CHAPTER 36 Role of the Kidneys in the Regulation of Acid-Base Balance
The concentration of H+ in body fluids is low in comparison to the concentration of other ions. For example, Na+ is present at a concentration some 3 million times greater than that of H+ ([Na+] = 140 mEq/L; [H+] = 40 nEq/L). Because of the low [H+] of body fluids, it is commonly expressed as the negative logarithm, or pH.
Virtually all cellular, tissue, and organ processes are sensitive to pH. Indeed, life cannot exist outside a range of body fluid pH from 6.8 to 7.8 (160 to 16 nEq/L of H+). Normally, the pH of extracellular fluid (ECF) is maintained between 7.35 and 7.45. As described in Chapter 2, the pH of intracellular fluid (ICF) is slightly lower (7.1 to 7.2), but also tightly regulated.
Each day, acid and alkali are ingested in the diet. In addition, cellular metabolism produces a number of substances that have an impact on the pH of body fluids. Without appropriate mechanisms to deal with this daily acid and alkali load and thereby maintain acid-base balance, many processes necessary for life could not occur. This chapter reviews the maintenance of whole-body acid-base balance. Although emphasis is on the role of the kidneys in this process, the role of the lungs and liver is also considered. Moreover, the impact of diet and cellular metabolism on acid-base balance is presented. Finally, disorders of acid-base balance are considered, primarily to illustrate the physiological processes involved. Throughout this chapter, acid is defined as any substance that adds H+ to body fluids, whereas alkali is defined as a substance that removes H+ from body fluids.
THE HCO3− BUFFER SYSTEM
Bicarbonate (HCO3−) is an important buffer of ECF. With a normal plasma [HCO3−] of 23 to 25 mEq/L and a volume of 14 L (for a 70-kg individual), ECF can potentially buffer 350 mEq of H+. The HCO3− buffer system differs from other buffer systems of the body (e.g., phosphate) in that it is regulated by both the lungs and the kidneys. This is best appreciated by considering the following reaction:
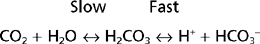
As indicated, the first reaction (hydration/dehydration of CO2) is the rate-limiting step. This normally slow reaction is greatly accelerated in the presence of carbonic anhydrase.* The second reaction, the ionization of H2CO3 to H+ and HCO3− is virtually instantaneous.
The Henderson-Hasselbalch equation (36-2) is used to quantitate how changes in CO2 and HCO3− affect pH.
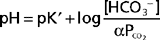
or

In these equations, the amount of CO2 is determined from the partial pressure of CO2 (PCO2) and its solubility (α) in solution. For plasma at 37° C, α has a value of 0.03. Also, pK′ is the negative logarithm of the overall dissociation constant for the reaction in Equation 36-1 and has a value of 6.1 for plasma at 37° C. Alternatively, the relationship between HCO3−, CO2, and [H+] can be expressed as follows:
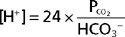
Inspection of Equations 36-3 and 36-4 show that pH and [H+] vary when either [HCO3−] or PCO2 is altered. Disturbances in acid-base balance that result from a change in [HCO3−] are termed metabolic acid-base disorders, whereas those resulting from a change in PCO2 are termed respiratory acid-base disorders. These disorders are considered in more detail in a subsequent section. The kidneys are primarily responsible for regulating [HCO3−] in ECF, whereas the lungs control PCO2.
OVERVIEW OF ACID-BASE BALANCE
The diet of humans contains many constituents that are either acid or alkali. In addition, cellular metabolism produces acid and alkali. Finally, alkali is normally lost each day in feces. As described later, the net effect of these processes is the addition of acid to body fluids. For acid-base balance to be maintained, acid must be excreted from the body at a rate equivalent to its addition. If addition of acid exceeds excretion, acidosis results. Conversely, if excretion of acid exceeds addition, alkalosis results.
The major constituents of the diet are carbohydrates and fats. When tissue perfusion is adequate, O2 is available to tissues, and insulin is present at normal levels, carbohydrates and fats are metabolized to CO2 and H2O. On a daily basis, 15 to 20 mol of CO2 is generated through this process. Normally, this large quantity of CO2 is effectively eliminated from the body by the lungs. Therefore, this metabolically derived CO2 has no impact on acid-base balance. CO2 is usually termed volatile acid because it has the potential to generate H+ after hydration with H2O (Equation 36-1). Acid not derived directly from the hydration of CO2 is termed nonvolatile acid (e.g., lactic acid).
The cellular metabolism of other dietary constituents also has an impact on acid-base balance. For example, cysteine and methionine, sulfur-containing amino acids, yield sulfuric acid when metabolized, whereas hydrochloric acid results from the metabolism of lysine, arginine, and histidine. A portion of this nonvolatile acid load is offset by the production of HCO3− through metabolism of the amino acids aspartate and glutamate. On average, the metabolism of dietary amino acids yields net nonvolatile acid production. The metabolism of certain organic anions (e.g., citrate) results in the production of HCO3−, which offsets the production of nonvolatile acid to some degree. Overall, in individuals ingesting a meatcontaining diet, acid production exceeds HCO3− production. In addition to the metabolically derived acids and alkalis, the foods ingested contain acid and alkali. For example, the presence of phosphate (H2PO4−) in ingested food increases the dietary acid load. Finally, during digestion, some HCO3− is normally lost in feces. This loss is equivalent to the addition of nonvolatile acid to the body. Together, dietary intake, cellular metabolism, and fecal HCO3− loss result in the addition of approximately 0.7 to 1.0 mEq/kg body weight of nonvolatile acid to the body each day (50 to 100 mEq/day for most adults).
Nonvolatile acids do not circulate throughout the body but are immediately neutralized by the HCO3− in ECF.
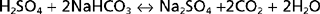
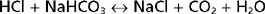
This neutralization process yields the Na+ salts of the strong acids and removes HCO3− from ECF. Thus, HCO3− minimizes the effect of these strong acids on the pH of ECF. As noted previously, ECF contains approximately 350 mEq of HCO3−. If this HCO3− were not replenished, the daily production of nonvolatile acids (≈70 mEq/day) would deplete the ECF of HCO3− within 5 days. To maintain acid-base balance the kidneys must replenish the HCO3− that is lost by neutralization of the nonvolatile acids.
When insulin levels are normal, carbohydrates and fats are completely metabolized to CO2 + H2O. However, if insulin levels are abnormally low (e.g., diabetes mellitus), metabolism of carbohydrates leads to the production of several organic keto acids (e.g., βhydroxybutyric acid).
In the absence of adequate levels of O2 (hypoxia), anaerobic metabolism by cells can also lead to the production of organic acids (e.g., lactic acid) rather than CO2 + H2O. This frequently occurs in normal individuals during vigorous exercise. Poor tissue perfusion, such as that occurring with reduced cardiac output, can also lead to anaerobic metabolism by cells and thus to acidosis. In these conditions organic acids accumulate, and the pH of body fluids decreases (acidosis). Treatment (e.g., administration of insulin in the case of diabetes) or improved delivery of adequate levels of O2 to tissues (e.g., in the case of poor tissue perfusion) results in the metabolism of these organic acids to CO2 + H2O, which consumes H+ and thereby helps correct the acid-base disorder.
NET ACID EXCRETION BY THE KIDNEYS
Under normal conditions the kidneys excrete an amount of acid equal to the production of nonvolatile acids and in so doing replenish the HCO3− that is lost by neutralization of the nonvolatile acids. In addition, the kidneys must prevent the loss of HCO3− in urine. This latter task is quantitatively more important because the filtered load of HCO3− is approximately 4320 mEq/day (24 mEq/L × 180 L/day = 4320 mEq/day), as compared with only 50 to 100 mEq/day needed to balance nonvolatile acid production.
