-controlled titration to response study in 100 normotensive subjects with BPH. Of the 41 evaluable subjects receiving doxazosin, 88% underwent titration to the maximal dose (8 mg). The group mean changes in PFR and symptom score were significantly greater in the doxazosin group compared with the placebo group (see Table 92–4). The magnitudes of these treatment-related effects are similar to those of terazosin. The systolic blood pressure changes in normotensive subjects are greater than those with terazosin. The treatment-related incidences of dizziness, fatigue, headache, somnolence, hypotension, and nausea were 20%, 8%, 8%, 6%, 8%, and 8%, respectively. The percentages of subjects withdrawing because of an adverse event were 14% and 2.1% in the doxazosin and placebo groups, respectively. The treatment-related incidence of adverse clinical events in this doxazosin study appears slightly higher than that of terazosin and may be a result of its greater effect on blood pressure.
Gillenwater and coworkers (1995) reported a multicenter, randomized, double-blind, placebo-controlled titration to fixed dose study comparing placebo versus 2, 4, 8, and 12 mg of doxazosin in 248 men with mild to moderate essential hypertension. The group mean changes in PFR and Boyarsky symptom score are summarized in Table 92–4 according to treatment groups. Because relatively small numbers of subjects were randomized into the individual treatment groups the failure to demonstrate statistical significance between placebo and some of the active treatment groups reflects the small sample size. The group mean improvement in PFR was dose dependent and statistically significant relative to placebo for all active treatment groups. The mean improvements in symptom scores relative to placebo for the group were statistically significant for the 4- and 8-mg doxazosin groups. Statistically and clinically significant changes in systolic blood pressure were observed between the placebo and the 4-, 8-, and 12-mg doxazosin groups. Lowering of blood pressure was a desirable outcome in these hypertensive patients. The overall treatment-related incidence of dizziness and fatigue was 15% and 10%, respectively. The percentages of subjects withdrawing because of an adverse event in the doxazosin versus placebo groups were 11.1% and 4.1%, respectively. Statistically significant changes in symptom scores and PFR relative to baseline have been reported in a long-term open-label doxazosin extension study (Lepor, 1995). The initial improvements in symptom scores and PFR in 450 subjects were maintained for up to 42 months. Kirby (1995) summarized the effects of doxazosin on blood pressure in normotensive and hypertensive men enrolled into two double-blind, placebo-controlled trials (Fawzy et al, 1995; Gillenwater et al, 1995). The treatment-related group mean reductions in sitting systolic blood pressure in the normotensive and hypertensive subjects were 3 and 17 mm Hg, respectively. The treatment-related group mean reductions in sitting diastolic blood pressure in the normotensive and hypertensive subjects were 4 and 3 mm Hg, respectively.
With the standard preparation of doxazosin, multiple titration steps are used to obtain optimal therapeutic response. Often doxazosin standard is started at 1 mg/day and titrated through 2 and 4 mg/day to 8 mg/day to obtain the optimal response. The controlled release gastrointestinal therapeutic system (GITS) formulation of doxazosin reduces the plasma peak-to-trough ratio to minimize the need for titration. To compare the two formulations in 795 men with BPH, doxazosin standard was initiated at 1 mg/day, titrated to 2 mg/day after 1 week to 4 mg/day at 3 weeks and 8 mg/day at 7 weeks if indicated. This regimen was compared with doxazosin GITS initiated at 4 mg once daily and titrated to 8 mg once daily after 7 weeks if indicated and to a placebo group over 13 weeks. The symptoms of BPH were measured with the IPSS, which has seven questions (covering frequency, nocturia, weak urinary stream, hesitancy, intermittence, incomplete emptying, and urgency) scored 0 (absent) to 5 (severe). On the IPSS there was an improvement of −8.4 and −8.0 with doxazosin standard and GITS, respectively, compared with −6.0 in the placebo group. Doxazosin standard and GITS produced clinically comparable increases in mean PFR, compared with placebo, with a greater improvement observed earlier following treatment with doxazosin GITS than with doxazosin standard. A similar number of patients in both doxazosin groups was titrated to the maximum dose of 8 mg for both formulations. The incidence of adverse effects was slightly higher with doxazosin standard than doxazosin GITS and placebo, which caused a similar incidence.
The previous study and another with 680 men were combined to further analyze the comparison between doxazosin standard and GITS (Kirby et al, 2003). In addition to confirming the results given earlier, in a subgroup reporting sexual dysfunction at baseline there was a modest clinical improvement in sexual function with both preparations of doxazosin.. Treatment-related adverse events occurred in 16.1% of patients on doxazosin GITS, 25.3% of patients on doxazosin standard, and 7.7% of placebo patients. Headache and dizziness occurred in 6.0% and 5.3% of doxazosin GITS patients, compared with 5.1% and 9.1% of doxazosin standard patients, respectively (placebo, 4.5% and 1.9%, respectively). Fewer patients on doxazosin GITS (5.7%) or placebo (2.6%) discontinued treatment because of adverse events than on doxazosin standard (7.2%). The reduction in blood pressure was not clinically significant in the normotensive patients, but there were clinically significant reductions in blood pressure with both preparations of doxazosin (placebo, 3.9/5.0; doxazosin standard, 7.4/6.1; doxazosin GITS, 9.4/6.8). A comparison of the nonconcurrent multicenter, randomized, double-blind, placebo-controlled studies of terazosin (see Table 92–2) and doxazosin (see Table 92–4) shows similar efficacy. Studies of doxazosin versus tamsulosin (Kirby et al, 2003) and doxazosin versus alfuzosin (de Reijke and Klarskov, 2004) revealed only minor differences in safety and efficacy.
α-Adrenergic blockers such as doxazosin may influence smooth muscle growth in the prostate. In BPH patients treated with α1-adrenoceptor antagonists there is a decreased expression of myosin heavy chain messenger RNA, a functional marker for the smooth muscle phenotype (Lin et al, 2001).
Biopsy and prostatectomy specimens from untreated and doxazosin-treated BPH patients suggest that doxazosin may induce apoptosis in both the epithelial and stromal cells with little effect on cell proliferation. The apoptosis was associated with a decrease in smooth muscle α-actin expression and stromal regression. Another study showed that the mean pretreatment baseline apoptosis was 1.9% and 1.0% for the epithelial and stromal prostate components. The mean apoptotic indexes increased after 3 months of doxazosin treatment for BPH to 6% in the glandular epithelial and 12% in the smooth muscle cells. By 12 months after treatment, epithelial apoptosis had decreased to constitutive levels while the apoptotic index of prostatic stromal cells remained high (Kyprianou et al, 1998).
In primary cultures of human prostate stroma cells, doxazosin increased apoptosis and decreased cell numbers. Transforming growth factor (TGF)-β1 also decreased cell numbers, and because doxazosin increased the levels of TGF-β1 in the cells it was suggested that the effect of doxazosin may be mediated through TGF-β1 (Ilio et al, 2001).
The ability of doxazosin to induce apoptosis may be shared with the other quinazoline-based α1-adrenergic receptor antagonists terazosin and prazosin, although it seems unlikely that this effect is α1-adrenergic receptor mediated (Gonzalez-Juanatey et al, 2003). The apoptotic effects of the quinazoline-based α1 antagonists may be linked to their ability to inhibit HERG potassium channels, which has been demonstrated using cloned channels expressed in Xenopus oocytes (Thomas et al, 2004).
Recent data also suggest that doxazosin may have a mildly beneficial impact on sexual function in men suffering from BPH (Kirby et al, 2005). The mechanism for this effect is uncertain but may be the result of a vasodilatory action within the corpora cavernosa.
Tamsulosin
Tamsulosin is currently the most widely employed α1 antagonist investigated for BPH (Foglar et al, 1995). One of the features of tamsulosin is that it exhibits some degree of specificity for the α1A-adrenergic receptor (Foglar et al, 1995). The efficacy and safety of tamsulosin has been investigated in four multicenter, randomized, double-blind, placebo-controlled studies (Kawabe et al, 1990; Abrams et al, 1995, Lepor et al, 1998; Narayan and Tewari, 1998) (Table 92–5).
Lepor and coworkers (1998) reported a multicenter, randomized, double-blind, placebo-controlled study of 756 American men with clinical BPH randomized to receive placebo or 0.4 or 0.8 mg of tamsulosin for 13 weeks. The mean changes in AUASI score, PFR, and adverse events are summarized in Table 92–5. The symptom score improvements were significantly greater in the 0.8-mg tamsulosin group compared with the 0.4-mg group. The treatment-related incidences of dizziness, asthenia, rhinitis, and abnormal ejaculation in the 4-mg group were 5%, 3%, 3%, and 6%, respectively, and in the 0.8-mg group they were 6%, 3%, 9%, and 18%, respectively. The mean changes in systolic and diastolic blood pressure in the placebo and tamsulosin groups were not significantly different for both hypertensive and normotensive subjects. In the subjects who were hypertensive and those whose blood pressure was uncontrolled, the systolic blood pressure changes in the placebo, 0.4-mg, and 0.8-mg groups were −8.4, −7.2, and −10.2 mm Hg, respectively. The advantage of not lowering blood pressure in men who are hypertensive at baseline is controversial.
Of the 618 subjects who completed the 13-week randomized study reported by Lepor and coworkers (1998), 418 (68%) continued into the 40-week extension study on the same double-blind medication and dose. The symptom and flow rate improvements observed at the end of the 13-week study were maintained throughout the 40-week extension study.
Narayan and associates (1998) reported the results of a randomized, double-blind, placebo-controlled trial comparing the safety and effectiveness of 0.4 and 0.8 mg of tamsulosin versus placebo. Seven hundred thirty-five men were randomized in the study. The active treatment was 13 weeks. The treatment-related improvements in the AUASI score and PFR were comparable with those reported by Lepor and coworkers (1998). The differences between 0.4 mg and 0.8 mg were not statistically significant; however, the study lacked statistical power to show clinically significant differences between the active treatment groups. The treatment-related incidences of asthenia, dizziness, rhinitis, and abnormal ejaculation observed for the 0.4-mg tamsulosin group were 2%, 5%, 3%, and 11%, respectively; and for the 0.8-mg tamsulosin group they were 3%, 8%, 9%, and 18%, respectively. The incidences of retrograde ejaculation and rhinitis were significantly greater in the 0.8-mg group compared with the 0.4-mg group. No statistically or clinically significant differences were observed for systolic blood pressure between any of the treatment groups.
A systematic review of tamsulosin therapy for BPH has been published (Wilt et al, 2002b). This included 14 studies with a total of 4122 patients. The mean change in symptom score was 12% for the 0.4-mg dosage and 16% with the 0.8-mg dosage. Improvements in flow rate were 1.1 mL/sec for both dosages. Adverse events were generally mild and included dizziness, rhinitis, and abnormal ejaculation. These increased in a dose-dependent manner with discontinuations due to such effects similar to placebo at 0.2 mg but increasing to 16% with the 0.8-mg/day dosage.
Alfuzosin
Jardin and colleagues (1991) reported the first large-scale, multicenter, randomized, placebo-controlled trial demonstrating that alfuzosin was safe and effective for the treatment of BPH (Table 92–6). A long-term open-label extension study showed that the effectiveness of alfuzosin was durable up to 30 months (Jardin et al, 1994). The primary limitation of alfuzosin was a requirement for multiple daily doses (2.5 mg three times a day or 5 mg twice a day). In the absence of any demonstrable advantage over the once-a-day drugs like terazosin, doxazosin, and tamsulosin there was no compelling reason to prescribe alfuzosin.
Extended-release or slow-release (SR) alfuzosin is a new formulation that allows for a once-daily dosing regimen without dose titration. Buzelin and coworkers (1997b) reported the first randomized, multicenter, placebo-controlled trial evaluating the safety and effectiveness of SR alfuzosin for the treatment of BPH. Three hundred and ninety subjects were randomized to once-daily 5 mg alfuzosin versus placebo for 12 weeks. The treatment-related improvements in the IPSS and PFR were −1.6 symptom unit and 1.3 mL/sec, respectively. The incidence of dropouts because of adverse events was 4.6% and 7.1% in the SR alfuzosin and placebo groups, respectively. The 2-mm Hg change in systolic and diastolic blood pressure was not significantly different from that in the placebo group. The incidences of dizziness and asthenia were similar in the SR alfuzosin and placebo groups.
SR alfuzosin (10 mg once a day) has been compared with IR alfuzosin (2.5 mg three times daily) and placebo (van Kerrebroeck et al, 2000). After a 1-month placebo lead-in, 447 patients were randomly assigned in equal proportions to the three treatment groups for 3 months. The improvement in the IPSS was 6.9, 6.4, and 4.9 in the alfuzosin 10-mg/day, alfuzosin 2.5-mg three times a day, and placebo groups, respectively. The symptom improvement observed in both active treatment groups was significantly greater than that in the placebo group. The improvements in the filling and voiding subscores and quality of life index were also significantly greater in the active treatment group relative to the placebo group. The improvement in the PFR was 2.3 mL/sec, 3.2 mL/sec, and 1.4 mL/sec in the SR alfuzosin, IR alfuzosin, and placebo groups, respectively. The modest improvements in the PFR were significantly greater in both active treatment groups compared with placebo. The incidences of dizziness were 2.1%, 4.7%, and 1.3%; and those for asthenia were 3.5%, 0.7%, and 2.6% in the SR alfuzosin, IR alfuzosin, and placebo groups, respectively. No sexual dysfunction was reported in the 10-mg/day alfuzosin group. There were no statistically or clinically significant treatment-related effects on blood pressure in normotensive or hypertensive subjects. Of those men who were hypertensive at baseline, the mean reductions in the standing blood pressure were 8.1, 8.6, and 5.8 mm Hg, respectively, in the SR alfuzosin, IR alfuzosin, and placebo groups.
Alfuzosin has been shown to have a beneficial effect on the quality of life of men suffering from BPH. In the ALFUS study (Roehrborn et al, 2001) the quality of life index improved by 18% in both active groups compared with 8% in the placebo study arm (P = .002). In an open extension of the ALFORTI trial there was a 35% improvement from baseline of quality of life (van Kerrebroeck et al, 2002). Some part of this effect may be the result of a mildly beneficial impact on sexual function (van Moorselaar et al, 2005).
Because of the lack of adverse effects and blood pressure changes, alfuzosin has been described as a uroselective drug (Kirby, 1998a). SR alfuzosin exhibits no pharmacologic uroselectivity for any of the α1 subtypes (Andersson et al, 1997). In-vivo studies in the conscious rat have shown that alfuzosin reduces urethral pressure without significantly altering blood pressure (Martin et al, 1995). This experimental observation does not prove clinical uroselectivity because terazosin and doxazosin do not alter blood pressure in normotensive subjects. Another explanation for the lack of adverse events has been the low penetration of alfuzosin into the brain (Rouquier et al, 1994). It is also important to consider that the better tolerance may simply be related to a lower level of α1 blockade because the treatment-related improvement of the 10 mg of alfuzosin appears to be less than that achieved with 10 mg of terazosin and 8 mg of doxazosin.
The long-term effectiveness of IR alfuzosin 2.5 mg three times a day is supported by an open-label prospective 3-year trial involving 3228 men with clinical BPH (Lukacs et al, 2000). The improvements in symptom score in BPH-specific health-related quality of life index observed at the 3-month visit were maintained throughout the 36 months of follow-up. A total of 20.1% of the men withdrew from the study. Only 4.2% of the men discontinued therapy because of an adverse event. The other reasons for withdrawal were death, 7.6%; loss of follow-up, 1.7%; lack of efficacy, 1.8%; study withdrawal owing to personal reasons, 0.8%; concomitant disease, 0.7%; and other reasons, 3.3%. Only 0.3% of men experienced AUR.
Silodosin
In Japan, silodosin, a selective α1-adrenergic receptor antagonist, is the BPH market leader and received U.S. Food and Drug Administration (FDA) approval in October 2008 (Watson Pharmaceuticals, Inc). In two phase 3 studies, 8 mg once-daily silodosin taken for 12 weeks resulted in significant and rapid improvement of the IPSS compared with placebo. Silodosin was shown to increase urine flow in 2 to 6 hours after the initial dose. Improvement of symptoms was realized in 3 to 4 days, with the majority of patients, including men on concomitant cardiovascular medications, achieving at least a 3-point improvement in IPSS score, regardless of age or severity of symptoms. It was associated with a low incidence of orthostatic hypotension and syncope, fainting, and dizziness. As the newest agent available silodosin has had to satisfy more rigorous cardiovascular requirements by the FDA than other, older agents. Silodosin demonstrated minimal effects on the cardiovascular system, without any meaningful prolongation of the QT interval. The most common drug-related side effect was retrograde ejaculation (better described as anejaculation). Rates of discontinuing therapy due to retrograde ejaculation were low. The second most commonly reported adverse event was dizziness, the rate of which being only slightly higher than in placebo-treated patients. There were no reported events of symptomatic orthostatic hypotension or dizziness when administered with a single dose of medications for erectile dysfunction (ED) in healthy male subjects.
