Medical Nutrition Therapy for Genetic Metabolic Disorders
Genetic metabolic disorders are inherited traits that result in the absence or reduced activity of a specific enzyme or cofactor. Most genetic metabolic disorders are inherited as autosomal-recessive traits; autosomal means that the gene is located on a chromosome other than the X or Y chromosomes. The treatment for many metabolic disorders is medical nutrition therapy (MNT), with medications specific to the disorder (e.g., phenylketonuria PKU). Here, the goals of MNT are to maintain biochemical equilibrium for the affected pathway, provide adequate nutrients to support typical growth and development, and support social and emotional development. Nutrition interventions are designed to circumvent the missing or inactive enzyme by (1) restricting the amount of substrate available, (2) supplementing the amount of product, (3) supplementing the enzymatic cofactor, or (4) combining any or all of these approaches. This chapter describes the primary conditions found more commonly in the United States. Table 44-1 outlines other disorders by the enzymatic defects, distinctive clinical and biochemical features, and current approaches to dietary therapy.
TABLE 44-1
Selected Genetic Metabolic Disorders That Respond To Dietary Treatment
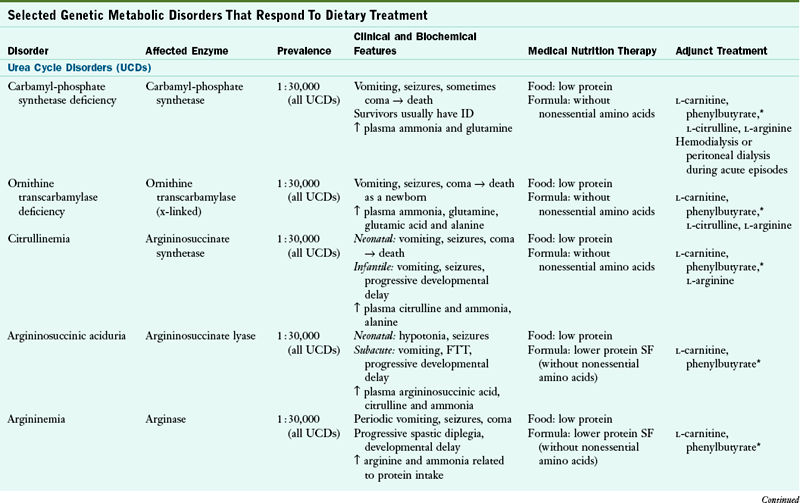
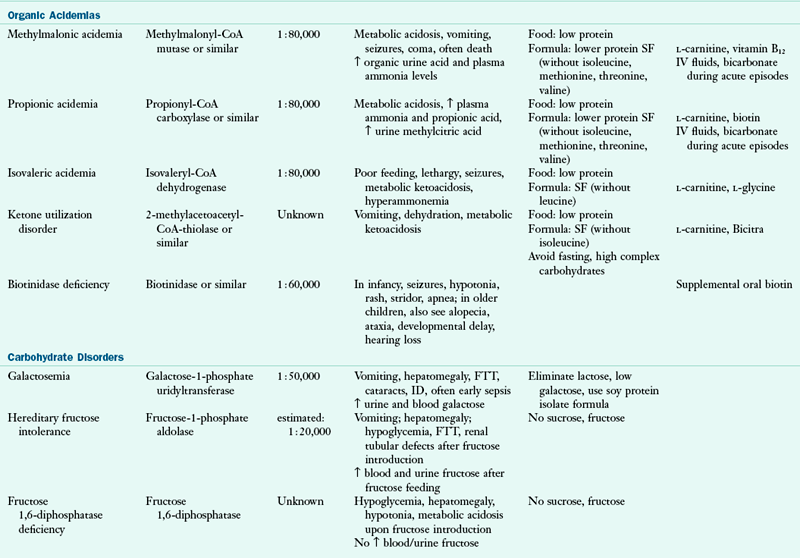
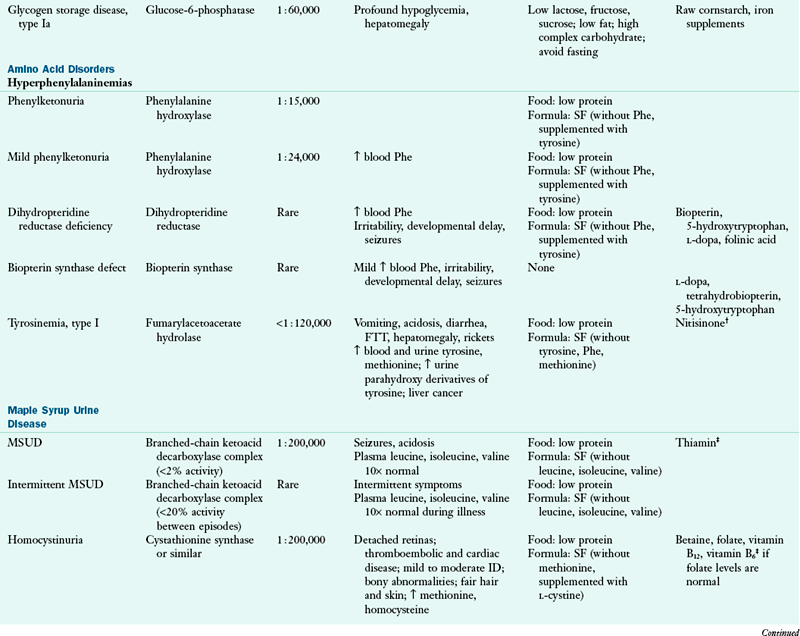
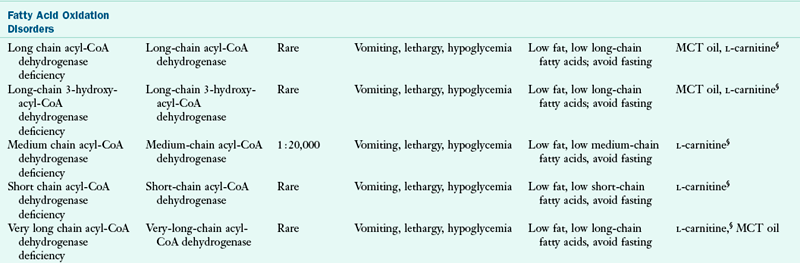
CoA, Coenzyme A; FTT, failure to thrive; ID, intellectual disability; IV, intravenous; MCT, medium-chain triglyceride; MSUD, maple syrup urine disease; Phe, phenylalanine; SF, specialized formulas are available for medical nutrition therapy for this disorder; UCD, urea cycle disorder.
*Phenylbutyrate is a chemical administered to enhance waste ammonia excretion; other compounds producing the same effect are also used.
†Nitisinone, formerly NTBC, 2-(2-nitro-4-trifluoro-methyl-benzoyl-1,3-cyclohexanedione, commercially available as Orfadin.®
‡Patient may or may not respond to the compound.
§Use depends on clinic.
In some instances, when treatment is initiated early in the newborn period and meticulously continued for a lifetime, the affected individual can be cognitively and physically normal. In conditions such as galactosemia, cognitive and physical damage can occur despite early and meticulous treatment. Biochemical disorders range from variations in enzyme activity that are benign, to severe manifestations that are incompatible with life. For many of them, significant questions related to diagnosis and treatment remain.
Newborn Screening
Most inherited metabolic disorders are associated with severe clinical illness that often appears soon after birth. Intellectual disability and severe neurologic involvement may be immediately apparent. Diagnosis of a specific disorder may be difficult, and appropriate treatment measures may be uncertain. Prenatal diagnosis is available for many metabolic disorders, but it usually requires the identification of a family at risk, which can be done only after the birth of an affected child. Effective newborn screening programs, as well as advanced diagnostic techniques and treatment modalities have improved the outcome for many of these infants.
Infants suspected of having a metabolic disorder should be afforded access to care offered by centers with expertise in treating these disorders. Infants who are afebrile for no apparent reason, lethargic, vomiting, in respiratory distress, or having seizures should be evaluated for an undiagnosed metabolic disorder. The initial assessment should include blood gas measurements, electrolyte values, glucose and ammonia tests, and a urine test for ketones.
Advances in newborn screening technology offer opportunities for earlier diagnosis, prevention of neurologic crisis, and improved intellectual and physical outcomes. When tandem mass spectrometry techniques are used in newborn screening laboratories, infants with a broader range of metabolic disorders can be identified, and the disorder can be identified earlier (see Focus On: Newborn Screening). See Fig. 44-1.

