CHAPTER 35 Potassium, Calcium, and Phosphate Homeostasis
K+ HOMEOSTASIS
Potassium (K+) is one of the most abundant cations in the body, and it is critical for many cell functions, including regulation of cell volume, regulation of intracellular pH, synthesis of DNA and protein, growth, enzyme function, resting membrane potential, and cardiac and neuromuscular activity. Despite wide fluctuations in dietary K+ intake, [K+] in cells and extracellular fluid (ECF) remains remarkably constant. Two sets of regulatory mechanisms safeguard K+ homeostasis. First, several mechanisms regulate [K+] in the ECF. Second, other mechanisms maintain the amount of K+ in the body constant by adjusting renal K+ excretion to match dietary K+ intake. It is the kidneys that regulate excretion of K+.
Total body [K+] is 50 mEq/kg of body weight, or 3500 mEq for a 70-kg individual. Ninety-eight percent of the K+ in the body is located within cells, where the average [K+] is 150 mEq/L. High intracellular [K+] is required for many cell functions, including cell growth and division and volume regulation. Only 2% of total body [K+] is located in the ECF, where its normal concentration is approximately 4 mEq/L. A [K+] in ECF that exceeds 5.0 mEq/L constitutes hyperkalemia. Conversely, a [K+] in ECF of less than 3.5 mEq/L constitutes hypokalemia.
Hypokalemia is one of the most common electrolyte disorders in clinical practice and can be observed in as many as 20% of hospitalized patients. The most frequent causes of hypokalemia include administration of diuretic drugs, surreptitious vomiting (e.g., bulimia), and severe diarrhea. Gitelman’s syndrome (a genetic defect in the Na+-Cl− symporter in the apical membrane of distal tubule cells) also causes hypokalemia (see Chapter 33, Table 33-3). Hyperkalemia is also a common electrolyte disorder and is seen in 1% to 10% of hospitalized patients. Hyperkalemia often occurs in patients with renal failure, in patients taking drugs, including angiotensin-converting enzyme (ACE) inhibitors and K+-sparing diuretics, in patients with hyperglycemia (i.e., high blood sugar), and in the elderly. Pseudohyperkalemia, a falsely high plasma [K+], is caused by traumatic lysis of red blood cells during blood drawing. Red blood cells, like all cells, contain K+, and lysis of red blood cells releases K+ into plasma, thereby artificially elevating plasma [K+].
The large concentration difference of K+ across cell membranes (approximately 146 mEq/L) is maintained by the operation of Na+,K+-ATPase. This [K+] gradient is important in maintaining the potential difference across cell membranes. Thus, K+ is critical for the excitability of nerve and muscle cells, as well as for the contractility of cardiac, skeletal, and smooth muscle cells (Fig. 35-1).
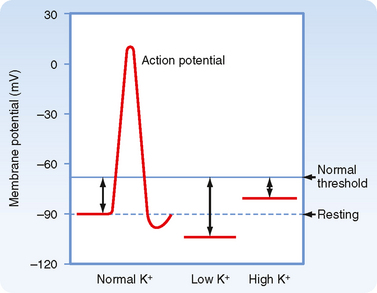
Figure 35-1 The effects of variations in plasma [K+] on the resting membrane potential of skeletal muscle. Hyperkalemia causes the membrane potential to become less negative, which decreases excitability by inactivating the fast Na+ channels responsible for the depolarizing phase of the action potential. Hypokalemia hyperpolarizes the membrane potential and thereby reduces excitability.
After a meal, the K+ absorbed by the gastrointestinal tract enters the ECF within minutes (see Fig. 35-3). If the K+ ingested during a normal meal (≈33 mEq) were to remain in the ECF compartment (14 L), plasma [K+] would increase by a potentially lethal 2.4 mEq/L (33 mEq added to 14 L of ECF):
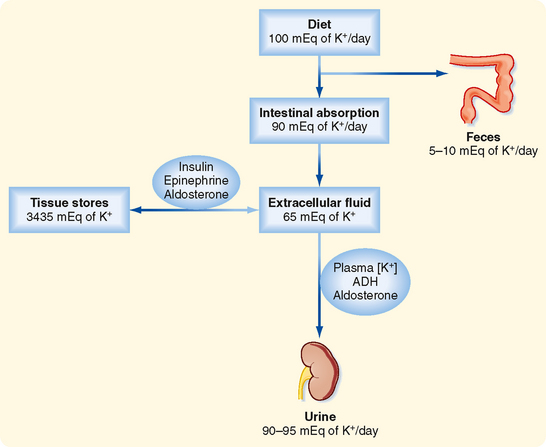
Figure 35-3 Overview of K+ homeostasis. An increase in plasma insulin, epinephrine, or aldosterone stimulates movement of K+ into cells and decreases plasma [K+], whereas a fall in the plasma concentration of these hormones increases plasma [K+]. The amount of K+ in the body is determined by the kidneys. An individual is in K+ balance when dietary intake and urinary output (plus output by the gastrointestinal tract) are equal. Excretion of K+ by the kidneys is regulated by plasma [K+], aldosterone, and ADH.
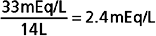
This rise in plasma [K+] is prevented by the rapid (minutes) uptake of K+ into cells. Because excretion of K+ by the kidneys after a meal is relatively slow (hours), uptake of K+ by cells is essential to prevent lifethreatening hyperkalemia. Maintaining total body [K+] constant requires that all the K+ absorbed by the gastrointestinal tract eventually be excreted by the kidneys. This process requires about 6 hours.
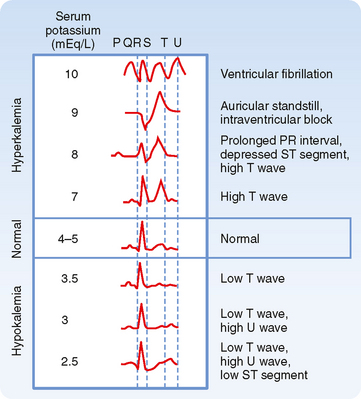
Cardiac arrhythmias are produced by both hypokalemia and hyperkalemia. The electrocardiogram (ECG; see Fig. 35-2 and Chapter 16) monitors the electrical activity of the heart and is a fast and easy way to determine whether changes in plasma [K+] influence the heart and other excitable cells. In contrast, measurement of plasma [K+] by the clinical laboratory requires a blood sample, and values are often not immediately available. The first sign of hyperkalemia is the appearance of tall, thin T waves on the ECG. Further increases in plasma [K+] prolong the PR interval, depress the ST segment, and lengthen the QRS interval of the ECG. Finally, as plasma [K+] approaches 10 mEq/L, the P wave disappears, the QRS interval broadens, the ECG appears as a sine wave, and the ventricles fibrillate (i.e., manifest rapid, uncoordinated contractions of muscle fibers). Hypokalemia prolongs the QT interval, inverts the T wave, and lowers the ST segment of the ECG.
REGULATION OF PLASMA [K+]
As illustrated in Figure 35-3 and Table 35-1, several hormones, including epinephrine, insulin, and aldosterone, increase uptake of K+ into skeletal muscle, liver, bone, and red blood cells by stimulating Na+,K+-ATPase, the 1Na+-1K+-2Cl− symporter, and the Na+-Cl− symporter in these cells. Acute stimulation of K+ uptake (i.e., within minutes) is mediated by an increased turnover rate of existing Na+,K+-ATPase, 1Na+-1K+-2Cl−, and Na+-Cl− transporters, whereas a chronic increase in K+ uptake (i.e., within hours to days) is mediated by an increase in the quantity of Na+,K+-ATPase. The rise in plasma [K+] that follows K+ absorption by the gastrointestinal tract stimulates secretion of insulin from the pancreas, release of aldosterone from the adrenal cortex, and secretion of epinephrine from the adrenal medulla. In contrast, a decrease in plasma [K+] inhibits the release of these hormones. Whereas insulin and epinephrine act within a few minutes, aldosterone requires about an hour to stimulate uptake of K+ into cells.
Table 35-1 Major Factors, Hormones, and Drugs Influencing the Distribution of K+ between the Intracellular and Extracellular Fluid Compartments
Epinephrine
Catecholamines affect the distribution of K+ across cell membranes by activating α- and β2-adrenergic receptors. Stimulation of α-adrenoceptors releases K+ from cells, especially in the liver, whereas stimulation of β2-adrenoceptors promotes K+ uptake by cells.
For example, activation of β2-adrenoceptors after exercise is important in preventing hyperkalemia. The rise in plasma [K+] after a K+-rich meal is greater if the patient has been pretreated with propranolol, a β2-adrenoceptor antagonist. Furthermore, the release of epinephrine during stress (e.g., myocardial ischemia) can rapidly lower plasma [K+].
