Chapter 6 Choosing the target
Introduction: the scope for new drug targets
The word ‘target’ in the context of drug discovery has several common meanings, including the market niche that the drug is intended to occupy, therapeutic indications for the drug, the biological mechanism that it will modify, the pharmacokinetic properties that it will possess, and – the definition addressed in this chapter – the molecular recognition site to which the drug will bind. For the great majority of existing drugs the target is a protein molecule, most commonly a receptor, an enzyme, a transport molecule or an ion channel, although other proteins such as tubulin and immunophilins are also represented. Some drugs, such as alkylating agents, bind to DNA, and others, such as bisphosphonates, to inorganic bone matrix constituents, but these are exceptions. The search for new drug targets is directed mainly at finding new proteins although approaches aimed at gene-silencing by antisense or siRNA have received much recent attention.
Since the 1950s, when the principle of drug discovery based on identified (then pharmacological or biochemical) targets became established, the pharmaceutical industry has recognized the importance of identifying new targets as the key to successful innovation. As the industry has become more confident in its ability to invent or discover candidate molecules once the target has been defined – a confidence, some would say, based more on hubris than history – the selection of novel targets, preferably exclusive and patentable ones, has assumed increasing importance in the quest for competitive advantage.
In this chapter we look at drug targets in more detail, and discuss old and new strategies for seeking out and validating them.
How many drug targets are there?
Even when we restrict our definition to defined protein targets, counting them is not simple. An obvious starting point is to estimate the number of targets addressed by existing therapeutic drugs, but even this is difficult. For many drugs, we are ignorant of the precise molecular target. For example, several antiepileptic drugs apparently work by blocking voltage-gated sodium channels, but there are many molecular subtypes of these, and we do not know which are relevant to the therapeutic effect. Similarly, antipsychotic drugs block receptors for several amine mediators (dopamine, serotonin, norepinephrine, acetylcholine), for each of which there are several molecular subtypes expressed in the brain. Again, we cannot pinpoint the relevant one or ones that represent the critical targets.
A compendium of therapeutic drugs licensed in the UK and/or US (Dollery, 1999) lists a total of 857 compounds (Figure 6.1), of which 626 are directed at human targets. Of the remainder, 142 are drugs used to treat infectious diseases, and are directed mainly at targets expressed by the infecting organism, and 89 are miscellaneous agents such as vitamins, oxygen, inorganic salts, plasma substitutes, etc., which are not target-directed in the conventional sense.
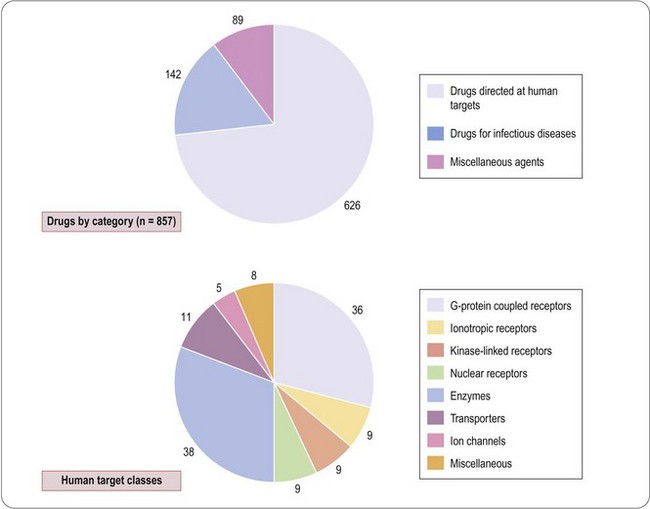
Drews and Ryser (1997) estimated that these drugs addressed approximately 500 distinct human targets, but this figure includes all of the molecular subtypes of the generic target (e.g. 14 serotonin receptor subtypes and seven opioid receptor subtypes, most of which are not known to be therapeutically relevant, or to be specifically targeted by existing drugs), as well as other putative targets, such as calcineurin, whose therapeutic relevance is unclear. Drews and Ryser estimated that the number of potential targets might be as high as 5000–10 000. Adopting a similar but more restrictive approach, Hopkins and Groom (2002) found that 120 drug targets accounted for the activities of compounds used therapeutically, and estimated that 600–1500 ‘druggable’ targets exist in the human genome. From an analysis of all prescribed drugs produced by the 10 largest pharmaceutical companies, Zambrowicz and Sands (2003) identified fewer than 100 targets; when this analysis was restricted to the 100 best-selling drugs on the market, the number of targets was only 43, reflecting the fact that more than half of the successful targets (i.e. those leading to therapeutically effective and safe drugs) failed to achieve a significant commercial impact. More recently, Imming et al. (2006) identified 218 targets of registered drugs, including those expressed by infectious agents, whereas Overington et al. (2006) found 324 (the difference depending on the exact definition of what constitutes a single identified target). How many targets remain is highly uncertain and Zambrowicz and Sands (2003) suggested that during the 1990s only two or three new targets were exploited by the 30 or so new drugs registered each year, most of which addressed targets that were already well known. The small target base of existing drugs, and the low rate of emergence of new targets, suggests that the number yet to be discovered may be considerably smaller than some optimistic forecasters have predicted. They estimated that 100–150 high-quality targets in the human genome might remain to be discovered. However, a recent analysis of 216 therapeutic drugs registered during the decade 2000–2009 (Rang unpublished) showed that 62 (29%) were first in class compounds directed at novel human targets. This is clearly an increase compared with the previous decade and may give grounds for optimism that many more new targets remain to be found. In the earlier analysis by Drews and Ryser (1997), about one-quarter of the targets identified were associated with drugs used to treat infectious diseases, and belonged to the parasite, rather than the human, genome. Their number is even harder to estimate. Most anti-infective drugs have come from natural products, reflecting the fact that organisms in the natural world have faced strong evolutionary pressure, operating over millions of years, to develop protection against parasites. It is likely, therefore, that the ‘druggable genome’ of parasitic microorganisms has already been heavily trawled – more so than that of humans, in whom many of the diseases of current concern are too recent for evolutionary counter-measures to have developed.
