4 Body fluid volume
By the end of the chapter you should be able to:
• Name the fluid compartments of the body and state their volume in a 70-kg male
• Explain the importance of sodium in the control of body fluid volume
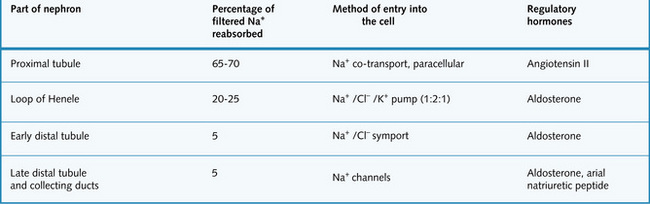
• State where in the nephron most Na+ and water are reabsorbed and state the mechanism
• Describe in detail the renin–angiotensin–aldosterone system and state the actions of angiotensin II
• Explain the renal responses to cardiac failure, liver failure and hypotension
• List six causes of hypertension and outline its management
• Understand the differences between the classes of diuretics and state which are considered the most powerful
• List the side effects of thiazide diuretics
• Describe the mechanism of action of potassium-sparing diuretics
• Understand the effects of ACE inhibitors in renal artery stenosis
Control of body fluid volume
Basic concepts
Body fluids
• Plasma – ECF within the vascular system, i.e. the non-cellular component of blood
• Interstitial fluid (ISF) – ECF outside the vascular system (and separated from plasma by the capillary endothelium)
• Transcellular fluid (TCF) – ECF (e.g. synovial fluid, aqueous and vitreous humour, cerebrospinal fluid) separated from plasma by the capillary endothelium and an additional epithelial layer that has specialized functions (Fig. 4.1).
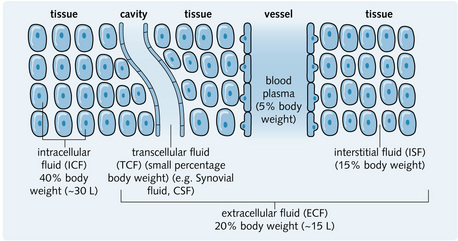
Water is a major component of the human body. Approximately 63% of an adult male and 52% of an adult female is water (i.e. 45 L in a 70-kg male, 36 L in a 70-kg female). This difference is due to the fact that females have a higher proportion of body fat, which has a low water content. One-third of total body water (TBW) is ECF (about 15 L in a 70-kg male) and two-thirds is ICF (about 30 L in a 70-kg male).
The volume of fluid that perfuses tissues is the effective circulating volume; this needs to be kept constant.
Sodium balance
Na+ is the major cation in the extracellular fluid with a normal concentration of 135–145 mmol/L. It therefore controls 90% of body fluid osmolality. As discussed in the previous chapter, osmolality of the plasma is carefully controlled by the loop of Henle and collecting duct. Thus, changes in the amount of Na+ in the ECF actually lead to changes in the ECF volume. For example, a rise in ECF Na+ results in increased osmolality, which leads to water retention and thirst (increased drinking of water). This increases ECF volume and normalizes osmolality.
Handling of sodium by the kidney
The concentration of Na+ in the Bowman's capsule is equal to the plasma level because Na+ is freely filtered. Virtually all the Na+ that is filtered into the nephron is reabsorbed back into the circulation, with only 1% or less of the filtered Na+ being excreted in the urine (Fig. 4.2).
Transport of sodium in the proximal tubule
A lot of Na+ is reabsorbed in the early proximal tubule but, as the cell junctions are leaky, the concentration gradient between the filtrate and the intercellular plasma is limited. Less reabsorption occurs in the late proximal tubule, but the cell junctions are tight so a better concentration gradient is established. The primary transporter Na+/K+ ATPase (Na+ pump) on the basolateral membrane actively transports Na+ out of the cell into the lateral intercellular spaces between adjacent cells (Fig. 4.3).
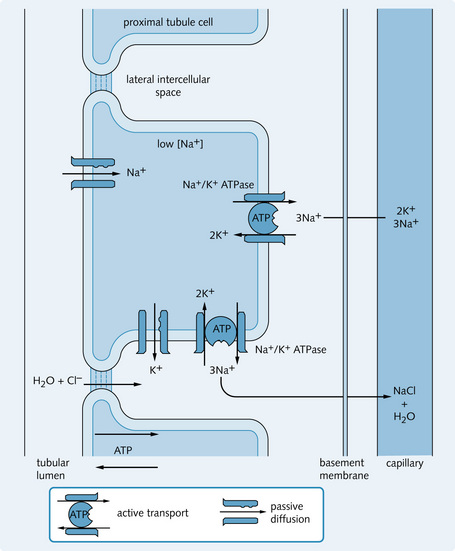
Fig. 4.3 Na+ transport processes in the proximal tubule. Sodium entry into the cell is driven by its concentration gradient set up by the Na+/K+ ATPase pump found on the basal membrane.
