3 The tubules and the interstitium
By the end of the chapter you should be able to:
• Define reabsorption, secretion and excretion
• Outline the difference between primary and secondary active transport
• State the normal pH range. Outline how and why it is tightly controlled
• Explain what a ‘buffer’ is, and name the main buffering system in the body and describe how it works
• Explain how metabolic acidosis and metabolic alkalosis are differentiated by arterial blood gas results and give an outline of how you would correct each of them
• Explain the relation between plasma calcium and phosphate
• Describe the influence of three factors involved in their regulation
• List five drugs used in the management of hyperkalaemia and state how they work
• Understand the countercurrent mechanism of the loop of Henle
• Briefly describe the synthesis, storage and function of antidiuretic hormone (ADH)
• Describe how the kidneys’ ability to concentrate or dilute urine is altered in disease
• List the causes and the clinical presentation of syndrome of inappropriate ADH secretion (SIADH)
• Distinguish the two types of diabetes insipidus and explain how they differ
• State how the tubules are damaged in ATN and describe rhabdomyolysis
• List the predisposing factors for pyelonephritis
• Give three mechanisms by which multiple myeloma causes damage to the kidney
Overview
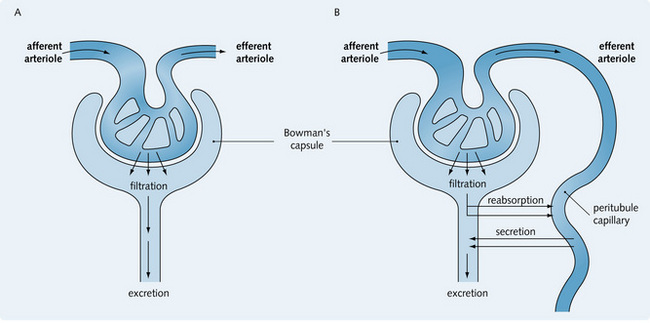
This chapter considers the convoluted tubules, loop of Henle, collecting duct and the interstitium between these tubules. The ultrafiltrate produced from the glomerular filter has a similar composition to plasma. The tubules and the interstitium are considered to act together, modifying the ultrafiltrate through reabsorption and secretion to produce the final urine. This process is important in maintaining a normal homeostasis of many ions in the body.
Transport processes in the renal tubule
Reabsorption, secretion and excretion are defined as follows:
• Reabsorption: the movement of a substance from the tubular fluid back into the circulation
• Secretion: the movement of substances from the blood into the tubular fluid via tubular cells (active transport) or intercellular spaces (passive process)
• Excretion: the removal of waste products from the blood and the net result of filtration, secretion and reabsorption of a substance.
Fig. 3.1 illustrates the processes that occur in the nephron and result in excretion of a substance. Two types of solute transport are involved:
1. Paracellular movement (between cells) across the tight junctions that connect the cells. This is driven by concentration and the electrical and osmotic gradients
2. Transcellular movement (through cells) via both the apical and basal membranes and the cell cytoplasm. Here, water follows the movement of solutes by osmosis.
Transport mechanisms
Diffusion
Diffusion is the movement of substances down their electrochemical gradient. It is a ‘passive’ process, as it does not require any metabolic energy or carrier molecules.
Facilitated diffusion
Like diffusion, this is also passive movement of substances along their electrochemical gradient, but it relies on a carrier molecule to transport substances across the membrane. Consequently, it is much faster than diffusion.
Primary active transport
This is an energy-dependent process in which substances cross cell membranes against their concentration and electrochemical gradients. It involves the hydrolysis of adenosine triphosphate (ATP), which provides chemical energy for the transport mechanism.
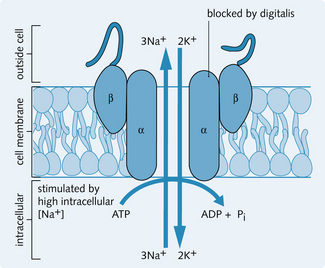
The most important active transporter is the Na+/K+ ATPase pump, which is found on the basal and basolateral membranes of tubular cells. It is involved in the active transport of Na+ from intracellular to extracellular spaces, allowing the nephron to reabsorb over 99% of the filtered Na+. This maintains a low Na+ concentration and a high K+ concentration in the cell (Fig. 3.2). The other primary active transporters on the tubular cell membrane are:
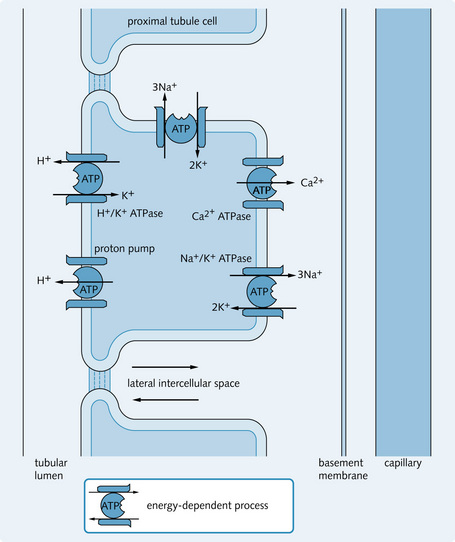
The ATP molecule is part of the protein structure in the primary active transporters. Energy is derived from the hydrolysis of the terminal phosphate bond of the ATP molecule to form adenosine diphosphate (ADP) and phosphate (Pi) (Fig. 3.3).
Secondary active transport
This process uses the energy produced from another process for transporting molecules (i.e. the transport of the solutes is coupled). The most important example of this mechanism involves the Na+/K+ ATPase pump as the driving force for the secretion and reabsorption of other solutes in which the energy is provided by the Na+ gradient.
The Na+/K+ ATPase pump creates an ionic gradient across the cell membrane, which allows the energy produced from the diffusion of Na+ into the cell as it moves along its electrochemical gradient to be used for active transport (i.e. against their electrochemical gradients) of other solutes.
Substances can move in two directions by the following processes:
• Symport: energy produced by the movement of Na+ is used to transport other substances in the same direction across the cell membrane, i.e. with the Na+ gradient (e.g. the Na+/K+/Cl– co-transporter in the thick ascending limb and the Na+/glucose in the cells of the proximal tubule cells)
• Antiport: movement of substances against their electrochemical gradient in the opposite direction to the Na+ gradient (e.g. the Ca2 +/Na+ and the H+/Na+ exchangers).
These processes are carried out by specific carrier proteins embedded in the cell membrane called transporters.
Ion channels
These are protein pores found on the epithelial cell membranes. They allow rapid transport of ions into the cell. Channels that are specific for Na+, K+ and Cl– are found on the apical membrane of all the cells lining the nephron. Although transport through these channels is very fast (106–108 ions/s) there are only about 100 channels per cell, compared with the slower (100 ions/s) but more numerous active transporters (107 transporters per cell).
Regulation of body fluid pH
Body fluid pH is tightly controlled because most enzyme reactions are sensitive to pH changes.
Buffers
Definition
A buffer is a mixture of a weak acid (HA) and a conjugate base. It undergoes minimal pH change when either an acid or a base is added to it:
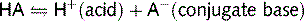
It can also be a mixture of a weak base (BH) and conjugate acid:
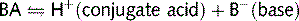
For example, if there is an increase in H+, the equations above shift to the left so that the extra H+ combines with the buffer and the H+ concentration in the body falls.
Henderson-Hasselbalch equation
This equation is used to determine the pH of body fluids from the buffer concentrations. The equations below show how it is derived mathematically. Ka is the acid dissociation constant, the equilibrium constant in terms of conjugate acids and bases, proton donators, and acceptors.
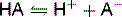
Equation 1. At equilibrium:
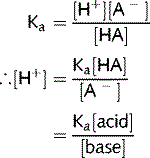
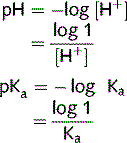
Equation 3. Combining equations 1 and 2 (the Henderson–Hasselbalch equation).
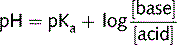
Physiological buffers
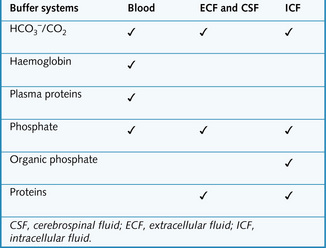
There are several buffer systems in the different body compartments (Fig. 3.4), of which the most important is the bicarbonate buffer system.
Bicarbonate buffer system
The bicarbonate buffer system is important in all body fluids. CO2 and H2O combine to form carbonic acid (H2CO3) in the presence of the enzyme carbonic anhydrase (CA). The H2CO3 then dissociates spontaneously to form bicarbonate ions (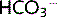 ) and H+:
) and H+:
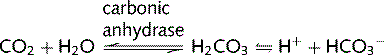
CO2 concentration is regulated by the lungs and 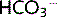 concentration is regulated by the kidneys. Therefore, pH regulation depends equally on both these organs. Substituting this equation in the Henderson–Hasselbalch equation, we get:
concentration is regulated by the kidneys. Therefore, pH regulation depends equally on both these organs. Substituting this equation in the Henderson–Hasselbalch equation, we get:
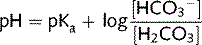
The acid dissociation constant, pKa, of the 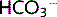 /pCO2 system = 6.1.
/pCO2 system = 6.1.
