Appendix IV Laboratory Techniques Commonly Used in Immunology
Many laboratory techniques that are routine in research and clinical settings are based on the use of antibodies. In addition, many of the techniques of modern molecular biology have provided invaluable information about the immune system. We have mentioned these techniques often throughout the book. In this appendix, we describe the principles underlying some of the most commonly used laboratory methods in immunology. In addition, we summarize how B and T lymphocyte responses are studied with use of laboratory techniques. Details of how to carry out various assays may be found in laboratory manuals.
Laboratory Methods Using Antibodies
The exquisite specificity of antibodies for particular antigens makes antibodies valuable reagents for detecting, purifying, and quantitating antigens. Because antibodies can be produced against virtually any type of macromolecule and small chemical, antibody-based techniques may be used to study virtually any type of molecule in solution or in cells. The method for producing monoclonal antibodies (see Chapter 5) has greatly increased our ability to generate antibodies of almost any desired specificity. Historically, many of the uses of antibody depended on the ability of antibody and specific antigen to form large immune complexes, either in solution or in gels, that could be detected by various optical methods. These methods were of great importance in early studies but have now been replaced almost entirely by simpler methods based on immobilized antibodies or antigens.
Quantitation of Antigen by Immunoassays
Immunologic methods of quantifying antigen concentration provide exquisite sensitivity and specificity and have become standard techniques for both research and clinical applications. All modern immunochemical methods of quantitation are based on having a pure antigen or antibody whose quantity can be measured by an indicator molecule (or a label). When the antigen or antibody is labeled with a radioisotope, as first introduced by Rosalyn Yalow and colleagues, it may be quantified by instruments that detect radioactive decay events; the assay is called a radioimmunoassay (RIA). When the antigen or antibody is covalently coupled to an enzyme, it may be quantified by determining with a spectrophotometer the rate at which the enzyme converts a clear substrate to a colored product; the assay is called an enzyme-linked immunosorbent assay (ELISA). Several variations of RIA and ELISA exist, but the most commonly used version is the sandwich assay (Fig. A-1). The sandwich assay uses two different antibodies reactive with different epitopes on the antigen whose concentration needs to be determined. A fixed quantity of one antibody is attached to a series of replicate solid supports, such as plastic microtiter wells. Test solutions containing antigen at an unknown concentration or a series of standard solutions with known concentrations of antigen are added to the wells and allowed to bind. Unbound antigen is removed by washing, and the second antibody, which is enzyme linked or radiolabeled, is allowed to bind. The antigen serves as a bridge, so the more antigen in the test or standard solutions, the more enzyme-linked or radiolabeled second antibody will bind. The results from the standard solutions are used to construct a binding curve for the second antibody as a function of antigen concentration, from which the quantities of antigen in the test solutions may be inferred. When this test is performed with two monoclonal antibodies, it is essential that these antibodies see nonoverlapping determinants on the antigen; otherwise, the second antibody cannot bind.
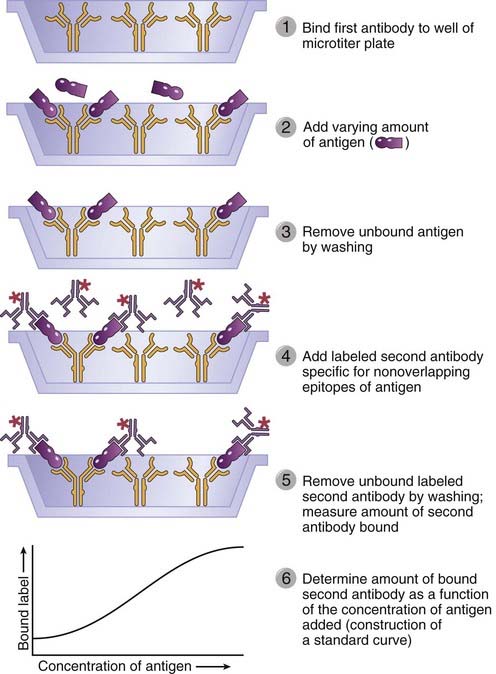
FIGURE A–1 Sandwich enzyme-linked immunosorbent assay or radioimmunoassay.
A fixed amount of one immobilized antibody is used to capture an antigen. The binding of a second, labeled antibody that recognizes a nonoverlapping determinant on the antigen will increase as the concentration of antigen increases and thus allow quantification of the antigen.
In an important clinical variant of immunobinding assays, samples from patients may be tested for the presence of antibodies that are specific for a microbial antigen (e.g., antibodies reactive with proteins from human immunodeficiency virus [HIV] or hepatitis B virus) as indicators of infection. In this case, a saturating quantity of antigen is added to replicate wells containing plate-bound antibody or the antigen is attached directly to the plate, and serial dilutions of the patient’s serum are then allowed to bind. The amount of the patient’s antibody bound to the immobilized antigen is determined by use of an enzyme-linked or radiolabeled second anti-human immunoglobulin (Ig) antibody.
Identification and Purification of Proteins
Antibodies can be used to identify and characterize proteins and to purify specific proteins from mixtures. Two commonly used methods to identify and purify proteins are immunoprecipitation and immuno–affinity chromatography. Western blotting is a widely used technique to determine the presence and size of a protein in a biologic sample.
Immunoprecipitation and Immuno–Affinity Chromatography
Immunoprecipitation is a technique in which an antibody specific for one protein antigen in a mixture of proteins is used to identify this specific antigen (Fig. A-2A). The antibody is typically added to a protein mixture (usually a detergent lysate of specific cells), and staphylococcal protein A (or protein G) covalently attached to agarose beads is added to the mixture. The Fab portions of the antibody bind to the target protein, and the Fc portion of the antibody is captured by the protein A or protein G on the beads. Unwanted proteins that do not bind to the antibody are then removed by washing the beads (by repeated detergent addition and centrifugation). The specific protein that is recognized by and now bound to the antibody may be eluted from the beads and dissociated from the antibody by use of a harsh denaturant (such as sodium dodecyl sulfate), and the proteins are separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins may be detected after electrophoresis by staining the polyacrylamide gel with a protein stain or by a Western blot analysis (described later). If the original mixture contained radioactively labeled proteins, specific proteins immunoprecipitated by the antibody may be revealed by autofluorography or autoradiography, protein bands being captured on x-ray film placed on the dried SDS–polyacrylamide gel containing separated proteins.
