Chapter 3 Bacterial physiology and genetics
Bacterial physiology
Growth
Bacteria, like all living organisms, require nutrients for metabolic purposes and for cell division, and grow best in an environment that satisfies these requirements. Chemically, bacteria are made up of polysaccharide, protein, lipid, nucleic acid and peptidoglycan, all of which must be manufactured for successful growth.
Nutritional requirements
Oxygen and hydrogen
Both oxygen and hydrogen are obtained from water; hence, water is essential for bacterial growth. In addition, the correct oxygen tension is necessary for balanced growth. While the growth of aerobic bacteria is limited by availability of oxygen, anaerobic bacteria may be inhibited by low oxygen tension.
Carbon
Inorganic ions
Nitrogen, sulphur, phosphate, magnesium, potassium and a number of trace elements are required for bacterial growth.
Reproduction
Bacteria reproduce by a process called binary fission, in which a parent cell divides to form a progeny of two cells. This results in a logarithmic growth rate – one bacterium will produce 16 bacteria after four generations. The doubling or mean generation time of bacteria may vary (e.g. 20 min for Escherichia coli, 24 h for Mycobacterium tuberculosis); the shorter the doubling time, the faster the multiplication rate. Other factors that affect the doubling time include the amount of nutrients, the temperature and the pH of the environment.
Bacterial growth cycle
The growth cycle of a bacterium has four main phases (Fig. 3.1):
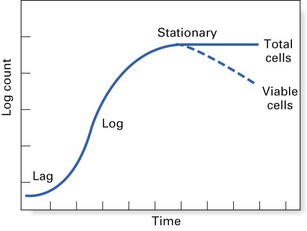
Growth regulation
Bacterial growth is essentially regulated by the nutritional environment. However, both intracellular and extracellular regulatory events can modify the growth rate. Intracellular factors include:
Extracellular factors that modify bacterial growth are:
 mesophiles, which grow well between 25 and 40°C, comprising most medically important bacteria (that grow best at body temperature)
mesophiles, which grow well between 25 and 40°C, comprising most medically important bacteria (that grow best at body temperature)Aerobic and anaerobic growth
A good supply of oxygen enhances the metabolism and growth of most bacteria. The oxygen acts as the hydrogen acceptor in the final steps of energy production and generates two molecules: hydrogen peroxide (H2O2) and the free radical superoxide (O2). Both of these are toxic and need to be destroyed. Two enzymes are used by bacteria to dispose of them: the first is superoxide dismutase, which catalyses the reaction:
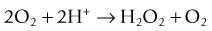
and the second is catalase, which converts hydrogen peroxide to water and oxygen:
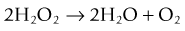
Bacteria can therefore be classified according to their ability to live in an oxygen-replete or an oxygen-free environment (Fig. 3.2, Table 3.1). This has important practical implications, as clinical specimens must be incubated in the laboratory under appropriate gaseous conditions for the pathogenic bacteria to grow. Thus, bacteria can be classified as follows:
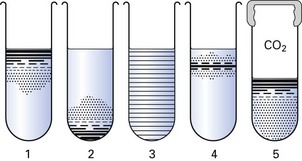
Fig. 3.2 Atmospheric requirements of bacteria, as demonstrated in agar shake cultures. (1) Obligate aerobe; (2) obligate anaerobe; (3) facultative anaerobe; (4) microaerophile; (5) capnophilic organism (growing in carbon dioxide-enriched atmosphere). (See also Table 3.1.)
Table 3.1 Effect of oxygen on the growth of bacteria
| Degree of oxygenation | Term | Example |
|---|---|---|
| Oxygen essential for growth | Obligate aerobe | Pseudomonas aeruginosa |
| Grows well under low oxygen concentration (5%) | Microaerophile | Campylobacter fetus |
| Grows in the presence or absence of oxygen | Facultative anaerobea | Streptococcus milleri |
| Only grows in the absence of oxygen | Obligate anaerobe | Porphyromonas gingivalis |
a Facultative anaerobes may be subgrouped as capnophiles or capnophilic organisms if they grow well in the presence of 8–10% carbon dioxide (e.g. Legionella pneumophila).
Bacterial genetics
Genetics is the study of inheritance and variation. All inherited characteristics are encoded in DNA, except in RNA viruses.