Both reabsorption of the filtered HCO3− and excretion of acid are accomplished via H+ secretion by nephrons. Thus, in a single day the nephrons must secrete approximately 4390 mEq of H+ into tubular fluid. Most of the secreted H+ serves to reabsorb the filtered load of HCO3−. Only 50 to 100 mEq of H+, an amount equivalent to the production of nonvolatile acids, is excreted in urine. As a result of this acid excretion, the urine is normally acidic.
The kidneys cannot excrete urine more acidic than pH 4.0 to 4.5. Even at a pH of 4.0 only 0.1 mEq/L of H+ can be excreted. Therefore, to excrete sufficient acid, the kidneys excrete H+ with urinary buffers such as phosphate (Pi).* Other constituents of urine can also serve as buffers (e.g., creatinine), although their role is less important than that of Pi. Collectively, the various urinary buffers are termed titratable acids. This term is derived from the method by which these buffers are quantitated in the laboratory. Typically, alkali (OH−) is added to a urine sample to titrate its pH to that of plasma (i.e., 7.4). The amount of alkali added is equal to the amount of H+ titrated by these urine buffers and is termed titratable acid.
Excretion of H+ as a titratable acid is insufficient to balance the daily nonvolatile acid load. An additional and important mechanism by which the kidneys contribute to the maintenance of acid-base balance is through the synthesis and excretion of ammonium (NH4+). The mechanisms involved in this process are discussed in more detail later in this chapter. With regard to the renal regulation of acid-base balance, each NH4+ excreted in urine results in the return of one HCO3− to the systemic circulation, which replenishes the HCO3− lost during neutralization of the nonvolatile acids. Thus, production plus excretion of NH4+, like the excretion of titratable acid, is equivalent to the excretion of acid by the kidneys.
In brief, the kidneys contribute to acid-base homeostasis by reabsorbing the filtered load of HCO3− and excreting an amount of acid equivalent to the amount of nonvolatile acid produced each day. This overall process is termed net acid excretion (NAE), and it can be quantitated as follows:
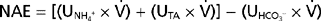
where (UNH4+ ×  ) and (UTA × ) are the rates of excretion (mEq/day) of NH4+ and titratable acid (TA) and (UHCO3− × ) is the amount of HCO3− lost in urine (equivalent to adding H+ to the body).* Again, maintenance of acid-base balance means that net acid excretion must equal nonvolatile acid production. Under most conditions, very little HCO3− is excreted in urine. Thus, net acid excretion essentially reflects titratable acid and NH4+ excretion. Quantitatively, titratable acid accounts for approximately a third and NH4+ for two thirds of net acid excretion.
) and (UTA × ) are the rates of excretion (mEq/day) of NH4+ and titratable acid (TA) and (UHCO3− × ) is the amount of HCO3− lost in urine (equivalent to adding H+ to the body).* Again, maintenance of acid-base balance means that net acid excretion must equal nonvolatile acid production. Under most conditions, very little HCO3− is excreted in urine. Thus, net acid excretion essentially reflects titratable acid and NH4+ excretion. Quantitatively, titratable acid accounts for approximately a third and NH4+ for two thirds of net acid excretion.
HCO3− Reabsorption along the Nephron
As indicated by Equation 36-7, net acid excretion is maximized when little or no HCO3− is excreted in urine. Indeed, under most circumstances, very little HCO3− appears in urine. Because HCO3− is freely filtered at the glomerulus, approximately 4320 mEq/day is delivered to the nephrons and then reabsorbed. Figure 36-1 summarizes the contribution of each nephron segment to reabsorption of the filtered HCO3−.
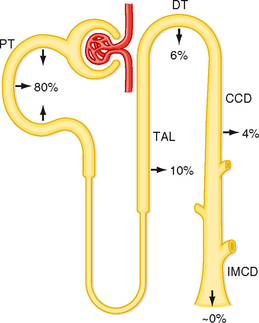
Figure 36-1 Segmental reabsorption of HCO3−. The fraction of the filtered load of HCO3− reabsorbed by the various segments of the nephron is shown. Normally, the entire filtered load of HCO3− is reabsorbed and little or no HCO3− appears in urine. CCD, cortical collecting duct; DT, distal tubule; IMCD, inner medullary collecting duct; PT, proximal tubule; TAL, thick ascending limb.
The proximal tubule reabsorbs the largest portion of the filtered load of HCO3−. Figure 36-2 summarizes the primary transport processes involved. H+ secretion across the apical membrane of the cell occurs by both an Na+-H+ antiporter and H+-ATPase. The Na+-H+ antiporter (NHE3) is the predominant pathway for H+ secretion and uses the lumen-to-cell [Na+] gradient to drive this process (i.e., secondary active secretion of H+). Within the cell, H+ and HCO3− are produced in a reaction catalyzed by carbonic anhydrase. The H+ is secreted into tubular fluid, whereas HCO3− exits the cell across the basolateral membrane and returns to the peritubular blood. Movement of HCO3− out of the cell across the basolateral membrane is coupled to other ions. The majority of HCO3− exits via a symporter that couples the efflux of 1Na+ with 3HCO3− (sodium bicarbonate cotransporter: NBC1). In addition, some of the HCO3− may exit in exchange for Cl− (via Na+-independent and/or Na+-dependent Cl−-HCO3− antiporters). As noted in Figure 36-2, carbonic anhydrase is also present in the brush border of the proximal tubule cells. This enzyme catalyzes the dehydration of H2CO3 in luminal fluid and thereby facilitates reabsorption of HCO3−.
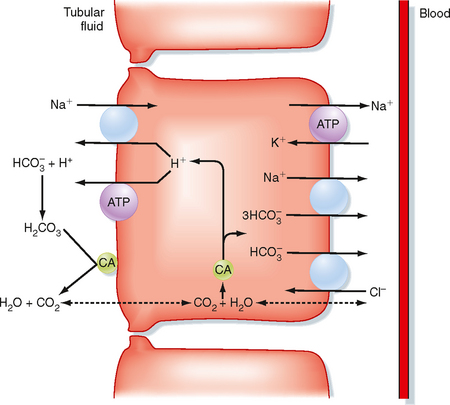
Figure 36-2 Cellular mechanism for the reabsorption of filtered HCO3− by cells of the proximal tubule. Only the primary H+ and HCO3− transporters are shown. CA, carbonic anhydrase.
Carbonic anhydrases are zinc-containing enzymes that catalyze the hydration of CO2 (see Equation 36-1). The isoform CA-I is found in red blood cells and is critical for these cells’ ability to carry CO2. Two isoforms, CA-II and CA-IV, play important roles in urine acidification. The CA-II isoform is localized to the cytoplasm of many cells along the nephron, including the proximal tubule, thick ascending limb of Henle’s loop, and intercalated cells of the distal tubule and collecting duct. The CA-IV isoform is membrane bound and exposed to the contents of the tubular fluid. It is found in the apical membrane of both the proximal tubule and thick ascending limb of Henle’s loop, where it facilitates reabsorption of the large amount of HCO3− reabsorbed by these segments. CA-IV has also been demonstrated in the basolateral membrane of the proximal tubule and thick ascending limb of Henle’s loop. Its function at this site is thought to facilitate the exit of HCO3− from the cell in some way.