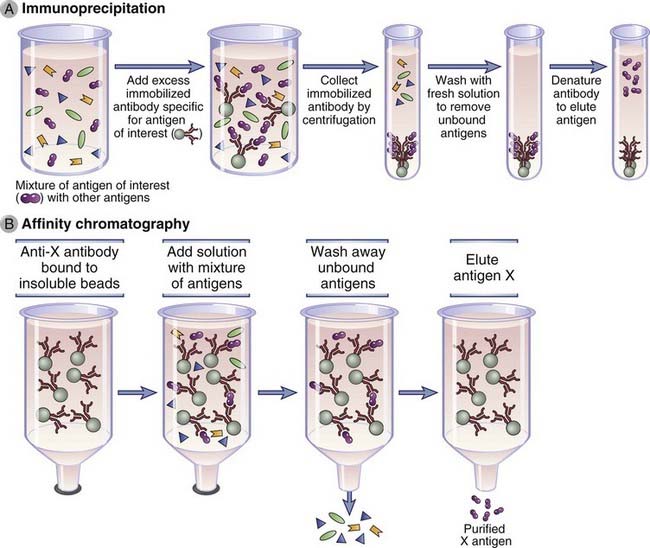
FIGURE A–2 Isolation of an antigen by immunoprecipitation or affinity chromatography.
A, A particular antigen can be purified from a mixture of antigens in serum or other solutions by adding antibodies specific to the antigen that are bound to insoluble beads. Unbound antigens are then washed away, and the desired antigen is recovered by changing the pH or ionic strength of the solution so that the affinity of antibody-antigen binding is lowered. Immunoprecipitation can be used as a means of purification, as a means of quantification, or as a means of identification of an antigen. Antigens purified by immunoprecipitation are often analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. B, Affinity chromatography is based on the same principle as immunoprecipitation, except that the antibody is fixed to an insoluble matrix or beads, usually in a column. The method is often used to isolate soluble antigens (shown) or antibodies specific for an immobilized antigen.
Immuno–affinity chromatography, a variant of affinity chromatography, is a purification method that relies on antibodies attached to an insoluble support to purify antigens from a solution (Fig. A-2B). Antibodies specific for the desired antigen are typically covalently attached to a solid support, such as agarose beads, and packed into a column. A complex mixture of antigens is passed through the beads to allow the antigen that is recognized by the antibody to bind. Unbound molecules are washed away, and the bound antigen is eluted by changing the pH or by exposure to high salt or other chaotropic conditions that break antigen-antibody interactions. A similar method may be used to purify antibodies from culture supernatants or natural fluids, such as serum, by first attaching the antigen to beads and passing the supernatants or serum through.
Western Blotting
Western blotting (Fig. A-3) is used to identify and determine the relative quantity and molecular weight of a protein within a mixture of proteins or other molecules. The mixture is first subjected to analytical separation, typically by SDS-PAGE, so that the final positions of different proteins in the gel are a function of their molecular size. The array of separated proteins is then transferred from the separating polyacrylamide gel to a support membrane by electrophoresis such that the membrane acquires a replica of the array of separated macromolecules present in the gel. SDS is displaced from the protein during the transfer process, and native antigenic determinants are often regained as the protein refolds. The position of the protein antigen on the membrane can then be detected by binding of an unlabeled antibody specific for that protein (the primary antibody) followed by a labeled second antibody that binds to the primary antibody. This approach provides information about antigen size and quantity. In general, second antibody probes are labeled with enzymes that generate chemiluminescent signals and leave images on photographic film. Near-infrared fluorophores can also be used to label antibodies, and light produced by the excitation of the fluorophore provides more accurate antigen quantitation compared with enzyme-linked second antibodies. The sensitivity and specificity of this technique can be increased by starting with immunoprecipitated proteins instead of crude protein mixtures. This sequential procedure is especially useful for detection of protein-protein interactions. For example, the physical association of two different proteins in the membrane of a lymphocyte can be established by immunoprecipitating a membrane extract by use of an antibody specific for one of the proteins and probing a Western blot of the immunoprecipitate using an antibody specific for the second protein that may have been co-immunoprecipitated along with the first protein. A variation of the Western blot technique is routinely used to detect the presence of anti-HIV antibodies in patients’ sera. In this case, a defined mixture of HIV proteins is separated by SDS-PAGE and blotted onto a membrane, and the membrane is incubated with dilutions of the test serum. The blot is then probed with a second labeled anti-human Ig to detect the presence of HIV-specific antibodies that were in the serum and bound to the HIV proteins.
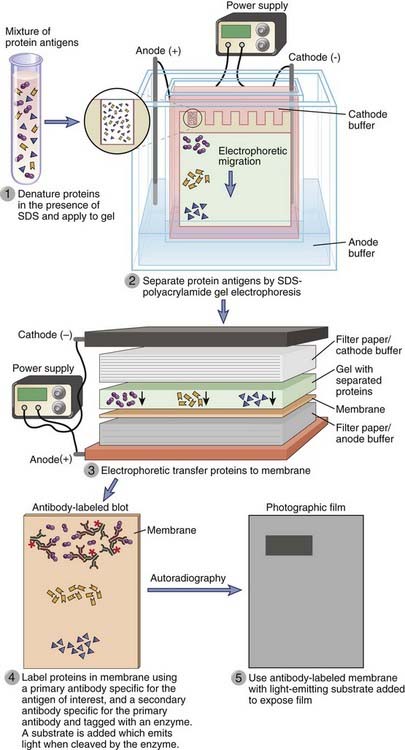
FIGURE A–3 Characterization of antigens by Western blotting.
