Chapter 7 The role of information, bioinformatics and genomics
The pharmaceutical industry as an information industry
As outlined in earlier chapters, making drugs that can affect the symptoms or causes of diseases in safe and beneficial ways has been a substantial challenge for the pharmaceutical industry (Munos, 2009). However, there are countless examples where safe and effective biologically active molecules have been generated. The bad news is that the vast majority of these are works of nature, and to produce the examples we see today across all biological organisms, nature has run a ‘trial-and-error’ genetic algorithm for some 4 billion years. By contrast, during the last 100 years or so, the pharmaceutical industry has been heroically developing handfuls of successful new drugs in 10–15 year timeframes. Even then, the overwhelming majority of drug discovery and development projects fail and, recently, the productivity of the pharmaceutical industry has been conjectured to be too low to sustain its current business model (Cockburn, 2007; Garnier, 2008).
A great deal of analysis and thought is now focused on ways to improve the productivity of the drug discovery and development process (see, for example, Paul et al., 2010). While streamlining the process and rebalancing the effort and expenditures across the various phases of drug discovery and development will likely increase productivity to some extent, the fact remains that what we need to know to optimize productivity in drug discovery and development far exceeds what we do know right now. To improve productivity substantially, the pharmaceutical industry must increasingly become what in a looser sense it always was, an information industry (Robson and Baek, 2009).
Initially, the present authors leaned towards extending this idea by highlighting the pharmaceutical process as an information flow, a flow seen as a probabilistic and information theoretic network, from the computations of probabilities from genetics and other sources, to the probability that a drug will play a useful role in the marketplace. As may easily be imagined, such a description is rich, complex, and evolving (and rather formal), and the chapter was rapidly reaching the size of a book while barely scratching the surface of such a description. It must suffice to concentrate on the nodes (old and new) of that network, and largely on those nodes that represent the sources and pools of data and information that are brought together in multiple ways.
This chapter begins with general concepts about information then focuses on information about biological molecules and on its relationship to drug discovery and development. Since most drugs act by binding to and modulating the function of proteins, it is reasonable for pharmaceutical industry scientists to want to have as much information as possible about the nature and behaviour of proteins in health and disease. Proteins are vast in numbers (compared to genes), have an enormous dynamic range of abundances in tissues and body fluids, and have complex variations that underlie specific functions. At present, the available information about proteins is limited by lack of technologies capable of cost-efficiently identifying, characterizing and quantifying the protein content of body fluids and tissues. So, this chapter deals primarily with information about genes and its role in drug discovery and development.
Innovation depends on information from multiple sources
Information from three diverse domains sparks innovation in drug discovery and development (http://dschool.stanford.edu/big_picture/multidisciplinary_approach.php):
• Feasibility (Is the product technically/scientifically possible?)
• Viability (Can such a product be brought cost-effectively to the marketplace?)
The viability and desirability domains include information concerning public health and medical need, physician judgment, patient viewpoints, market research, healthcare strategies, healthcare economics and intellectual property. The feasibility domain includes information about biology, chemistry, physics, statistics and mathematics. Currently, the totality of available information can be obtained from diverse, unconnected (’siloed’) public or private sources, such as the brains of human beings, books, journals, patents, general literature, databases available via the Internet (see, for example, Fig. 7.1) and results (proprietary or otherwise) of studies undertaken as part of drug discovery and development. However, due to its siloed nature, discreet sets of information from only one domain are often applied, suboptimally, in isolation to particular aspects of drug discovery and development. One promising approach for optimizing the utility of available, but diverse, information across all aspects of drug discovery and development is to connect apparently disparate information sets using a common format and a collection of rules (a language) that relate elements of the content to each other. Such connections make it possible for users to access the information in any domain or set then traverse across information in multiple sets or domains in a fashion that sparks innovation. This promising approach is embodied in the Semantic Web, a vision for the connection of not just web pages but of data and information (see http://en.wikipedia.org/wiki/Semantic_Web and http://www.w3.org/2001/sw/), which is already beginning to impact the life sciences, generally, and drug discovery and development, in particular (Neumann and Quan, 2006; Stephens et al., 2006; Ruttenberg et al., 2009).
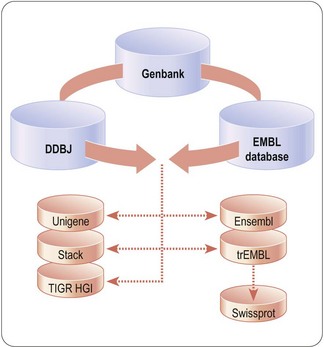
Fig. 7.1 Flow of public sequence data between major sequence repositories. Shown in blue are the components of the International Nucleotide Sequence Database Collaboration (INSDC) comprising Genbank (USA), the European Molecular Biology Laboratory (EMBL) Database (Europe) and the DNA Data Bank of Japan (DDBJ).
Bioinformatics
The term bioinformatics for the creation, analysis and management of information about living organisms, and particularly nucleotide and protein sequences, was probably first coined in 1984, in the announcement of a funding programme by the European Economic Community (EC COM84 Final). The programme was in response to a memo from the White House that Europe was falling behind America and Japan in biotechnology.
The overall task of bioinformatics as it was defined in that announcement is to generate information from biological data (bioinformation) and to make that information accessible to humans and machines that are in need of information to advance toward the achievement of an objective. Handling all the data and information about life forms, even the currently available information on nucleotide and protein sequences, is not trivial. Overviews of established basic procedures and databases in bioinformatics, and the ability to try one’s hand at them, are provided by a number of high-quality sources many of which have links to other sources, for example:
• The European Bioinformatics Institute (EMBL-EBI), which is part of the European Molecular Biology Laboratory (EMBL) (http://www.ebi.ac.uk/2can/home.html)
• The National Center for Biotechnology Information of the National Institutes of Health (http://www.ncbi.nlm.nih.gov/Tools/)
• The Biology Workbench at the University of San Diego (http://workbench.sdsc.edu/).
Rather than give a detailed account of specific tools for bioinformatics, our focus will be on the general ways by which bioinformation is generated by bioinformatics, and the principles used to analyse and interpret information.
Bioinformatics is the management and data analytics of bioinformation. In genetics and molecular biology applications, that includes everything from classical statistics and specialized applications of statistics or probability theory, such as the Hardy–Weinberg equilibrium law and linkage analysis, to modelling interactions between the products of gene expression and subsequent biological interpretation (for interpretation tool examples see www.ingenuity.com and www.genego.com). According to taste, one may either say that a new part of data analytics is bioinformatics, or that bioinformatics embraces most if not all of data analytics as applied to genes and proteins and the consequences of them. Here, we embrace much as bioinformatics, but also refer to the more general field of data analytics for techniques on which bioinformatics draws and will continue to borrow.
The term bioinformatics does have the merit of addressing not only the analysis but also the management of data using information technology. Interestingly, it is not always easy to distinguish data management from the processing and analysis of data. This is not least because computers often handle both. Sometimes, efficiency and insight can be gained by not segregating the two aspects of bioinformatics.
Bioinformatics as data mining and inference
Data mining includes also analysis of market, business, communications, medical, meteorological, ecological, astronomical, military and security data, but its tools have been implicit and ubiquitous in bioinformatics from the outset, even if the term ‘data mining’ has only fairly recently been used in that context. All the principles described below are relevant to a major portion of traditional bioinformatics. The same data mining programme used by one of the authors has been applied to both joint market trends in South America and the relationship of protein sequences to their secondary structure and immunological properties. In the broader language of data analytics, bioinformatics can be seen as having two major modes of application in the way it obtains information from data – query or data mining. In the query mode (directed analysis), for example, a nucleotide sequence might be used to find its occurrence in other gene sequences, thus ‘pulling them from the file’. That is similar to a Google search and also somewhat analogous to testing a hypothesis in classical statistics, in that one specific question is asked and tested as to the hypothesis that it exists in the data.