[H2CO3] is determined by dissolved CO2: [H2CO3] = 0.23 × pCO2 (0.23 is the CO2 solubility coefficient at 37 °C; pCO2 is the pressure of CO2 in the lungs in kPa)
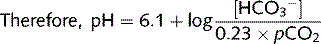
This is the key equation that can be used to relate pH, [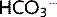 ] and pCO2. Normal values are:
] and pCO2. Normal values are:
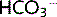 ]: 22–26 mmol/L
]: 22–26 mmol/L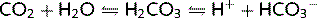
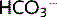 ] rises, [H+] will become lower, hence pH will rise. If pCO2 rises, the pH will become lower. This relationship is calculated precisely by the Henderson-Hasselbalch equation.
] rises, [H+] will become lower, hence pH will rise. If pCO2 rises, the pH will become lower. This relationship is calculated precisely by the Henderson-Hasselbalch equation.Renal regulation of plasma 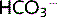
The kidneys control the concentration of 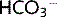 in the plasma. They can only reabsorb up to a certain amount of
in the plasma. They can only reabsorb up to a certain amount of 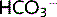 per second (Tm). If plasma
per second (Tm). If plasma 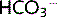 increases, Tm is exceeded and the kidneys cannot reabsorb all of it, some
increases, Tm is exceeded and the kidneys cannot reabsorb all of it, some 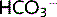 is excreted until the plasma level returns to normal.
is excreted until the plasma level returns to normal.
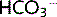 reabsorption by the proximal tubule
reabsorption by the proximal tubule
On the apical membrane Na+ reabsorption down its concentration gradient drives H+ secretion into the lumen. H+ combines with 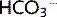 ions to form H2CO3 (carbonic acid). Carbonic anhydrase (CA) on the brush border of the cells catalyses the dissociation of H2CO3 to H2O + CO2 within the tubular lumen. Both H2O and CO2 diffuse freely into the cell, where they re-form H2CO3, this process being catalysed by intracellular CA.
ions to form H2CO3 (carbonic acid). Carbonic anhydrase (CA) on the brush border of the cells catalyses the dissociation of H2CO3 to H2O + CO2 within the tubular lumen. Both H2O and CO2 diffuse freely into the cell, where they re-form H2CO3, this process being catalysed by intracellular CA. 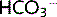 and Na+ are actively transported out of the cell across the basolateral membrane. H+ is secreted out of the cell into the tubular lumen and recycled to allow continuation of this cycle (Fig. 3.5). CA inhibitors suppress H+ secretion, leading to a fall in Na+ and
and Na+ are actively transported out of the cell across the basolateral membrane. H+ is secreted out of the cell into the tubular lumen and recycled to allow continuation of this cycle (Fig. 3.5). CA inhibitors suppress H+ secretion, leading to a fall in Na+ and 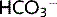 absorption, and thus act as a weak diuretic.
absorption, and thus act as a weak diuretic.
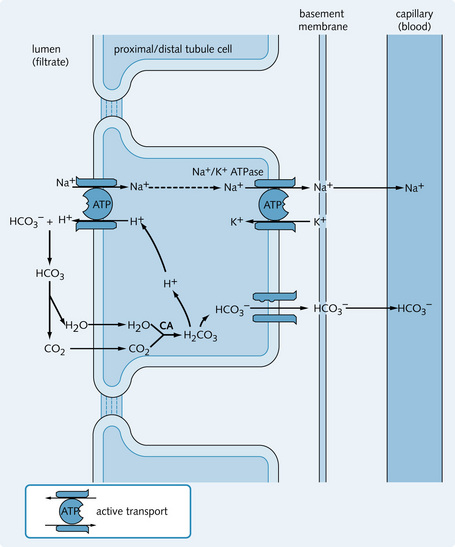
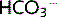 reabsorption in the proximal tubule cells. Secreted H+ combines with
reabsorption in the proximal tubule cells. Secreted H+ combines with 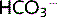 to form carbonic acid. This is broken down by carbonic anhydrase (CA) in the brush border to CO2 and H2O, which diffuse freely into the cell. The process is reversed inside the cell to re-form
to form carbonic acid. This is broken down by carbonic anhydrase (CA) in the brush border to CO2 and H2O, which diffuse freely into the cell. The process is reversed inside the cell to re-form 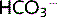 .
.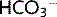 reabsorption by the distal tubule
reabsorption by the distal tubule
Only a small amount of 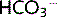 can be reabsorbed by the intercalated cells of the distal tubule. The Na+gradient is insufficient so H+ is pumped into the lumen using H+-ATPase. Little CO2 is produced in the lumen to enter the cell so CO2 from the cell's own metabolism is used to create
can be reabsorbed by the intercalated cells of the distal tubule. The Na+gradient is insufficient so H+ is pumped into the lumen using H+-ATPase. Little CO2 is produced in the lumen to enter the cell so CO2 from the cell's own metabolism is used to create 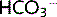 , which is moved into the plasma.
, which is moved into the plasma.
Conversion of alkaline phosphate to acid phosphate – luminal buffer
Alkaline phosphate Na2HPO4 and acid phosphate NaH2PO4 are present in the plasma in the ratio of 4:1. Both are filtered at the glomerulus. Alkaline phosphate is converted to acid phosphate, mainly in the distal tubule but also in the proximal tubule. This generates Na+ and binds H+ in the lumen. This feeds the system already described, generating 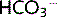 for the plasma (Fig. 3.5). The buffer is more effective at a lower pH.
for the plasma (Fig. 3.5). The buffer is more effective at a lower pH.
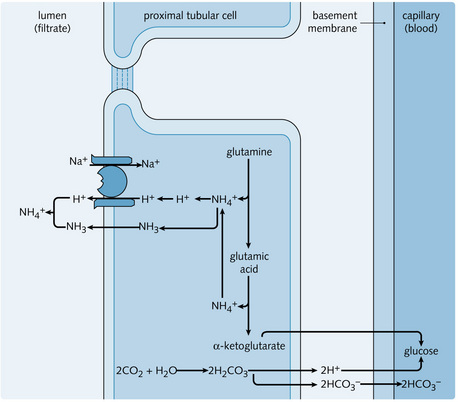
Ammonia secretion – luminal buffer
Deamination of glutamine in the proximal tubule yields ammonium ions (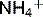 ) and
) and 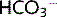 . This
. This 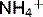 is secreted into the lumen and the
is secreted into the lumen and the 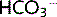 enters the plasma (Fig. 3.6). Fifty percent of
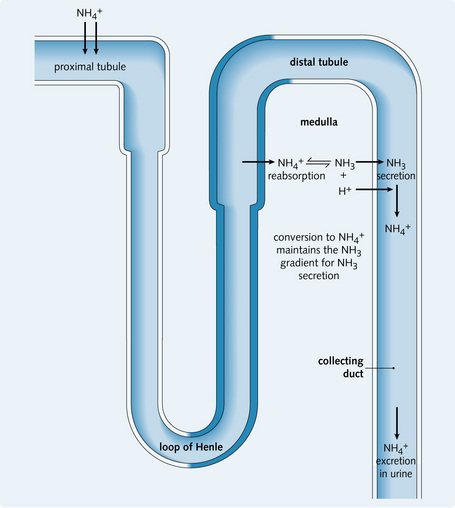
enters the plasma (Fig. 3.6). Fifty percent of 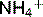 secreted into the proximal tubule is actually reabsorbed by the thick ascending limb of the loop of Henle and accumulates in the cells of the medullary interstitium (Fig. 3.7).
secreted into the proximal tubule is actually reabsorbed by the thick ascending limb of the loop of Henle and accumulates in the cells of the medullary interstitium (Fig. 3.7).
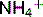 by nephrons.
by nephrons.Acidosis increases 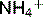 excretion because:
excretion because:
• Acidosis stimulates enzymes that deaminate glutamine, thereby increasing 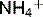 synthesis
synthesis
• Increased H+ secretion results in NH3 production, which in turn results in increased 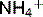 in the collecting tubules. The conversion of NH3 to
in the collecting tubules. The conversion of NH3 to 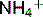 maintains a gradient for NH3 secretion. In this way, excess NH3/
maintains a gradient for NH3 secretion. In this way, excess NH3/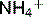 is removed from the medulla.
is removed from the medulla.
Acid–base disturbances
If the pH of the blood is too high, this is called alkalaemia, which is dangerous principally because it reduces the solubility of calcium salts. The resulting hypocalcaemia causes excitation of nerves leading to tetany.
If the pH of the blood is too low, this is called acidaemia, which can lead to potassium ions being pumped out of cells in exchange for hydrogen ions through exchange channels. The resulting hyperkalaemia can cause cardiac arrhythmias.
The processes that tend to cause alkalaemia or acidaemia are respectively called alkalosis and acidosis. There are four main types:
Metabolic disturbances result from changes in cellular metabolism or diet, so are independent of pCO2 changes. If the body fluid pH alters, the buffering system mechanism is activated. Thus, overall, there might be very little change in arterial pH despite acid–base imbalance.
• Compensation is the restoration of normal pH even when acid–base imbalance is still present
• Correction is the restoration of both the pH and acid–base imbalance to normal.