The bacterial chromosome
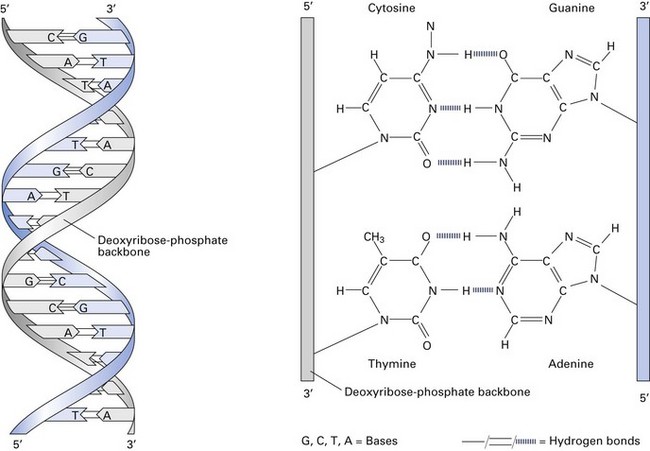
The bacterial chromosome contains the genetic information that defines all the characteristics of the organism. It is a single, continuous strand of DNA (Fig. 3.3) with a closed, circular structure attached to the cell membrane of the organism. The ‘average’ bacterial chromosome has a molecular weight of 2 × 109.
Replication
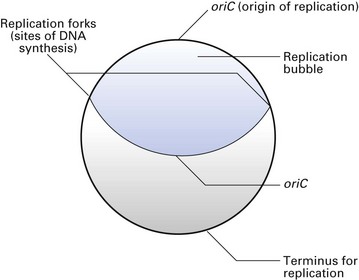
Chromosome replication is an accurate process that ensures that the progeny cells receive identical copies from the mother cell. The replication process is initiated at a specific site on the chromosome (oriC site) where the two DNA strands are locally denatured. A complex of proteins binds to this site, opens up the helix and initiates replication. Each strand then serves as a template for a complete round of DNA synthesis, which occurs in both directions (bidirectional) and on both strands, creating a replication bubble (Fig. 3.4). The two sites at which the replication occurs are called the replication forks. As replication proceeds, the replication forks move around the molecule in opposite directions opening up the DNA strands, synthesizing two new complementary strands until the two replication forks meet at a termination site. Of the four DNA strands now available, each daughter cell receives a parental strand and a newly synthesized strand. This process is called semiconservative replication. Such chromosomal replication is synchronous with cell division, so that each cell receives a full complement of DNA from the mother cell.
The main enzyme that mediates DNA replication is DNA-dependent DNA polymerase, although a number of others take part in this process. When errors occur during DNA replication, repair mechanisms excise incorrect nucleotide sequences with nucleases, replace them with the correct nucleotides and religate the sequence.
Bacteria have evolved mechanisms to delete foreign nucleotides from their genomes. Restriction enzymes are mainly used for this purpose, and they cleave double-stranded DNA at specific sequences. The DNA fragments produced by restriction enzymes vary in their molecular weight and can be demonstrated in the laboratory by gel electrophoresis. Hence, these restriction enzymes are used in many clinical analytical techniques to cleave DNA and to characterize both bacteria and viruses (see below).
Genes
The genetic code of bacteria is contained in a series of units called genes. As the normal bacterial chromosome has only one copy of each gene, bacteria are called haploid organisms (as opposed to higher organisms, which contain two copies of the gene and hence are diploid).
A gene is a chain of purine and pyrimidine nucleotides. The genetic information is coded in triple nucleotide groups or codons. Each codon or triplet nucleotide codes for a specific amino acid or a regulatory sequence, e.g. start and stop codons. In this way, the structural genes determine the sequence of amino acids that form the protein, which is the gene product.
The genetic material of a typical bacterium (e.g. E. coli) comprises a single circular DNA with a molecular weight of about 2 × 109 and composed of approximately 5 × 106 base pairs, which in turn can code for about 2000 proteins.
Genetic variation in bacteria
Genetic variation can occur as a result of mutation or gene transfer.
Mutation
A mutation is a change in the base sequence of DNA, as a consequence of which different amino acids are incorporated into a protein, resulting in an altered phenotype. Mutations result from three types of molecular change, as follows.