Protein antigens, separated by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and transferred to a membrane, can be detected by an antibody that is in turn revealed by a second antibody that may be conjugated to an enzyme such as horseradish peroxidase or to a fluorophore.
The technique of transferring proteins from a gel to a membrane is called Western blotting as a biochemist’s joke. Southern is the last name of the scientist who first blotted DNA from a separating gel to a membrane by capillary transfer, a technique since called Southern blotting. By analogy, Northern blotting was the term applied to the technique of transferring RNA from a gel to a membrane, and Western blotting is the term used to describe the transfer of proteins to a membrane.
Labeling and Detection of Antigens in Cells and Tissues
Antibodies specific for antigens expressed on or in particular cell types are commonly used to identify these cells in tissues or cell suspensions and to separate these cells from mixed populations. In these methods, the antibody can be radiolabeled, enzyme linked, or, most commonly, fluorescently labeled, and a detection system is used that can identify the bound antibody. Antibodies attached to magnetic beads can be used to physically isolate cells expressing specific antigens.
Flow Cytometry and Fluorescence-Activated Cell Sorting
The tissue lineage, maturation stage, or activation status of a cell can often be determined by analyzing the cell surface or intracellular expression of different molecules. This technique is commonly done by staining the cell with fluorescently labeled probes that are specific for those molecules and measuring the quantity of fluorescence emitted by the cell (Fig. A-4). The flow cytometer is a specialized instrument that can detect fluorescence on individual cells in a suspension and thereby determine the number of cells expressing the molecule to which a fluorescent probe binds. Suspensions of cells are incubated with fluorescently labeled probes, and the amount of probe bound by each cell in the population is measured by passing the cells one at a time through a fluorimeter with a laser-generated incident beam. The relative amounts of a particular molecule on different cell populations can be compared by staining each population with the same probe and determining the amount of fluorescence emitted. In preparation for flow cytometric analysis, cell suspensions are stained with the fluorescent probes of choice. Most often, these probes are fluorochrome-labeled antibodies specific for a cell surface molecule. Alternatively, cytoplasmic molecules can be stained by temporarily permeabilizing cells and permitting the labeled antibodies to enter through the plasma membrane. In addition to antibodies, various fluorescent indicators of cytoplasmic ion concentrations and reduction-oxidation potential can be detected by flow cytometry. Cell cycle studies can be performed by flow cytometric analysis of cells stained with fluorescent DNA-binding probes such as propidium iodide. Apoptotic cells can be identified with fluorescent probes, such as annexin V, that bind to abnormally exposed phospholipids on the surface of the dying cells. Modern flow cytometers can routinely detect three or more different-colored fluorescent signals, each attached to a different antibody or other probe. This technique permits simultaneous analysis of the expression of many different combinations of molecules by a cell. In addition to detecting fluorescent signals, flow cytometers also measure the forward and side light-scattering properties of cells, which reflect cell size and internal complexity, respectively. This information is often used to distinguish different cell types. For example, compared with lymphocytes, neutrophils cause greater side scatter because of their cytoplasmic granules, and monocytes cause greater forward scatter because of their size.
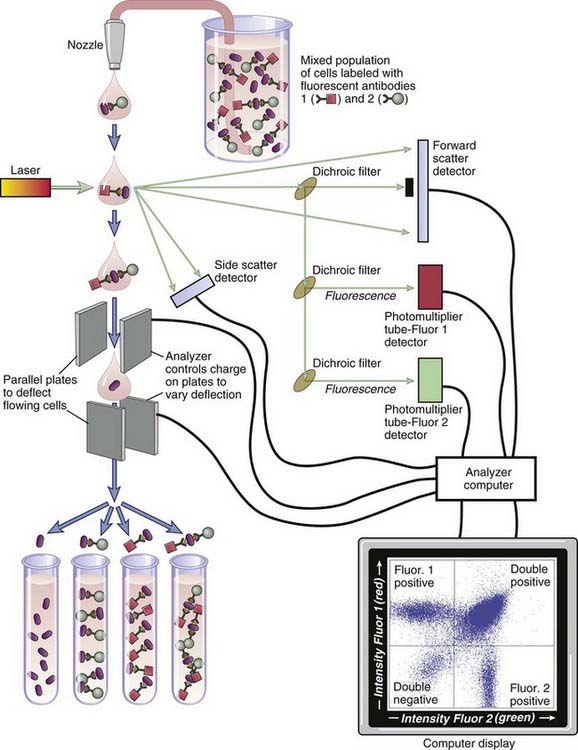
FIGURE A–4 Principle of flow cytometry and fluorescence-activated cell sorting.
The incident laser beam is of a designated wavelength, and the light that emerges from the sample is analyzed for forward and side scatter as well as fluorescent light of two or more wavelengths that depend on the fluorochrome labels attached to the antibodies. The separation depicted here is based on two antigenic markers (two-color sorting). Modern instruments can routinely analyze and separate cell populations on the basis of three or more different-colored probes.
Purification of Cells
A fluorescent-activated cell sorter is an adaptation of the flow cytometer that allows one to separate cell populations according to which and how much fluorescent probe they bind. This technique is accomplished by differentially deflecting the cells with electromagnetic fields whose strength and direction are varied according to the measured intensity of the fluorescent signal (see Fig. A-4). The cells may be labeled with fluorescently tagged antibodies ex vivo, or, in the case of experimental animal studies, labeling may be accomplished in vivo by expression of transgenes that encode fluorescent proteins, such as green fluorescent protein. (Transgenic technology is described later in this appendix.)
Another commonly used technique to purify cells with a particular phenotype relies on antibodies that are attached to magnetic beads. These “immunomagnetic reagents” will bind to certain cells, depending on the specificity of the antibody used, and the bound cells can then be pulled out of suspension by a strong magnet.