This movement of Na+ out of the cell maintains a very low concentration of Na+ within the proximal tubule cells. This drives Na+ ions to move along their concentration gradients into the cells from the tubular fluid via carrier molecules on the apical membrane. In the early proximal tubule, movement of other substances, e.g. glucose, amino acids and 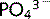 , is coupled with Na+ transport in and out of tubule cells (discussed in the previous chapter).
, is coupled with Na+ transport in and out of tubule cells (discussed in the previous chapter).
The fluid leaving the proximal tubule is isosmotic because both ions and water move out of the filtrate together, i.e. it has no concentrating capacity.
The distal tubule and loop of Henle handling sodium
The complicated mechanism by which the loop of Henle handles sodium is dealt with in the previous chapter. This is mainly to create a concentration gradient to allow control of osmolality.
The distal convoluted tubule reabsorbs only around 10% of the filtered Na+ but this is amount is adjustable and important in the control of body fluid volume. Sodium leaves the basolateral side through the Na+/K+ ATPase and enters the cell from the lumen through a Na+/Cl– co-transporter, down its concentration gradient.
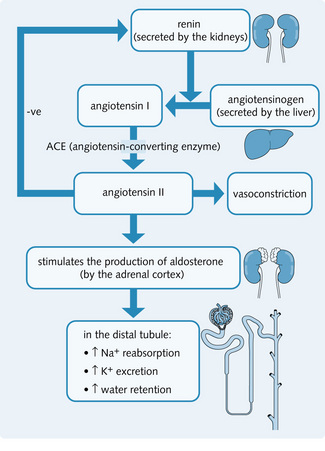
Renin–angiotensin–aldosterone system
The renin–angiotensin–aldosterone system (Fig. 4.4) maintains Na+ balance.
Renin
Renin is an enzyme that is synthesized and stored in the JGA in the kidneys. A fall in plasma Na+ leads to a fall in ECF volume, causing the release of renin by:
• Increased sympathetic innervation: a fall in ECF volume results in a fall in blood pressure. This is detected by baroreceptors in carotid arteries and causes increased sympathetic activity. Granular cells of the JGA are innervated by the sympathetic system, so an increase in sympathetic activity leads to an increase in renin release. The process is mediated by ß-adrenergic receptors
• The wall tension in afferent arterioles falls: decreased ECF volume reduces blood pressure, which in turn decreases perfusion pressure to the kidneys. Changes in the blood pressure decrease wall tension at granular cells, which stimulates renin release
• Decreased Na+ to the macula densa: if less NaCl reaches the macula densa, the macula densa is stimulated to secrete the prostaglandin PGI2. This acts on the granular cells to cause renin release.
Conversion of angiotensinogen to angiotensin
Renin acts on angiotensinogen (α2-globulin), which splits off angiotensin I (a decapeptide). Angiotensin-converting enzyme (ACE) in the lungs then removes two amino acids to produce angiotensin II (an octapeptide). Angiotensin II:
• Stimulates the zona glomerulosa of the adrenal cortex to release aldosterone
• Directly vasoconstricts arterioles within the kidney (efferent > afferent)
• Directly increases Na+ reabsorption from the proximal tubule
• Provides negative feedback to the JGA cells, and therefore affects renin release.
In addition to the generation of circulating angiotensin II, the local generation of angiotensin II by ACE (within the tissues) might have an important pathogenic role. ACE inhibitors are used to treat high blood pressure. They decrease the production of angiotensin II and consequently:
Aldosterone
Aldosterone is synthesized by zona glomerulosa cells in the adrenal cortex. Its release (Fig. 4.5) is controlled by:
• ECF volume: if circulating Na+ falls, effective circulating volume also falls. This stimulates aldosterone release via the renin–angiotensin–aldosterone system
• Na+ concentration: via direct aldosterone release from the adrenal cortex, as well as through the renin–angiotensin–aldosterone system
• K+ concentration: a rise in circulating K+ stimulates direct release of aldosterone from the adrenal cortex. This returns K+ to normal by increasing distal tubular secretion of K+.