In the data mining mode (undirected or unsupervised analysis), one is seeking to discover anything interesting in the data, such as a hidden pattern. Ultimately, finding likely (and effectively testing) hypotheses for combinations of N symbols or factors (states, events, measurements, etc.) is equivalent to making 2N-1 queries or classical statistical tests of hypotheses. For N = 100, this is 1030. If such an activity involves continuous variables of interest to an error of e percent (and e is usually much less than 50%) then this escalates to (100/e)N. Clearly, therefore, data mining is a strategy, not a guaranteed solution, but, equally clearly, delivers a lot more than one query. Insomuch as issuing one query is simply a limiting case of the ultimate in highly constrained data mining, both modes can be referred to as data mining.
Both querying and data mining seem a far cry from predicting what regions of DNA are likely to be a gene, or the role a pattern of gene variants might play in disease or drug response, or the structure of a protein, and so on. As it happens, however, prediction of what regions of DNA are genes or control points, what a gene’s function is, of protein secondary and tertiary structure, of segments in protein primary structure that will serve as a basis for a synthetic diagnostic or vaccine, are all longstanding examples of what we might today call data mining followed by its application to prediction. In those activities, the mining of data, as a training set, is used to generate probabilistic parameters often called ‘rules’. These rules are preferably validated in an independent test set. The further step required is some process for using the validated rules to draw a conclusion based on new data or data sets, i.e. formally the process of inference, as a prediction.
An Expert System also uses rules and inference except the rules and their probabilities are drawn from human experts at the rate of 2–5 a day (and, by definition, are essentially anecdotal and likely biased). Computer-based data mining can generate hundreds of thousands of unbiased probabilistic rules in the order of minutes to hours (which is essentially in the spirit of evidence based medicine’s best evidence). In the early days of bioinformatics, pursuits like predicting protein sequence, signal polypeptide sequences, immunological epitopes, DNA consensus sequences with special meaning, and so forth, were often basically like Expert Systems using rules and recipes devised by experts in the field. Most of those pursuits have now succumbed to use rules provided from computer-based mining of increasingly larger amounts of data, and those rules bear little resemblance to the original expert rules.
General principles for data mining
Data mining is usually undertaken on a sample dataset. Several difficulties have dogged the field. At one end of the spectrum is the counterintuitive concern of ‘too much (relevant) information’. Ideally, to make use of maximum data available for testing a method and quality of the rules, it quickly became clear that one should use the jackknife method. For example, in predicting something about each and every accessible gene or protein in order to test a method, that gene or protein is removed from the sample data set used to generate the rules for its prediction. So for a comprehensive test of predictive power, the rules are regenerated afresh for every gene or protein in the database, or more correctly put, for the absence of each in the database. The reason is that probabilistic rules are really terms in a probabilistic or information theoretic expansion that, when brought together correctly, can predict something with close to 100% accuracy, if the gene or protein was in the set used to derive the rules. That has practical applications, but for most purposes would be ‘cheating’ and certainly misleading. Once the accuracy is established as acceptable, the rules are generated from all genes or proteins available, because they will typically be applied to new genes or proteins that emerge and which were not present in the data. On the other hand, once these become ‘old news’, they are added to the source data, and at intervals the rules are updated from it.
At the other end of the scale are the concerns of too little relevant information. For example, data may be too sparse for rules with many parameters or factors (the so-called ‘curse of high dimensionality’), and this includes the case where no observations are available at all. Insight or predictions may then be incorrect because many relevant rules may need to come together to make a final pronouncement. It is rare that a single probabilistic rule will say all that needs to be said. Perhaps the greatest current concern, related to the above, is that the information obtained will only be of general utility if the sample dataset is a sufficient representation of the entire population. This is a key consideration in any data mining activity and likely underlies many disputes in the literature and elsewhere about the validity of the results of various studies involving data mining. To generate useful information from any such study, it is essential to pay particular attention to the design of the study and to replicate and validate the results using other sample datasets from the entire population. The term data dredging is often used in reference to preliminary data mining activities on sample datasets that are too small to generate results of sufficient statistical power to likely be valid but can generate hypotheses for exploration using large sample datasets.
A further concern is that sparse events in data can be particularly important precisely because they are sparse. What matters is not the support for a rule in terms of the amount and quality of data concerning it, but (in many approaches at least) whether the event occurred with an abundance much more, or much less, than expected, say on a chance basis. Negative associations are of great importance in medicine when we want to prevent something, and we want a negative relationship between a therapy and disease. The so-called unicorn events about observations never seen at all are hard to handle. A simple pedagogic example is the absence of pregnant males in a medical database. Whilst this particular example may only be of interest to a Martian, most complex rules that are not deducible from simpler ones (that are a kind of subset to them) might be of this type. Of particular concern is that drugs A, B, and C might work 100% used alone, and so might AB, BC, and AC, but ABC might be a useless (or, perhaps, lethal) combination. Yet, traditionally, unicorn events are not even seen to justify consideration, and in computing terms they are not even ‘existentially qualified’, no variables may even be created to consider them. If we do force creation of them, then the number of things to allow for because they just might be, in principle, can be astronomical.
Data mining algorithms can yield information about:
• the presence of subgroups of samples within a sample dataset that are similar on the basis of patterns of characteristics in the variables for each sample (clustering samples into classes)
• the variables that can be used (with weightings reflecting the importance of each variable) to classify a new sample into a specified class (classification)
• mathematical or logical functions that model the data (for example, regression analysis) or
• relationships between variables that can be used to explore the behaviour of the system of variables as a whole and to reveal which variables provide either novel (unique) or redundant information (association or correlation).
Some general principles for data mining in medical applications are exemplified in the mining of 667 000 patient records in Virginia by Mullins et al. (2006). In that study, which did not include any genomic data, three main types of data mining were used (pattern discovery, predictive analysis and association analysis). A brief description of each method follows.
Pattern discovery
Pattern discovery is a data mining technique which seeks to find a pattern, not necessarily an exact match, that occurs in a dataset more than a specified number of times k. It is itself a ‘number of times’, say n(A & B & C &…). Pattern discovery is not pattern recognition, which is the use of these results. In the pure form of the method, there is no normalization to a probability or to an association measure as for the other two data mining types discussed below. Pattern discovery is excellent at picking up complex patterns with multiple factors A, B, C, etc., that tax the next two methods. The A, B, C, etc. may be nucleotides (A, T, G, C) or amino acid residues (in IUPAC one letter code), but not necessarily contiguous, or any other variables. In practice, due to the problems typically addressed and the nature of natural gene sequences, pattern discovery for nucleotides tends to focus on runs of, say, 5–20 symbols. Moreover, the patterns found can be quantified in the same terms as either of the other two methods below. However, if one takes this route alone, important relationships are missed. It is easy to see that this technique cannot pick up a pattern occurring less than k times, and k = 1 makes no sense (everything is a pattern). It must also miss alerting to patterns that statistically ought to occur but do not, i.e. the so-called unicorn events. Pattern discovery is an excellent example of an application that does have an analogue at operating system level, the use of the regular expression for partial pattern matching (http://en.wikipedia.org/wiki/Regular_expression), but it is most efficiently done by an algorithm in a domain-specific application (the domain in this case is fairly broad, however, since it has many other applications, not just in bioinformatics, proteomics, and biology generally). An efficient pattern discovery algorithm developed by IBM is available as Teireisis (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC169027/).