Base substitution
This occurs during DNA replication when one base is inserted in place of another. When the base substitution results in a codon that instructs a different amino acid to be inserted, the mutation is called a missense mutation; when the base substitution generates a termination codon that stops protein synthesis prematurely, the mutation is called a nonsense mutation. The latter always destroys protein function.
Frame shift mutation
A frame shift mutation occurs when one or more base pairs are added or deleted, which shifts the reading frame on the ribosome and results in the incorporation of the wrong amino acids ‘downstream’ from the mutation and in the production of an inactive protein.
Insertion
The insertion of additional pieces of DNA (e.g. transposons) or an additional base can cause profound changes in the reading frames of the DNA and in adjacent genes (Fig. 3.5).
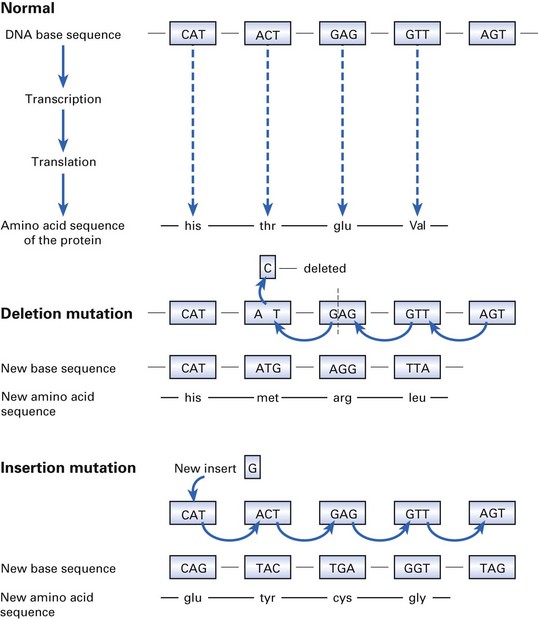
Fig. 3.5 Events that entail mutation: the effect of the deletion and insertion of a single base on the amino acid sequence (and the quality of the protein thus produced) is shown.
Mutations can be induced by chemicals, radiation or viruses.
Gene transfer
The transfer of genetic information can occur by:
Clinically, the most important consequence of DNA transfer is that antibiotic-resistant genes are spread from one bacterium to another.
Conjugation
This is the mating of two bacteria, during which DNA is transferred from the donor to the recipient cell (Fig. 3.6A). The mating process is controlled by an F (fertility) plasmid, which carries the genes for the proteins required for mating, including the protein pilin, which forms the sex pilus (conjugation tube). During mating, the pilus of the donor (male) bacterium carrying the F factor (F+) attaches to a receptor on the surface of the recipient (female) bacterium. The latter is devoid of an F plasmid (F−). The cells are then brought into direct contact with each other by ‘reeling in’ of the sex pilus. Then the F factor DNA is cleaved enzymatically, and one strand is transferred across the bridge into the female cell. The process is completed by synthesis of the complementary strand to form a double-stranded F plasmid in both the donor and recipient cells. The recipient now becomes an F+ male cell that has the ability to transmit the plasmid further. The new DNA can integrate into the recipient’s DNA and become a stable component of its genetic material. Complete transfer of the bacterial DNA takes about 100 min.
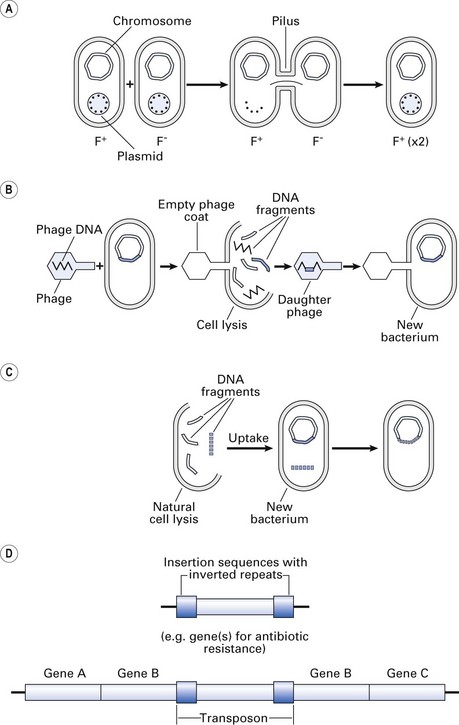
Fig. 3.6 Gene transfer. (A) Conjugation: transfer of a plasmid gene by conjugation (see text); (B) transduction: phage-mediated gene transfer from one bacterium to another; (C) transformation: gene transfer by uptake of exogenous bacterial DNA by another bacterium in the vicinity (not mediated by plasmid or phage); (D) transposition: transposons (jumping genes) can move from one DNA site to another, thereby inactivating the recipient gene and conferring new traits such as drug resistance.