Insulin
Insulin also stimulates uptake of K+ into cells. The importance of insulin is illustrated by two observations. First, the rise in plasma [K+] after a K+-rich meal is greater in patients with diabetes mellitus (i.e., insulin deficiency) than in normal people. Second, insulin (and glucose to prevent insulin-induced hypoglycemia) can be infused to correct hyperkalemia. Insulin is the most important hormone that shifts K+ into cells after the ingestion of K+ in a meal.
Aldosterone
Aldosterone, like catecholamines and insulin, also promotes uptake of K+ into cells. A rise in aldosterone levels (e.g., primary aldosteronism) causes hypokalemia, whereas a fall in aldosterone levels (e.g., Addison’s disease) causes hyperkalemia. As discussed later, aldosterone also stimulates urinary K+ excretion. Thus, aldosterone alters plasma [K+] by acting on uptake of K+ into cells and altering urinary K+ excretion.
ALTERATIONS IN PLASMA [K+]
Several factors can alter plasma [K+] (Table 35-1). These factors are not involved in the regulation of plasma [K+] but rather alter the movement of K+ between the intracellular fluid (ICF) and ECF and thus cause the development of hypokalemia or hyperkalemia.
Acid-Base Balance
Metabolic acidosis increases the plasma [K+], whereas metabolic alkalosis and respiratory alkalosis decreases it. In contrast, respiratory acidosis has little or no effect on the plasma [K+]. Metabolic acidosis produced by the addition of inorganic acids (e.g., HCl, H2SO4) increases plasma [K+] much more than an equivalent acidosis produced by the accumulation of organic acids (e.g., lactic acid, acetic acid, keto acids). The reduced pH (i.e., increased [H+]) promotes the movement of H+ into cells and the reciprocal movement of K+ out of cells to maintain electroneutrality. This effect of acidosis occurs in part because acidosis inhibits the transporters that accumulate K+ inside cells, including Na+,K+-ATPase and the 1Na+-1K+-2Cl− symporter. In addition, movement of H+ into cells occurs as the cells buffer changes in [H+] of the ECF (see Chapter 36). As H+ moves across the cell membranes, K+ moves in the opposite direction, and thus cations are neither gained nor lost across cell membranes. Metabolic alkalosis has the opposite effect; plasma [K+] decreases as K+ moves into cells and H+ exits.
Although organic acids produce a metabolic acidosis, they do not cause significant hyperkalemia. Two explanations have been suggested for the reduced ability of organic acids to cause hyperkalemia. First, the organic anion may enter the cell with H+ and thereby eliminate the need for K+-H+ exchange across the membrane. Second, organic anions may stimulate insulin secretion, which moves K+ into cells. This movement may counteract the direct effect of the acidosis, which moves K+ out of cells.
Plasma Osmolality
The osmolality of plasma also influences the distribution of K+ across cell membranes. An increase in the osmolality of ECF enhances the release of K+ by cells and thus increases extracellular [K+]. Plasma [K+] may increase by 0.4 to 0.8 mEq/L with a 10 mOsm/kg H2O elevation in plasma osmolality. In patients with diabetes mellitus who do not take insulin, plasma [K+] is often elevated, in part because of the lack of insulin and in part because of the increase in plasma [glucose] (i.e., from a normal value of ≈100 mg/dL to as high as ≈1200 mg/dL), which increases plasma osmolality. Hypoosmolality has the opposite action. The alterations in plasma [K+] associated with changes in osmolality are related to changes in cell volume. For example, as plasma osmolality increases, water leaves cells because of the osmotic gradient across the plasma membrane (see Chapter 1). Water leaves cells until the intracellular osmolality equals that of the ECF. This loss of water shrinks cells and causes [K+] in cells to rise. The rise in intracellular [K+] provides a driving force for the exit of K+ from cells. This sequence increases plasma [K+]. A fall in plasma osmolality has the opposite effect.
Cell Lysis
Cell lysis causes hyperkalemia as a result of the addition of intracellular K+ to the ECF. Severe trauma (e.g., burns) and some conditions such as tumor lysis syndrome (i.e., chemotherapy-induced destruction of tumor cells) and rhabdomyolysis (i.e., destruction of skeletal muscle) destroy cells and release K+ and other cell solutes into the ECF. In addition, gastric ulcers may cause seepage of red blood cells into the gastrointestinal tract. The blood cells are digested, and the K+ released from the cells is absorbed and can cause hyperkalemia.
K+ EXCRETION BY THE KIDNEYS
The kidneys play a major role in maintaining K+ balance. As illustrated in Figure 35-3, the kidneys excrete 90% to 95% of the K+ ingested in the diet. Excretion equals intake even when intake increases by as much as 10-fold. This balance in urinary excretion and dietary intake underscores the importance of the kidneys in maintaining K+ homeostasis. Although small amounts of K+ are lost each day in feces and sweat (approximately 5% to 10% of the K+ ingested in the diet), this amount is essentially constant, is not regulated, and therefore is relatively less important than the K+ excreted by the kidneys. K+ secretion from blood into tubular fluid by cells of the distal tubule and collecting duct system is the key factor in determining urinary K+ excretion (Fig. 35-4).
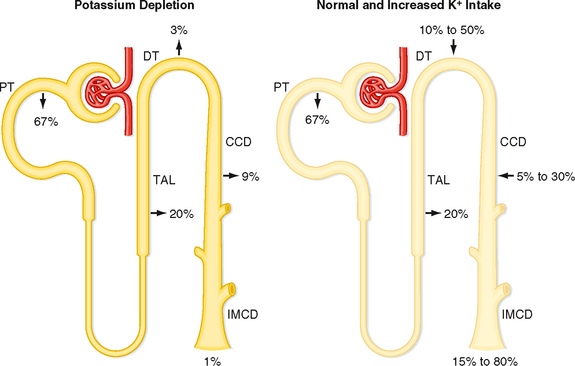
Figure 35-4 K+ transport along the nephron. Excretion of K+ depends on the rate and direction of K+ transport by the distal tubule and collecting duct. Percentages refer to the amount of filtered K+ reabsorbed or secreted by each nephron segment. Left, Dietary K+ depletion. An amount of K+ equal to 1% of the filtered load of K+ is excreted. Right, Normal and increased dietary K+ intake. An amount of K+ equal to 15% to 80% of the filtered load is excreted. CCD, cortical collecting duct; DT, distal tubule; IMCD, inner medullary collecting duct; PT, proximal tubule; TAL, thick ascending limb.
Exercise-induced changes in plasma [K+] do not usually produce symptoms and are reversed after several minutes of rest. However, exercise can lead to life-threatening hyperkalemia in individuals (1) who have endocrine disorders that affect the release of insulin, epinephrine, or aldosterone; (2) whose ability to excrete K+ is impaired (e.g., renal failure); or (3) who take certain medications, such as β2-adrenergic blockers. For example, during exercise, plasma [K+] may increase by at least 2 to 4 mEq/L in individuals who take β2adrenergic receptor antagonists for hypertension.
Because acid-base balance, plasma osmolality, cell lysis, and exercise do not maintain plasma [K+] at a normal value, they do not contribute to K+ homeostasis (Table 35-1). The extent to which these pathophysiological states alter plasma [K+] depends on the integrity of the homeostatic mechanisms that regulate plasma [K+] (e.g., secretion of epinephrine, insulin, and aldosterone).
Because K+ is not bound to plasma proteins, it is freely filtered by the glomerulus. When individuals ingest 100 mEq of K+ per day, urinary K+ excretion is about 15% of the amount filtered. Accordingly, K+ must be reabsorbed along the nephron. When dietary K+ intake increases, however, K+ excretion can exceed the amount filtered. Thus, K+ can also be secreted.
The proximal tubule reabsorbs about 67% of the filtered K+ under most conditions. Approximately 20% of the filtered K+ is reabsorbed by the loop of Henle, and as with the proximal tubule, the amount reabsorbed is a constant fraction of the amount filtered. In contrast to these segments, which can only reabsorb K+, the distal tubule and collecting duct are able to reabsorb or secrete K+. The rate of K+ reabsorption or secretion by the distal tubule and collecting duct depends on a variety of hormones and factors. When 100 mEq/day of K+ is ingested, it is secreted by these nephron segments. A rise in dietary K+ intake increases K+ secretion. K+ secretion can increase the amount of K+ that appears in urine so that it approaches 80% of the amount filtered (Fig. 35-4). In contrast, a low-K+ diet activates K+ reabsorption along the distal tubule and collecting duct so that urinary excretion falls to about 1% of the K+ filtered by the glomerulus (Fig. 35-4). The kidneys cannot reduce K+ excretion to the same low levels as they can for Na+ (i.e., 0.2%). Therefore, hypokalemia can develop in individuals placed on a K+-deficient diet. Because the magnitude and direction of K+ transport by the distal tubule and collecting duct are variable, the overall rate of urinary K+ excretion is determined by these tubular segments.