Arterial blood gases (ABGs) measure the pH, pO2 and pCO2 in an arterial blood sample. This can be used to diagnose an acid–base imbalance. The Davenport diagram is a graph of the plasma 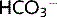 versus plasma pH, used to classify acid–base disturbances (Fig. 3.8). For example, if pH = 7.2 and pCO2 is 9.3 kPa, by looking at Fig. 3.8 it can be seen that [
versus plasma pH, used to classify acid–base disturbances (Fig. 3.8). For example, if pH = 7.2 and pCO2 is 9.3 kPa, by looking at Fig. 3.8 it can be seen that [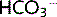 ] will be raised. This corresponds to a respiratory acidosis.
] will be raised. This corresponds to a respiratory acidosis.
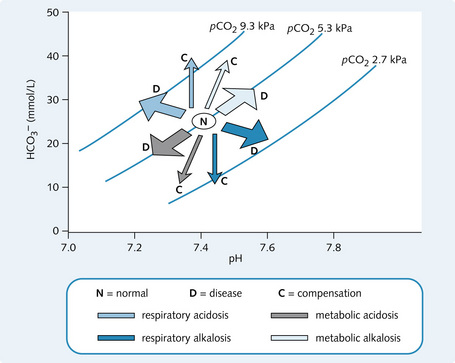
Examples of acid–base disturbances
Respiratory acidosis
The causes of respiratory acidosis are:
• Chronic obstructive pulmonary disease (COPD)
• Obstruction of the airway (e.g. tumour, foreign body)
• Drugs: general anaesthetic, morphine, barbiturates (respiratory centre depressant)
• Injuries and infections to the respiratory centre in the brainstem.
ABG results show pCO2 > 6.0 kPa and decreased pH. Clinically, the respiratory system cannot remove enough CO2, so CO2 increases together with pCO2. Therefore the following equation is shifted to the right:

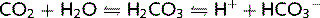
This results in elevated [H+] and [ ]. The extra H+ results in increased H+ secretion and increased
]. The extra H+ results in increased H+ secretion and increased  reabsorption. This restores pH, acting as a compensatory response. The acid–base disturbance is not corrected because the pCO2 and [
reabsorption. This restores pH, acting as a compensatory response. The acid–base disturbance is not corrected because the pCO2 and [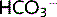 ] are still high. Correction requires hyperventilation to decrease pCO2.
] are still high. Correction requires hyperventilation to decrease pCO2.
Respiratory alkalosis
The causes of respiratory alkalosis are:
• Decreased pO2, which is detected by chemoreceptors in the carotid body resulting in hyperventilation and decreased pCO2
The ABG results show a pCO2 < 4.7 kPa and an elevated pH. Clinically too much CO2 is removed by the respiratory system. Therefore the following equation is shifted to the left:

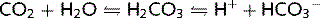
This causes [H+] to fall and hence an increased pH and a slight decrease in [HCO3-]. The compensatory response involves reduced H+ secretion, increased HCO3- excretion and decreased HCO3- reabsorption, thus restoring pH. Correction involves rectification of the underlying respiratory defect.
Metabolic acidosis
The causes of metabolic acidosis are:
• Excess metabolic production of H+ (e.g. lactate acidosis, diabetic ketoacidosis)
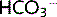 (e.g. severe diarrhoea, drainage from fistulae)
(e.g. severe diarrhoea, drainage from fistulae)ABG results show a normal pCO2 and decreased pH. There is an increase in [H+]. Therefore, the following equation is shifted to the left:
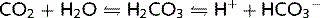
Consequently, [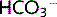 ] falls as it is used to ‘mop up’ the excess H+ ions.
] falls as it is used to ‘mop up’ the excess H+ ions.
The decreased pH stimulates respiration to cause hyperventilation. This respiratory compensation decreases pCO2 and returns the pH to normal, although 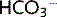 falls further. The fall in
falls further. The fall in 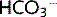 hinders the compensatory response of the kidneys, which is to increase
hinders the compensatory response of the kidneys, which is to increase 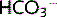 reabsorption and produce titratable acid.
reabsorption and produce titratable acid.
The anion gap represents the difference between plasma anions and cations and represents unaccounted anions (e.g. phosphates, ketones, lactate):
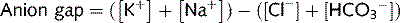
The normal range is 8–16 mmol/L. Changes in the anion gap help define the cause of a metabolic acidosis. Metabolic acidosis due to diarrhoea or renal tubular acidosis does not alter the anion gap. Acidosis caused by renal failure, diabetes or lactic acidosis increases the anion gap.
A primary decrease in plasma bicarbonate and a slight reduction in pH indicate a metabolic acidosis. This may be caused by failure of the kidneys to excrete H+ or reabsorb HCO3. This may be caused by renal tubular acidosis types I, II and IV. It is important to note that the acidosis refers to the plasma pH, not that of the tubular fluid and that the urine can be alkaline. Alkali is usually administered to correct the acidosis.
Metabolic alkalosis
The causes of metabolic alkalosis are:
• Loss of acid (e.g. vomiting, diarrhoea)
• Ingestion of alkali (e.g. antacid ingestion)
• Depleted ECF (e.g. haemorrhage, burns, excess diuretic use, contraction alkalosis).
ABG results show a normal pCO2 and an elevated pH because of a rise in plasma [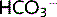 ]. Therefore, the following equation is shifted to the right:
]. Therefore, the following equation is shifted to the right:
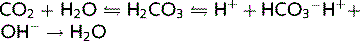
Consequently, [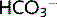 ] is increased.
] is increased.
Respiratory compensation occurs. The increase in pH acts on chemoreceptors, which reduce the ventilatory rate and so increase pCO2. The equation therefore shifts to the right and the pH returns to normal, but 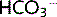 increases further. This hinders correction.
increases further. This hinders correction.
Fig. 3.9 summarizes acid–base disturbances.
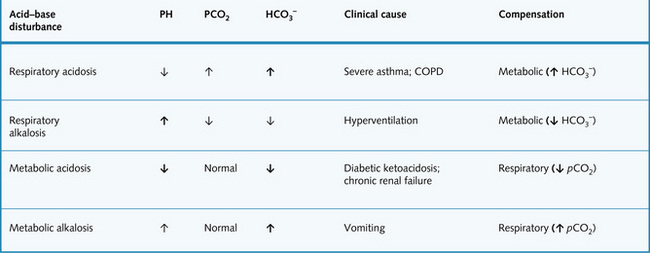
Regulation of calcium and phosphate
Calcium
Ca2 + is present mainly in bone but has an important extraskeletal function. The threshold potential of cell membranes of nerve and muscle for action potentials varies inversely with plasma calcium concentration. Thus it is important to keep calcium levels constant.
Calcium and phosphate homeostasis
Ca2 + and 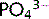 precipitate to form insoluble calcium phosphate and their concentrations in the blood are close to the saturation point. Adding more of one of the ions results in the precipitation of some calcium phosphate thus some of the other ion is removed from the solution. Hence, Ca2 + and
precipitate to form insoluble calcium phosphate and their concentrations in the blood are close to the saturation point. Adding more of one of the ions results in the precipitation of some calcium phosphate thus some of the other ion is removed from the solution. Hence, Ca2 + and 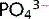 concentrations are inversely proportional.
concentrations are inversely proportional.
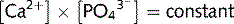
Therefore, a rise in Ca2 + leads to a decrease in 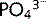 whereas a fall in Ca2 + stimulates an increase in
whereas a fall in Ca2 + stimulates an increase in 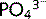 concentration, and vice versa.
concentration, and vice versa.
Ca2 + and 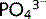 enter the ECF via the intestine (diet) and bone stores. They leave the ECF via the kidneys (urine) and move into the bone.
enter the ECF via the intestine (diet) and bone stores. They leave the ECF via the kidneys (urine) and move into the bone.
Parathyroid hormone
PTH is a polypeptide secreted by the parathyroid gland when there is a fall in plasma Ca2 +. 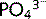 also affects PTH release, both directly and secondary to changes in Ca2 + levels. Fig 3.10 illustrates mechanisms of Ca2 + and
also affects PTH release, both directly and secondary to changes in Ca2 + levels. Fig 3.10 illustrates mechanisms of Ca2 + and 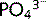 homeostasis. Vitamin D can also affect PTH release because it alters sensitivity of the parathyroid gland to Ca2 +.
homeostasis. Vitamin D can also affect PTH release because it alters sensitivity of the parathyroid gland to Ca2 +.
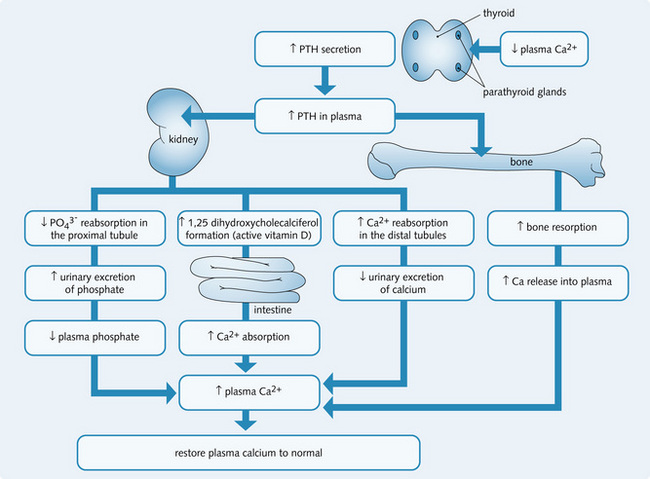
Vitamin D
Vitamin D refers to a group of closely related sterols obtained from the diet or by the action of ultraviolet light on certain provitamins. It is metabolized to 1,25-dihydroxycholecalciferol by the liver and kidney. This causes an increase in Ca2 + and 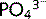 by:
by:
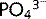 excretion.
excretion.Calcitonin
Calcitonin is a peptide produced by the parafollicular cells of the thyroid. It has the opposite effect to PTH, reducing Ca2 + release from bone and causing a decrease in ECF Ca2 + concentration.