Transduction
Transduction is a process of DNA transfer by means of a bacterial virus – a bacteriophage (phage). During the replication of the phage, a piece of bacterial DNA is incorporated, accidentally, into the phage particle and is carried into the recipient cell at the time of infection (Fig. 3.6B). There are two types of transduction:
Plasmid DNA can also be transferred to another bacterium by transduction. However, the donated plasmid can function independently without recombining with bacterial DNA. The ability to produce an enzyme that destroys penicillin (β-lactamase) is mediated by plasmids that are transferred between staphylococci by transduction.
Transformation
This is the transfer of exogenous bacterial DNA from one cell to another. It occurs in nature when dying bacteria release their DNA, which is then taken up by recipient cells and recombined with the recipient cell DNA. This process appears to play an insignificant role in disease (Fig. 3.6C).
Transposition
This occurs when transposable elements (transposons; see below) move from one DNA site to another within the genome of the same organism (e.g. E. coli). The simplest transposable elements, called ‘insertion sequences’, are less than 2 kilobases in length and encode enzymes (transposase) required for ‘jumping’ from one site to another (Fig. 3.6D).
Recombination
When the DNA is transferred from the donor to the recipient cell by one of the above mechanisms, it is integrated into the host genome by a process called recombination. There are two types of recombination:
Plasmids
Plasmids are extrachromosomal, double-stranded circular DNA molecules within the size range 1–200 MDa. They are capable of replicating independently of the bacterial chromosome (i.e. they are replicons). Plasmids occur in both Gram-positive and Gram-negative bacteria, and several different plasmids can often coexist in one cell.
Transmissible plasmids can be transferred from cell to cell by conjugation. They contain about 10–12 genes responsible for synthesis of the sex pilus and for the enzymes required for transfer; because of their large size, they are usually present in a few (one to three) copies per cell.
Non-transmissible plasmids are small and do not contain the transfer genes. However, they can be mobilized by co-resident plasmids that do contain the transfer gene. Many copies (up to 60 per cell) of these small plasmids may be present.
Clinical relevance of plasmids
A number of medically important functions of bacteria are attributable to plasmids (i.e. are plasmid-coded). The plasmid-coded bacterial attributes include:
Transposons
Transposons, also called jumping genes, are pieces of DNA that move readily from one site to another, either within or between the DNAs of bacteria, plasmids and bacteriophages. In this manner, plasmid genes can become part of the chromosomal complement of genes. Interestingly, when transposons transfer to a new site, it is usually a copy of the transposon that moves, while the original remains in situ (like photocopying). For their insertion, transposons do not require extensive homology between the terminal repeat sequences of the transposon (which mediate integration) and the site of insertion in the recipient DNA.
Transposons can code for metabolic or drug resistance enzymes and toxins. They may also cause mutations in the gene into which they insert, or alter the expression of nearby genes.
In contrast to plasmids or bacterial viruses, transposons cannot replicate independently of the recipient DNA. More than one transposon can be located in the DNA; for example, a plasmid can contain several transposons carrying drug resistance genes. Thus, transposons can jump from:
Recombinant DNA technology in microbiology
By definition, every classified species must have somewhere on its genome a unique DNA or RNA sequence that distinguishes it from another species. In diagnostic microbiology, this attribute is used to identify microbes where the DNA sequence of the offending pathogen can be identified by means of a number of clever techniques, using clinical samples from the patient.
Gene cloning
Gene cloning is the artificial incorporation of one or more genes into the genome of a new host cell by various genetic recombination techniques.
The candidate DNA is first extracted from the source, purified and cut or cleaved into small fragments by restriction enzymes – leaving ‘sticky ends’. These are then inserted into a vector DNA, first by cutting the vector DNA with the same enzyme so as to produce complementary sticky ends. The sticky ends of the vector and the candidate DNA are then tied or ligated together using enzymes called ‘DNA ligases’ to produce a recombinant DNA molecule. This process can also be used for cloning RNA, when complementary copies of DNA are produced by reverse transcription using reverse transcriptase enzymes. The vector used for gene transfer is usually a plasmid or a virus.