In individuals with advanced renal disease, the kidneys are unable to eliminate K+ from the body. Therefore, plasma [K+] rises. The resulting hyperkalemia reduces the resting membrane potential (i.e., the voltage becomes less negative), and this reduced potential decreases the excitability of neurons, cardiac cells, and muscle cells by inactivating fast Na+ channels, which are critical for the depolarization phase of the action potential (Fig. 35-1). Severe, rapid increases in plasma [K+] can lead to cardiac arrest and death. In contrast, in patients taking diuretic drugs for hypertension, urinary K+ excretion often exceeds dietary K+ intake. Accordingly, K+ balance is negative, and hypokalemia develops. This decline in extracellular [K+] hyperpolarizes the resting cell membrane (i.e., the voltage becomes more negative) and reduces the excitability of neurons, cardiac cells, and muscle cells. Severe hypokalemia can lead to paralysis, cardiac arrhythmias, and death. Hypokalemia can also impair the ability of the kidneys to concentrate the urine and can stimulate the renal production of NH4+, which affects acid-base balance (see Chapter 36). Therefore, maintenance of high intracellular [K+], low extracellular [K+], and a high [K+] gradient across cell membranes is essential for a number of cellular functions.
CELLULAR MECHANISM OF K+ SECRETION BY PRINCIPAL CELLS IN THE DISTAL TUBULE AND COLLECTING DUCT
Figure 35-5 illustrates the cellular mechanisms of K+ secretion by principal cells in the distal tubule and collecting duct. Secretion from blood into the tubule lumen is a two-step process: (1) uptake of K+ from blood across the basolateral membrane by Na+,K+-ATPase and (2) diffusion of K+ from the cell into tubular fluid via K+ channels. Na+,K+-ATPase creates a high intracellular [K+] that provides the chemical driving force for exit of K+ across the apical membrane through K+ channels. Although K+ channels are also present in the basolateral membrane, K+ preferentially leaves the cell across the apical membrane and enters the tubular fluid. K+ transport follows this route for two reasons. First, the electrochemical gradient of K+ across the apical membrane favors its downhill movement into tubular fluid. Second, the permeability of the apical membrane to K+ is greater than that of the basolateral membrane. Therefore K+ preferentially diffuses across the apical membrane into tubular fluid. The three major factors that control the rate of K+ secretion by the distal tubule and the collecting duct are
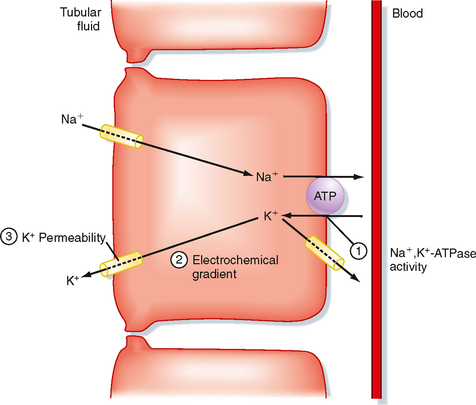
Figure 35-5 Cellular mechanism of K+ secretion by a principal cell in the distal tubule and collecting duct. The numbers indicate the sites where K+ secretion is regulated. 1, Na+,K+-ATPase; 2, electrochemical gradient of K+ across the apical membrane; 3, permeability of the apical membrane to K+.
Every change in K+ secretion results from an alteration in one or more of these factors.
Intercalated cells reabsorb K+ via an H+,K+-ATPase transport mechanism located in the apical membrane (see Chapter 36). This transporter mediates uptake of K+ in exchange for H+. The pathway for exit of K+ from intercalated cells into blood is unknown. Reabsorption of K+ is activated by a low K+-diet.
REGULATION OF K+ SECRETION BY THE DISTAL TUBULE AND COLLECTING DUCT
Regulation of K+ excretion is achieved mainly by alterations in K+ secretion by principal cells of the distal tubule and collecting duct. Plasma [K+] and aldosterone are the major physiological regulators of K+ secretion. Antidiuretic hormone (ADH) also stimulates K+ secretion; however, it is less important than plasma [K+] and aldosterone. Other factors, including the flow rate of tubular fluid and acid-base balance, influence secretion of K+ by the distal tubule and collecting duct. However, they are not homeostatic mechanisms because they disturb K+ balance (Table 35-2).
Table 35-2 Major Factors and Hormones Influencing K+ Excretion
Plasma [K+]
Plasma [K+] is an important determinant of K+ secretion by the distal tubule and collecting duct. Hyperkalemia (e.g., resulting from a high-K+ diet or from rhabdomyolysis) stimulates secretion of K+ within minutes. Several mechanisms are involved. First, hyperkalemia stimulates Na+,K+-ATPase and thereby increases K+ uptake across the basolateral membrane. This uptake raises intracellular [K+] and increases the electrochemical driving force for exit of K+ across the apical membrane. Second, hyperkalemia also increases the permeability of the apical membrane to K+. Third, hyperkalemia stimulates secretion of aldosterone by the adrenal cortex, which as discussed later, acts synergistically with plasma [K+] to stimulate secretion of K+. Fourth, hyperkalemia also increases the flow rate of tubular fluid, which as discussed later, stimulates secretion of K+ by the distal tubule and collecting duct.
Hypokalemia (e.g., caused by a low-K+ diet or loss of K+ in diarrhea fluid) decreases K+ secretion via actions opposite those described for hyperkalemia. Hence, hypokalemia inhibits Na+,K+-ATPase, decreases the electrochemical driving force for efflux of K+ across the apical membrane, reduces permeability of the apical membrane to K+, and decreases plasma aldosterone levels.
Chronic hypokalemia (plasma [K+] <3.5 mEq/L) occurs most often in patients who receive diuretics for hypertension. Hypokalemia also occurs in patients who vomit, undergo nasogastric suction, have diarrhea, abuse laxatives, or have hyperaldosteronism. Hypokalemia occurs because excretion of K+ by the kidneys exceeds the dietary intake of K+. Vomiting, nasogastric suction, diuretics, and diarrhea can all decrease ECF volume, which in turn stimulates secretion of aldosterone (see Chapter 34). Because aldosterone stimulates excretion of K+ by the kidneys, its action contributes to the development of hypokalemia.
Chronic hyperkalemia (plasma [K+] >5.0 mEq/L) occurs most frequently in individuals with reduced urine flow, low plasma aldosterone levels, and renal disease in which the glomerular filtration rate falls below 20% of normal. In these individuals, hyperkalemia occurs because the excretion of K+ by the kidneys is less than the dietary intake of K+. Less common causes of hyperkalemia occur in people with deficiencies in insulin, epinephrine, and aldosterone secretion or in people with metabolic acidosis caused by inorganic acids.
Aldosterone
Chronically (i.e., ≥24 hours) elevated plasma aldosterone levels enhance secretion of K+ across principal cells in the distal tubule and collecting duct via five mechanisms (Fig. 35-6): (1) by increasing the amount of Na+, K+-ATPase in the basolateral membrane; (2) by increasing expression of the epithelial sodium channel (ENaC) in the apical cell membrane; (3) by elevating SGK1 (serum glucocorticoid-stimulated kinase) levels, which also increases expression of ENaC in the apical membrane and activates K+ channels; (4) by stimulating CAP1 (channel-activating protease, also called prostatin), which directly activates ENaC; and (5) by stimulating the permeability of the apical membrane to K+. The cellular mechanisms by which aldosterone affects the expression and activity of Na+,K+-ATPase and ENaC (actions 1 to 5 just listed) have been described (see Chapter 33). Aldosterone increases the permeability of the apical membrane to K+ by increasing the number of K+ channels in the membrane. However, the cellular mechanisms involved in this response are not completely known. Increased expression of Na+,K+-ATPase facilitates uptake of K+ across the basolateral membrane into cells and thereby elevates intracellular [K+]. The increase in the number and activity of Na+ channels enhances entry of Na+ into the cell from tubular fluid, an effect that depolarizes the apical membrane voltage. Depolarization of the apical membrane and increased intracellular [K+] enhance the electrochemical driving force for secretion of K+ from the cell into the tubule fluid. Taken together, these actions increase uptake of K+ into the cell across the basolateral membrane and enhance exit of K+ from the cell across the apical membrane. Secretion of aldosterone is increased by hyperkalemia and by angiotensin II (after activation of the renin-angiotensin system). Secretion of aldosterone is decreased by hypokalemia and natriuretic peptides released from the heart.
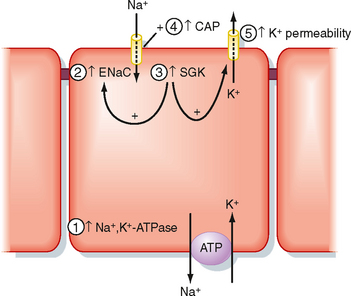
Figure 35-6 Effects of aldosterone on secretion of K+ by principal cells in the collecting duct. Numbers refer to the five effects of aldosterone discussed in text.