Immunofluorescence and Immunohistochemistry
Antibodies can be used to identify the anatomic distribution of an antigen within a tissue or within compartments of a cell. To do so, the tissue or cell is incubated with an antibody that is labeled with a fluorochrome or enzyme, and the position of the label, determined with a suitable microscope, is used to infer the position of the antigen. In the earliest version of this method, called immunofluorescence, the antibody was labeled with a fluorescent dye and allowed to bind to a monolayer of cells or to a frozen section of a tissue. The stained cells or tissues were examined with a fluorescence microscope to locate the antibody. Although sensitive, the fluorescence microscope is not an ideal tool to identify the detailed structures of the cell or tissue because of a low signal-to-noise ratio. This problem has been overcome by new technologies including confocal microscopy, which uses optical sectioning technology to filter out unfocused fluorescent light, and two-photon microscopy, which prevents out-of-focus light from forming. Alternatively, antibodies may be coupled to enzymes that convert colorless substrates to colored insoluble substances that precipitate at the position of the enzyme. A conventional light microscope may then be used to localize the antibody in a stained cell or tissue. The most common variant of this method uses the enzyme horseradish peroxidase, and the method is commonly referred to as the immunoperoxidase technique. Another commonly used enzyme is alkaline phosphatase. Different antibodies coupled to different enzymes may be used in conjunction to produce simultaneous two-color localizations of different antigens. In other variations, antibody can be coupled to an electron-dense probe such as colloidal gold, and the location of antibody can be determined subcellularly by means of an electron microscope, a technique called immunoelectron microscopy. Different-sized gold particles have been used for simultaneous localization of different antigens at the ultrastructural level.
In all immunomicroscopic methods, signals may be enhanced by use of sandwich techniques. For example, instead of attaching horseradish peroxidase to a specific mouse antibody directed against the antigen of interest, it can be attached to a second anti-antibody (e.g., rabbit anti-mouse Ig antibody) that is used to bind to the first, unlabeled antibody. When the label is attached directly to the specific, primary antibody, the method is referred to as direct; when the label is attached to a secondary or even tertiary antibody, the method is indirect. In some cases, molecules other than antibody can be used in indirect methods. For example, staphylococcal protein A, which binds to IgG, or avidin, which binds to primary antibodies labeled with biotin, can be coupled to fluorochromes or enzymes.
Measurement of Antigen-Antibody Interactions
In many situations, it is important to know the affinity of an antibody for an antigen. For example, the usefulness of a monoclonal antibody as an experimental or therapeutic reagent depends on its affinity. Antibody affinities for antigen can be measured directly for small antigens (e.g., haptens) by a method called equilibrium dialysis (Fig. A-5). In this method, a solution of antibody is confined within a “semipermeable” membrane of porous cellulose and immersed in a solution containing the antigen. (Semipermeable in this context means that small molecules, such as antigen, can pass freely through the membrane pores but that macromolecules, such as antibody, cannot.) If no antibody is present within the membrane-bound compartment, the antigen in the bathing solution enters until the concentration of antigen within the membrane-bound compartment becomes exactly the same as that outside. Another way to view the system is that at dynamic equilibrium, antigen enters and leaves the membrane-bound compartment at exactly the same rate. However, when antibody is present inside the membrane, the net amount of antigen inside the membrane at equilibrium increases by the quantity that is bound to antibody. This phenomenon occurs because only unbound antigen can diffuse across the membrane, and at equilibrium, it is the unbound concentration of antigen that must be identical inside and outside the membrane. The extent of the increase in antigen inside the membrane depends on the antigen concentration, on the antibody concentration, and on the dissociation constant (Kd) of the binding interaction. By measurement of the antigen and antibody concentrations, by spectroscopy or by other means, Kd can be calculated.
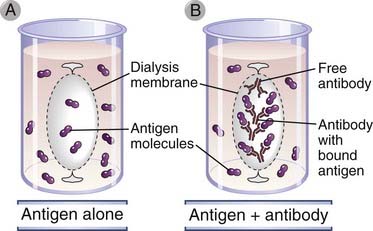
FIGURE A–5 Analysis of antigen-antibody binding by equilibrium dialysis.
In the presence of antibody (B), the amount of antigen within the dialysis membrane is increased compared with the absence of antibody (A). As described in the text, this difference, caused by antibody binding of antigen, can be used to measure the affinity of the antibody for the antigen. This experiment can be performed only when the antigen is a small molecule (e.g., a hapten) capable of freely crossing the dialysis membrane.
An alternative way to determine Kd is by measurement of the rates of antigen-antibody complex formation and dissociation. These rates depend, in part, on the concentrations of antibody and antigen and on the affinity of the interaction. All parameters except the concentrations can be summarized as rate constants, and both the on-rate constant (Kon) and the off-rate constant (Koff) can be calculated experimentally by determining the concentrations and the actual rates of association or dissociation, respectively. The ratio of Koff/Kon allows one to cancel out all the parameters not related to affinity and is exactly equal to the dissociation constant Kd. Thus, one can measure Kd at equilibrium by equilibrium dialysis or calculate Kd from rate constants measured under nonequilibrium conditions.
Another method, more commonly used today, to measure the kinetics of antigen-antibody interactions depends on surface plasmon resonance. In this method, a specialized biosensing instrument (such as the Biacore) uses an optical approach to measure the affinity of an antibody that is passed over an antigen that is immobilized over a metal film. A light source is focused on this film through a prism at a specific angle (resonance), and the reflected light provides a surface plasmon resonance readout. Adsorption of an antibody to the antigen alters the surface plasmon resonance readout, and this alteration can provide information on affinity.
Transgenic Mice and Gene Targeting
Three important and related methods for studying the functional effects of specific gene products in vivo are the creation of conventional transgenic mice that ectopically express a particular gene in a defined tissue; the creation of gene “knockout” mice, in which a targeted disruption is used to ablate the function of a particular gene; and the generation of “knockin” mice, in which an existing gene in the germline is replaced with a modified version of the same. A knockin approach could either replace a normal version of a gene with a mutant version or, in principle, “correct” an existing mutant gene with a “normal” version. These techniques involving genetically engineered mice have been widely used to analyze many biologic phenomena, including the development, activation, and tolerance of lymphocytes.