The vector with the integrated DNA has to be inserted into a cell in order to obtain multiple copies of the organism that express the selected gene. This can be done by:
The insertion of the vector containing the recombinant DNA does not necessarily mean that all the progeny bacteria will contain the inserted element, because the vector integration process is somewhat random. In order to select the clone of bacteria that expresses the recombinant gene, other devious manoeuvres have to be adopted. For instance, one can choose a plasmid vector that carries resistance to antibiotics A and B. If the foreign DNA is inserted in the middle of gene A that confers resistance to antibiotic A, then this gene will be inactivated as a consequence. In this manner, bacteria with the cloned foreign DNA can be selected and are called the gene library.
Gene probes
DNA probes
Used extensively in diagnostic microbiology, gene probes are pieces of DNA that are labelled radioactively or with a chemiluminescent marker. The probes carry a single strand of DNA analogous to the pathogen that is sought in the clinical sample. There are different types of DNA probe:
Oligonucleotide probes
Oligonucleotide probes are based on the variable region of the 16S ribosomal RNA (rRNA) genes. The nucleotide sequences of the latter gene of a number of microbes have been well characterized, and are known to be well preserved across species, except for several small variable regions. This property is helpful in the construction of specific oligonucleotide probes of about 18–30 bases, which are much more specific than the DNA probes described above.
RNA probes
Cellular protein synthesis is dependent on rRNA, and any mutation of the rRNA leads to cell death. Further, rRNA is highly species-specific, and this property is exploited to produce RNA probes that are useful for both diagnostic microbiology and taxonomic studies. The most commonly used are the 5S, 16S and 23S probes.
DNA/RNA probes and oral microbiology
Cultivation of the complex mixture of bacteria residing in the oral cavity is fraught with problems, and it is now recognized that a number of bacterial genera are difficult or almost impossible to culture. The introduction of DNA and RNA probes has helped us to obtain a more complete picture of the oral flora. For example, commercially available probes can now be used in diagnostic laboratories not only to identify but also to quantify periodontopathic flora in subgingival plaque samples obtained from a periodontal pocket (Fig. 3.7). Further, the samples, say in paper points, could be simply sent by post to distant laboratories for identification without the fear of death of organisms and the associated cumbersome culture procedures.
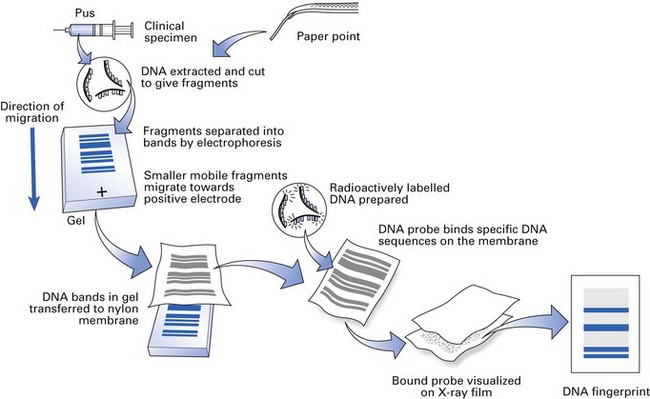
Polymerase chain reaction
Gene-cloning techniques revolutionized the molecular biological advances in the 1970s. The analogous event that took place in the late 1980s was the invention of the PCR. It is a simple technique in which a short region of a DNA molecule, a single gene, for instance, is copied repetitiously by a DNA polymerase enzyme (Fig. 3.8). This technique, in combination with a number of others described below, is used to identify unculturable bacteria from the oral cavity and other body sites (Fig. 3.9).