Although an acute (e.g., within hours) increase in aldosterone levels enhances the activity of Na+,K+-ATPase, K+ excretion does not increase. The reason for this relates to the effect of aldosterone on Na+ reabsorption and tubular flow. Aldosterone stimulates reabsorption of Na+ and water and thus decreases tubular flow. The reduction in flow in turn decreases K+ secretion (as discussed in more detail later). However, chronic stimulation of Na+ reabsorption expands the ECF and thereby returns tubular flow to normal. These actions allow a direct stimulatory effect of aldosterone on the distal tubule and collecting duct to enhance K+ excretion.
Antidiuretic Hormone
Although ADH does not affect urinary K+ excretion, this hormone does stimulate secretion of K+ by the distal tubule and collecting duct (Fig. 35-7). ADH increases the electrochemical driving force for exit of K+ across the apical membrane of principal cells by stimulating uptake of Na+ across the apical membrane of these cells. The increased Na+ uptake reduces the electrical potential difference across the apical membrane (i.e., the interior of the cell becomes less negatively charged). Despite this effect, ADH does not change K+ secretion by these nephron segments. The reason for this relates to the effect of ADH on tubular fluid flow. ADH decreases flow of tubular fluid by stimulating water reabsorption. The decrease in tubular flow in turn reduces secretion of K+ (explained later). The inhibitory effect of decreased flow of tubular fluid offsets the stimulatory effect of ADH on the electrochemical driving force for exit of K+ across the apical membrane (Fig. 35-7). If ADH did not increase the electrochemical gradient favoring K+ secretion, urinary K+ excretion would fall as ADH levels increased and urinary flow rates decreased. Hence, K+ balance would change in response to alterations in water balance. Thus, the effects of ADH on the electrochemical driving force for exit of K+ across the apical membrane and on tubule flow enable urinary K+ excretion to be maintained constant despite wide fluctuations in water excretion.
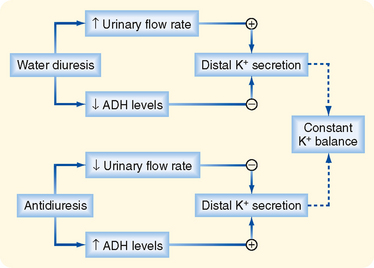
Figure 35-7 Opposing effects of ADH on secretion of K+ by the distal tubule and cortical collecting duct. Secretion is stimulated by an increase in the electrochemical gradient for K+ across the apical membrane and by an increase in the permeability of the apical membrane to K+. In contrast, secretion is reduced by a fall in the flow rate of tubular fluid. Because these effects oppose each other, net K+ secretion is not affected by ADH.
FACTORS THAT PERTURB K+ EXCRETION
Although plasma [K+], aldosterone, and ADH play important roles in regulating K+ balance, the factors and hormones discussed next perturb K+ balance (Table 35-2).
Flow of Tubular Fluid
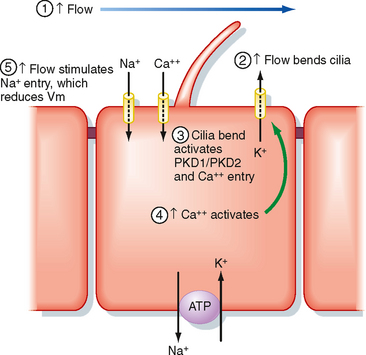
A rise in the flow of tubular fluid (e.g., with diuretic treatment, ECF volume expansion) stimulates secretion of K+ within minutes, whereas a fall (e.g., ECF volume contraction caused by hemorrhage, severe vomiting, or diarrhea) reduces secretion of K+ by the distal tubule and collecting duct. Increments in tubular fluid flow are more effective in stimulating secretion of K+ as dietary K+ intake is increased. Recent studies on the primary cilium in principal cells have elucidated some of the mechanisms whereby increased flow stimulates secretion of K+ (Fig. 35-8). Increased flow bends the primary cilium in principal cells, which activates the PKD1/PKD2 Ca++ conducting channel complex. This allows more Ca++ to enter principal cells and increases intracellular [Ca++]. The increase in [Ca++] activates K+ channels in the apical plasma membrane, which enhances secretion of K+ from the cell into tubule fluid. Increased flow may also stimulate secretion of K+ by other mechanisms. As flow increases, such as after the administration of diuretics or as the result of an increase in ECF volume, so does the [Na+] of tubule fluid. This increase in [Na+] facilitates entry of Na+ across the apical membrane of distal tubule and collecting duct cells, thereby decreasing the cells’ interior negative membrane potential. This depolarization of the cell membrane potential increases the electrochemical driving force that promotes secretion of K+ across the apical cell membrane into tubule fluid. In addition, increased uptake of Na+ into cells activates the Na+,K+-ATPase in the basolateral membrane, thereby increasing uptake of K+ across the basolateral membrane and consequently elevating [K+]. However, it is important to note that an increase in flow rate during a water diuresis does not have a significant effect on excretion of K+, most likely because during a water diuresis the [Na+] of tubule fluid does not increase as flow rises.
Acid-Base Balance
Another factor that modulates secretion of K+ is the [H+] of ECF. Acute alterations (within minutes to hours) in the pH of plasma influences secretion of K+ by the distal tubule and collecting duct. Alkalosis (i.e., plasma pH above normal) increases secretion of K+, whereas acidosis (i.e., plasma pH below normal) decreases it. An acute acidosis reduces K+ secretion via two mechanisms: (1) it inhibits Na+,K+-ATPase and thereby reduces cell [K+] and the electrochemical driving force for exit of K+ across the apical membrane, and (2) it reduces the permeability of the apical membrane to K+. Alkalosis has the opposite effects.
ROMK (KCNJ1) is the primary channel in the apical membrane responsible for secretion of K+. Four ROMK subunits make up a single channel. In addition, a maxi-K+ channel (rbsol1), which is activated by elevations in intracellular [Ca++], is also expressed in the apical membrane. The maxi-K+ channel mediates the flow-dependent increase in K+ secretion, as discussed earlier. Interestingly, knockout of the KCNJ1 gene (ROMK) causes increased excretion of NaCl and K+ by the kidneys, thereby leading to reduced ECF volume and hypokalemia. Although this effect is somewhat perplexing, it should be noted that ROMK is also expressed in the apical membrane of the thick ascending limb of Henle’s loop, where it plays a very important role in recycling of K+ across the apical membrane, an effect that is critical for operation of the Na+-K+-2Cl− symporter (see Chapter 33). In the absence of ROMK, reabsorption of NaCl by the thick ascending limb is reduced, which leads to loss of NaCl in urine. Reduction of NaCl reabsorption by the thick ascending limb also reduces the positive transepithelial luminal voltage, which is the driving force for reabsorption of K+ by this nephron segment. Thus, the reduction in paracellular K+ reabsorption by the thick ascending limb increases urinary K+ excretion, even when the cortical collecting duct is unable to secrete the normal amount of K+ because of a lack of ROMK channels. The cortical collecting duct, however, does secrete K+ even in ROMK knockout mice via the flow- and Ca++-dependent maxi-K+ channels and possibly by the operation of a K+-Cl− symporter expressed in the apical membrane of principal cells.
The effect of metabolic acidosis on excretion of K+ is time dependent. When metabolic acidosis lasts for several days, urinary K+ excretion is stimulated (Fig. 35-9). This occurs because chronic metabolic acidosis decreases the reabsorption of water and solutes (e.g., NaCl) by the proximal tubule by inhibiting Na+,K+-ATPase. Hence, the flow of tubular fluid is augmented along the distal tubule and collecting duct. The inhibition of water and NaCl reabsorption by the proximal tubule also decreases ECF volume and thereby stimulates secretion of aldosterone. In addition, chronic acidosis, caused by inorganic acids, increases plasma [K+], which stimulates secretion of aldosterone. The rise in tubular fluid flow, plasma [K+], and aldosterone levels offsets the effects of acidosis on cell [K+] and apical membrane permeability, and K+ secretion rises. Thus, metabolic acidosis may either inhibit or stimulate excretion of K+, depending on the duration of the disturbance. Renal K+ excretion remains elevated during chronic metabolic acidosis and may even increase further, depending on the cause of the acidosis.