Predictive analysis
Predictive analysis encompasses a variety of techniques from statistics and game theory that analyse current and historical facts to predict future events (http://en.wikipedia.org/wiki/Predictive_analytics). ‘Predictive analysis’ appears to be the term most frequently used for the part of data mining that involves normalizing n(A & B & C) to conditional probabilities, e.g.
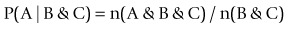 (1)
(1)In usual practice, predictive analysis tends to focus on 2–3 symbols or factors. This approach is an excellent form for inference of the type that uses ‘If B & C then A’, but takes no account of the fact that A could occur by chance anyway. In practice, predictive analysis usually rests heavily on the commonest idea of support, i.e. if the pattern is not seen a significant number of times, the conditional probability of it is not a good estimate. So again, it will not detect important negative associations and unicorn events. Recently, this approach has probably been the most popular kind of data mining for the general business domain.
Association analysis
Association analysis is often formulated in log form as Fano’s mutual information, e.g.
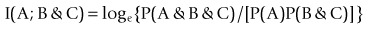 (2)
(2)(Robson, 2003, 2004, 2005, 2008; Robson and Mushlin, 2004; Robson and Vaithiligam, 2010). Clearly it does take account of the occurrence of A. This opens up the full power of information theory, of instinctive interest to the pharmaceutical industry as an information industry, and of course to other mutual information measures such as:
 (3)
(3)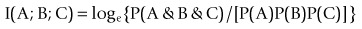 (4)
(4)The last atomic form is of interest since other measures can be calculated from it (see below). Clearly it can also be calculated from the conditional probabilities of predictive analysis. To show relationship with other fields such as evidence based medicine and epidemiology,
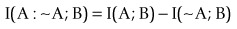 (5)
(5)where ~A is a negative of complementary state or event such that
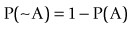 (6)
(6)is familiar as log predictive odds, while
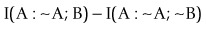 (7)
(7)The association analysis approach handles positive, zero, and negative associations including treatment sparse joint events. To that end, it may use the more general definition of information in terms of zeta functions, ζ. Unlike predictive analysis, the approach used in this way returns expected information, basically building into the final value the idea of support. In the Virginia study, using the above ‘zeta approach’, one could detect patterns of 2–7 symbols or factors, the limit being the sparseness of data for many such. As data increase, I(males; tall) = ζ(1, observed[males, tall]) − ζ(1, expected[males, tall]) will rapidly approach loge (observed[males, tall]) − loge (expected[males, tall]), but unlike log ratios, ζ(1, observed[males, pregnant]) − ζ(1, expected[males, pregnant]) works appropriately with the data for the terms that are very small or zero. To handle unicorn events still requires variables to be created in the programme, but the overall approach is more natural.
The above seem to miss out various forms of data mining such as time series analysis and clustering analysis, although ultimately these can be expressed in the above terms. What seems to require an additional comment is correlation. While biostatistics courses often use association and correlation synonymously, data miners do not. Association relates to the extent to the number of times things are observed together more, or less, than on a chance basis in a ‘presence or absence’ fashion (such as the association between a categorical SNP genotype and a categorical phenotype). This is reminiscent of the classical chi square test, but revealing the individual contributions to non-randomness within the data grid (as well as the positive or negative nature of the association). In contrast, correlation relates to trends in values of potentially continuous variables (independence between the variances), classically exemplified by use of Pearson’s correlation. Correlation is important in gene expression analysis, in proteomics and in metabolomics, since a gene transcript (mRNA) or a protein or a small molecule metabolite, in general, has a level of abundance in any sample rather than a quantized presence/absence. Despite the apparent differences, however, the implied comparison of covariance with what is expected on independent, i.e. chance, basis is essentially the same general idea as for association. Hence results can be expressed in mutual information format, based on a kind of fuzzy logic reasoning (Robson and Mushlin, 2004).
Much of the above may not seem like bioinformatics, but only because the jargon is different. That this ‘barrier’ is progressively coming down is important, as each discipline has valuable techniques less well known in the other. Where they do seem to be bioinformatics it is essentially due to the fact that they come packaged in distinct suites of applications targeted at bioinformatics users, and where they do not seem to be bioinformatics, they do not come simply packaged for bioinformatics users.
Genomics
The genome and its offspring ‘-omes’
In contrast to bioinformatics, the term genome is much older, first believed to be used in 1920 by Professor Hans Winkler at the University of Hamburg, as describing the world or system within the discipline of biology and within each cell of an organism that addresses the inherited executable information. The word genome (Gk: 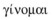 ) means I become, I am born, to come into being, and the Oxford English Dictionary gives its aetiology as being from gene and chromosome. This aetiology may not be entirely correct.
) means I become, I am born, to come into being, and the Oxford English Dictionary gives its aetiology as being from gene and chromosome. This aetiology may not be entirely correct.
In this chapter, genomes of organisms are in computer science jargon the ‘primary objects’ on which bioinformatics ‘acts’. Their daughter molecular objects, such as the corresponding transcriptomes, proteomes and metabolomes, should indeed be considered in their own right but may also be seen as subsets or derivatives of the genome concept. The remainder of this chapter is largely devoted to genomic information and its use in drug discovery and development.
While the term genome has recently spawned many offspring ‘-omes’ relating to the disciplines that address various matters downstream from inherited information in DNA, e.g. the proteome, these popular -ome words have an even earlier origin in the 20th century (e.g. biome and rhizome). Adding the plural ‘-ics’ suffix seems recent. The use of ‘omics’ as a suffix is more like an analogue of the earlier ‘-netics’ and ‘-onics’ in engineering. The current rising hierarchy of ‘-omes’ is shown in Table 7.1, and these and others are discussed by Robson and Baek (2009). There are constant additions to the ‘-omes’.
Table 7.1 Gene to function is paved with ‘-omes’
| Commonly used terms | ||
| Genome | Full complement of genetic information (i.e. DNA sequence, including coding and non-coding regions) | Static |
| Transcriptome | Population of mRNA molecules in a cell under defined conditions at a given time | Dynamic |
| Proteome | Either: the complement of proteins (including post-translational modifications) encoded by the genome | Static |
| or: the set of proteins and their post-translational modifications expressed in a cell or tissue under defined conditions at a specific time (also sometimes referred to as the translatome) | Dynamic | |
| Terms occasionally encountered (to be interpreted with caution) | ||
| Secretome | Population of secreted proteins produced by a cell | Dynamic |
| Metabolome | Small molecule content of a cell | Dynamic |
| Interactome | Grouping of interactions between proteins in a cell | Dynamic |
| Glycome | Population of carbohydrate molecules in a cell | Dynamic |
| Foldome | Population of gene products classified by tertiary structure | Dynamic |
| Phenome | Population of observable phenotypes describing variations of form and function in a given species | Dynamic |
The new ‘-omes’ are not as simple as a genome. For one thing, there is typically no convenient common file format or coding scheme for them comparable with the linear DNA and protein sequence format provided by nature. Whereas the genome contains a static compilation of the sequence of the four DNA bases, the subject matters of the other ‘-omes’ are dynamic and much more complex, including the interactions with each other and the surrounding ecosystem. It is these shifting, adapting pools of cellular components that determine health and pathology. Each of them as a discipline is currently a ‘mini’ (or, perhaps more correctly, ‘maxi’) genome project, albeit with ground rules that are much less well defined. Some people have worried that ‘-omics’ will prove to be a spiral of knowing less and less about more and more. Systems biology seeks to integrate the ‘-omics’ and simulate the detailed molecular and macroscopic processes under the constraint of as much real-world data as possible (van der Greef and McBurney, 2005; van der Greef et al., 2007).