For the creation of conventional transgenic mice, foreign DNA sequences, called transgenes, are introduced into the pronuclei of fertilized mouse eggs, and the eggs are implanted into the oviducts of pseudopregnant females. Usually, if a few hundred copies of a gene are injected into pronuclei, about 25% of the mice that are born are transgenic. One to 50 copies of the transgene insert in tandem into a random site of breakage in a chromosome and are subsequently inherited as a simple mendelian trait. Because integration usually occurs before DNA replication, most (about 75%) of the transgenic pups carry the transgene in all their cells, including germ cells. In most cases, integration of the foreign DNA does not disrupt endogenous gene function. Also, each founder mouse carrying the transgene is a heterozygote, from which homozygous lines can be bred.
The great value of transgenic technology is that it can be used to express genes in particular tissues by attaching coding sequences of the gene to regulatory sequences that normally drive the expression of genes selectively in that tissue. For instance, lymphoid promoters and enhancers can be used to overexpress genes, such as rearranged antigen receptor genes, in lymphocytes, and the insulin promoter can be used to express genes in the β cells of pancreatic islets. Examples of the utility of these methods for study of the immune system are mentioned in many chapters of this book. Transgenes can also be expressed under the control of promoter elements that respond to drugs or hormones, such as tetracycline or estrogens. In these cases, transcription of the transgene can be controlled at will by administration of the inducing agent.
A powerful method for development of animal models of single-gene disorders, and the most definitive way to establish the obligatory function of a gene in vivo, is the creation of knockout mice by targeted mutation or disruption of the gene. This technique relies on the phenomenon of homologous recombination. If an exogenous gene is inserted into a cell, for instance, by electroporation, it can integrate randomly into the cell’s genome. However, if the gene contains sequences that are homologous to an endogenous gene, it will preferentially recombine with and replace endogenous sequences. To select for cells that have undergone homologous recombination, a drug-based selection strategy is used. The fragment of homologous DNA to be inserted into a cell is placed in a vector typically containing a neomycin resistance gene and a viral thymidine kinase (tk) gene (Fig. A-6A). This targeting vector is constructed in such a way that the neomycin resistance gene is always inserted into the chromosomal DNA, but the tk gene is lost whenever homologous recombination (as opposed to random insertion) occurs. The vector is introduced into cells, and the cells are grown in neomycin and ganciclovir, a drug that is metabolized by thymidine kinase to generate a lethal product. Cells in which the gene is integrated randomly will be resistant to neomycin but will be killed by ganciclovir, whereas cells in which homologous recombination has occurred will be resistant to both drugs because the tk gene will not be incorporated. This positive-negative selection ensures that the inserted gene in surviving cells has undergone homologous recombination with endogenous sequences. The presence of the inserted DNA in the middle of an endogenous gene usually disrupts the coding sequences and ablates expression or function of that gene. In addition, targeting vectors can be designed such that homologous recombination will lead to the deletion of one or more exons of the endogenous gene.
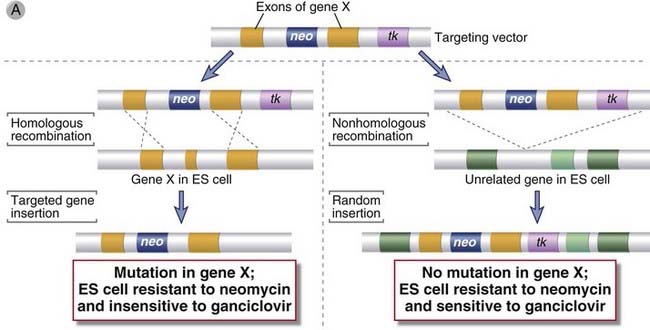
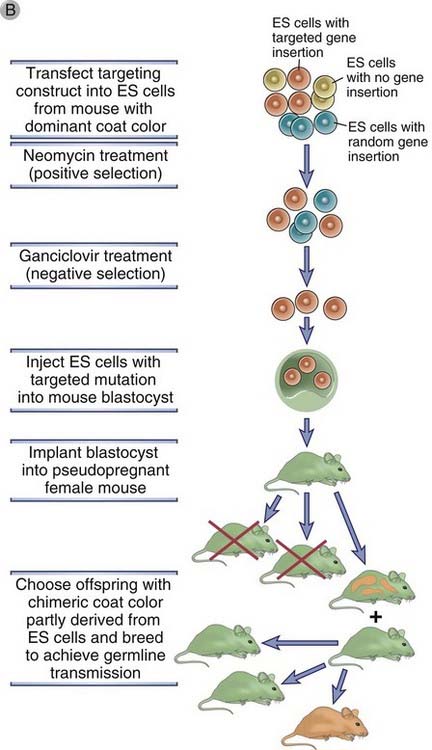
FIGURE A–6 Generation of gene knockout.
A, The disruption of gene X in an embryonic stem (ES) cell is accomplished by homologous recombination. A population of ES cells is transfected with a targeting vector that contains sequences homologous to two exons of gene X flanking a neomycin resistance (neo) gene. The neo gene replaces or disrupts one of the exons of gene X on homologous recombination. The thymidine kinase (tk) gene in the vector will be inserted into the genome only if random, nonhomologous recombination occurs. B, The ES cells that were transfected by the targeting vector are selected by neomycin and ganciclovir so that only those cells with targeted insertion (homologous recombination) survive. These cells are then injected into a blastocyst, which is then implanted into the uterus of a pseudopregnant mouse. A chimeric mouse will develop in which some of the tissues are derived from the ES cell carrying the targeted mutation in gene X. These chimeric mice are identified by a mixed-color coat, including the color of the mouse strain from which the ES cells were derived and the color of the mouse strain from which the blastocyst was derived. If the mutation is present in germ cells, it can be propagated by further breeding.