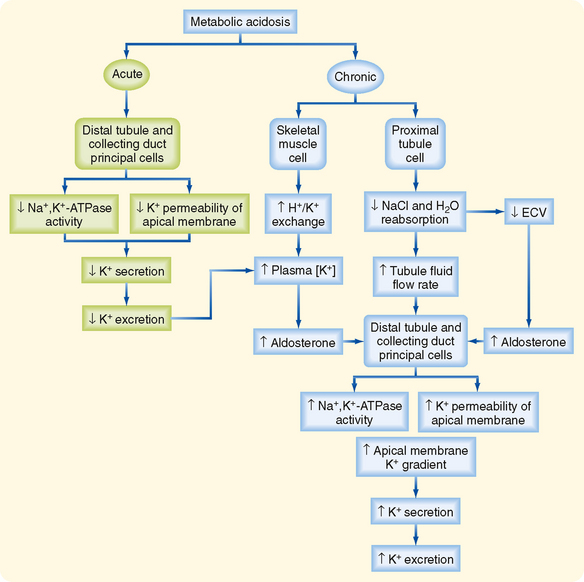
Figure 35-9 Acute versus chronic effect of metabolic acidosis on excretion of K+. See text for details. ECV, effective circulating volume.
As noted, acute metabolic alkalosis stimulates excretion of K+. Chronic metabolic alkalosis, especially in association with ECF volume contraction, significantly increases renal K+ excretion because of the associated increased levels of aldosterone.
Glucocorticoids
Glucocorticoids increase urinary K+ excretion. This effect is mediated in part by an increase in the glomerular filtration rate, which enhances the urinary flow rate, a potent stimulus of K+ excretion, and by stimulation of SGK1 activity (see earlier).
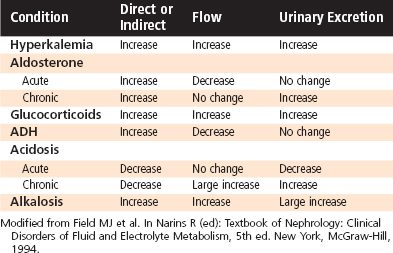
As discussed earlier, the rate of urinary K+ excretion is frequently determined by simultaneous changes in hormone levels, acid-base balance, or the flow rate of tubule fluid (Table 35-3). The powerful effect of flow often enhances or opposes the response of the distal tubule and collecting duct to hormones and changes in acid-base balance. This interaction can be beneficial in the case of hyperkalemia, in which the change in flow enhances excretion of K+ and thereby restores K+ homeostasis. However, this interaction can also be detrimental, as in the case of alkalosis, in which changes in flow and acid-base status alter K+ homeostasis.
Table 35-3 Net Effects of Hormones and Other Factors on K+ Secretion by the Distal Tubule and Collecting Duct
The cellular mechanisms whereby changes in the K+ content of the diet and acid-base balance regulate secretion of K+ by the distal tubule and collecting duct have recently been elucidated. Elevated K+ intake increases secretion of K+ by several mechanisms, all related to increased serum [K+]. Hyperkalemia increases the activity of the ROMK channel in the apical plasma membrane of principal cells. Moreover, hyperkalemia inhibits reabsorption of NaCl and water by the proximal tubule, thereby increasing the distal tubule and collecting duct flow rate, a potent stimulus to secretion of K+. Hyperkalemia also enhances [aldosterone], which increases K+ secretion by three mechanisms. First, aldosterone increases the number of K+ channels in the apical plasma membrane. Second, aldosterone stimulates uptake of K+ across the basolateral membrane by increasing the number of Na+,K+-ATPase pumps, thereby enhancing the electrochemical gradient driving secretion of K+ across the apical membrane. Third, aldosterone increases movement of Na+ across the apical membrane, which depolarizes the apical plasma membrane voltage and thus increases the electrochemical gradient promoting secretion of K+.
A low-K+ diet dramatically reduces secretion of K+ by the distal tubule and collecting duct by increasing the activity of protein tyrosine kinase, which causes ROMK channels to be endocytosed from the apical plasma membrane, thereby reducing K+ secretion.
Acidosis decreases secretion of K+ by inhibiting the activity of ROMK channels, whereas alkalosis stimulates secretion of K+ by enhancing ROMK channel activity.
OVERVIEW OF CALCIUM AND INORGANIC PHOSPHATE HOMEOSTASIS
Ca++ and inorganic phosphate (Pi)* are multivalent ions that subserve many complex and vital functions. Ca++ is an important cofactor in many enzymatic reactions; it is a key second messenger in numerous signaling pathways; it plays an important role in neural transduction, blood clotting, and muscle contraction; and it is a critical component of the extracellular matrix, cartilage, teeth, and bone. Pi, like Ca++, is a key component of bone. Pi is essential for metabolic processes, including the formation of ATP, and it is an important component of nucleic acids. Phosphorylation of proteins is an important mechanism of cellular signaling, and Pi is an important buffer in cells, plasma, and urine.
In a normal adult, renal excretion of Ca++ and Pi is balanced by gastrointestinal absorption. If the plasma concentrations of Ca++ and Pi decline substantially, gastrointestinal absorption, bone resorption (i.e., loss of Ca++ and Pi from bone), and renal tubular reabsorption increase and return plasma concentrations of Ca++ and Pi to normal levels. During growth and pregnancy, intestinal absorption exceeds urinary excretion, and these ions accumulate in newly formed fetal tissue and bone. In contrast, bone disease (e.g., osteoporosis) or a decline in lean body mass increases urinary multivalent ion loss without a change in intestinal absorption. These conditions produce a net loss of Ca++ and Pi from the body.
This brief introduction reveals that the kidneys, in conjunction with the gastrointestinal tract and bone, play a major role in maintaining plasma Ca++ and Pi levels, as well as Ca++ and Pi balance (see also Chapter 39). Accordingly, this section of the chapter discusses Ca++ and Pi handling by the kidneys with an emphasis on the hormones and factors that regulate urinary excretion.
Calcium
Cellular processes in which Ca++ plays a part include bone formation, cell division and growth, blood coagulation, hormone-response coupling, and electrical stimulus–response coupling (e.g., muscle contraction, neurotransmitter release). Ninety-nine percent of Ca++ is stored in bone, approximately 1% is found in intracellular fluid (ICF), and 0.1% is located in the ECF. The total [Ca++] in plasma is 10 mg/dL (2.5 mM or 5 mEq/L), and its concentration is normally maintained within very narrow limits. A low ionized plasma [Ca++] (hypocalcemia) increases the excitability of nerve and muscle cells and can lead to hypocalcemic tetany, which is characterized by skeletal muscle spasms. The association of hypocalcemia with tetany is due to the fact that hypocalcemia causes the threshold potential to shift to more negative values (i.e., closer to the resting membrane voltage). Elevated ionized plasma [Ca++] (hypercalcemia) may decrease neuromuscular excitability or produce cardiac arrhythmias, lethargy, disorientation, and even death. This hypercalcemic effect occurs because hypercalcemia causes the threshold potential to shift to less negative values (i.e., further from the resting membrane voltage). Within cells, Ca++ is sequestered in the endoplasmic reticulum and mitochondria, or it is bound to proteins. Thus, the free intracellular [Ca++] is very low (≈100 nM). The large concentration gradient for [Ca++] across cell membranes is maintained by a Ca++-ATPase pump (PMCa1b) in all cells and by a 3Na+-1Ca++ antiporter (NCX1) in some cells.
Overview of Calcium Homeostasis
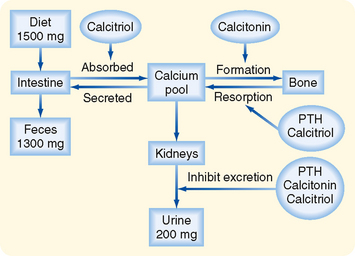
Ca++ homeostasis depends on two factors: (1) the total amount of Ca++ in the body and (2) the distribution of Ca++ between bone and ECF. Total body [Ca++] is determined by the relative amounts of Ca++ absorbed by the gastrointestinal tract and excreted by the kidneys (Fig. 35-10). The gastrointestinal tract absorbs Ca++ through an active, carrier-mediated transport mechanism that is stimulated by calcitriol, a metabolite of vitamin D3. Net Ca++ absorption is normally 200 mg/day, but it can increase to 600 mg/day when calcitriol levels rise. In adults, Ca++ excretion by the kidneys equals the amount absorbed by the gastrointestinal tract (200 mg/day), and it changes in parallel with reabsorption of Ca++ by the gastrointestinal tract. Thus, in adults, Ca++ balance is maintained because the amount of Ca++ ingested in an average diet (1500 mg/day) equals the amount lost in feces (1300 mg/day, the amount that escapes absorption by the gastrointestinal tract) plus the amount excreted in urine (200 mg/day).
The second factor that controls Ca++ homeostasis is the distribution of Ca++ between bone and ECF. Three hormones (parathyroid hormone [PTH], calcitriol, and calcitonin) regulate the distribution of Ca++ between bone and ECF and thereby regulate plasma [Ca++].
PTH is secreted by the parathyroid glands, and its secretion is regulated by the [Ca++] in ECF. The plasma membrane of the chief cells of the parathyroid glands contains the calcium-sensing receptor (CaSR), which monitors the [Ca++] in ECF. A decrease in [Ca++] (i.e., hypocalcemia) increases PTH gene expression and release by the chief cells. In contrast, an increase in [Ca++] (i.e., hypercalcemia) decreases PTH release by the chief cells.