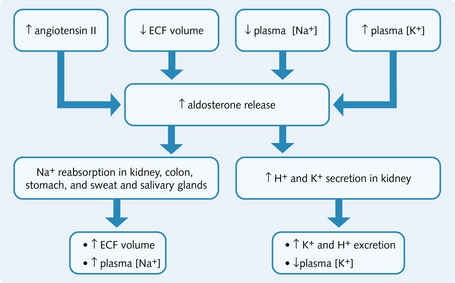
Fig. 4.5 Factors causing aldosterone release and the effects of aldosterone. ECF, extracellular fluid.
Aldosterone primarily regulates sodium concentration. It acts within cells to:
Adrenal insufficiency is a condition where the adrenal glands fail to secrete the required hormone levels. It normally has a non-specific presentation unless the patient is having an acute adrenal crisis, which is typically life-threatening. On investigation, the patient may have abnormally high potassium levels (hyperkalaemia) and low sodium levels (hyponatraemia). These findings result from reduced aldosterone secretion and resultant renal Na+ wasting and reduced K+ excretion.
Other factors affecting Na+ reabsorption
Starling's forces in the proximal tubule
The amount of Na+ and water reabsorbed into the peritubular capillaries from the proximal tubule depends on the rate and amount of uptake from the lateral intercellular spaces into the capillaries.
Changes in the body fluid volume alter plasma hydrostatic and oncotic pressure, for example, increased NaCl intake is mirrored by a rise in ECF volume. This in turn increases hydrostatic pressure and decreases oncotic pressure, so NaCl and water reabsorption by the proximal tubule cells decreases.
Sympathetic drive from the renal nerves
The arterial baroreceptors regulate renal sympathetic nerve activity, for example, a fall in ECF volume decreases blood pressure, which is sensed by baroreceptors, and results in an increase in sympathetic activity. This stimulates Na+ retention and an increase in peripheral resistance, thus restoring ECF volume and blood pressure.
A rise in sympathetic nerve activity to the kidney stimulates renin release either directly or as a result of renal vasoconstriction (this activates the JGA) (Fig. 4.6). Catecholamines from sympathetic nerve endings also stimulate Na+ reabsorption by the proximal tubule, but it is unclear if this is a direct effect or secondary to altered peritubular forces.
Prostaglandins
A decrease in the effective circulating volume stimulates cortical prostaglandin (PG) synthesis. In the kidney, PG synthesis occurs in the:
Several prostaglandins exist: PGE2 (medullary), PGI2 (cortical), PGF2α, PGD2 and thromboxane A2 (TXA2). The main functions of each are as follows:
• PGE2, PGI2: vasodilators, preventing excessive vasoconstriction
• PGI2 (prostacyclin): renin release
• PGE2 (medullary): promotes water (diuretic) and sodium (natriuretic) excretion within the collecting tubules and thus overrides the antidiuretic action of ADH. PGE2 protects the medullary tubule cells from excessive hypoxia when the ECF volume decreases
• TXA2: a vasoconstrictor, which is synthesized after repeated kidney damage (e.g. ureteral obstruction). It reduces the amount of blood available for filtration by a poorly functioning kidney.
Atrial natriuretic peptide (ANP)
ANP is a peptide produced by cardiac atrial cells in response to an increase in ECF volume. It is found in the atrial cells and released into the plasma. ANP binds to specific cell surface receptors, resulting in increased cyclic guanosine monosulphate (cGMP). ANP acts to:
• Inhibit Na+/K+ ATPase and close Na+ channels of the collecting ducts, reducing Na+ reabsorption. Na+ reabsorption is also reduced in the proximal tubules. Thus, Na+ and water excretion by the kidney is increased
• Vasodilate afferent arterioles, thereby increasing GFR
Renal responses to systemic disorders
Congestive cardiac failure
Congestive cardiac failure (CCF) occurs when the heart muscle pump cannot cope with its work load. The cardiac output falls and fails to perfuse the tissues adequately. Hypoperfusion of the tissues results in sodium and water retention by the kidneys, leading to oedema. CCF is a common end result of all types of severe heart disease.
Renal hypoperfusion following a fall in cardiac output is sensed by the kidney as hypovolaemia resulting in compensation by retaining NaCl and water to increase the circulating volume (Fig. 4.7). As the kidney attempts to increase the circulating fluid volume, peripheral oedema develops. This increase in pulmonary venous pressure, results in fluid transudation from the capillaries in the lungs and in pulmonary oedema.
