63 Hepatitis Viruses
A 43-year-old woman complained of fatigue, nausea, and abdominal discomfort. She had a slight fever, her urine was dark yellow, and her abdomen was distended and tender. Serologic assays demonstrated the presence of immunoglobulin M (IgM) antibody to the hepatitis B core antigen (HBcAg) and the presence of the hepatitis B surface antigen (HBsAg) and the hepatitis Be antigen (HBeAg). She also had IgG to hepatitis A virus.
1. Which aspects are common to hepatitis disease and which are specific to hepatitis B virus (HBV)?
A 41-year-old intravenous drug abuser complained of fatigue, nausea, and abdominal discomfort. He had a slight fever, his urine was dark yellow, and his abdomen was distended and tender. Serologic assays demonstrated the presence of IgG antibody to the HBsAg but no hepatitis antigens nor other anti-HBV antibodies. Reverse transcriptase polymerase chain reaction (RT-PCR) analysis of his serum detected the hepatitis C virus genome.
4. Is this person infected with HBV? Has this person ever been infected with HBV?
5. What is the most likely disease outcome for this patient?
Answers
1. Nausea and abdominal discomfort, slight fever, dark yellow urine, and distended and tender abdomen. The time course of disease, possibility for chronic infection (unlikely for HAV, very likely for HCV), and the serology of infection are different for HBV.
2. HBV is transmitted in contaminated blood products, tissues, and semen.
3. Infection with HBV can be prevented by screening the blood supply to prevent transmission by this route, by safe sex, by not sharing or reusing syringe needles, and by abiding by universal blood precautions. Vaccination is the best means of prevention. HBV disease can be treated with reverse transcriptase inhibitors, such as lamivudine, entecavir, or tenofovir.
4. No, this person has never been infected but has been immunized and developed antibodies to HBsAg in the vaccine for HBV.
5. Chronic infection is the most likely outcome for this patient.
6. Treatment includes pegylated interferon, ribavirin with a new protease inhibitor.
The hepatitis alphabet of viruses includes at least six viruses, A through E, and G (Table 63-1). Although the target organ for each of these viruses is the liver and the basic hepatitis symptoms are similar, they differ greatly in their structure, mode of replication, mode of transmission, and in the time course and sequelae of the disease they cause. Hepatitis A virus (HAV) and hepatitis B virus (HBV) are the classic hepatitis viruses, and hepatitis C, G, E, and hepatitis D virus (HDV), the delta agent, are called non-A, non-B hepatitis (NANBH) viruses. Other viruses can also cause hepatitis.
Each of the hepatitis viruses infects and damages the liver, causing the classic icteric symptoms of jaundice and the release of liver enzymes. The specific virus causing the disease can be distinguished by the course, nature, and serology of the disease. These viruses are readily spread because infected people are contagious before, or even without, showing symptoms.
Hepatitis A, which is sometimes known as infectious hepatitis, (1) is caused by a picornavirus, a ribonucleic acid (RNA) virus; (2) is spread by the fecal-oral route; (3) has an incubation period of approximately 1 month, after which icteric symptoms start abruptly; (4) does not cause chronic liver disease; and (5) rarely causes fatal disease.
Hepatitis B, previously known as serum hepatitis, (1) is caused by a hepadnavirus with a deoxyribonucleic acid (DNA) genome; (2) is spread parenterally by blood or needles, by sexual contact, and perinatally; (3) has a median incubation period of approximately 3 months, after which icteric symptoms start insidiously; (4) is followed by chronic hepatitis in 5% to 10% of patients; and (5) is causally associated with primary hepatocellular carcinoma (PHC). More than one third of the world’s population has been infected with HBV, resulting in 1 to 2 million deaths per year. The incidence of HBV is decreasing, however, especially in infants, because of the development and use of the HBV subunit vaccine.
Hepatitis C virus (HCV) is also widely prevalent, with more than 170 million carriers of the disease. HCV is spread by the same routes as HBV but is more prone to cause chronic disease. HCV also increases risk for PHC. HCV is a flavivirus with an RNA genome. Hepatitis G virus (HGV) is also a flavivirus and causes chronic infections. Hepatitis E virus (HEV) is an enteric encapsidated virus with an RNA genome in its own family, and its disease resembles HAV.
Hepatitis D, or delta hepatitis, is unique in that it requires actively replicating HBV as a “helper virus” and occurs only in patients who have active HBV infection. HBV provides an envelope for HDV RNA and its antigens. HDV exacerbates the symptoms caused by HBV.
Hepatitis A Virus
HAV causes infectious hepatitis and is spread by the fecal-oral route. HAV infections often result from consumption of contaminated water, shellfish, or other food. HAV is a picornavirus and was formerly called enterovirus 72, but it has been placed into a new genus, Heparnavirus, on the basis of its unique genome.
Structure
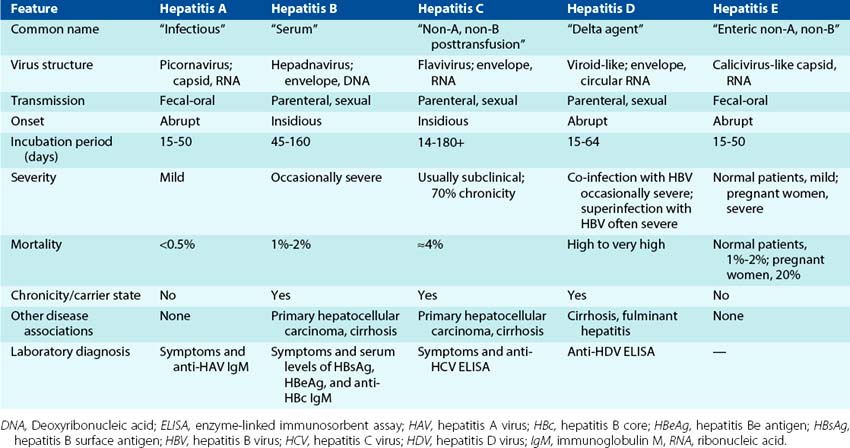
HAV has a 27-nm, naked, icosahedral capsid surrounding a positive-sense, single-stranded RNA genome consisting of approximately 7470 nucleotides (Figure 63-1). The HAV genome has a VPg protein attached to the 5′ end and a polyadenylate sequence attached to the 3′ end. The capsid is even more stable than other picornaviruses to acid and other treatments (Box 63-1). There is only one serotype of HAV.
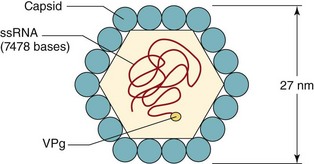
Figure 63-1 The picornavirus structure of hepatitis A virus. The icosahedral capsid is made up of four viral polypeptides (VP1 to VP4). Inside the capsid is a single-stranded, positive-sense ribonucleic acid (ssRNA) that has a genomic viral protein (VPg) on the 5′ end.
Replication
HAV replicates like other picornaviruses (see Chapter 54). It interacts specifically with the HAV cell receptor 1 glycoprotein (HAVCR-1, also known as T-cell immunoglobuluin and mucin domain protein [TIM-1]) expressed on liver cells and T cells. The structure of HAVCR-1 can vary for different individuals, and specific forms correlate with severity of disease. Unlike other picornaviruses, however, HAV is not cytolytic and is released by exocytosis. Laboratory isolates of HAV have been adapted to grow in primary and continuous monkey kidney cell lines, but clinical isolates are difficult to grow in cell culture.