FIGURE 44-1 Tandem mass spectrometer. This technology makes screening for a broad range of metabolic disorders feasible. Blood levels of a number of organic acids are measured. (Courtesy Washington State Newborn Screening Program.)
 Focus On
Focus On
Newborn Screening (NBS)
Since the 1960s, states across the United States have adopted mandatory newborn screening (NBS) as law (Waisbren, 2006). These programs were developed as a result of the efficacy of the Guthrie bacterial inhibition assay, in which dried blood spots were assayed. This simple, sensitive, and inexpensive screening test became the basis for population-based screening systems for newborns. Hemoglobinopathies, endocrine disorders, metabolic disorders, and some infectious disorders can be effectively identified in this way.
In the 1990s tandem mass spectrometry began to be used in NBS and is now used across the United States (Therrell, 2006). This technology makes it possible to identify multiple disorders from dried blood spots. The number of disorders screened for varies widely by state, and expanded screening is also offered by private, for-profit companies. Follow-up programs also vary; some states have a centralized program, whereas follow-up in other states is less coordinated. Successful early NBS programs include screening for congenital hypothyroidism, phenylketonuria, congenital adrenal hyperplasia, galactosemia, sickle cell disease, and maple syrup urine disease (Brosco et al., 2006).
The Maternal and Child Health Bureau (MCHB) of the U.S. Health Resources and Services Administration commissioned a report from the American College of Medical Genetics (ACMG). This expert panel identified 29 conditions for which newborn screening should be mandated and 25 secondary conditions that may be detected incidentally (MCHB, 2007). The ACMG developed a series of ACTion (ACT) sheets and confirmatory algorithms for disorders that are identified by NBS screening. The ACT sheets describe the steps health professionals should follow in communicating with the family and determining follow-up. They are available at http://www.acmg.net/AM/Template.cfm?Section=NBS_ACT_Sheets_and_Algorithms_Table&Template=/CM/HTMLDisplay.cfm&ContentID=5649.
Other groups, including the World Health Organization, March of Dimes, and Massachusetts Newborn Screening Advisory Committee, have also issued recommendations.
Providers who may be involved in the care and follow-up of families identified by NBS should have a good understanding of their state’s system, as well as the factors that may affect results. Communication among families, primary health care providers, and tertiary clinics is critical to timely identification and treatment. Follow-up, including referral to the appropriate specialists, is important for any family who receives NBS test results. NBS fact sheets from the Committee on Genetics of the American Academy of Pediatrics describe (1) newborn testing; (2) follow-up of abnormal screening results to facilitate timely diagnostic testing and management; (3) diagnostic testing; (4) disease management, which requires coordination with the medical home and genetic counseling; and (5) continuous evaluation and improvement of the NBS system (Kaye et al., 2006).
Disorders of Amino Acid Metabolism
Nutrition therapy for amino acid disorders most commonly consists of substrate restriction, which involves limiting one or more essential amino acids to the minimum requirement while providing adequate energy and nutrients to promote typical growth and development (e.g., restricting phenylalanine [Phe] in phenylketonuria PKU). An inadequate intake of an essential amino acid is often as detrimental as excess. Supplementation of the product of the specific enzymatic reaction is usually required in nutrition therapy for amino acid disorders; for example, tyrosine (Tyr) is supplemented in formulas for treatment of PKU.
Requirements for individual amino acids are difficult to determine because typical growth and development can be achieved over a wide range of intake. The data of Holt and Snyderman (1967) are often used as the basis for prescribing amino acid intakes (Table 44-2). Careful and frequent monitoring is required to ensure the adequacy of the nutritional prescription. Although nitrogen studies are the most precise, weight gain in infants is a sensitive and easily monitored index of well being and nutrition adequacy.
TABLE 44-2
Approximate Daily Requirements for Selected Dietary Components and Amino Acids in Infancy and Childhood

Compiled from amino acid data of Holt and Snyderman. Information on amino acid requirements of infants and children at different ages is limited; the figures given here are in excess of minimum requirements. Consequently, this table should be used only as a guide and should not be regarded as an authoritative statement to which individual patients must conform.
*More phenylalanine (>800 mg) is required in the absence of tyrosine.
†Total phenylalanine plus tyrosine should be considered in the prescription because most phenylalanine is converted to tyrosine.
‡More methionine is required in the absence of cyst(e)ine.
§More cyst(e)ine is required in presence of a blocked trans-sulfuration outflow pathway for methionine metabolism.
Modified from Committee on Nutrition, American Academy of Pediatrics: Special diets for infants with inborn errors of metabolism, Pediatrics 57:783, 1976.
Phenylketonuria
Phenylketonuria (PKU) is the most common of the hyperphenylalaninemias. In this disorder phenylalanine is not metabolized to Tyr because of a deficiency or inactivity of phenylalanine hydroxylase as shown in Figure 44-2. Of the amino acid disorders, PKU provides a reasonable model for detailed discussion because it (1) occurs relatively frequently and most neonates are screened for it; (2) has a successful MNT treatment; and (3) has a predictable course, with available documentation of “natural” and “intervention” history (see Focus On: Time Line of Events in the Diagnosis and Treatment of Phenylketonuria).
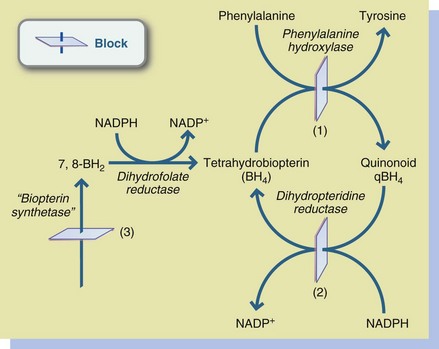
FIGURE 44-2 Hyperphenylalaninemias. 1, Phenylalanine hydroxylase deficiency; 2, Dihydropteridine reductase deficiency; 3, Biopterin synthetase deficiency. NADPH, Nicotinamide-adenine dinucleotide phosphate (reduced form); NADP+, nicotinamide-adenine dinucleotide phosphate (oxidized form).
Focus On
Time Line of Events in the Diagnosis and Treatment of Phenylketonuria
1934: A. Folling identifies phenylpyruvic acid in the urine of mentally retarded siblings.
1950s: G. Jervis demonstrates a deficiency of phenylalanine oxidation in the liver tissue of an affected patient. H. Bickel demonstrates that dietary phenylalanine restrictions lower the blood concentration of phenylalanine.
1960s: R. Guthrie develops a bacterial inhibition assay for measuring blood phenylalanine levels.
Mid-1960s: Semisynthetic formulas restricted in phenylalanine content become commercially available.
1965-1970: States adopt newborn screening programs to detect phenylketonuria (PKU).
1967-1980: Collaborative Study of Children Treated for Phenylketonuria is conducted. Data from this study form the basis for treatment protocols for PKU clinics in the United States.
Late-1970s: Detrimental effects of maternal PKU are recognized as a significant public health problem.
1980s: Lifelong restriction of phenylalanine intake becomes the standard of care in PKU clinics in the United States.
1983: The Maternal PKU Collaborative Study begins to study the effects of treatment on the pregnancy outcome of women with phenylketonuria.
1987: Techniques for carrier detection and prenatal diagnosis of PKU are developed.
Late-1980s: The gene for phenylalanine hydroxylase deficiency (MIM No. 261600) is located on chromosome 12q22-q24.1. Deoxyribonucleic acid mutation analysis can be accomplished with peripheral leukocytes.
1990s: Phenylalanine level of 1-6 mg/dL (60-360 mmol/L), lower than the previous level of less than 10 mg/dL (600 mmol/L), becomes the new standard of care for treatment of PKU.
2000: Tetrahydrobiopterin-responsive forms of PKU are recognized, especially those with mild mutations.
2010: Continued research into alternative and adjunct therapies such as the use of large neutral amino acids, BH4 supplementation, enzyme substitution, and somatic gene therapy.
Data from Maternal Child Health Bureau: Newborn screening: toward a uniform screening panel and system, Genet Med 8(Suppl1):1S, 2006; Saugstad LF: From genetics to epigenetics, Nutr Health, 18:285, 2006; Mitchell JJ, Scriver CR: Phenylalanine hydroxylase deficiency. In Pago RA et al., editors: GeneReviews [Internet]. Seattle, 1993-2000, University of Washington, Seattle [updated November 5, 2010].