To generate a mouse carrying a targeted gene disruption or mutation, a targeting vector is used to first disrupt the gene in a murine embryonic stem (ES) cell line. ES cells are pluripotent cells derived from mouse embryos that can be propagated and induced to differentiate in culture or that can be incorporated into a mouse blastocyst, which may be implanted in a pseudopregnant mother and carried to term. Importantly, the progeny of the ES cells develop normally into mature tissues that will express the exogenous genes that have been transfected into the ES cells. Thus, the targeting vector designed to disrupt a particular gene is inserted into ES cells, and colonies in which homologous recombination has occurred (on one chromosome) are selected with drugs, as described before (Fig. A-6B). The presence of the desired recombination is verified by analysis of DNA with techniques such as Southern blot hybridization or polymerase chain reaction. The selected ES cells are injected into blastocysts, which are implanted into pseudopregnant females. Mice that develop will be chimeric for a heterozygous disruption or mutation, that is, some of the tissues will be derived from the ES cells and others from the remainder of the normal blastocyst. The germ cells are also usually chimeric, but because these cells are haploid, only some will contain the chromosome copy with the disrupted (mutated) gene. If chimeric mice are mated with normal (wild-type) animals and either sperm or eggs containing the chromosome with the mutation fuse with the wild-type partner, all cells in the offspring derived from such a zygote will be heterozygous for the mutation (so-called germline transmission). Such heterozygous mice can be mated to yield animals that will be homozygous for the mutation with a frequency that is predictable by simple mendelian segregation. Such knockout mice are deficient in expression of the targeted gene.
Homologous recombination can also be used to replace a normal gene sequence with a modified version of the same gene (or of another gene), thereby creating a knockin mouse strain. Knockin mice can be used to assess the biologic consequences of a change in a single base, for instance, as opposed to the deletion of a gene. A knockin approach could in principle also be used to replace a defective gene with a normal one. In certain circumstances, a different gene may be placed at a defined site in the genome by use of a knockin strategy rather than in a random site as in conventional transgenic mice. Knockin approaches are used when it is desirable to have the expression of the transgene regulated by certain endogenous DNA sequences, such as a particular enhancer or promoter region. In this case, the targeting vector contains an exogenous gene encoding a desired product as well as sequences homologous to an endogenous gene that are needed to target the site of recombination.
Although the conventional gene-targeting strategy has proved to be of great usefulness in immunology research, the approach has some limitations. First, the mutation of one gene during development may be compensated for by altered expression of other gene products, and therefore the function of the targeted gene may be obscured. Second, in a conventional gene knockout mouse, the importance of a gene in only one tissue or at only one time during development cannot be easily assessed. Third, a functional selection marker gene, such as the neomycin resistance gene, is permanently introduced into the animal genome, and this alteration may have unpredictable results on the phenotype of the animal. An important refinement of gene knockout technology that can overcome many of these drawbacks is a “conditional” targeting approach. A commonly used conditional strategy takes advantage of the bacteriophage-derived Cre/loxP recombination system. The Cre enzyme is a DNA recombinase that recognizes a 34-bp sequence motif called loxP, and the enzyme mediates the deletion of gene segments flanked by two loxP sites in the same orientation. To generate mice with loxP-tagged genes, targeting vectors are constructed with one loxP site flanking the neomycin resistance gene at one end and a second loxP site flanking the sequences homologous to the target at the other end. These vectors are transfected into ES cells, and mice carrying the loxP-flanked but still functional target gene are generated as described for conventional knockout mice. A second strain of mice carrying a cre transgene is then bred with the strain carrying the loxP-flanked target gene. In the offspring, expression of Cre recombinase will mediate deletion of the target gene. Both the normal gene sequences and the neomycin resistance gene will be deleted. Importantly, expression of the cre gene, and therefore deletion of the targeted gene, can be restricted to certain tissues or specified times by the use of cre transgene constructs with different promoters. For example, selective deletion of a gene only in helper T cells can be accomplished by using a cre transgenic mouse in which cre is driven by a CD4 promoter. Alternatively, a steroid-inducible promoter can be used so that Cre expression and subsequent gene deletion occur only after mice are given a dose of dexamethasone. Many other variations on this technology have been devised to create conditional mutants. Cre/loxP technology can also be used to create knockin mice. In this case, loxP sites are placed in the targeting vector to flank the neomycin resistance gene and the homologous sequences, but they do not flank the replacement (knockin) gene sequences. Therefore, after cre-mediated deletion, the exogenous gene remains in the genome at the targeted site.
Methods for Studying T Lymphocyte Responses
Our current knowledge of the cellular events in T cell activation is based on a variety of experimental techniques in which different populations of T cells are activated by defined stimuli and functional responses are measured. In vitro experiments have provided a great deal of information on the changes that occur in a T cell when it is stimulated by antigen. More recently, several techniques have been developed to study T cell proliferation, cytokine expression, and anatomic redistribution in response to antigen activation in vivo. The new experimental approaches have been particularly useful for the study of naive T cell activation and the localization of antigen-specific memory T cells after an immune response has waned.
Polyclonal Activation of T Cells
Polyclonal activators of T cells bind to many or all T cell receptor (TCR) complexes regardless of specificity and activate the T cells in ways similar to peptide-MHC complexes on antigen-presenting cells (APCs). Polyclonal activators are mostly used in vitro to activate T cells isolated from human blood or the lymphoid tissues of experimental animals. Polyclonal activators can also be used to activate T cells with unknown antigen specificities, and they can evoke a detectable response from mixed populations of naive T cells, even though the frequency of cells specific for any one antigen would be too low to elicit a detectable response. The polymeric carbohydrate-binding plant proteins called lectins, such as concanavalin-A and phytohemagglutinin, are one commonly used group of polyclonal T cell activator. These lectins bind specifically to certain sugar residues on T cell surface glycoproteins, including the TCR and CD3 proteins, and thereby stimulate the T cells. Antibodies specific for invariant framework epitopes on TCR or CD3 proteins also function as polyclonal activators of T cells. Often, these antibodies need to be immobilized on solid surfaces or beads or cross-linked with secondary anti-antibodies to induce optimal activation responses. Because soluble polyclonal activators do not provide costimulatory signals that are normally provided by APCs, they are often used together with stimulatory antibodies to receptors for costimulators, such as anti-CD28 or anti-CD2. Superantigens, another kind of polyclonal stimulus, bind to and activate all T cells that express particular types of TCR β chain (see Chapter 15, Fig. 15-2). T cells of any antigen specificity can also be stimulated with pharmacologic reagents, such as the combination of the phorbol ester PMA and the calcium ionophore ionomycin, that mimic signals generated by the TCR complex.