Pathogenesis
HAV is ingested and probably enters the bloodstream through the epithelial lining of the oropharynx or the intestines to reach its target, the parenchymal cells of the liver (Figure 63-2). The virus replicates in hepatocytes and Kupffer cells. Virus is produced in these cells and is released into the bile and from there into the stool. Virus is shed in large quantity into the stool approximately 10 days before symptoms of jaundice appear or antibody can be detected.
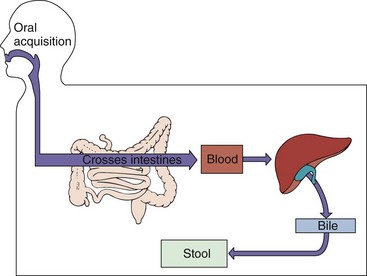
HAV replicates slowly in the liver without producing apparent cytopathic effects. Although interferon limits viral replication, natural killer cells and cytotoxic T cells are required to eliminate infected cells. Antibody, complement, and antibody-dependent cellular cytotoxicity also facilitate clearance of the virus and induction of immunopathology. Icterus, resulting from damage to the liver, occurs when cell-mediated immune responses and antibody to the virus can be detected. Antibody protection against reinfection is lifelong.
The liver pathology caused by HAV infection is indistinguishable histologically from that caused by HBV. It is most likely caused by immunopathology and not virus-induced cytopathology. However, unlike HBV, HAV cannot initiate a chronic infection and is not associated with hepatic cancer.
Epidemiology
Approximately 40% of acute cases of hepatitis are caused by HAV (Box 63-2). The virus spreads readily in a community because most infected people are contagious 10 to 14 days before symptoms occur, and 90% of infected children and 25% to 50% of infected adults have inapparent but productive infections.
Box 63-2
Epidemiology of Hepatitis A Virus (HAV) and Hepatitis E Virus (HEV)
The virus is released into stool in high concentrations and is spread via the fecal-oral route. Virus is spread in contaminated water, in food, and by dirty hands. HAV is resistant to detergents, acid (pH of 1), and temperatures as high as 60° C, and it can survive for many months in fresh water and salt water. Raw or improperly treated sewage can taint the water supply and contaminate shellfish. Shellfish, especially clams, oysters, and mussels, are important sources of the virus because they are efficient filter feeders and can therefore concentrate the viral particles, even from dilute solutions. This is exemplified by an epidemic of HAV that occurred in Shanghai, China in 1988, when 300,000 people were infected with the virus as the result of eating clams obtained from a polluted river.
HAV outbreaks usually originate from a common source (e.g., water supply, restaurant, day-care center). Asymptomatic shedding and a long (15 to 40 days) incubation period make it difficult to identify the source. Day-care settings are a major source for spread of the virus among classmates and their parents. A further problem is posed by the fact that because the children and personnel in day-care centers may be transient, the number of contacts at risk for HAV infection from a single day-care center can be great.
A relatively high incidence of HAV infection is directly related to poor hygienic conditions and overcrowding. Most people infected with HAV in developing countries are children who have mild illness and then lifelong immune protection against reinfection. In the populations of more highly developed countries, infection occurs later in life. The seropositivity rate of adults ranges from a low of 13% of the adult population in Sweden to highs of 88% in Taiwan and 97% in Yugoslavia, with a 41% to 44% rate in the United States.
Clinical Syndromes
The symptoms caused by HAV are very similar to those caused by HBV and stem from immune-mediated damage to the liver. As already noted, disease in children is generally milder than that in adults and is usually asymptomatic. The symptoms occur abruptly 15 to 50 days after exposure and intensify for 4 to 6 days before the icteric (jaundice) phase (Figure 63-3). Initial symptoms include fever, fatigue, nausea, loss of appetite, and abdominal pain. Dark urine (bilirubinuria), pale stool, and then jaundice may be accompanied by abdominal pain and itch. Jaundice is observed in 70% to 80% of adults but in only 10% of children (<6 years of age). Symptoms generally wane during the jaundice period. Viral shedding in the stool precedes the onset of symptoms by approximately 14 days but stops before the cessation of symptoms. Complete recovery occurs 99% of the time within 2 to 4 weeks of onset.
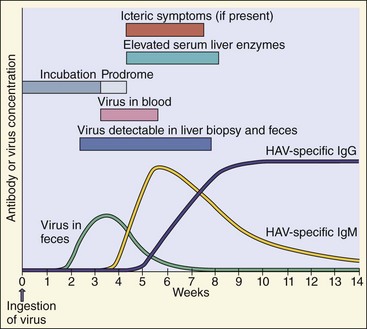
Figure 63-3 Time course of hepatitis A virus (HAV) infection. IgG, Immunoglobulin G; IgM, immunoglobulin M.
Fulminant hepatitis in HAV infection occurs in one to three persons per 1000 and is associated with an 80% mortality rate. Unlike HBV, immune complex–related symptoms (e.g., arthritis, rash) rarely occur in people with HAV disease.
Laboratory Diagnosis
The diagnosis of HAV infection is generally made on the basis of the time course of the clinical symptoms, the identification of a known infected source, and most reliably, the results yielded by specific serologic tests. The best way to demonstrate an acute HAV infection is by finding anti-HAV IgM, as measured by an enzyme-linked immunosorbent assay (ELISA) or radioimmunoassay. Virus isolation is not performed because efficient tissue culture systems for growing the virus are not available.
Treatment, Prevention, and Control
The spread of HAV is reduced by interrupting the fecal-oral spread of the virus. This is accomplished by avoiding potentially contaminated water or food, especially uncooked shellfish. Proper hand washing, especially in day-care centers, mental hospitals, and other care facilities, is vitally important. Chlorine treatment of drinking water is generally sufficient to kill the virus.
Prophylaxis with immune serum globulin given before or early in the incubation period (i.e., less than 2 weeks after exposure) is 80% to 90% effective in preventing clinical illness.
Killed HAV vaccines have been approved by the U.S. Food and Drug Administration (FDA) and are available for all children and for adults at high risk for infection, especially travelers to endemic regions. The vaccine is administered to infants at 2 years of age and can be administered with the HBV vaccine to adults. A live HAV vaccine is in use in China. There is only one serotype of HAV, and HAV infects only humans, factors that help ensure the success of an immunization program.
Hepatitis B Virus
HBV is the major member of the hepadnaviruses. Other members of this family (Box 63-3) include woodchuck, ground squirrel, and duck hepatitis viruses. These viruses have limited tissue tropisms and host ranges. HBV infects the liver and, to a lesser extent, the kidneys and pancreas of only humans and chimpanzees. Advances in molecular biology have made it possible to study HBV despite the limited host range of the virus and difficult cell-culture systems in which to grow it.
Box 63-3
Unique Features of Hepadnaviruses
Virus has enveloped virion containing partially double-stranded, circular DNA genome.
Replication is through a circular RNA intermediate.
Virus encodes and carries a reverse transcriptase.
Virus encodes several proteins (HBsAg [L, M, S]; HBe/HBc antigens) that share genetic sequences but with different in-frame start codons.
HBV has a strict tissue tropism to the liver.
HBV-infected cells produce and release large amounts of HBsAg particles lacking DNA.
DNA, Deoxyribonucleic acid; HBc, hepatitis B core antigen; HBe, hepatitis Be antigen; HBsAg, hepatitis B surface antigen; HBV, hepatitis B virus; RNA, ribonucleic acid.