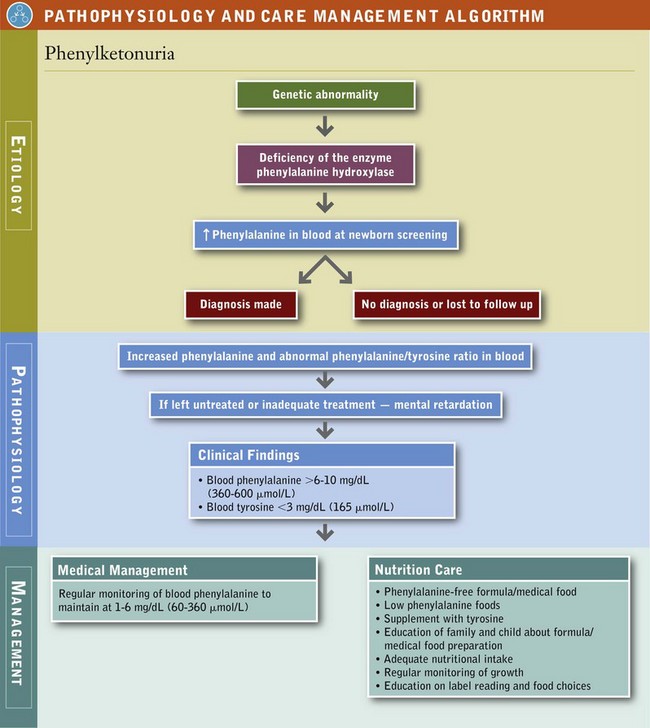
Nutritional treatment involves restricting the substrate (Phe) and supplementing the product (Tyr) (see Pathophysiology and Care Management Algorithm: Phenylketonuria). Most affected infants exhibit phenylalanine hydroxylase deficiency; the remainder (less than 3%) have defects in associated pathways. Low phenylalanine nutrition therapy does not prevent the neurologic deterioration present in the disorders of associated pathways.
Medical Treatment
All states have newborn screening programs for PKU and other metabolic disorders. Diagnostic criteria for PKU include blood concentrations of Phe that consistently exceed 6 to 10 mg/dL (360 to 600 mmol/L) and Tyr concentrations of less than 3 mg/dL (165 mmol/L). The diagnostic process should also include evaluation for hyperphenylalaninemia that results from the deficiency of enzymes other than phenylalanine hydroxylase. An effective newborn screening program and access to an organized follow-up program are critical to early identification and treatment of infants with PKU.
The advantage of rigorous nutrition therapy has been demonstrated by measurements of intellectual function. Individuals who do not receive diet therapy have severe intellectual disability (mean intelligence quotient [IQ] of approximately 40), whereas individuals who are on therapy from the early neonatal period have normal IQs. Outcome, measured as intellectual function, depends on the age of the infant at diagnosis and start of nutrition therapy, as well as the individual’s biochemical control over time.
Tetrahydrobiopterin (BH4) has been studied to evaluate its effectiveness as an alternative treatment to severe dietary Phe restriction. BH4 is a cofactor needed for proper activity of the enzyme. Although supplementation with BH4 holds promise as an adjunct therapy for some milder mutations, observations on long-term outcome are needed (Lee et al., 2005). Those individuals who respond have what is called BH4-responsive PKU. Other studies have examined the possibility of enzyme substitution with phenylalanine ammonia lyase to degrade Phe, or gene therapy to restore phenylalanine hydroxylase activity (Blau et al., 2010).
Blood Phenylalanine Control: Blood Phe concentration must be checked regularly, depending on the age and health status of the child, to be sure it remains within the range of 2 to 6 mg/dL or 120 to 360 mmol/L. Phe-containing foods are offered as tolerated as long as the blood concentration of Phe remains in the range of good biochemical control. The child’s rate of growth and mental development must be carefully monitored.
Effective management requires a team approach in which the child, parents, registered dietitian, pediatrician, psychologist, social worker, and nurse work together to achieve and maintain biochemical control in an atmosphere promoting normal mental and emotional development. An essential management tool for parents, children, and clinicians is the food diary used to monitor Phe intake. Daily record keeping supports compliance with treatment and builds self-management skills. An accurate record of food and formula intake for at least the 3 days before a laboratory specimen is obtained is mandatory for accurate interpretation of the results and subsequent adjustment of the Phe prescription.
Elevations in blood Phe concentration are generally caused by either excessive Phe intake or tissue catabolism. Intake of Phe in excess of the amount required for growth accumulates in the blood. Deficient energy intake or the stress of illness or infection can result in protein breakdown and the release of amino acids, including Phe, into the blood. In general, the anorexia of illness limits energy intake. Preventing tissue catabolism by maintaining intake of the formula/medical food as much as possible is essential. Although it may occasionally be necessary to offer only clear liquids during an illness, the Phe-free formula/medical food should be reintroduced as soon as it is feasible. It can be used as a tube feeding if oral intake is not possible.
The necessity of continuing the restricted-Phe dietary therapy beyond adolescence is a consideration. Progressively decreasing IQs, learning difficulties, poor attention span, and behavioral difficulties have been reported in children who have discontinued the dietary regimen. Children who maintain well-controlled blood Phe levels demonstrate comparatively higher intellectual achievement than those who do not. Good dietary control of blood Phe concentrations is the best predictor of IQ, whereas “off-diet” blood Phe concentrations of greater than 20 mg/dL (1200 mmol/L) are the best predictors of IQ loss. Subtle deficits in higher-level cognitive function may persist even at blood Phe levels of 6 to 10 mg/dL (360 to 600 mmol/L); thus most clinics recommend treatment blood levels of 1 to 6 mg/dL (60 to 360 mmol/L). Restricted-Phe therapy should be continued for life to maintain normal cognitive function (Waisbren et al., 2007).
Medical Nutrition Therapy
Formula: For PKU dietary therapy is planned around the use of a formula/medical food with Phe removed from the protein. Formulas or medical foods provide a major portion of the daily protein and energy needs for affected infants, children, and adults. In general, the protein source in the formula/medical food is L-amino acids, with the critical amino acid (i.e., Phe) omitted. Carbohydrate sources are corn syrup solids, modified tapioca starch, sucrose, and hydrolyzed cornstarch. Fat is provided by a variety of oils.
Some formula/medical foods contain no fat or carbohydrate; therefore these components must be provided from other sources. If prescribing formulas without fat, sources of essential fatty acids must be provided. Essential fatty acid deficiencies have been noted among individuals consuming fat-free formulas (Cleary et al., 2006; Rose, et al., 2005). Most formulas and medical foods contain calcium, iron, and all other necessary vitamins and minerals and are a reliable source of these nutrients. When others are devoid of these nutrients, supplementation is needed to ensure nutritional adequacy.
Phe-free formula is supplemented with regular infant formula or breast milk during infancy and cow’s milk in early childhood to provide high–biologic value protein, nonessential amino acids, and sufficient Phe to meet the individualized requirements of the growing child. The optimal amount of protein substitute depends on the individual’s age (and thus requirements for growth) and enzyme activity; thus it must be individually prescribed. Because the protein in specialized formulas is synthetic, it is provided in amounts greater than the Dietary Reference Intake (DRI).
The Phe-free formula and milk mixture should provide approximately 90% of the protein and 80% of the energy needed by infants and toddlers. A method for calculating the appropriate quantities of a Phe-free formula is shown in Table 44-3. It must be stressed that calculations should provide adequate but not excessive energy for the infant, as well as appropriate fluid to maintain hydration. To support metabolic control effectively, formula/medical foods must be consumed in three or four nearly equal portions throughout the day.
TABLE 44-3
Guidelines for Low-Phenylalanine Food Pattern Calculations


*A phenylalanine intake of 60 mg/kg/day is chosen as a moderate intake level. The prescription for phenylalanine must be adapted to individual needs as judged by growth and blood levels.
†Although these intakes are higher than the recommended dietary allowance, they are the intakes found by the Collaborative Study to promote normal growth with consumption of protein hydrolysate-based formula (Acosta, 1996).
‡Total energy intake must be adjusted to meet individual needs, and an excess must be avoided.
Low-Phenylalanine Foods: Foods of moderate- or low-Phe content are used as a supplement to the formula or medical food mixture. These foods are offered at the appropriate ages to support developmental readiness and to meet energy needs. Puréed foods from a spoon might be introduced at 5 to 6 months of age, finger foods at 7 to 8 months, and the cup at 8 to 9 months, using the same timing and progression of texture recommended for typical children. Table 44-4 suggests typical low-Phe food patterns for young children.