A randomized, double-blind, placebo-controlled study comparing silodosin to tamsulosin or placebo study was conducted at 88 centers in Japan (Kawabe et al, 2006). Men aged 50 years or older with an IPSS of 8 or higher, a quality of life score of 3 or more, a PFR of less than 15 mL/sec, a prostate volume of 20 mL or more, and a PVR urine volume of less than 100 mL were eligible for enrollment. Patients were randomized to receive silodosin 4 mg twice daily, tamsulosin 0.2 mg once daily (i.e., a lower dose than used in the United States or Europe), or placebo, for 12 weeks: 457 men were randomized (silodosin, 176; tamsulosin, 192; and placebo, 89). The change in the total IPSS from baseline in the silodosin, tamsulosin, and placebo groups was −8.3, −6.8, and −5.3, respectively. There was a significant decrease in the IPSS versus placebo in the silodosin group from 1 week. In the early-stage comparison, silodosin showed a significant decrease in IPSS versus tamsulosin at 2 weeks. The change in quality of life score from baseline was −1.7, −1.4, and −1.1 in the silodosin, tamsulosin, and placebo groups, respectively. Silodosin showed a significant improvement in quality of life score versus placebo. In the subgroup of patients with severe symptoms (IPSS ≥ 20) silodosin also gave a significantly better improvement than placebo (−12.4 vs. −8.7). The incidence rates of adverse events and drug-related adverse events were, respectively, 88.6%, 82.3%, and 71.6% and 69.7%, 47.4%, and 36.4%, respectively. The most common adverse event in the silodosin group was abnormal ejaculation, which occurred more often in the silodosin than in the tamsulosin group (22.3% vs. 1.6%). However, only 5 men (2.9%) discontinued treatment for abnormal ejaculation. It is unclear how men in other populations will respond to this effect. Measurement by Noguchi and colleagues (2008) of intraurethral pressure in the prostatic urethra and intraluminal pressure in the vas deferens in anesthetized male dogs given the α1-adrenergic receptor agonist phenylephrine and then a series of α1-adrenergic receptor antagonists showed that silodosin had the highest selectivity for the vas deferens (7.5-fold), followed by naftopidil (4.3-fold), alfuzosin (3.8-fold), tamsulosin (2.6-fold), and prazosin (2.5-fold). These results suggest that high tissue selectivity for the vas deferens over the urethra may contribute to the incidence of abnormal ejaculation. These results (and U.S. data presented at the AUA meeting in 2009) do, however, suggest that silodosin is clinically useful for treating LUTS associated with BPH.
Naftopidil
Naftopidil, a relative selective α1D-adrenergic receptor antagonist, seems more effective for patients showing a dominant expression of the α1D-adrenergic receptor subtype. The α1 adrenergic receptor antagonists tamsulosin and naftopidil were administered to 96 patients with BPH for 8 weeks in a crossover study. With the administration of both drugs, the IPSS significantly decreased and the maximum urinary flow significantly increased. Whereas naftopidil monotherapy decreased the IPSS for storage symptoms, tamsulosin monotherapy decreased the IPSS for voiding symptoms (Ikemoto et al, 2003). It remains to be seen how clinically important this new agent will turn out to be.
Effects of α-Adrenergic Blockers on Bladder Outlet Obstruction
The primary objective of medical therapy is to improve urinary symptoms. The relevance of urodynamic studies for assessing the clinical use of medical therapy for BPH is controversial. A drug that improves urodynamic parameters of BOO without relieving LUTS would be of limited clinical utility. Conversely, a drug that relieves LUTS without improving urodynamic parameters of BOO would be of great clinical importance. There are relatively few randomized, placebo-controlled studies examining the effects of α-adrenergic blockers on pressure-flow parameters. One of the limitations to designing these urodynamic studies is the definition of a clinically significant outcome.
Martorana and colleagues (1997) reported a randomized, double-blind, placebo-controlled study examining the effect of 1 month of alfuzosin, 2.5 mg three times a day, on pressure-flow urodynamic parameters. The changes in Pdet at maximum flow, detrusor opening pressure, and maximum Pdet were significantly greater in the alfuzosin-treatment group compared with the placebo group. There were no significant differences between the effects of alfuzosin and those of placebo on PFR.
α-Adrenergic Blockers and Sexual Function and Sexual Side Effects
Sexual function is complex and includes multiple domains such as sexual desire (libido), erectile function, and ejaculatory function. Erectile and ejaculatory functions are frequently reduced in patients with LUTS/BPH and can impact on their quality of life. Therefore the treatment of BPH should aim to maintain or even restore sexual function. α1-Adrenergic receptor antagonists lack major effects on sexual desire in placebo-controlled studies (van Dijk et al, 2006). Reports on erectile function are inconsistent, with both beneficial and adverse effects being reported, but ED can occur in some patients without clear differences between drugs. Ejaculatory dysfunction during treatment may represent (relative) anejaculation. It occurs more frequently with tamsulosin and even more so for silodosin than with other drugs of this class.
α-Adrenergic Blockers in the Elderly
The adverse events associated with α-adrenergic blockers that may be particularly problematic in the elderly are dizziness and orthostatic hypertension. Kaplan and colleagues (1997) reviewed a personal series of 36 men with BPH older than age 80 years treated with terazosin or doxazosin. α-Adrenergic blockers were well tolerated, and no serious adverse events were observed. This experience was not of adequate size to address the incidence of falls. Pooled analysis of multicenter, randomized, placebo-controlled studies of terazosin (Zhang and Manski, 1998), doxazosin (Kaplan and D’Alisera, 1998), and tamsulosin (Chapple et al, 1997) have reported that the incidences of adverse events are not age dependent. It is important to emphasize that men enrolled into these studies were highly selected and so the tolerability and safety findings are not generalizable to all elderly men.
Data from population studies such as the 5872 participants in the Osteoporotic Fractures in Men study and a population-based case-control study using data from a managed care organization in southern California with more than 3 million members suggest that moderate and severe LUTS independently increase the 1-year risk of falls, particularly recurrent falls, in community-dwelling older men (Parsons et al, 2009) and that there was a modest increase in risk associated with exposure to α-adrenergic blockers that requires further investigation (Jacobsen et al, 2008).
α-Adrenergic Blocker Therapy and Coexisting Hypertension
The α-adrenergic blockers terazosin and doxazosin are established agents for the treatment of hypertension. The overwhelming clinical evidence demonstrates that terazosin and doxazosin lower blood pressure primarily in hypertensive men and that the blood pressure lowering is clinically significant (Fawzy et al, 1995; Gillenwater et al, 1995; Kirby, 1995, 1998b; Lepor et al, 1997; Lowe et al, 1999). Approximately 30% of men treated for BPH have coexisting hypertension (Boyle and Napalkov, 1995). It is reasonable to advocate the use of α-adrenergic blockers as the treatment of choice for men with hypertension and BPH.
An interim analysis of the Anti-Hypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) questioned the use of doxazosin in men at risk for developing congestive heart failure (ALLHAT Collaborative Research Group, 2000). In this study of 24,335 subjects with hypertension and at least one other coronary risk factor, men were randomized to chlorthalidone, doxazosin, amlodipine, or lisinopril. A significant increased risk of congestive heart failure in the doxazosin group relative to the chlorthalidone group was the basis for the decision to discontinue the doxazosin arm of the antihypertensive trial. There was no significant increase in congestive heart failure between doxazosin and the other antihypertensive agents. It is interesting that comparable levels of blood pressure reduction were achieved by both drugs. The ALLHAT study questions the role of doxazosin as first-line therapy for the treatment of hypertension. The study does not assess the relative risks and benefits of doxazosin in men with BPH and hypertension, nor does it have any bearing on the use of doxazosin in combination with other antihypertensive agents. Doxazosin remains an acceptable agent to treat BPH that coexists with hypertension. It should be the discretion of the physician managing the hypertension to add additional agents for treating the hypertension.
Mechanism of Adverse Events Associated with α-Adrenergic Blockade
Dizziness and asthenia are the adverse events most commonly associated with α-adrenergic blocker therapy. Elucidating the mechanism of action for these adverse events is essential for α1 subtype drug development programs. It has been assumed that dizziness and possibly asthenia were caused by cardiovascular effects. Lepor and colleagues (2000) correlated the incidence of adverse events associated with terazosin relative to blood pressure changes. Men experiencing dizziness and asthenia did not exhibit greater changes in blood pressure while on terazosin therapy. Only postural hypotension was associated with greater changes in blood pressure. α1-Adrenergic-mediated dizziness and asthenia are likely due to effects at the level of the central nervous system. Therefore it cannot be assumed that developing an α-adrenergic blocker that eliminates effects on blood pressure will significantly improve the tolerability of α-adrenergic blockers.
α-Adrenergic Blockers and Intraoperative Floppy Iris Syndrome
Intraoperative floppy iris syndrome (IFIS) complicates approximately 2% of cataract surgery cases. The clinical manifestations of IFIS are poor preoperative pupil dilation, iris billowing and prolapse, and progressive intraoperative miosis. Surgical complications are increased when IFIS is not anticipated or recognized by the surgeon. Since IFIS was first described in 2005 (Chang et al, 2005), its association with the α1-adrenergic antagonist tamsulosin has become well established. It appears that α1A is the predominant receptor subtype in the iris dilator muscle as well. The persistence of IFIS long after the discontinuation of tamsulosin suggests a semipermanent muscular atrophy and loss of tone. It is not clear how long one must take tamsulosin before experiencing these chronic muscular changes. Anecdotal reports suggest that IFIS does not occur until patients have been on tamsulosin therapy for 4 to 6 months. IFIS can also occur up to several years after discontinuation of tamsulosin. IFIS has also been reported but less commonly with non–subtype-specific α1-adrenergic antagonists, such as terazosin, doxazosin, and alfuzosin. According to an online survey, most members of the American Society of Cataract and Refractive Surgery believe that tamsulosin makes cataract surgery more difficult (95%) and increases the risks of surgery (77%).
Being able to elicit a prior history of tamsulosin use now enables cataract surgeons to anticipate IFIS and to employ alternative methods of managing the complication. Prevention of IFIS by withdrawing tamsulosin preoperatively has not shown consistent benefit. Therefore, in a patient with a known diagnosis of cataract, prescribing physicians may wish to consider involving the patient’s cataract surgeon before initiating tamsulosin or α-adrenergic blocker treatment. Patients should also be encouraged to report any prior or current history of α1 antagonist use to their ophthalmic surgeon before undergoing any eye surgery.
Comparison of α-Adrenergic Blockers
Because the therapeutic effect and adverse events associated with α blockers are both dose dependent, the effectiveness and tolerability of two different α blockers can be determined only in a randomized, double-blind, placebo-controlled trial. It is imperative that these studies be appropriately powered to show statistically significant effects for effectiveness and tolerability.
Buzelin and coworkers (1997a) reported a randomized, placebo-controlled study comparing α blockers (IR alfuzosin, 2.5 mg three times a day, vs. tamsulosin, 0.4 mg/day). The improvements in Boyarsky symptom score and PFR and the incidences of dizziness and asthenia were not significantly different between the two treatment groups. The effects of alfuzosin and tamsulosin on systolic and diastolic supine or standing blood pressures in the hypertensive patients were also not significantly different. This study suggests that IR alfuzosin and tamsulosin have equivalent effectiveness and tolerability. The obvious benefit of tamsulosin is that the dose does not have to be titrated.
The recommended daily doses of terazosin, doxazosin, tamsulosin, and SR alfuzosin are 10 mg, 8 mg, 0.4 mg, and 10 mg, respectively. The clinical data suggest that terazosin, 10 mg, and doxazosin, 8 mg, are more effective than tamsulosin, 0.4 mg, and alfuzosin, 10 mg (see Tables 92-2 and 92-4 to 92-6). The incidences of asthenia and dizziness appear to be higher for terazosin and doxazosin. The apparent better tolerability of tamsulosin and SR alfuzosin may simply be because of degree of α1 blockade and not uroselectivity. Assessing the relative efficacy and tolerability of daily terazosin, 10 mg, or doxazosin, 8 mg, versus tamsulosin, 0.4 mg, or SR alfuzosin, 10 mg, would address this issue. In the absence of these studies, nonconcurrent studies can be compared, recognizing the potential impact of differences in study design, patient selection, recording of adverse events, and dosing.
Terazosin and doxazosin exhibit very similar pharmacologic and pharmacokinetic properties. It is therefore not surprising that the effectiveness and tolerability of these two agents are also comparable. The effectiveness of terazosin and doxazosin are both dose dependent, with the greatest recorded improvements in symptom scores observed at the 10-mg and 8-mg daily doses, respectively. These doses have both been shown to be significantly more effective than lower doses. Although the incidence of adverse events is dose dependent, the 10-mg and 8-mg doses of terazosin and doxazosin are generally well tolerated.
Terazosin is unit priced so that there is no financial disincentive to titrate up to the 10-mg dose. There is no significant cost advantage between 10 mg of terazosin and 8 mg of doxazosin. Thus, until data from randomized, double-blind comparative trials demonstrate the contrary, 10 mg of terazosin and 8 mg of doxazosin should be considered equivalent. Although the price of doxazosin therapy can be reduced by dividing the 8-mg tablet, this comes at the expense of significant loss of effectiveness.
Tamsulosin and SR alfuzosin have been positioned as uroselective α1 blockers. Randomized studies have shown that 0.8 mg of tamsulosin is significantly more effective at relieving symptoms than the 0.4-mg dose (Lepor et al, 1998). No dose-ranging studies have been performed with SR alfuzosin. Unfortunately, an 0.8-mg tamsulosin dose has not been manufactured. Whereas the 0.8-mg tamsulosin dose appears to have less asthenia than terazosin and doxazosin, the incidence of dizziness is comparable and rhinitis and abnormal ejaculation are markedly greater. The most appropriate dose of tamsulosin is 0.4 mg, owing to the cost and adverse events associated with the 0.8-mg dosage. The advantage of 0.4 mg of tamsulosin is that this clinically effective dose can be administered without dose titration.
The major advantage of 0.4 mg of tamsulosin and SR alfuzosin is the lack of requirement for dose titration. For men presenting in urinary retention, tamsulosin and SR alfuzosin will likely decrease the time to voiding trial because of the lack of titration to an effective dose. The data suggest that tamsulosin and SR alfuzosin exhibit less effect on blood pressure in hypertensive men compared with terazosin and doxazosin.
Summary
Multicenter, randomized, double-blind, placebo-controlled studies have unequivocally demonstrated the safety and efficacy of α-adrenergic blockers for the treatment of BPH. The clinical response is rapid and dose dependent. Long-term open-label studies have demonstrated that the clinical response is durable. The long-acting α1 blockers are well tolerated. The blockers are safe in the elderly, diminish BOO, and reduce the risk of urinary retention. Their effect on falling needs further research. Terazosin and doxazosin significantly lower blood pressure only in hypertensive subjects, allowing treatment of coexisting BPH and hypertension. Direct comparison studies of the α-adrenergic blockers are sparse and involve small numbers of patients; therefore any claims of superiority cannot be justified.
Androgen Manipulation
Rationale
The rationale for androgen suppression is based on the observation that the embryonic development of the prostate is dependent on the androgen dihydrotestosterone (DHT) (Shapiro, 1990). Testosterone is converted to DHT by the enzyme 5α-reductase. The genetic deficiency of 5α-reductase in men results in a rudimentary prostate and in feminized external genitalia (Walsh et al, 1974). The development of BPH is also an androgen-dependent process (Coffey and Walsh, 1990). Castration and pharmacologic agents suppressing testosterone and DHT synthesis or action have been shown to reduce prostate volume in men with established BPH (McConnell, 1990). Peters and Walsh (1987) demonstrated that androgen suppression causes regression primarily of the epithelial elements of the prostate. Reducing prostate volume is thought to decrease the static component of BOO resulting from BPH. The primary limitation of the androgen suppression hypothesis is that the pathophysiology of clinical BPH and LUTS are not sufficiently dependent on prostate size.