More recent estimates of the number of possible drug targets (Overington et al., 2006; Imming et al., 2006) are in line with the figure of 600–1500 suggested by Hopkins and Groom (2002). The increased use of gene deletion mutant mice and si RNA for gene knock down coupled with a bioinformatic approach to protein characterization is starting to increase the reliability of target identification (Imming et al., 2006; Wishart et al., 2008; Bakheet and Doig, 2009). New approaches, in which networks of genes are associated with particular disease states, are likely to throw up additional targets that may not have been discovered otherwise (e.g. see Emilsson et al., 2008).
The nature of existing drug targets
The human targets of the 626 drugs, where they are known, are summarized in Figure 6.1. Of the 125 known targets, the largest groups are enzymes and G-protein-coupled receptors, each accounting for about 30%, the remainder being transporters, other receptor classes, and ion channels. This analysis gives only a crude idea of present-day therapeutics, and underestimates the number of molecular targets currently addressed, since it fails to take into account the many subtypes of these targets that have been identified by molecular cloning. In most cases we do not know which particular subtypes are responsible for the therapeutic effect, and the number of distinct targets will certainly increase as these details become known. Also there are many drugs whose targets we do not yet know. There are a growing number of examples where drug targets consist, not of a single protein, but of an oligomeric assembly of different proteins. This is well established for ion channels, most of which are hetero-oligomers, and recent studies show that G-protein-coupled receptors (GPCRs) may form functional dimers (Bouvier, 2001) or may associate with accessory proteins known as RAMPs (McLatchie et al., 1998; Morphis et al., 2003), which strongly influence their pharmacological characteristics. The human genome is thought to contain about 1000 genes in the GPCR family, of which about one-third could be odorant receptors. Excluding the latter, and without taking into account the possible diversity factors mentioned, the 40 or so GPCR targets for existing drugs represent only about 7–10% of the total. Roughly one-third of cloned GPCRs are classed as ‘orphan’ receptors, for which no endogenous ligand has yet been identified, and these could certainly emerge as attractive drug targets when more is known about their physiological role. Broadly similar conclusions apply to other major classes of drug target, such as nuclear receptors, ion channels and kinases.
The above discussion relates to drug targets expressed by human cells, and similar arguments apply to those of infective organisms. The therapy of infectious diseases, ranging from viruses to multicellular parasites, is one of medicine’s greatest challenges. Current antibacterial drugs – the largest class – originate mainly from natural products (with a few, e.g. sulfonamides and oxazolidinediones, coming from synthetic compounds) first identified through screening on bacterial cultures, and analysis of their biochemical mechanism and site of action came later. In many cases this knowledge remains incomplete, and the molecular targets are still unclear. Since the pioneering work of Hitchings and Elion (see Chapter 1), the strategy of target-directed drug discovery has rarely been applied in this field1; instead, the ‘antibiotic’ approach, originating with the discovery of penicillin, has held sway. For the 142 current anti-infective drugs (Figure 6.1), which include antiviral and antiparasitic as well as antibacterial drugs, we can identify approximately 40 targets (mainly enzymes and structural proteins) at the biochemical level, only about half of which have been cloned. The urgent need for drugs that act on new targets arises because of the major problem of drug resistance, with which the traditional chemistry-led strategies are failing to keep up. Consequently, as with other therapeutic classes, genomic technologies are being increasingly applied to the problem of finding new antimicrobial drug targets (Rosamond and Allsop, 2000; Buysse, 2001). In some ways the problem appears simpler than finding human drug targets. Microbial genomes are being sequenced at a high rate, and identifying prokaryotic genes that are essential for survival, replication or pathogenicity is generally easier than identifying genes that are involved in specific regulatory mechanisms in eukaryotes.