Low-protein pastas, breads, and baked goods made from wheat starch add variety to the food pattern and allow children to eat some foods “to appetite.” A variety of low-protein pastas, rice, baked goods, egg replacers, and other foods are available. Wheat starch and a variety of low-protein baking mixes for breads, cakes, and cookies are also available. Table 44-5 compares low-protein and regular food items.
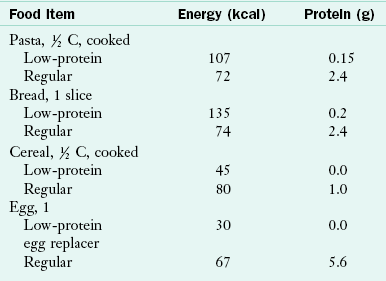
In many cases parents create recipes or adapt family favorites to meet the needs of their children. These recipes offer the children a variety of textures and food choices, allowing them to participate in family meals. Families are also able to meet the energy and Phe needs of their children without resorting to excessive intakes of sugars and concentrated sweets.
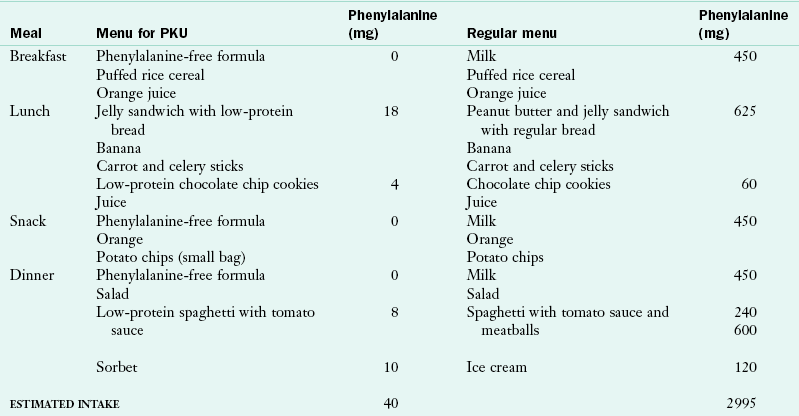
A formula or medical food that is free of Phe and has a more appropriate amino acid, vitamin, and mineral composition for an older child is generally introduced in the toddler or preschool period. The criteria for introduction of the “next-step” formulas are that the child accept the food pattern and formula well and reliably consume a wide variety of foods from the low-Phe food list. Successful management with consistently low blood Phe levels is based on habit (i.e., the formula/medical food is offered and consumed without negotiation or threat). Children respond favorably to the regularity of the time of ingestion of the formula/medical food and the familiarity of its taste and presentation. Table 44-6 compares a restricted Phe food pattern with a typical food pattern for a child of the same age.
Education about Therapy Management: The energy needs and amino acid requirements of children with PKU do not differ appreciably from those of children in general. With proper management, typical growth can be expected (Figure 44-3). However, parents may tend to offer excessive energy as sweets because they feel the child is being deprived of food experiences. Health care providers should support families in recognizing that children with PKU are healthy children who must make careful food choices for themselves, not chronically ill children who require food indulgences.
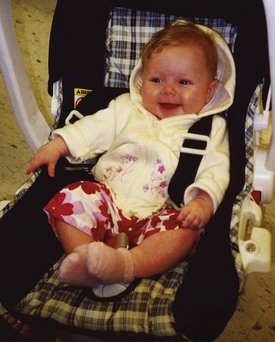
FIGURE 44-3 Infant with phenylketonuria, who was identified by a newborn screening program and started on treatment by 7 days of age, demonstrates typical growth and development. (Courtesy Cristine M. Trahms, Seattle.)
Appropriate clinical interaction with family members provides them with the information and skills to differentiate between food behaviors that are typical for the age and developmental level of the child and those related specifically to PKU (Ievers-Landis et al., 2005). To avoid power struggles and conflicts over food, it is advisable to involve the child in choosing appropriate foods at an early age. Children who are 2 to 3 years old can master the concept of appropriate choices when foods are categorized as “yes foods” and “no foods.” The concept of an appropriate quantity of a food can be introduced to a 3- or 4-year-old child in terms of “how many” by counting crackers or raisins and then in terms of “how much” by weighing or measuring foods such as cereal or fruit. The child then moves to more complex tasks (e.g., formula and food preparation) and planning of meals (e.g., breakfast or a packed lunch). Responsibility for planning a full day’s menu by calculating the quantity of Phe in portions of food and compiling the daily total is the ultimate goal. These age-related tasks are shown in Table 44-7.
TABLE 44-7
Tasks Expected of Children with Phenylketonuria by Age Level
| Age (yr) | School Level | Task |
| 2-3 | Preschool | Distinguishing between “yes” and “no” foods |
| 3-4 | Preschool | Counting: how many? |
| 4-5 | Preschool | Measuring: how much? |
| 5-6 | Kindergarten | Preparing own formula; using scale |
| 6-7 | Grade 1-2 | Writing basic notes in food diary |
| 7-8 | Grade 2 | Making some decisions on after-school snack |
| 8-9 | Grade 3 | Preparing breakfast |
| 9-10 | Grade 4 | Packing lunches |
| 10-14 | Middle school | Managing food choices with increasing independence |
| 14-18 | High school | Independently managing phenylketonuria |
Psychosocial Development: The necessity of carefully controlling food intake may prompt parents to overprotect their children and perhaps to restrict their social activities. The children, in turn, may react negatively to their parents and to their nutrition therapy. The ability of the family to respond to the stresses of PKU, as reflected by adaptability and cohesion scores, is demonstrated by improved blood Phe concentrations and the positive coping behaviors of older children with PKU. Thus continuing nutrition therapy beyond early childhood requires that children become knowledgeable about and responsible for managing their own food choices. The health care team becomes responsible for working with families and children to provide strategies that enable children and adolescents to participate in social and school activities, interact with peers, and progress through the typical developmental stages with self-confidence and self-esteem (Ievers-Landis et al., 2005).
Children require parental and professional support as they assume responsibility for their food management. Self-management of food choices is a strategy to prevent the child using dietary noncompliance as a wedge against parental restrictions. Normal intellectual development is a laudable goal of management of PKU, but to be entirely successful children with PKU concomitantly need to develop self-assurance and a strong self-image. This can be achieved in part by fostering self-management, problem-solving skills, independence, and a typical lifestyle.
Maternal PKU
A pregnant woman with elevated blood Phe concentrations endangers her fetus because of the amplified transport of amino acids across the placenta. The fetus is exposed to approximately twice the Phe level contained in normal maternal blood. Babies whose mothers have elevated blood Phe concentrations have an increased occurrence of cardiac defects, retarded growth, microcephaly, and intellectual disability, as presented in Table 44-8. The fetus appears to be at risk of damage even with minor elevations in maternal blood Phe levels, and the higher the level, the more severe the effect. Strict control of maternal Phe levels before conception and throughout pregnancy offers the best opportunity for normal fetal development (Koch et al., 2010).
TABLE 44-8
Frequency of Abnormalities in Children Born to Mothers with Phenylketonuria
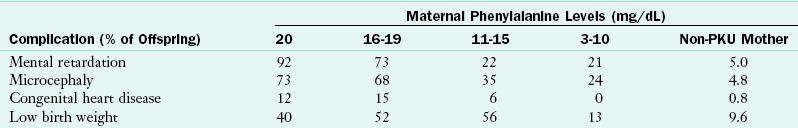
Modified from Lenke RR, Levy HL: Maternal phenylketonuria and hyper-phenylalaninemia: an international survey of the outcome of untreated and treated pregnancies, N Engl J Med 303:1202, 1980.
Nutrition management for pregnant women with hyperphenylalaninemia is complex. The changing physiology of pregnancy and fluctuating nutritional needs are difficult to monitor with the precision required to maintain appropriately low blood-Phe concentrations. Even with meticulous attention to Phe intake, blood concentrations, and the nutritional requirements of pregnancy, a woman cannot be assured of a normal infant (Lee et al., 2005). The risks of abnormal development of the fetus, even with therapeutic dietary management and maintenance of blood Phe concentrations at 1 to 5 mg/dL (60 to 300 mmol/L), are an important consideration for young women with PKU considering pregnancy (Waisbren and Azen, 2003).
Nutritional management during pregnancy is challenging, even for women who have consistently followed a low-Phe dietary regimen since infancy. Women who have discontinued Phe dietary treatment find that reinstituting medical food consumption and limiting food choices can be difficult and overwhelming. Inadequate maternal nourishment (i.e., inadequate intakes of total protein, fat, and energy) may contribute to poor fetal development and should be avoided. Adherence to nutrition therapy during pregnancy for even the well-motivated woman requires family and professional support, as well as frequent monitoring of biochemical and nutritional aspects of both pregnancy and PKU.
