CHAPTER 15 Immunity to Microbes
In the preceding chapters, we have described the components of the immune system and the generation and functions of immune responses. Throughout, we have referred to protection against infections as the major physiologic function of the immune system, and discussed immune responses in the context of responses to microbes. In this chapter, we will integrate this information and discuss the main features of immunity to different types of pathogenic microorganisms as well as the mechanisms microbes use to resist immune defenses.
The development of an infectious disease in an individual involves complex interactions between the microbe and the host. The key events during infection include entry of the microbe, invasion and colonization of host tissues, evasion of host immunity, and tissue injury or functional impairment. Microbes produce disease by killing host cells or by liberating toxins that can cause tissue damage and functional derangements even without extensive colonization of host tissues. In some infections, the host response is the culprit, being the main cause of tissue injury and disease. Many features of microorganisms determine their virulence, and many diverse mechanisms contribute to the pathogenesis of infectious diseases. The topic of microbial pathogenesis is beyond the scope of this book and will not be discussed here. Rather, our discussion focuses on host immune responses to pathogenic microorganisms.
General Features of Immune Responses to Microbes
Although antimicrobial host defense reactions are numerous and varied, there are several important general features of immunity to microbes.
This chapter considers the main features of immunity to five major categories of pathogenic microorganisms: extracellular bacteria, intracellular bacteria, fungi, viruses, and protozoan and multicellular parasites (Table 15-1). Our discussion of the immune responses to these microbes illustrates the diversity of antimicrobial immunity and the physiologic significance of the effector functions of lymphocytes discussed in earlier chapters.
TABLE 15–1 Examples of Pathogenic Microbes
| Microbe | Examples of Human Diseases | Mechanisms of Pathogenicity |
|---|---|---|
| Extracellular bacteria | ||
| Staphylococcus aureus | Skin and soft tissue infections, lung abscess Systemic: toxic shock syndrome, food poisoning |
Skin infections: acute inflammation induced by toxins; cell death caused by pore-forming toxins Systemic: enterotoxin (“superantigen”)-induced cytokine production by T cells causing skin necrosis, shock, diarrhea |
| Streptococcus pyogenes (group A) | Pharyngitis Skin infections: impetigo, erysipelas; cellulitis Systemic: scarlet fever |
Acute inflammation induced by various toxins, e.g., streptolysin O damages cell membranes |
| Streptococcus pyogenes (pneumococcus) | Pneumonia, meningitis | Acute inflammation induced by cell wall constituents; pneumolysin is similar to streptolysin O |
| Escherichia coli | Urinary tract infections, gastroenteritis, septic shock | Toxins act on intestinal epithelium chloride and water secretion; endotoxin (LPS) stimulates cytokine secretion by macrophages |
| Vibrio cholerae | Diarrhea (cholera) | Cholera toxin ADP ribosylates G protein subunit, which leads to increased cyclic AMP in intestinal epithelial cells and results in chloride secretion and water loss |
| Clostridium tetani | Tetanus | Tetanus toxin binds to the motor end plate at neuromuscular junctions and causes irreversible muscle contraction |
| Neisseria meningitidis (meningococcus) | Meningitis | Acute inflammation and systemic disease caused by potent endotoxin |
| Corynebacterium diphtheriae | Diphtheria | Diphtheria toxin ADP ribosylates elongation factor 2 and inhibits protein synthesis |
| Intracellular Bacteria | ||
| Mycobacteria | Tuberculosis, leprosy | Macrophage activation resulting in granulomatous inflammation and tissue destruction |
| Listeria monocytogenes | Listeriosis | Listeriolysin damages cell membranes |
| Legionella pneumophila | Legionnaires’ disease | Cytotoxin lyses cells and causes lung injury and inflammation |
| Fungi | ||
| Candida albicans | Candidiasis | Unknown; binds complement proteins |
| Aspergillus fumigatus | Aspergillosis | Invasion and thrombosis of blood vessels causing ischemic necrosis and cell injury |
| Histoplasma capsulatum | Histoplasmosis | Lung infection caused by granulomatous inflammation |
| Viruses | ||
| Polio | Poliomyelitis | Inhibits host cell protein synthesis (tropism for motor neurons in the anterior horn of the spinal cord) |
| Influenza | Influenza pneumonia | Inhibits host cell protein synthesis (tropism for peripheral nerves) |
| Rabies | Rabies encephalitis | Inhibits host cell protein synthesis (tropism for ciliated peripheral nerves) |
| Herpes simplex | Various herpes infections (skin, systemic) | Inhibits host cell protein synthesis; functional impairment of immune cells |
| Hepatitis B | Viral hepatitis | Host CTL response to infected hepatocytes |
| Epstein-Barr virus | Infectious mononucleosis; B cell proliferation, lymphomas | Acute infection: cell lysis (tropism for B lymphocytes) Latent infection: stimulates B cell proliferation |
| Human immunodeficiency virus (HIV) | Acquired immunodeficiency syndrome (AIDS) | Multiple: killing of CD4+ T cells, functional impairment of immune cells (see Chapter 20) |
Examples of pathogenic microbes of different classes are listed, with brief summaries of known or postulated mechanisms of tissue injury and disease. Examples of parasites are listed in Table 15-4. ADP, adenosine diphosphate; AMP, adenosine monophosphate; CTL, cytotoxic T lymphocyte; LPS, lipopolysaccharide.
This table was compiled with the assistance of Dr. Arlene Sharpe, Department of Pathology, Harvard Medical School and Brigham and Women’s Hospital, Boston, Massachusetts.
Immunity to Extracellular Bacteria
Extracellular bacteria are capable of replicating outside host cells, for example, in the blood, in connective tissues, and in tissue spaces such as the lumens of the airways and gastrointestinal tract. Many different species of extracellular bacteria are pathogenic, and disease is caused by two principal mechanisms. First, these bacteria induce inflammation, which results in tissue destruction at the site of infection. Second, many of these bacteria produce toxins, which have diverse pathologic effects. The toxins may be endotoxins, which are components of bacterial cell walls, or exotoxins, which are actively secreted by the bacteria. The endotoxin of gram-negative bacteria, also called lipopolysaccharide (LPS), has been mentioned in earlier chapters as a potent activator of macrophages and dendritic cells. Many exotoxins are cytotoxic, and they kill cells by various biochemical mechanisms. Other exotoxins interfere with normal cellular functions without killing cells, and yet other exotoxins stimulate the production of cytokines that cause disease.
Innate Immunity to Extracellular Bacteria
The principal mechanisms of innate immunity to extracellular bacteria are complement activation, phagocytosis, and the inflammatory response.
Adaptive Immunity to Extracellular Bacteria
Humoral immunity is a major protective immune response against extracellular bacteria, and it functions to block infection, to eliminate the microbes, and to neutralize their toxins (Fig. 15-1A). Antibody responses against extracellular bacteria are directed against cell wall antigens and secreted and cell-associated toxins, which may be polysaccharides or proteins. The polysaccharides are prototypic thymus-independent antigens, and humoral immunity is the principal mechanism of defense against polysaccharide-rich encapsulated bacteria. The effector mechanisms used by antibodies to combat these infections include neutralization, opsonization and phagocytosis, and activation of complement by the classical pathway (see Chapter 12). Neutralization is mediated by high-affinity IgG, IgM, and IgA isotypes, the latter mainly in the lumens of mucosal organs; opsonization by some subclasses of IgG; and complement activation by IgM and subclasses of IgG.
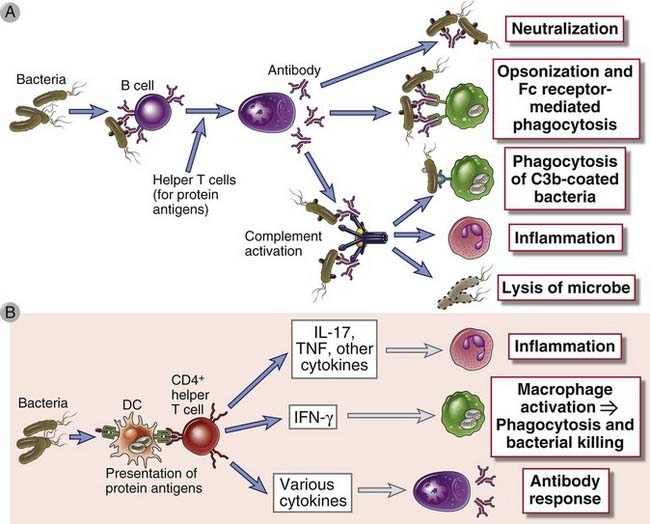
FIGURE 15–1 Adaptive immune responses to extracellular microbes.