Classification of Pharmacologic Agents
Surgical castration was reported to be an effective treatment for enlarged prostates in the 1890s (White, 1895; Cabot, 1896). Scott and Wade (1969) reported the first study investigating androgen suppression (medical castration) for BPH. Cyproterone acetate, an antiandrogen, was reported to decrease symptoms and increase PFR in the majority of treated subjects. The pharmacologic strategies of androgen suppression that have been investigated for BPH over the past 3 decades are summarized in Table 92–7.
Table 92–7 Androgen Suppression: Classification of Pharmacologic Agents and Dosages
| DRUGS | DOSE | REFERENCE |
|---|---|---|
| GnRH Analogues | ||
| Leuprolide | 3.75 mg IM qd mo | Schroeder et al, 1986 |
| Keane et al, 1988 | ||
| Eri and Tveter, 1993b | ||
| Nafarelin acetate | 400 mg SQ qd | Peters and Walsh, 1987 |
| Cetrorelix | 1 mg SQ qd ± loading dose | Lepor et al, 1997 |
| 3 dosing regimens | Debruyne et al, 2008 | |
| Progestational Agents | ||
| 17α-Hydroxycortisone | 200 mg IM weekly | Meiraz et al, 1977 |
| Megestrol | 250 mg PO tid | Donkervoort et al, 1975 |
| 40 mg PO tid | Geller et al, 1979 | |
| Antiandrogens | ||
| Flutamide | 100 mg tid | Caine et al, 1975 |
| 250 mg tid | Stone, 1989 | |
| Oxandolone | 200 mg IM weekly | Ostri et al, 1989 |
| Bicalutamide | 50 mg qd | Eri and Tveter, 1993 |
| Zanoterone | 100-800 mg qd | Berger et al, 1995 |
| 5α-Reductase Inhibitors | ||
| Finasteride | 5 mg PO qd | Gormley et al, 1992 |
| 5 mg PO qd | Finasteride Study Group, 1993 | |
| 5 mg PO qd | Andersen et al, 1995 | |
| 5 mg PO qd | Marberger et al, 1998 | |
| 5 mg PO qd | McConnell et al, 1998 | |
| Dutasteride | 0.5 mg PO qd | Roehrborn et al, 2002 |
| 0.5 mg PO qd | Debruyne et al, 2004 | |
| 0.5 mg PO qd | Roehrborn et al, 2004 |
Caveats Related to Interpreting Androgen Manipulation Studies
The efficacy of androgen suppression in BPH is presumed to be mediated by reduction of prostate volume. Maximal reduction of prostate volume after initiation of androgen suppression is achieved within 6 months (Peters and Walsh, 1987; Gormley et al, 1992). Therefore randomized treatment must be at least 6 months to capture maximal therapeutic effect.
Because the mechanism of action is by means of reduction of prostate volume, it is reasonable to assume that subjects with larger prostates achieve the greatest therapeutic benefit. The majority of randomized, double-blind, placebo-controlled clinical trials evaluating androgen suppression enrolled subjects with larger prostates (Gormley et al, 1992 [finasteride]; Eri and Tveter, 1993a [Casodex], 1993b [leuprolide]; McConnell et al, 1998 [finasteride], and Roehrborn et al, 2008b [dutasteride]). The results of studies enrolling a disproportionate number of men with large prostates may not be generalizable to the “typical” patient with clinical BPH.
Review of the Literature
Several excellent reviews of androgen suppression for BPH have been published (Vaughan and Lepor, 1996; McConnell, 1998). The overwhelming majority of drug studies evaluating androgen suppression were not randomized, enrolled small numbers of subjects, and utilized qualitative outcome measures. This section reviews only multicenter, randomized, double-blind, placebo-controlled trials. Finasteride, a type 2 5α-reductase inhibitor, and dutasteride, a dual inhibitor of both type 1 and type 2 5α-reductase, represent the paradigm for androgen suppression and are emphasized in this section.
Finasteride
Finasteride is a competitive inhibitor of the enzyme 5α-reductase (Vermeulen et al, 1989). Finasteride lowers serum and intraprostatic DHT levels. At least two isozymes (types 1 and 2) of 5α-reductase exist (Jenkins et al, 1992). Finasteride is a selective inhibitor of the type 2 isozyme. Finasteride does not reduce DHT levels to castrate levels because circulating testosterone is converted to DHT by type 1 isozymes that exist in skin and liver (Thigpen et al, 1993). Gormley and coworkers (1992) reported the first multicenter, randomized, double-blind, placebo-controlled trial investigating the safety and efficacy of finasteride in 895 men with BPH. The subjects were randomized to receive placebo or 1 or 5 mg of finasteride for 1 year. This study is often referred to as the North American Finasteride Trial.
The mean baseline prostate volumes in the placebo and 1- and 5-mg finasteride groups were 61, 61, and 59 cm3, respectively. The primary outcome measures were group mean changes in a modified Boyarsky symptom score (maximum score, 36) and PFR (Figs. 92-9 and 92-10). The group mean changes in symptom score, PFR, and prostate volume are shown in Table 92–8. The group mean percentage changes in symptom score at 12 months in the placebo and 1- and 5-mg finasteride groups were −2%, 9%, and 21%, respectively. The mean percentage changes in PFR were 8%, 23%, and 22% in the placebo and 1- and 5-mg finasteride groups, respectively. The mean percentage changes in prostate volume were −3%, −18%, and −19% in the placebo and 1- and 5-mg finasteride groups, respectively. The difference between the mean changes in PFR and prostate volume was statistically significant for both the 1- and the 5-mg finasteride groups versus the placebo group. The difference between the mean changes in symptom scores was statistically significant only for placebo versus 5 mg of finasteride. The dose-dependent symptom response was not associated with a dose-dependent prostate volume or PFR response. The changes in prostate volume were not directly related to the magnitude of the clinical response to finasteride. These observations suggest that the efficacy of finasteride may not be mediated exclusively by reduction of prostate volume. The incidences of decreased libido, ejaculatory disorder, and impotence were significantly greater in the finasteride groups compared with the placebo group. The treatment-related incidences of decreased libido, ejaculatory disorder, and impotence in the 1- and 5-mg treatment groups were 4.7%, 2.7%, and 3.7%, respectively, and 3.4%, 2.7%, and 1.7%, respectively. The percentage of subjects withdrawing because of an adverse clinical event was equivalent in the three treatment groups. Prostate volume regression was maximal at 6 months. The greatest change in symptom scores and PFR occurred within the first 2 months of initiating active treatment.
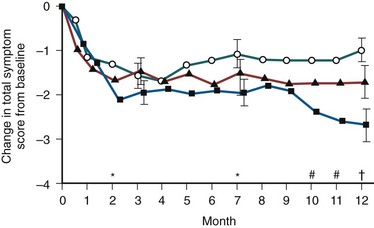
Figure 92–9 Mean (±SE) change in the total symptom score in men with benign prostatic hyperplasia during treatment with placebo (circles), 1 mg of finasteride (triangles), or 5 mg of finasteride (squares). The asterisks (P ≤ .05), the pound symbols (P < .01), and the dagger (P < .001) indicate significant differences between the finasteride-treated groups and the placebo group. Month 0 represents the baseline.
(From Gormley GJ, Stoner E, Brusekewitz RC, et al. The effect of finasteride in men with benign prostatic hyperplasia. N Engl J Med 1992;327:1185–91.
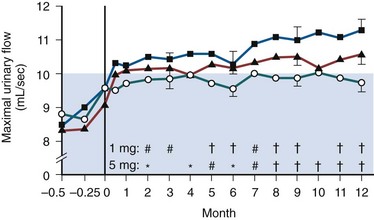
Figure 92–10 Mean (±SE) maximal urinary flow rates in men with benign prostatic hyperplasia during treatment with placebo (circles), 1 mg of finasteride (triangles), or 5 mg of finasteride (squares). The blue area indicates the range in which urinary flow was considered to be obstructed. Month 0 represents the baseline. Values before month 0 were obtained during the 2-week placebo run-in period. The asterisks (P < .05), pound symbols (P < .001), and daggers (P < .01) indicate significant differences between the finasteride-treated groups and the placebo group.
(From Gormley GJ, Stoner E, Brusekewitz RC, et al. The effect of finasteride in men with benign prostatic hyperplasia. N Engl J Med 1992;327:1185–91.
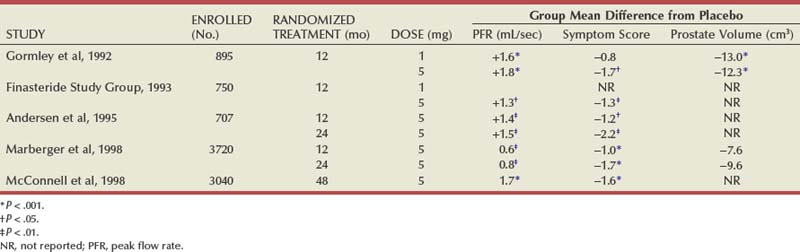
The Finasteride Study Group (1993) reported another multicenter, randomized, double-blind, placebo-controlled clinical trial that is referred to as the International Finasteride Study. The effect of finasteride on symptom scores, PFR, and prostate volume is in agreement with the findings of the North American Finasteride Trial. Andersen and associates (1995) reported the results of a multicenter, randomized, double-blind, placebo-controlled study investigating the safety and efficiency of finasteride in 707 Scandinavian subjects maintained on randomized treatment for 2 years. The mean baseline prostate volumes in the placebo and finasteride groups were 41.7 and 40.6 cm3, respectively. A selection bias for enrolling large prostates did not exist in this study. A modified Boyarsky symptom score was used to capture changes in symptom score. The differences in the group mean changes in the symptom score and PFR between finasteride and placebo after 12 and 24 months on active treatment are summarized in Table 92–8. The difference between the group mean changes in symptom scores and PFR after 1 year of randomized treatment is slightly less than that in the North American Finasteride trial? This may be attributed to the smaller baseline prostate volumes.
Although the proportion of subjects experiencing any adverse clinical event and the number of withdrawals from adverse clinical events were similar to those in the finasteride and placebo groups there were more patients with sexual dysfunction in the finasteride versus placebo groups (19% vs. 10%). The time-dependent symptom score changes demonstrate that the placebo response returns to baseline between 1 to 2 years whereas the finasteride response remains stable. The authors interpret this to show that finasteride halts or alters the natural history of the disease. The mean differences between the symptom scores at 12 and 24 months in the finasteride and placebo groups were only −0.3 and 0.6 symptom unit, respectively.
Marberger and coworkers (1998) reported the results of a 2-year randomized, placebo-controlled trial of 3270 men receiving finasteride versus placebo that are comparable with those from the report of Andersen and associates (1995). The mean baseline prostate volumes in the placebo and finasteride groups were 39.2 and 38.7 cm3, respectively. The baseline prostate volume is the lowest of all the finasteride trials. The treatment-related improvements in the quasi-AUASI at 1 and 2 years were 1.0 and 1.7 symptom units, respectively. The incidences of AUR were 1.0% and 2.5% in the finasteride and placebo groups, respectively.
Stoner and associates (1994) reported on the safety and efficacy of 3 years of therapy with finasteride. Subjects participating in the North American Finasteride Study and International Finasteride Study were offered the opportunity to participate in an open-label extension study after completing 1 year of randomized therapy. The long-term (3 years) efficacy and safety analysis was limited to the 543 subjects randomized to 5 mg. Of the 543 subjects, 297 (55%) completed 3 years of treatment and were evaluable. Of the 246 unevaluable patients, 178 were dropouts and 68 were placed in a category indicating insufficient data. Inspection of the group mean changes in outcome measures reveals that the most precipitous changes in symptom scores, PFR, and prostate volume occurred between the 12- and the 18-month assessment, which coincides with transferring from blinded to unblinded treatment. After 18 months the time-dependent changes were stable, suggesting durability of response. A subsequent report of the open-label extension study demonstrated the durability of finasteride effective up to 5 years (Hudson et al, 1999).
Boyle and coworkers (1996) reported a meta-analysis of six randomized, placebo-controlled clinical trials with finasteride. The mean group changes in symptom scores and PFR correlated with the mean baseline prostate volume. This observation accounts for the variability of treatment effect observed in the different studies.
The Proscar Long-Term Efficacy and Safety Study (PLESS) represented at the time the longest-duration multicenter, randomized, double-blind, placebo-controlled study reported in the medical therapy of BPH literature (McConnell et al, 1998). The 3040 men with moderate to severe urinary symptoms were randomized to receive daily finasteride, 5 mg, versus placebo for 4 years. A quasi-AUASI was used. The baseline prostate volume in the study population was approximately 55 cm3, indicating a bias for enrolling men with markedly enlarged prostates. The mean group changes in symptom score, PFR, and prostate volume throughout the study are shown for the finasteride and placebo in Figure 92–11. The treatment-related effects of finasteride on symptom score, PFR, and prostate volume were 2.0 symptom units, 1.7 mL/sec, and 32% size reduction for those subjects on active treatment at the end of the study. The symptom and PFR improvement was modest and consistent with findings of prior finasteride trials. The PLESS demonstrated the durability of symptom and flow improvements by finasteride and very modest progression of these end points in the placebo group.
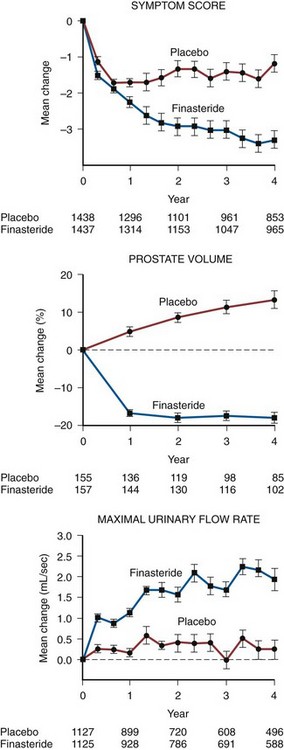
Figure 92–11 The effect of finasteride or placebo on symptom scores (on the quasi-AUASI), prostate volume, and maximal urinary flow rate over time. Values are mean (±SE) changes from baseline. The numbers below the panels show the numbers of patients with valid data who remained in the study.
(From McConnell JD, Bruskewitz R, Walsh P, et al. The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. N Engl J Med 1998;338:557–63.)
The unique findings of the PLESS were related to incidences of both AUR and surgical intervention for BPH (Fig. 92–12). The cumulative incidences of AUR at 4 years in the finasteride and placebo groups were 7% and 3%, respectively (57% risk reduction). The cumulative risk of undergoing BPH-related surgery was 10% and 5% in the placebo and finasteride groups, respectively (55% risk reduction). In those men with prostate volumes greater than 55 cm3, the risk reduction of AUR and/or surgical intervention for finasteride was 70%. The risk reduction of AUR and BPH-related surgery was clinically relevant, especially in men with very large prostates. Men with significantly enlarged prostates and LUTS should therefore be advised of their significant risk of urinary retention and the beneficial effects of finasteride.
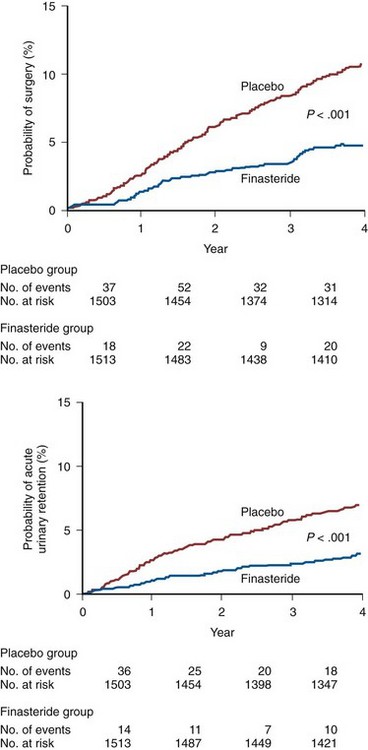
Figure 92–12 Probability of undergoing surgery for benign prostatic hyperplasia or the development of acute urinary retention during the 4-year study period in the placebo and finasteride groups. The figure shows life-table analyses of the proportion of men with each outcome. The numbers of events shown below the graphs are those that occurred during each 1-year interval. The numbers of men at risk are those at the beginning of the respective 1-year intervals. Data on men who died, were given a diagnosis of prostate cancer, or were lost to follow-up were censored at the time of death, diagnosis of prostate cancer, or discontinuation of therapy.