Adults Living with Phenylketonuria
Many adults with PKU have had the benefits of early diagnosis and treatment and are less likely to be affected by neurologic damage. However, among those who have had some degree of intellectual disability, hyperactivity and self-abuse are often major concerns. Not all patients have responded to late initiation of treatment with improved behavioral or intellectual function. For the difficult-to-manage older patient, a trial of a low-Phe food pattern is recommended. If successful, continued Phe restriction therapy may facilitate behavioral management.
Reinstituting a Phe-restricted food pattern is difficult after the eating pattern has been liberalized. However, the current recommendation of most clinics is effective management of blood Phe concentration throughout a lifetime. This recommendation is based on reports of declining intellectual capabilities and changes in the brain after prolonged, significant elevation of Phe concentrations (Waisbren and Azen, 2003). The efficacy of continued treatment throughout adulthood has been documented by reports of improved intellectual performance and problem-solving abilities when blood Phe levels are kept low. Dietary management of PKU throughout the life span is similar to that of other chronic disorders, and prudent MNT results in a normal quality of life (Bosch et al., 2006).
Maple Syrup Urine Disease
Maple syrup urine disease (MSUD), or branched-chain ketoaciduria, results from a defect in enzymatic activity, specifically the branched chain α-ketoacid dehydrogenase complex. It is an autosomal-recessive disorder. Infants appear normal at birth, but by 4 or 5 days of age they demonstrate poor feeding, vomiting, lethargy, and periodic hypertonia. A characteristic sweet, malty odor from the urine and perspiration can be noted toward the end of the first week of life.
Pathophysiology
The decarboxylation defect of MSUD prevents metabolism of the branched-chain amino acids (BCAAs) leucine, isoleucine, and valine (Figure 44-4). Leucine tends to be more problematic than the others. The precise mechanism for the complete decarboxylase reaction and the resultant neurologic damage is not known. Neither is the reason why leucine metabolism is significantly more abnormal than that of the other two BCAAs.
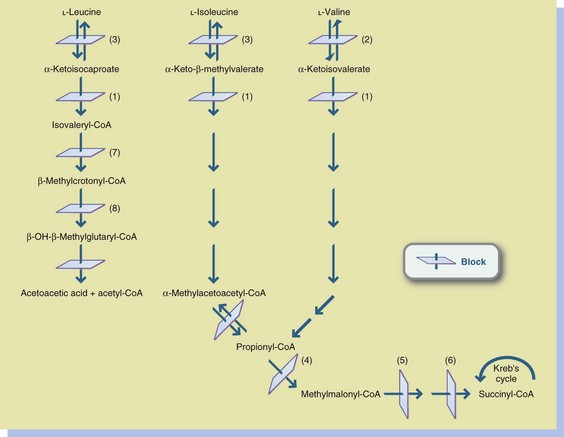
FIGURE 44-4 Organic acidemias and maple syrup urine disease (MSUD). 1, Branched-chain ketoacid decarboxylase (MSUD); 2, Valine aminotransferase; 3, Leucine-isoleucine aminotransferase; 4, Propionyl-CoA carboxylase (propionic acidemia); 5, Methylmalonyl-CoA racemase (methylmalonic aciduria); 6, Methylmalonyl-CoA mutase (methylmalonic aciduria); 7, Isovaleryl-CoA dehydrogenase (isovaleric acidemia); 8, β-methylcrotonyl-CoA carboxylase (biotin-responsive multiple carboxylase deficiency). CoA, Coenzyme A.
Medical Treatment
Failure to treat this condition leads to acidosis, neurologic deterioration, seizures, and coma, proceeding eventually to death. Management of acute disease often requires peritoneal dialysis and hydration (see Chapter 36).
Depending on the severity of the enzyme defect, early intervention and meticulous biochemical control can provide a more hopeful prognosis for infants and children with MSUD. Reasonable growth and intellectual development in the normal-to-low-normal range have been described. Diagnosis before 7 days of age and long-term metabolic control are critical factors in long-term normalization of intellectual development. Maintenance of plasma leucine concentrations in infants and preschool children should be as close to physiologically normal as possible (Hoffman, 2006). Concentrations above 10 mg/dL (760 mmol/L) are often associated with α-ketoacidemia and neurologic symptoms.
Because the liver is the central site of metabolic control for amino acids and other compounds that cause acute degeneration of the brain during illness, therapeutic liver transplantation has been proposed as an option in MSUD. Studies are underway to assess the long-term effects of this procedure on stabilization of biochemical and neurologic status (Strauss et al., 2006).
Medical Nutrition Therapy
Nutrition therapy requires very careful monitoring of blood concentrations of leucine, isoleucine, and valine as well as growth and general nutritional adequacy. Several formulas specifically designed for the treatment of this disorder are available to provide a reasonable amino acid and vitamin mixture. These are generally supplemented with a small quantity of standard infant formula or cow’s milk to provide the BCAAs needed to support growth and development. Some infants and children may require additional supplementation with L-valine or L-isoleucine to maintain biochemical balance.
BCAAs may be introduced gradually into the diet when plasma leucine concentrations are sufficiently decreased (Chuang and Shih, 2010). Clinical relapse is most often related to the degree of elevation of leucine concentrations, and these relapses often are related to infection. Acute infections represent life-threatening medical emergencies in this group of children. If the plasma leucine concentration increases rapidly during illness, BCAAs should be removed from the diet immediately and intravenous therapy started.
Disorders of Organic Acid Metabolism
The organic acid disorders are a group of disorders characterized by the accumulation in the blood of nonamino acid organic acids. Most of the organic acids are efficiently excreted in the urine. Diagnosis is based on excretion of compounds not normally present or the presence of abnormally high amounts of other compounds in the urine. The clinical course can vary but is generally marked by vomiting, lethargy, hypotonia, dehydration, seizures, and coma. Survivors often have permanent neurologic damage.
Pathophysiology
Propionic acidemia is a defect of propionyl–coenzyme A (CoA) carboxylase in the pathway of propionyl-CoA to methylmalonyl-CoA, as illustrated in Figure 44-4. Metabolic acidosis with a marked anion gap and hyperammonemia is characteristic. Long-chain ketonuria may also be present.
At least five separate enzyme deficiencies have been identified that result in methylmalonic acidemia or aciduria. The defect of methylmalonyl-CoA mutase apoenzyme is the most frequently identified. In methylmalonic academia, the clinical features are similar to those of propionic acidemia. Acidosis is common, and diagnosis is confirmed by the presence of large amounts of methylmalonic acid in blood and urine. Other findings include hypoglycemia, ketonuria, and elevation of plasma ammonia and lactate levels.
Ketone utilization disorders (mitochondrial 2-methylacetoacetyl-CoA thiolase deficiency or similar enzyme defect) are disorders of isoleucine and ketone body metabolism. Affected individuals are usually older infants or toddlers who present with ketoacidosis, vomiting, and lethargy with secondary dehydration and sometimes coma. This event often is preceded by febrile illness or fasting.
Medical Treatment
Some patients with propionic acidemia may respond to pharmacologic doses of biotin. Long-term outcome in propionic acidemia is variable; hypotonia and cognitive delay may result even in children who are diagnosed early and who receive rigorous treatment. Liver damage and cardiomyopathy are possible sequelae. Liver transplantation may limit intellectual disability and cardiac changes (de Baulny et al., 2005).
Methylmalonic acidemia patients may respond to pharmacologic doses of vitamin B12. Responsiveness should be determined as part of the diagnostic process (Venditti, 2007). Progressive renal insufficiency is often a long-term outcome. Developmental delay is often caused by early and or prolonged hyperammonemia.
For ketone utilization use disorders, the treatment is dietary protein restriction (usually 1.5 g/kg of body weight per day); supplementation with L-carnitine, a carrier of fatty acids across the mitochondrial membranes; avoidance of fasting by providing small, frequent meals that consist primarily of complex carbohydrates; and the use of Bicitra (sodium citrate-citric acid) to treat ketoacidosis.
Medical Nutrition Therapy
The goals of managing acute episodes of propionic acidemia and methylmalonic acidemia are to achieve and maintain normal nutrient intake and biochemical balance. Maintenance of energy and fluid intake is important to prevent tissue catabolism and dehydration. Intravenous fluids correct electrolyte imbalances, and abnormal metabolites are removed through urinary excretion, promoted by a high fluid intake. Relapses of metabolic acidosis may result from excessive protein intake, infection, constipation, or unidentified factors. Parents become skilled at identifying early signs of illness. Treatment for these episodes must be rapid because coma and death can occur quickly.