Structure
HBV is a small, enveloped DNA virus with several unusual properties (Figure 63-4). Specifically, the genome is a small, circular, partly double-stranded DNA of only 3200 bases. Although a DNA virus, it encodes a reverse transcriptase and replicates through an RNA intermediate.
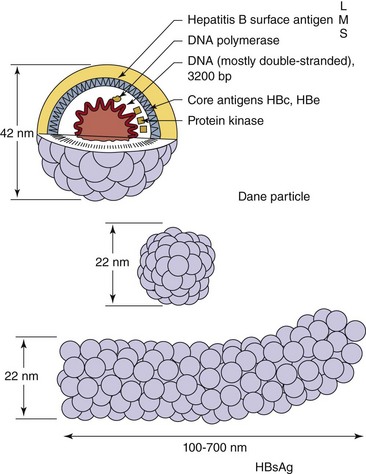
Figure 63-4 Hepatitis B virus (Dane particle) and hepatitis B surface antigen (HBsAg) particles. The spheric HBsAg consists mainly of the S form of HBsAg, with some M. The filamentous HBsAg has S, M, and L forms. bp, Base pair; DNA, deoxyribonucleic acid; L, gp42; M, gp36; S, gp27.
The virion, also called the Dane particle, is 42 nm in diameter. The virions are unusually stable for an enveloped virus. They resist treatment with ether, low pH, freezing, and moderate heating. These characteristics assist transmission from one person to another and hamper disinfection.
The HBV virion includes a protein kinase and a polymerase with reverse transcriptase and ribonuclease H activity, as well as a P protein attached to the genome. All of this is surrounded by an icosahedral capsid formed by the hepatitis B core antigen (HBcAg) and an envelope containing three forms of the glycoprotein hepatitis B surface antigen (HBsAg). A hepatitis Be antigen (HBeAg) protein shares most of its protein sequence with HBcAg but is processed differently by the cell, is primarily secreted into serum, does not self-assemble (like a capsid antigen), and expresses different antigenic determinants.
HBsAg-containing particles are released into the serum of infected people and outnumber the actual virions. These particles can be spheric (but smaller than the Dane particle) or filamentous (see Figure 63-4). They are immunogenic and were processed into the first commercial vaccine against HBV.
HBsAg, originally termed the Australia antigen, includes three glycoproteins (L, M, and S) encoded by the same gene and read in the same frame but translated into protein from different AUG (adenine, uracil, guanine) start codons. The S (gp27; 24 to 27 kDa) glycoprotein is completely contained in the M (gp36; 33 to 36 kDa) glycoprotein, which is contained in the L (gp42; 39 to 42 kDa) glycoprotein; all share the same C-terminal amino acid sequences. All three forms of HBsAg are found in the virion. The S glycoprotein is the major component of HBsAg particles; it self-associates into 22-nm spheric particles that are released from the cells. The filamentous particles of HBsAg found in serum contain mostly S but also small amounts of the M and L glycoproteins and other proteins and lipids. The glycoproteins of HBsAg contain the group-specific (termed a) and type-specific determinants of HBV (termed d or y and w or r). Combinations of these antigens (e.g., ady and adw) result in eight subtypes of HBV that are useful epidemiologic markers.
Replication
The replication of HBV is unique for several reasons (see Box 63-1). First, HBV has a distinctly defined tropism for the liver. Its small genome also necessitates economy, as illustrated by the pattern of its transcription and translation. In addition, HBV replicates through an RNA intermediate and produces and releases antigenic decoy particles (HBsAg) (Figure 63-5).
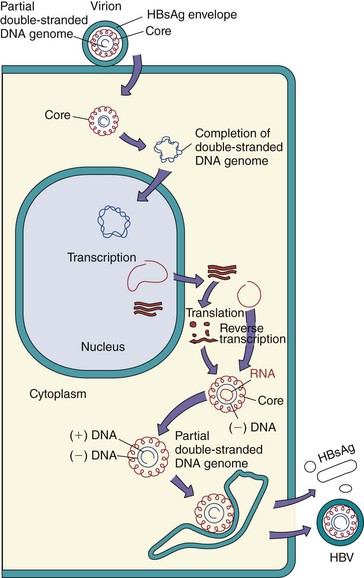
Figure 63-5 Replication of hepatitis B virus (HBV). After entry into the hepatocyte and uncoating of the nucleocapsid core, the partially double-stranded deoxyribonucleic acid (DNA) genome is completed by enzymes in the core and then delivered to the nucleus. Transcription of the genome produces four messenger RNAs (mRNAs), including an mRNA larger than the genome (3500 bases). The mRNA then moves to the cytoplasm and is translated into protein. Core proteins assemble around the 3500-base mRNA, and negative-sense DNA is synthesized by a reverse transcriptase activity in the core. The ribonucleic acid (RNA) is then degraded as a positive-sense (+) DNA is synthesized. The core is enveloped before completion of the positive-sense DNA and then released by exocytosis. HBsAg, hepatitis B surface antigen.
The attachment of HBV to hepatocytes is mediated by the HBsAg glycoproteins. Several liver cell receptors have been suggested, including the transferrin receptor, the asialoglycoprotein receptor, and human liver annexin V. The mechanism of entry is not known, but HBsAg binds to polymerized human serum albumin and other serum proteins, and binding and uptake of these proteins may facilitate virus uptake by the liver.
On penetration into the cell, the partial DNA strand of the genome is completed by being formed into a complete double-stranded DNA circle, and the genome is delivered to the nucleus. Transcription of the genome is controlled by cellular transcription elements found in hepatocytes. The DNA is transcribed from different starting points on the circle but have the same 3′ end. There are three major classes (2100, 2400, and 3500 bases) and two minor classes (900 bases) of overlapping messenger RNAs (mRNAs) (Figure 63-6). The 3500-base mRNA is larger than the genome. It encodes the HBc and HBe antigens, the polymerase, and a protein primer for DNA replication and acts as the template for replication of the genome. The HBe and HBc are related proteins that are translated from different in-phase start codons of closely related mRNA. This causes differences in their processing and structure, with shedding of the HBe and incorporation of HBc into the virion. Similarly, the 2100-base mRNA encodes the small and medium glycoproteins from different in-phase start codons. The 2400-base mRNA, which encodes the large glycoprotein, overlaps the 2100-base mRNA. The 900-base mRNA encodes the X protein, which promotes viral replication as a transactivator of transcription and as a protein kinase.
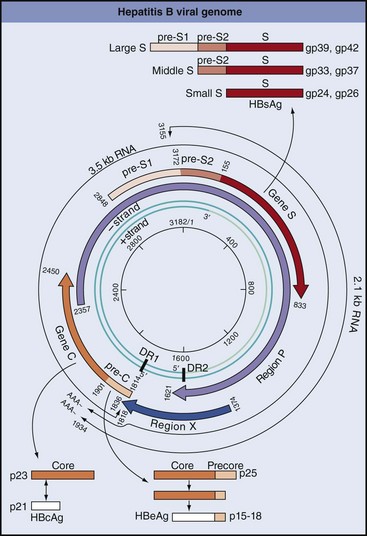
Figure 63-6 DNA, RNA, mRNA, and proteins of hepatitis B virus. The inner green circles represent the DNA genome with the nucleotide number at the center. DR1 and DR2 are direct repeat sequences of DNA and are important for replication and integration of the genome. The 3500-base transcript (outer black thin-line circle) is larger than the genome and is the template for replication of the genome. Bold arcs represent mRNA for viral proteins. Note that several proteins are translated from the same mRNA but from different AUG codons and that different mRNAs overlap. AAA, 3′ polyA (polyadenylate) at end of mRNA; AUG, adenine, uracil, guanine; C, C mRNA (hepatitis B core antigen [HBcAg]); E, E mRNA (hepatitis Be antigen [HBeAg]); HBsAg, hepatitis B surface antigen; l, large glycoprotein; m, medium glycoprotein; P, polymerase-protein primer for replication; s, small glycoprotein; S, S mRNA (HBsAg); X, X mRNA.