Ca2 + transport by the kidney
Only ionized Ca2 + is filtered through the glomerulus (approximately 50% plasma Ca2 +). Reabsorption proceeds as follows:
• Proximal tubule: 70% is reabsorbed by diffusion, Ca2 +-activated ATPase and the Ca2 +/Na+ counter-transport system
• Thick ascending loop of Henle: 20–25% is reabsorbed passively
• Distal convoluted tubule: 5–10% is reabsorbed against an electrochemical gradient
• Collecting tubule: less than 0.5% is reabsorbed against an electrochemical gradient.
Phosphate
Phosphate (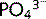 ) salts are essential for the structure of bones and teeth. Eighty per cent of the body's
) salts are essential for the structure of bones and teeth. Eighty per cent of the body's 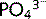 content is in bone and 20% is in the intracellular fluid (ICF). It is filtered easily at the glomerulus; 80% is then reabsorbed in the proximal tubule and the remaining 20% is excreted in urine.
content is in bone and 20% is in the intracellular fluid (ICF). It is filtered easily at the glomerulus; 80% is then reabsorbed in the proximal tubule and the remaining 20% is excreted in urine.
The kidneys play an important role in the regulation of 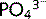 . Reabsorption of
. Reabsorption of 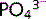 occurs with Na+ ions (two Na+ for every
occurs with Na+ ions (two Na+ for every 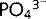 ion) at the apical membrane of the tubular cells. Any increase in plasma
ion) at the apical membrane of the tubular cells. Any increase in plasma 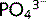 concentration (> 1.2 mmol/L) leads to an increase in the amount filtered and excreted, which is how plasma
concentration (> 1.2 mmol/L) leads to an increase in the amount filtered and excreted, which is how plasma 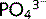 levels are controlled. A fall in GFR will result in increased plasma
levels are controlled. A fall in GFR will result in increased plasma 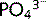 concentration. This hyperphosphataemia is a common cause of itching in CKD.
concentration. This hyperphosphataemia is a common cause of itching in CKD.
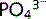 is an important urinary buffer for H+ and its excretion is influenced by:
is an important urinary buffer for H+ and its excretion is influenced by:
Clinical features and causes of Ca2 + disturbances
Hypocalcaemia
Decreased Ca2 + results in neuromuscular excitability leading to tetany with convulsions, hand and feet muscle cramps and cardiac arrhythmias.
• Chronic kidney disease, due to hyperphosphataemia (if 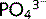 rises, Ca2 + must fall proportionally) and low levels of activated vitamin D
rises, Ca2 + must fall proportionally) and low levels of activated vitamin D
• Rickets and osteomalacia (low vitamin D)
• Alkalosis, which reduces the amount of H+ available to bind to protein, so more Ca2 + can bind to protein. This results in decreased ionized Ca2 +, although total Ca2 + remains the same.
Treatment is with oral or intravenous calcium and patients with chronic kidney disease will benefit from alfacalcidol, a vitamin D analogue.
Hypercalcaemia
• Sudden acidosis, resulting in the release of bound calcium, which becomes ionized Ca2 +
• Increased intestinal absorption due to excess vitamin D or ingestion of calcium (milk–alkali syndrome)
• Bone destruction resulting in increased Ca2 + release from bone – usually caused by secondary deposits from malignancy or myeloma
• Production of humoral hypercalcaemic agents by tumours
• Granulomatous disease (sarcoid)
Symptoms and signs of hypercalcaemia are:
Treatment is of the underlying cause, with fluids for rehydration and bisphosphonates.
Regulation of potassium and magnesium
Potassium
Transport of potassium
Approximately 70% of K+ is reabsorbed in the proximal tubule, mostly by passive paracellular reabsorption across the tight junctions between tubular cells. K+ can be secreted or reabsorbed in the nephron. Excretion of the filtered K+ can vary from 1% to 110% depending on:
K+ reabsorption occurs mainly in the thick ascending loop of Henle by co-transport of Na+/K+/Cl– on the luminal membrane. Reabsorption of K+ occurs in the distal tubule during severe dietary depletion of K+.
Potassium (K+) is the main intracellular cation. The intracellular and extracellular [K+] is very important in the function of excitable tissues (e.g. nerves and muscles) as it determines the resting potentials of these tissues. Therefore, a constant [K+] is critical for survival. Concentration is as follows:
K+ transport by the kidney
K+ is filtered freely in the glomerulus. The proximal tubule reabsorbs 80–90%:
• K+ reabsorption and leakage back are approximately equal in the early distal tubule
• The late distal tubule and collecting ducts secrete K+ into the urinary filtrate (passively via an electrochemical gradient) according to the body's needs – increased cellular K+ concentration results in increased secretion and vice versa.
Changes in the distal tubular lumen also influence the rate of K+ secretion. Fig. 3.11 illustrates K+ transport in the kidney.
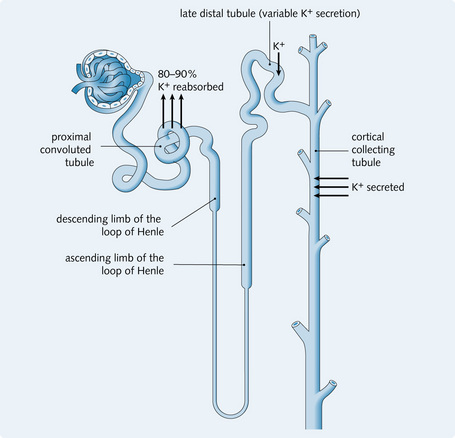
ADH stimulates the secretion of K+ by the collecting ducts by enhancing Na+ reabsorption. Aldosterone increases K+ secretion. Increased plasma K+ concentration stimulates aldosterone production by the adrenal cortex, so plasma aldosterone concentration rises. This in turn increases K+ secretion and therefore K+ excretion.
Hypokalaemia
Causes of a decreased K+ concentration are:
Hypokalaemia is asymptomatic until K+ concentration falls below 2–2.5 mmol/L. The low K+ concentration results in a decreased resting potential (more negative) so the nerve and muscle cells become hyperpolarized. This means that cells are less sensitive to depolarizing stimuli and therefore less excitable, so fewer action potentials are generated and paralysis ensues. Clinical effects of hypokalaemia are:
• Muscle weakness, cramps and tetany, which starts in the lower extremities and progresses upwards (death is usually by paralysis of respiratory muscles)
• Impaired liver conversion of glucose to glycogen
• Vasoconstriction and cardiac arrhythmias
• Impaired ADH action, causing thirst and polyuria and no concentration of urine
• Metabolic alkalosis due to an increase in intracellular H+ concentration.
Treatment involves treating the underlying cause, and calculated oral or intravenous administration of potassium salt may be required.
Hyperkalaemia
Hyperkalaemia can result from:
• Reduced renal excretion: due to acute kidney injury or chronic kidney disease, mineralocorticoid deficiency (e.g. Addison's disease), potassium-sparing diuretics or renal tubular defects
• Increased plasma load: due to dietary changes or cellular tissue breakdown
• Transcellular shift of potassium out of cells: due to metabolic acidosis, insulin deficiency, exercise or drugs (e.g. digoxin)
• Pseudohyperkalaemia, an artefact: due to haemolysis during venepuncture or storage of the sample, a high white cell or high platelet count.
Clinical features
Very similarly to hypokalaemia, it may be asymptomatic or cause muscle weakness. Importantly it can cause cardiac arrhythmias by influencing myocardial excitability (Fig. 3.12).
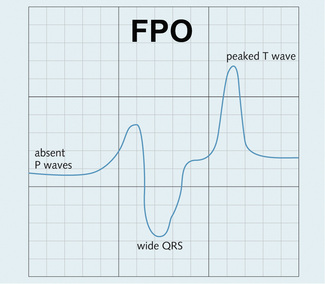
Fig. 3.12 ECG changes in hyperkalaemia. There are peaked T waves, absent P waves and broad QRS complexes. Eventually the pattern looks sinusoidal and can cause arrhythmias.
Treatment
• Calcium gluconate: This does not alter the potassium concentration but Ca2 + stabilizes the myocardium, preventing arrhythmias
• Insulin: This acts to drive potassium into cells, thus lowers plasma [K+] but does not remove potassium from the body. It is given with glucose to avoid hypoglycaemia
• Salbutamol: This also drives potassium into cells when given nebulized or i.v. but should not be used in patients with ischaemic heart disease or arrhythmias
• Sodium bicarbonate: Correction of acidosis would also drive potassium into cells. Not used in patients at risk of fluid overload
• Calcium resonium: The removes K+ by increasing its excretion from the bowels, given orally or by enema. This is the only way to remove K+ from the body, apart from renal replacement therapy
• Renal replacement therapy: Dialysis or haemofiltration are used if medical therapies fail to correct hyperkalaemia.