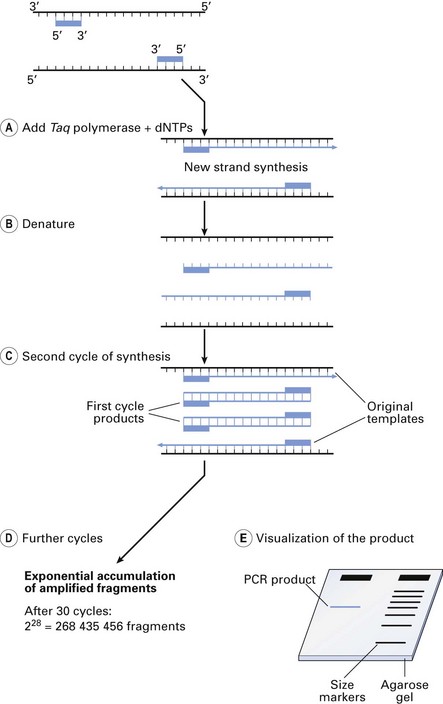
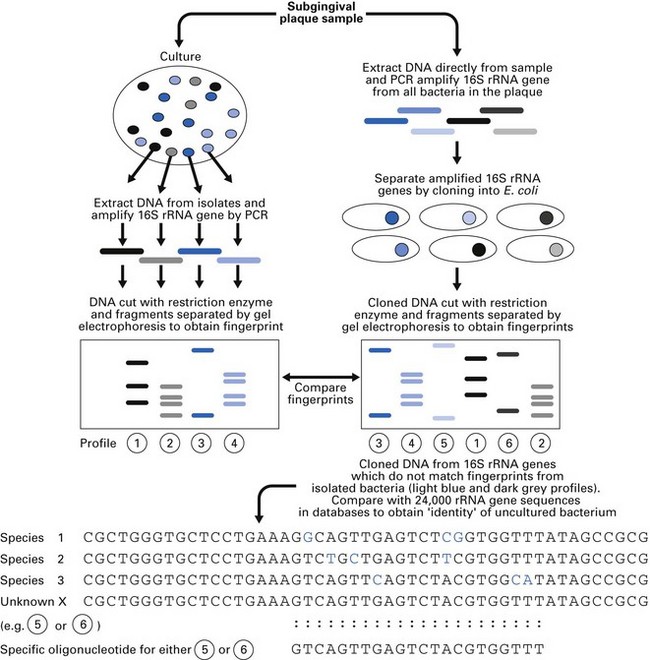
Fig. 3.9 Use of polymerase chain reaction (PCR) technology to identify unculturable bacteria obtained from a subgingival plaque sample.
(Modified from Jenkinson, H and Dymock, D. (1999). Dental Update 26: 191–197, by permission of George Warman Publications (UK) Ltd.)
Materials
Method
PCR and its variations
The basic PCR methodology is now modified to provide sophisticated analytical tools. The main features of three commonly used variations of PCR, namely nested, mutiplex and real-time PCR, are given below.
Nested PCR
Here, two sets of primers are used: the first set is used for the primary amplification round. The second primer set, specifically chosen to anneal with an internal sequence of the amplicon, re-amplifies the latter ‘specific’ sequence; nested PCR has increased sensitivity than the conventional PCR.
Multiplex PCR
In this method, more than one locus of the nucleotide is simultaneously amplified using multiple sets of primers, thus saving time and resources; mutiplex PCR has increased specificity and can identify organisms more accurately.
Real-time PCR
Conventional PCR requires gel electrophoresis for analysis of the amplicons. In real-time PCR, this step is automatically performed in real time, and the target sequence is identified within a closed system, using either labelled fluorophores or other similar labelled probes. Further advantages are the versatility of the system, enabling (1) analysis of multiple amplicons at specific time sequences during a reaction period, (2) semiquantitative estimation of the yield, and (3) multiplex evaluation of the products (see above). The disadvantage is the relatively expensive technology.
Why is PCR so widely used?
Some reasons why the use of PCR is so widespread:
Other techniques for genetic typing of microorganisms
Restriction enzyme analysis
A genetic ‘fingerprint’ of the organism is obtained by extracting its DNA and cutting or cleaving the DNA at specific points by restriction endonucleases. The DNA fragments so generated are run on an agarose electrophoresis gel and viewed under ultraviolet illumination after staining with ethidium bromide. The profiles of the bands produced on the gel (the ‘fingerprints’) can be compared or contrasted with those from other strains. This was the original molecular method used for genotyping organisms, but has been supplanted by newer methods that are more discriminatory.
Restriction fragment length polymorphism
In restriction fragment length polymorphism (RFLP), the DNA is first cleaved using restriction endonucleases and separated on the agarose gel. Afterwards, the separated fragments are transferred by blotting on to a nitrocellulose or nylon membrane by a method called Southern blotting, and DNA probes constructed from genes of known organisms (species or strains) are then hybridized to the membrane; these will bind to complementary sequences in the DNA fragments on the membrane, revealing the species or strain identity.