(Modified from Armstrong D, Cohen J: Infectious diseases, St Louis, 1999, Mosby.)
Replication of the genome utilizes the larger-than-genome 3500-base mRNA. This is packaged into the core nucleocapsid that contains the RNA-dependent DNA polymerase (P protein). This polymerase has reverse transcriptase and ribonuclease H activity, but HBV lacks the integrase activity of the retroviruses. The 3500-base RNA acts as a template, and negative-strand DNA is synthesized using a protein primer from the P protein, which remains covalently attached to the 5′ end. After this, the RNA is degraded by the ribonuclease H activity as the positive-strand DNA is synthesized from the negative-sense DNA template. However, this process is interrupted by envelopment of the nucleocapsid at HBsAg-containing intracellular membranes, thereby capturing genomes containing RNA-DNA circles with different lengths of RNA. Continued degradation of the remainder of the RNA in the virion yields a partly double-stranded DNA genome. The virion is then released from the hepatocyte by exocytosis without killing the cell, not by cell lysis.
The entire genome can also be integrated into the host cell chromatin. HBsAg, but not other proteins, can often be detected in the cytoplasm of cells containing integrated HBV DNA. The significance of the integrated DNA in the replication of the virus is not known, but integrated viral DNA has been found in hepatocellular carcinomas.
Pathogenesis and Immunity
HBV can cause acute or chronic, symptomatic or asymptomatic disease. Which of these occurs seems to be determined by the person’s immune response to the infection (Figure 63-7). Detection of both the HBsAg and the HBeAg components of the virion in the blood indicates the existence of an ongoing active infection. HBsAg particles continue to be released into the blood, even after virion release has ended and until the infection is resolved.
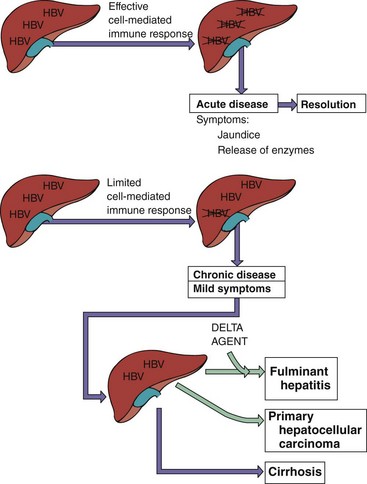
Figure 63-7 Major determinants of acute and chronic hepatitis B virus (HBV) infection. HBV infects the liver but does not cause direct cytopathology. Cell-mediated immune lysis of infected cells produces the symptoms and resolves the infection. Insufficient immunity can lead to chronic disease. Chronic HBV disease predisposes a person to more serious outcomes. Purple arrows indicate symptoms; green arrows indicate a possible outcome.
The major source of infectious virus is blood, but HBV can be found in semen, saliva, milk, vaginal and menstrual secretions, and amniotic fluid. The most efficient way to acquire HBV is through injection of the virus into the bloodstream (Figure 63-8). Common but less efficient routes of infection are sexual contact and birth.
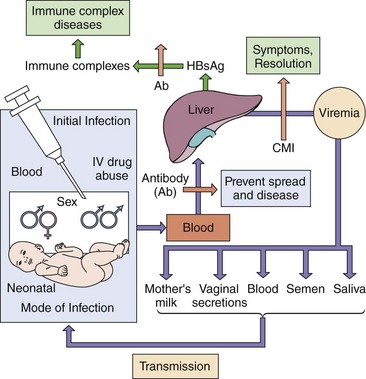
Figure 63-8 Spread of hepatitis B virus (HBV) in the body. Initial infection with HBV occurs through injection, heterosexual and homosexual sex, and birth. The virus then spreads to the liver, replicates, induces a viremia, and is transmitted in various body secretions in addition to blood to start the cycle again. Symptoms are caused by cell-mediated immunity (CMI) and immune complexes between antibody and hepatitis B surface antigen (HBsAg). IV, Intravenous.
The virus starts to replicate in the liver within 3 days of its acquisition, but as already noted, symptoms may not be observed for 45 days or longer, depending on the infectious dose, the route of infection, and the person. The virus replicates in hepatocytes with minimal cytopathic effect. Infection proceeds for a relatively long time without causing liver damage (i.e., elevation of liver enzyme levels) or symptoms. During this time, copies of the HBV genome integrate into the hepatocyte chromatin and remain latent. Intracellular buildup of filamentous forms of HBsAg can produce the ground-glass hepatocyte cytopathology characteristic of HBV infection.
Cell-mediated immunity and inflammation are responsible for causing the symptoms and effecting resolution of the HBV infection by eliminating the infected hepatocyte. Epitopes from the HBc antigen are prominent T-cell antigens. An insufficient T-cell response to the infection generally results in the occurrence of mild symptoms, an inability to resolve the infection, and the development of chronic hepatitis (see Figure 63-7). Chronic infection also exhausts CD8 T cells preventing them from killing infected cells. Antibody (as generated by vaccination) can protect against initial infection by preventing delivery of the virus to the liver. Later in the infection, the large amount of HBsAg in serum binds to and blocks the action of neutralizing antibody, which limits the antibody’s capacity to resolve an infection. Immune complexes formed between HBsAg and anti-HBs contribute to the development of hypersensitivity reactions (type III), leading to problems such as vasculitis, arthralgia, rash, and renal damage.
Antibody to HBc is present in serum but is nonprotective. The HBeAg protein, like HBsAg, is released into serum, and, during its production, anti-HBeAg is bound to the antigen and undetectable.
Infants and young children have an immature cell-mediated immune response and are less able to resolve the infection, but they suffer less tissue damage and milder symptoms. As many as 90% of infants infected perinatally become chronic carriers. Viral replication persists in these people for long periods.
During the acute phase of infection, the liver parenchyma shows degenerative changes consisting of cellular swelling and necrosis, especially in hepatocytes surrounding the central vein of a hepatic lobule. The inflammatory cell infiltrate is mainly composed of lymphocytes. Resolution of the infection allows the parenchyma to regenerate. Fulminant infections, activation of chronic infections, or co-infection with the delta agent can lead to permanent liver damage and cirrhosis.
Epidemiology
In the United States, more than 12 million people have been infected with HBV (1 out of 20), with 5000 deaths per year. In the world, one out of three people have been infected with HBV, with approximately a million deaths per year. More than 350 million people worldwide have chronic HBV infection. In developing nations, as many as 15% of the population may be infected during birth or childhood. High rates of seropositivity are observed in Italy, Greece, Africa, and Southeast Asia (Figure 63-9). In some areas of the world (southern Africa and southeastern Asia), the seroconversion rate is as high as 50%. PHC, a long-term sequela of the infection, is also endemic in these regions.