Magnesium
Magnesium (Mg2 +) is an intracellular cation that:
• Controls mitochondrial oxidative metabolism and so regulates energy production
The plasma concentration of magnesium is 2.12–2.65 mmol/L; about 20% is protein bound.
Renal handling of Mg2 +
Ionized Mg2 + is filtered at the glomerulus. 15% is reabsorbed in the proximal tubule and 60% in the thick ascending loop of Henle.
The Tm for Mg2 + absorption is equal to the concentration of Mg2 + filtered. Therefore, an increase in Mg2 + results in increased filtering, which therefore exceeds the Tm, resulting in increased excretion.
There is intrinsic regulation by cells of the thick ascending loop of Henle – if Mg2 + decreases, cell transport of Mg2 + increases. PTH increases reabsorption of Mg2 + in the thick ascending loop of Henle.
Transport of other solutes in the tubules
Glucose
Normal plasma glucose concentration is 2.5–5.5 mmol/L. Usually, 0.2–0.5 mmol of glucose is filtered every minute. An increase in the plasma glucose concentration results in a proportional increase in the amount of glucose filtered. Virtually all filtered glucose is reabsorbed in the proximal tubule, unless the amount of filtered glucose exceeds the resorptive capacity of the cells. Glucose is transported into the proximal tubule cells by symport against its concentration gradient. It is driven by the energy released from the transport of Na+ down its electrochemical gradient because the Na+/K+ ATPase pump on the basolateral membrane maintains a low Na+ concentration and negative potential within the cell (Fig. 3.13). This is an example of secondary active transport. The transport ratio is:
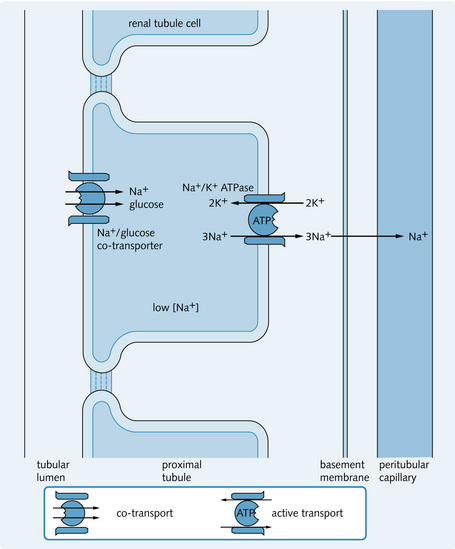
Fig. 3.13 Active transport of glucose in the proximal tubule (pars convoluta). This occurs against a concentration gradient.
Tm is the maximum tubular resorptive capacity for a solute (i.e. the point of saturation for the carriers), and this value can be calculated for glucose. There is a limited number of Na+/glucose carrier molecules, so glucose reabsorption is Tm limited. Fig. 3.14 shows that the lowest renal threshold of glucose is at a plasma glucose concentration of 10 mmol/L. At this level, filtered glucose will begin to be excreted in the urine (glycosuria). If the plasma glucose concentration increases further even those nephrons with highest resorptive capacity become saturated, and glucose is excreted. Urinary glucose increases in parallel with plasma glucose.
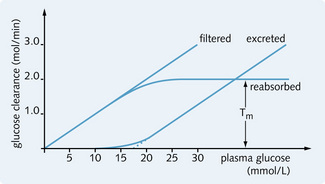
Fig. 3.14 Relation between plasma concentration, filtration, reabsorption and excretion of glucose (glomerular filtration rate = 100 mL/min). Tm is exceeded in nephrons with plasma glucose > 10 mmol/L and for all nephrons when plasma glucose > 20 mmol/L (nephron heterogeneity gives rise to ‘splay’ on the curve).
If plasma glucose rises above 10 mmol/L (as in diabetes), glycosuria will develop. However, glycosuria may also occur in non-diabetic people with normal blood sugar levels as a result of certain inherited renal tubule disorders. This is called renal glycosuria. Renal glycosuria also happens in pregnancy because the Tm for glucose falls, and glucose is excreted in the urine. A glucose tolerance test may be required to differentiate renal glycosuria from diabetes.
Amino acids
Amino acids are the basic unit of proteins and are absorbed constantly from the gut. The plasma concentration of amino acids is 2.5–3.5 mmol/L. They are small molecules that filter easily through the glomerulus, with most reabsorption occurring in the proximal tubule. The transport is a secondary active process (by symport with Na+) and is driven by Na+/K+ ATPase, as with glucose. There are at least five different transport systems coupled with Na+ and these are responsible for the movement of different types of amino acid residue. This is a Tm-limited process, so amino aciduria results if the reabsorption mechanism is saturated or if the reabsorption mechanism is defective (e.g. in Fanconi's syndrome).
Urea
Urea is the end-product of protein metabolism, which occurs in the liver. Urea is transported to the kidneys via the blood. It is a small molecule that is filtered freely at the glomerulus. The normal plasma concentration of urea is 2.5–7.5 mmol/L. Urea concentration increases in the filtrate as a result of Na+, Cl– and water reabsorption. This allows passive reabsorption of 40–50% of urea along its concentration gradient; 50–60% of the filtered urea is excreted in the urine. ADH increases the permeability of the inner medullary collecting ducts to urea. The distal tubule and the outer medullary ducts are impermeable to urea.
The loop of henle
Role of the loop of Henle
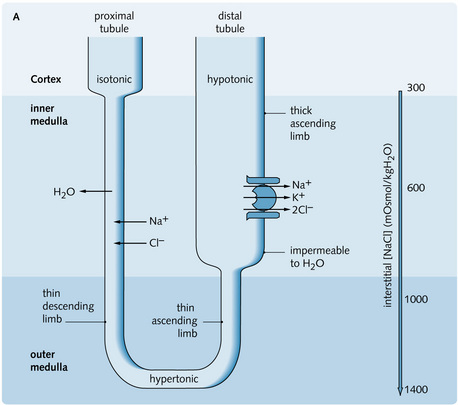
The loop of Henle reabsorbs 20% of the filtered Na+ and 15% of tubular water. As filtrate flows through the loop of Henle, reabsorption of NaCl in the thick ascending limb produces a hypertonic interstitial fluid in the surrounding medulla. This creates a concentration gradient and water moves passively out of the thin descending limb.
The tubular fluid is isotonic to the plasma on entering the loop of Henle; however, by the time it leaves the loop it is hypotonic because ion reabsorption occurs within the loop. This mechanism allows urine to be concentrated, using the least amount of energy, because water is then reabsorbed passively from the collecting ducts into the hypertonic interstitium of the medulla.
Structure of the loop of Henle
The different components of the loop are functionally separate units, each with its own specific properties.
Thin descending limb
The thin descending limb is lined by thin, flat cells that have minimal cytoplasmic specialization. It is permeable to water, Na+ and Cl–. Water is reabsorbed passively down a concentration gradient caused by the hypertonic interstitium of the medulla. NaCl moves into the lumen and water moves out of the lumen into the interstitium, allowing the tubular fluid to come into equilibrium with the interstitium.
Thin ascending limb
The thin ascending limb has a similar structure to the thin descending limb but is impermeable to water and has minimal NaCl transport occurring within the cells.
Thick ascending limb
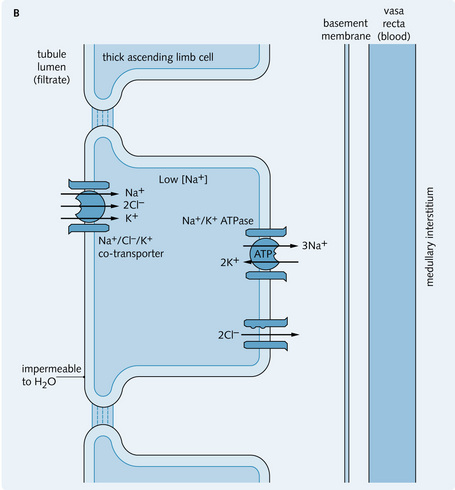
The thick ascending limb consists of large cells with mitochondria, which generate energy for the active transport of Na+ (20% of filtered Na+ is reabsorbed in the loop of Henle) and Cl– ions from the tubular fluid into the interstitium. As a result, the filtrate becomes progressively diluted (this part of the tubule is impermeable to water). There is co-transport (symport) of Na+, Cl– and K+ (in the ratio 1:2:1 – so the pump is electrochemically neutral) on the apical membrane. This transport process is driven by the Na+ gradient across the cell membrane. Na+ is removed from the cell by the Na+/K+ ATPase pump on the basolateral membrane and K+ and Cl– diffuse passively out as a result of Na+ movement; however, most of the K+ leaks back into the cell and tubular lumen. Overall, NaCl accumulates in the medullary interstitium. Fig. 3.15 shows the transport processes in the loop of Henle; the inset shows the transport of ions in the cells in the thick ascending limb of the loop of Henle.
Countercurrent multiplication
Any mechanism that will concentrate urine must be able to reabsorb water from the tubular fluid as it passes through the collecting ducts. The loop of Henle, which acts as a countercurrent multiplier, produces a hypertonic medulla by pooling NaCl in the interstitium, which favours the subsequent movement of water out of the collecting ducts (under the regulation of ADH). Each portion of the loop contributes to the effectiveness of this system.