(From McConnell JD, Bruskewitz R, Walsh P, et al. The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. N Engl J Med 1998;338:557–63.
The PLESS study also provided insights into the impact of finasteride on the detection of prostate cancer. The decision to pursue the diagnosis of prostate cancer in the study was left at the discretion of the principal investigator and therefore represents standard practice. The detection of prostate cancer was not significantly different in the placebo and finasteride group, suggesting that finasteride does not mask the diagnosis of prostate cancer (Andriole et al, 1998).
Tammela and Kontturi (1993) reported a randomized, double-blind, placebo-controlled study examining the effects of finasteride on BOO in 36 men on a routine waiting list for prostatectomy. The mean prostate volumes in the finasteride and placebo study groups were 50 and 48 cm3, respectively. The mean baseline Pdet at maximum PFR in the placebo and finasteride groups were 115 and 126 cm H2O, respectively, indicating that the subjects were severely obstructed. The group mean changes in Pdet at PFR was +3 and −39 cm H2O in the placebo and finasteride groups, respectively. Although the difference between placebo and finasteride was highly statistically significant, the overwhelming majority of the finasteride-treated subjects remained obstructed after treatment. The marked treatment-related changes in Pdet were not associated with statistically significant changes in symptom scores. The authors did not comment on whether the changes in Pdet correlated with changes in prostate volume. Of the subjects participating in the randomized, double-blind study, 27 completed a 4-year open-label extension study (Tammela and Kontturi, 1995). The Pdet at PFR showed further improvements over time.
PSA has become widely accepted as a screening instrument for prostate cancer (Tchetgen and Oesterling, 1995). An elevated or significantly rising PSA value is often an indication for prostatic biopsy. Finasteride reduces group mean serum PSA levels approximately 50% (Guess et al, 1993). The effect of finasteride on individual serum PSA levels is highly variable. Because of the variable effect on PSA, men who are candidates for early detection of prostate cancer should have their PSA level determined before beginning finasteride therapy. A biopsy should be performed if the PSA level is elevated. A repeat biopsy should be performed for progressively rising PSA levels after initiation of therapy.
The Prostate Cancer Prevention Trial (PCPT) (Thompson et al, 2003) showed that finasteride reduced the incidence of diagnosed prostate cancer (vs. placebo). There was an increased incidence of high-grade disease, probably owing to technical pathologic reasons.
Gross hematuria is a relatively rare, yet troublesome, manifestation of BPH. Puchner and Miller (1995) reported an uncontrolled personal experience of 18 BPH patients treated with finasteride for refractory gross hematuria secondary to BPH. In the 18 reported cases, 12 of the patients had undergone a prior prostatectomy. Finasteride was very effective in relieving the postprostatectomy-associated gross hematuria. Miller and Puchner (1998) reported a follow-up series that demonstrated the long-term effectiveness of finasteride for the treatment of hematuria from BPH. Carlin and coworkers (1997) reported resolution of gross hematuria in 12 men treated with finasteride. These preliminary observations have been confirmed by a randomized, double-blind, placebo-controlled study by Foley and associates (2000) demonstrating that finasteride prevents recurrent gross hematuria secondary to BPH. Gross refractory hematuria recurred within 1 year for 63% and 14% of men randomized to placebo and finasteride, respectively.
Dutasteride
Dutasteride is a dual inhibitor of 5α-reductase types 1 and 2 and therefore has a greater impact on suppressing serum DHT levels (Clark et al, 2004). In a randomized controlled trial of 4325 men (2951 completed) Roehrborn and colleagues (2002) reported that serum DHT was reduced by 90.2%. The symptom score was improved by 4.5 points (21.4%) (P < .001), and the maximal flow rate improved significantly by 2.2 mL/sec (P < .001) at 24 months. The risk reduction of AUR was 57% and the risk reduction of BPH-related surgery was 48% compared with placebo. Debruyne and coworkers (2004) reported the pooled results of a 2-year open-label extension study in which both dutasteride- and placebo-treated groups received dutasteride 0.5 mg/day. Significant improvements in symptom scores and PFRs were observed in both study groups. It was concluded that long-term treatment with dutasteride results in continuing improvements in both symptoms and PFR and that the risk reduction of AUR and BPH-related surgery was durable over 4 years. Similar to finasteride, the principal side effects were loss of libido and ED, but these were most frequently seen at the start of therapy and declined over time with treatment. Roehrborn and associates (2004) reported similar results and confirmed 93% suppression of DHT at 4 years. Recently, the results of the REDUCE study of dutasteride versus placebo in terms of their ability to prevent prostate cancer have been reported by Andriole and associates (2009). A 23% reduction in prostate cancer was seen in the dutasteride study arm, with no increase in less well-differentiated cancers in the dutasteride arm. Beneficial effects on symptoms, PFR, and risk of progression were also seen in this group of men at high risk for BPH progression as well as for a prostate cancer diagnosis.
Further re-analysis has suggested some risk of high-grade cancers, and in December 2010 the FDA Oncologic Drugs Advisory Committee voted against use of dutasteride for reduction in the risk of prostate cancer. The manufacturer has withdrawn this application.
Zanoterone
Zanoterone is a steroidal competitive androgen receptor antagonist (Juniewicz et al, 1993). Berger and coworkers (1995) reported a multicenter, randomized, double-blind, placebo-controlled study in 463 subjects receiving placebo and 100, 200, 400, or 800 mg of zanoterone for 6 months. The group mean changes in AUASI score were not reported; however, the differences between placebo and all of the zanoterone groups were not statistically significant. The mean baseline prostate volumes ranged between 37.7 and 42.2 cm3 in the five treatment groups. The differences between the percent group mean changes in prostate volume in the placebo (−6%) and active treatment groups (−4% to −8%) were not significantly different. Interestingly, the differences between the percent group mean changes in serum PSA in all of the active treatment groups were significantly greater than those in the placebo group, despite the lack of an apparent drug effect on prostate volume. The difference between the group mean changes in PFR for placebo and the 200-mg dose was 0.7 mL/sec. Fifty-six percent and 22% of all zanoterone subjects reported breast pain and gynecomastia, respectively. The incidence and severity of adverse clinical events and the equivocal efficacy precluded further development of this drug for BPH.
Flutamide
Flutamide is an orally administered nonsteroidal antiandrogen that inhibits the binding of androgen to its receptor (Sufrin and Coffey, 1973). The first reported randomized, double-blind, placebo-controlled trial in BPH examined the safety and efficacy of flutamide in 31 men with symptomatic BPH (Caine et al, 1975). Statistically significant differences were not observed between placebo and 300 mg of flutamide for symptoms, prostate size, PVR, and PFR. Stone (1989) reported a multicenter, randomized, double-blind study comparing flutamide and placebo in men with BPH. Eighty-four patients were randomized to receive 24 weeks of either flutamide, 250 mg three times a day, or placebo. Of the 84 patients, 58 (69%) and 12 (14%) were evaluable 12 and 24 weeks on double-blind treatment. The small sample size at 24 weeks precludes any meaningful conclusions. The between-group comparisons of group mean changes from baseline for placebo versus flutamide were not statistically significant at any time point. The incidences of breast tenderness and diarrhea in the flutamide group were 53% and 11%, respectively. Although the interim analysis was reported in 1989, a subsequent report of the multicenter study has not been published.
Cetrorelix
Cetrorelix is a gonadotropin-releasing hormone antagonist that has been investigated for BPH. A potential advantage of a gonadotropin-releasing hormone antagonist over the luteinizing hormone–releasing hormone agonists in the treatment of BPH is the ability to titrate the level of androgen suppression. This would be clinically relevant if different levels of androgen suppression mediate prostate size reduction and adverse events (hot flashes, decreased libido, ED). An open-label study of 11 men demonstrated that cetrorelix reduced prostate volume and improved LUTS without significant adverse events. Lepor and coworkers (1997) reported a proof of concept randomized, double-blind, placebo-controlled study of cetrorelix in men with BPH. After an 8-day placebo lead in, men received daily subcutaneous injections of placebo or 1 mg of cetrorelix (group C01) for 27 days. One group received loading doses of 10 mg of cetrorelix on the first 4 days of active treatment (group C10). Maximal lowering of testosterone was observed within 24 hours. The testosterone level was reduced to castrate levels in the C10 group and to intermediate testosterone suppression level in the non–loading-dose group (C01) (approximately 20 ng/dL). Men exhibiting a clinical effect were observed for 1 year. In the C10 and C01 groups, the treatment-related improvement in AUASI score was 3.0 and 2.0 symptom units, respectively. In both the C10 and the C01 groups the treatment-related improvement in PFR was 2.0 mL/sec. The treatment-related reductions of prostate volume were 5.5 and 3.0 cm3, respectively. The treatment-related incidence of hot flashes and sexual dysfunction in the C01 group was negligible. During the open-label extension, prostate volume did not return to baseline, suggesting a prolonged effect on the disease. Because of problems with the drug formulation, phase 3 studies with cetrorelix were not pursued until recently (Debruyne et al, 2008).
In this dose-finding study three dosing regimens were explored: one or two injections weekly for 4 weeks. In all groups a rapid improvement in mean IPSS was obtained, with a peak effect of −5.4 to −5.9 (placebo: −2.8). Changes from baseline and differences to placebo were statistically significant up to week 20. Placebo response was less sustained than usually seen with oral preparations.
The primary disadvantage of cetrorelix and other gonadotropin-releasing hormone antagonists will be the requirement for an injection and the cost. If single-injection therapy provides desirable clinical response (over, for example, 6 months) with minimal adverse events there may exist a role for these antagonists in the treatment of BPH.
Aromatase Inhibitors
The rationale for aromatase inhibition is that estrogens may be involved in the pathogenesis of BPH. The estrogenic effect most likely mediates stromal-epithelial interactions that regulate the proliferative activity of the prostate. Several observations support the role of stroma in the development of BPH and the influence of estrogens on prostatic stroma. The inductive potential of prostatic mesenchyme (stroma) is supported by the observation of Cunha and colleagues (1980) in a mouse embryonic animal model. Coffey and Walsh (1990) reported that estrogen treatment of castrated beagles produced a threefold to fourfold increase in the total amount of prostatic stroma. Estrogens also greatly enhanced the ability of androgens to induce BPH in a canine model (Walsh and Wilson, 1976; DeKlerk et al, 1979). This synergistic effect may be mediated by the ability of estrogens to upregulate prostatic androgen receptor content. Stromal hyperplasia can be induced in the prostates of dogs and monkeys treated with aromatizable androgens and prevented by aromatase inhibitors such as atamestane (Habenicht et al, 1987; Habenicht and El Etreby, 1989).
Atamestane is a highly selective aromatase inhibitor that lowers both serum and intraprostatic levels of estradiol and estrone (El Etreby et al, 1991). Gingell and coworkers (1995) reported a multicenter, randomized, double-blind, placebo-controlled study comparing placebo and 400 mg of atamestane in 160 subjects with clinical BPH. Atamestane resulted in a statistically significant decrease in serum estradiol and estrone levels and a statistically significant increase in serum testosterone. No statistically significant group mean differences were observed for changes in Boyarsky symptom score, PFR, or prostate volume between the atamestane and the placebo groups. One of the explanations contributing to the failure of atamestane to achieve clinical efficacy was the increase in testosterone. The development of atamestane for BPH was suspended because of these negative clinical findings. The failure to demonstrate that atamestane causes regression of established BPH or clinical improvement does not negate the influence of estrogens in the pathogenesis of BPH.
Finasteride and dutasteride are the only drugs available that achieve androgen suppression with acceptable tolerability. Both symptoms and flow rates are improved, especially in men with larger glands. Impotence and decreased ejaculatory volume are the primary treatment-related adverse experiences. The literature also suggests finasteride and dutasteride may be offered to men with hematuria secondary to friable prostatic tissue and in those men with LUTS and enlarged prostates who elect to reduce their risk of developing urinary retention.
Summary
Finasteride represents the androgen suppression drug that has been most extensively characterized in BPH. Multicenter, randomized, double-blind, placebo-controlled studies support its role in the treatment of BPH. Finasteride reduces prostate volume approximately 20%. The overall treatment-related improvements in symptom score (approximately 1.0 symptom unit) and PFR (approximately 1.5 mL/sec) relative to placebo are modest. Long-term safety and durability of efficacy has been demonstrated for finasteride. The adverse clinical events associated with finasteride are minimal and are related primarily to sexual function. Finasteride is effective in the management of gross hematuria associated with BPH, especially in the presence of friable prostate tissue. Dutasteride is an inhibitor of both type 1 and type 2 5α-reductase and has similar efficacy and side effects to finasteride. Finasteride and dutasteride alter the natural history of urinary retention in men with LUTS and enlarged prostates. In the recently reported REDUCE study, dutasteride reduced the incidence of prostate cancer by 23%. Antiandrogens have also been investigated for BPH. These studies failed to demonstrate statistically significant treatment-related efficacy. The equivocal efficacy and problematic toxicity of antiandrogens limited the enthusiasm for marketing these drugs for the treatment of BPH. The role of gonadotropin-releasing hormone antagonists requires further study.
Combination Therapy with α-Adrenergic Blockers and 5α-Reductase Inhibitors
Lepor and coworkers (1996) reported the first multicenter, randomized, double-blind trial comparing placebo, finasteride, terazosin, and combination therapy (finasteride and + terazosin) in 1229 U.S. veterans with clinical BPH. All subjects randomized to finasteride received a daily dose of 5 mg. The dose of terazosin was titrated in all patients up to 10 mg, providing adverse clinical events were not encountered. The dose of terazosin was reduced at the discretion of the investigators to 5 mg for adverse events. Of the 1229 subjects randomized, 1007 (81.9%) completed the 1-year randomized treatment on assigned study medication.
The AUASI score (Fig. 92–13) and PFRs (Fig. 92–14) are shown for the four treatment groups throughout the study. The group mean changes between baseline and final study visit for the relevant primary and secondary outcome measures are summarized in Table 92–9. The mean group differences between finasteride and placebo were not statistically significant for AUASI, Symptom Problem Index, and BPH Impact Index scores and PFR. The mean group differences between terazosin versus placebo and terazosin versus finasteride for all of the outcome measures other than prostate volume were highly statistically significant. The group mean differences between combination therapy and terazosin for all of the outcome measures other than prostate volume were not statistically significant, owing to the lack of treatment-related efficacy of finasteride. The Veterans Affairs study demonstrated the superiority of α-adrenergic blockade over androgen suppression for the treatment of clinical BPH over a 1-year interval. Prostate volume decreased approximately 20% in the finasteride and combination groups. The numbers of subjects withdrawing from the study because of adverse clinical events in the finasteride, terazosin, and combination groups were similar.
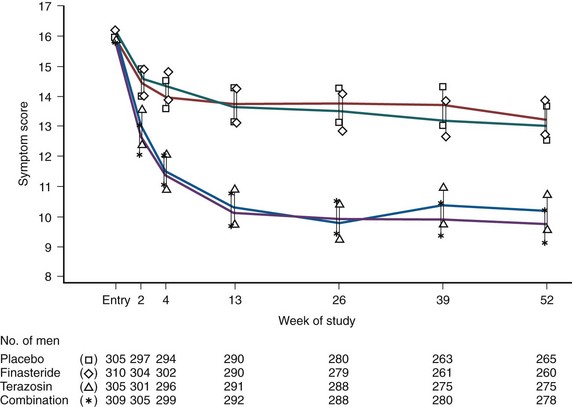
Figure 92–13 AUA Symptom Index in men with benign prostatic hyperplasia, according to treatment group. Scores are expressed as adjusted means and 95% confidence intervals. The results of primary pairwise comparisons (with Bonferroni’s adjustment) are as follows: finasteride and terazosin, P < .001; finasteride and combination therapy, P < .001; and terazosin and combination therapy, P = 1.00. The results of secondary pairwise treatment comparisons are as follows: finasteride and placebo, P =.63; terazosin and placebo, P < .001; and combination therapy and placebo, P < .001.
(From Lepor H, Williford WO, Barry MJ, et al. The efficacy of terazosin, finasteride or both in benign prostatic hypertrophy. N Engl J Med 1996;335:533–9.