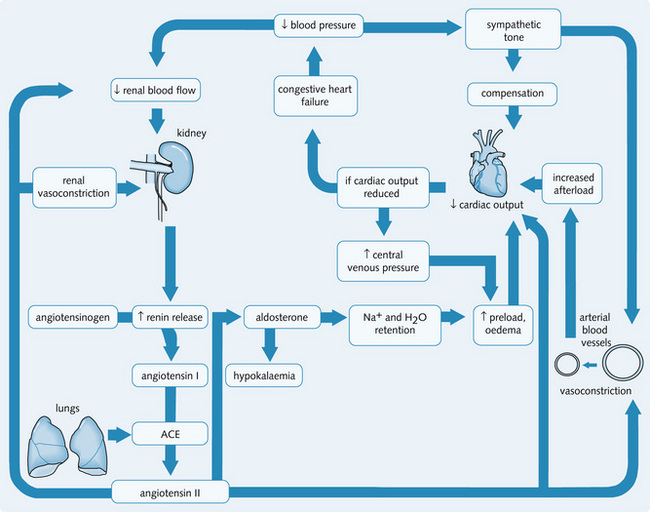
Treatment and management
Management involves reducing the fluid load within the body and thereby decreasing the workload of the heart.
• Diuretics: produce symptomatic relief from pulmonary oedema
• ACE inhibitors: act as vasodilators (by reducing the synthesis of angiotensin II) and as diuretics (by decreasing aldosterone synthesis)
Prognosis depends on the overall clinical picture, and the extent of cardiovascular disease. For further information see Crash course: Cardiovascular system.
Hypovolaemia and shock
Shock is a medical emergency in which the vital organs are inadequately perfused. As the amount of oxygen and nutrients delivered to the cells is inadequate, the resulting hypoxic state within the cells leads to anaerobic metabolism and there is inefficient clearance of the metabolites, which build up in the cell. Hypovolaemia and mild shock cause tiredness, dizziness and a feeling of thirst. A severe decrease in the circulating volume stimulates sympathetic activity to maintain the blood pressure by:
Vasodilation occurs in the vital organs (heart, lungs, brain) to maintain blood supply, but this is at the expense of perfusion to other organs. If there is inadequate compensation, tissue hypoxia and necrosis can occur in vulnerable organs (e.g. acute tubular necrosis in the kidneys).
Cardiogenic shock
This occurs when the heart fails to maintain cardiac output acutely (e.g. ischaemic heart disease, arrhythmias). As a result, tissue perfusion decreases dramatically. Venous pressure increases, causing pulmonary or peripheral oedema (as described above). Prognosis is poor (90% mortality). Massive pulmonary embolisms and pericardial tamponade are also causes of shock.
Vasodilated shock
Sepsis, anaphylaxis and spinal trauma decrease the total systemic resistance and can reduce the blood pressure sufficiently to cause shock.
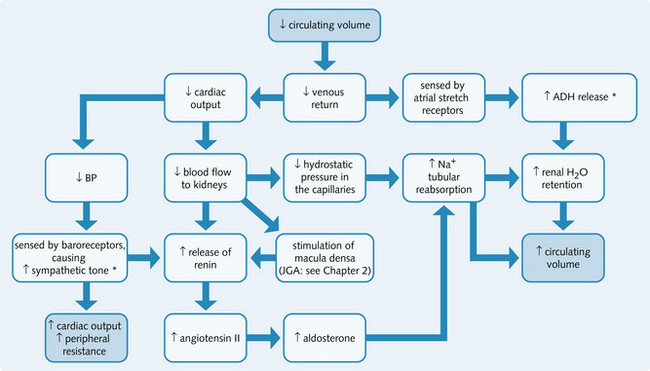
Hypovolaemic shock
This occurs when there is an acute reduction in effective circulating blood volume from blood loss (haemorrhage), loss of plasma (e.g. burns) or loss of water and electrolytes (e.g. diarrhoea and vomiting).
Figure 4.8 shows the response to a fall in circulating fluid volume. To counteract excessive vasoconstriction as a result of sympathetic activity in the kidneys, more vasodilating prostaglandins (PGE2 and PGI2) are secreted within the kidneys. This maintains adequate blood flow through the kidney to allow sufficient glomerular filtration, unless the shock is severe. The loss of large amounts of fluid has two major consequences:
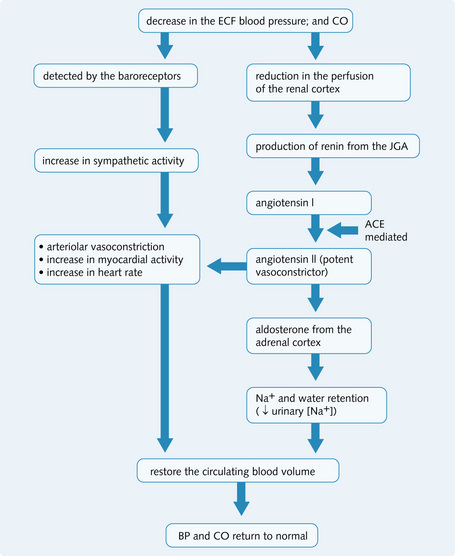
Fig. 4.8 Response to a fall in circulating fluid volume. ACE, angiotensin-converting enzyme; BP, blood pressure; CO, cardiac output; ECF, extracellular fluid; JGA, juxtaglomerular apparatus.