The cellular mechanism for reabsorption of HCO3− by the thick ascending limb of the loop of Henle is very similar to that in the proximal tubule. H+ is secreted by an Na+-H+ antiporter and H+-ATPase. As in the proximal tubule, the Na+-H+ antiporter is the predominant pathway for secretion of H+. Exit of HCO3− from the cell involves both a 1Na+-3HCO3− symporter (although the isoform is different from that in the proximal tubule), and a Cl−-HCO3− antiporter (anion exchanger: AE-2). A K+-HCO3− symporter in the basolateral membrane may also contribute to exit of HCO3− from the cell.
The distal tubule* and collecting duct reabsorb the small amount of HCO3− that escapes reabsorption by the proximal tubule and loop of Henle. Figure 36-3 shows the cellular mechanism of H+/HCO3− transport by intercalated cells located within these segments (see Chapter 32).
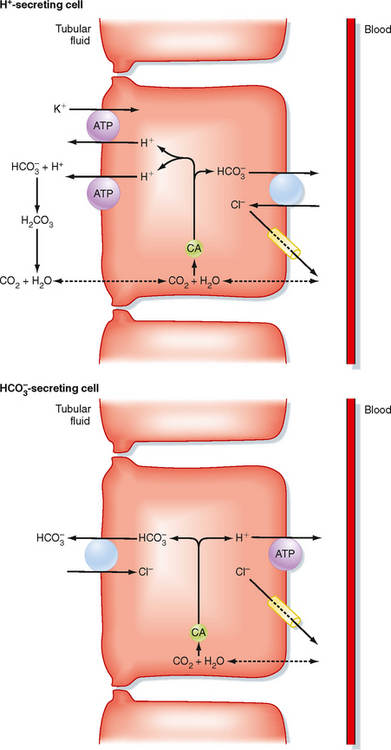
Figure 36-3 Cellular mechanisms for the reabsorption and secretion of HCO3− by intercalated cells of the collecting duct. Only the primary H+ and HCO3− transporters are shown. CA, carbonic anhydrase.
One type of intercalated cell secretes H+ (reabsorbs HCO3−) and is called the A- or α-intercalated cell. Within this cell, H+ and HCO3− are produced by the hydration of CO2; this reaction is catalyzed by carbonic anhydrase. H+ is secreted into tubular fluid via two mechanisms. The first involves an apical membrane H+-ATPase. The second couples the secretion of H+ with the reabsorption of K+ via an H+,K+-ATPase similar to that found in the stomach. The HCO3− exits the cell across the basolateral membrane in exchange for Cl− (via a Cl−-HCO3− antiporter: AE-1) and enters the peritubular capillary blood. Other HCO3− transporters have been localized to this cell. However, their role in H+ secretion (HCO3− reabsorption) has not been completely defined.
A second population of intercalated cells secrete HCO3− rather than H+ into the tubular fluid (also called B- or β-intercalated cells).† In these cells, the H+-ATPase is located in the basolateral membrane, and the Cl−-HCO3− antiporter is located in the apical membrane (Fig. 36-3). However, the apical membrane Cl−-HCO3− antiporter is different from the one found in the basolateral membrane of the H+-secreting intercalated cells and has been identified as pendrin. Other HCO3− transporters have been localized to the HCO3−-secreting intercalated cell, but their precise role in the function of the cell has not been defined. The activity of the HCO3−-secreting intercalated cell is increased during metabolic alkalosis, when the kidneys must excrete excess HCO3−. However, under most conditions (i.e., ingestion of a meat-containing diet), H+ secretion predominates in these segments.
The apical membrane of collecting duct cells is not very permeable to H+, and thus the pH of tubular fluid can become quite acidic. Indeed, the most acidic tubular fluid along the nephron (pH of 4.0 to 4.5) is produced there. In comparison, the permeability of the proximal tubule to H+ and HCO3− is much higher, and tubular fluid pH falls to only 6.5 in this segment. As explained later, the ability of the collecting duct to lower the pH of tubular fluid is critically important for the excretion of urinary titratable acids and NH4+.
Regulation of H+ Secretion
A number of factors regulate secretion of H+ and thus reabsorption of HCO3− by cells of the nephron (Table 36-1). From a physiological perspective, the primary factor that regulates H+ secretion by the nephron is a change in systemic acid-base balance. Thus, acidosis stimulates H+ secretion, whereas H+ secretion is reduced during alkalosis. The response of the kidneys to changes in acid-base balance includes both immediate changes in the activity or number of transporters in the membrane (or both) and longer-term changes in the synthesis of transporters. For example, with metabolic acidosis, whether produced by a decrease in ECF [HCO3−] or by an increase in the partial pressure of carbon dioxide (PCO2), the pH of cells of the nephron decreases. This will stimulate H+ secretion by multiple mechanisms, depending on the particular nephron segment. First, the decrease in intracellular pH will create a more favorable cell-to–tubular fluid [H+] gradient and thereby make the secretion of H+ across the apical membrane more energetically favorable. Second, the decrease in pH may lead to allosteric changes in transport proteins, thereby altering their kinetics. This has been reported for the Na+-H+ antiporter (NHE3) in the proximal tubule. Finally, transporters may be shuttled to the membrane from intracellular vesicles. This mechanism occurs in both the intercalated cells of the collecting duct, where acidosis stimulates the exocytotic insertion of H+-ATPase into the apical membrane, and in the proximal tubule, where insertion of the Na+-H+ antiporter and H+-ATPase into the apical membrane occurs. With long-term acidosis, the abundance of transporters increases, either by increased transcription of appropriate transporter genes or by increased translation of transporter mRNA. Examples include the Na+-H+ antiporter and the 1Na+-3HCO3− symporter in the proximal tubule and H+-ATPase in the intercalated cell.
Table 36-1 Factors Regulating H+ Secretion (HCO3− Reabsorption) by the Nephron
| Factor | Primary Site of Action |
|---|---|
| Increased H+ Secretion | |
| Primary | |
| Decrease in ECF [HCO3−] (↓pH) | Entire nephron |
| Increase in arterial PCO2 | Entire nephron |
| Cortisol | Proximal tubule* |
| Endothelin | Proximal tubule* |
| Secondary | |
| Increase in the filtered load of HCO3− | Proximal tubule |
| ECF volume contraction | Proximal tubule |
| Angiotensin II | Proximal and distal tubules |
| Aldosterone | Distal tubule and collecting duct |
| Hypokalemia | Proximal tubule |
| PTH (chronic) | Thick ascending limb; distal tubule |
| Decreased H+ Secretion | |
| Primary | |
| Increase in ECF [HCO3−] (↑pH) | Entire nephron |
| Decrease in arterial PCO2 | Entire nephron |
| Secondary | |
| Decrease in the filtered load of HCO3− | Proximal tubule |
| ECF volume expansion | Proximal tubule |
| Hypoaldosteronism | Distal tubule and collecting duct |
| Hyperkalemia | Proximal tubule |
| PTH (acute) | Proximal tubule |
* Effect on the proximal tubule is established. It may also regulate H+ secretion in other nephron segments.
Although some of the effects just described may be directly attributable to the decrease in intracellular pH, most of these changes in cellular H+ transport are mediated by hormones or other factors. Two important mediators of the renal response to acidosis are endothelin and cortisol. Endothelin-1 (ET-1) is produced by endothelial and proximal tubule cells, and thus it exerts its effects via autocrine and paracrine mechanisms. With acidosis, secretion of ET-1 is enhanced. In the proximal tubule, ET-1 stimulates the phosphorylation and subsequent insertion of the Na+-H+ antiporter into the apical membrane and insertion of the 1Na+-3HCO3− symporter into the basolateral membrane. ET-1 may mediate the response to acidosis in other nephron segments as well. Acidosis also stimulates secretion of the glucocorticoid hormone cortisol by the adrenal cortex. Cortisol in turn acts on the kidneys to increase transcription of the Na+-H+ antiporter and 1Na+-3HCO3− symporter genes in the proximal tubule, as well as increase translation of the mRNA of these transporters.