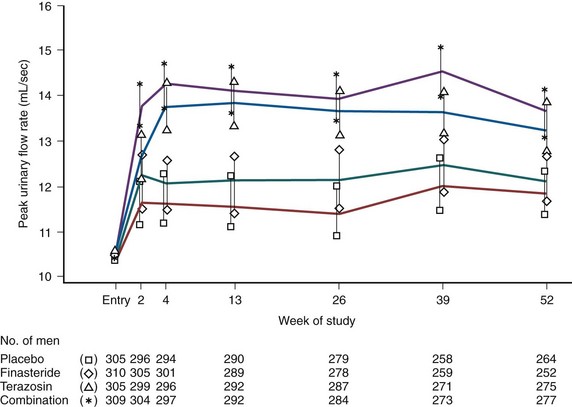
Figure 92–14 Peak urinary flow rates in men with benign prostatic hyperplasia, according to treatment group. Rates are expressed as adjusted means and 95% confidence intervals. The results of primary pairwise comparisons (with Bonferroni’s adjustment) are as follows: finasteride and terazosin, P < .001; finasteride and combination therapy, P < .001; and terazosin and combination therapy, P = .15. The results of secondary pairwise treatment comparisons are as follows: finasteride and placebo, P = .07; terazosin and placebo, P < .001; and combination therapy and placebo, P < .001.
(From Lepor H, Williford WO, Barry MJ, et al. The efficacy of terazosin, finasteride or both in benign prostatic hypertrophy. N Engl J Med 1996;335:533–9.
Table 92–9 Comparison of Placebo, Finasteride, Doxazosin, and Combination Therapy: Veterans Affairs Study
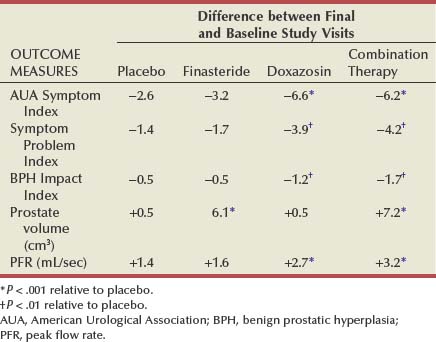
A multicenter, randomized, double-blind, placebo-controlled study comparing placebo, doxazosin, finasteride, and combination therapy confirmed the findings of the Veterans Affairs cooperative study (Kirby et al, 2003). In the Prospective European Doxazosin and Combination Therapy Trial (PREDICT), 1089 men were randomized in equal proportions to one of the just discussed four treatment groups for 1 year. The daily dose of doxazosin was titrated up to 8 mg. The baseline prostate volume was approximately 36 cm3. The group mean improvement of AUASI score and PFR and change in prostate volume between baseline and final study visit are shown in Table 92–10.
Table 92–10 Comparison of Placebo, Finasteride, Doxazosin, and Combination Therapy in the PREDICT Study
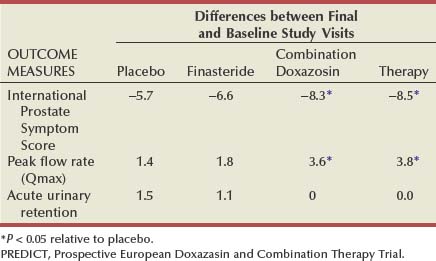
A multicenter, double-blind study compared SR alfuzosin (5 mg), finasteride (5 mg), and combination therapy in 1051 men receiving active treatment for 6 months (Debruyne et al, 1998). The improvement in IPSS was not significantly different in the alfuzosin versus combination groups. At 6 months there were no significant differences between PFR among any of the treatment groups.
Medical Therapy of Prostatic Symptoms (MTOPS) Trial
The results of all three large-scale studies just described appear to suggest that the role of 5α-reductase inhibitors as generalized monotherapy for LUTS may be somewhat limited, a finding at variance with some of the original studies on finasteride. An additional, longer-term trial, which has been progressing in parallel to these studies, has generated important new information on both the clinical potential of 5α-reductase inhibitors as monotherapy and the potential of drug combinations. The MTOPS trial, a prospective, randomized, double-blind, multicenter, placebo-controlled trial, was established to determine whether medical therapy can prevent or delay the progression of BPH in the long term. Further elucidation of the natural history of BPH, determining baseline factors associated with more rapid disease progression, was a secondary aim of the study.
In 18 academic centers across the United States a total of 3047 patients were recruited and randomized to receive doxazosin, finasteride, a combination of both, or placebo. Mean age of participants was 62.6 years, most were white (82.6%), with 8.8% black and 7.2% Hispanic participants. The inclusion/exclusion criteria allowed men with all prostate sizes to be enrolled, as long as the serum PSA value was less than 10 ng/mL. This resulted in a wide distribution of prostate sizes and serum PSA values, allowing for stratified analyses of subsets based on these criteria (McConnell et al, 2003).
Disease progression was defined as a worsening of BPH symptoms according to the AUASI. Progression was deemed to have occurred in the case of one of the following: a 4-point rise in the AUASI score, confirmed by a second visit within 4 weeks; a 50% increase in creatinine relative to baseline levels; AUR; two or more UTIs within 1 year or a single episode of urosepsis due to BOO; and socially unacceptable incontinence. The first occurrence of any of these events indicated BPH progression. Progression as an end point represented a novel concept at the time of the initiation of the MTOPS study, although the PLESS as well as the dutasteride studies later on utilized AUR and surgery as end points in their study designs (McConnell et al, 1998; Roehrborn et al, 2002). Entirely novel was the concept of utilizing a threshold to define symptom progression. Based on data from the Veterans Affairs cooperative study, in which men perceived general improvement in their symptom status once the AUASI score improved by more than 3 points, a threshold of 4 points was chosen—to be confirmed within 4 weeks—to indicate global subjective worsening of symptom status.
To assess the natural history of BPH, PFR, prostate volume, sexual function, and quality of life were regularly recorded with respect to BPH symptoms. Transrectal ultrasonography and DRE were used to evaluate prostate volume, the Sexual Function Inventory Questionnaire evaluated sexuality, and the Short Form-36 Health Survey instrument recorded quality of life scores. Prostatic biopsies were obtained at baseline and at 5 years (or at primary end point) in 37% of study participants who volunteered to take part in a biopsy substudy. Patients were randomized to receive 5 mg finasteride and doxazosin placebo, 5 mg finasteride and doxazosin titrated to up to 8 mg, titrated doxazosin and finasteride placebo, or two placebo drugs.
The results of the MTOPS trial suggest that the combination of doxazosin and finasteride exerts a clinically relevant, positive effect on rates of disease progression. Men who received combination therapy were significantly less likely to experience BPH progression than those receiving either monotherapy or placebo, with risk reduction rates of 39% for doxazosin, 34% for finasteride, and 67% for combination therapy compared with placebo.
Invasive therapy and AUR risk were significantly reduced by finasteride and combination therapy (by 69% and 64%, and by 79% and 67%, respectively), whereas all treatment regimens (placebo, doxazosin, finasteride, and combination) brought about a significant improvement in AUASI score (4.0, 6.0, 5.0, and 7.0, respectively) and PFR (1.4, 2.5, 2.2, and 3.7 mL/sec, respectively) at 4 years. AUASI score and PFR improved significantly more in the combination therapy group compared with the monotherapy groups, whereas adverse events were similar to previously reported studies.
In addition to indicating the potential benefits of combination therapy, MTOPS provided important data regarding the natural history of untreated BPH and the prediction of BPH patients who will respond most effectively to medical treatment. Although the patients receiving finasteride alone or in combination experienced the expected decrease in prostate volume, patients on placebo or doxazosin alone experienced an increase in prostate volume from a baseline of 34.0 mL by 9.3 (30.3%) (placebo) and from 36.4 mL by 9.9 (31.4%) (doxazosin), respectively. Stratified by PSA quartiles, total prostate volume in both placebo and doxazosin-treated patients increased from 4.9 mL (24.9%) to 16.2 mL (34.5%) from the lowest to the highest quartile for an annualized growth of 1.1 to 3.6 mL/yr. These findings suggest that doxazosin, despite its apoptotic effect (Glassman et al, 2001), does not interfere with the natural growth tendency of the prostate gland and that baseline PSA is a useful predictor of future prostate growth in men with LUTS and BPH.
Examination of baseline measures and the disease outcomes of 737 patients treated with placebo revealed that PSA, PFR, PVR, and prostate volume at baseline correlated with clinical progression of the disease and the need for BPH-related surgery (P = .03 to < .001). Age was linked to clinical progression (P < .001) and AUASI score correlated with need for surgery (P = .002). Baseline PSA and prostate volume correlated with risk of AUR (P = .03 to .003). Risk of progression, BPH-related surgery, and AUR increased alongside levels of serum PSA. In medically treated patients, however, baseline values were variably predictive of BPH outcome. In doxazosin-treated patients, for example, PSA, PFR, and prostate volume were predictive of outcome; however, this was not true of patients treated with finasteride alone or combined therapy.
The number needed to treat (NNT) to prevent a case of BPH progression as defined in MTOPS in the overall population was 8.4 for the combination therapy group and 13.7 and 15.0, respectively, for the doxazosin- and finasteride-treated patients. For those men treated with combination therapy who had a baseline PSA of more than 4.0 ng/mL, however, the NNT was 4.7, and for those with a prostate volume over 40 mL it was 4.9, suggesting that combination therapy becomes an economically stronger option in patients at higher risk for progression.
The link between sexual dysfunction and severity of LUTS was also confirmed by the MTOPS data. A correlation was observed between LUTS and five domains of sexual dysfunction (libido, sexual function, ejaculatory function, the patient’s assessment of his sexual problems, and overall satisfaction). In addition, men with larger prostates were more likely to have low libido, low overall sexual functioning, reduced ejaculatory function, and greater sexual problems.
Another study of combination therapy in BPH employing dutasteride and tamsulosin has been reported by Barkin and colleagues (2003). Three hundred twenty-seven patients were treated with both drugs for 24 weeks and then the α-adrenergic blocker was withdrawn for a further 12 weeks; of those patients with an IPSS less than 20, 84% continued without noticeable deterioration of their symptoms. In contrast, in the 27% of patients with more severe symptoms (IPSS > 20), 42.7% reported a worsening of their symptoms compared with 14% of those who remained on combination therapy. It was concluded that a 5α-reductase inhibitor can be used in combination with an α-adrenergic blocker to achieve rapid onset of symptom relief in patients at risk of underlying disease progression and the α-adrenergic blocker can then be discontinued. In patients with severe symptoms, combination therapy should be continued for a longer term.
Combination versus Avodart or Tamsulosin (CombAT) Trial
Recently, Roehrborn and colleagues (2008b) have reported the 2-year results of the CombAT study. In these men with a pretreatment PSA between 1.5 and 10 ng/mL, combination of dutasteride and tamsulosin was more effective than either drug alone. Interestingly, in men with larger prostates, although the tamsulosin effect was rapid, over time dutasteride was the more effective agent. Superiority of combination therapy versus dutasteride was seen from month 3 and versus tamsulosin from month 9 and was maintained for the study duration (P < .001).
The full 4-year data will be published soon and are important because longer-term studies are clearly needed to fully evaluate the efficacy of 5α-reductase inhibitor therapy in BPH.
Anticholinergic (Antimuscarinic) Receptor Blockers
There is an overlap between symptoms traditionally regarded as being due to BPH and those ascribed to the syndrome of OAB. OAB symptoms may coexist with BPH or BOO and may be either secondary to that obstruction or unrelated. Telephone surveys in the United States (Stewart et al, 2003) and in Europe (Milsom et al, 2001) show that 12% to 16% of the adult population admit to OAB symptoms. In Europe 60% of those with symptoms had consulted a medical practitioner about the symptoms and two thirds reported that they had an effect on daily living. Traditionally, in the treatment of OAB symptoms the use of antimuscarinic (commonly called anticholinergic) agents is often employed in women. However, in men there is always anxiety that these antimuscarinic agents decrease detrusor contractility and could, in theory, increase the risk of urinary retention, particularly in a man with significant obstruction. If not leading to AUR, then the risk of increasing PVR might lead to other complications, such as infection.
There have been small studies and anecdotal evidence showing that antimuscarinic agents can be used judiciously with efficacy and with minimal side effects. More recently, newer antimuscarinic agents have been introduced. Tolterodine studies have shown symptomatic benefit without increased AUR in men with OAB with proven BOO (Abrams et al, 2006) and suggest that antimuscarinic agents can be safely administered in men with BOO. Abrams and associates (2006) investigated a total of 222 men older than age 40 years with BOO and detrusor overactivity confirmed by pressure-flow studies who were enrolled and were randomized to tolterodine (2 mg twice daily in 149) or placebo (in 72) for 12 weeks: 87% completed the trial. Primary end points were PFR and Pdet at PFR. Median treatment differences in PFR (−0.7 mL/sec, 95% CI −1.6 to 0.4) and Pdet at PFR (−7 cm H2O, 95% CI −3 to 11) were comparable. Tolterodine significantly reduced the BOO index versus placebo (−9 vs. 0, P < .02), although the urodynamic significance of these changes has been questioned. There were significant treatment differences in volume to first detrusor contraction (+59 mL, 95% CI 19 to 100) and maximum cystometric capacity (+67 mL, 95% CI 35 to 103), favoring tolterodine over placebo (P < .003). Change in PVR was significantly greater among patients treated with tolterodine (+25 mL) than placebo (0 mL, P < .004). But there were no significant between-group differences in the incidence of adverse events. Urinary retention was reported by 1 patient treated with placebo. Tolterodine did not appear to adversely affect urinary function in men with OAB and BOO. Urinary flow rate was unaltered, and there was no evidence of clinically meaningful changes in voiding pressure and PVR or urinary retention. Tolterodine was also well tolerated. These results suggest that antimuscarinic agents could be safely administered in men with BOO.
Blake-James and coworkers (2007) performed a systematic review of the available literature then and, when data were sufficient, performed a meta-analysis to assess the safety and efficacy of antimuscarinic therapy in men with LUTS suggestive of BPH. They analyzed 5 randomized controlled trials and 15 observational studies of good quality (Table 92–11). Minimal changes were reported in PFR or IPSS overall, although storage symptoms scores did appear to improve in 1 trial. PVR did increase, but minimally, and there was no convincing increase in AUR rates. The authors concluded that, although antimuscarinic therapy was safe, more studies were required to define the efficacy of this therapy for men with LUTS/BPH.
Table 92–11 Summary of Outcomes of Randomized Controlled Trials of Antimuscarinic (Anticholinergic) Treatment
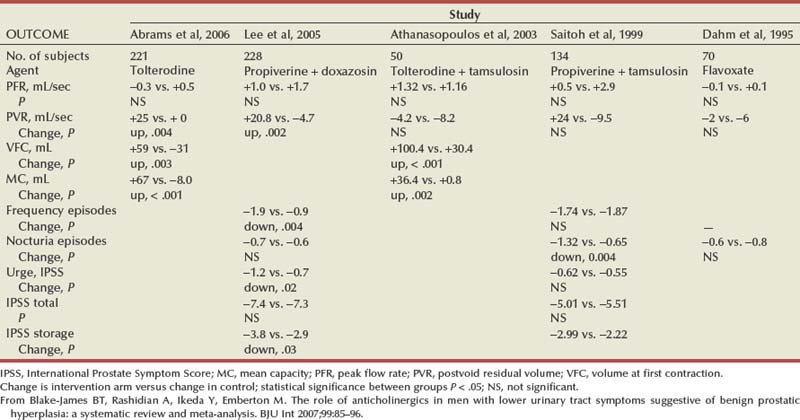
Combination Therapy: α-Adrenergic Blockers and Anticholinergic (Antimuscarinic) Receptor Blockers
Response of the OAB symptoms seen in men with LUTS/BPH raises the possibility that a combination with α-adrenergic blocker therapy could both reduce the risk of retention or deteriorating bladder function and also add to the treatment of the remaining obstructive symptoms of LUTS/BPH.