For the purposes of this chapter, we follow the United States Food and Drug Administration (FDA) and European Medicines Evaluation Agency (EMEA) agreed definition of ‘genomics’ as captured in the definition of a ‘genomic biomarker’, which is defined as: A measurable DNA and/or RNA characteristic that is an indicator of normal biologic processes, pathogenic processes, and/or response to therapeutic or other interventions (see http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073162.pdf). A genomic biomarker can consist of one or more deoxyribonucleic acid (DNA) and/or ribonucleic acid (RNA) characteristics.
DNA characteristics include, but are not limited to:
• Single nucleotide polymorphisms (SNPs);
• Variability of short sequence repeats
• Deletions or insertions of (a) single nucleotide(s)
• Cytogenetic rearrangements, e.g., translocations, duplications, deletions, or inversions.
RNA characteristics include, but are not limited to:
The definition of a genomic biomarker does not include the measurement and characterization of proteins or small molecule metabolites and, therefore, to limit the scope of this chapter, the roles of proteomics and metabolomics in drug discovery and development will not be discussed.
It remains that the genome currently rules in the sense of being the basic instruction set from which various subsets of gene products are derived (Greenbaum et al., 2001), and hence largely responsible for the manifestation of all the ‘-omes’, albeit the details of that manifestation are contingent upon the environment of the cell and organism (see below). The coded information is a strength in terms of simplicity, and as an essentially invariant feature of a patient, while all else on the lifelong ‘health record’ may change. And whereas the downstream ‘-omes’ seen can have a huge role in interpreting the genome, inspection of features in the genome alone will often inform what is not possible, or likely to be a malfunction. Although it will ultimately be important to know all of the possible gene products that a species is capable of producing and to understand all the mechanistic details concerning the behaviour of an organism, understanding which ones are altered in disease or play a role in drug responses is most relevant to drug discovery and development. Furthermore, while detailed mechanisms may never be fully understood, physicians can certainly make use of validated relationships between genome features and the efficacy or safety outcomes of various treatments to tailor treatment strategies for individual patients, often referred to as personalized medicine.
A few genome details
A great deal of technology had to be invented to accomplish the sequencing of genomes (Cantor and Smith, 1999). Recent years have seen dramatic progress. By the middle of 2010, the sequences of the genomes of more than 2400 organisms were either complete (770 prokaryotes; 37 eukaryotes), in draft assembly (568 prokaryotes; 240 eukaryotes) or in progress (551 prokaryotes; 266 eukaryotes) (http://www.ncbi.nlm.nih.gov/sites/genome). Most sequenced genomes are for bacteria, but the eukaryote genomes sequenced include Homo sapiens (Lander et al., 2001; Venter et al., 2001) and a wide variety of animals that play a role in the discovery of disease mechanisms or in drug discovery and development, such as the fruit fly (Drosophila simulans (melanogaster)), flatworm (Caenorhabditis elegans), guinea pig (Cavia porcellus), mouse (Mus musculus), rat (Rattus norvegicus), dog (Canis lupus) and monkey (Macaca mulatta). The massive amount of information generated in the various genome projects and gene mapping studies has to be stored, disseminated and analysed electronically, relying heavily on bioinformatics software systems and the Internet (see, for example, Fig. 7.2). Here we may note that defining the nucleotide sequence is only the starting point of the genomic approach.
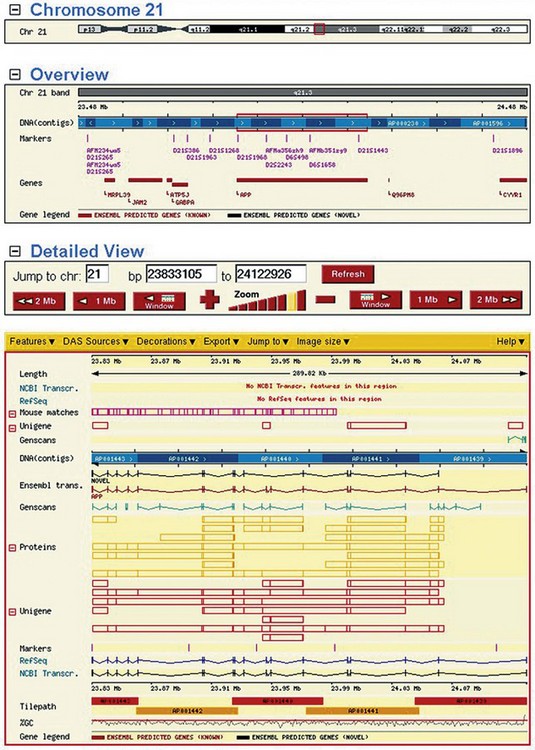
Fig. 7.2 Screenshot from the Ensembl website. Shown is a chromosomal overview and a regional view of chromosome 21 surrounding the gene APP, the Alzheimer’s disease amyloid A4 protein precursor. In the detailed view the precise exon–intron structure of the gene is shown, including sequence homology matches to a variety of databases or de novo gene predictions. The viewer is highly customizable and users can easily navigate along the genomic axis.
Reproduced with kind permission of European Bioinformatics Institute and Wellcome Trust Sanger Institute.
While the percentage of human DNA spanned by genes is 25.5–37.8%, only about 2% of the human genome is nucleotide sequence that actually codes for protein, or indeed for structural or catalytic RNA. The other 98%, initially considered to be ‘junk’ DNA, may indeed include evolution’s junk, i.e. fossil relics of evolution or, more kindly put, non-functional genes that might serve as the basis of evolution in the longer term. However, information about this ‘junk’ DNA is not uninteresting or useless. Even truly non-functional DNA may have significant medical and forensic value. Significant human individual differences represented by genomic biomarkers are an exploding opportunity for improving healthcare and transforming the activities of the pharmaceutical industry (Svinte et al., 2007), but these genomic biomarkers frequently are found not to lie in protein coding regions. They have often travelled with the genes about which they carry information, in the course of the migrations of human prehistory and history. The so-called ‘junk’, including the introns discussed below and control segments, is involved in chromatin structure, recombination templating, and is now seen as representing hotspots for sponsoring heterogeneity (see comment on epigenetics below).
There was, for a time, a surprising reduction in the promised amount of what constitutes really interesting genomic information in humans. Although identifying genes within the genome sequence is not straightforward, earlier estimates of about 100 000 genes in the human genome based on the variety of proteins produced came down, rather startlingly, to about 35 000 when the genome was determined, and later (International Human Genome Sequencing Consortium, 2004) to about 25 000. A clue as to how this can be so was already well known in the late 1970s and 1980s – that genes in complex organisms are suspiciously much larger than would be expected from the number of amino acid residues in the proteins for which they code. It was found that only the regions of a gene called exons actually code for proteins, and the intervening introns are removed by RNA splicing at the messenger RNA (mRNA) level. The average human gene has some 30 000 nucleotides and 7 exons. The largest known human gene, for titin, has 363 introns.