Adaptive immune responses to extracellular microbes such as bacteria and their toxins consist of antibody production (A) and the activation of CD4+ helper T cells (B). Antibodies neutralize and eliminate microbes and toxins by several mechanisms. Helper T cells produce cytokines that stimulate inflammation, macrophage activation, and B cell responses. DC, dendritic cell.
The protein antigens of extracellular bacteria also activate CD4+ helper T cells, which produce cytokines that induce local inflammation, enhance the phagocytic and microbicidal activities of macrophages and neutrophils, and stimulate antibody production (Fig. 15-1B). TH17 responses induced by these microbes recruit neutrophils and monocytes and thus promote local inflammation at sites of bacterial infection. Defective TH17 responses are associated with increased susceptibility to bacterial and fungal infections, with formation of multiple skin abscesses (localized infections). One cause of this disorder is mutations affecting the transcription factor STAT3, which is required for the development of TH17 cells. This inherited disease is called Job’s syndrome (because patients develop skin abscesses, thought to resemble the pestilence visited on Job in the biblical story) or hyper-IgE syndrome (because patients have increased levels of serum IgE, for unknown reasons). Bacteria also induce TH1 responses, and interferon-γ (IFN-γ) produced by TH1 cells activates macrophages to destroy phagocytosed microbes and may stimulate production of opsonizing and complement-binding antibody isotypes.
Injurious Effects of Immune Responses
The principal injurious consequences of host responses to extracellular bacteria are inflammation and septic shock. The same reactions of neutrophils and macrophages that function to eradicate the infection also cause tissue damage by local production of reactive oxygen species and lysosomal enzymes. These inflammatory reactions are usually self-limited and controlled. Cytokines secreted by leukocytes in response to bacterial products also stimulate the production of acute-phase proteins and cause the systemic manifestations of the infection (see Chapter 4). Septic shock is a severe pathologic consequence of disseminated infection by some gram-negative and gram-positive bacteria. It is a syndrome characterized by circulatory collapse and disseminated intravascular coagulation. The early phase of septic shock is caused by cytokines produced by macrophages that are activated by microbial components, including LPS and bacterial peptidoglycans. Tumor necrosis factor (TNF), IL-6, and IL-1 are the principal cytokine mediators of septic shock, but IFN-γ and interleukin-12 (IL-12) may also contribute (see Chapter 4). This early burst of large amounts of cytokines is sometimes called a cytokine storm. There is some evidence that the progression of septic shock is associated with defective immune responses, perhaps related to depletion or suppression of T cells, resulting in unchecked microbial spread.
Certain bacterial toxins stimulate all the T cells in an individual that express a particular family of Vβ T cell receptor (TCR) genes. Such toxins are called superantigens because they resemble antigens in that they bind to TCRs and to class II MHC molecules (although not to the peptide-binding clefts) but activate many more T cells than do conventional peptide antigens (Fig. 15-2). Their importance lies in their ability to activate many T cells, with the subsequent production of large amounts of cytokines that can also cause a systemic inflammatory syndrome.
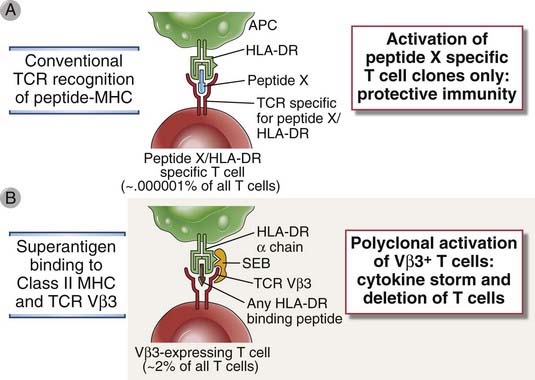
FIGURE 15–2 Polyclonal activation of T cells by bacterial superantigens.
A, Conventional microbial T cell antigens, composed of a peptide bound to the peptide-binding groove of an MHC molecule, are recognized by a very small fraction of T cells in any one individual, and only these T cells are activated to become effector T cells that protect against the microbe. B, In contrast, a superantigen binds to class II MHC molecules outside the peptide-binding groove and simultaneously binds to the variable region of any TCR β chain, as long as it belongs to a particular Vβ family, regardless of the peptide-MHC specificity of the TCR. In this way, superantigens activate T cells to secrete cytokines and also induce apoptosis of T cells. Different superantigens bind to TCRs of different Vβ families. Because thousands of clones of T cells will express a TCR β chain from a particular Vβ family, superantigens can induce massive cytokine release (cytokine storm) and cause deletion of many T cells. In the example shown, staphylococcal enterotoxin B (SEB) is the superantigen, which binds mainly to HLA-DR and the Vβ segments of TCRs belonging to the Vβ3 family. APC, antigen-presenting cell.
A late complication of the humoral immune response to bacterial infection may be the generation of disease-producing antibodies. The best defined examples are two rare sequelae of streptococcal infections of the throat or skin that are manifested weeks or even months after the infections are controlled. Rheumatic fever is a sequel to pharyngeal infection with some serologic types of β-hemolytic streptococci. Infection leads to the production of antibodies against a bacterial cell wall protein (M protein). Some of these antibodies cross-react with myocardial proteins and are deposited in the heart and subsequently cause inflammation (carditis). Poststreptococcal glomerulonephritis is a sequel to infection of the skin or throat with other serotypes of β-hemolytic streptococci. Antibodies produced against these bacteria form complexes with bacterial antigen, which may be deposited in kidney glomeruli and cause nephritis.
Immune Evasion by Extracellular Bacteria
The virulence of extracellular bacteria has been linked to a number of mechanisms that resist innate immunity (Table 15-2), including antiphagocytic mechanisms and inhibition of complement or inactivation of complement products. Bacteria with polysaccharide-rich capsules resist phagocytosis and are therefore much more virulent than homologous strains lacking a capsule. The capsules of many pathogenic gram-positive and gram-negative bacteria contain sialic acid residues that inhibit complement activation by the alternative pathway.
TABLE 15–2 Mechanisms of Immune Evasion by Bacteria
| Mechanism of Immune Evasion | Examples |
|---|---|
| Extracellular bacteria | |
| Antigenic variation | Neisseria gonorrhoeae, Escherichia coli, Salmonella typhimurium |
| Inhibition of complement activation | Many bacteria |
| Resistance to phagocytosis | Pneumococcus |
| Scavenging of reactive oxygen species | Catalase-positive staphylococci |
| Intracellular bacteria | |
| Inhibition of phagolysosome formation | Mycobacterium tuberculosis, Legionella pneumophila |
| Inactivation of reactive oxygen and nitrogen species | Mycobacterium leprae (phenolic glycolipid) |
| Disruption of phagosome membrane, escape into cytoplasm | Listeria monocytogenes (hemolysin protein) |
A mechanism used by bacteria to evade humoral immunity is genetic variation of surface antigens. Some surface antigens of bacteria such as gonococci and Escherichia coli are contained in their pili, which are the structures responsible for bacterial adhesion to host cells. The major antigen of the pili is a protein called pilin. The pilin genes of gonococci undergo extensive gene conversion, because of which the progeny of one organism can produce up to 106 antigenically distinct pilin molecules. This ability to alter antigens helps the bacteria evade attack by pilin-specific antibodies, although its principal significance for the bacteria may be to select for pili that are more adherent to host cells so that the bacteria are more virulent. In other bacteria, such as Haemophilus influenzae, changes in the production of glycosidases lead to chemical alterations in surface LPS and other polysaccharides, which enable the bacteria to evade humoral immune responses against these antigens.
Immunity to Intracellular Bacteria
A characteristic of facultative intracellular bacteria is their ability to survive and even to replicate within phagocytes. Because these microbes are able to find a niche where they are inaccessible to circulating antibodies, their elimination requires the mechanisms of cell-mediated immunity (Fig. 15-3). As we shall discuss later in this section, in many intracellular bacterial infections the host response also causes tissue injury.