As Na+ is involved in the co-transport of H+, K+ and Cl–, the acid–base balance is disturbed because Na+ is retained. Cl– is reabsorbed in equal quantities but, initially, there is increased secretion of H+ and K+, resulting in metabolic alkalosis (contraction alkalosis) and hypokalaemia. This is balanced by the shift to anaerobic metabolism as a result of hypoxia in the tissues, which eventually prevails to cause a metabolic acidosis. This is further potentiated as hypovolaemia becomes more severe, as less urine is excreted and H+ is no longer excreted.
Hepatorenal syndrome
Patients with liver disease can have a reduced urine flow (oliguria). This is especially so in patients with portal hypertension and ascites. Portal hypertension results from an increase in resistance to blood flow from the gut and spleen, resulting in venous congestion. Possibly due to the release of nitric oxide, there is peripheral vasodilatation. The resulting decrease in blood pressure causes sympathetic activation and activation of the renin–angiotensin–aldosterone system, leading to intense renal vessel vasoconstriction which leads to oliguira and renal failure.
The circulating blood volume can also be reduced in liver disease from the formation of ascites and oedema. These result from portal hypertension and the impaired synthesis of albumin, decreasing the oncotic (colloid osmotic) pressure in the capillaries, favouring fluid movement out of the vsculature. These shifts in fluid out of the vasculature can contribute to the acute kidney injury.
Hypertension
Blood pressure (BP) is influenced by the interaction of genetic and environmental factors, which regulate cardiac output (CO) and total peripheral resistance (TPR):
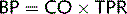
The kidneys influence blood pressure by regulating the volume of extracellular fluid (ECF). They also release vasoactive substances:
Renal autoregulation maintains renal function despite variations in systolic blood pressure. Any change in the ECF will affect the blood pressure. The kidney compensates for these changes by controlling Na+ and water excretion. If this mechanism is disturbed there will be uncontrolled Na+ and water retention, resulting in hypertension. Hypertension is defined by the World Health Organization as a sustained blood pressure of 140/90 mmHg or above.
Essential hypertension
This accounts for about 95% of all cases of hypertension and the cause is unknown. Initially, there is an increase in cardiac output as a result of sympathetic overactivity. In the later stages the increase in blood pressure is maintained by an increase in the total peripheral resistance, but cardiac output is normal. Hypertensive changes seen in the kidney include:
• Arteriosclerosis of the major renal arteries
• Hyalinization of the small vessels with intimal thickening.
This can lead to chronic renal damage (hypertensive nephrosclerosis) and a reduction in the size of the kidneys.
Malignant or ‘accelerated’ hypertension is a rare and rapidly progressing form of severe hypertension. It is characterized by fibrinoid necrosis of the blood vessel walls, and ischaemic damage to the brain and kidney. This can lead to acute renal failure or heart failure, requiring urgent treatment.
Secondary hypertension
This is caused by renal (80%) and endocrine diseases, and occasionally drugs (ciclosporin).
Renal causes
Renal mechanisms causing hypertension include:
Most diseases of the kidney disease can result in hypertension, e.g. diabetic nephropathy, any cause of glomerulonephritis, chronic pyelonephritis and polycystic kidney disease. Renal artery stenosis (discussed below) causes reduced perfusion of the kidney and therefore excessive activation of the renin–angiotensin system.
Endocrine causes
In primary hyperaldosteronism there is chronic excessive secretion of aldosterone because of an adrenal cortical adenoma or hyperplasia (Fig. 4.9). Patients present with hypertension and hypokalaemia. Diagnosis is made based on the triad of:
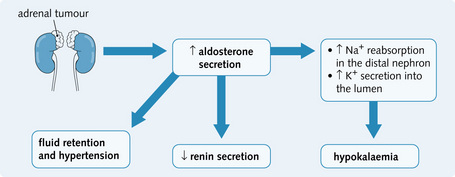
Treatment is by surgical removal of the adenoma, with a cure rate of 60% on aldosterone antagonists (spironolactone). Recent studies have suggested that hyperaldosteronism may be a more common cause of hypertension than previously realized.