Alkalosis, caused by an increase in ECF [HCO3−] or a decrease in PCO2, inhibits secretion of H+ secondary to an increase in the intracellular pH of nephron cells. Thus, the responses just described for the renal adaptation to acidosis are reversed.
Table 36-1 also lists other factors that influence secretion of H+ by cells of the nephron. However, these factors are not directly related to the maintenance of acid-base balance. Because H+ secretion in the proximal tubule and thick ascending limb of the loop of Henle is linked to the reabsorption of Na+ (via the Na+-H+ antiporter), factors that alter Na+ reabsorption secondarily affect H+ secretion. For example, the process of glomerulotubular balance ensures that the reabsorption rate of the proximal tubule is matched to the glomerular filtration rate (GFR) (see Chapter 33). Thus, when the GFR is increased, the filtered load to the proximal tubule is increased, and more fluid (including HCO3−) is reabsorbed. Conversely, a decrease in the filtered load results in decreased reabsorption of fluid and thus HCO3−.
Alterations in Na+ balance, through changes in ECF volume, also have an impact on H+ secretion. With volume contraction (negative Na+ balance), secretion of H+ is enhanced. This occurs via several mechanisms. One mechanism involves the renin-angiotensinaldosterone system, which is activated by volume contraction and leads to enhanced reabsorption of Na+ by the nephron (see Chapter 34). Angiotensin II acts on the proximal tubule to stimulate the apical membrane Na+-H+ antiporter, as well as the basolateral 1Na+-3HCO3− symporter. This stimulatory effect includes increased activity of the transporters and exocytotic insertion of transporters into the membrane. To a lesser degree, angiotensin II stimulates H+ secretion in the early portion of the distal tubule, a process also mediated by the Na+-H+ antiporter. Aldosterone’s primary action on the distal tubule and collecting duct is to stimulate Na+ reabsorption by principal cells (see Chapter 33). However, it also stimulates intercalated cells in these segments to secrete H+. This effect is both indirect and direct. By stimulating Na+ reabsorption by principal cells, aldosterone hyperpolarizes the transepithelial voltage (i.e., the lumen becomes more electrically negative). This change in transepithelial voltage then facilitates the secretion of H+ by the intercalated cells. In addition to this indirect effect, aldosterone acts directly on intercalated cells to stimulate H+ secretion. The precise mechanism or mechanisms for this stimulatory effect are not fully understood.
Another mechanism by which ECF volume contraction enhances H+ secretion (HCO3− reabsorption) is via changes in peritubular capillary Starling forces. As described in Chapters 33 and 34, ECF volume contraction alters the peritubular capillary Starling forces such that overall proximal tubule reabsorption is enhanced. With this enhanced reabsorption, more of the filtered load of HCO3− is reabsorbed.
With volume expansion (positive Na+ balance), secretion of H+ is reduced because of low levels of angiotensin II and aldosterone, as well as alterations in peritubular Starling forces that reduce overall proximal tubule reabsorption.
Parathyroid hormone (PTH) has both inhibitory and stimulatory effect on renal H+ secretion. Acutely, PTH inhibits H+ secretion by the proximal tubule by inhibiting the activity of the Na+-H+ antiporter and by also causing the antiporter to be endocytosed from the apical membrane. Long-term, PTH stimulates renal acid excretion by acting on the thick ascending limb of Henle’s loop and the distal tubule. Because secretion of PTH is increased during acidosis, this long-term stimulatory effect on renal acid excretion is a component of the renal response to acidosis. The stimulatory effect of PTH on acid excretion is due in part to the delivery of increased amounts of Pi to more distal nephron sites, where it is then titrated and excreted as titratable acid.*
Finally, K+ balance influences secretion of H+ by the proximal tubule. Hypokalemia stimulates and hyperkalemia inhibits H+ secretion. It is thought that K+-induced changes in intracellular pH are responsible, at least in part, for this effect, with hypokalemia acidifying and hyperkalemia alkalinizing the cells. Hypokalemia also stimulates H+ secretion by the collecting duct. This occurs as a result of increased expression of H+-K+-ATPase in the intercalated cells.
Formation of New HCO3−
As discussed previously, reabsorption of the filtered load of HCO3− is important for maximizing net acid excretion. However, HCO3− reabsorption alone does not replenish the HCO3− lost during neutralization of the nonvolatile acids produced during metabolism. To maintain acid-base balance, the kidneys must replace this lost HCO3− with new HCO3−. Generation of new HCO3− is achieved by the excretion of titratable acid and by the synthesis and excretion of NH4+.
The production of new HCO3− as a result of excretion of titratable acid is depicted in Figure 36-4. Because of HCO3− reabsorption by the proximal tubule and loop of Henle, fluid reaching the distal tubule and collecting duct normally contains little HCO3−. Thus, when H+ is secreted, it will combine with non-HCO3− buffers (primarily Pi) and be excreted as titratable acid. Because the H+ was produced inside the cell from the hydration of CO2, HCO3− is also produced. This HCO3− is returned to the ECF as new HCO3−. As noted, Pi excretion increases with acidosis. However, even with increased Pi available for the formation of titratable acid, this response is insufficient to generate the required amount of new HCO3−. The remainder of new HCO3− generation occurs as a result of NH4+ production and excretion.
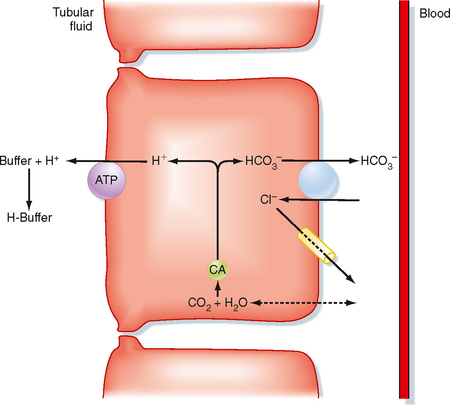
Figure 36-4 General scheme for the excretion of H+ with non-HCO3− urinary buffers (titratable acid). The primary urinary buffer is phosphate (HPO4−2). An H+-secreting intercalated cell is shown. For simplicity, only the H+-ATPase is depicted. H+ secretion by H+-K+-ATPase also titrates luminal buffers. CA, carbonic anhydrase.
NH4+ is produced by the kidneys, and its synthesis and subsequent excretion add HCO3− to the ECF. Importantly, this process is regulated in response to the acid-base requirements of the body.
NH4+ is produced in the kidneys via the metabolism of glutamine. Essentially, the kidneys metabolize glutamine, excrete NH4+, and add HCO3− to the body. However, the formation of new HCO3− via this process depends on the kidneys’ ability to excrete NH4+ in urine. If NH4+ is not excreted in urine but enters the systemic circulation instead, it is converted to urea by the liver. This conversion process generates H+, which is then buffered by HCO3−. Thus, production of urea from renally generated NH4+ consumes HCO3− and negates the formation of HCO3− through the synthesis and excretion of NH4+ by the kidneys.