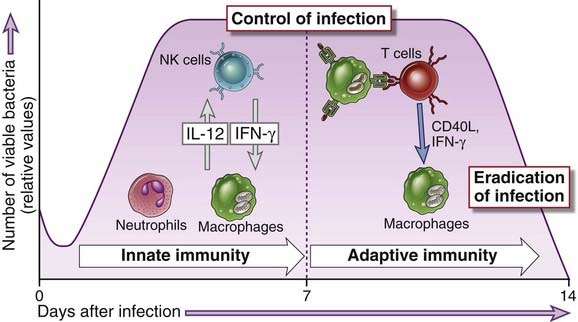
FIGURE 15–3 Innate and adaptive immunity to intracellular bacteria.
The innate immune response to intracellular bacteria consists of phagocytes and NK cells, interactions among which are mediated by cytokines (IL-12 and IFN-γ). The typical adaptive immune response to these microbes is cell-mediated immunity, in which T cells activate phagocytes to eliminate the microbes. Innate immunity may control bacterial growth, but elimination of the bacteria requires adaptive immunity. These principles are based largely on analysis of Listeria monocytogenes infection in mice; the numbers of viable bacteria shown on the y-axis are relative values of bacterial colonies that can be grown from the tissues of infected mice.
(Data from Unanue ER. Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunological Reviews 158:11-25, 1997.)
Innate Immunity to Intracellular Bacteria
The innate immune response to intracellular bacteria is mediated mainly by phagocytes and natural killer (NK) cells. Phagocytes, initially neutrophils and later macrophages, ingest and attempt to destroy these microbes, but pathogenic intracellular bacteria are resistant to degradation within phagocytes. Products of these bacteria are recognized by TLRs and cytoplasmic proteins of the NOD-like receptor (NLR) family, resulting in activation of the phagocytes (see Chapter 4). Intracellular bacteria activate NK cells by inducing expression of NK cell–activating ligands on infected cells and by stimulating dendritic cell and macrophage production of IL-12 and IL-15, both of which are NK cell–activating cytokines. The NK cells produce IFN-γ, which in turn activates macrophages and promotes killing of the phagocytosed bacteria. Thus, NK cells provide an early defense against these microbes, before the development of adaptive immunity. In fact, mice with severe combined immunodeficiency, which lack T and B cells, are able to transiently control infection with the intracellular bacterium Listeria monocytogenes by NK cell–derived IFN-γ production. However, innate immunity usually fails to eradicate these infections, and eradication requires adaptive cell-mediated immunity.
Adaptive Immunity to Intracellular Bacteria
The major protective immune response against intracellular bacteria is T cell–mediated immunity. Individuals with deficient cell-mediated immunity, such as patients with acquired immunodeficiency syndrome (AIDS), are extremely susceptible to infections with intracellular bacteria (and viruses). The mechanisms of cell-mediated immunity were studied in the 1950s in mice, in examining protection against the intracellular bacterium L. monocytogenes. This form of immunity could be adoptively transferred to naive animals with lymphoid cells but not with serum from infected or immunized animals (see Chapter 10, Fig. 10-6).
As we discussed in Chapter 10, cell-mediated immunity consists of two types of reactions: CD4+ T cells recruit phagocytes and activate them through the actions of CD40 ligand and IFN-γ, resulting in killing of phagocytosed microbes, and CD8+ cytotoxic T lymphocytes (CTLs) kill infected cells. Both CD4+ T cells and CD8+ T cells respond to protein antigens of phagocytosed microbes, which are displayed as peptides associated with class II and class I major histocompatibility complex (MHC) molecules, respectively. CD4+ T cells differentiate into TH1 effectors under the influence of IL-12, which is produced by macrophages and dendritic cells. The T cells express CD40 ligand and secrete IFN-γ, and these two stimuli activate macrophages to produce several microbicidal substances, including reactive oxygen species, nitric oxide, and lysosomal enzymes. IFN-γ also stimulates the production of antibody isotypes (e.g., IgG2a in mice) that activate complement and opsonize bacteria for phagocytosis, thus aiding the effector functions of macrophages. The stimuli for the production of these antibodies in humans are not as well defined. The importance of IL-12 and IFN-γ in immunity to intracellular bacteria has been demonstrated in experimental models and in congenital immunodeficiencies. For instance, individuals with inherited mutations in receptors for IFN-γ or IL-12 are highly susceptible to infections with atypical mycobacteria.
Phagocytosed bacteria stimulate CD8+ T cell responses if bacterial antigens are transported from phagosomes into the cytosol or if the bacteria escape from phagosomes and enter the cytoplasm of infected cells. In the cytoplasm, the microbes are no longer susceptible to the microbicidal mechanisms of phagocytes, and for eradication of the infection, the infected cells have to be killed by CTLs. Thus, the effectors of cell-mediated immunity, namely, CD4+ T cells that activate macrophages and CD8+ CTLs, function cooperatively in defense against intracellular bacteria (Fig. 15-4).
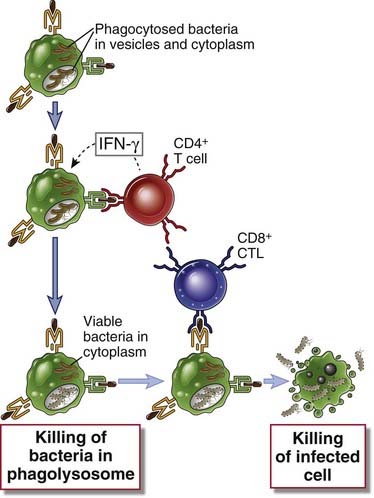
FIGURE 15–4 Cooperation of CD4+ and CD8+ T cells in defense against intracellular microbes.
Intracellular bacteria such as L. monocytogenes are phagocytosed by macrophages and may survive in phagosomes and escape into the cytoplasm. CD4+ T cells respond to class II MHC–associated peptide antigens derived from the intravesicular bacteria. These T cells produce IFN-γ, which activates macrophages to destroy the microbes in phagosomes. CD8+ T cells respond to class I–associated peptides derived from cytosolic antigens and kill the infected cells.
The macrophage activation that occurs in response to intracellular microbes is capable of causing tissue injury. This injury may be the result of delayed-type hypersensitivity (DTH) reactions to microbial protein antigens (see Chapter 18). Because intracellular bacteria have evolved to resist killing within phagocytes, they often persist for long periods and cause chronic antigenic stimulation and T cell and macrophage activation, which may result in the formation of granulomas surrounding the microbes (see Chapter 18, Fig. 18-8). The histologic hallmark of infection with some intracellular bacteria is granulomatous inflammation. This type of inflammatory reaction may serve to localize and prevent spread of the microbes, but it is also associated with severe functional impairment caused by tissue necrosis and fibrosis.
Tuberculosis is an example of an infection with an intracellular bacterium in which protective immunity and pathologic hypersensitivity coexist, and the host response contributes significantly to the pathology. In a primary infection with M. tuberculosis, bacilli multiply slowly in the lungs and cause only mild inflammation. The infection is contained by alveolar macrophages (and probably dendritic cells). More than 90% of infected patients remain asymptomatic, but bacteria survive in the lungs, mainly in macrophages. By 6 to 8 weeks after infection, the macrophages have traveled to the draining lymph nodes, and CD4+ T cells are activated; CD8+ T cells may also be activated later. These T cells produce IFN-γ, which activates macrophages and enhances their ability to kill phagocytosed bacilli. TNF produced by T cells and macrophages also plays a role in local inflammation and macrophage activation. The T cell reaction is adequate to control bacterial spread. However, M. tuberculosis is capable of surviving within macrophages because components of its cell wall inhibit the fusion of phagocytic vacuoles with lysosomes. Continuing T cell activation leads to the formation of granulomas, which attempt to wall off the bacteria and are often associated with central necrosis, called caseous necrosis, which is caused by macrophage products such as lysosomal enzymes and reactive oxygen species. Necrotizing granulomas and the fibrosis (scarring) that accompanies granulomatous inflammation are the principal causes of tissue injury and clinical disease in tuberculosis. Previously infected persons show cutaneous DTH reactions to skin challenge with a bacterial antigen preparation (purified protein derivative, or PPD). Bacilli may survive for many years and are contained without any pathologic consequences but may be reactivated at any time, especially if the immune response becomes unable to control the infection.