Restricted protein intake is an essential component of the treatment of organic acid disorders. A daily protein intake of 1 to 1.5 g/kg of body weight is often an effective treatment modality for infants who have a mild form of the disorder. This can be supplied by diluting standard infant formula to decrease the protein content and adding a protein-free formula to meet nutrient needs. Specialized formulas that limit threonine and isoleucine and omit methionine and valine are used, as clinically indicated, to support an adequate protein intake and growth.
Requirements for the limited amino acids may vary widely. Growth rate, state of health, residual enzyme activity, and overall protein and energy intakes must be monitored carefully and correlated with plasma amino acid levels. Adequate hydration is critical to maintain metabolic equilibrium. Food refusal and lack of appetite may complicate nutrition therapy, which compromises medical management.
Disorders of Urea Cycle Metabolism
All urea cycle defects result in an accumulation of ammonia in the blood. The clinical signs of elevated ammonia are vomiting and lethargy, which may progress to seizures, coma, and ultimately death. In infants the adverse effects of elevated ammonia levels are rapid and devastating. In older children symptoms of elevated ammonia may be preceded by hyperactivity and irritability. Neurologic damage may result from frequent and severe episodes of hyperammonemia. The severity and variation of the clinical courses of some urea cycle defects may be related to the degree of residual enzyme activity (Brusilow and Horwich, 2010). The common urea cycle defects are discussed in a progression that proceeds around the urea cycle, as shown in Figure 44-5.
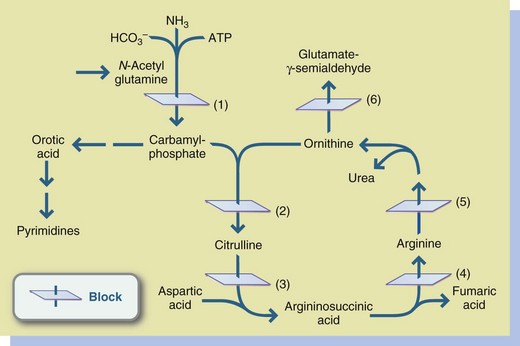
FIGURE 44-5 Urea cycle disorders. 1, Carbamyl-phosphate synthetase deficiency; 2, Ornithine carbamyl transferase deficiency; 3, Argininosuccinic acid synthetase (citrullinemia); 4, Argininosuccinic acid lyase (argininosuccinic aciduria); 5, Arginase deficiency (arginemia); 6, Adenosine triphosphate.
Pathophysiology
Ornithine transcarbamylase (OTC) deficiency is an X-linked recessive disorder marked by blockage in the conversion of ornithine and carbamyl phosphate to citrulline. OTC deficiency is identified by hyperammonemia and increased urinary orotic acid, with normal levels of citrulline, argininosuccinic acid, and arginine. Severe OTC deficiency is usually lethal in males. Heterozygous females with various degrees of enzyme activity may not demonstrate symptoms until they are induced by stress, as from an infection, or a significant increase in protein intake.
Citrullinemia is the result of a deficiency of argininosuccinic acid synthetase in the metabolism of citrulline to argininosuccinic acid. Citrullinemia is identified by markedly elevated citrulline levels in the urine and blood. Symptoms may be present in the neonatal period, or they may develop gradually in early infancy. Poor feeding and recurrent vomiting occur which, without immediate treatment, progress to seizures, neurologic abnormalities, and coma.
Argininosuccinic aciduria (ASA) results from deficiency of argininosuccinate lyase, which is involved in the metabolism of argininosuccinic acid to arginine. ASA is identified by the presence of argininosuccinic acid in urine and blood. L-Arginine must be supplemented to provide an alternative pathway for waste nitrogen excretion.
Carbamyl-phosphate synthetase (CPS) deficiency is the result of deficient activity of CPS. The onset is usually in the early neonatal period, with vomiting, irritability, marked hyperammonemia, respiratory distress, altered muscle tone, lethargy, and often coma. Specific laboratory findings usually include low plasma levels of citrulline and arginine and normal orotic acid levels in urine.
Medical Treatment
Acute episodes of illness are managed by discontinuing protein intake and administering intravenous fluids and glucose to correct dehydration and provide energy. If hyperammonemia is severe, peritoneal dialysis, hemodialysis, or exchange transfusion may be required. Intravenous sodium benzoate or other alternative pathway compounds have been beneficial in reducing the hyperammonemia.
Neurologic outcome and intellectual development in individuals with urea cycle disorders vary, with a range from normal IQ and motor function to severe intellectual disability and cerebral palsy. Although information on long-term follow-up is limited, the use of alternative pathways for waste nitrogen excretion and a protein-restricted food pattern may improve the outcome.
Medical Nutrition Therapy
Nutritional management of patients who have urea cycle disorders is a challenging task (Singh et al., 2005). The aim of therapy for the urea cycle disorders is to prevent or decrease hyperammonemia and the detrimental neurologic consequences associated with it. Treatment is similar for all of the disorders. For mildly affected infants a standard infant formula can be diluted to provide protein at 1 to 1.5 g/kg body weight per day. The energy, vitamin, and mineral concentrations can be brought up to recommended intake levels with the addition of a protein-free formula. However, for most individuals, specialized formulas are needed to adjust protein composition in an effort to limit ammonia production.
The amount of protein tolerated is affected by variables such as specific enzyme defect, age-related growth rate, health status, level of physical activity, amount of free amino acids administered, energy needs, residual enzyme function, and the use of nitrogen-scavenging medications. Recommendations must take family lifestyle and the individual’s eating behaviors into consideration (Singh et al., 2005). Long-term therapy consists of restricting dietary protein to 1 to 2 g/kg/day, depending on individual tolerance. For most infants and children with these disorders, except for arginase deficiency, L-arginine supplements are required to prevent arginine deficiency and assist in waste nitrogen excretion. L-Arginine is supplemented based on individual needs, except in the case of arginase deficiency (Brusilow and Howich, 2010). Phenylbutyrate or other compounds that enhance alternative metabolic pathways are usually required to normalize ammonia levels.
Protein-Restricted Diets
Infants and children with urea cycle defects or organic acidemias generally require restricted-protein intakes and specialized formulas. The amount of protein prescribed is based on the individual’s tolerance or residual enzyme activity, age, and projected growth rate. The highest protein level tolerated should be given to ensure adequate growth and a margin of nutritional safety. The steps for effective planning of a low-protein food pattern are shown in Box 44-1.
In general, low-protein or restricted-protein food patterns can be formulated from readily available, lower-protein infant, toddler, and table foods. Special low-protein foods (see Table 44-5) can be used to provide energy, texture, and variety in the food pattern without appreciably increasing the protein load. The prescribed protein level can be met by adding a protein-free or specialized formula product to standard infant formula. Supplementing carbohydrate and fat makes up the resultant energy deficit.
Specialty formulas are available when needed. The appropriate choice depends on the level of protein restriction, age, and condition of the child. The usual recommendations for energy density and vitamin and mineral composition are generally appropriate to support growth for the infant or child. Osmolarity of the formula must be considered; feedings of no more than 400 mOsm/L of solution have been recommended.
Disorders of Carbohydrate Metabolism
Disorders of carbohydrate metabolism vary in presentation, clinical course, and outcome. Galactosemia may present in the early newborn period as life-threatening seizures and sepsis. Hereditary fructose intolerance may present during midinfancy when solids that contain offending ingredients are introduced. Glycogen storage diseases (GSDs) may present at the time when feedings are spaced and subsequent hypoglycemia appears. All of these disorders require early and aggressive nutritional therapy.
Galactosemia
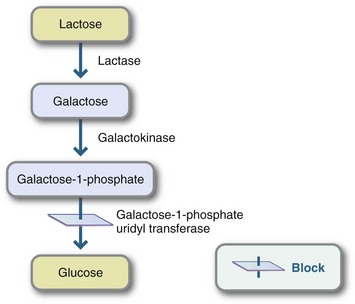
Galactosemia, a high level of plasma galactose-1-phosphate combined with galactosuria, is found in two autosomal-recessive metabolic disorders: galactokinase deficiency and galactose-1-phosphate uridyltransferase deficiency, which is also called “classic galactosemia.” Illness generally occurs within the first 2 weeks of life. Symptoms are vomiting, diarrhea, lethargy, failure to thrive, jaundice, hepatomegaly, and cataracts. Infants with galactosemia may be hypoglycemic and susceptible to infection from gram-negative organisms. If the condition is not treated, death frequently ensues secondarily to septicemia.