In a Korean randomized study (Lee et al, 2005), 211 men with OAB symptoms and urodynamically proven BOO were randomized (1 : 2) into two groups; one group was given doxazosin (4 mg once daily) only and the other was given propiverine (20 mg once daily) plus doxazosin for an 8-week trial. Significant improvements were noted in each group after treatment in urinary frequency, maximum flow rate, average micturition volume, and IPSS. Improvement in urinary frequency (23.5% vs. 14.3%, P = .004), average micturition volume (32.3% vs. 19.2%, P = .004), and storage (41.3% vs. 32.6%, P = .029) and urgency (P = .019) symptoms were more significant in group 2 given propiverine plus doxazosin. PVR urine was found to be significantly increased only in group 2, but this was not accompanied by urinary retention. Patient satisfaction rates were found to be significantly higher in group 2 than in group 1 (P = .002). Overall adverse event rates were higher in group 2 (P = .002), although discontinuation rates and discontinuation rates due to adverse events were not different between the two groups, suggesting that combination therapy consisting of α1-adrenoceptor antagonists with antimuscarinic agents represents an effective and relatively safe treatment modality in select patients with OAB coexisting with benign prostatic obstruction.
Kaplan and coworkers (2006) in a double-blind placebo-controlled trial in 95 centers in the United States enrolled 879 men with the usual LUTS/BPH parameters often found in trials but who also had proven frequency (minimum 8 times a day) and urgency (at least three episodes per 24 hours as recorded in voiding diaries) and randomized them between tolterodine 4 mg daily, tamsulosin 0.4 mg daily, a combination of tolterodine and tamsulosin, and placebo over a 12-week period. The combination group showed a significant benefit over placebo in response to a patient-reported single-item question determining the perception of benefit (80% vs. 62%). This was the primary end point. Data supplied suggest an NNT (number needed to treat) of 5, which is reasonable. Interestingly, neither drug used alone was significantly better than placebo in terms of patient perception of benefit, although the tamsulosin study arm showed IPSS improvement (Fig. 92–15). There were only minor adverse events, which were equally distributed in the groups.
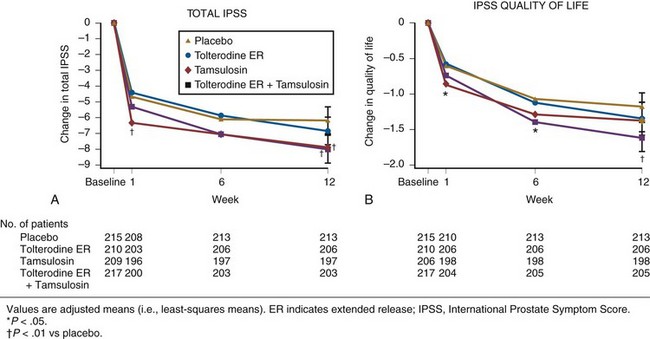
Figure 92–15 A and B, Response of lower urinary tract symptoms to tolterodine, tamsulosin, or combination vs placebo.
(From Kaplan SA, Roehrborn CG, et al. Tolterodine and tamsulosin for treatment of men with lower urinary tract symptoms and overactive bladder: a randomized controlled trial. JAMA 2006;296[19]:2319–28.)
The data suggest that in this population of men with LUTS but enriched by recruiting men with OAB symptoms (24% reported episodes of incontinence at baseline, for instance) that a treatment strategy targeting both the bladder and the prostate was needed to achieve maximal benefit. They postulate that the best candidates for this form of combination therapy might be those men not immediately responding adequately to α-adrenergic blocker therapy alone.
Once again, the added expenses of combination therapy need to be considered, although the NNT rate is encouraging. There is minimal available evidence on the long-term outcome of medical therapy of mixed OAB and BOO due to BPH. The short-term data suggest that combination of antimuscarinic and α-adrenergic blocker therapy is safe with minimal risk of retention or AUR in carefully selected men. It would seem advisable to avoid treating men with a substantial residual urine (200 mL or more in the study), and men on this therapy who are reporting increased hesitancy or showing signs of increasing PVR or clinical evidence of retention should be warned to stop the antimuscarinic element of the combination therapy immediately. Men with significant obstruction and large, persistent residual urine volumes should be considered for surgical therapy rather than the addition of antimuscarinic agents.
Key Points
Antimuscarinic Therapy
Phosphodiesterase Inhibitors
The rationale for the use of PDEIs in the treatment of LUTS/BPH was initially based on demographic data showing the frequent occurrence of both ED and LUTS in men as they age (Table 92–12). This raised the possibility of a common underlying mechanism at least contributing to both processes. This, in turn, raised the possibility of new treatment options that might impact on both processes.
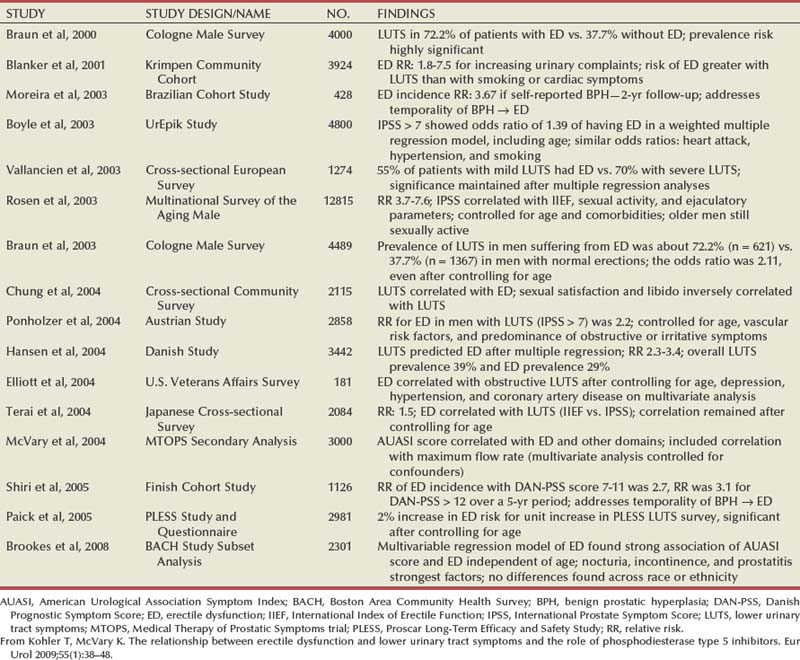
After the first clinical report in 2002 of improvement in LUTS in men given sildenafil (Viagra) for their ED (Sairam et al, 2002) there has been an explosion of interest in the role of PDEIs as treatments for LUTS and, in particular, for the management of the many men with both conditions whom we commonly see in clinics. The scientific basis is rapidly becoming stronger and there is now good level 1 evidence from (currently) four clinical trials clearly showing improvement of LUTS after treatment with PDEIs (McVary et al, 2007a, 2007b; Roehrborn et al, 2008b; Stief et al, 2008) (Table 92–13).
Table 92–13 Clinical Trials Showing Improvement of LUTS after Treatment with PDEIs: Summary of Level 1 Evidence from Four Randomized Controlled Trials Investigating PDEIs (Sildenafil, Vardenafil, and Tadalafil)
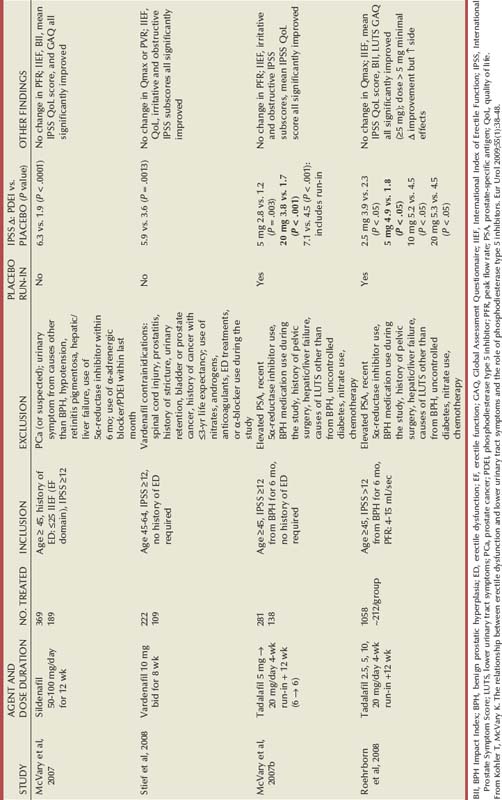
However, in the studies so far available to us there are no significant changes in PFR, suggesting that the effects of PDEIs may be either more focused on bladder muscle function than on prostatic tissue or are more profound on storage symptoms than on bladder outflow obstruction itself. The relationship between the management of LUTS and ED has been well reviewed (Kohler and McVary, 2009).
ED is commonly found in the male population who are at risk for LUTS. This association is found throughout the Western world as well as in Asian studies. The pathophysiologic link between these conditions is not yet clear, but several theories have been described with various levels of supporting data (Fig. 92–16). It is likely that there is an overlap between the roles of each of these candidate mechanisms, and an ultimate effect leading to smooth muscle relaxation in prostatic, bladder neck, or erectile tissues appears to be crucial. The candidate mechanisms include the following:
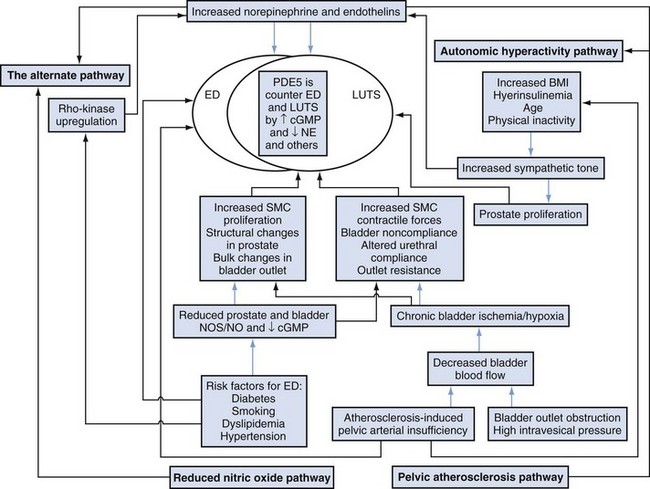
Figure 92–16 Theories linking pathophysiology of erectile dysfunction (ED) and lower urinary tract symptoms (LUTS). BMI, body mass index; cGMP, cyclic guanosine monophosphate; NOS/NO, nitric oxide synthase/nitric oxide; PDE5, phosphodiesterase type 5; SMC, smooth muscle cell.
(From Kohler T, McVary K. The relationship between erectile dysfunction and lower urinary tract symptoms and the role of phosphodiesterase type 5 inhibitors. Eur Urol 2009;55[1]:38–48.)
The pelvic atherosclerosis theory suggests that just as penile ischemia leads to smooth muscle loss in the penis so smooth muscle damage in the bladder would decrease compliance and predispose to replacement of bladder smooth muscle with collagen and fibrosis. Reduced nitric oxide synthase (NOS) expression is seen with ischemia. Furthermore, conditions promoting pelvic atherosclerosis such as hypertension, smoking, hypocholesterolemia, and diabetes mellitus are implicated in both ED and LUTS.
Autonomic hyperactivity and particularly increased sympathetic tone are seen in various components of the metabolic syndrome and have been implicated in the development of LUTS (McVary, 2005).
Abnormal activation of the Rho/Rho-kinase pathway is involved in the pathogenesis of hypertension, vasospasm, and arteriosclerosis and is a potent target of new therapies for these diseases. RhoA/Rho-kinase may suppress endothelial nitric oxide synthase (eNOS). Rho-kinase appears to play a key role in the regulation of force and velocity of actomyosin cross-bridging in smooth muscle by inhibiting myosin phosphatase–mediated dephosphorylation of the regulatory chain of myosin II. This calcium-independent pathway leads to smooth muscle contraction, probably by creating mediators of α-adrenergic (norepinephrine) and endothelin-1 (ET-1) promoted smooth muscle contraction. Abnormal Rho-kinase activation or upregulation contributes to a lack of smooth muscle relaxation in the urinary tract and thus to changes in bladder compliance and to LUTS.
Nitric oxide is a nonadrenergic noncholinergic mediator of smooth muscle activity. Burnett and coworkers (1995) reported NOS activity in the prostate. Takeda and associates (1995) showed that prostate smooth muscle tension is mediated by NO.
NO activates soluble guanylate cyclase of smooth muscle, cells, which in turn increases cyclic guanosine monophosphate (cGMP) levels. That, in turn, is responsible for smooth muscle cell relaxation and penile erection, for instance. This process is most likely to involve activation of potassium channels by cGMP, leading to hyperpolarization and closure of voltage-dependent calcium channels. Intracellular levels of calcium are reduced with an effect on myosin and its detachment from actin and muscle relaxation.
Clinical investigation really began in 2000 with a study of 112 men being investigated for their response to sildenafil but whose urinary symptoms were also prospectively assessed by IPSS and measurement of PFR over a 3-month period (Sairam et al, 2002). This open-label descriptive study was the first study to measure parameters of both ED and urinary symptoms and showed an improvement in LUTS as well as ED in response to sildenafil. Similar smaller studies were followed by four randomized, placebo-controlled studies using sildenafil, tadalafil (Cialis), and vardenafil (Levitra) (McVary et al, 2007a, 2007b; Roehrborn et al, 2008b; Stief et al, 2008). These trials are summarized in Table 92–13.
The first randomized controlled trial published to elucidate the impact of PDEIs on LUTS was by McVary and coworkers (2007a). This was the first proper randomized controlled study and was a 12-week double-blind, placebo-controlled investigation performed in 41 centers in the United States between March 2004 and May 2005. Men with both ED (scoring < 25 on the International Index of Erectile Function [IIEF]) and LUTS (scoring > 12 on IPSS) with no evidence of prostate cancer were enrolled: 369 men in total were randomized between sildenafil (189 men) and placebo (180 men). They were assessed according to a range of other parameters (BPH Impact Index, quality of life score, self-esteem and relationship questionnaire, etc) and, as expected, men’s ED improved. However, their IPSS and other urinary symptoms scores improved as well. IPSS improved by 6.32 points compared with 1.93 for placebo. The BPH Impact Index improved by 2 points compared with 0.9 for placebo, and the quality of life score improved by 0.97 compared with 0.29 for placebo. Interestingly, men with more severe LUTS improved more (by 8.6 compared with 2.4 in the less severely symptomatic men). There was no significant difference in PFR, which led the authors to suggest that possibly other mechanisms were involved than simply smooth muscle relaxation in the bladder and prostate. They pointed out that other phosphodiesterases were present in the prostate, particularly types 4 and 11, and that these may have a role to play.
In answer to questions raised about their study, more information was subsequently provided regarding baseline IPSS scores. These data showed baseline IPSS scores of 20.76 (±5.6) for those randomized to sildenafil and 20.55 (±5.5) for those randomized to placebo. The sildenafil group improved by 6.32 points compared with 1.93 for the placebo study arm. The subsequent data also indicated that the improvement in IPSS score was greater in those men whose ED also responded, that is, an IPSS improvement of 7 versus 3.2 in those whose ED did not respond (P < .0001).
The second randomized controlled trial (McVary et al, 2007b) investigated tadalafil. This was designed to establish a proof of principle prior to larger and more definitive trials. It was a double-blind, placebo-controlled, randomized multicenter study in 21 centers in the United States. It included first a 4-week washout phase but also a 4-week single-blinded placebo run-in period before determination of baseline values and compliance to reduce the placebo effect and the reversion to the mean phenomenon. Then followed the double-blind phase randomizing men between tadalafil and placebo. Men in this study were chosen on the basis of their urinary symptoms (men older than 45 with LUTS by IPSS of 13 and higher and a PFR of 4 to 15 mL/sec, PSA < 10, and PVR of < 200 mL). They were randomized between placebo or tadalafil at 5 mg for 6 weeks, after which the dose was escalated to 20 mg daily for a total of 12 weeks. The study ran between November 2004 and July 2005. As before, a range of parameters were measured. Of 281 men, 138 were randomized to tadalafil and 143 to placebo. There was a moderate response at 6 weeks but a greater difference between the 2 arms by 12 weeks. Baseline IPSS scores in the tadalafil arm improved from 17.5 to 13.3, that is, by 3.8 points, whereas the placebo arm baseline IPSS of 18.3 improved to 16.1 (or 1.7 points; p < .001), thus indicating a net improvement of 2.1 points for the active study arm. Whether a 2-point change is clinically significant or easily perceived by men is uncertain, but it is comparable to the response to α-adrenergic blocker treatment in the meta-analysis performed by the AUA BPH guideline update panel (American Urological Association, 2006). If the placebo run-in period is included, then the active study arm response of 7.1 points on IPSS (±0.6) is similar to that in the sildenafil study (6.32 points). More convincingly, men in the tadalafil group were more likely to show a 3-point or greater improvement in IPSS by 12 weeks than men in the placebo group (60.9% vs. 42.7%), indicating an NNT of 5.5, which is seen as reasonable and representing effective therapy (Bandolier, 2011).