Differences among individuals in the patterns of T cell responses to intracellular microbes are important determinants of disease progression and clinical outcome (Fig. 15-5). An example of this relationship between the type of T cell response and disease outcome is leprosy, which is caused by Mycobacterium leprae. There are two polar forms of leprosy, the lepromatous and tuberculoid forms, although many patients fall into less clear intermediate groups. In lepromatous leprosy, patients have high specific antibody titers but weak cell-mediated responses to M. leprae antigens. Mycobacteria proliferate within macrophages and are detectable in large numbers. The bacterial growth and persistent but inadequate macrophage activation result in destructive lesions in the skin and underlying tissue. In contrast, patients with tuberculoid leprosy have strong cell-mediated immunity but low antibody levels. This pattern of immunity is reflected in granulomas that form around nerves and produce peripheral sensory nerve defects and secondary traumatic skin lesions but less with tissue destruction and a paucity of bacteria in the lesions. One possible reason for the differences in these two forms of disease caused by the same organism may be that there are different patterns of T cell differentiation and cytokine production in individuals. Some studies indicate that patients with the tuberculoid form of the disease produce IFN-γ and IL-2 in lesions (indicative of TH1 cell activation), whereas patients with lepromatous leprosy produce less IFN-γ and perhaps more IL-4 and IL-10 (suggestive of TH2 cells). In lepromatous leprosy, both the deficiency of IFN-γ and the macrophage-suppressive effects of IL-10 and possibly IL-4 may result in weak cell-mediated immunity and failure to control bacterial spread. The role of TH1- and TH2-derived cytokines in determining the outcome of infection has been most clearly demonstrated in infection by the protozoan parasite Leishmania major in different strains of inbred mice (discussed later in this chapter).
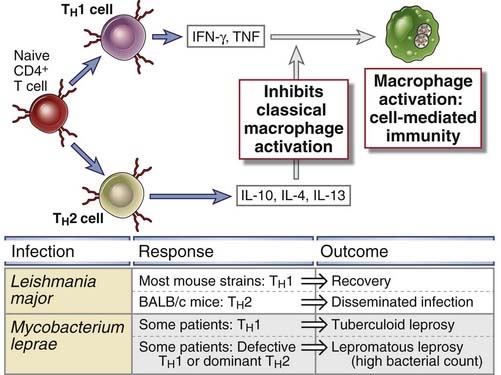
FIGURE 15–5 Role of T cells and cytokines in determining the outcome of infections.
Naive CD4+ T lymphocytes may differentiate into TH1 cells, which activate phagocytes to kill ingested microbes, and TH2 cells, which inhibit this classical pathway of macrophage activation. The balance between these two T cell subsets may influence the outcome of infections, as illustrated by Leishmania infection in mice and mycobacterium leprae in humans.
Immune Evasion by Intracellular Bacteria
Different intracellular bacteria have developed various strategies to resist elimination by phagocytes (see Table 15-2). These include inhibiting phagolysosome fusion or escaping into the cytosol, thus hiding from the microbicidal mechanisms of lysosomes, and directly scavenging or inactivating microbicidal substances such as reactive oxygen species. The outcome of infection by these organisms often depends on whether the T cell–stimulated antimicrobial mechanisms of macrophages or microbial resistance to killing gains the upper hand. Resistance to phagocyte-mediated elimination is also the reason that such bacteria tend to cause chronic infections that may last for years, often recur after apparent cure, and are difficult to eradicate.
Immunity to Fungi
Fungal infections, also called mycoses, are important causes of morbidity and mortality in humans. Some fungal infections are endemic, and these infections are usually caused by fungi that are present in the environment and whose spores enter humans. Other fungal infections are said to be opportunistic because the causative agents cause mild or no disease in healthy individuals but may infect and cause severe disease in immunodeficient persons. Compromised immunity is the most important predisposing factor for clinically significant fungal infections. Neutrophil deficiency as a result of bone marrow suppression or damage is frequently associated with such infections. A recent increase has been noted in opportunistic fungal infections secondary to an increase in immunodeficiency disease caused mainly by HIV and by therapy for disseminated cancer and transplant rejection. A serious opportunistic fungal infection associated with AIDS is Pneumocystis jiroveci pneumonia, but many others contribute to the morbidity and mortality caused by immune deficiencies.
Different fungi infect humans and may live in extracellular tissues and within phagocytes. Therefore, the immune responses to these microbes are often combinations of the responses to extracellular and intracellular bacteria. However, less is known about antifungal immunity than about immunity against bacteria and viruses. This lack of knowledge is partly due to the paucity of animal models for mycoses and partly due to the fact that these infections typically occur in individuals who are incapable of mounting effective immune responses.
Innate and Adaptive Immunity to Fungi
The principal mediators of innate immunity against fungi are neutrophils and macrophages. Patients with neutropenia are extremely susceptible to opportunistic fungal infections. Phagocytes and dendritic cells sense fungal organisms by TLRs and lectin-like receptors called dectins (see Chapter 4). Neutrophils presumably liberate fungicidal substances, such as reactive oxygen species and lysosomal enzymes, and phagocytose fungi for intracellular killing. Virulent strains of Cryptococcus neoformans inhibit the production of cytokines such as TNF and IL-12 by macrophages and stimulate production of IL-10, thus inhibiting macrophage activation.
Cell-mediated immunity is the major mechanism of adaptive immunity against fungal infections. Histoplasma capsulatum, a facultative intracellular parasite that lives in macrophages, is eliminated by the same cellular mechanisms that are effective against intracellular bacteria. CD4+ and CD8+ T cells cooperate to eliminate the yeast forms of C. neoformans, which tend to colonize the lungs and brain in immunodeficient hosts. Many extracellular fungi elicit strong TH17 responses, which are driven in part by the activation of dendritic cells by fungal glucans binding to dectin-1, a receptor for this fungal polysaccharide, and this results in the production of TH17-inducing cytokines (IL-6, IL-23) from the dendritic cells (see Chapter 9). The TH17 cells stimulate inflammation, and the recruited neutrophils and monocytes destroy the fungi. Candida infections often start at mucosal surfaces, and cell-mediated immunity is believed to prevent spread of the fungi into tissues. TH1 responses are protective in intracellular fungal infections, such as histoplasmosis, but these responses may elicit granulomatous inflammation, which is an important cause of host tissue injury in these infections (see Chapter 18). Fungi also elicit specific antibody responses that are of protective value.
Immunity to Viruses
Viruses are obligatory intracellular microorganisms that live inside cells, using components of the nucleic acid and protein synthetic machinery of the host to replicate and spread. Viruses typically infect various cell types by using normal cell surface molecules as receptors to enter the cells. After entering cells, viruses can cause tissue injury and disease by any of several mechanisms. Viral replication interferes with normal cellular protein synthesis and function and leads to injury and ultimately death of the infected cell. This result is one type of cytopathic effect of viruses, and the infection is said to be lytic because the infected cell is lysed. Viruses may also cause latent infections, discussed later.
Innate and adaptive immune responses to viruses are aimed at blocking infection and eliminating infected cells (Fig. 15-6). Infection is prevented by type I interferons as part of innate immunity and neutralizing antibodies contributing to adaptive immunity. Once infection is established, infected cells are eliminated by NK cells in the innate response and CTLs in the adaptive response.
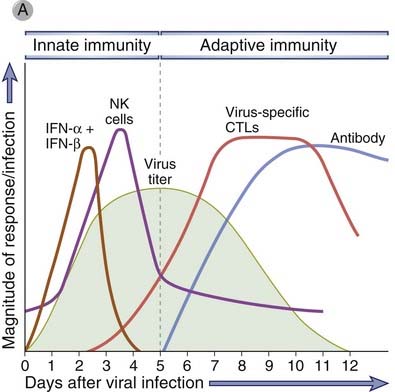
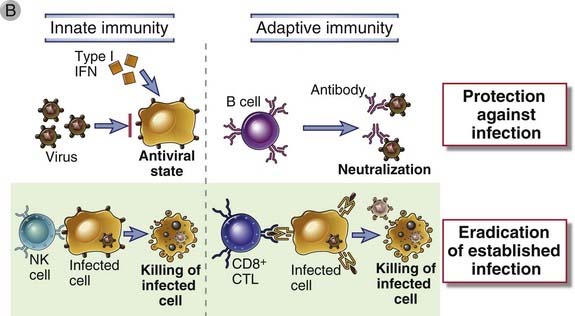
FIGURE 15–6 Innate and adaptive immune responses against viruses.