PTH increases plasma [Ca++] by (1) stimulating bone resorption, (2) increasing Ca++ reabsorption by the kidneys, and (3) stimulating the production of calcitriol, which in turn increases Ca++ absorption by the gastrointestinal tract and facilitates PTH-mediated bone resorption.
The production of calcitriol, a metabolite of vitamin D3 produced in the proximal tubule of the kidney, is stimulated by hypocalcemia and hypophosphatemia. In addition, hypocalcemia stimulates secretion of PTH, which also stimulates production of vitamin D3 by the proximal tubule cells. Calcitriol increases plasma [Ca++] primarily by stimulating absorption of Ca++ from the gastrointestinal tract. It also facilitates the action of PTH on bone and increases the expression of key Ca++ transport and binding proteins in the kidneys.
Calcitonin is secreted by thyroid C cells (a.k.a., parafollicular cells), and its secretion is stimulated by hypercalcemia. Calcitonin decreases plasma [Ca++] mainly by stimulating bone formation (i.e., deposition of Ca++ in bone). Figure 35-11 illustrates the relationship between plasma [Ca++] and plasma levels of PTH and calcitonin. Although calcitonin plays an important role in Ca++ homeostasis in lower vertebrates, it plays only a minor role in normal Ca++ homeostasis in humans.
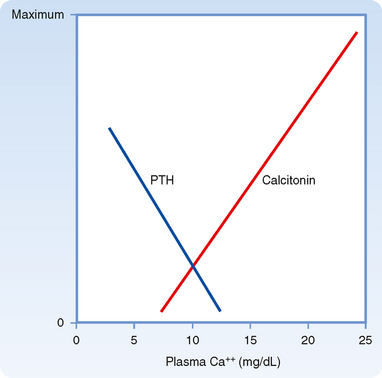
Figure 35-11 Effect of plasma [Ca++] on plasma levels of PTH and calcitonin.
(Modified from Azria M: The Calcitonins: Physiology and Pharmacology. Basel, Karger, 1989.)
Approximately 50% of the Ca++ in plasma is ionized, 45% is bound to plasma proteins (mainly albumin), and 5% is complexed to several anions, including HCO3−, citrate, Pi, and SO42−. The pH of plasma influences this distribution. The increase in [H+] in patients with metabolic acidosis causes more H+ to bind to plasma proteins, HCO3−, citrate, Pi, and SO42−, thereby displacing Ca++. This displacement increases the plasma concentration of ionized Ca++. In alkalosis, the [H+] of plasma decreases. Some H+ ions dissociate from plasma proteins, HCO3−, citrate, Pi, and SO42− in exchange for Ca++, thereby decreasing the plasma concentration of ionized Ca++. In addition, the plasma albumin concentration also affects ionized plasma [Ca++]. Hypoalbuminemia increases ionized [Ca++], whereas hyperalbuminemia decreases ionized plasma [Ca++]. Under both conditions the total plasma [Ca++] may not reflect the total ionized [Ca++], which is the physiologically relevant measure of Ca++ homeostasis. The Ca++ available for glomerular filtration consists of the ionized fraction and the amount complexed with anions. Thus, about 55% of the Ca++ in plasma is available for glomerular filtration.
Conditions that lower PTH levels (i.e., hypoparathyroidism after parathyroidectomy for an adenoma) reduce plasma [Ca++], which can cause hypocalcemic tetany (intermittent muscular contractions). In severe cases, hypocalcemic tetany can cause death by asphyxiation. Hypercalcemia can also cause lethal cardiac arrhythmias and decreased neuromuscular excitability. Clinically, the most common causes of hypercalcemia are primary hyperparathyroidism and malignancyassociated hypercalcemia. Primary hyperparathyroidism results from the overproduction of PTH by the parathyroid glands. In contrast, malignancy-associated hypercalcemia, which occurs in 10% to 20% of all patients with cancer, is caused by the secretion of parathyroid hormone–related peptide (PTHrP), a PTH-like hormone secreted by carcinomas in various organs. Increased levels of PTH and PTHrP cause hypercalcemia and hypercalciuria.
Calcium Transport along the Nephron
Normally, 99% of the filtered Ca++ (i.e., ionized and complexed) is reabsorbed by the nephron. The proximal tubule reabsorbs about 70% of the filtered Ca++. Another 20% is reabsorbed in the loop of Henle (mainly the cortical portion of the thick ascending limb), about 9% is reabsorbed by the distal tubule, and less than 1% is reabsorbed by the collecting duct. Approximately 1% (200 mg/day) is excreted in urine. This fraction is equal to the net amount absorbed daily in the gastrointestinal tract. Figure 35-12 summarizes the handling of Ca++ by the different portions of the nephron.
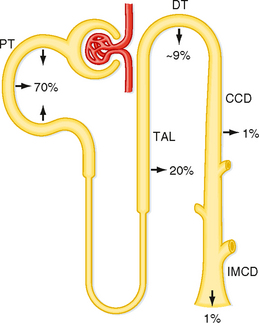
Figure 35-12 Ca++ transport along the nephron. Percentages refer to the amount of filtered Ca++ reabsorbed by each segment. Approximately 1% of the filtered Ca++ is excreted. CCD, cortical collecting duct; DT, distal tubule; IMCD, inner medullary collecting duct; PT, proximal tubule; TAL, thick ascending limb.
Reabsorption of Ca++ by the proximal tubule occurs via two pathways: transcellular and paracellular (Fig. 35-13). Ca++ reabsorption via the transcellular pathway accounts for 20% of proximal reabsorption. Reabsorption of Ca++ through the cell is an active process that occurs in two steps. First, Ca++ diffuses down its electrochemical gradient across the apical membrane through Ca++ channels and into the cell. Second, at the basolateral membrane, Ca++ is extruded from the cell against its electrochemical gradient by a Ca++-ATPase. In contrast, 80% of Ca++ is reabsorbed between cells across the tight junctions (i.e., the paracellular pathway). This passive, paracellular reabsorption of Ca++ occurs via solvent drag along the entire length of the proximal tubule and is also driven by the positive luminal voltage in the second half of the proximal tubule (i.e., diffusion). Thus, approximately 80% of Ca++ reabsorption is paracellular, and approximately 20% is transcellular in the proximal tubule.
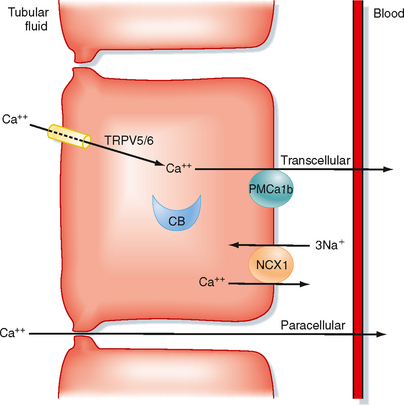
Figure 35-13 Cellular mechanisms for reabsorption of Ca++ by the transcellular and cellular pathways. Note that all transport mechanisms are not expressed in every nephron segment. In distal tubule cells, Ca++ enters the cells across the apical membrane via Ca++-permeable ion channels (TRPV5 and TRPV6). Inside distal tubule cells, Ca++ binds to calbindin (calbindin-D28K and calbindin-D9K, CB), and the Ca++-calbindin complex diffuses across the cell to deliver Ca++ to the basolateral membrane. Ca++ is transported across the basolateral membrane by a 3Na+-1Ca++ antiporter (NCX1) and Ca++-ATPase (PMCa1b). In the proximal tubule, reabsorption of Ca++ involves uptake across the brush border membrane via a Ca++-permeable ion channel and exit across the basolateral membrane via Ca++-ATPase. A large portion of proximal tubule Ca++ reabsorption occurs via the paracellular pathway. This component of proximal tubule Ca++ reabsorption is driven by solvent drag. Reabsorption of Ca++ via the paracellular pathway in the thick ascending limb of Henle’s loop is driven by the transepithelial electrochemical gradient for Ca++. The tight junction protein claudin-16 (CLDCN16) regulates the paracellular diffusion of Ca++ (see the molecular box on claudin-16). Ca++ reabsorption in the distal tubule occurs exclusively by the transcellular pathway.