Pulsed-field gel electrophoresis
Pulsed-field gel electrophoresis (PFGE) is similar to RFLP. Here, the chromosomal DNA of an organism is cut into relatively large pieces by restriction enzymes and the resultant fragments are separated in an agarose gel with the help of a pulsed electric field, in which the polarity is regularly reversed. Large pieces of chromosomes usually do not separate in conventional agarose gels, hence the necessity of the pulsed/reversed electric field.
Pyrosequencing
Pyrosequencing is one of the most novel and reliable techniques of DNA sequencing. It is based on the ‘sequencing by synthesis’ principle. So called as it relies on the detection of pyrophosphate release on nucleotide incorporation, rather than chain termination with dideoxynucleotides used in PCR techniques. It uses chemiluminescence enzyme reactions and photodetection techniques that are highly automated, rapid and sensitive.
The era of ‘-omics’
With the advent of the new millennium, there has been an explosion of digital and computer technology, the use of which has led to a parallel advancement of the knowledge of our biosphere. This in turn has led to focal developments of sub-disciplines such as genomics, proteomics and metabolomics – the so-called ‘-omics’ era. These new technologies have had a significant impact on the identification of microbes, particularly those that could not be cultured in the laboratory (unculturable bacteria), and on the elucidation of their pathogenic mechanisms such as resistance to antibiotics. A brief introduction to the various -omics domains are given below:
Genomics
This refers to the study of the identity of all genes within the chromosome of a cell. The human animal and microbial genome sequencing projects have thus far provided a rich genetic resource to better understand human diseases including oral diseases. As mentioned in Chapter 2, the development of technologies such as microarray analysis have helped microbiologists to explore patterns of gene expression in various infectious diseases, and their pathogenic mechanisms, for example, in periodontal disease. The subcategory of functional genomics deals with the organization of the genes and their expression patterns under defined conditions.
The development of computer models for high throughput analyses of genomic data has simplified the exploration of gene expression profiles in both eukaryotes and prokaryotes. Furthermore, DNA microarray technologies help investigators evaluate gene expression on a genome-wide basis, providing a ‘global’ perspective of how an organism responds to a specific stress, drug or toxin.
Proteomics
This is defined as the study of the myriad of proteins expressed by the genome of either an organism, cell or tissue type. Proteomics builds on and complements the knowledge gained from genomics by revealing the levels, activities, regulation and interactions of every protein in an organism or a cell. Study of the proteome is more complex than that of the genome as the number of proteins in an organism/cell is considered many orders of magnitude greater than that of the number of genes.
Such complexity is further confounded by the dynamic changes in the proteome in response to the environment and also the multiple possible interactive combinations among proteins. Protein chips that can simultaneously identify large numbers of proteins are helpful in unravelling such complexity.
Transcriptomics
This is a related branch of molecular biology that deals with the study of messenger RNA molecules produced in an individual or population of a particular cell type.
Metabolomics
This is defined as the scientific study of chemical processes involving metabolites of a cell or an organism. While proteomic analyses do not tell the whole story of what might be happening in a cell, metabolic profiling can give an instantaneous snapshot of the physiology of that organism. This has led to the development of a further domain known as interactomics. The latter is defined as a discipline involving the intersection of bioinformatics and biology that deals with studying both the interactions and the consequences of those interactions between and among proteins, and other molecules within an organism. The network of all such interactions is called the interactome. In essence, interactomics aims to compare networks of interactions (i.e. interactomes) between and within species in order to elucidate how the traits of such networks are either preserved or varied.
One of the current challenges of science is to integrate proteomic, transcriptomic, metabolic and interactomic data to provide a more complete picture of living organisms.
Key facts
Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. The molecular biology of the cell, 5th ed. New York: Garland; 2007.
Beebee T., Burke J. Gene structure and transcription, 2nd ed. Oxford: IRL Press/Oxford University Press; 1992.
. Topley and Wilson’s microbiology and microbial infections. Collier L.H., editor, 9th ed. London: Edward Arnold. 1998.
Moat A.G., Foster J.W., Spector M.P. Microbial physiology. New York: Wiley-Liss; 2002.
Review questions (answers on p. 351)
Please indicate which answers are true, and which are false.