A, Kinetics of innate and adaptive immune responses to a virus infection. B, Mechanisms by which innate and adaptive immunity prevent and eradicate virus infections. Innate immunity is mediated by type I interferons, which prevent infection, and NK cells, which eliminate infected cells. Adaptive immunity is mediated by antibodies and CTLs, which also block infection and kill infected cells, respectively.
Innate Immunity to Viruses
The principal mechanisms of innate immunity against viruses are inhibition of infection by type I interferons and NK cell–mediated killing of infected cells. Infection by many viruses is associated with production of type I interferons by infected cells, especially dendritic cells of the plasmacytoid type (see Chapter 4). Several biochemical pathways trigger interferon production (Fig. 15-7). These include recognition of viral RNA and DNA by endosomal TLRs and activation of cytoplasmic RIG-like receptors by viral RNA. These pathways converge on the activation of protein kinases, which in turn activate the IRF transcription factors that stimulate interferon gene transcription. Type I interferons function to inhibit viral replication in both infected and uninfected cells by inducing an “antiviral state.” The mechanisms by which interferons block viral replication were discussed in Chapter 4 (see Fig. 4-15).
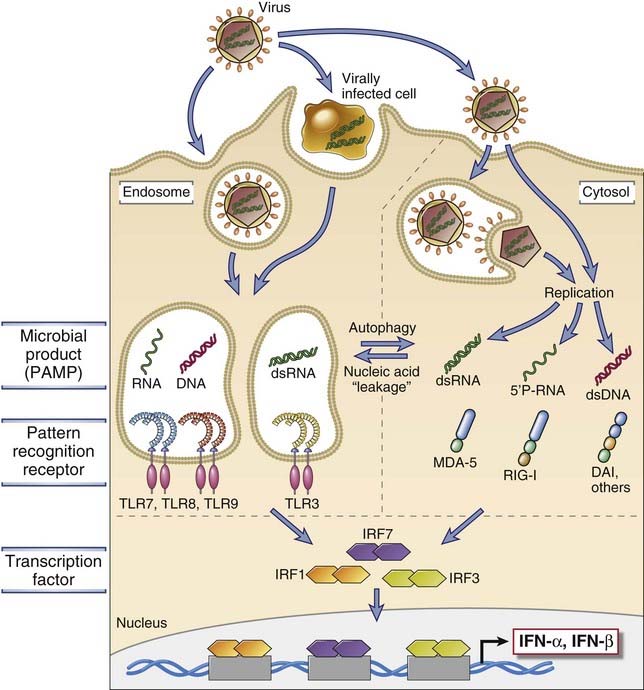
FIGURE 15–7 Mechanisms of induction of type I interferons by viruses.
Viral nucleic acids and proteins are recognized by several cellular receptor families (TLRs and the family of cytosolic RIG-like receptors, or RLRs, which include MDA-5, RIG-I, DAI and others), which activate transcription factors (the IRF proteins) that stimulate the production of type I interferons, IFN-α and IFN-β. This process and the actions of interferons are described in more detail in Chapter 4.
NK cells kill cells infected with a variety of viruses and are an important mechanism of immunity against viruses early in the course of infection, before adaptive immune responses have developed. NK cells also recognize infected cells in which the virus has shut off class I MHC expression as an escape mechanism from CTLs because the absence of class I releases NK cells from a normal state of inhibition (see Fig. 4-6, Chapter 4).
Adaptive Immunity to Viruses
Adaptive immunity against viral infections is mediated by antibodies, which block virus binding and entry into host cells, and by CTLs, which eliminate the infection by killing infected cells (see Fig. 15-6). The most effective antibodies are high-affinity antibodies produced in T-dependent germinal center reactions (see Chapter 11). Antibodies are effective against viruses only during the extracellular stage of the lives of these microbes. Viruses may be extracellular early in the course of infection, before they infect host cells, or when they are released from infected cells by virus budding or if the cells are killed. Antiviral antibodies bind to viral envelope or capsid antigens and function mainly as neutralizing antibodies to prevent virus attachment and entry into host cells. Thus, antibodies prevent both initial infection and cell-to-cell spread. Secreted antibodies of the IgA isotype are important for neutralizing viruses within the respiratory and intestinal tracts. Oral immunization against poliomyelitis works by inducing mucosal immunity. In addition to neutralization, antibodies may opsonize viral particles and promote their clearance by phagocytes. Complement activation may also participate in antibody-mediated viral immunity, mainly by promoting phagocytosis and possibly by direct lysis of viruses with lipid envelopes.
The importance of humoral immunity in defense against viral infections is supported by the observation that resistance to a particular virus, induced by either infection or vaccination, is often specific for the serologic (antibody-defined) type of the virus. An example is influenza virus, in which exposure to one serologic type does not confer resistance to other serotypes of the virus. Neutralizing antibodies block viral infection of cells and spread of viruses from cell to cell, but once the viruses enter cells and begin to replicate intracellularly, they are inaccessible to antibodies. Therefore, humoral immunity induced by previous infection or vaccination is able to protect individuals from viral infection but cannot by itself eradicate established infection.
Elimination of viruses that reside within cells is mediated by CTLs, which kill the infected cells. As we have mentioned in previous chapters, the principal physiologic function of CTLs is surveillance against viral infection. Most virus-specific CTLs are CD8+ T cells that recognize cytosolic, usually endogenously synthesized, viral peptides presented by class I MHC molecules. If the infected cell is a tissue cell and not a professional antigen-presenting cell (APC), such as a dendritic cell, the infected cell may be phagocytosed by the dendritic cell, which processes the viral antigens and presents them to naive CD8+ T cells. This process of cross-presentation, or cross-priming, was described in Chapter 6 (see Fig. 6-20). Full differentiation of CD8+ CTLs often requires cytokines produced by CD4+ helper cells or costimulators expressed on infected cells (see Chapter 9). As discussed in Chapter 9, CD8+ T cells undergo massive proliferation during viral infection, and most of the proliferating cells are specific for a few viral peptides. Some of the activated T cells differentiate into effector CTLs, which can kill any infected nucleated cell. The antiviral effects of CTLs are mainly due to killing of infected cells, but other mechanisms include activation of nucleases within infected cells that degrade viral genomes and secretion of cytokines such as IFN-γ, which activates phagocytes and may have some antiviral activity.
The importance of CTLs in defense against viral infection is demonstrated by the increased susceptibility to such infections seen in patients and animals deficient in T lymphocytes and by the experimental observation that mice can be protected against some virus infections by adoptive transfer of virus-specific, class I–restricted CTLs. Furthermore, many viruses are able to alter their surface antigens, such as envelope glycoproteins, and thus escape attack by antibodies. However, infected cells may produce some viral proteins that are invariant, so that CTL-mediated defense remains effective against such viruses.
In latent infections, viral DNA persists in host cells but the virus does not replicate or kill infected cells. Latency is often a state of balance between infection and the immune response. CTLs are generated in response to the virus that can control the infection but not eradicate it. As a result, the virus persists in infected cells, sometimes for the life of the individual. Any deficiency in the host immune response can result in reactivation of the latent infection, with expression of viral genes that are responsible for cytopathic effects and for spread of the virus. These cytopathic effects may include lysis of infected cells or uncontrolled proliferation of the cells. Such latent infections are common with Epstein-Barr virus and several other DNA viruses of the herpesvirus family.
In some viral infections, tissue injury may be caused by CTLs. An experimental model of a disease in which the pathology is due to the host immune response is lymphocytic choriomeningitis virus (LCMV) infection in mice, which induces inflammation of the spinal cord meninges. LCMV infects meningeal cells, but it is noncytopathic and does not injure the infected cells directly. The virus stimulates the development of virus-specific CTLs that kill infected meningeal cells during a physiologic attempt to eradicate the infection. Therefore, meningitis develops in normal mice with intact immune systems, but T cell-deficient mice do not develop disease and instead become carriers of the virus. This observation appears to contradict the usual situation, in which immunodeficient individuals are more susceptible to infectious diseases than normal individuals are. Hepatitis B virus infection in humans shows some similarities to murine LCMV in that immunodeficient persons who become infected do not develop the disease but become carriers who can transmit the infection to otherwise healthy persons. The livers of patients with acute and chronic active hepatitis contain large numbers of CD8+ T cells, and hepatitis virus–specific, class I MHC–restricted CTLs can be isolated from liver biopsy specimens and propagated in vitro.