Genome variability and individual differences
Personalized medicine in the clinical setting is an emerging field. Human individuality underpins susceptibility to disease and response to treatment. Fundamental to human individuality is the content of each individual’s genome. All humans have 99.5–99.9% of nucleotide sequence identity in their genomes, depending upon whether one takes into account just SNPs or all forms of genetic variation (see http://www.genome.gov/19016904 and also Freeman et al., 2006). However, the 0.1% difference in SNPs represents 6 million bases in a genome of 6 billion bases – plenty of scope for individuality. Even monozygotic twins can develop individuality at the genome (epigenome) level in individual body cells or tissues through ‘life experience’. Understanding human individuality at the genomic level has the potential to enable great advances in drug discovery and development. Pharmacogenomics addresses the matter of whether the same drug will cure, do little, or harm according to the unique genomics of the patient.
The epigenome
In addition to the linear sequence code of DNA that contains the information necessary to generate RNA and, ultimately, the amino acid sequences of proteins, there is an additional level of information in the DNA structure in the form of the complex nucleoprotein entity chromatin. Genetic information is encoded not only by the linear sequence of DNA nucleotides but by modifications of chromatin structure which influence gene expression. Epigenetics as a field of study is defined as ‘… the study of changes in gene function … that do not entail a change in DNA sequence’ (Wu and Morris, 2001). The word was coined by Conrad Waddington in 1939 (Waddington, 1939) in recognition of relationship between genetics and developmental biology and of the sum of all mechanisms necessary for the unfolding of the programme for development of an organism.
The modifications of chromatin structure that can influence gene expression comprise histone variants, post-translational modifications of amino acids on the amino-terminal tail of histones, and covalent modifications of DNA bases – most notably methylation of cytosine bases at CpG sequences. CpG-rich regions are not evenly distributed in the genome, but are concentrated in the promoter regions and first exons of certain genes (Larsen et al., 1992). If DNA is methylated, the methyl groups protrude from the cytosine nucleotides into the major groove of the DNA and inhibit the binding of transcription factors that promote gene expression (Hark et al., 2000). Changes in DNA methylation can explain the fact that genes switch on or off during development and the pattern of methylation is heritable (first proposed by Holliday and Pugh, 1975). It has been shown that DNA methylation can be influenced by external stressors, environmental toxins, and aging (Dolinoy et al., 2007; Hanson and Gluckman, 2008), providing a key link between the environment and life experience and the regulation of gene expression in specific cell types.
A key finding in epigenetics was reported by Fraga et al. in 2005. In a large cohort of monozygotic twins, DNA methylation was found to increase over time within different tissues and cell types. Importantly, monozygotic twins were found to be indistinguishable in terms of DNA methylation early in life but were found to exhibit substantial differences in DNA methylation with advancing age. The authors of the landmark paper stated that ‘distinct profiles of DNA methylation and histone acetylation patterns that among different tissues arise during the lifetime of monozygotic twins may contribute to the explanation of some of their phenotypic discordances and underlie their differential frequency/onset of common disease.’
The transcriptome
The fact that the cell must cut out the introns from mRNA and splice together the ends of the remaining exons is the ‘loophole’ that allows somatic diversity of proteins, because the RNA coding for exons can be spliced back together in different ways. Alternative splicing might therefore reasonably be called recombinant splicing, though that term offers some confusion with the term ‘recombinant’ as used in classical genetics. That some 100 000 genes were expected on the basis of protein diversity and about 25 000 are found does not mean that the diversity so generated is simply a matter of about four proteins made from each gene on average. Recent work has shown that alternative splicing can be associated with thousands of important signals.
The above is one aspect of the transcriptome, i.e. the world of RNA produced from DNA. Whereas one may defer discussion of other ‘omes’ (e.g. of proteins, except as the ‘ultimate’ purpose of many genes), the above exemplifies that one makes little further progress at this stage by ignoring the transcriptome. A primary technology for measuring differences in mRNA levels between tissue samples is the microarray, illustrated in Figs. 7.3 & 7.4.
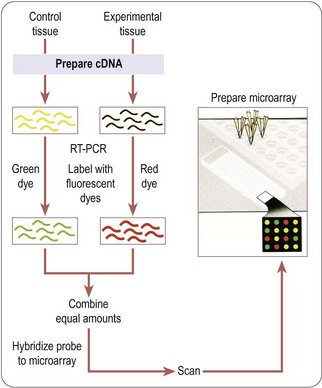

Fig. 7.4 Cluster analysis of gene expression experiment. Expression levels of 54 genes (listed on right) in eight control and eight multiple sclerosis brain samples (listed above). Relative expression levels are indicated in shades of red for high values and green for low values. A computer algorithm was used to calculate similarity between the expression pattern of each gene for all subjects, and the expression pattern for each subject for all genes, and to display the results as a dendrogram. The top dendrogram shows that MS and control samples are clearly distinguishable, the intragroup distance being much greater than the intersample difference within each group. The left-hand dendrogram shows the grouping of genes into clusters that behave similarly. A group of genes with highly similar expression patterns is outlined in yellow.
Adapted, with permission, from Baranzini et al., 2000.
The RNA world now intermediates between the genome and the proteome, including carrying transcripts of DNA in order to determine the amino acid sequences of proteins, but is believed to be the more ancient genomic world before DNA developed as a sophisticated central archive. Not least, since unlike a protein any RNA is a direct copy of regions of DNA (except for U replacing T), much of an account of the transcriptome looks like and involves an account of the genome, and vice versa.
To be certain, the transcriptome does go beyond that. The RNA produced by DNA can perform many complex roles, of which mRNA and its alternative splicing provide just one example. Many of the sites for producing these RNAs lie not in introns but outside the span of a gene, i.e. in the intergenic regions. Like elucidation of the exon/intron structure within genes, intergenic regions are similarly yielding to more detailed analysis. The longest known intergenic length is about 3 million nucleotides. While 3% of DNA is short repeats and 5% is duplicated large pieces, 45% of intergenic DNA comprises four classes of ‘parasitic elements’ arising from reverse transcription. In fact it may be that this DNA is ‘a boiling foam’ of RNA transcription and reverse transcription. It now appears that both repeats and boiling foam alike appear to harbour many control features.
At first, the fact that organisms such as most bacteria have 75–85% of coding sequences, and that even the puffer fish has little intergenic DNA and few repeats, may bring into question the idea that the intergenic DNA and its transcriptome can be important. However, there is evidently a significant difference in sophistication between humans and such organisms. This thinking has now altered somewhat the original use of the term genome. The human genome now refers to all nuclear DNA (and ideally mitochondrial DNA as well). That is, the word has been increasingly interpreted as ‘the full complement of genetic information’ coding for proteins or not. The implication is that ‘genome’ is all DNA, including that which does not appear to carry biological information, on the basis of the growing suspicion that it does. As it happens, the term ‘coding’ even traditionally sometimes included genes that code for tRNA and rRNA to provide the RNA-based protein-synthesizing machinery, since these are long known. The key difference may therefore mainly lie in departure from the older idea that RNA serves universal housekeeping features of the cell as ‘a given’ that do not reflect the variety of cell structure and function, and differentiation. That, in contrast, much RNA serves contingency functions relating to the embryonic development of organisms is discussed below.
There are still many unidentified functionalities of RNA coding regions in the ‘junk DNA’, but one that is very topical and beginning to be understood is microRNA (or miRNA). miRNAs are short lengths of RNA, just some 20–25 nucleotides, which may base pair with RNA and in some cases DNA – possibly to stop it binding and competing with its own production, but certainly to enable control process that hold the miRNAs in check.