The mechanism of the countercurrent multiplier is illustrated in Fig. 3.16. The thick, ascending limb can maintain a difference of 200 mOsmol/kg H2O between the tubular fluid and the interstitium at any point along its length. The maximum osmolality of the interstitium is 1400 mOsmol/kg H2O (normal plasma osmolality is 300 mOsmol/kg H2O) at the tip of the loop. The fluid leaving the loop of Henle is hypotonic (100 mOsmol/kg H2O).
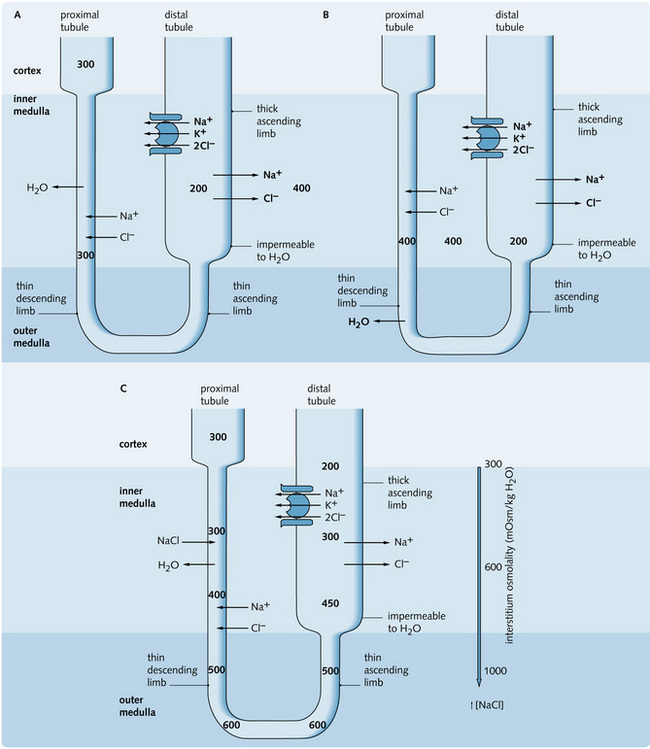
Fig. 3.16 (A) Active reabsorption occurs in the thick ascending limb, increasing the osmolality of the medulla (400 mOsmol/kg H2O). The tubular fluid therefore decreases in osmolality (200 mOsmol/kg H2O). (B) The increase in interstitial osmolality stimulates H2O to leave the descending limb into the medulla. At the same time, increased interstitial osmolality results in passive diffusion of NaCl out of the medulla into the tubule until equilibrium is reached (400 mOsmol/kg H2O). (C) The fluid in the tubule is progressively concentrated in descending the tubule as it comes into equilibrium with its surroundings (maximum value of 600 mOsmol/kg H2O at the tip of the loop) and hence progressively diluted as it ascends the loop. This is all due to active NaCl reabsorption in the thick ascending limb (and some passive movement of NaCl in the ascending limb). Therefore a longitudinal gradient is set up, with greatest osmolality in the lower medulla and least in the cortex.
Role of vasa recta and urea
The countercurrent mechanism requires an environment in which the waste products and water are cleared without disturbing the solutes that maintain the medullary hypertonicity. This exchange is provided by the vasa recta capillary system derived from the efferent arterioles of the longer juxtaglomerular nephrons. It does not require metabolic energy.
The capillaries have a hairpin arrangement surrounding the loop of Henle and are permeable to water and solutes. As the descending vessels pass through the medulla they absorb solutes such as Na+, urea and Cl–. Water moves along its osmotic gradient out of the capillaries. At the tip of the loop the capillary blood has the same osmolality as the interstitium, and an osmotic equilibrium is reached. The capillaries that ascend with the corresponding loop of Henle contain very viscous concentrated blood as a result of the earlier loss of water from the capillaries. A consequent increase in oncotic pressure because of the concentration of plasma proteins favours the movement of water back into the blood vessel from the interstitium. However, most of the NaCl is retained in the interstitium to maintain the hypertonic medullary environment.
The collecting tubules pass through the cortex and medulla. They consist of two functionally different parts:
Both parts are impermeable to NaCl. The permeability to water and urea (only in the inner medullary collecting ducts) varies according to the presence of ADH. ADH increases the permeability to water and thus controls the concentration of the urine produced. ADH acts to increase water uptake in the cortical collecting tubules resulting in the production of a more concentrated urine.
The water reabsorbed in the medullary part of the collecting ducts is taken up by the vasa recta to prevent dilution of the medullary interstitium, which is crucial to the function of the distal nephron and the concentration of urine.
Although about 20% of the initial glomerular filtrate enters the distal nephron, only 5% enters the medullary collecting ducts. This is mainly due to water reabsorption in the cortical tubules.
The structure, location and function of the loop of Henle has a central role in the development of a hypertonic gradient in the medulla. This allows urine to be concentrated as it passes through the collecting tubules.
Fig. 3.17 shows the countercurrent exchanger and the collecting duct as it passes through the medulla.
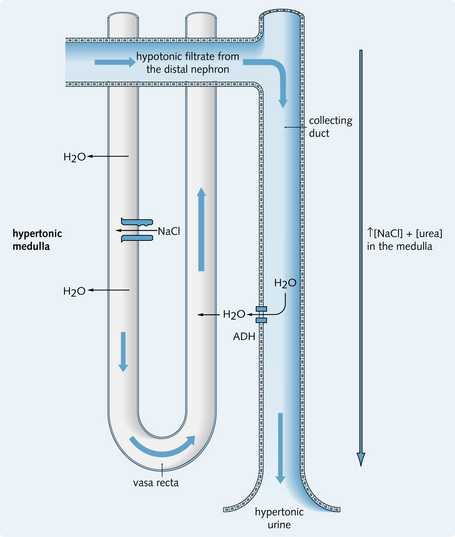
Fig. 3.17 Countercurrent exchanger as it passes through the medulla. The descending vessels of the vasa recta lose water as they pass through the hypertonic medulla. As a result of increasing oncotic pressure in the ascending vessels, water is reabsorbed passively back into the blood vessels from the interstitium as water uptake occurs in the collecting ducts under the influence of antidiuretic hormone (ADH). Because of this uptake of water by the vasa recta, the high osmolality of the medullary interstitium is maintained and this hypertonic environment allows continued concentration of the tubular fluid in the collecting duct.
Although urea is impermeable in the cortical collecting tubules, ADH affects the permeability of urea within the medullary cortical tubules. Urea, along with NaCl, helps maintain medullary hypertonicity as follows
• 50% of the filtered urea is reabsorbed in the proximal tubule with Na+
• The tubular concentration of urea increases as it diffuses out of the medullary interstitium into the lumen down its concentration gradient
• The remaining urea becomes further concentrated within the tubular lumen as water and other solutes are reabsorbed into the cells of the distal tubule and the cortical collecting tubules, a process aided by the fact that these parts of the nephron are impermeable to urea
• The concentration of urea in the medullary collecting tubules is so high that it diffuses out of the lumen into the interstitium, thus increasing the concentration of urea in the medulla and recycling it. This occurs in the presence of ADH.
A high-protein diet increases the amount of urea in the blood for excretion as a result of increased metabolism. Consequently, there is more urea in the medullary interstitium, resulting in a higher urine osmolality.
Body fluid osmolality
Concepts of osmolality
The normal plasma osmolality (Posm) is 285–295 mOsmol/kg H2O. This is strictly regulated and an increase or decrease of 3 mOsmol/kg H2O will stimulate the body's osmolality regulation mechanism.
Osmoreceptors
Osmoreceptors detect changes in the plasma osmolality and are located in the supraoptic and paraventricular areas of the anterior hypothalamus. Their blood supply is the internal carotid artery. They have two functions:
1. To regulate the release of antidiuretic hormone (ADH, also known as vasopressin)
2. To regulate thirst (this also depends on other osmoreceptors in the lateral preoptic area of the hypothalamus).
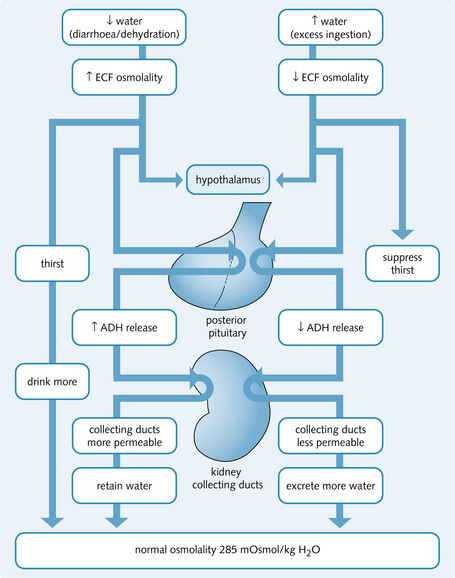
Fig. 3.18 illustrates the role of ADH in maintaining osmolality.
Sensitivity of osmoreceptors to osmotic changes caused by different solutes
Na+ and other associated anions are the main constituents that determine plasma osmolality. Water loss alters the Na+ concentration. Other solutes without the addition or loss of water can also change the osmolality. Not all solutes stimulate the osmoreceptors to the same degree – this depends on how easily they can cross the cell membrane (i.e. their ability to cause cellular dehydration).
Antidiuretic hormone (vasopressin)
Synthesis and storage
ADH is a peptide hormone synthesized in the supraoptic nucleus of the hypothalamus as a large precursor molecule. It is transported to the posterior pituitary gland, where its synthesis is completed and it is stored until release (Fig. 3.19).