Antigen-Induced Activation of Polyclonal T Cell Populations
Polyclonal populations of normal T cells that are enriched for T cells specific for a particular antigen can be derived from the blood and peripheral lymphoid organs of individuals after immunization with the antigen. The immunization serves to expand the number of antigen-specific T cells, which can then be restimulated in vitro by adding antigen and MHC-matched APCs to the T cells. This approach can be used to study antigen-induced activation of a mixed population of previously activated (“primed”) T cells expressing many different TCRs, but the method does not permit analysis of responses of naive T cells.
Antigen-Induced Activation of T Cell Populations with a Single Antigen Specificity
Monoclonal populations of T cells, which express identical TCRs, have been useful for functional, biochemical, and molecular analyses. The limitation of these monoclonal populations is that they are maintained as long-term tissue culture lines and therefore may have phenotypically diverged from normal T cells in vivo. One type of monoclonal T cell population that is frequently used in experimental immunology is an antigen-specific T cell clone. Such clones are derived by isolating T cells from immunized individuals, as described for polyclonal T cells, followed by repetitive in vitro stimulation with the immunizing antigen plus MHC-matched APCs and cloning of single antigen–responsive cells in semisolid media or in liquid media by limiting dilution. Antigen-specific responses can easily be measured in these populations because all the cells in a cloned cell line have the same receptors and have been selected for growth in response to a known antigen-MHC complex. Both helper and cytotoxic T lymphocyte clones have been established from mice and humans. Other monoclonal T cell populations used in the study of T cell activation include antigen-specific T cell hybridomas, which are produced like B cell hybridomas (see Fig. 5-9, Chapter 5), and tumor lines derived from T cells have been established in vitro after removal of malignant T cells from animals or humans with T cell leukemias or lymphomas. Although some tumor-derived lines express functional TCR complexes, their antigen specificities are not known, and the cells are usually stimulated with polyclonal activators for experimental purposes. The Jurkat line, derived from a human T cell leukemia cell, is an example of a tumor line that is widely used as a model to study T cell signal transduction.
TCR transgenic mice are a source of homogeneous, phenotypically normal T cells with identical antigen specificities that are widely used for in vitro and in vivo experimental analyses. If the rearranged α and β chain genes of a single TCR of known specificity are expressed as a transgene in mice, a majority of the mature T cells in the mice will express that TCR. If the TCR transgene is crossed onto a RAG-1– or RAG-2–deficient background, no endogenous TCR gene expression occurs and 100% of the T cells will express only the transgenic TCR. TCR transgenic T cells can be activated in vitro or in vivo with a single peptide antigen, and they can be identified by antibodies specific for the transgenic TCR. One of the unique advantages of TCR transgenic mice is that they permit the isolation of sufficient numbers of naive T cells of defined specificity to allow one to study functional responses to the first exposure to antigen. This advantage has allowed investigators to study the in vitro conditions under which antigen activation of naive T cells leads to differentiation into functional subsets such as TH1 and TH2 cells (see Chapter 9). Naive T cells from TCR transgenic mice can also be injected into normal syngeneic recipient mice, where they home to lymphoid tissues. The recipient mouse is then exposed to the antigen for which the transgenic TCR is specific. By use of antibodies that label the TCR transgenic T cells, it is possible to follow their expansion and differentiation in vivo and to isolate them for analysis of recall (secondary) responses to antigen ex vivo.
Methods to Enumerate and Study Functional Responses of T Cells
Proliferation assays for T lymphocytes, like those of other cells, are conducted in vitro by determining the amount of 3H-labeled thymidine incorporated into the replicating DNA of cultured cells. Thymidine incorporation provides a quantitative measure of the rate of DNA synthesis, which is usually directly proportional to the rate of cell division. Cellular proliferation in vivo can be measured by injecting the thymidine analogue bromodeoxyuridine (BrdU) into animals and staining cells with anti-BrdU antibody to identify and enumerate nuclei that have incorporated BrdU into their DNA during DNA replication.
Fluorescent dyes can be used to study proliferation of T cells in vivo. T cells are first labeled with chemically reactive lipophilic fluorescent esters and then adoptively transferred into experimental animals. The dyes enter cells, form covalent bonds with cytoplasmic proteins, and then cannot leave the cells. One commonly used dye of this type is 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE), which can be detected in cells by standard flow cytometric techniques. Every time a T cell divides, its dye content is halved, and therefore it is possible to determine whether the adoptively transferred T cells present in lymphoid tissues of the recipient mouse have divided in vivo and to estimate the number of doublings each T cell has gone through.
Peptide-MHC tetramers are used to enumerate T cells with a single antigen specificity isolated from blood or lymphoid tissues of experimental animals or humans. These tetramers contain four of the peptide-MHC complexes that the T cell would normally recognize on the surface of APCs. The tetramer is made by producing a class I MHC molecule to which is attached a small molecule called biotin by use of recombinant DNA technology. Biotin binds with high affinity to a protein called avidin, and each avidin molecule binds four biotin molecules. Thus, avidin forms a substrate for assembly of four biotin-conjugated MHC proteins. The MHC molecules can be loaded with a peptide of interest and thus stabilized, and the avidin molecule is labeled with a fluorochrome, such as FITC. This tetramer binds to T cells specific for the peptide-MHC complex with high enough avidity to label the T cells even in suspension. This method is the only feasible approach for identification of antigen-specific T cells in humans. For instance, it is possible to identify and enumerate circulating HLA-A2–restricted T cells specific for an HIV peptide by staining blood cells with a tetramer of HLA-A2 molecules loaded with the peptide. The same technique is being used to enumerate and isolate T cells specific for self antigens in normal individuals and in patients with autoimmune diseases. Peptide-MHC tetramers that bind to a particular transgenic TCR can also be used to quantify the transgenic T cells in different tissues after adoptive transfer and antigen stimulation. The technique is now widely used with class I MHC molecules; in class I molecules, only one polypeptide is polymorphic, and stable molecules can be produced in vitro. This is more difficult for class II molecules because both chains are polymorphic and required for proper assembly, but class II–peptide tetramers are also being produced.