The maturation of miRNAs is quite complicated. Two complementary regions in the immature and much larger miRNA associate to form a hairpin, and a nuclear enzyme Drosha cleaves at the base of the hairpin. The enzyme Dicer then extracts the mature miRNA from its larger parent. A typical known function of a mature miRNA is as an antisense inhibitor, i.e. binding messenger mRNA, and then initiating its degradation, so that no protein molecule can be made from the mRNA. Note that in the above, one could think largely of recognition regions in the proto-miRNA transcript as simply mapping to patterns of nucleotides in DNA. But it would be impossible to meaningfully discuss, or at least not be insightful, without consideration of the biochemical processes downstream of the first transcription step.
In defence of the genome
The complexities contained in the discussion above challenge the authority of the genome as ‘king’. At present, the genome rules much because of our ignorance. DNA is potentially no more, and no less, important than the magnetic tapes, floppy disks, CDs, and Internet downloads that we feed into our computers, indispensible and primary, but cold and lifeless without all the rest. While genomic data now represent a huge amount of information that is relevant to the pharmaceutical industry, it cannot be denied that complete pharmaceutical utility lies in understanding how the information in DNA, including interaction between many genes and gene products, translates into function at the organism level. Not least, drugs influence that translation process. The rise of systems biology reflects this awareness of the organism as a dynamic system dwelling in three dimensions of space and one of time.
The problem is that the child ‘-omes’ are still immature and rather weak. We are assembling a very rich lexicon from the genomes, but are left with the questions of when, how, and why, the patterns of life, molecular and macroscopic, unfold. And one may say fold too, since the folding of proteins, the key step at which linear information in DNA becomes three-dimensional biology, remains largely unsolved. Perhaps for all such reasons, the earlier pharmaceutical industry hopes for genomics and bioinformatics (Li and Robson, 2000) have not yet been realized (Drews, 2003).
But low-hanging fruits have been picked to keep the pharmaceutical industry buoyant. Biotechnology companies drink of biological knowledge much closer to the genome than pharmaceutical companies usually do, and they largely enabled the methodologies that made the Human Genome Project a reality. Thus, they currently position themselves as leaders in managing and tapping this vast new information resource. Companies specializing in genomic technology, bioinformatics, and related fields were the fastest expanding sector of the biotechnology industry during the 1990s, aspiring to lead the pharmaceutical industry into an era of rapid identification and exploitation of novel targets (Chandra and Caldwell, 2003).
Not the least because the biotechnology industry has provided proof of concept, there is still consensus that the Human Genome Project will be worth to the pharmaceutical industry the billions of dollars and millions of man-hours that nations have invested. But it still remains unclear as to exactly how all the available and future information can be used cost-effectively to optimize drug discovery and development.
So just how prominent should genome-based strategies be, right now, in the research portfolio of pharmaceutical companies, and what should those strategies be? The principle of using genomic information as the starting point for identifying novel drug targets was described in Chapter 6. Following is an overview of the ways in which genomic information can, or might, impact almost every aspect of the drug discovery and development process.
Genomic information in drug discovery and development
Drug discovery and development is mankind’s most complicated engineering process, integrating a wide variety of scientific and business activities, some of which are presented in Table 7.2.
Table 7.2 Steps in the drug discovery and development process
With regard to the relevance of genomic information in the drug discovery and development process, a key consideration is how best (cost-effectively) to use the information that underpins human individuality to improve the process such that:
• Diseases are characterized, or defined, by molecular means rather than by symptoms – a key advance since pharmaceutical scientists make drug candidate molecules which interact with other molecules not with symptoms
• Diseases are detected by molecular means earlier than currently possible, at a time when the underlying pathologies are reversible
• Subgroups of patients with similar molecular characteristics can be identified within the larger group of patients with similar symptoms, enabling clinical trials to be focused on those subgroups of patients most likely to benefit from a particular pharmacological approach
• The best drug targets are selected for specific diseases or subgroups of patients
• Individual human variation in drug exposure or drug response in terms of efficacy or side effects can be anticipated or explained in the drug discovery and development process, with the resulting reduction in the frequency of clinical trials that fail because of unanticipated ‘responder versus non-responder’ treatment outcomes and overall increase in productivity of drug discovery and development.
Following are comments on the impact that genomics and genomic information can, or will, have on some of the steps in Table 7.2.
Understanding human disease intrinsic mechanisms
Many common human diseases are thought to result from the interaction of genetic risk profiles with life style and environmental risk factors. Over the past few decades, large scale studies of healthy humans and human suffering from diseases have associated specific gene variants with diseases. In many cases, these studies have revealed only relatively weak associations between many individual gene variants and diseases (Robinson, 2010). To date, this area of investigation has not returned the immediate benefit hoped for, and perhaps ‘hyped for’, by the genomic community. Nevertheless, insights have been gained into intrinsic biochemical mechanisms underlying certain diseases.
Insight into the biochemical mechanisms of common diseases has definitely been gained from the study of inherited monogenic diseases in special families (Peltonen et al., 2006). In these cases, the association between the gene variant and disease phenotype is very strong, although the application of the information to common diseases must be undertaken with caution.
Understanding the biology of infectious agents
The availability of complete genome sequences for many infectious agents has provided a great deal of insight into their biology (Pucci, 2007). Furthermore, such genetic information has also revealed the mutations in the genetic material of these organisms that are responsible for drug resistance (Kidgell and Winzeler, 2006) – of great interest to those involved in the discovery of new drugs to treat major infections. There is a strong interplay with the human genome, especially the HLA genes, which determine whether the T cell system will recognize certain features of proteins expressed by pathogens.
Identifying potential drug targets
Studies of the association of gene variants with the phenotypes of common diseases have revealed, in some cases, hundreds of gene variants that can predispose humans to certain diseases. When the proteins coded for by the genes associated with a specific disease are mapped onto known biochemical pathways (see, for example, www.ingenuity.com or www.genego.com), it is sometimes possible to gain an overall picture of likely disease mechanisms and to identify and prioritize potential drug targets for the disease (see Chapter 6).
Validating drug targets
Transgenic animals, in which the gene coding for the drug target protein can be knocked out or its expression down-regulated, can provide validation for the pharmacological effect of an inhibitor drug prior to availability of any candidate drug molecules (Sacca et al., 2010).
Assay development for selected drug targets
An analysis of variation in the gene coding for the drug target across a diverse human population can identify potential drug response variability in patients (Sadee et al., 2001; Niu et al., 2010; Zhang et al., 2010). For example, at SNP locations, the relative frequencies of occurrence of each of the genotypes (for example, AA, AG or GG) can provide information on likely differences in the abundance or structure of the drug target across the human population. Different protein isoforms with even slightly different structures could be associated with altered drug binding or activation/inhibition of downstream biochemical mechanisms. To avoid the discovery of drug candidates which interact effectively with only one isoform of the protein drug target or, alternatively, to focus on discovering such drug candidates, multiple assays based on different protein isoforms expressed from variants of each drug target gene could be established. These parallel assays could be used for screening chemical libraries to select hits which were selective for one isoform of the drug target protein, for enabling personalized medicine, or were able to interact effectively with more than one isoform, for enabling population-focused medicine.