The process by which the kidneys excrete NH4+ is complex. Figure 36-5 illustrates the essential features of this process. NH4+ is produced from glutamine in the cells of the proximal tubule, a process termed ammoniagenesis. Each glutamine molecule produces two molecules of NH4+ and the divalent anion 2-oxoglutarate−2. Metabolism of this anion ultimately provides two molecules of HCO3−. The HCO3− exits the cell across the basolateral membrane and enters the peritubular blood as new HCO3−. NH4+ exits the cell across the apical membrane and enters the tubular fluid. The primary mechanism for secretion of NH4+ into tubular fluid involves the Na+-H+ antiporter, with NH4+ substituting for H+. In addition, NH3 can diffuse out of the cell across the plasma membrane into tubular fluid, where it is protonated to NH4+.
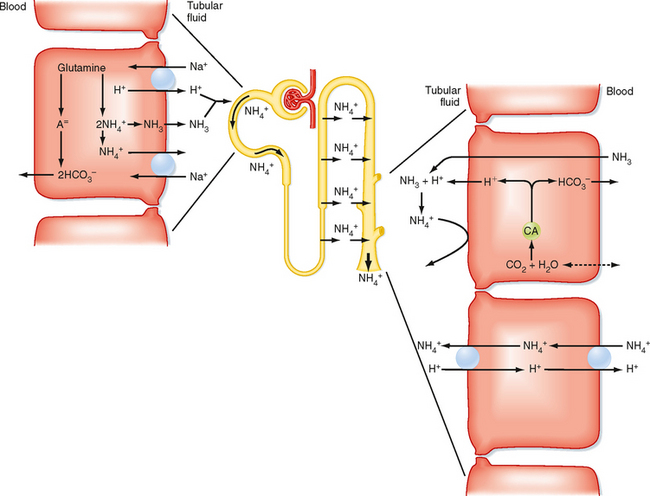
Figure 36-5 Production, transport, and excretion of NH4+ by the nephron. Glutamine is metabolized to NH4+ and HCO3− in the proximal tubule. The NH4+ is secreted into the lumen, and the HCO3− enters the blood. The secreted NH4+ is reabsorbed in Henle’s loop primarily by the thick ascending limb and accumulates in the medullary interstitium. NH4+ is secreted by the collecting duct via nonionic diffusion and diffusion trapping, as well as by NH4+ antiporters. Both secretory processes require secretion of H+ by the collecting duct. For each molecule of NH4+ excreted in urine, a molecule of “new” HCO3− is added back to the ECF. CA, carbonic anhydrase.
A significant proportion of the NH4+ secreted by the proximal tubule is reabsorbed by the loop of Henle. The thick ascending limb is the primary site of this NH4+ reabsorption, with NH4+ substituting for K+ on the 1Na+-1K+-2Cl− symporter. In addition, the positive transepithelial luminal voltage in this segment drives the paracellular reabsorption of NH4+.
The NH4+ reabsorbed by the thick ascending limb of the loop of Henle accumulates in the medullary interstitium. From there it is then secreted into tubular fluid by the collecting duct. Two mechanisms for secretion of NH4+ by the collecting duct have been identified. The first is nonionic diffusion and diffusion trapping. By this mechanism, NH3 diffuses from the medullary interstitium into the lumen of the collecting duct. As previously described, H+ secretion by the intercalated cells of the collecting duct acidifies the luminal fluid (a luminal fluid pH as low as 4.0 to 4.5 can be achieved). Consequently, NH3 diffusing from the medullary interstitium into the collecting duct lumen (nonionic diffusion) is protonated to NH4+ by the acidic tubular fluid. Because the collecting duct is less permeable to NH4+ than to NH3, NH4+ is trapped in the tubule lumen (diffusion trapping) and eliminated from the body in urine. The second mechanism involves NH4+-H+ antiporters located in the basolateral and apical membranes of the collecting duct cells (see Fig. 36-5). Because acidification of the tubular fluid drives both nonionic diffusion and diffusion trapping, as well as secretion of NH4+ across the apical membrane by the NH4+-H+ antiporter, the relative role of each mechanism to overall NH4+ secretion is not known.
H+ secretion by the collecting duct is critical for the excretion of NH4+. If collecting duct H+ secretion is inhibited, the NH4+ reabsorbed by the thick ascending limb of Henle’s loop will not be excreted in urine. Instead, it will be returned to the systemic circulation, where as described previously, it will be converted to urea by the liver and consume HCO3− in the process. Thus, new HCO3− is produced during the metabolism of glutamine by cells of the proximal tubule. However, the overall process is not complete until the NH4+ is excreted (i.e., production of urea from NH4+ by the liver is prevented). Therefore, NH4+ excretion in urine can be used as a “marker” of glutamine metabolism in the proximal tubule. In the net result, one new HCO3− is returned to the systemic circulation for each NH4+ excreted in urine.
An important feature of the renal NH4+ system is that it can be regulated by systemic acid-base balance. An alteration in the pH of the ECF, by affecting the pH of the ICF, changes glutamine metabolism in the cells of the proximal tubule. In addition, as already noted, cortisol levels increase during acidosis and cortisol stimulates ammoniagenesis (i.e., NH4+ production from glutamine). During systemic acidosis, the enzymes in the proximal tubule cell that are responsible for the metabolism of glutamine are stimulated. This involves the synthesis of new enzyme and requires several days for complete adaptation. With increased levels of these enzymes, NH4+ production is increased, thereby allowing enhanced production of new HCO3−. Conversely, glutamine metabolism is reduced with alkalosis.
The NH4+ transporters (RhBG and RhCG) are termed rhesus glycoproteins for their homology to the rhesus proteins found on the surface of erythrocytes that are responsible for hemolytic diseases and blood transfusion reactions. These transporters have been localized to the late portion of the distal tubule and the collecting duct. RhBG is localized to the basolateral membrane, whereas RhCG is localized to the apical membrane (in some species, RhCG is also found in the basolateral membrane). Both transporters appear to function as NH4+-H+ antiporters.
Acidosis also increases the abundance of RhCG in the medullary portion of the collecting duct. Thus, the ability to secrete NH4+ is enhanced.
Other factors also influence ammoniagenesis. Both angiotensin II and PTH stimulate ammoniagenesis, whereas ammoniagenesis is inhibited by prostaglandins. Because PTH levels are increased with acidosis, it may play a role in mediating the renal response, which as noted, includes increased production and excretion of NH4+. Finally, the [K+] of ECF also alters NH4+ production. When hyperkalemia exists, NH4+ production is inhibited, whereas hypokalemia stimulates NH4+ production. The mechanism by which plasma [K+] alters NH4+ production is not fully understood. Alterations in plasma [K+] may change the intracellular pH of the proximal tubule cells, and the change in intracellular pH may then control glutamine metabolism. Via this mechanism, hyperkalemia would raise intracellular pH and thereby inhibit glutamine metabolism. The opposite would occur during hypokalemia.
Assessing NH4+ excretion by the kidneys is done indirectly because assays of urine NH4+ are not routinely available. Consider, for example, the situation of metabolic acidosis. In metabolic acidosis, the appropriate renal response is to increase net acid excretion. Accordingly, little or no HCO3− will appear in urine, the urine will be acidic, and NH4+ excretion will be increased. To assess this and especially the amount of NH4+ excreted, the “urinary net charge” or “urine anion gap” can be calculated by measuring the urinary concentrations of Na+, K+, and Cl−.
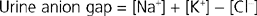
The concept of urine anion gap during metabolic acidosis assumes that the major cations in urine are Na+, K+, and NH4+ and that the major anion is Cl− (with urine pH <6.5, virtually no HCO3− is present). As a result, the urine anion gap will yield a negative value when adequate amounts of NH4+ are being excreted. Indeed, the absence of a urine anion gap or the existence of a positive value indicates a renal defect in NH4+ production and excretion.