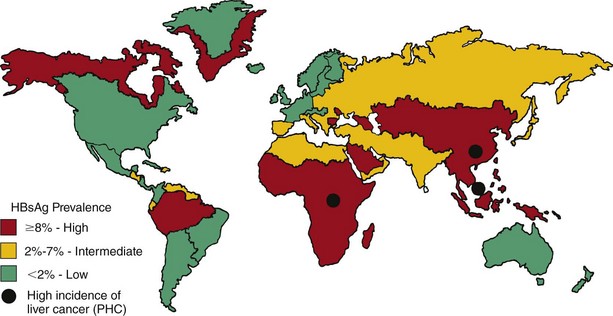
Figure 63-9 Worldwide prevalence of hepatitis B carriers and primary hepatocellular carcinoma (PHC). HBsAg, Hepatitis B surface antigen.
(Courtesy Centers for Disease Control and Prevention, Atlanta.)
The many asymptomatic chronic carriers with virus in blood and other body secretions foster the spread of the virus. In the United States, 0.1% to 0.5% of the general population are chronic carriers, but this is very low in comparison with many areas of the world. Carrier status may be lifelong.
The virus is spread by sexual, parenteral, and perinatal routes. Transmission occurs through contaminated blood and blood components by transfusion, needle sharing, acupuncture, ear piercing, or tattooing, and through very close personal contact involving the exchange of semen, saliva, and vaginal secretions (e.g., sex, childbirth) (see Figure 63-8). Medical personnel are at risk in accidents involving needlesticks or sharp instruments. People at particular risk are listed in Box 63-4. Sexual promiscuity and drug abuse are major risk factors for HBV infection. HBV can be transmitted to babies through contact with the mother’s blood at birth and in the mother’s milk. Babies born to chronic HBV-positive mothers are at highest risk for infection. Serologic screening of donor units in blood banks has greatly reduced the risk of acquisition of the virus from contaminated blood or blood products. Safer sex habits adopted to prevent human immunodeficiency virus (HIV) transmission and the administration of the HBV vaccine have also been responsible for decreasing the transmission of HBV.
Box 63-4
High-Risk Groups for Hepatitis B Virus Infection
People from endemic regions (i.e., China, parts of Africa, Alaska, Pacific Islands)
Babies of mothers with chronic hepatitis B virus
People with multiple sex partners, homosexual and heterosexual
Hemophiliacs and other patients requiring blood and blood product treatments
Health care personnel who have contact with blood
Residents and staff members of institutions for the mentally retarded
One of the major concerns about HBV is its association with PHC. This type of carcinoma probably accounts for 250,000 to 1 million deaths per year worldwide; in the United States, approximately 5000 deaths per year are attributed to PHC.
Clinical Syndromes
Acute Infection
As already noted, the clinical presentation of HBV in children is less severe than that in adults, and infection may even be asymptomatic. Clinically apparent illness occurs in as many as 25% of those infected with HBV (Figures 63-10 to 63-12).
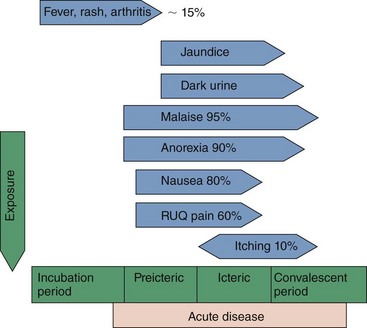
Figure 63-10 Symptoms of typical acute viral hepatitis B infection are correlated with the four clinical periods of this disease. RUQ, Right upper quadrant.
(Modified from Hoofnagle JH: Type A and type B hepatitis, Lab Med 14:705–716, 1983.)
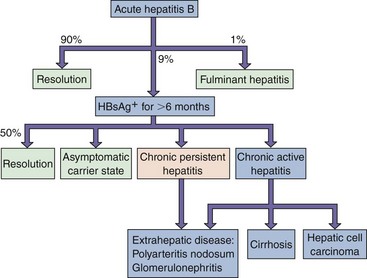
Figure 63-11 Clinical outcomes of acute hepatitis B infection. HBsAg, Hepatitis B surface antigen.
(Modified from White DO, Fenner F: Medical virology, ed 3, New York, 1986, Academic.)
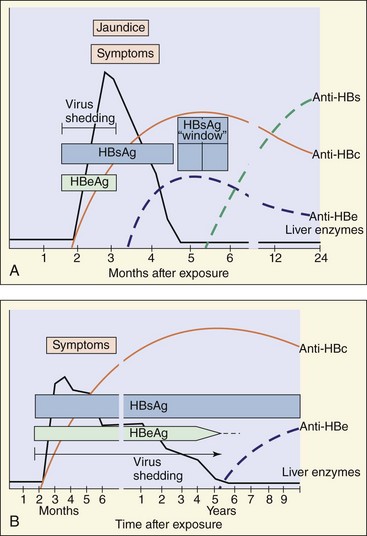
Figure 63-12 A, The serologic events associated with the typical course of acute hepatitis B disease. B, Development of the chronic hepatitis B virus carrier state. Routine serodiagnosis is difficult during the hepatitis B surface antigen (HBsAg) window, when HBs and anti-HBs are undetectable. Anti-HBc, Antibody to hepatitis B core antigen [HBcAg]; Anti-HBe, antibody to hepatitis Be antigen [HBeAg]; Anti-HBs, antibody to HBsAg.
(Modified from Hoofnagle JH: Serologic markers of hepatitis B virus infection, Annu Rev Med 32:1–11, 1981.)
HBV infection is characterized by a long incubation period and an insidious onset. Symptoms during the prodromal period may include fever, malaise, and anorexia, followed by nausea, vomiting, abdominal discomfort, and chills. The classic icteric symptoms of liver damage (e.g., jaundice, dark urine, pale stools) follow soon thereafter. Recovery is indicated by a decline in the fever and renewed appetite.
Fulminant hepatitis occurs in approximately 1% of icteric patients and may be fatal. It is marked by more severe symptoms and indications of severe liver damage, such as ascites and bleeding.
HBV infection can promote hypersensitivity reactions that are caused by immune complexes of HBsAg and antibody. These may produce rash, polyarthritis, fever, acute necrotizing vasculitis, and glomerulonephritis.
Chronic Infection
Chronic hepatitis occurs in 5% to 10% of people with HBV infections, usually after mild or inapparent initial disease. Approximately one third of these people have chronic active hepatitis with continued destruction of the liver leading to scarring of the liver, cirrhosis, liver failure, or PHC. The other two thirds have chronic passive hepatitis and are less likely to have problems. Chronic hepatitis may be detected accidentally by finding elevated liver enzyme levels on a routine blood chemistry profile. Chronically infected people are the major source for spread of the virus and are at risk for fulminant disease if they become co-infected with HDV.
Primary Hepatocellular Carcinoma
The World Health Organization estimates that 80% of all cases of PHC can be attributed to chronic HBV infections. The HBV genome is integrated into these PHC cells, and the cells express HBV antigens. PHC is usually fatal and is one of the three most common causes of cancer mortality in the world. In Taiwan, at least 15% of the population are carriers of HBV, and nearly half die of PHC or cirrhosis. PHC, like cervical cancer, is a vaccine-preventable human cancer.
HBV may induce PHC by promoting continued liver repair and cell growth in response to inflammation and tissue damage or by integrating into the host chromosome and stimulating cell growth directly. Such integration could stimulate genetic rearrangements or juxtapose viral promoters next to cellular growth-controlling genes. Alternatively, a protein encoded by the HBV X gene may transactivate (turn on) the transcription of cellular proteins and stimulate cell growth. The presence of the HBV genome may allow a subsequent mutation to promote carcinogenesis. The latency period between HBV infection and PHC may be as short as 9 years or as long as 35 years.