Pathophysiology
Galactosemia results from a disturbance in the conversion of galactose to glucose because of the absence or inactivity of one of the enzymes shown in Figure 44-6. The enzyme deficiency causes an accumulation of galactose, or galactose and galactose-1-phosphate, in body tissues. In addition, expanded newborn screening programs have identified many newborns with Duarte galactosemia. These infants have one allele for galactosemia and one for “Duarte” galactosemia and are often said to have “DG/G galactosemia.” The Duarte allele produces approximately 5% to 20% of the GALT enzyme. Little is known about the natural history of DG galactosemia; apparently infants and children develop normally without medical complications.
Medical Treatment
When diagnosis and therapy are delayed, intellectual disability can result (Waisbren, 2006). With early diagnosis and treatment, physical and motor development should proceed normally. However, intellectual achievement may be depressed. Patients often have IQs of 85 to 100; visual-perceptual and speech difficulties are common (Kaufman et al., 1995). Ovarian failure affects approximately 95% of women with galactosemia (Forges et al., 2006).
Medical Nutrition Therapy
Galactosemia is treated by lifelong galactose restriction. Although galactose is required for the production of galactolipids and cerebrosides, it can be produced by an alternative pathway if galactose is omitted from the diet. Galactose restriction mandates strict avoidance of all milk and milk products and lactose-containing foods because lactose is hydrolyzed into galactose and glucose. Effective galactose restriction requires careful reading of food product labels. Milk is added to many products, and lactose often appears in the coating of the tablet form of medications. Infants are fed soy-based formula. Some fruits and vegetables contain significant amounts of galactose. Whether these sources of galactose contribute to the pathophysiology characteristics of the disorder is unclear. Table 44-9 presents a low-galactose food pattern.
TABLE 44-9
Food Lists for Low-Galactose Food Pattern
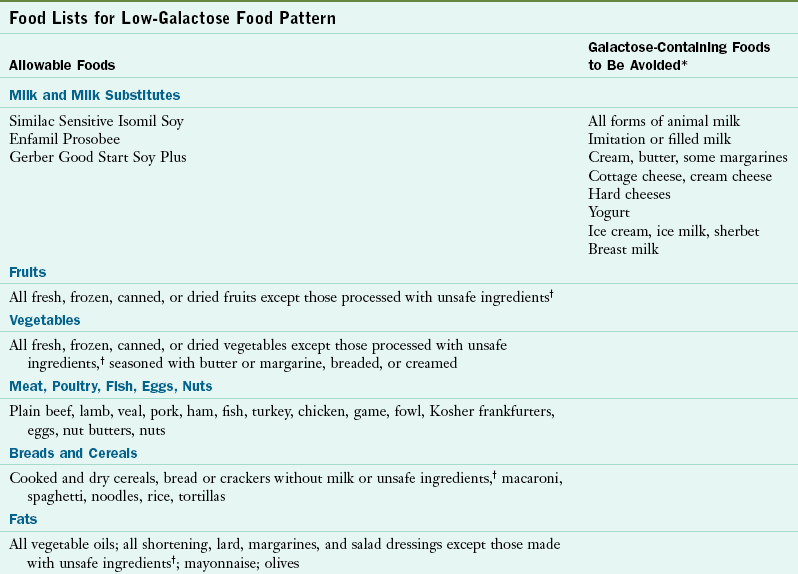
*Lactose is often used as a pharmaceutical bulking agent, filler, or excipient; thus tablets, tinctures, and vitamin and mineral mixtures should be evaluated carefully for galactose content. The Physician’s Desk Reference now lists active and inactive ingredients in medications, as well as manufacturers’ telephone numbers.
†Unsafe ingredients include milk, buttermilk, cream, lactose, galactose, casein, caseinate, whey, dry milk solids, or curds. Labels should be checked regularly and carefully because formulations of products change often.
Medical opinions differ about the intensity and duration of treatment for Duarte galactosemia. Many centers eliminate galactose from the diets of these children for the first year of life; other centers do not.
Glycogen Storage Diseases
Glycogen storage diseases (GSDs) reflect an inability to metabolize glycogen to glucose. There are a number of possible enzyme defects along the pathway. The most common of the GSD disorders are types I and III. Their symptoms are poor physical growth, hypoglycemia, hepatomegaly, and abnormal biochemical parameters, especially for cholesterol and triglycerides. Advances in the treatment of GSDs may improve the quality of life for affected children (Bali and Chen, 2010).
Pathophysiology
GSD type Ia is a defect in the enzyme glucose-1-6-phosphatase, which impairs the formation of new glucose (gluconeogenesis) and the breakdown of glycogen from storage (glycogenolysis). The affected person is unable to metabolize glycogen stored in the liver. Severe hypoglycemia can result and cause irreparable damage.
Amylo-1, 6-glucosidase deficiency (GSD III or debrancher enzyme deficiency) prevents glycogen breakdown beyond branch points. This disorder is similar to GSD I in that glycogenolysis is inefficient but gluconeogenesis is amplified to help maintain glucose production. The symptoms of GSD III are usually less severe and range from hepatomegaly to severe hypoglycemia (Dagli et al., 2010).
Medical Treatment
The outcome of treatment has been good. The hazard of severe hypoglycemic episodes is diminished, physical growth is improved, and liver size is decreased. The risk of progressive renal dysfunction is not entirely eliminated by current treatment, but liver transplantation for some types of GSD (e.g., type Ib) is sometimes an option. Treatment protocols for the GSDs are still evolving. The protocols include various kinds of carbohydrates at various doses during the day and night. Individual tolerance, body weight, state of health, ambient temperature, and physical activity all play important roles in designing the specific pattern of carbohydrate administration. The goal for all of the protocols remains the same: normalization of blood glucose levels.
Medical Nutrition Therapy
The rationale for intervention is to maintain plasma glucose in a normal range and prevent hypoglycemia by providing a constant supply of exogenous glucose. Administration of raw cornstarch at regular intervals and a high–complex carbohydrate, low-fat dietary pattern are advocated to prevent hypoglycemia. Some infants and children do very well with oral cornstarch administration, whereas others require glucose polymers administered via continuous-drip gastric feedings to prevent hypoglycemic episodes during the night (Bali and Chen, 2010; Dagli et al., 2010). The dose of cornstarch should be individualized; doses of 1.6 to 2.5 g/kg at 4- to 6-hour intervals are generally effective for young children with GSD I (Bali and Chen, 2010. The glucose vehicle suggested is a lactose-free formula. Iron supplementation is required to maintain adequate hematologic status because cornstarch interferes with iron absorption.
Disorders of Fatty Acid Oxidation
Recent laboratory advancements have enabled identification of fatty acid oxidation disorders such as medium-chain acyl-CoA dehydrogenase deficiency (MCAD) and long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) (Figure 44-7). Children who are not identified by newborn screening usually present during periods of fasting or clinical illness with symptoms of variable severity, including failure to thrive, episodic vomiting, and hypotonia.
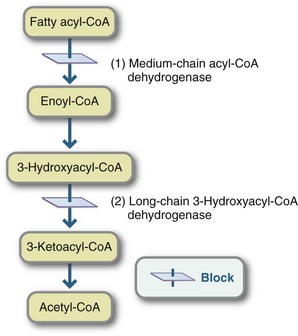
FIGURE 44-7 Mitochondrial fatty acid oxidation disorders: 1, Medium-chain acyl-Coenzyme A dehydrogenase deficiency, the most common fatty acid oxidation disorder. 2, Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency.
Pathophysiology
Children with MCAD who present clinically typically have hypoglycemia without urine ketones, lethargy, seizures, and coma. Children with LCHAD become hypoglycemic and demonstrate abnormal liver function, reduced or absent ketones in the urine, and often secondary carnitine deficiency. They may also have hepatomegaly and acute liver disease. Hypoglycemia can progress quickly and be fatal (Matern and Rinaldo, 2005; Roe and Ding, 2010).