Roehrborn and colleagues (2008a) built on this proof of principle experience with a “dose finding study” in a larger study of 1058 men, performed in 92 centers in 10 countries. Men were randomized between placebo and either a 2.5-, 5-, 10-, or 20-mg tadalafil once-daily dose. This randomized, double-blind, placebo-controlled 12-week study also included a 4-week single-blind placebo run-in period (during which period 67 men’s IPSS “improved” from the “moderate/severe” range to the “mild” category) and generally similar inclusion criteria and outcome assessments, with the IPSS response to the 5-mg dose of tadalafil being the primary outcome.
Daily dosing with 5 mg of tadalafil led to a statistically significant net improvement of 2.6 points in IPSS compared with placebo (4.87 vs. 2.27), again similar to responses to α-adrenergic blockers. Most of the response was seen by 8 weeks, and there was minimal further improvement at the higher doses (Fig. 92–17). There was no significant change to PFR at any dose, nor changes to PSA or PVR.
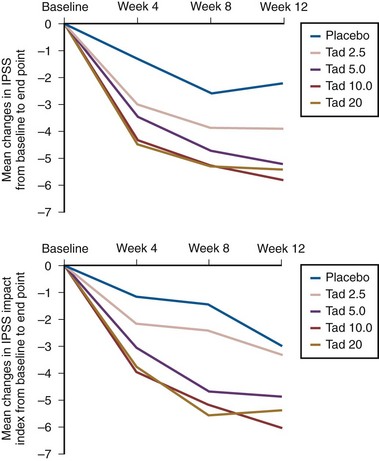
Figure 92–17 Response of lower urinary tract symptoms to tadalafil. Mean change in IPSS and BPH impact index after treatment with placebo or tadalafil (Tad) at 5, 10, and 20 mg/day.
(From Roehrborn CG, McVary KT, Elion-Mboussa A, Viktrup L. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a dose finding study. J Urol 2008a;180[4]:1228–34.)
Stief and associates (2008) investigated vardenafil in a similar fashion in a randomized, double-blind, placebo-controlled phase 2b study undertaken in 16 centers in Germany between October 2005 and June 2006. Men were randomized between vardenafil 10 mg twice daily (109 men) and placebo (113 men) and assessed at 4 and 8 weeks. This was another proof of concept study and did not include a placebo run-in period. Eventually, 105 men took vardenafil and 110 took a placebo. Vardenafil was associated with a significant improvement in IPSS versus placebo. IPSS score improved from 16.8 to 11 (or by approximately 5.9) on vardenafil compared with 16.8 to 13.2 (3.6) on placebo, that is, a mean difference of 2.3 points, again similar to an α-adrenergic blocker response to alfuzosin or tamsulosin. There were small but not significant improvements in PFR (15.9 mL/sec to 17.5). This was a relatively young cohort and had reasonable PFR values at baseline, so a study with older men and lower flow rates might more readily assess whether there is any effect of PDEIs on the flow aspects of BPH/LUTS.
Combination Therapy: α-Adrenergic Blockers and Phosphodiesterase Inhibitors
Most recently there is published evidence that PDEIs and α-adrenergic blockers in combination reduce adrenergic tone in both prostate and cavernosal smooth muscle tissue in organ bath studies (Oger et al, 2009). It is logical to consider combining PDEIs and α-adrenergic blocker medication. α-Adrenergic blockers are established treatments, and from the previous discussion it is likely that PDEIs are effective at reducing LUTS (although not by altering flow rate) and are likely to become more important options for the management of men with BPH/LUTS and ED in future.
So far evidence for combination therapy with α-adrenergic blockers and PDEIs is limited. Kaplan and coworkers (2007) in a 12-week open-label single-center pilot study randomized 62 men between 25 mg of sildenafil (21 men), 10 mg of alfuzosin (20 men), and a combination of both agents (21 men). IPSS improved by 11.8% on sildenafil (i.e., decreased from 16.9 to 14.9), by 15.6% on alfuzosin, and by 24.1% on a combination of sildenafil and alfuzosin, suggesting that there may be a synergistic benefit from the use of both agents.
Improvement in PFR was observed in all groups, but patients receiving combination therapy had greater improvement (29.6%) than patients receiving either only alfuzosin (21.7%) or only tadalafil (9.5%). IPSS was significantly improved in the alfuzosin group (27.2%) and was more marked in the combination therapy group (41.6%). There was a small nonsignificant increase in IPSS in the tadalafil-only group (8.4%).
Liguori and colleagues (2009) in a randomized, open-label, three-arm study gave 66 men complaining of ED and LUTS tadalafil or alfuzosin or tadalafil and alfuzosin and found that the IIEF sexual function scores did improve with alfuzosin alone (+15%). As expected, sexual function scores were more obviously improved with tadalafil alone (+36.3%) but the greatest improvement was experienced with combination therapy (+37.6%).
Improvement in PFR was observed in all groups, but patients receiving combination therapy had greater improvement (29.6%) than patients receiving either only alfuzosin (21.7%) or only tadalafil (9.5%). IPSS was significantly improved in the alfuzosin group (27.2%) and was more marked with the combination therapy (41.6%), and a small increase, although not statistically significant, was also observed with tadalafil (8.4%).
Finally, a new PDEI agent, udenafil (Zydena), has been administered to 120 men with BPH and ED. Men whose conditions were stable on α-adrenergic blocker therapy for their LUTS/BPH were given 100 mg of udenafil for 8 weeks. LUTS and ED improved significantly compared with baseline (IPSS improved from 14.3 to 11.5 and IIEF improved from 11.95 to 18.32). No significant change in blood pressure or heart rate was reported in this study. This is an important issue because the concomitant use of α-adrenergic blockers and PDEIs may lead to symptomatic hypotension in some patients because both are vasodilators.
It seems reasonable to suggest that concomitant treatment with PDEIs should only be initiated once the patient’s condition has been stabilized on α-adrenergic blocker therapy. In these patients PDEIs should be initiated at the lowest recommended starting dose. In those men already taking an optimized dose of a PDEI, α-adrenergic blocker therapy should be initiated at the lowest dose. Any stepwise increase in α-blocker dose may be associated with further lowering of blood pressure in patients taking PDEIs.
Studies so far suggest that vardenafil may be administered at any time with tamsulosin whereas men given vardenafil and terazosin at the same time were more prone to hypotension. This effect was minimized by giving vardenafil and terazosin at doses separated by a time interval of 6 hours. The coadministration of doxazosin (4 and 8 mg daily) and tadalafil (5 mg/day or 20 mg as a single dose intermittently) leads to further lowering of blood pressure, and this combination is not recommended by the manufacturers.
Summary
PDEIs may well have a role in therapy for BPH/LUTS. There is now good level 1 evidence of a beneficial effect of PDEIs on urinary symptoms. The mechanisms of effect are still unclear but the subject of extensive research because so many other body systems are also affected. It is likely that PDEI treatment will be of value, especially for men with LUTS and significant ED. Because recent U.S. data indicate the proportion of men reporting moderate to severe LUTS ranges from 8% in those 30 to 39 years old to 26% in those 70 to 79 years old and the prevalence of ED is also high and increases dramatically with age with 10% of 30- to 39-year-old and 59% of 70- to 79-year-old men reporting mild to moderate or moderate to severe symptoms, this is clearly a substantial number of men who may request treatment (Brookes et al, 2008).
Further data on safety and cost-effectiveness, especially for combination therapy, will be needed. Currently, PDEIs are more expensive than α-adrenergic blockerr or 5α-reductase inhibitors, which are now off patent protection in many countries and health care systems. Studies are needed to determine if costs may be reduced by using combination therapy initially followed by later withdrawal of the more expensive agent.
Phytotherapy
The pharmacologic use of plants and herbs (phytotherapy) for the treatment of LUTS associated with BPH is common. Phytotherapeutic agents for LUTS/BPH have gained widespread use since about 1990 (Lowe and Fagelman, 1999). These agents are popular in Europe, particularly in France, Austria, and Germany, where they are often prescribed and their costs reimbursed (Lowe et al, 1998). Recent European data (Fourcade et al, 2008) show that although α-adrenergic blocker monotherapy was the most frequently prescribed treatment (62.5% overall, 87.1% in Germany, 46.1% in France), phytotherapy was very popular (23.5%), followed by 5α-reductase inhibitor monotherapy (3.75%). Combination therapy was rare. Treatment varied according to the severity of the symptoms (P = .008), with phytotherapy being given to patients with the lowest IPSS and combination therapy given to those with the highest IPSS. However, use in the United States has escalated. It has been estimated that over $1 billion was spent in the United States alone for these products (Lowe and Fagelman, 1999). These agents have been marketed to “promote prostatic health,” and therefore it is not surprising that many men try them. Additional factors that contribute to their widespread use include being “natural” products (not “medications”), presumed safety, ease of accessibility (no prescription necessary), avoidance of prostate surgery, and prevention of prostate cancer (falsely assumed). The widespread availability of these products in health food stores, vitamin shops, traditional pharmacies, and supermarkets, as well as at numerous websites on the Internet, has contributed to their use and reflects the demand for these phytotherapeutic agents.
Origin of Phytotherapeutic Agents
Phytotherapeutic products are not the actual plant but are extracts derived from either the roots, the seeds, the bark, or the fruits of the various plants used (Table 92–14). Although monopreparations (single plant only) are available, many companies manufacture combination products (two or more plant extracts) in an attempt to provide “enhanced” efficacy (never proven), to improve marketability, and to provide their own “unique” product that can be registered because there is no patent protection for these products. However, to determine mechanisms of action and efficacy, only the monopreparations are evaluated and reviewed here.
Table 92–14 Origin of Plant Extracts
| SPECIES | COMMON NAME |
|---|---|
| Serenoa repens, Sabal serrulata | Saw palmetto berry/American dwarf palm |
| Hypoxis rooperi | South African star grass |
| Pygeum africanum | African plum tree |
| Urtica dioica | Stinging nettle |
| Secale cereale | Rye pollen |
| Cucurbita pepo | Pumpkin seed |
| Opuntia | Cactus flower |
| Pinus | Pine flower |
| Picea | Spruce |
Composition of the Phytotherapy Extracts
The composition of plant extracts is very complex. They contain a wide variety of chemical compounds, which include phytosterols, plant oils, fatty acids, and phytoestrogens (Table 92–15). Which of these is the exact “active” component is not definitely known. Both the free fatty acids and the sitosterols have been thought to be the active components.
Table 92–15 Components of Plant Extracts
| PHYTOSTEROLS | PHYTOESTROGENS | TERPENOIDS |
|---|---|---|
Most of the plant extracts are unique. First, the plants are not identical, owing to natural variability. Second, the extraction processes used by the various manufacturers are frequently different and use various substrates for the process. Thus, even if the phytotherapeutic compounds produced by two different companies contain the same plant, the exact composition of the final products is probably different and the content of the “active” component in each preparation may also be different. For example, analysis by the National Consumer Lab of free fatty acid content in 27 different saw palmetto products showed variability from 0% to 95%, with only 17 containing more than the presumed standard amount of 85% (http://www.ConsumerLab.com, March 2000).
Mechanism of Action
The mechanisms of action of the phytotherapeutic agents are generally unknown (Lowe et al, 1998). Many in-vitro experimental studies have been undertaken to elucidate this; thus there are numerous proposed mechanisms of action (Table 92–16). Almost all these studies use supraphysiologic doses that are many times higher than the standard doses used clinically. The biologic effects are typically examined in tissue culture, which might not be an accurate reflection of in-vivo effects (Lowe and Ku, 1996). The three mechanisms of action that have received the greatest attention are anti-inflammatory effects, 5α-reductase inhibition, and growth factor alteration.
Table 92–16 Suggested Mechanisms of Action of Plant Extracts
The anti-inflammatory effects are modulated by effects on prostaglandin synthesis. Plant flavonoids are inhibitors of both cyclooxygenase and lipoxygenase enzymes (Bach, 1982; Buck, 1996). Flavone, a phytoestrogen commonly found in plants and herbs, has been shown to be a strong inhibitor of cyclooxygenase (Mower et al, 1984; Alcaraz and Ferrandiz, 1987). Serenoa repens (Permixon) has been shown to inhibit phospholipase A2 activity, thereby decreasing arachidonic acid metabolites and prostaglandin E2 synthesis (Plosker and Brogden, 1996). Additionally, in two different studies, Paubert-Braquet and colleagues (1994, 1997) demonstrated inhibition of the production of lipoxygenase metabolites and leukotrienes by neutrophils by S. repens and Pygeum africanum (Tadenan).
The most widely suggested mechanism of action of S. repens is as a 5α-reductase inhibitor (Plosker and Brogden, 1996), thus reducing the conversion of testosterone to DHT and potentially leading to reduction of prostate volume (Gormley et al, 1992).
Although inhibition of 5α-reductase activity by S. repens was found in experimental models using foreskin fibroblasts (Sultan et al, 1984), transfected Sf9 insect cells (Iehle et al, 1995), DU145 cancer cell line (Delos et al, 1994), primary cultures of human BPH and adenocarcinoma epithelial and fibroblast cells (Delos et al, 1995), and cocultures of human epithelial and fibroblast cells (Scaglione et al 2008), other in-vitro and in-vivo data have not confirmed this effect (Rhodes et al, 1993; Weisser et al, 1996).
Two ex-vivo experiments have demonstrated conflicting results. Pretreatment with S. repens for 3 months before suprapubic prostatectomy demonstrated a decrease in prostatic DHT and an increase in prostatic testosterone concentrations compared with controls, which suggests inhibition of 5α-reductase activity (DiSilverio et al, 1998). In a similar pretreatment study using Sabal serrulata (IDS-89) for 3 months, prostate tissue levels of 5α-reductase, 3α-hydroxysteroid, and 3β-hydroxysteroid oxidoreductase were not different compared with placebo (Weisser et al, 1996). An in-vivo experiment of healthy male volunteers demonstrated a reduction of serum DHT levels with finasteride but not with S. repens (Strauch et al, 1994). Clinically, in a large multicenter trial comparing S. repens with finasteride, no effect on PSA levels and only 6% reduction in prostate volume were noted for S. repens–treated patients, whereas the finasteride-treated patients had reduction of PSA levels by 41% and prostate volume by 18% (Carraro et al, 1996).
It has been subsequently postulated that S. repens inhibits intracellular 5α-reductase while having little or no effect on other androgen-dependent processes (e.g., PSA production) that rely on binding of androgens to their receptor. S. repens causes disruption of nuclear membrane in cell culture but has no effect on the integrity of the cell membrane (Bayne et al, 1999); therefore, the intracellular theory has been promulgated. Whether this supraphysiologic cell culture experiment actually reflects what is happening in vivo is uncertain and unproved.
Plant extracts are also thought to act by altering growth factor–induced growth and proliferation. In-vitro studies with P. africanum demonstrated an inhibitory effect on both basic fibroblastic growth factor (bFGF)– and epidermal growth factor (EGF)–induced human and rat prostate fibroblast proliferation (Paubert-Braquet et al, 1994; Yablonsky et al, 1997). Subsequent experiments with S. repens have also shown inhibition of bFGF- and EGF-induced proliferation of human BPH prostate cells from biopsy specimens (Paubert-Braquet et al, 1998). Additionally, an ex-vivo experiment in men treated with S. repens before prostatectomy demonstrated reduced levels of tissue EGF, particularly in periurethral tissue (DiSilverio et al, 1998).
Although experimental data have suggested numerous possible mechanisms of actions for the phytotherapeutic agents it is uncertain which, if any, of these proposed mechanisms is responsible for the clinical responses.
Serenoa Repens (Saw Palmetto Berry)
The extract of the berry of the American saw palmetto, or dwarf palm plant, Serenoa repens (also known by its botanical name of Sabal serrulatum), is the most popular phytotherapeutic agent available for the treatment of BPH. Although numerous clinical trials with saw palmetto berry extracts have been published, many were uncontrolled open-label studies, thus providing little useful information in determining the efficacy of these phytotherapies. Even though placebo-controlled studies have been published (Table 92–17), most of them are flawed and few of them would meet the generally accepted criteria developed by the International Consultation on BPH for assessing treatment results in men with LUTS (McConnell, 1998). The studies are of limited value because of their small numbers of patients, short duration (only 1 to 3 months), and lack of use of standardized symptoms scores.