Laboratory Diagnosis
The initial diagnosis of hepatitis can be made on the basis of the clinical symptoms and the presence of liver enzymes in the blood (see Figure 63-12). However, the serology of HBV infection describes the course and the nature of the disease (Table 63-2). Acute and chronic HBV infections can be distinguished by the presence of HBsAg and HBeAg in the serum and the pattern of antibodies to the individual HBV antigens.
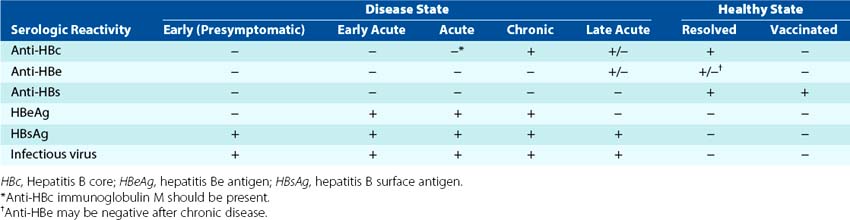
HBsAg and HBeAg are secreted into the blood during viral replication. The detection of HBeAg is the best correlate to the presence of infectious virus. A chronic infection can be distinguished by the continued finding of HBeAg, HBsAg, or both, and a lack of detectable antibody to these antigens. Antibody to HBsAg indicates resolution of infection or vaccination.
Antibody to HBcAg indicates current or prior infection by HBV and IgM anti-HBc is the best way to diagnose a recent acute infection, especially during the period when neither HBsAg nor anti-HBs can be detected (the window). Detection of antibodies to HBeAg and HBsAg is obscured during infection because the antibody is complexed with antigen in the serum.
The amount of virus in blood can be determined by quantitative genome assays using polymerase chain reaction (PCR) and related techniques. Knowing the virus load can help in following the course of chronic HBV infection and antiviral drug efficacy.
Treatment, Prevention, and Control
Hepatitis B immune globulin may be administered within a week of exposure and to newborn infants of HBsAg-positive mothers to prevent and ameliorate disease. Chronic HBV infection can be treated with drugs targeted at the polymerase—for example, lamivudine (2′3′dideoxy-3′-thiacytidine), which is also an HIV reverse transcriptase inhibitor—or the nucleoside analogues, adefovir dipivoxil and famciclovir. These FDA-approved treatments are taken for 1 year. Unfortunately, antiviral drug resistance can develop. Pegylated interferon-α can also be effective and is taken for at least 4 months.
Transmission of HBV in blood or blood products has been greatly reduced by screening donated blood for the presence of HBsAg and anti-HBc. Additional efforts to prevent transmission of HBV consist of avoiding sex with a carrier of HBV and avoiding the lifestyles that facilitate spread of the virus. Household contacts and sexual partners of HBV carriers are at increased risk, as are patients undergoing hemodialysis, recipients of pooled plasma products, health care workers exposed to blood, and babies born to HBV-carrier mothers.
Vaccination is recommended for infants, children, and especially people in high-risk groups (see Box 63-4). For newborns of HBsAg-positive mothers and people accidentally exposed either percutaneously or permucosally to blood or secretions from an HBsAg-positive person, vaccination is useful even after exposure. Immunization of mothers should decrease the incidence of transmission to babies and older children, also reducing the number of chronic HBV carriers. Prevention of chronic HBV will reduce the incidence of PHC.
The HBV vaccines form virus-like particles. The initial HBV vaccine was derived from the 22-nm HBsAg particles in human plasma obtained from chronically infected people. The current vaccine was genetically engineered by the insertion of a plasmid containing the S gene for HBsAg into a yeast, Saccharomyces cerevisiae. The protein self-assembles into particles, which enhances its immunogenicity.
The vaccine must be given in a series of three injections, with the second and third given 1 and 6 months after the first. The single serotype and limited host range (humans) help ensure the success of an immunization program.
Universal blood and body fluid precautions are used to limit exposure to HBV. It is assumed that all patients are infected. Gloves are required for handling blood and body fluids; wearing protective clothing and eye protection may also be necessary. Special care should be taken with needles and sharp instruments. HBV-contaminated materials can be disinfected with 10% bleach solutions, but unlike most enveloped viruses, HBV is not readily inactivated by detergents.
Hepatitis C and G Viruses
HCV was identified in 1989 after isolation of a viral RNA from a chimpanzee infected with blood from a person with NANBH. The viral RNA obtained from blood was converted to DNA with reverse transcriptase, its proteins were expressed, and antibodies from people with NANBH were then used to detect the viral proteins. These studies led to the development of ELISA and genomic and other tests for detection of the virus, which still cannot be grown in tissue culture.
HCV is the predominant cause of NANBH virus infections and was the major cause of posttransfusion hepatitis before routine screening of the blood supply for HCV. There are more than 170 million carriers of HCV in the world and more than 4 million in the United States. HCV is transmitted by means similar to HBV but has an even greater potential for establishing persistent, chronic hepatitis. Many HCV infected individuals are also infected with HBV or HIV. The chronic hepatitis often leads to cirrhosis and potentially to hepatocellular carcinoma. The significance of the HCV epidemic has become more apparent with the development of laboratory screening procedures.
Structure and Replication
HCV is the only member of the Hepacivirus genus of the Flaviviridae family. It is 30 to 60 nm in diameter, has a positive-sense RNA genome, and is enveloped. The genome of HCV (9100 nucleotides) encodes 10 proteins, including two glycoproteins (E1, E2). The viral RNA-dependent RNA polymerase is error prone and generates mutations in the glycoprotein and other genes. This generates antigenic variability. Such variability makes the development of a vaccine very difficult. There are six major groups of variants (clades), which differ in their worldwide distribution.
HCV infects only humans and chimpanzees. HCV binds to CD81 (tetraspanin) surface receptors, which is expressed on hepatocytes and B lymphocytes, and can also coat itself with low-density lipoprotein or very-low-density lipoprotein and then use the lipoprotein receptor to facilitate uptake into hepatocytes. The virus replicates like other flaviviruses. The virion assembles at and buds into the endoplasmic reticulum, becoming cell associated. HCV proteins inhibit apoptosis and interferon-α action by binding to the tumor necrosis factor receptor and to protein kinase R. These actions prevent the death of the host cell and promote persistent infection.
Pathogenesis
The ability of HCV to remain cell associated and prevent host cell death promotes persistent infection but results in liver disease later in life. Cell-mediated immune responses are responsible for both the resolution of infection and tissue damage. As for HBV, chronic infection can exhaust CD8 cytotoxic T cells to prevent resolution of infection. The extent of lymphocytic infiltration, inflammation, portal and periportal fibrosis, and lobular necrosis in liver biopsies can be used to grade the severity of disease. It has been suggested that the cytokines of inflammation and continual liver repair and induction of cell growth occurring during chronic HCV infection are predisposing factors in the development of PHC. Antibody to HCV is not protective.
Epidemiology
HCV is transmitted primarily in infected blood and sexually. Intravenous drug abusers, tattoo recipients, transfusion and organ recipients, and hemophiliacs receiving factors VIII or IX are at highest risk for infection (Box 63-5). Almost all (>90%) HIV-infected people who are or were intravenous drug users are infected with HCV. HCV is especially prevalent in southern Italy, Spain, central Europe, Japan, and parts of the Middle East (e.g., almost 20% of Egyptian blood donors are HCV positive). The high incidence of chronic asymptomatic infections promotes the spread of the virus in the population. Screening procedures have led to a reduction in the levels of transmission by blood transfusion and organ donation, but transmission by other routes is prevalent.