Renal tubule acidosis (RTA) refers to conditions in which net acid excretion by the kidneys is impaired. Under these conditions the kidneys are unable to excrete a sufficient amount of net acid to balance nonvolatile acid production, and acidosis results. RTA can be caused by a defect in H+ secretion in the proximal tubule (proximal RTA) or distal tubule (distal RTA) or by inadequate production and excretion of NH4+.
Proximal RTA can be caused by a variety of hereditary and acquired conditions (e.g., cystinosis, Fanconi’s syndrome, administration of carbonic anhydrase inhibitors). The majority of cases of proximal RTA are acquired and reflect generalized tubule dysfunction rather than a selective defect in one of the proximal tubule acid-base transporters. However, autosomal recessive and autosomal dominant forms of proximal RTA have been identified. An autosomal recessive form of proximal RTA results from a defect in the 1Na+-3HCO3− symporter (NBC1). Because this transporter is also expressed in the eye, these patients have ocular abnormalities as well. Another autosomal recessive form of proximal RTA occurs in individuals who lack carbonic anhydrase (CA-II). Because CA-II is required for normal distal acidification, this defect also includes a distal RTA component. Finally, an autosomal dominant form of proximal RTA has been identified. However, the transporter involved has not been identified. Regardless of the cause, if H+ secretion by cells of the proximal tubule is impaired, reabsorption of the filtered load of HCO3− is decreased. Consequently, HCO3− is lost in urine, plasma [HCO3−] decreases, and acidosis ensues.
Distal RTA also occurs in a number of hereditary and acquired conditions (e.g., medullary sponge kidney, certain drugs such as amphotericin B, and conditions secondary to urinary obstruction). Like the inherited forms of proximal RTA, the inherited forms of distal RTA are rare. Both autosomal dominant and autosomal recessive forms of distal RTA have been identified. An autosomal dominant form results from mutations in the gene coding for the Cl−-HCO3− antiporter (AE-1) in the basolateral membrane of the acid-secreting intercalated cell. Autosomal recessive forms are caused by mutations in various subunits of H+-ATPase. In some patients with Sjögren’s syndrome, an autoimmune disease, distal RTA develops as a result of antibodies directed against H+-ATPase. Finally, H+ secretion by the distal tubule and collecting duct may be normal, but the permeability of the cells to H+ is increased. This occurs with the antifungal drug amphotericin B, administration of which also leads to the development of distal RTA. Regardless of the cause of distal RTA, the ability to acidify tubular fluid in the distal tubule and collecting duct is impaired. Consequently, excretion of titratable acid and NH4+ is reduced. This in turn decreases net acid excretion, with the subsequent development of acidosis.
Failure to produce and excrete sufficient quantities of NH4+ can also reduce net acid excretion by the kidneys. This situation occurs as a result of generalized dysfunction of the distal tubule and collecting duct with impaired secretion of H+, NH4+, and K+. Generalized distal nephron dysfunction is seen in individuals with mutations in the epithelial Na+ channel (ENaC), which is inherited in an autosomal recessive pattern. An autosomal dominant form is also seen with mutations in the mineralocorticoid receptor. More commonly, NH4+ production and excretion are impaired in patients with hyporeninemic hypoaldosteronism. These patients typically have moderate degrees of renal failure with reduced levels of renin and thus aldosterone. As a result, distal tubule and collecting duct function are impaired. Finally, a number of drugs can also result in distal tubule and collecting duct dysfunction, including drugs that block the Na+ channel (e.g., amiloride), block the production or action of angiotensin II (angiotensin-converting enzyme inhibitors), or block the action of aldosterone (e.g., spironolactone). Regardless of the cause, the impaired function of the distal tubule and collecting duct results in the development of hyperkalemia, which in turn impairs ammoniagenesis by the proximal tubule. H+ secretion by the distal tubule and collecting duct and thus NH4+ secretion are also impaired by these drugs. Thus, net acid excretion is less than net acid production, and metabolic acidosis develops.
If the acidosis that results from any of these forms of RTA is severe, individuals must ingest alkali (e.g., baking soda or a citrate-containing solution*) to maintain acid-base balance. In this way the HCO3− lost each day in the buffering of nonvolatile acid is replenished by the extra HCO3− ingested in the diet.
* One of the byproducts of citrate metabolism is HCO3−. Ingestion of citrate-containing drinks is often more palatable to patients than ingesting baking soda.
RESPONSE TO ACID-BASE DISORDERS
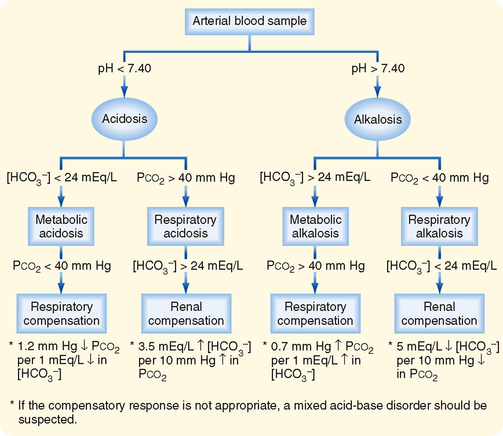
The pH of ECF is maintained within a very narrow range (7.35 to 7.45).* Inspection of Equation 36-3 shows that the pH of ECF varies when either [HCO3−] or PCO2 is altered. As already noted, disturbances in acid-base balance that result from a change in ECF [HCO3−] are termed metabolic acid-base disorders, whereas those resulting from a change in PCO2 are termed respiratory acid-base disorders. The kidneys are primarily responsible for regulating [HCO3−], whereas the lungs regulate PCO2.
When an acid-base disturbance develops, the body uses a series of mechanisms to defend against the change in pH of the ECF. These defense mechanisms do not correct the acid-base disturbance but merely minimize the change in pH imposed by the disturbance. Restoration of blood pH to its normal value requires correction of the underlying process or processes that produced the acid-base disorder. The body has three general mechanisms to compensate for or defend against changes in body fluid pH produced by acid-base disturbances: (1) extracellular and intracellular buffering, (2) adjustments in blood PCO2 via alterations in the ventilatory rate of the lungs, and (3) adjustments in renal net acid excretion.
Extracellular and Intracellular Buffers
The first line of defense against acid-base disorders is extracellular and intracellular buffering. The response of the extracellular buffers is virtually instantaneous, whereas the response to intracellular buffering is slower and can take several minutes.
Metabolic disorders that result from the addition of nonvolatile acid or alkali to body fluids are buffered in both the ECF and ICF compartments. The HCO3− buffer system is the principal ECF buffer. When nonvolatile acid is added to body fluids (or alkali is lost from the body), HCO3− is consumed during the process of neutralizing the acid load, and the [HCO3−] of ECF is reduced. Conversely, when nonvolatile alkali is added to body fluids (or acid is lost from the body), H+ is consumed, which causes more HCO3− to be produced from the dissociation of H2CO3. Consequently, [HCO3−] increases.
Although the HCO3− buffer system is the principal ECF buffer, Pi and plasma proteins provide additional extracellular buffering. The combined action of the buffering processes for HCO3−, Pi, and plasma protein accounts for approximately 50% of the buffering of a nonvolatile acid load and 70% of a nonvolatile alkali load. The remainder of the buffering under these two conditions occurs intracellularly. Intracellular buffering involves the movement of H+ into cells (during buffering of nonvolatile acid) or the movement of H+ out of cells (during buffering of nonvolatile alkali). H+ is titrated inside the cell by HCO3−, Pi, and the histidine groups on proteins.