Cytokine secretion assays can be used to quantify cytokine-secreting effector T cells within lymphoid tissues. The most commonly used methods are cytoplasmic staining of cytokines and single-cell enzyme-linked immunosorbent assays (ELISpot). In these types of studies, antigen-induced activation and differentiation of T cells take place in vivo, and then T cells are isolated and tested for cytokine expression in vitro. Cytoplasmic staining of cytokines requires permeabilizing of the cells so that fluorochrome-labeled antibodies specific for a particular cytokine can gain entry into the cell, and the stained cells are analyzed by flow cytometry. Cytokine expression by T cells specific for a particular antigen can be determined by additionally staining T cells with peptide-MHC tetramers or, in the case of TCR transgenic T cells, antibodies specific for the transgenic TCR. By use of a combination of CFSE and anticytokine antibodies, it is possible to examine the relationship between cell division and cytokine expression. In the ELISpot assay, T cells freshly isolated from blood or lymphoid tissues are cultured in plastic wells coated with antibody specific for a particular cytokine. As cytokines are secreted from individual T cells, they bind to the antibodies in discrete spots corresponding to the location of individual T cells. The spots are visualized by adding secondary enzyme-linked anti-Ig, as in a standard ELISA (see earlier), and the number of spots is counted to determine the number of cytokine-secreting T cells.
Methods for Studying B Lymphocyte Responses
Activation of Polyclonal B Cell Populations
It is technically difficult to study the effects of antigens on normal B cells because, as the clonal selection hypothesis predicted, very few lymphocytes in an individual are specific for any one antigen. An approach to circumventing this problem is to use anti-Ig antibodies as analogues of antigens, with the assumption that anti-Ig will bind to constant (C) regions of membrane Ig molecules on all B cells and will have the same biologic effects as an antigen that binds to the hypervariable regions of membrane Ig molecules on only the antigen-specific B cells. To the extent that precise comparisons are feasible, this assumption appears generally correct, indicating that anti-Ig antibody is a valid model for antigens. Thus, anti-Ig antibody is frequently used as a polyclonal activator of B lymphocytes, similar to the use of anti-CD3 antibodies as polyclonal activators of T lymphocytes discussed earlier.
Antigen-Induced Activation of B Cell Populations with a Single Antigen Specificity
To examine the effects of antigen binding to B cells, investigators have attempted to isolate antigen-specific B cells from complex populations of normal lymphocytes or to produce cloned B cell lines with defined antigenic specificities. These efforts have met with little success. However, transgenic mice have been developed in which virtually all B cells express a transgenic Ig of known specificity, so that most of the B cells in these mice respond to the same antigen. A somewhat more sophisticated approach has been to generate antigen receptor knockin mice, in which rearranged Ig H and L chain genes have been homologously recombined into their endogenous loci. Such knockin animals have proved particularly useful in the examination of receptor editing.
Assays to Measure B Cell Proliferation and Antibody Production
Much of our knowledge of B cell activation is based on in vitro experiments, in which different stimuli are used to activate B cells and their proliferation and differentiation can be measured accurately. The same assays may be done with B cells recovered from mice exposed to different antigens or with homogeneous B cells expressing transgene-encoded antigen receptors.
B cell proliferation is measured by use of CFSE labeling or 3H-labeled thymidine incorporation in vitro and BrdU labeling in vivo, as described for T cell proliferation before.
Antibody production is measured in two different ways: with assays for cumulative Ig secretion, which measure the amount of Ig that accumulates in the supernatant of cultured lymphocytes or in the serum of an immunized individual; and with single-cell assays, which determine the number of cells in an immune population that secrete Ig of a particular specificity or isotype. The most accurate, quantitative, and widely used technique to measure the total amount of Ig in a culture supernatant or serum sample is ELISA. By use of antigens bound to solid supports, it is possible to use ELISA to quantify the amount of antibody in a sample specific for a particular antigen. In addition, the availability of anti-Ig antibodies that detect Igs of different heavy or light chain classes allows measurement of the quantities of different isotypes in a sample. Other techniques to measure antibody levels include hemagglutination for anti-erythrocyte antibodies and complement-dependent lysis for antibodies specific for known cell types. Both assays are based on the demonstration that if the amount of antigen (i.e., cells) is constant, the concentration of antibody determines the amount of antibody bound to cells, and this is reflected in the degree of cell agglutination or subsequent binding of complement and cell lysis. Results from these assays are usually expressed as antibody titers, which are the dilution of the sample giving half-maximal effects or the dilution at which the endpoint of the assay is reached.
A single-cell assay for antibody secretion is the ELISpot assay. In this method, antigen is bound to the bottom of a well, antibody-secreting cells are added, and antibodies that have been secreted and are bound to the antigen are detected by an enzyme-linked anti-Ig antibody, as in an ELISA, in a semisolid medium. Each spot represents the location of an antibody-secreting cell. Single-cell assays provide a measure of the numbers of Ig-secreting cells, but they cannot accurately quantify the amount of Ig secreted by each cell or by the total population. The ELISA and ELISpot techniques can be adapted to assess affinity of antibodies, by the use of antigens with differing numbers of hapten moieties. In this way, affinity maturation can be assessed by testing serum or B cells sampled at different times during an immune response.