Phase 0 clinical studies – understanding from compounds in low concentration
This relatively recent term relates to trials which are safe because they need such low doses of a new chemical entity being tested. Usually this applies to testing the very low concentrations of radioactively labelled compounds for molecular imaging; concentrations still are sufficient to see how they travel, where they go, where they accumulate, and how they are metabolized. A compound can be labelled at several points to track its components and derivatives formed in metabolism. It is inevitable that many differences in transport and metabolism will occur in individuals, so genomics will play a strong supporting role. It is likely that these kinds of trial will increasingly frequently be applied not just to develop a research agent, but to gather data for the compound to be used in the following trials.
Phase I clinical studies – pharmacokinetics and safety
Preclinical drug metabolism studies conducted on drug candidates in line with the guidance provided by the US FDA (see http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf) can provide a wealth of information on the likely routes of metabolism of the drug, the enzymes and transporters involved in the metabolism, and the drug metabolites generated. Enzymes of particular interest are the cytochromes P450 (CYPs) which introduce a polar functional group into the parent molecule in the first phase of biotransformation of a drug.
An analysis of the genotypes of Phase I human subjects with respect to drug metabolizing enzymes can provide explanations for unusual pharmacokinetics and even adverse drug effects in certain subjects (Crettol et al., 2010). In particular, subjects with a CYP2D6 gene variant associated with lower than normal enzyme expression or activity (referred to as ‘poor metabolizers’) might have an exaggerated response to a standard dose of the drug. In those subjects, drug exposure might reach toxic levels and trigger adverse effects. On the other hand, subjects with a different CYP2D6 variant, associated with higher than normal enzyme expression or activity (referred to as ‘rapid metabolizers’ or ‘ultra rapid metabolizers’) might not exhibit any drug effects whatsoever. Clearly, information concerning each individual’s genotype, with respect to drug metabolizing enzymes, in concert with information about the routes of metabolism of a drug candidate, can be used to interpret the response to drug dosing in Phase I studies but, more comprehensively, can potentially be used to optimize the probability of success for the development of the drug and even the strategies for using an approved drug in medical practice.
Phase II and III clinical studies – efficacy and safety
In clinical studies to determine the efficacy and safety of drug candidates in patients, it is important to recognize and take steps to deal with the likelihood that the different genotypes across the clinical trial population could influence overall trial outcome. A not uncommon feature of clinical trials is that, with respect to treatment outcome (efficacy or safety), patients fall into two broad groups – ‘responders’ and ‘non-responders’. The balance between these two groups of patients can have a profound effect on the overall results of a trial. While variants in the genes encoding drug metabolizing enzymes might explain some such results, as mentioned above, other genetic variations across the population could also play a role in diverse responses to drug treatment in a clinical trial population. For example, drug treatment response could be altered in patients with variants in individual genes encoding the drug receptors or with variants in individual genes encoding associated signalling proteins or with patterns of variants in the genes encoding multiple proteins in relevant biochemical signalling pathways.
At present there are relatively few clear examples of the influence of genomic variation on clinical trial outcome (see http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm). Nevertheless, this area of investigation has a substantial place in drug discovery and development strategies in the future. All clinical studies should now incorporate DNA collection from every subject and patient. The costs of whole genome analysis are constantly diminishing and bioinformatic methods for data mining to discover associations between genomic variables and treatment–response variables are continually improving.
A research effort undertaken in Phase I and Phase II clinical studies to discover information about genetic variants which influence the response to a drug candidate can contribute substantially to the planning for pivotal Phase III trials and the interpretation of the results of those trials, enhancing the overall prospects for successful completion of the development programme for a drug candidate. More information on clinical trials can be found later in the book.
Genomic information and drug regulatory authorities
Successful drug discovery and development culminates in the approval for marketing of a product containing the drug. Approval for marketing in a specific country or region is based on a review by the appropriate drug regulatory authority of a substantial amount of information submitted to the regulatory authority by the sponsor (e.g. pharmaceutical company) – on manufacturing, efficacy and safety of the product.
How has the growing availability of vast amounts of genomic information affected the drug approval process? What changes are taking place at the regulatory authorities? What new types of information do pharmaceutical and biotechnology companies need to provide in the regulatory submissions associated with the drug discovery and development process?
Over the past 10 years, in response to the changing landscape of genomics information and its potential to improve the drug discovery and development process and the cost-effectiveness of new medicines, there has been a steep learning curve around the world within regulatory authorities and pharmaceutical/biotechnology companies. Fortunately, representatives of the regulatory authorities and the companies have joined together to explore and define the role of genomic information in drug discovery and development, and marketing approval.
Critical Path Initiative and the EMEA’s equivalent programme
In the early 2000s, the US Food and Drug Administration (FDA) and the European Medicines Evaluation Agency (EMEA) separately initiated efforts to improve the drug discovery and development process. In part, these efforts were stimulated by a trend in the preceding years of fewer and fewer drug approvals each year. Additionally, there was an understanding at these regulatory agencies that new developments in scientific and medical research, such as the explosion of genomic information, were not being incorporated into the drug evaluation processes. The FDA kicked-off its initiative in March 2004 with an insightful white paper entitled ‘Innovation/Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products’ (http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/CriticalPathInitiative/CriticalPathOpportunitiesReports/ucm113411.pdf) and the initiative became known as the Critical Path Initiative. Also in 2004, the EMEA set up the EMA/CHMP Think-Tank Group on Innovative Drug Development (http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/general/general_content_000339.jsp&murl=menus/special_topics/special_topics.jsp&mid=WC0b01ac05800baed8&jsenabled=true).
Voluntary exploratory data submissions and guidance documents
In November 2003, the FDA published a draft guidance document on Pharmacogenomic Data Submissions (final guidance published in March 2005, http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm126957.pdf) and introduced a programme to encourage pharmaceutical/biotechnology companies to make Voluntary Genomic Data Submissions (VGDS) concerning aspects of their drug discovery or development projects, under ‘safe harbour’ conditions without immediate regulatory impact. The goal of the VGDS programme was to provide ‘real-world’ examples around which to create dialogue between the FDA and pharmaceutical/biotechnology companies about the potential roles of genomic information in the drug development process and to educate both regulators and drug developers. Within a short time of its initiation, the programme was expanded to include other molecular data types and is now known as the Voluntary Exploratory Data Submissions (VXDS) programme and also involves collaboration with the EMEA. A recent article (Goodsaid et al., 2010) provides an overview of the first 5 years of operation of the programme and presents a number of selected case studies.
Overall the VXDS programme has been of mutual benefit to regulators, pharmaceutical companies, technology providers and academic researchers. As envisioned by the FDA’s Critical Path Initiative, the VXDS programme will continue to shape the ways that pharmaceutical companies incorporate, and regulators review, genomic information in the drug discovery, development and approval process.
The FDA and EMEA guidance documents and meeting reports are presented in Table 7.3.
Table 7.3 EMEA and FDA Genomics guidance documents, concept papers and policies
Conclusion
The biopharmaceutical industry depends upon vast amounts of information from diverse sources to support the discovery, development and marketing approval of its products. In recent years, the sustainability of the industry’s business model has been questioned because the frequency of successful drug discovery and development projects, in terms of approved products, is considered too low in light of the associated costs – particularly in a social context where healthcare costs are spiralling upwards and ‘out of control’.
The lack of successful outcome for the overwhelming majority of drug discovery and development projects results from the fact that we simply do not know enough about biology in general, and human biology in particular, to avoid unanticipated ‘roadblocks’ or ‘minefields’ in the path to drug approval. In short, we do not have enough bioinformation.