For example, nocturia was the only symptom available for analysis in all the studies reviewed in two meta-analyses (Wilt et al, 1998; Boyle et al, 2000). In the Wilt and colleagues (1998) meta-analysis of 18 trials involving 2939 patients using various S. repens single and combination preparations, the mean weighted difference for nocturia between patients and subjects taking a placebo was −0.76 times per evening (−1.22 to −0.32) for 10 trials. In the Boyle and coworkers (2000) meta-analysis of 13 trials involving 2859 patients using only the Permixon brand of S. repens, the attributable reduction of nocturnal urinations was 0.50 (±0.01) times per night. Wilt and coworkers conducted a systematic review, first published in 1998 and updated in 2002 and again in 2009, to evaluate the efficacy and adverse events of S. repens (Tacklind et al, 2009). Initially their analysis was mildly supportive of the efficacy of S. repens. In this update 9 new trials involving 2053 additional men (a 64.8% increase) were included. For the main comparison—S. repens versus placebo—3 trials were added with 419 subjects and three end points (IPSS, peak urine flow, prostate size). Overall, 5222 subjects from 30 randomized trials lasting from 4 to 60 weeks were assessed. Twenty-six trials were double blinded, and treatment allocation concealment was adequate in 18 studies. With these extra data and especially the double-blind, placebo-controlled trial reported by Bent and associates (2006), they concluded that S. repens was not superior to placebo in improving IPSS or PFR or for reducing prostate size. For nocturia, S. repens was significantly better than placebo (weighted mean difference −0.78 nocturnal visits, 95% CI −1.34 to −0.22, P < 0.05; 9 trials), but with the caveat of significant heterogeneity within studies. A sensitivity analysis, utilizing higher-quality, larger trials demonstrated no significant difference. S. repens was also not superior to finasteride or to tamsulosin. Overall, it was concluded that S. repens was not more effective than placebo for treatment of urinary symptoms consistent with BPH.
Despite inherent weaknesses in meta-analyses (Table 92–18) these analyses attempt to maximize the information available from clinical trials using S. repens in the treatment of symptomatic BPH/LUTS and to provide the best information available until the appropriate placebo-controlled, randomized clinical studies are conducted. However, approximately 35% of the prior meta-analyses performed are not accurately predicted by the subsequent randomized controlled trials (LeLorier et al, 1997). Therefore, whereas the efficacy of S. repens has not been completely determined, current meta-analysis suggests that S. repens is not more effective than placebo for treatment of urinary symptoms consistent with BPH.
Table 92–18 Inherent Weaknesses of Meta-Analyses
Pygeum africanum (African Plum)
In addition to the proposed mechanisms of actions previously discussed, P. africanum (Tadenan) has been postulated to have additional beneficial effects on LUTS by having a protective effect on the obstructed bladder. Using a rabbit model with partial BOO, Levin and associates (1996) demonstrated that changes in bladder mass, decrease in compliance, and alterations in contractile response to various forms of stimulation could be tempered by pretreatment with P. africanum.
A review of the published experience with P. africanum identified 2262 patients treated with this extract: 1810 in open-label studies and 452 in comparative trials (Andro and Riffaud, 1995). Twelve double-blind, placebo-controlled studies were completed between 1972 and 1990. Only one study enrolled more than 100 subjects, and none was longer than 12 weeks or used standardized symptom scores. In a 2-month open-label study (Breza et al, 1998) and a 2-month comparison trial of 50 mg twice daily versus 100 mg daily (Chatelain et al, 1999) both trials demonstrated a reduction in IPSS score of approximately 40% (5.7 and 6.4 units) and an improvement in PFRs of approximately 18% (2.1 and 1.7 mL/sec) over baseline. However, without a placebo study arm, the actual magnitude of drug effect cannot be determined. Thus, without at least the data from the Tadenan-IPSS study, the efficacy of P. africanum cannot be determined. Additionally, there are few other significant data available with regard to any of the other P. africanum extracts.
Thus none of those trials meets the guidelines recommended by the International Consultation Conferences on BPH. Another meta-analysis for the Cochrane collaboration (Wilt et al, 2002a) concluded that an effect was possible but not proven. Therefore the data concerning the efficacy of P. africanum are not conclusive.
Hypoxis rooperi (South African Star Grass)
Hypoxis rooperi (Harzol) has been studied in both a 6-month double-blind, placebo-controlled trial of 200 patients (Berges et al, 1995) and, subsequently, an open-label follow-up (Berges et al, 2000). In the initial study, statistically significant improvements were documented for symptom scores (IPSS), quality of life, PFRs, and PVRs (Berges et al, 1995). The placebo group showed appropriate mild improvement in these parameters. The IPSS improved by 7.4 units for β-sitosterol patients and 2.3 units for placebo. Similarly, peak urinary flow rates improved by 5.2 mL/sec for treated patients compared with 1.1 mL/sec for placebo. This magnitude of improvement has not been observed with any other medical therapy previously evaluated for BPH.
During the follow-up study, patients were able to remain on or switch over to Harzol therapy. For those 38 patients who continued Harzol therapy, IPSS improvements were maintained. The 27 patients who received placebo initially and were then treated with Harzol demonstrated similar levels of improvement in IPSS scores and PFRs. Surprisingly, the 14 patients who stopped therapy still maintained similar levels of improvement over the next 12 months. This suggests that intermittent therapy could be an option.
Another product (Azuprostat) contains primarily β-sitosterols from H. rooperi as well as from Pinus (pine) and Picea (spruce). Although this is a combination preparation of extracts, its proposed active ingredient β-sitosterol is common to all three. This product (Azuprostat) was evaluated in a 6-month randomized, placebo-controlled trial with 177 patients (Klippel et al, 1997). Significant improvements in IPSS scores, PFRs, and PVRs were found. IPSS scores improved by 8.2 units for treated patients and 2.8 units for placebo. PFRs improved by 8.9 mL/sec for treated patients and 4.4 mL/sec for placebo. This magnitude of improvement has not been reported for any type of medical therapy for BPH. It is hard to believe these results in which the mean post-treatment PFRs are 19.4 mL/sec, which is a normal value for younger men and not for the typical age of men in the study. If these results are reproducible, they would rival surgical intervention.
Wilt and associates (1999) also produced a meta-analysis for β-sitosterol products. It included four trials (two mentioned earlier and two earlier suboptimal ones) encompassing three different products: Harzol, Azuprostat, and WA184, all of which have different amounts of β-sitosterol. WA184 did not improve PFR (Kadow et al, 1986). Again, this meta-analysis is tempered by the same factors mentioned previously. Their conclusion was that β-sitosterol does improve urologic symptoms and urinary flow rates, but its long-term effectiveness, safety, and ability to prevent the complications of BPH are unknown (Wilt et al, 1999).
Other Extracts
The other extracts listed in Table 92–14—Urtica dioica, Cucurbita pepo, Secale cereale, and Opuntia—have even fewer relevant clinical studies published than the aforementioned ones. Of these extracts, Secale cereale (Cernilton) had two placebo-controlled trials published over a decade ago that did not have standardized scores, used different dosages of the preparation, lasted 12 and 24 weeks, and enrolled only 103 and 60 subjects (Buck et al, 1990). Wilt and associates (1999) in a systematic review and meta-analysis of Cernilton concluded that it modestly improves overall urologic symptoms including nocturia, but additional trials are needed to evaluate clinical effectiveness. More recently Zhang and coworkers (2008) have reported a randomized, double-blind, placebo-controlled clinical trial of a flaxseed lignan extract containing 33% secoisolariciresinol diglucoside (SDG). SDG was evaluated for its ability to alleviate LUTS in 87 subjects with BPH with repeated measurements conducted over a 4-month period using treatment dosages of 0 (placebo), 300, or 600 mg/day SDG. After 4 months of treatment, 78 of the 87 subjects completed the study. For the 0, 300, and 600 mg/day SDG groups, respectively, the IPSS decreased −3.67 ± 1.56, −7.33 ± 1.18, and −6.88 ± 1.43 (mean ± SE, P = .100, < .001, and < .001 compared with baseline), the quality of life score improved by −0.71 ± 0.23, −1.48 ± 0.24, and −1.75 ± 0.25 (mean ± SE, P = .163 and .012 compared with placebo and P = .103, < .001, and < .001 compared with baseline), and the number of subjects whose LUTS grade changed from “moderate to severe” to “mild” increased by 3, 6, and 10 (P = .188, .032, and .012 compared with baseline). PFR insignificantly increased 0.43 ± 1.57, 1.86 ± 1.08, and 2.7 ± 1.93 mL/sec (mean ± SE, no statistical significance reached), and postvoid urine volume decreased insignificantly by −29.4 ± 20.46, −19.2 ± 16.91, and −55.62 ± 36.45 mL (mean ± SE, no statistical significance reached). The observed decreases in IPSS and quality of life score were correlated with the concentrations of plasma total lignans. It was concluded that dietary flaxseed lignan extract appreciably improved LUTS in BPH subjects and that the therapeutic efficacy appeared comparable to that of α1-adrenergic receptor blockers and 5α-reductase inhibitors.
Summary
Most phytotherapeutic preparations are plant extracts with different components manufactured by different extraction procedures, which prevents comparison of the preparations. Although numerous in-vitro experiments have been conducted to determine their possible mechanisms of pharmaceutical action, it is uncertain which of the actions demonstrated in vitro might be responsible for clinical responses in vivo. Appropriate randomized placebo-controlled clinical trials monitored by an outside agency are needed to ascertain and to confirm the efficacy of these products. Phytotherapies for BPH should be scrutinized and subjected to trials just as are all regulated drugs.
Acute Urinary Retention
Acute urinary retention is the most common urologic emergency managed by most urologists worldwide. Furthermore, it will be encountered by most physicians whatever their specialty and is commonly witnessed on surgical and elderly care wards. AUR may be “spontaneous” where it is usually associated with previous LUTS suggestive of BPH. Alternatively, it may be “precipitated” by some other factor, such as the effects of various medications, particularly anticholinergic or sympathicomimetic agents, which are commonly found in cough and cold remedies. Urinary infection, excessive fluid intake, and the consequences of surgery (postoperative pain or the effects of anesthesia or analgesia or loss of mobility) may precipitate AUR. Obviously there can be an overlap between these two rather artificial divisions and AUR in a man with symptomatic or silently progressing BPH may well be precipitated by one of these other factors.
Population-based cohort studies from the United States (Jacobsen et al, 1997; Meigs et al, 1999), from Holland (Verhamme et al, 2005), and also from the United Kingdom (Cathcart et al, 2006) defined the incidence, although it varies among populations. Verhamme and coworkers (2005), in a study based on Dutch general practitioner records covering essentially the whole male population of Holland, reported an incidence of 2.2 AUR events per thousand man-years, of which 40% were precipitated (Fig. 92–18). AUR was the first symptom of BPH in 49% of men designated as suffering from LUTS/BPH, and in this group the risk of AUR was 11 times higher (18.3 per 1000 man-years). They concluded that AUR was of low incidence in the general population but substantial in the LUTS/BPH population. Cathcart and colleagues (2006), reviewing national figures from the United Kingdom, reported 3.1 AUR episodes per 1000 man-years.
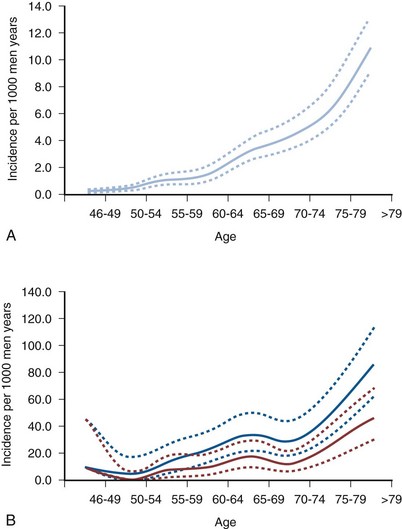
Figure 92–18 Incidence of acute urinary retention in the general population of Dutch men (A) and in the lower urinary tract symptom/benign prostatic hyperplasia population (B).
(From Verhamme KM, Dieleman JP, van Wijk MA, et al: Low incidence of acute urinary retention in the general male population: the triumph project. Eur Urol 2005;47[4]:494–8.)
These figures are lower than the two U.S. cohort studies. Jacobsen and associates (1997) found an incidence of 6.8 events per 1000 man-years in the Olmsted County Study of Urinary Symptoms and Health Status Among Men, and the Health Professionals Follow-up Study (Meigs et al, 1999) reported a rate of 4.5 per 1000 man-years. There may be selection bias here because the prevalence of BPH/LUTS in these U.S. studies is much higher than in the European studies (33% of the Olmsted County men had an IPSS of greater than 8, 30% of the men in the Health Professionals Follow-up Study had a diagnosis of BPH/LUTS compared with just 8% in the Dutch men. Analysis of the placebo arms of a series of large studies such as the PLESS evaluation of finasteride, MTOPS, and the CombAT study indicate that increasing age, the presence of LUTS, a low PFR, and prostatic enlargement and/or raised PSA increase the risk of AUR.
When AUR develops, most men are catheterized for a period or taught intermittent self-catheterization. At some variable point the catheter will usually be removed and a trial without catheter performed (Emberton and Fitzpatrick, 2008). It is reasonable to speculate that urinary retention is caused in part by dynamic as opposed to static outflow obstruction because a significant proportion of men void spontaneously after catheter placement (Taube and Gajraj, 1989). If urinary retention is caused by increased sympathetic activity at the level of the prostatic smooth muscle, an α-adrenergic blocker should increase the likelihood of spontaneous voiding after catheter removal.
The chance of successful voiding after a period of AUR was improved by the use of alfuzosin (McNeill et al, 2005) and tamsulosin (Lucas et al, 2005). In a multicenter study a total of 360 men with AUR underwent emergency catheterization and were blindly randomized to alfuzosin 10 mg/day or placebo for 3 days (first phase). All patients with successful trial without catheterization, regardless of treatment, were then again blindly randomized to alfuzosin 10 mg/day or placebo for 6 months (second phase). Alfuzosin significantly increased the successful trial without catheterization rate (146 of 236, 61.9%) compared with placebo (58 of 121, 47.9%; P = .012). In the second phase, 14 (17.1%) of the 82 alfuzosin-treated patients versus 20 (24.1%) of the 83 placebo-treated patients required BPH surgery, 5 (36%) of 14 versus 13 (65%) of 20 within 1 month, and 8 (57%) of 14 versus 17 (85%) of 20 within 3 months of treatment. Emergency surgery because of AUR relapse was the main cause of failure in both groups (11 [78.6%] of 14 in the alfuzosin group and 16 [80.0%] of 20 in the placebo group). Compared with placebo, alfuzosin improved the Kaplan-Meier survival rates by 9.6% (P = .04), 11.4% (P = .04), and 8.3% (P = .20), with surgical risk reductions of 61%, 52%, and 29% at 1, 3, and 6 months of treatment, respectively. High PSA values and the residual urine volume after the trial without catheterization significantly increased the risk of AUR relapse and BPH surgery (McNeill et al, 2005).
An economic analysis (Annemans et al, 2005) suggests that treatment with alfuzosin before and after a successful trial without catheterization reduces treatment costs in the first 6 months. Unfortunately, even those who succeed will have a high rate of subsequent failure to void and 80% of those who will experience this failure do so within 6 months (Cathcart et al, 2006). At that point surgery and/or pressure-flow studies are indicated.
Medical Therapy for the Prevention of Acute Urinary Retention
Large-scale, randomized, double-blind, placebo-controlled trials of long-term duration are necessary to determine whether a medical therapy prevents a relatively uncommon event such as AUR. In the MTOPS trial, finasteride or combination therapy of finasteride with doxazosin, but not doxazosin alone, reduced the incidence of AUR (McConnell et al, 2003). Debruyne and associates (2004) reported data for dutasteride suggesting similar results. Because men with large prostates have on average a threefold greater chance of developing urinary retention (Jacobsen et al, 1997), enrolling men with large prostates would enhance the probability of observing an effect on AUR. The 3-year open-label prospective study of alfuzosin supported a 0.3% risk of retention (Lukacs et al, 2000). This is markedly lower than the predicted risk of developing urinary retention in an age-matched cohort of men (Jacobsen et al, 1997) but may be more a delay rather than prevention. The trial data suggest a role for 5α-reductase drugs in the prevention of AUR.