Immune responses to viral infections may be involved in producing disease in other ways. A consequence of persistent infection with some viruses, such as hepatitis B, is the formation of circulating immune complexes composed of viral antigens and specific antibodies (see Chapter 18). These complexes are deposited in blood vessels and lead to systemic vasculitis. Some viral proteins contain amino acid sequences that are also present in some self antigens. It has been postulated that because of this “molecular mimicry,” antiviral immunity can lead to immune responses against self antigens.
Immune Evasion by Viruses
Viruses have evolved numerous mechanisms for evading host immunity (Table 15-3).
TABLE 15–3 Mechanisms of Immune Evasion by Viruses
| Mechanism of Immune Evasion | Examples |
|---|---|
| Antigenic variation | Influenza, rhinovirus, HIV |
| Inhibition of antigen processing | |
| Blockade of TAP transporter | Herpes simplex |
| Removal of class I molecules from the ER | Cytomegalovirus |
| Production of cytokine receptor homologues | Vaccinia, poxviruses (IL-1, IFN-γ) Cytomegalovirus (chemokine) |
| Production of immunosuppressive cytokine | Epstein-Barr (IL-10) |
| Infection and death or functional impairment of immune cells | HIV |
Representative examples of different mechanisms used by viruses to resist host immunity are listed. ER, endoplasmic reticulum; HIV, human immunodeficiency virus; TAP, transporter associated with antigen processing.
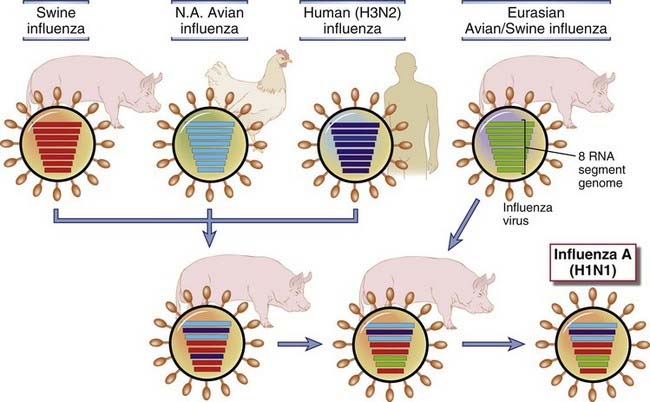
FIGURE 15–8 Generation of new influenza virus strains by genetic recombination (antigenic shift).
The genome of the influenza virus is composed of eight separate RNA strands, which allows genetic recombination by reassortment of the segments in various hosts, such as a pig, bird, or humans, that are simultaneously infected with two different strains. These genetic reassortments create new viruses that are antigenically distinct from their precursors and thus are able to evade immune detection in large numbers of newly infected hosts. In the example shown, H1N1 influenza virus, which was responsible for the pandemic of 2009, was generated by reassortment of swine, avian, and human viruses in pigs and then passed back to humans.
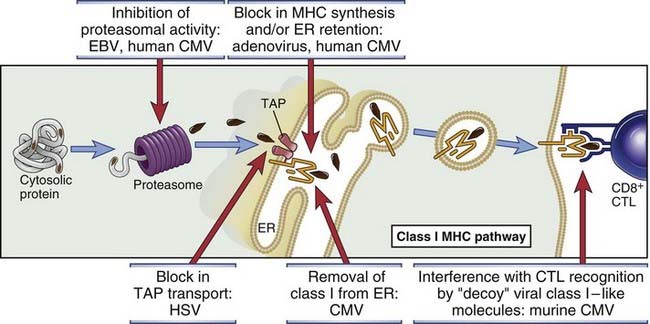
FIGURE 15–9 Mechanisms by which viruses inhibit antigen processing and presentation.
The pathway of class I MHC–associated antigen presentation is shown, with examples of viruses that block different steps in this pathway. CMV, cytomegalovirus; EBV, Epstein-Barr virus; ER, endoplasmic reticulum; HSV, herpes simplex virus; TAP, transporter associated with antigen processing.
Immunity to Parasites
In infectious disease terminology, parasitic infection refers to infection with animal parasites such as protozoa, helminths, and ectoparasites (e.g., ticks and mites). Such parasites currently account for greater morbidity and mortality than any other class of infectious organisms, particularly in developing countries. It is estimated that about 30% of the world’s population suffers from parasitic infestations. Malaria alone affects more than 100 million people worldwide and is responsible for 1 to 2 million deaths annually. The magnitude of this public health problem is the principal reason for the great interest in immunity to parasites and for the development of immunoparasitology as a distinct branch of immunology.
Most parasites go through complex life cycles, part of which occurs in humans (or other vertebrates) and part of which occurs in intermediate hosts, such as flies, ticks, and snails. Humans are usually infected by bites from infected intermediate hosts or by sharing a particular habitat with an intermediate host. For instance, malaria and trypanosomiasis are transmitted by insect bites, and schistosomiasis is transmitted by exposure to water in which infected snails reside. Most parasitic infections are chronic because of weak innate immunity and the ability of parasites to evade or resist elimination by adaptive immune responses. Furthermore, many antiparasite drugs are not effective at killing the organisms. Individuals living in endemic areas require repeated chemotherapy because of continued exposure, and such treatment is often not possible because of expense and logistic problems. Therefore, the development of prophylactic vaccines for parasites has long been considered an important goal for developing countries.
Innate Immunity to Parasites
Although different protozoan and helminthic parasites have been shown to activate different mechanisms of innate immunity, these organisms are often able to survive and replicate in their hosts because they are well adapted to resisting host defenses. The principal innate immune response to protozoa is phagocytosis, but many of these parasites are resistant to phagocytic killing and may even replicate within macrophages. Some protozoa express surface molecules that are recognized by TLRs and activate phagocytes. Plasmodium species (the protozoa that are responsible for malaria), Toxoplasma gondii (the agent that causes toxoplasmosis), and Cryptosporidium species (the major parasite that causes diarrhea in HIV-infected patients) all express glycosyl phosphatidylinositol lipids that can activate TLR2 and TLR4. Phagocytes may also attack helminthic parasites and secrete microbicidal substances to kill organisms that are too large to be phagocytosed. However, many helminths have thick teguments that make them resistant to the cytocidal mechanisms of neutrophils and macrophages, and they are too large to be ingested by phagocytes. Some helminths may activate the alternative pathway of complement, although as we shall discuss later, parasites recovered from infected hosts appear to have developed resistance to complement-mediated lysis.
Adaptive Immunity to Parasites
Different protozoa and helminths vary greatly in their structural and biochemical properties, life cycles, and pathogenic mechanisms. It is therefore not surprising that different parasites elicit distinct adaptive immune responses (Table 15-4). Some pathogenic protozoa have evolved to survive within host cells, so protective immunity against these organisms is mediated by mechanisms similar to those that eliminate intracellular bacteria and viruses. In contrast, metazoa such as helminths survive in extracellular tissues, and their elimination is often dependent on special types of antibody responses.
TABLE 15–4 Immune Responses to Disease-Causing Parasites
| Parasite | Diseases | Principal Mechanisms of Protective Immunity |
|---|---|---|
| Protozoa | ||
| Plasmodium species | Malaria | Antibodies and CD8+ CTLs |
| Leishmania donovani | Leishmaniasis (mucocutaneous disseminated) | CD4+ TH1 cells activate macrophages to kill phagocytosed parasites |
| Trypanosoma brucei | African trypanosomiasis | Antibodies |
| Entamoeba histolytica | Amebiasis | Antibodies, phagocytosis |
| Metazoa | ||
| Schistosoma species | Schistosomiasis | Killing by eosinophils, macrophages |
| Filaria, e.g., Wuchereria bancrofti | Filariasis | Cell-mediated immunity; role of antibodies? |
Selected examples of parasites and immune responses to them are listed.