Reabsorption of Ca++ by the loop of Henle is restricted to the cortical portion of the thick ascending limb. Ca++ is reabsorbed by the cellular and paracellular routes via mechanisms similar to those described for the proximal tubule but with one difference (Fig. 35-13): Ca++ is not reabsorbed by solvent drag in this segment. (The thick ascending limb is impermeable to water.) In the thick ascending limb, reabsorption of Ca++ and Na+ occurs in parallel. These processes are parallel because of the significant component of Ca++ reabsorption that occurs via passive paracellular mechanisms secondary to reabsorption of Na+ and via generation of positive transepithelial voltage in the lumen. Loop diuretics inhibit reabsorption of Na+ by the thick ascending limb of the loop of Henle and in so doing reduce the magnitude of the positive transepithelial luminal voltage (see Chapter 33). This action in turn inhibits reabsorption of Ca++ via the paracellular pathway. Thus, loop diuretics are used to increase renal Ca++ excretion in patients with hypercalcemia. Therefore, reabsorption of Na+ also changes in parallel with reabsorption of Ca++ by both the proximal tubule and the thick ascending limb of the loop of Henle.
Mutations in the tight junction protein, claudin-16, alters the diffusive movement of Ca++ across tight junctions in the thick ascending limb of Henle’s loop (TAL). Familial hypomagnesemic hypercalciuria is caused by mutations in claudin-16, a protein that is a component of the tight junctions in TAL cells. This disorder is characterized by enhanced excretion of Ca++ and magnesium (Mg++) because of a fall in the passive reabsorption of these ions across the paracellular pathway in the TAL. The mutation in the claudin-16 gene reduces the permeability of the paracellular pathway to Ca++ and Mg++, thereby reducing passive, paracellular reabsorption of both ions.
In the distal tubule, where the voltage in the tubule lumen is electrically negative with respect to blood, reabsorption of Ca++ is entirely active because Ca++ is reabsorbed against its electrochemical gradient (Fig. 35-13). Reabsorption of Ca++ by the distal tubule is exclusively transcellular. Calcium enters the cell across the apical membrane through Ca++-permeable epithelial ion channels (TRPV5/TRPV6). Inside the cell, Ca++ binds to calbindin. The calbindin-Ca++ complex carries Ca++ across the cell and delivers Ca++ to the basolateral membrane, where it is extruded from the cell by either Ca++-ATPase (PMCA1b) or the 3Na+-1Ca++ antiporter (NCX1). Urinary Na+ and Ca++ excretion usually changes in parallel. However, excretion of these ions does not always change in parallel because reabsorption of Ca++ and Na+ by the distal tubule is independent and differentially regulated. For example, thiazide diuretics inhibit reabsorption of Na+ by the distal tubule and stimulate reabsorption of Ca++ by this segment. Accordingly, the net effects of thiazide diuretics are to increase urinary Na+ excretion and reduce urinary Ca++ excretion.
Regulation of Urinary Calcium Excretion
Several hormones and factors influence urinary Ca++ excretion (Table 35-4). Of these, PTH exerts the most powerful control on renal Ca++ excretion, and it is responsible for maintaining Ca++ homeostasis. Overall, this hormone stimulates reabsorption of Ca++ by the kidneys (i.e., reduces Ca++ excretion). Although PTH inhibits reabsorption of NaCl and fluid and therefore reabsorption of Ca++ by the proximal tubule, PTH stimulates reabsorption of Ca++ by the thick ascending limb of the loop of Henle and the distal tubule. In humans, this effect is greater in the distal tubule. Changes in [Ca++] in ECF also regulate urinary Ca++ excretion, with hypercalcemia increasing excretion and hypocalcemia decreasing it. Hypercalcemia increases urinary Ca++ excretion by (1) reducing proximal tubule Ca++ reabsorption (reduced paracellular reabsorption because of increased interstitial fluid [Ca++]); (2) inhibiting Ca++ reabsorption by the thick ascending limb of the loop of Henle, an effect mediated by the CaSR located in the basolateral membrane of these cells (the activity of the 1Na+-1K+-2Cl− symporter is decreased, thereby reducing the magnitude of the positive transepithelial luminal voltage); and (3) suppressing reabsorption of Ca++ by the distal tubule by reducing PTH levels. As a result, urinary Ca++ excretion increases. The opposite effect occurs with hypocalcemia.
Calcitonin stimulates reabsorption of Ca++ by the thick ascending limb and distal tubule, but it is less effective than PTH, and it is not known how important this effect is in humans. Calcitriol either directly or indirectly enhances Ca++ reabsorption by the distal tubule, but it is also less effective than PTH.
Several factors disturb Ca++ excretion. An increase in plasma [Pi] (e.g., caused by increased dietary intake of Pi) elevates PTH levels and thereby decreases Ca++ excretion. A decline in plasma [Pi] (e.g., caused by dietary Pi depletion) has the opposite effect. Changes in ECF volume alter excretion of Ca++ mainly by affecting reabsorption of NaCl and fluid in the proximal tubule. Volume contraction increases NaCl and water reabsorption by the proximal tubule and thereby enhances reabsorption of Ca++. Accordingly, urinary Ca++ excretion declines. Volume expansion has the opposite effect. Acidosis increases Ca++ excretion, whereas alkalosis decreases it. Regulation of Ca++ reabsorption by pH occurs in the distal tubule. Alkalosis stimulates the apical membrane Ca++ channel (TRPV5), thereby increasing reabsorption of Ca++. By contrast, acidosis inhibits the same channel, thereby reducing reabsorption of Ca++.
Calcium-Sensing Receptor
The CaSR is a receptor expressed in the plasma membrane of cells involved in regulating Ca++ homeostasis. The CaSR senses small changes in extracellular [Ca++]. Ca++ binds to CaSRs in PTH-secreting cells of the parathyroid gland, calcitonin-secreting parafollicular cells in the thyroid gland, and calcitriol-producing cells of the proximal tubule. Activation of the receptor by an increase in plasma [Ca++] results in inhibition of PTH secretion and calcitriol production and stimulation of calcitonin secretion. Moreover, the reduction in PTH secretion also contributes to decreased production of calcitriol because PTH is a potent stimulus of calcitriol synthesis. By contrast, a fall in plasma [Ca++] has the opposite effect on PTH, calcitriol, and calcitonin secretion. These three hormones act on the kidneys, intestine, and bone to regulate plasma [Ca++] by mechanisms described elsewhere in this chapter.
Mutations in the gene coding for CaSR cause disorders in Ca++ homeostasis. Familial hypocalciuric hypercalcemia (FHH) is an autosomal dominant disease caused by an inactivating mutation of CaSR. The hypercalcemia is caused by deranged Ca++-regulated PTH secretion (i.e., PTH levels are elevated at any level of plasma [Ca++]). Hypocalciuria is caused by enhanced Ca++ reabsorption in the thick ascending limb and distal tubule as a result of elevated PTH levels and defective CaSR regulation of Ca++ transport in the kidneys. Autosomal dominant hypocalcemia is caused by an activating mutation in CaSR. Activation of CaSRs causes deranged Ca++-regulated PTH secretion (i.e., PTH levels are decreased at any level of plasma [Ca++]). Hypercalciuria results and is caused by decreased PTH levels and defective CaSR-regulated Ca++ transport in the kidneys.
The CaSR also maintains Ca++ homeostasis by directly regulating excretion of Ca++ by the kidneys. CaSRs in the thick ascending limb and distal tubule respond directly to changes in plasma [Ca++] and regulate Ca++ absorption by these nephron segments. An increase in plasma [Ca++] activates CaSRs in the thick ascending limb and distal tubule and inhibits Ca++ absorption in these nephron segments, thereby stimulating urinary Ca++ excretion. By contrast, a fall in plasma [Ca++] leads to an increase in Ca++ absorption by the thick ascending limb and distal tubule and a corresponding decrease in urinary Ca++ excretion. Thus, the direct effect of plasma [Ca++] on CaSRs in the thick ascending limb and distal tubule acts in concert with changes in PTH to regulate urinary Ca++ excretion and thereby maintain Ca++ homeostasis.
Phosphate
Pi is an important component of many organic molecules, including DNA, RNA, ATP, and intermediates of metabolic pathways. It is also a major constituent of bone. Its concentration in plasma is an important determinant of bone formation and resorption. In addition, urinary Pi is an important buffer (titratable acid) for the maintenance of acid-base balance (see Chapter 36). Eighty-six percent of Pi is located in bone, approximately 14% in ICF, and 0.03% in ECF. The normal plasma [Pi] is 4 mg/dL. Approximately 10% of the Pi in plasma is protein bound and therefore unavailable for ultrafiltration by the glomerulus. Accordingly, the [Pi] in the ultrafiltrate is 10% less than that in plasma.
Overview of Phosphate Homeostasis
A general scheme of Pi homeostasis is shown in Figure 35-14. Maintenance of Pi homeostasis depends on two factors: (1) the amount of Pi in the body and (2) the distribution of Pi between the ICF and ECF compartments. Total body [Pi] is determined by the relative amount of Pi absorbed by the gastrointestinal tract versus the amount excreted by the kidneys. Absorption of Pi by the gastrointestinal tract occurs via active and passive mechanisms; Pi absorption increases as dietary Pi rises, and it is stimulated by calcitriol. Despite variations in Pi intake between 800 and 1500 mg/day, the kidneys keep total body Pi balance constant by excreting an amount of Pi in urine equal to the amount absorbed by the gastrointestinal tract. Thus, renal Pi excretion is the primary mechanism by which the body regulates Pi balance and thereby Pi homeostasis.