Management of hypertension
It is difficult to detect and treat hypertension because it is often asymptomatic and many patients are reluctant to take medication if they feel well. It is very important to exclude an underlying cause of hypertension.
Hypertension is an important risk factor for strokes, cardiac failure, myocardial infarction and chronic kidney disease. Effective treatment will improve the prognosis for each of these conditions. NICE guidelines are available for the treatment of hypertension.
Drug treatment of hypertension involves:
Angiotensin-converting enzyme inhibitors (ACE i)
These inhibit ACE, and so block formation of angiotensin II. Angiotensin II is a potent vasoconstrictor and promotes sodium reabsorption in the tubule (Fig. 4.10). ACE inhibitors (e.g. captopril, ramipril) lower blood pressure by:
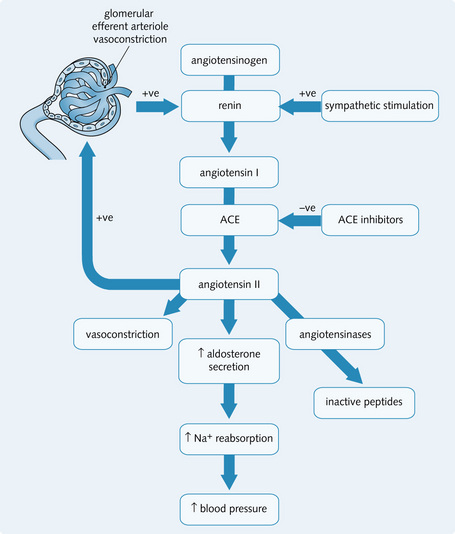
Fig. 4.10 Effects of ACE (angiotensin-converting enzyme) inhibitors. + ve, positive feedback; –ve, negative feedback.
ACE inhibitors also reduce proteinuria and delay the progress of renal disease in patients with diabetic nephropathy and patients with proteinuric non-diabetic renal disease. They are also used to treat CCF.
The side-effects of ACE inhibitors include:
• Allergic reactions or rashes
• Changes in the sensation of taste
• Severe hypotension especially in patients who are hypovolaemic
• Acute renal failure in patients with renal artery stenosis (renal function should be checked after giving ACE inhibitors)
ACE inhibitors are contraindicated in pregnancy because of the risk of:
Diuretics
Diuretics increase the volume of urine produced by increasing renal sodium excretion (natriuresis), which is followed passively by water. Each type of diuretic has specific actions on the normal physiology of a particular segment (Fig. 4.11):
• Act on the membrane transport proteins found on the luminal surface
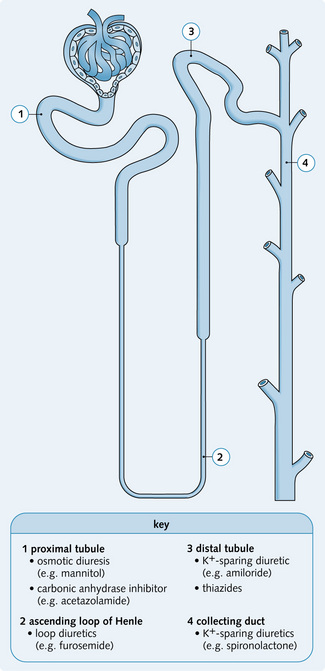
Osmotic diuretics
Osmotic diuresis can be induced by an inert substance that is not reabsorbed in the tubule. The proximal tubule and the descending limb of the loop of Henle allow free movement of water molecules. If an agent such as mannitol is introduced into the tubular fluid, it is not absorbed and thus reduces water reabsorption. There is increased urine flow through the nephrons resulting in reduced sodium reabsorption. Osmotic diuretics are used to:
• Increase urine volume when renal haemodynamics are compromised, and thus prevent anuria
Excessive use of osmotic diuretics without adequate fluid replacement can cause dehydration and hypernatraemia.
Loop diuretics
These are the most powerful diuretics, causing up to 20% of filtered Na+ to be excreted. They inhibit sodium transport out of the thick ascending limb of the loop of Henle into the medullary interstitium. Examples include furosemide and bumetanide. Loop diuretics act by inhibiting the Na+/K+/2Cl– co-transporter on the luminal membrane of the cells. This inhibits Na+ reabsorption, thereby diluting the osmotic gradient in the medulla. This results in increased Na+ and water excretion. Positive lumen potential falls as cations are retained, causing an increase in Ca2 + and Mg2 + excretion. As a higher [Na+] reaches the distal tubule, there is increased K+ secretion, so loop diuretics can be used to reduce total body K+.