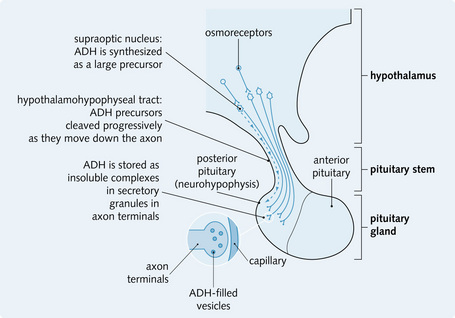
Release
A rise in plasma osmolality triggers ADH release. Action potentials in the neurons from the hypothalamus (which contains ADH) depolarize the axon membrane, resulting in Ca2 + influx, fusion of secretory granules with the axon membrane and the release of ADH and neurophysin into the bloodstream.
Aquaporins
When present on the peritubular side of the collecting tubule cell (Fig. 3.20), ADH causes intracellular water channels (aquaporins) to fuse with the luminal membrane. There are at present 11 known members of the mammalian aquaporin gene family which encode for proteins involved in the transport of water or small molecules. In the kidney, aquaporin 2 (AQP2) resides in intracellular vesicles and is trafficked to the luminal membrane on stimulation. ADH triggers this by binding to V2 receptors on the basal membrane. These are G-protein-coupled receptors, which on activation cause fusion of the inactive vesicles with the luminal membrane. The relation between urine osmolality (mOsmol/kg) and plasma ADH concentration is shown in Fig. 3.21.
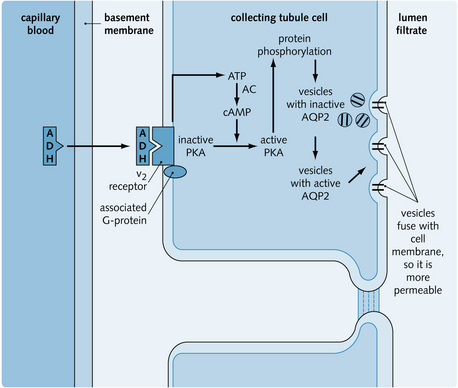
Fig. 3.20 Actions of antidiuretic hormone (ADH) in the collecting tubule. AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase; AQP2, aquaporin 2 water channel.
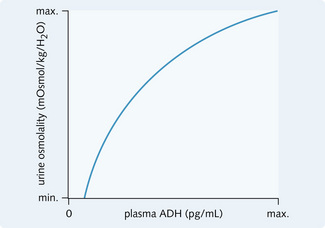
Water clearance and reabsorption
Dehydration leads to a rise in plasma osmolality. Thus, the kidneys reabsorb ‘solute-free’ water from the tubules. This produces a more dilute plasma and a concentrated urine.
Excessive water intake lowers plasma osmolality. Thus, the kidneys excrete ‘solute-free’ water from the tubules, producing dilute urine. Dilute urine has a lower osmolality than plasma, concentrated urine has a higher osmolality than plasma, isotonic urine has the same osmolality as plasma. The osmotic clearance (Cosm) (Fig. 3.22) is the rate at which osmotically active substances are cleared from the plasma. If urine is isotonic, Cosm = urine flow.

Effect of solute output on urine volume
The concentrating ability of the kidneys is limited, with a maximum urinary osmolality of 1400 mOsmol/kg. Thus, the amount of urine excreted per day depends on the:
At maximum ADH concentration, large amounts of solutes can still cause a dieresis.
Mannitol is an osmotic diuretic that cannot be reabsorbed. It alters the kidney's concentrating ability and produces isotonic urine. In diabetes mellitus, the excess blood glucose causes an osmotic diuresis.
In children younger than 5 years, bed wetting may be normal. However, repeated bedwetting in children over the age of 5 years may indicate a pathological cause and is called nocturnal enuresis. It occurs in about 10% of children aged 10, affecting boys more than girls. One cause is a reduction in circulating nocturnal antidiuretic hormone (ADH) levels. This may be managed by many methods, including the use of an ADH analogue called desmopressin, administered by a nasal spray.
Disorders of osmolality
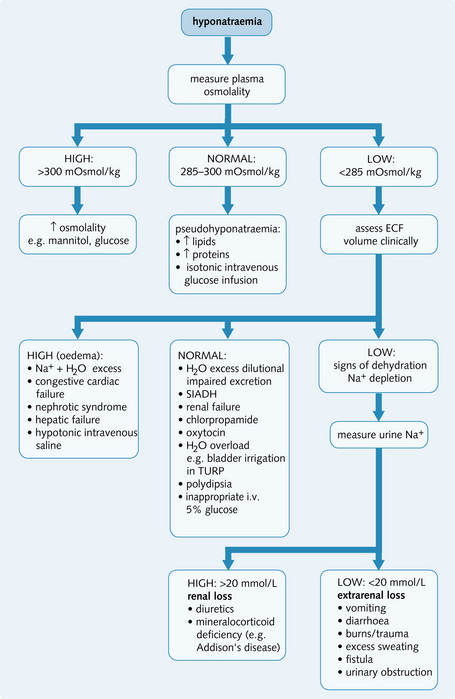
Hyponatraemia
In hyponatraemia, plasma [Na+] is < 130 mmol/L and there is a decreased solute: water ratio in the extracellular fluid.
Causes
The causes of hyponatraemia are:
• Diuretics (mainly thiazides)
• Increased antidiuretic hormone (ADH) secretion
• Increased plasma osmolarity (e.g. caused by mannitol, glucose)
• Increased protein or lipids (pseudohyponatraemia). Here, there is less sodium relative to the increase in protein or lipids, giving the impression of a low sodium.
The diagnostic approach to hyponatraemia is shown in Fig. 3.23.
Syndrome of inappropriate ADH secretion (SIADH)
Occasionally ADH is secreted inappropriately by the pituitary or other areas in the body. Causes are given in Fig. 3.24. Signs and symptoms are:
• Hyponatraemia (< 125 mmol/L) and low plasma osmolality (< 260 mmol/L)
• Inappropriate urine osmolality: the urine concentration is higher than normal (i.e. not maximally diluted)
• Inappropriate Na+ excretion: urinary [Na+] is greater than 20 mmol/L despite a decrease in plasma Na+ concentration because the plasma volume is maintained by water retention (unless volume contracted or sodium restricted, which can decrease urinary Na+).
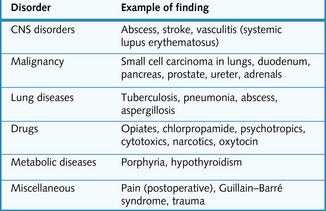
The diagnosis should be considered in hyponatraemic patients in the absence of hypovolaemia, oedema, endocrine dysfunction, renal failure and drugs, all of which can impair water excretion.
Hypernatraemia
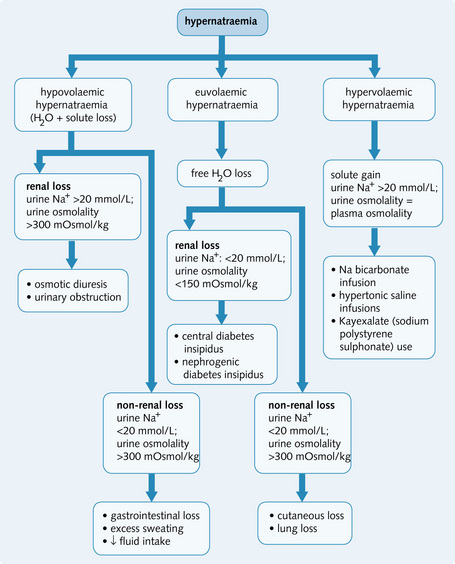
In hypernatraemia the serum sodium is > 140 mmol/L and there is an increase in solute to water ratio in body fluids and increased serum osmolality (> 300 mOsmol/kg).
Causes
The causes of hypernatraemia are:
• Osmotic diuresis (e.g. uncontrolled diabetes)
• Fluid loss without replacement (sweating, burns, vomiting)
• Diabetes insipidus (suspect if lots of dilute urine is produced)
• Incorrect intravenous fluid replacement (i.e. hypertonic fluids)
A diagnostic algorithm is given in Fig. 3.25.
Diabetes insipidus
This is the inability to reabsorb water from the distal part of the nephron, due to the failure of secretion or action of ADH. Symptoms are:
The causes of diabetes insipidus are:
• Neurogenic/central: impaired ADH synthesis or secretion by the hypothalamus, which might be congenital, caused by hypothalamic damage or due to pituitary tumours. It can be treated by administering ADH
• Nephrogenic: failure of the kidneys to respond to circulating ADH, which could be caused by mutations in the gene coding for V2 receptors, chronic pyelonephritis, polycystic kidneys or drugs such as lithium. Plasma ADH levels are normal. There is no current treatment to correct the deficit.
Diabetes insipidus can be mistaken for psychogenic polydipsia, in which large volumes of dilute urine are produced secondary to compulsive water drinking. This causes a decrease in the urine-concentrating ability because of loss of medullary tonicity.
Diseases of the tubules and interstitium
Overview
The tubules and interstitium are affected by several diseases. Typically, tubules become obstructed (this reduces glomerular filtration) or their transport functions become impaired (reduces water and solute reabsorption). Damage can be acute or chronic.