Box 63-5
Epidemiology of Hepatitis B, C, and D Viruses
Who Is at Risk?
Children: mild asymptomatic disease with establishment of chronic infection
Adults: insidious onset of hepatitis
HBV-infected people co-infected or superinfected with HDV: abrupt, more severe symptoms with possible fulminant disease
Adults with chronic HBV or HCV: at high risk for cirrhosis and primary hepatocellular carcinoma
Clinical Syndromes (Clinical Case 63-1)
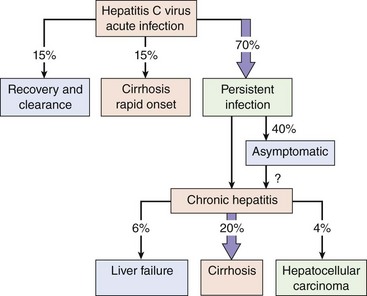
HCV causes three types of disease (Figure 63-13): (1) acute hepatitis with resolution of the infection and recovery in 15% of cases, (2) chronic persistent infection with possible progression to disease much later in life for 70% of infected persons, and (3) severe rapid progression to cirrhosis in 15% of patients. A viremia can be detected within 1 to 3 weeks of a transfusion of HCV-contaminated blood. The viremia lasts 4 to 6 months in people with an acute infection and longer than 10 years in those with a persistent infection. In its acute form, HCV infection is similar to acute HAV and HBV infection, but the inflammatory response is less intense, and the symptoms are usually milder. More commonly (>70% of cases), the initial disease is asymptomatic but establishes chronic persistent disease. The predominant symptom is chronic fatigue. The chronic, persistent disease often progresses to chronic active hepatitis within 10 to 15 years and to cirrhosis (20% of chronic cases) and liver failure (20% of cirrhotic cases) after 20 years. HCV-induced liver damage may be exacerbated by alcohol, certain medications, and other hepatitis viruses to promote cirrhosis. HCV promotes the development of hepatocellular carcinoma after 30 years in up to 5% of chronically infected patients.
Clinical Case 63-1
Hepatitis C Virus (HCV)
In a case reported by Morsica and associates (Scand J Infect Dis 33:116–120, 2001), a 35-year-old woman was admitted with malaise and jaundice. Elevated blood levels of bilirubin (71.8 µmol/l (normal value <17 µmol/l) and aspartate amino transferase (ALT) 410 IU/l (normal value <30 IU/l) indicated liver damage. Serology was negative for antibodies to hepatitis A, hepatitis B, hepatitis C, Epstein-Barr virus, cytomegalovirus, and HIV-1. However, HCV genomic RNA sequences were detected by reverse transcriptase polymerase chain reaction analysis. ALT levels peaked on the third week after admission and returned to normal by the eighth week. HCV genomes in blood were undetectable by the eighth week. Anti-HCV antibody was also detected by the eighth week. It was suspected that she was infected by her sexual partner, and this was confirmed by genotyping virus obtained from both individuals. Confirmation was provided by partial sequence analysis of the E2 gene from the two viral isolates. The 5% genetic divergence detected between the isolates was less than the ≈20% divergence expected for unrelated strains. Before the analysis, the sexual partner was unaware of his chronic HCV infection. Even more than HBV, which is also transmitted by sexual and parenteral means, HCV causes inapparent and chronic infections. Inapparent transmission of the virus, as in this case, enhances the spread of the virus. The molecular analysis demonstrates the genetic instability of the HCV genome, a possible mechanism for facilitating its chronic infection by changing its antigenic appearance to promote escape from the immune response.
Laboratory Diagnosis
The diagnosis and detection of HCV infection are based on ELISA recognition of anti-HCV antibody or detection of the RNA genome. Seroconversion occurs within 7 to 31 weeks of infection. ELISA is used for screening the blood supply from normal donors. As for HIV, results can be confirmed by Western immunoblot procedures. Antibody is not always detectable in viremic people, in immunocompromised patients, or in those receiving hemodialysis. Genome detection and quantitation by RT-PCR, branched-chain DNA, and related techniques is the gold standard for confirming a diagnosis of HCV and for following the success of antiviral drug therapy. Genetic assays are less strain specific and can detect HCV RNA in seronegative people.
Treatment, Prevention, and Control
Recombinant interferon-α or pegylated interferon (treated with polyethylene glycol to enhance its biologic lifetime), alone or with ribavirin, were the only known treatments for HCV. Treatment with pegylated interferon and ribavirin for extended periods may yield up to 50% recovery rates. This therapy can now be supplemented with either of two protease inhibitors, boceprevir or telaprevir. As for HIV, the addition of a protease inhibitor to the previous antiviral protocol is expected to make a significant difference in therapeutic efficacy.
The precautions for transmission of HCV are similar to that of HBV and other blood-borne pathogens. The blood supply and organ donors are screened for HCV. Persons with HCV should not share any personal care items and syringe needles that may get contaminated with blood and should practice safe sex. Alcohol drinking should be limited because it exacerbates the liver damage caused by HCV.
Hepatitis G Virus
HGV (also known as GB virus-C [GBV-C]) resembles HCV in many ways. HGV is a flavivirus, is transmitted in blood and has a predilection for chronic hepatitis infection. It is identified by detection of the genome by RT-PCR or other RNA detection methods.
Hepatitis D Virus
Approximately 15 million people in the world are infected with HDV (delta agent), and the virus is responsible for causing 40% of fulminant hepatitis infections. HDV is unique in that it uses HBV and target cell proteins to replicate and produce its one protein. It is a viral parasite, proving that “even fleas have fleas.” HBsAg is essential for packaging the virus. The delta agent resembles plant virus satellite agents and viroids in its size, genomic structure, and requirement for a helper virus for replication (Figure 63-14).
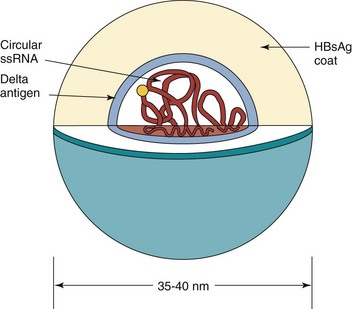
Figure 63-14 The delta hepatitis virion. HBsAg, Hepatitis B surface antigen; ssRNA, single-stranded RNA.
Structure and Replication
The HDV RNA genome is very small (approximately 1700 nucleotides), and unlike other viruses, the single-stranded RNA is circular and forms a rod shape as a result of its extensive base pairing. The virion is approximately the same size as the HBV virion (35 to 37 nm in diameter). The genome is surrounded by the delta antigen core, which in turn is surrounded by an HBsAg-containing envelope. The delta antigen exists as a small (24 kDa) or large (27 kDa) form; the small form is predominant.
The delta agent binds to and is internalized by hepatocytes in the same manner as HBV because it has HBsAg in its envelope. The transcription and replication processes of the HDV genome are unusual. The host cell’s RNA polymerase II makes an RNA copy to replicate the genome. The genome then forms an RNA structure called a ribozyme, which cleaves the RNA circle to produce an mRNA for the small delta antigen. The gene for the delta antigen is mutated by a cellular enzyme (double-stranded RNA-activated adenosine deaminase) during infection, thereby allowing production of the large delta antigen. Production of this antigen limits replication of the virus but also promotes association of the genome with HBsAg to form a virion, and the virus is then released from the cell.