Thiazide diuretics
These reduce active Na+ reabsorption in the early distal tubule by inhibiting the Na+/Cl– co-transporter. As there is more reabsorption of Na+ in the loop of Henle, the loop diuretics are more potent than thiazide diuretics. Thiazides help reduce peripheral vascular resistance, and consequently are used to manage hypertension. They are also used in CCF and nephrogenic diabetes insipidus.
Potassium-sparing diuretics
These diuretics are K+-sparing and act:
• In the collecting ducts (e.g. spironolactone)
• By inhibiting the uptake of Na+ in the cells of the distal nephron (e.g. amiloride and triamterene).
Aldosterone is a mineralocorticoid that increases the activity of the Na+/K+ ATPase, potassium and sodium channels, resulting in Na+ absorption and K+ secretion.
Spironolactone (a mineralocorticoid analogue) competes with aldosterone for the receptor site. This reduces sodium reabsorption in the distal nephron and decreases K+ secretion (potassium-sparing activity).
Potassium-sparing diuretics are used if there is mineralocorticoid excess such as primary aldosteronism (Conn's syndrome) or ectopic ACTH production. They are also used in secondary aldosteronism where salt and water retention have occurred (e.g. CCF, nephrotic syndrome, liver disease and hypovolaemia). They are fairly weak, often used with loop diuretics or thiazides to prevent K+ loss.
• Hyperkalaemia: this results from an increase in H+ – and therefore K+ – retention as Na+ absorption falls and ranges from mild to life-threatening
• Endocrine effects with spironolactone (e.g. gynaecomastia).
Potassium-sparing diuretics are contraindicated in patients with chronic renal insufficiency.
Carbonic anhydrase inhibitors
Carbonic anhydrase (CA) is found in many places in the nephron but primarily on the brush border of the luminal membrane of the proximal tubule cells. CA catalyses the dehydration of H2CO3:
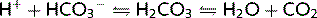
This reaction is driven by H+ secretion into the lumen, by cotransport with Na+. Once in the cell, H2CO3 is reformed under the influence of intracellular CA, the 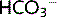 ions are reabsorbed, and H+ is secreted back into the lumen (see Fig. 3.5).
ions are reabsorbed, and H+ is secreted back into the lumen (see Fig. 3.5).
CA inhibitors interfere with the action of carbonic anhydrase and inhibit 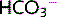 reabsorption. The presence of
reabsorption. The presence of 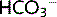 in the lumen reduces Na+ reabsorption, which continues into the distal nephron where it enhances K+ secretion.
in the lumen reduces Na+ reabsorption, which continues into the distal nephron where it enhances K+ secretion.
CA inhibitors such as acetazolamide are weak diuretics, which cause the excretion of only about 5–10% of the filtered Na+ and water. Their main clinical use is to treat acute and chronic glaucoma by reducing intraocular pressure (the production of aqueous humour in the eye involves secretion of 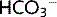 by the ciliary body in a process similar to that in the proximal tubule).
by the ciliary body in a process similar to that in the proximal tubule).
The side-effects of CA inhibitors include:
CA inhibitors should be avoided in patients with liver disease or late-stage CKD.
Diseases of the renal blood vessels
Renal artery stenosis
Between 2% and 5% of hypertensive patients have hypertension secondary to narrowing of one or both renal arteries. This reduces the pressure in the afferent arterioles, which stimulates the juxtaglomerular apparatus to secrete renin. The affected, ischaemic kidney is small. There are two types:
• Atherosclerotic renovascular disease: Atherosclerosis accounts for 70% of renal artery stenosis. It may be suspected when there is other evidence of vascular disease e.g. femoral or aortic bruits, coronary artery disease, peripheral vascular disease, aortic aneurysms. It may be asymptomatic
• Fibromuscular dysplasia: This is usually seen in young women and can often be cured with renal artery angioplasty and stenting (Fig. 4.12).