Acute tubular necrosis
Acute tubular necrosis (ATN) is the result of acute tubular cell damage by ischaemia or toxins. It can be oliguric (< 400 mL/day urine) or non-oliguric. Hyperkalaemia can develop as a result of K+ retention and this can trigger cardiac arrhythmias, which can be life-threatening. Uraemia develops because there is a significant fall in GFR – this could be due to haemodynamic changes and intratubular obstruction. Recovery is accompanied by a diuretic phase that occurs because of failure to concentrate urine (this can cause hypokalaemia).
ATN is a cause of acute kidney injury (see Chapter 7). Mortality is high but full recovery is possible with prompt treatment – fluid and electrolyte therapy and dialysis if necessary.
The tubular cells are capable of regenerating rapidly to allow complete recovery.
Ischaemic acute tubular necrosis
This is caused by hypotension and hypovolaemic shock following trauma, infections, burns or haemorrhage. There is a rapid fall in blood pressure, which causes hypoperfusion of the peritubular capillaries with consequent tubular necrosis along the entire length of the nephron. The kidneys appear pale and swollen. Histological examination reveals:
• Infiltration of inflammatory cells and the tubular cells
• Flattened and vacuolated tubular cells
• Cellular debris and protein casts in the distal tubule and the collecting ducts.
Non-steroidal anti-inflammatory drugs (NSAIDs) can increase the risk of ATN following other renal insults by preventing the synthesis of prostaglandins (PGs). PGs are vasodilators, which protect the kidney from ischaemic injury by dilating blood vessels and increasing blood flow.
Toxic acute tubular necrosis
This disorder is caused by agents with specific nephrotoxic activity causing damage to the epithelial cells. Such substances include:
• Organic solvents: carbon tetrachloride (CCl4) in dry-cleaning fluid
These substances cause the cells to come away from the basement membrane and consequently collect in and obstruct the tubular lumen. The effect is limited because there is regeneration of the epithelial cells in 10–20 days, which permits clinical recovery and is confirmed by the presence of mitotic figures on biopsy. Damage by nephrotoxic substances is limited to the proximal tubules. The kidneys appear swollen and red.
Rhabdomyolysis
Muscle breakdown leads to the release of myoglobin into the blood. This is filtered freely by the glomerulus. If the filtrate is acidic myoglobin precipitates to form casts which block the normal flow of urine through the tubules. The muscle damage can be caused by:
Investigations will reveal a very high creatine kinase (> 10 000) and hyperkalaemia from release from muscle cells. The urine will be dark and give a false positive for blood on a dipstick test.
Treatment involves stopping the damage to muscle tissue if possible. Intravenous fluids are given to promote high urine production alongside alkalinisation of the urine with sodium bicarbonate to help to decrease the precipitation of myoglobin in the tubules.
Tubulointerstitial nephritis
Pyelonephritis
This is a bacterial infection of the kidney and results in inflammation and damage to the renal calyces, parenchyma and pelvis. It can be acute or chronic.
Acute pyelonephritis
This occurs because of infection in the kidney and is spread via two routes:
1. Ascending infection: bacteria from the gut enter the kidney from the lower urinary tract if there is an incompetent vesicoureteric valve. This permits vesicoureteric reflux (VUR) and results in ascending transmission of infection
2. Haematogenous spread: seen in patients with septicaemia or infective endocarditis. The pathogens include fungi, bacteria (staphylococci and Escherichia coli) and viruses. The kidney is often affected in septicaemic diseases because of its large blood supply.
The predisposing factors of acute pyelonephritis are:
• Urinary tract obstruction (congenital and acquired)
• Instrumentation of the urinary tract
• Immunosuppression (human immunodeficiency virus infection, lymphoma and transplants).
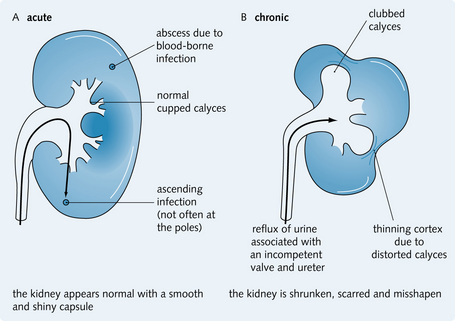
Patients present with general malaise, fever, loin pain, tenderness and often rigors with or without symptoms of lower UTI. Infection spreads into the renal pelvis and papillae and causes abscess formation throughout the cortex and medulla.
With retrograde ureteric spread the kidney characteristically contains areas of wedge-shaped suppuration especially at the upper and lower poles. In septicaemia there is haematogenous seeding within the kidney and minute abscesses are distributed randomly in the cortex. On histological examination there is:
Uncomplicated cases resolve with antibiotic treatment and high fluid intake. The important complications of acute pyelonephritis are:
Chronic pyelonephritis
This condition is characterized by long-standing parenchymal scarring, which develops from tubulointerstitial inflammation. It is the end-result of various pathological processes. There are two main types:
1. Obstructive: chronic obstruction (stones, tumours or congenital abnormalities) prevents pelvicalyceal drainage and increases the risk of renal infection. Chronic pyelonephritis develops because of recurrent infection
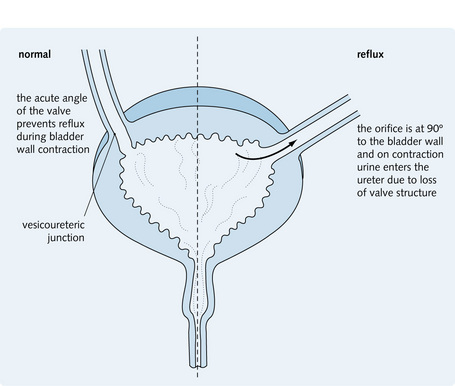
2. Reflux nephropathy: this is the most common cause of chronic pyelonephritis. It is associated with VUR, which is congenital. The organisms enter the ascending portion of the ureter with refluxed urine as the valvular orifice is held open on contraction of the bladder during micturition. Reflux results from the abnormal angle at which the ureter enters the bladder wall (Fig. 3.26).
The disease process usually begins in childhood and has a silent, insidious onset. Reflux of urine into the renal pelvis occurs during micturition and this increases the pressure in the major calyces. The high intrapelvic pressure forces urine into the collecting ducts with intraparenchymal reflux further distorting the internal structure. This is most predominant at the poles of the kidney and results in deep irregular scars on the cortical surface. The tubulointerstitial inflammation heals with the formation of corticomedullary scars that overlie the deformed and dilated calyces, which are characteristic of chronic pyelonephritis (Fig. 3.27).
On histological examination there is interstitial fibrosis and dilated tubules containing eosinophilic casts; 10–20% of patients requiring dialysis have chronic pyelonephritis.
Ultrasonography is used to diagnose chronic pyelonephritis and may show distortion of the calyceal system and contraction of the kidney because of cortical scarring. Intravenous pyelography may be more sensitive but requires exposure to X-rays which should be avoided, especially in children.
Toxin- and drug-induced tubulointerstitial nephritis
Heavy metals (mercury, gold, lead) and drugs (ampicillin, rifampicin, NSAIDs) can cause T-cell-mediated inflammation in the interstitium. This reaction usually occurs 2–40 days after exposure to the toxin. Clinical features include fever, skin rash, haematuria, proteinuria and ARF. Withdrawal of the causative agent leads to recovery.
On histological examination there is interstitial oedema and tubular degeneration with eosinophil infiltration. In chronic analgesic abuse with phenacetin, and to a lesser extent aspirin, PG synthesis is inhibited, causing ischaemia (as described on p. 50 for ischaemic ATN). This causes papillary necrosis and a secondary tubulonephritis (analgesic nephropathy). It is associated with an increased risk of developing transitional cell carcinomas with chronic analgesic abuse.
Acute urate nephropathy
If there is an increased blood urate concentration, urate crystals are precipitated in the acidic environment of the collecting ducts, causing inflammatory obstruction and dilatation of the tubules. This is called acute urate nephropathy and causes acute kidney injury. A typical cause is tumour lysis syndrome. This involves rapid cell turnover in those patients with haematological or lymphatic malignancy who are receiving chemotherapy. There is excess cell breakdown and release of nucleic acids, which results in acute urate nephropathy and presents as Acute Kidney Injury.
Hypercalcaemia and nephrocalcinosis
A persistently high blood Ca2 + level causes Ca2 + deposition in the kidneys. The hypercalcaemia can be due to:
Renal insufficiency occurs in these patients because of stones (nephrolithiasis) or focal calcification in the renal parenchyma (nephrocalcinosis).
In nephrocalcinosis the Ca2 + accumulates in the tubular cells and the basement membrane, resulting in interstitial fibrosis and inflammation. Hypercalcaemia also causes a renal concentrating defect, which leads to polyuria, nocturia and dehydration.
Multiple myeloma
Approximately 50% of patients with multiple myeloma develop renal insufficiency, which can cause AKI or CKD. Histological changes include:
• Bence Jones proteins (light chains) enter the urine and these are toxic to the tubular epithelial cells. They combine with Tamm-Horsfall protein to precipitate as casts in the tubules, causing inflammation and obstruction to the tubular cells
• Amyloid lambda (λ) or kappa (κ) light-chain fragments (paraproteins) are deposited in the renal blood vessels, glomeruli and tubules