Pathogenesis
Similar to HBV, the delta agent is spread in blood, semen, and vaginal secretions. However, it can replicate and cause disease only in people with active HBV infections. Because the two agents are transmitted by the same routes, a person can be co-infected with HBV and the delta agent. A person with chronic HBV can also be superinfected with the delta agent. More rapid, severe progression occurs in HBV carriers superinfected with HDV than in people co-infected with HBV and the delta agent because, during co-infection, HBV must first establish its infection before HDV can replicate (Figure 63-15), whereas superinfection of an HBV-infected person allows the delta agent to replicate immediately.
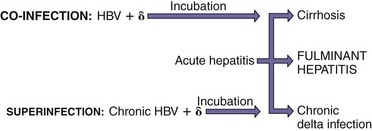
Figure 63-15 Consequences of delta virus infection. Delta virus (δ) requires the presence of hepatitis B virus (HBV) infection. Superinfection of a person already infected with HBV (carrier) causes more rapid, severe progression than co-infection (shorter arrow).
Replication of the delta agent results in cytotoxicity and liver damage. Persistent delta agent infection is often established in HBV carriers. Although antibodies are elicited against the delta agent, protection probably stems from the immune response to HBsAg because it is the external antigen and viral attachment protein for HDV. Unlike HBV disease, damage to the liver occurs as a result of the direct cytopathic effect of the delta agent combined with the underlying immunopathology of the HBV disease.
Epidemiology
The delta agent infects children and adults with underlying HBV infection (see Box 63-5), and people who are persistently infected with both HBV and HDV are a source for the virus. The agent has a worldwide distribution, infecting approximately 5% of the 3 × 108 HBV carriers and is endemic in southern Italy, the Amazon Basin, parts of Africa, and the Middle East. Epidemics of HDV infection occur in North America and Western Europe, usually in illicit drug users. HDV is spread by the same routes as HBV, and the same groups are at risk for infection, with parenteral drug abusers, hemophiliacs, and others receiving blood products at highest risk. Screening of the blood supply has reduced the risk for recipients of blood products.
Clinical Syndromes (Box 63-6)
The delta agent increases the severity of HBV infections. Fulminant hepatitis is more likely to develop in people infected with the delta agent than in those infected with the other hepatitis viruses. This very severe form of hepatitis causes altered brain function (hepatic encephalopathy), extensive jaundice, and massive hepatic necrosis, which is fatal in 80% of cases. Chronic infection with the delta agent can occur in people with chronic HBV.
Box 63-6
Clinical Summaries
Hepatitis A: A 37-year-old man develops fever, chills, headache, and fatigue 4 weeks after eating at a greasy-spoon diner. Within 2 days, he develops anorexia, vomiting, and right upper quadrant abdominal pain, followed by jaundice and dark-colored urine and stools persisting for 12 days. Then symptoms decrease.
Hepatitis B: A 27-year-old intravenous (IV) drug user develops symptoms of hepatitis 60 days after using a dirty needle.
Hepatitis B and D: A different IV drug user develops symptoms of hepatitis, altered mental capacity, and massive hepatic necrosis and then dies.
Hepatitis C: Elevated liver enzymes were detected in an individual during a physical examination. Hepatitis C virus in the blood was detected by enzyme-linked immunosorbent assay. Ten years later, cirrhosis and liver failure developed, requiring a liver transplant.
Laboratory Diagnosis
The presence of the agent can be noted by detecting the RNA genome, the delta antigen, or anti-HDV antibodies. ELISA and radioimmunoassay procedures are available for detection. The delta antigen can be detected in the blood during the acute phase of disease in a detergent-treated serum sample. RT-PCR techniques can be used to detect the virion genome in blood.
Treatment, Prevention, and Control
There is no known specific treatment for HDV hepatitis. Because the delta agent depends on HBV for replication and is spread by the same routes, prevention of infection with HBV prevents HDV infection. Immunization with HBV vaccine protects against subsequent delta virus infection. If a person has already acquired HBV, delta agent infection may be prevented by halting illicit intravenous drug use and avoiding HDV-contaminated blood products.
Hepatitis E Virus
HEV (E-NANBH) (the E stands for enteric or epidemic) is predominantly spread by the fecal-oral route, especially in contaminated water (see Box 63-2). HEV is unique but resembles the caliciviruses, based on its size (27 to 34 nm) and structure. Although HEV is found throughout the world, it is most problematic in developing countries. Epidemics have been reported in India, Pakistan, Nepal, Burma, North Africa, and Mexico.
The symptoms and course of HEV disease are similar to those of HAV disease; it causes only acute disease. However, the symptoms for HEV may occur later than those of HAV disease. The mortality rate associated with HEV disease is 1% to 2%, approximately 10 times that associated with HAV disease. HEV infection is especially serious in pregnant women (mortality rate of approximately 20%).
A 55-year-old man (patient A) was admitted to the hospital with fatigue, nausea, and abdominal discomfort. He had a slight fever, his urine was dark yellow, and his abdomen was distended and tender. He had returned from a trip to Thailand within the previous month.
A 28-year-old woman (patient B) was admitted to the hospital complaining of vomiting, abdominal discomfort, nausea, anorexia, dark urine, and jaundice. She admitted that she was a former heroin addict and that she had shared needles. In addition, she was 3 months pregnant.
A 65-year-old man (patient C) was admitted with jaundice, nausea, and vomiting 6 months after undergoing coronary artery bypass grafting.
1. What clinical or epidemiologic clues would have assisted in the diagnosis of hepatitis A, B, and C?
2. What laboratory tests would have been helpful in distinguishing the different hepatitis infections?
3. What was the most likely means of viral acquisition in each case?
4. What personal and public health precautions should have been taken to prevent the transmission of virus in each case?
5. Which of the patients was susceptible to chronic disease?
6. What laboratory tests distinguish acute from chronic HBV disease?
1. In each case, the time course and nature of the onset of disease would help in distinguishing the hepatitis viruses. Hepatitis A has an acute onset of disease, whereas the onset of hepatitis B and C are slower and more insidious.
2. Serologic tests would be helpful to determine recent exposure for all three hepatitis viruses and the stage of disease for hepatitis B. Genomic assays for HBV and HCV can also be performed (PCR [HBV], RT-PCR [HCV]).
3. Patient A probably has hepatitis A infection obtained from food. Patient B may have hepatitis B or C infection acquired from sharing contaminated syringe needles. Patient C is likely to have obtained HCV (but possibly HBV) from a blood transfusion obtained prior to the screening of the blood supply.
4. Disease from hepatitis A or B can be prevented by immunization of the individual. The risk for infection by hepatitis B and C can be reduced by careful screening of the blood supply and use of new syringe and needles and carefully sterilized surgical equipment. Attention to proper hygiene for food service workers and others, and properly disinfected water supplies, are important to limit HAV and HEV dissemination.
5. Patient B (HBV), and especially patient C (HCV), are susceptible to chronic disease. Most individuals infected with HCV experience chronic infection.
6. Acute and chronic HBV disease are discriminated serologically. The presence of HBsAg and hepatitis E antigen combined with the inability to detect antibodies to anti-HBsAg and anti-HBeAg are good indicators of chronic HBV.
7. HBV infection can be prevented by proper blood handling procedures, by not sharing needles when taking drugs, and by practicing safe, protected sex.