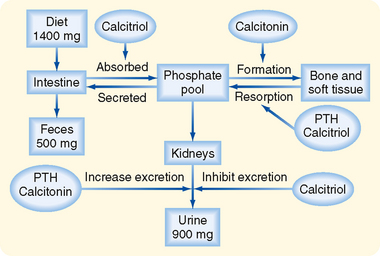
The second factor that maintains Pi homeostasis is the distribution of Pi among bone and the ICF and ECF compartments. PTH, calcitriol, and calcitonin regulate the distribution of Pi between bone and the ECF. As with Ca++ homeostasis, calcitonin is the least important of the hormones involved in Pi homeostasis in humans. Release of Pi from bone is stimulated by the same hormones (i.e., PTH, calcitriol) that release Ca++ from this pool. Thus, release of Pi is always accompanied by a release of Ca++. In contrast, calcitonin increases bone formation and thereby decreases plasma [Pi].
The kidneys also make an important contribution to the regulation of plasma [Pi]. A small rise in plasma [Pi] increases the amount of Pi filtered by the glomerulus. Because the kidneys normally reabsorb Pi at a maximum rate, any increase in the amount filtered leads to a rise in urinary Pi excretion. In fact, an increase in the amount of Pi filtered enhances urinary Pi excretion to a value greater than the rate of Pi absorption by the gastrointestinal tract. This process results in net loss of Pi from the body and decreases plasma [Pi]. In this way the kidneys regulate plasma [Pi]. The maximum reabsorptive rate for Pi varies and is regulated by dietary Pi intake. A diet high in Pi decreases the maximum reabsorptive rate of Pi by the kidneys, and a diet low in Pi increases it. This effect is independent of changes in PTH levels.
Phosphate Transport along the Nephron
Figure 35-15 summarizes Pi transport by the various portions of the nephron. The proximal tubule reabsorbs 80% of the Pi filtered by the glomerulus, and the distal tubule reabsorbs 10%. In contrast, the loop of Henle and the collecting duct reabsorb negligible amounts of Pi. Therefore approximately 10% of the filtered load of Pi is excreted.
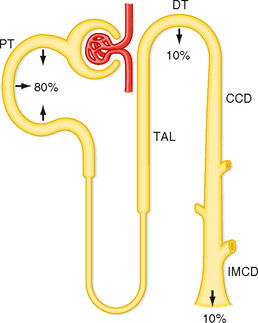
Figure 35-15 Pi transport along the nephron. Pi is reabsorbed primarily by the proximal tubule. Percentages refer to the amount of the filtered Pi reabsorbed by each nephron segment. Approximately 10% of the filtered Pi is excreted. CCD, cortical collecting duct; DT, distal tubule; IMCD, inner medullary collecting duct; PT, proximal tubule; TAL, thick ascending limb.
Reabsorption of Pi by the proximal tubule occurs mainly, if not exclusively, by means of a transcellular route. Uptake of Pi across the apical membrane occurs via Na+-Pi symport mechanisms (NPT). Three symporters have been identified: one transports 2Na+ with each Pi (NPT1), whereas the other two transport 3Na+ with each Pi (NPT2 and NPT3). NPT2 is the most important symporter involved in reabsorption of Pi by the proximal tubule (Fig. 35-16). Pi exits across the basolateral membrane by a Pi–inorganic anion antiporter. The cellular mechanism of Pi reabsorption by the distal tubule has not been characterized.
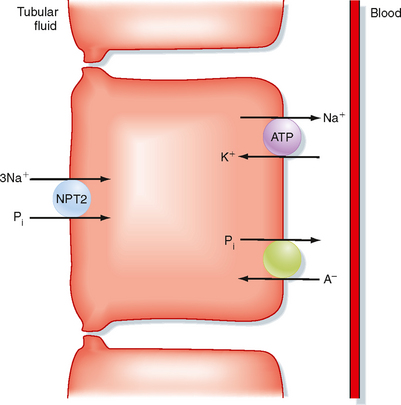
Figure 35-16 Cellular mechanisms of Pi reabsorption by the proximal tubule. The apical transport pathway operates primarily as a 3Na+-1Pi symporter (NPT2). Pi leaves the cell across the basolateral membrane by a Pi-anion antiporter. A− indicates an anion.
In patients with chronic renal failure, the kidneys cannot excrete Pi. Because of continued Pi absorption by the gastrointestinal tract, Pi accumulates in the body, and plasma [Pi] rises. The excess Pi complexes with Ca++ and reduces plasma [Ca++]. Accumulation of Pi also decreases the production of calcitriol. This response reduces absorption of Ca++ by the intestine, an effect that further reduces plasma [Ca++]. This reduction in plasma [Ca++] increases PTH secretion and Ca++ release from bone. These actions result in osteitis fibrosa cystica (i.e., increased bone resorption with replacement by fibrous tissue, which renders bone more susceptible to fracture). Chronic hyperparathyroidism (i.e., elevated PTH levels because of the fall in plasma [Ca++]) during chronic renal failure can lead to metastatic calcifications in which Ca++ and Pi precipitate in arteries, soft tissues, and viscera. Deposition of Ca++ and Pi in heart and lung tissue may cause myocardial failure and pulmonary insufficiency, respectively. Prevention and treatment of hyperparathyroidism and Pi retention include a low-Pi diet or the administration of a “phosphate binder” (i.e., an agent that forms insoluble Pi salts and thereby renders Pi unavailable for absorption by the gastrointestinal tract). Supplemental Ca++ and calcitriol are also prescribed.
Regulation of Urinary Phosphate Excretion
Several hormones and factors regulate urinary Pi excretion (Table 35-5). PTH, the most important hormone that controls Pi excretion, inhibits reabsorption of Pi by the proximal tubule and thereby increases Pi excretion. PTH reduces Pi reabsorption by stimulating the endocytic removal of NPT2 from the brush border membrane of the proximal tubule. Dietary Pi intake also regulates Pi excretion by mechanisms unrelated to changes in PTH levels. Pi loading increases excretion, whereas Pi depletion decreases it. Changes in dietary Pi intake modulate Pi transport by altering the transport rate of each NPT2 symporter and the number of transporters.
Table 35-5 Summary of Hormones and Factors Affecting Pi Reabsorption by the Proximal Tubule
| Factor/Hormone | Rate of Occurrence | Proximal Tubule Reabsorption |
|---|---|---|
| Volume expansion | Decrease | |
| Hypercalcemia | Acute | Increase |
| Hypercalcemia | Chronic | Decrease |
| Phosphate loading | Decrease | |
| Phosphate depletion | Increase | |
| Metabolic acidosis | Chronic | Decrease |
| Metabolic alkalosis | Chronic | Increase |
| PTH | Decrease | |
| Vitamin D | Acute | Increase |
| Vitamin D | Chronic | Decrease |
| Growth hormone | Increase | |
| FGF-23/FGF-24 | Decrease | |
| Glucocorticoids | Decrease |
ECF volume also affects Pi excretion. Volume expansion increases excretion, and volume contraction decreases it. The effect of ECF volume on Pi excretion is indirect and may involve changes in levels of hormones other than PTH. Acid-base balance also influences Pi excretion; acidosis increases Pi excretion, whereas alkalosis decreases it. Glucocorticoids increase the excretion of Pi. Glucocorticoids increase delivery of Pi to the distal tubule and collecting duct by inhibiting reabsorption of Pi by the proximal tubule. This inhibition enables the distal tubule and collecting duct to secrete more H+ and generate more HCO3− because Pi is an important urinary buffer (see Chapter 36). Finally, growth hormone decreases excretion of Pi. Several phosphaturic factors, also called phosphatonins, including fibroblast growth factor 23 (FGF-23) and frizzled-related protein 4 (FRP-4), are hormones produced by tumors in patients with osteomalacia that inhibit renal Pi reabsorption. An increase in dietary Pi enhances plasma FGF-23 levels, which by reducing NPT2 expression in the apical membrane of the proximal tubule, enhances urinary Pi excretion and also decreases calcitriol levels. Prolonged increases in plasma [Pi] are associated with increased tissue calcification and a reduced life span.
In the absence of glucocorticoids (e.g., in Addison’s disease), excretion of Pi is depressed, as is the ability of the kidneys to excrete titratable acid and to generate new HCO3− (see Chapter 36). Growth hormone also has an important effect on Pi homeostasis. Growth hormone increases reabsorption of Pi by the proximal tubule. As a result, growing children have higher plasma [Pi] than adults do, and this elevated [Pi] is important for the formation of bone.