Bone represents an additional source of extracellular buffering. With acidosis, buffering by bone results in its demineralization because Ca++ is released from bone as Ca++-containing salts bind H+ in exchange for Ca++.
When respiratory acid-base disorders occur, the pH of body fluid changes as a result of alterations in PCO2. Virtually all buffering in respiratory acid-base disorders occurs intracellularly. When PCO2 rises (respiratory acidosis), CO2 moves into the cell, where it combines with H2O to form H2CO3, which then dissociates to H+ and HCO3−. Some of the H+ is buffered by cellular protein, and HCO3− exits the cell and raises ECF [HCO3−] (ECF [H+] is also increased). This process is reversed when PCO2 is reduced (respiratory alkalosis). Under this condition, the hydration reaction (H2O + CO2 ↔ H2CO3) is shifted to the left by the decrease in PCO2. As a result, the dissociation reaction (H2CO3 ↔ H+ + HCO3−) also shifts to the left, thereby reducing ECF [HCO3−] (ECF [H+] is also decreased). Thus, the CO2-associated changes in ECF [HCO3−] minimize the change in pH.
Respiratory Compensation
The lungs are the second line of defense against acid-base disorders. As indicated by the Henderson-Hasselbalch equation (Equation 36-3), changes in PCO2 alter blood pH: a rise decreases pH, and a reduction increases pH.
The ventilatory rate determines PCO2. Increased ventilation decreases PCO2, whereas decreased ventilation increases it. Blood PCO2 and pH are important regulators of the ventilatory rate. Chemoreceptors located in the brainstem (ventral surface of the medulla) and periphery (carotid and aortic bodies) sense changes in PCO2 and [H+] and alter the ventilatory rate appropriately. Thus, when metabolic acidosis occurs, a rise in [H+] (decrease in pH) increases the ventilatory rate. Conversely, during metabolic alkalosis, a decrease in [H+] (increase in pH) leads to a reduced ventilatory rate. With maximal hyperventilation, PCO2 can be reduced to approximately 10 mm Hg. Because hypoxia, a potent stimulator of ventilation, also develops with hypoventilation, the degree to which PCO2 can be increased is limited. In an otherwise normal individual, hypoventilation cannot raise PCO2 above 60 mm Hg. The respiratory response to metabolic acid-base disturbances may be initiated within minutes but could require several hours to complete.
Renal Compensation
The third and final line of defense against acid-base disorders is the kidneys. In response to an alteration in plasma pH and PCO2, the kidneys make appropriate adjustments in the excretion of HCO3− and net acid. The renal response may require several days to reach completion because it takes hours to days to increase the synthesis and activity of the proximal tubule enzymes involved in NH4+ production. In the case of acidosis (increased [H+] or PCO2), secretion of H+ by the nephron is stimulated, and the entire filtered load of HCO3− is reabsorbed. Excretion of titratable acid is increased, production and excretion of NH4+ are also stimulated, and net acid excretion by the kidneys is thus increased (Equation 36-7). The new HCO3− generated during the process of net acid excretion is added to the body, and plasma [HCO3−] increases.
Metabolic acidosis can develop in insulin-dependent diabetic patients secondary to the production of keto acids if insulin dosages are not adequate. As a compensatory response to this acidosis, deep and rapid breathing develops. This breathing pattern is termed Kussmaul respiration. With prolonged Kussmaul respiration, the muscles involved can become fatigued. When fatigue occurs, respiratory compensation is impaired and the acidosis can become more severe.
Loss of gastric contents from the body (i.e., vomiting, nasogastric suction) produces metabolic alkalosis secondary to the loss of HCl. If the loss of gastric fluid is significant, ECF volume contraction occurs. Under this condition, the kidneys cannot excrete sufficient quantities of HCO3− to compensate for the metabolic alkalosis. Excretion of HCO3− is impaired because ECF volume contraction reduces the filtered load of HCO3− (GFR is decreased) and stimulates HCO3− reabsorption by the nephron. ECF volume contraction stimulates HCO3− reabsorption because of the need for the kidneys to reduce Na+ excretion (see Chapter 34). Thus, in response to ECF volume contraction, Na+ reabsorption by the proximal tubule is enhanced and aldosterone levels are increased. These responses in turn limit HCO3− excretion because a significant amount of Na+ reabsorption in the proximal tubule is coupled to H+ secretion via the Na+-H+ antiporter. As a result, HCO3− is reabsorbed because of the need to reduce Na+ excretion. In addition, the elevated aldosterone levels stimulate not only Na+ reabsorption but also H+ secretion by the distal tubule and collecting duct. Thus, in individuals who lose gastric contents, the metabolic alkalosis is seen in the setting of a paradoxically acidic urine. Correction of the alkalosis occurs only when euvolemia is reestablished. With restoration of euvolemia, the filtered load of HCO3− increases (GFR increases), and HCO3− reabsorption by the proximal tubule decreases, as does H+ secretion by the distal tubule and collecting duct. As a result, HCO3− excretion increases, and ECF [HCO3−] returns to normal.
When alkalosis exists (decreased [H+] or PCO2), the filtered load of HCO3− is increased (plasma [HCO3−] is elevated), and secretion of H+ by the nephron is inhibited. As a result, HCO3− excretion is increased, and the excretion of both titratable acid and NH4+ is decreased. Thus, net acid excretion is decreased and HCO3− appears in urine. In addition, some HCO3− is secreted into urine by the HCO3−-secreting intercalated cells of the distal tubule and collecting duct. With enhanced excretion of HCO3−, plasma [HCO3−] decreases.
SIMPLE ACID-BASE DISORDERS
Table 36-2 summarizes the primary alterations and the subsequent compensatory or defense mechanisms of the various simple acid-base disorders. In all acid-base disorders the compensatory response does not correct the underlying disorder but simply reduces the magnitude of the change in pH. Correction of the acid-base disorder requires treatment of its cause.
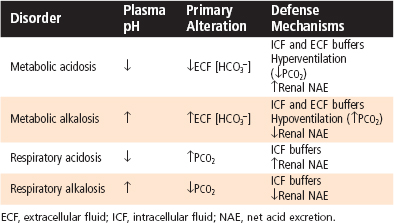
Types of Acid-Base Disorders
Metabolic Acidosis
Metabolic acidosis is characterized by decreased ECF [HCO3−] and pH. It can develop via the addition of nonvolatile acid to the body (e.g., diabetic ketoacidosis), loss of nonvolatile base (e.g., HCO3− loss caused by diarrhea), or failure of the kidneys to excrete sufficient net acid to replenish the HCO3− used to neutralize nonvolatile acids (e.g., renal tubular acidosis, renal failure). As previously described, buffering of H+ occurs in both the ECF and ICF compartments. When pH falls, the respiratory centers are stimulated, and the ventilatory rate is increased (respiratory compensation). This reduces PCO2, which further minimizes the fall in plasma pH. In general, there is a 1.2–mm Hg decrease in PCO2 for every 1-mEq/L fall in ECF [HCO3−]. Thus, if [HCO3−] were reduced to 14 mEq/L from a normal value of 24 mEq/L, the expected decrease in PCO2 would be 12 mm Hg and the measured PCO2 would fall to 28 mm Hg (normal PCO2 = 40 mm Hg).
Finally, in metabolic acidosis, renal net acid excretion is increased. This occurs via the elimination of all HCO3− from urine (enhanced reabsorption of filtered HCO3−) and via increased excretion of titratable acid and NH4+ (enhanced production of new HCO3−). If the process that initiated the acid-base disturbance is corrected, the enhanced net acid excretion by the kidneys will ultimately return the pH and [HCO3−] to normal. After correction of the pH, the ventilatory rate also returns to normal.