Most drugs interact with protein targets, so it would be natural for pharmaceutical scientists to want comprehensive information about the nature and behaviour of proteins in health and disease. Unfortunately, because of the complexity of protein species, availability proteomic technologies have not yet been able to reveal such comprehensive information about proteins – although substantial advances have been made over the past few decades.
However, as a result of tremendous advances in genomic and bioinformatic technologies during the second half of the 20th century and over the past decade, comprehensive information about the genomes of many organisms is becoming available to the biomedical research community, including pharmaceutical scientists. Genomic information is the primary type of bioinformation and the nature and behaviour of the genome of an organism underpins all of its activities. It is also ‘relatively’ simple in structure compared to proteomic or metabolomic information.
Genomic information, by itself, has the potential to transform the drug discovery and development process, to enhance the probability of success of any project and to reduce overall costs. Such a transformation will be brought about through new drug discovery and development strategies focused on understanding molecular subtypes underpinning disease symptoms, discovering drug targets relevant to particular disease molecular subtypes and undertaking clinical studies informed by the knowledge of how human genetic individuality can influence treatment outcome. Onward!
Baranzini SL, Elfstrom C, Chang SY, et al. Transcriptional analysis of multiple sclerosis brain lesions reveals a complex pattern of cytokine expression. Journal of Immunology. 2000;165:6576–6582.
Cantor CR, Smith CL. Genomics. The science and technology behind the Human Genome Project. New York: John Wiley and Sons; 1999.
Chandra SK, Caldwell JS. Fulfilling the promise: drug discovery in the post-genomic era. Drug Discovery Today. 2003;8:168–174.
Cockburn IM. Is the pharmaceutical industry in a productivity crisis? Innovation Policy and the Economy. 2007;7:1–32.
Crettol S, Petrovic N, Murray M. Pharmacogenetics of Phase I and Phase II drug metabolism. Current Pharmaceutical Design. 2010;16:204–219.
Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proceedings of the National Academy of Sciences of the USA. 2007;104:13056–13061.
Drews J. Strategic trends in the drug industry. Drug Discovery Today. 2003;8:411–420.
Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the USA. 2005;102:10604–10609.
Freeman JL, Perry GH, Feuk L, et al. Copy number variation: new insights in genome diversity. Genome Research. 2006;16:949–961.
Garnier JP. Rebuilding the R&D engine in big pharma. Harvard Business Review. 2008;86:68–70. 72–6, 128
Goodsaid FM, Amur S, Aubrecht J, et al. Voluntary exploratory data submissions to the US FDA and the EMA: experience and impact. Nature Reviews Drug Discovery. 2010;9:435–445.
Greenbaum D, Luscombe NM, Jansen R, et al. Interrelating different types of genomic data, from proteome to secretome: ‘oming in on function. Genome Research. 2001;11:1463–1468.
Hanson MA, Gluckman PD. Developmental origins of health and disease: new insights. Basic and Clinical Pharmacology and Toxicology. 2008;102:90–93.
Hark AT, Schoenherr CJ, Katz DJ, et al. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489.
Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232.
International Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:915–916.
Kidgell C, Winzeler EA. Using the genome to dissect the molecular basis of drug resistance. Future Microbiology. 2006;1:185–199.
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
Larsen F, Gundersen G, Lopez R, et al. CpG islands as gene markers in the human genome. Genomics. 1992;13:1095–1107.
Li J, Robson B. Bioinformatics and computational chemistry in molecular design. Recent advances and their application. In: Peptide and protein drug analysis. NY: Marcel Dekker; 2000:285–307.
Mullins IM, Siadaty MS, Lyman J, et al. Data mining and clinical data repositories: insights from a 667,000 patient data set. Computers in Biology and Medicine. 2006;36:1351–1377.
Munos BH. Lessons from 60 years of pharmaceutical innovation. Nature Reviews Drug Discovery. 2009;8:959–968.
Neumann EK, Quan D. Biodash: a semantic web dashboard for drug development. Pacific Symposium on Biocomputing. 2006;11:176–187.
Niu Y, Gong Y, Langaee TY, et al. Genetic variation in the β2 subunit of the voltage-gated calcium channel and pharmacogenetic association with adverse cardiovascular outcomes in the International VErapamil SR-Trandolapril STudy GENEtic Substudy (INVEST-GENES). Circulation. Cardiovascular Genetics. 2010;3:548–555.
Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature Reviews Drug Discovery. 2010;9:203–214.
Peltonen L, Perola M, Naukkarinen J, et al. Lessons from studying monogenic disease for common disease. Human Molecular Genetics. 2006;15:R67–R74.
Pucci MJ. Novel genetic techniques and approaches in the microbial genomics era: identification and/or validation of targets for the discovery of new antibacterial agents. Drugs R&D. 2007;8:201–212.
Robinson R. Common disease, multiple rare (and distant) variants. PLoS Biology. 8(1), 2010. e1000293
Robson B. Clinical and pharmacogenomic data mining. 1. The generalized theory of expected information and application to the development of tools. Journal of Proteome Research. 2003;2:283–301.
Robson B. The dragon on the gold: Myths and realities for data mining in biotechnology using digital and molecular libraries. Journal of Proteome Research. 2004;3:1113–1119.
Robson B. Clinical and pharmacogenomics data mining. 3 Zeta theory as a general tactic for clinical bioinformatics. Journal of Proteome Research. 2005;4:445–455.
Robson B. Clinical and phamacogenomic data mining. 4. The FANO program and command set as an example of tools for biomedical discovery and evidence based medicine. Journal of Proteome Research. 2008;7:3922–3947.
Robson B, Baek OK. The engines of Hippocrates: from medicine’s early dawn to medical and pharmaceutical informatics. New York: John Wiley & Sons; 2009.
Robson B, Mushlin R. Clinical and pharmacogenomic data mining. 2. A simple method for the combination of information from associations and multivariances to facilitate analysis, decision and design in clinical research and practice. Journal of Proteome Research. 2004;3:697–711.
Robson B, Vaithiligam A. Drug gold and data dragons: Myths and realities of data mining in the pharmaceutical industry. In: Balakin KV, ed. Pharmaceutical Data Mining. John Wiley & Sons; 2010:25–85.
Ruttenberg A, Rees JA, Samwald M, et al. Life sciences on the semantic web: the neurocommons and beyond. Briefings in Bioinformatics. 2009;10:193–204.
Sacca R, Engle SJ, Qin W, et al. Genetically engineered mouse models in drug discovery research. Methods in Molecular Biology. 2010;602:37–54.
Sadee W, Hoeg E, Lucas J, et al. Genetic variations in human g protein-coupled receptors: implications for drug therapy. AAPS PharmSci. 2001;3:E22.
Stephens S, Morales A, Quinlan M. Applying semantic web technologies to drug safety determination. IEEE Intelligent Systems. 2006;21:82–86.
Svinte M, Robson B, Hehenberger M. Biomarkers in drug development and patient care. Burrill 2007 Person. Medical Report. 2007;6:3114–3126.
van der Greef J, McBurney RN. Rescuing drug discovery: in vivo systems pathology and systems pharmacology. Nature Reviews Drug Discovery. 2005;4:961–967.
van der Greef J, Martin S, Juhasz P, et al. The art and practice of systems biology in medicine: mapping patterns of relationships. Journal of Proteome Research. 2007;4:1540–1559.
Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351.
Waddington CH. Introduction to modern genetics. London: Allen and Unwin; 1939.
Wu CT, Morris JR. Genes, genetics, and epigenetics: a correspondence. Science. 2001;293:1103–1105.
Zhang JP, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. American Journal of Psychiatry. 2010;167:763–772.