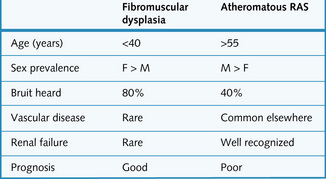
It may present with hypertension, suddenly decreasing renal function with ACE inhibitors, a renal bruit or recurrent flash pulmonary oedema from fluid retention. Treatment options include:
• Drugs to control the blood pressure and vascular risk factors: If the blood pressure is left uncontrolled the contralateral kidney may become damaged by hypertension. ACE inhibitors can be used cautiously to reduce the effect of the renin–angiotensin–aldosterone system, but renal function must be monitored carefully. It is essential to control vascular risk factors and aspirin and a statin should be considered and smoking cessation advised
• Angioplasty: to dilate the stenotic region. This can be supplemented with stenting to decrease the risk of restenosis. This is most successful for fibromuscular dysplasia but the benefit in atherosclerotic renal artery disease is less clear.
Thrombotic microangiopathies
This is a group of diseases that are all characterized by necrosis and thickening of the renal vessel walls and thrombosis in the interlobular arterioles, afferent arterioles and glomeruli. All clinically present with the triad of:
The main two microangiopathies are:
Haemolytic uraemic syndrome
This is characterized by the triad of:
• Idiopathic: this is more common in adults, and has a worse prognosis
• Secondary: this can be associated with gastroenteritis (e.g. Escherichia coli 0157 toxin), drugs (oestrogen, ciclosporin, cytotoxic therapy) or malignancy. HUS can also be caused by accelerated hypertension. There may also be a genetic cause.
Clinical features include sudden onset of oliguria with haematuria – occasionally with melaena or haematemesis (usually if gastroenteritis is the cause) – and jaundice. Hypertension is seen in 50% of patients.
Treatment involves early supportive therapy with dialysis for renal failure. Fresh frozen plasma or plasma exchange can be useful. Approximately 50% of patients later develop hypertension, and a few go on to develop CKD. Mortality ranges from 5% to 30%.
Thrombotic thrombocytopenic purpura
This is a rare and idiopathic condition that is more common in females (usually < 40 years) than in males. The features are fever, neurological signs (central nervous system (CNS) involvement), haemolytic anaemia and thrombocytopenia. TTP has a similar disease process to HUS, but affects different sites. Renal involvement occurs in only 50% of cases, and presents with:
The majority of cases have a dominant CNS component, with thrombosis leading to ischaemia in the brain.
Histological examination shows thrombi consisting of fibrin and platelets in the terminal interlobular arteries, the afferent arterioles and glomerular capillaries.
Treatment involves corticosteroid therapy and plasma exchange.
Renal infarction
Embolic infarction
The emboli lodge in the small renal vessels and cause narrowing of the arterioles and focal areas of ischaemic injury. It can be asymptomatic, or present with haematuria and loin tenderness. The areas of infarction appear pale and are characteristically wedge-shaped.
Diffuse cortical necrosis
Diffuse cortical necrosis is caused by profound hypotension. Typical causes are sepsis or hypovolaemia following severe blood loss. It presents with anuria and carries a very poor renal prognosis.
Sickle-cell disease nephropathy
Thrombotic occlusion by deformed sickle-shaped red cells causes papillary necrosis. It is precipitated by cold, dehydration, infection and exercise. Presentation is with pain, haematuria and polyuria. Management involves analgesia, warmth and rehydration, blood transfusions and antibiotics (if infection is suspected).
Benign nephrosclerosis
This is the term given to the changes in renal vasculature in response to longstanding essential (benign) hypertension. The changes consist of hyaline arteriolosclerosis, which is characterized by thickening (due to hyperplasia of smooth muscle) and hyalinization (protein deposition) of the arteriolar wall. This causes narrowing of the lumen of the interlobular arteries, which functionally impairs the smaller branches. The changes are more severe in patients with systemic diseases that affect the renal vessels (e.g. diabetes). The vascular wall lesions gradually reduce the blood supply to the kidney, which leads to ischaemic atrophy of the nephrons. This accounts for the small, contracted and granular appearance of the kidneys seen in advanced cases of untreated essential hypertension. Renal function may be well-preserved initially, although proteinuria is sometimes detected.
Malignant nephrosclerosis
This is associated with accelerated hypertension, with an increase in diastolic pressure to over 130 mmHg. There are fibrin deposits in the vessel wall, causing necrosis, especially in the distal part of the interlobular arteries and the afferent arterioles.
Renal function is impaired because of the ischaemia that results from severe arterial damage. Patients have proteinuria and haematuria, which can occasionally be massive. Acute kidney injury develops if untreated (in contrast to benign hypertension). Papilloedema is often present. The 5-year survival rate with treatment is more than 50%.
The trigger for the abrupt and rapid rise in blood pressure is unknown but might be associated with endothelial dysfunction. These patients also have increased plasma levels of renin, aldosterone and angiotensin.