The principal defense mechanism against protozoa that survive within macrophages is cell-mediated immunity, particularly macrophage activation by TH1 cell–derived cytokines. Infection of mice with Leishmania major, a protozoan that survives within the endosomes of macrophages, is the best documented example of how dominance of TH1 or TH2 responses determines disease resistance or susceptibility (see Fig. 15-5). Resistance to the infection is associated with activation of Leishmania-specific TH1 CD4+ T cells, which produce IFN-γ and thereby activate macrophages to destroy intracellular parasites. Conversely, activation of TH2 cells by the protozoa results in increased parasite survival and exacerbation of lesions because of the macrophage-suppressive actions of TH2 cytokines, notably IL-4. A good example of this difference is seen in Leishmania infections in different inbred mouse strains. Most inbred strains of mice are resistant to infection with L. major, but inbred BALB/c and some related strains of mice are highly susceptible and die if they are infected with large numbers of parasites. After infection, the resistant strains produce large amounts of IFN-γ in response to leishmanial antigens, whereas the strains that are susceptible to fatal leishmaniasis produce more IL-4 in response to the parasite. Promoting the TH1 response or inhibiting the TH2 response in susceptible strains increases their resistance to the infection. Multiple genes appear to control the balance of protective and harmful immune responses to intracellular parasites in inbred mice and presumably in humans as well. Attempts to identify these genes are ongoing in many laboratories.
Protozoa that replicate inside various host cells and lyse these cells stimulate specific antibody and CTL responses, similar to cytopathic viruses. An example of such an organism is the malaria parasite, which resides mainly in red blood cells and in hepatocytes during its life cycle. It was thought for many years that antibodies were the major protective mechanism against malaria, and early attempts at vaccinating against this infection focused on generating antibodies. It is now apparent that the CTL response against parasites residing in hepatocytes is an important defense against the spread of this intracellular protozoan. The cytokine IFN-γ has been shown to be protective in many protozoal infections, including malaria, toxoplasmosis, and cryptosporidiosis.
Defense against many helminthic infections is mediated by the activation of TH2 cells, which results in production of IgE antibodies and activation of eosinophils. Helminths stimulate differentiation of naive CD4+ helper T cells to the TH2 subset of effector cells, which secrete IL-4 and IL-5. IL-4 stimulates the production of IgE, which binds to the Fcε receptor of eosinophils and mast cells, and IL-5, which stimulates the development of eosinophils and activates eosinophils. IgE, mast cell and eosinophil-mediated effector mechanisms are described in Chapter 19. The combined actions of mast cells and eosinophils also contribute to expulsion of the parasites from the intestine, so-called barrier immunity (see Chapter 10, Fig. 10-9). The expulsion of some intestinal nematodes may be due to IL-4–dependent mechanisms that do not require IgE, such as increased peristalsis.
Adaptive immune responses to parasites can also contribute to tissue injury. Some parasites and their products induce granulomatous responses with concomitant fibrosis. Schistosoma mansoni eggs deposited in the liver stimulate CD4+ T cells, which in turn activate macrophages and induce DTH reactions. DTH reactions result in the formation of granulomas around the eggs; an unusual feature of these granulomas, especially in mice, is their association with TH2 responses. (Granulomas are generally induced by TH1 responses against persistent antigens; see Chapter 18.) Such TH2-induced granulomas may result from the process of “alternative macrophage activation” that is induced by IL-4 and IL-13 (see Chapter 10). The granulomas serve to contain the schistosome eggs, but severe fibrosis associated with this chronic cell-mediated immune response leads to cirrhosis, disruption of venous blood flow in the liver, and portal hypertension. In lymphatic filariasis, lodging of the parasites in lymphatic vessels leads to chronic cell-mediated immune reactions and ultimately to fibrosis. Fibrosis results in lymphatic obstruction and severe lymphedema. Chronic and persistent parasitic infestations are often associated with the formation of complexes of parasite antigens and specific antibodies. The complexes can be deposited in blood vessels and kidney glomeruli and produce vasculitis and nephritis, respectively (see Chapter 18). Immune complex disease is a complication of schistosomiasis and malaria.
Immune Evasion by Parasites
Parasites evade protective immunity by reducing their immunogenicity and by inhibiting host immune responses. Different parasites have developed remarkably effective ways of resisting immunity (Table 15-5).
TABLE 15–5 Mechanisms of Immune Evasion by Parasites
| Mechanism of Immune Evasion | Examples |
|---|---|
| Antigenic variation | Trypanosomes, Plasmodium |
| Acquired resistance to complement, CTLs | Schistosomes |
| Inhibition of host immune responses | Filaria (secondary to lymphatic obstruction), trypanosomes |
| Antigen shedding | Entamoeba |
CTL, cytotoxic T lymphocyte.
The consequences of parasitic infestations for health and economic development are devastating. Attempts to develop effective vaccines against these infections have been actively pursued for many years. Although the progress has been slower than one would have hoped, elucidation of the fundamental mechanisms of immune responses to and immune evasion by parasites holds promise for the future.
Strategies for Vaccine Development
The birth of immunology as a science dates from Edward Jenner’s successful vaccination against smallpox in 1796. The importance of prophylactic immunization against infectious diseases is best illustrated by the fact that worldwide programs of vaccination have led to the complete or nearly complete eradication of many of these diseases in developed countries (see Chapter 1, Table 1-1). The fundamental principle of vaccination is to administer a killed or attenuated form of an infectious agent, or a component of a microbe, that does not cause disease but elicits an immune response that provides protection against infection by the live, pathogenic microbe.
The success of vaccination in eradicating infectious disease is dependent on several properties of the microbes. Vaccines are effective if the infectious agent does not establish latency, if it does not undergo much or any antigenic variation, and if it does not interfere with the host immune response. It is difficult to effectively vaccinate against microbes such as HIV, which establishes latent infection, is highly variable, and disables key components of the immune system. Vaccines are most effective against infections that are limited to human hosts and do not have animal reservoirs.
Most vaccines in use today work by inducing humoral immunity. Antibodies are the only immune mechanism that prevents infections, by neutralizing and clearing microbes before they gain their foothold in the host. The best vaccines are those that stimulate the development of long-lived plasma cells that produce high-affinity antibodies as well as memory B cells. These aspects of humoral immune responses are best induced by the germinal center reaction (see Chapter 11), which requires help provided by protein antigen-specific CD4+ T cells.
In the following section, we summarize the approaches to vaccination that have been tried (Table 15-6) and their major value and limitations.
| Type of Vaccine | Examples |
|---|---|
| Live attenuated or killed bacteria | Bacillus Calmette-Guérin, cholera |
| Live attenuated viruses | Polio, rabies |
| Subunit (antigen) vaccines | Tetanus toxoid, diphtheria toxoid |
| Conjugate vaccines | Haemophilus influenzae, pneumococcus |
| Synthetic vaccines | Hepatitis (recombinant proteins) |
| Viral vectors | Clinical trials of HIV antigens in canarypox vector |
| DNA vaccines | Clinical trials ongoing for several infections |
Attenuated and Inactivated Bacterial and Viral Vaccines
Vaccines composed of intact nonpathogenic microbes are made by treating the microbes in such a way that they can no longer cause disease (i.e., their virulence is attenuated) or by killing the microbes while retaining their immunogenicity. The great advantage of attenuated microbial vaccines is that they elicit all the innate and adaptive immune responses (both humoral and cell mediated) that the pathogenic microbe would, and they are therefore the ideal way of inducing protective immunity. Live, attenuated bacteria were first shown by Louis Pasteur to confer specific immunity. The attenuated or killed bacterial vaccines in use today generally induce limited protection and are effective for only short periods. Live, attenuated viral vaccines are usually more effective; polio, measles, and yellow fever are three good examples. The most frequently used approach for producing such attenuated viruses is repeated passage in cell culture. More recently, temperature-sensitive and deletion mutants have been generated to achieve the same goal. Viral vaccines often induce long-lasting specific immunity, so immunization of children is sufficient for lifelong protection. Some attenuated viral vaccines (e.g., polio) may cause disease in immune-compromised hosts, and for this reason inactivated poliovirus vaccines are now more commonly used. The major concern with attenuated viral or bacterial vaccines is safety.
A widely used inactivated vaccine of considerable public health importance is the influenza vaccine. Influenza viruses grown in chicken eggs are used in two types of vaccines. The most common vaccine is a trivalent inactivated (killed) vaccine that is used in the “flu shot” that is given intramuscularly. Three of the most frequently encountered influenza strains are selected every year and incorporated in this vaccine. A second type of influenza vaccine involves the same three strains, but the vaccine is made up of live attenuated viruses and is used as a nasal spray.