Metabolic Alkalosis
Metabolic alkalosis is characterized by increased ECF [HCO3−] and pH. It can occur via the addition of nonvolatile base to the body (e.g., ingestion of antacids), as a result of volume contraction (e.g., hemorrhage), or more commonly, from the loss of nonvolatile acid (e.g., loss of gastric HCl because of prolonged vomiting). Buffering occurs predominantly in the ECF compartment and to a lesser degree in the ICF compartment. The increase in pH inhibits the respiratory centers, the ventilatory rate is reduced, and thus PCO2 is elevated (respiratory compensation). With appropriate respiratory compensation, a 0.7–mm Hg increase in PCO2 is expected for every 1-mEq/L rise in ECF [HCO3−].
The primary renal compensatory response to metabolic alkalosis is to increase the excretion of HCO3− by reducing its reabsorption along the nephron. Excretion of titratable acid and NH4+ is also reduced. Normally, this occurs quite rapidly (minutes to hours) and effectively. However, as already noted, when alkalosis occurs with ECF volume contraction (e.g., vomiting in which fluid loss occurs with H+ loss), HCO3− excretion is impaired. In individuals with ECF volume contraction, renal excretion of HCO3− is enhanced, and the alkalosis is corrected only with restoration of euvolemia. Enhanced renal excretion of HCO3− eventually returns the pH and [HCO3−] to normal, provided that the underlying cause of the initial acid-base disturbance is corrected. When the pH is corrected, the ventilatory rate also returns to normal.
When nonvolatile acid is added to body fluids, as in diabetic ketoacidosis, [H+] increases (pH decreases), and [HCO3−] decreases. In addition, the concentration of the anion associated with the nonvolatile acid increases. This change in anion concentration provides a convenient way of analyzing the cause of a metabolic acidosis by calculating what is termed the anion gap. The anion gap represents the difference between the concentration of the major ECF cation (Na+) and the major ECF anions (Cl− and HCO3−):
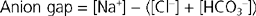
Under normal conditions the anion gap ranges from 8 to 16 mEq/L. It is important to recognize that an anion gap does not actually exist. All cations are balanced by anions. The gap simply reflects the parameters that are measured. In reality,
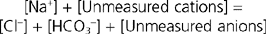
If the anion of the nonvolatile acid is Cl−, the anion gap will be normal. (That is, the decrease in [HCO3−] is matched by an increase in [Cl−].) The metabolic acidosis associated with diarrhea or renal tubular acidosis has a normal anion gap. In contrast, if the anion of the nonvolatile acid is not Cl− (e.g., lactate, β-hydroxybutyrate), the anion gap will increase (i.e., the decrease in [HCO3−] is not matched by an increase in [Cl−] but rather by an increase in concentration of the unmeasured anion). The anion gap is increased in metabolic acidosis associated with renal failure, diabetes mellitus (ketoacidosis), lactic acidosis, and the ingestion of large quantities of aspirin. Thus, calculation of the anion gap is a useful way of identifying the cause of metabolic acidosis in the clinical setting.
Respiratory Acidosis
Respiratory acidosis is characterized by an elevated PCO2 and reduced ECF pH. It results from decreased gas exchange across the alveoli as a result of either inadequate ventilation (e.g., drug-induced depression of the respiratory centers) or impaired gas diffusion (e.g., pulmonary edema, such as occurs in cardiovascular or lung disease). In contrast to the metabolic disorders, buffering during respiratory acidosis occurs almost entirely in the ICF compartment. The increase in PCO2 and the decrease in pH stimulate both reabsorption of HCO3− by the nephron and excretion of titratable acid and NH4+ (renal compensation). Together, these responses increase net acid excretion and generate new HCO3−. The renal compensatory response takes several days to occur. Consequently, respiratory acid-base disorders are commonly divided into acute and chronic phases. In the acute phase, the time needed for the renal compensatory response to take effect is not sufficient, and the body relies on ICF buffering to minimize the change in pH. During this phase and because of this buffering there is a 1-mEq/L increase in ECF [HCO3−] for every 10–mm Hg rise in PCO2. In the chronic phase, renal compensation takes place, and a 3.5-mEq/L increase in ECF [HCO3−] occurs for each 10–mm Hg rise in PCO2. Correction of the underlying disorder returns the PCO2 to normal, and renal net acid excretion decreases to its initial level.
Respiratory Alkalosis
Respiratory alkalosis is characterized by reduced PCO2 and increased ECF pH. It results from increased gas exchange in the lungs, usually caused by increased ventilation from stimulation of the respiratory centers (e.g., via drugs or disorders of the central nervous system). Hyperventilation also occurs at high altitude and as a result of anxiety, pain, or fear. As noted, buffering primarily takes place in the ICF compartment. As with respiratory acidosis, respiratory alkalosis has both acute and chronic phases reflecting the time required for renal compensation to occur. In the acute phase of respiratory alkalosis, which reflects intracellular buffering, ECF [HCO3−] decreases 2 mEq/L for every 10–mm Hg fall in PCO2. With renal compensation, the elevated pH and reduced PCO2 inhibit reabsorption of HCO3− by the nephron and reduce excretion of titratable acid and NH4+. As a result of these two effects, net acid excretion is reduced. With complete renal compensation there is an expected 5-mEq/L decrease in ECF [HCO3−] for every 10–mm Hg reduction in PCO2. Correction of the underlying disorder returns the PCO2 to normal, and renal excretion of acid then increases to its initial level.
Analysis of Acid-Base Disorders
Analysis of an acid-base disorder is directed at identifying the underlying cause so that appropriate therapy can be initiated. The patient’s medical history and associated physical findings often provide valuable clues about the nature and origin of an acid-base disorder. In addition, analysis of an arterial blood sample is frequently required. Such analysis is straightforward if approached systematically. For example, consider the following data:
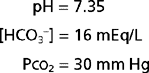
The acid-base disorder represented by these values, or any other set of values, can be determined by using the following three-step approach (Fig. 36-6):
If the appropriate compensatory response is not present, a mixed acid-base disorder should be suspected. Such a disorder reflects the presence of two or more underlying causes of the acid-base disturbance. A mixed disorder should be suspected when arterial blood gas analysis indicates that appropriate compensation has not occurred. For example, consider the following data:
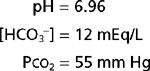
When the three-step approach is followed, it is evident that the disturbance is an acidosis that has both a metabolic component (ECF [HCO3−] <24 mEq/L) and a respiratory component (PCO2 >40 mm Hg). Thus, this disorder is mixed. Mixed acid-base disorders can occur, for example, in an individual who has a history of a chronic pulmonary disease such as emphysema (i.e., chronic respiratory acidosis) and in whom an acute gastrointestinal illness with diarrhea develops. Because diarrhea fluid contains HCO3−, its loss from the body results in the development of a metabolic acidosis.
A mixed acid-base disorder is also indicated when a patient has abnormal PCO2 and ECF [HCO3−] values but a normal pH. Such a condition can develop in a patient who has ingested a large quantity of aspirin. The salicylic acid (active ingredient in aspirin) produces metabolic acidosis, and at the same time it stimulates the respiratory centers and causes hyperventilation and respiratory alkalosis. Thus, the patient has a reduced ECF [HCO3−] and a reduced PCO2. (Note: The PCO2 is lower than would occur with normal respiratory compensation of a metabolic acidosis).
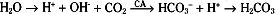 .
.