Medical Nutrition Therapy
The concept underlying effective treatment for fatty acid oxidation disorders is straightforward: avoidance of fasting. This is accomplished by the regularly spaced intake of foods that provide an adequate energy intake and are high in carbohydrates. A low-fat diet is advocated because fats are not effectively metabolized. Consumption of not more than 30% of energy as fat has been recommended; some individuals require more restriction. Supplementation with L-carnitine, a substance that functions as a carrier of fatty acids across the mitochondrial membranes, is recommended by some centers. Children often do very well with three meals and three snacks offered at regular intervals. Most children may require additional carbohydrate, either a complex carbohydrate snack or uncooked cornstarch before bed, based on individual ability to maintain blood glucose levels throughout the night (Matern and Rinaldo, 2005). Depending on the disorder, supplementation with specific fatty acids (e.g., medium-chain fats for disorders that involve blocks of long-chain metabolism) may be indicated.
Role of the Nutritionist in Genetic Metabolic Disorders
The role of the pediatric nutrition specialist in the treatment of metabolic disorders is a complex one that requires expertise in MNT for the specific disorder. Preparation and competency requires access to detailed information about the disorders and treatment modalities. A family-centered counseling approach, knowledge of feeding-skill development, and understanding of behavior modification techniques, as well as the support and counsel of a team of health care providers involved in the care of the patient, are also required. Nutrition intervention is often a lifelong consideration. Specific objectives of nutrition care are shown in Box 44-2.
 Clinical Scenario
Clinical Scenario
The 1-day newborn screening test result for phenylalanine for a 7-lb, 4-oz male child was 3 mg/dL (180 mmol/L). The infant was breast-fed with no supplemental formula. A repeat sample was requested to further document the phenylalanine concentration in the infant’s blood. The result from this sample, collected on day 5 of life, was 24 mg/dL (1440 mmol/L). To confirm the diagnosis for this child, which was considered to be “presumptive positive,” a quantitative sample was obtained, and phenylalanine and tyrosine levels were measured. On day 9 of life the serum phenylalanine concentration was 25.5 mg/dL (1530 mmol/L), and the tyrosine level was 1.1 mg/dL (60.5 mmol/L); phenylalanine to tyrosine ratio was 23 : 2.
To provide adequate protein and energy intake and at the same time decrease the serum phenylalanine concentration, a phenylalanine-free formula was introduced at standard dilution without a phenylalanine supplement. Within 24 hours the infant’s serum phenylalanine concentration had decreased to 16.5 mg/dL (990 mmol/L) while the infant was being provided an intake of 16 oz of formula. Within 48 hours the level was 8.8 mg/dL (528 mmol/L), with an intake of 18 oz of formula. At this point, standard infant formula was added to bring the calculated phenylalanine intake to approximately 60 mg/kg and to maintain a generous protein and energy intake for this 3.6-kg infant.
Phenylalanine concentrations were measured on alternate days for 4 days, and the levels were 7.6 mg/dL (456 mmol/L) and 5.6 mg/dL (336 mmol/L), respectively. In subsequent weeks growth and serum phenylalanine concentrations continued to be monitored carefully, and energy and phenylalanine intakes were adjusted as necessary to maintain blood phenylalanine concentrations between 1 and 6 mg/dL (60-360 mmol/L) and to maintain growth in appropriate channels. The parents are apprehensive about preparing formula.
Nutrition Diagnostic Statement
Nutrition-related knowledge deficit related to the need for and preparation of a specialized formula (used for treatment of phenylketonuria in infants), as evidenced by a new diagnosis and the parents’ asking for information
1. What is the expected energy requirement for this infant with phenylketonuria?
2. What baseline formula would you use for this infant to provide phenylalanine at 60 mg/kg, formula at 20 kcal/oz, and protein and energy intakes at recommended levels?
3. What are the growth expectations for this infant?
4. What steps would you take if the plasma phenylalanine concentration exceeded 6 mg/dL (360 mmol/L) on subsequent measurements?
American College of Medical Genetics (ACMG)
Genetic Metabolic Dietitians International (GMDI)
MedlinePlus: Metabolic Disorders
http://www.nlm.nih.gov/medlineplus/metabolicdisorders.html
National Newborn Screening and Genetics Resource Center
References
Acosta, PB. Recommendations for protein and energy intakes by patients with phenylketonuria. Eur J Pediatr. 1996;155:S121.
Bali, S, Chen, YT. Glycogen storage disease type 1. Gene reviews. http://www.geneclinics.org, 2010. [Accessed 17 March 2011 from].
Blau, N, et al. Optimizing the use of sapropterin (BH4) in the management of phenylketonuria. Molec Genet Metab. 2010;96:158.
Bosch, AM, et al. The course of life and quality of life of early and continuously treated Dutch patients with phenylketonuria. J Inherit Metab Dis. 2006;29:576.
Brosco, JP, et al. Universal newborn screening and adverse medical outcomes: a historical note. Ment Retard Dev Disabil Res Rev. 2006;12:262.
Brusilow, SW, Horwich, AL. Urea cycle enzymes. In: Valle D, et al, eds. The online metabolic and molecular bases of inherited disease. New York: McGraw Hill, 2010.
Chuang, DT, Shih, VE. Maple syrup urine disease (branched-chain ketoaciduria). In: Valle D, et al, eds. The online metabolic and molecular bases of inherited disease. New York: McGraw Hill, 2010.
Cleary, MA, et al. Randomised controlled trial of essential fatty acid supplementation in phenylketonuria. Eur J Clin Nutr. 2006;60:915.
Dagli, A, et al. Glycogen storage disease type III. GeneReviews. http://www.geneclinics.org, 2010. [Accessed 1 August 2010 from].
de Baulny, HO, et al. Methylmalonic and propionic acidaemias: management and outcome. J Inherit Metab Dis. 2005;28:415.
Forges, T, et al. Pathophysiology of impaired ovarian function in galactosaemia. Human Reprod Update. 2006;12:573.
Hoffman, B. Impact of longitudinal plasma leucine levels on the intellectual outcome in patients with classic MSUD. Pediatr Res. 2006;59:17.
Holt, LE, Snyderman, SE. The amino acid requirements of children. In: Nyhan WL, ed. Amino acid metabolism and genetic variation. New York: McGraw-Hill, 1967.
Ievers-Landis, CE, et al. Situational analysis of dietary challenges of the treatment regimen for children and adolescents with phenylketonuria and their primary caregivers. J Dev Behav Pediatr. 2005;26:186.
Kaufman, FR, et al. Cognitive functioning, neurologic status and brain imaging in classical galacotsemia. Eur J Pediatr. 1995;154(7 Suppl 2):S2.
Kaye, CI, et al. Newborn screening fact sheets. Pediatrics. 2006;118:e934.
Koch, R, et al. Psychosocial issues and outcomes in maternal PKU. Mol Genet Metab. 2010;99:S68.
Lee, PJ, et al. Maternal phenylketonuria: report from the United Kingdom Registry 1978-1997. Arch Dis Child. 2005;90:143.
Matern, D, Rinaldo, P. Medium-chain acyl-coenzyme A dehydrogenase deficiency. GeneReviews. http://www.geneclinics.org, 2005. [Accessed 1 August 2010 from].
Maternal Child Health Bureau (MCHB). Advisory Committee on Heritable Disorders and Genetic Diseases in Newborns and Children. http://www.hrsa.gov/heritabledisorderscommittee/default.htm, 2007. [Accessed 1 August 2010 from].
Maternal Child Health Bureau. Newborn screening: toward a uniform screening panel and system. Genet Med. 2006;8(suppl 1):1S.
Roe, CR, Ding, J. Mitochondrial fatty acid oxidation disorders. In: Valle D, et al, eds. The online metabolic and molecular bases of inherited disease. New York: McGraw Hill, 2010.
Rose, HJ, et al. Fat intakes of children with PKU on low phenylalanine diets. J Hum Nutr Diet. 2005;18:395.
Saugstad, LF. From genetics to epigenetics. Nutr Health. 2006;18:285.
Singh, RH, et al. Nutritional management of urea cycle disorders. Crit Care Clin. 2005;21:S27.
Strauss, KA, et al. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6:557.
Therrell, BL, et al. Status of newborn screening programs in the United States. Pediatrics. 2006;117:S212.
Venditti, CP. Methylmalonic acidemia. GeneReviews. http://www.geneclinics.org, 2007. [Accessed 1 August 2010 from].
Waisbren, SE. Newborn screening for metabolic disorders. JAMA. 2006;296:993.
Waisbren, SE, Azen, C. Cognitive and behavioral development in maternal phenylketonuria offspring. Pediatrics. 2003;112:1544.
Waisbren, SE, et al. Phenylalanine blood levels and clinical outcomes in phenylketonuria: a systemic literature review and meta-analyisis. Mol Genet Metab. 2007;92:63.