Purified Antigen (Subunit) Vaccines
Subunit vaccines are composed of antigens purified from microbes or inactivated toxins and are usually administered with an adjuvant. One effective use of purified antigens as vaccines is for the prevention of diseases caused by bacterial toxins. Toxins can be rendered harmless without loss of immunogenicity, and such “toxoids” induce strong antibody responses. Diphtheria and tetanus are two infections whose life-threatening consequences have been largely controlled because of immunization of children with toxoid preparations. Vaccines composed of bacterial polysaccharide antigens are used against pneumococcus and H. influenzae. Because polysaccharides are T-independent antigens, they tend to elicit low-affinity antibody responses and may be poorly immunogenic in infants (who do not mount strong T cell–independent antibody responses). High-affinity antibody responses may be generated against polysaccharide antigens even in infants by coupling the polysaccharides to proteins to form conjugate vaccines. Such vaccines work like hapten-carrier conjugates and are a practical application of the principle of T-B cell cooperation (see Chapter 11). The currently used H. influenzae, pneumococcal, and meningococcal vaccines are conjugate vaccines. Purified protein vaccines stimulate helper T cells and antibody responses, but they do not generate potent CTLs. The reason for poor CTL development is that, unlike with attenuated microbial vaccines, exogenous proteins (and peptides) are inefficient at entering the class I MHC pathway of antigen presentation and cannot readily displace peptides from surface class I molecules. As a result, protein vaccines are not recognized efficiently by class I–restricted CD8+ T cells.
Synthetic Antigen Vaccines
A goal of vaccine research has been to identify the most immunogenic microbial antigens or epitopes, to synthesize these in the laboratory, and to use the synthetic antigens as vaccines. It is possible to deduce the protein sequences of microbial antigens from nucleotide sequence data and to prepare large quantities of proteins by recombinant DNA technology. Vaccines made of recombinant DNA-derived antigens are now in use for hepatitis virus, herpes simplex virus, foot-and-mouth disease virus (a major pathogen for livestock), human papillomavirus, and rotavirus. In the case of the most widely used human papillomavirus vaccine, recombinant viral proteins from four viral strains (HPV 6, 11, 16, and 18) are made in yeast and combined with an adjuvant. HPV 6 and 11 are common causes of warts, and HPV 16 and 18 are the most common HPV strains linked to cervical cancer. This antiviral vaccine is therefore also a preventive cancer vaccine.
Live Viral Vaccines Involving Recombinant Viruses
Another approach for vaccine development is to introduce genes encoding microbial antigens into a noncytopathic virus and to infect individuals with this virus. Thus, the virus serves as a source of the antigen in an inoculated individual. The great advantage of viral vectors is that they, like other live viruses, induce the full complement of immune responses, including strong CTL responses. This technique has been used most commonly with vaccinia virus vectors. Inoculation of such recombinant viruses into many species of animals induces both humoral and cell-mediated immunity against the antigen produced by the foreign gene (and, of course, against vaccinia virus antigens as well). A potential problem with recombinant viruses is that the viruses may infect host cells, and even though they are not pathogenic, they may produce antigens that stimulate CTL responses that kill the infected host cells. These and other safety concerns have limited widespread use of viral vectors for vaccine delivery.
DNA Vaccines
An interesting method of vaccination was developed on the basis of an unexpected observation. Inoculation of a plasmid containing complementary DNA (cDNA) encoding a protein antigen leads to strong and long-lived humoral and cell-mediated immune responses to the antigen. It is likely that APCs, such as dendritic cells, are transfected by the plasmid and the cDNA is transcribed and translated into immunogenic protein that elicits specific responses. The unique feature of DNA vaccines is that they provide the only approach, other than live viruses, for eliciting strong CTL responses because the DNA-encoded proteins are synthesized in the cytosol of transfected cells. Furthermore, bacterial plasmids are rich in unmethylated CpG nucleotides and are recognized by a TLR (TLR9) on dendritic cells and other cells, thereby eliciting an innate immune response that enhances adaptive immunity (see Chapter 4). Therefore, plasmid DNA vaccines could be effective even when administered without adjuvants. The ease of manipulating cDNAs to express many diverse antigens, the ability to store DNA without refrigeration for use in the field, and the ability to coexpress other proteins that may enhance immune responses (such as cytokines and costimulators) make this technique promising. However, DNA vaccines have not been as effective as hoped in clinical trials, and the factors that determine the efficacy of these vaccines, especially in humans, are still not fully defined.
Adjuvants and Immunomodulators
The initiation of T cell–dependent immune responses against protein antigens requires that the antigens be administered with adjuvants. Most adjuvants elicit innate immune responses, with increased expression of costimulators and production of cytokines such as IL-12 that stimulate T cell growth and differentiation. Heat-killed bacteria are powerful adjuvants that are commonly used in experimental animals. However, the severe local inflammation that such adjuvants trigger precludes their use in humans. Much effort is currently being devoted to development of safe and effective adjuvants for use in humans. Several are in clinical practice, including aluminum hydroxide gel (which appears to promote B cell responses) and lipid formulations that are ingested by phagocytes. An alternative to adjuvants is to administer natural substances that stimulate T cell responses together with antigens. For instance, IL-12 incorporated in vaccines promotes strong cell-mediated immunity. As mentioned, plasmid DNA has intrinsic adjuvant-like activities, and it is possible to incorporate costimulators (e.g., B7 molecules) or cytokines into plasmid DNA vaccines. These interesting ideas remain experimental.
Passive Immunization
Protective immunity can also be conferred by passive immunization, for instance, by transfer of specific antibodies. In the clinical situation, passive immunization is most commonly used for rapid treatment of potentially fatal diseases caused by toxins, such as tetanus, and for protection from rabies and hepatitis. Antibodies against snake venom can be lifesaving when administered after poisonous snakebites. Passive immunity is short-lived because the host does not respond to the immunization, and protection lasts only as long as the injected antibody persists. Moreover, passive immunization does not induce memory, so an immunized individual is not protected against subsequent exposure to the toxin or microbe.
Summary
Alcais A, Abel L, Casanova J-L. Human genetics of infectious diseases: between proof of principle and paradigm. Journal of Clinical Investigation. 2009;119:2506-2514.
Dorhol A, Kaufmann SH. Fine-tuning T cell responses during infection. Current Opinion in Immunology. 2009;21:367-377.
Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767-782.
Immunity to Extracellular and Intracellular Bacteria
Brodsky IE, Medzhitov R. Targeting of immune signaling networks by bacterial pathogens. Nature Cell Biology. 2009;11:521-526.
Cooper AM. Cell-mediated immune responses in tuberculosis. Annual Review of Immunology. 2009;27:393-422.
Curtis MM, Way SS. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology. 2009;126:177-185.
Kaufmann SHE. Tuberculosis: back on the immunologists’ agenda. Immunity. 2006;24:351-357.
Antoniou AN, Powis SJ. Pathogen evasion strategies for the major histocompatibility complex class I assembly pathway. Immunology. 2008;124:1-12.
Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nature Immunology. 2005;6:873-879.
Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Research. 2005;15:407-422.
Rouse BT, Seherwat S. Immunity and immunopathology to viruses: what decides the outcome? Nature Reviews. Immunology. 2010;10:514-526.
Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30-50.
Good MF, Xu H, Wykes M, Engwerda CR. Development and regulation of cell-mediated immune responses to the blood stages of malaria: implications for vaccine research. Annual Review of Immunology. 2005;23:69-99.
Langhorne J, Ndungu FM, Sponaas A-M, Marsh K. Immunity to malaria: more questions than answers. Nature Immunology. 2008;9:725-732.
Maizels RM, Pearce EJ, Artis D, Yazdanbaksh M, Wynn TA. Regulation of pathogenesis and immunity in helminth infections. Journal of Experimental Medicine. 2009;201:2059-2066.
McCulloch R. Antigenic variation in African trypanosomes: monitoring progress. Trends in Parasitology. 2004;20:117-121.
Brunner R, Jensen-Jarolim E, Pali-Scholl I. The ABC of clinical and experimental adjuvants—a brief overview. Immunology Letters. 2010;128:29-35.
Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. Journal of Immunology. 2005;175:633-639.
Harris J, Sharp FA, Lavelle EC. The role of inflammasomes in the immunostimulatory effects of particulate vaccine adjuvants. European Journal of Immunology. 2010;40:634-638.