Conventional strategies for finding new drug targets
Two main routes have been followed so far:
There are numerous examples where the elucidation of pathophysiological pathways has pointed to the existence of novel targets that have subsequently resulted in successful drugs, and this strategy is still adopted by most pharmaceutical companies. To many scientists it seems the safe and logical way to proceed – first understand the pathway leading from the primary disturbance to the appearance of the disease phenotype, then identify particular biochemical steps amenable to therapeutic intervention, then select key molecules as targets. The pioneers in developing this approach were undoubtedly Hitchings and Elion (see Chapter 1), who unravelled the steps in purine and pyrimidine biosynthesis and selected the enzyme dihydrofolate reductase as a suitable target. This biochemical approach led to a remarkable series of therapeutic breakthroughs in antibacterial, anticancer and immunosuppressant drugs. The work of Black and his colleagues, based on mediators and receptors, also described in Chapter 1, was another early and highly successful example of this approach, and there have been many others (Table 6.1a). In some cases the target has emerged from pharmacological rather than pathophysiological studies. The 5HT3 receptor was identified from pharmacological studies and chosen as a potential drug target for the development of antagonists, but its role in pathophysiology was at the time far from clear. Eventually animal and clinical studies revealed the antiemetic effect of drugs such as ondansetron, and such drugs were developed mainly to control the nausea and vomiting associated with cancer chemotherapy. Similarly, the GABAB receptor (target for relaxant drugs such as baclofen) was discovered by analysis of the pharmacological effects of GABA, and only later exploited therapeutically to treat muscle spasm.
Table 6.1 Examples of drug targets identified, (a) by analysis of pathophysiology, and (b) by analysis of existing drugs
| (a) Targets identified via pathophysiology | ||
| Disease indication | Target identified | Drugs developed |
| AIDS | Reverse transcriptase HIV protease | Zidovudine Saquinavir |
| Asthma | Cysteinyl leukotriene receptor | Zafirlukast |
| Bacterial infections | Dihydrofolate reductase | Trimethoprim |
| Malignant disease | Dihydrofolate reductase | 6-mercaptopurine Methotrexate |
| Depression | 5HT transporter | Fluoxetine |
| Hypertension | Angiotensin-converting enzyme Type 5 phosphodiesterase Angiotensin-2 receptor |
Captopril Sildenafil Losartan |
| Inflammatory disease | COX-2 | Celecoxib Rofecoxib |
| Alzheimer’s disease | Acetylcholinesterase | Donepezil |
| Breast cancer | Oestrogen receptor | Tamoxifen Herceptin |
| Chronic myeloid leukemia | Abl kinase | Imatinib |
| Parkinson’s disease | Dopamine synthesis MAO-B |
Levodopa Selegiline |
| Depression | MAO-A | Moclobemide |
| (b) Targets identified via drug effects | ||
| Drug | Disease | Target |
| Benzodiazepines | Anxiety, sleep disorders | BDZ binding site on GABAA receptor |
| Aspirin-like drugs | Inflammation, pain | COX enzymes |
| Ciclosporin, FK506 | Transplant rejection | Immunophilins |
| Vinca alkaloids | Cancers | Tubulin |
| Dihydropyridines | Cardiovascular disease | L-type calcium channels |
| Sulfonylureas | Diabetes | KATP channels |
| Classic antipsychotic drugs | Schizophrenia | Dopamine D2 receptor |
| Tricyclic antidepressants | Depression | Monoamine transporters |
| Fibrates | Raised blood cholesterol | PPARα |
The identification of drug targets by the ‘backwards’ approach – involving analysis of the mechanism of action of empirically discovered therapeutic agents – has produced some major breakthroughs in the past (see examples in Table 6.1b). Its relevance is likely to decline as drug discovery becomes more target focused, though natural product pharmacology will probably continue to reveal novel drug targets.
It is worth reminding ourselves that there remain several important drug classes whose mechanism of action we still do not fully understand (e.g. acetaminophen (paracetamol)2, valproate). Whether their targets remain elusive because their effects depend on a cocktail of interactions at several different sites, or whether novel targets will emerge for such drugs, remains uncertain.
New strategies for identifying drug targets
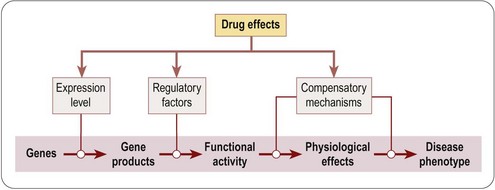
Figure 6.2 summarizes the main points at which drugs may intervene along the pathway from genotype to phenotype, namely by altering gene expression, by altering the functional activity of gene products, or by activating compensatory mechanisms. This is of course an oversimplification, as changes in gene expression or the activation of compensatory mechanisms are themselves indirect effects, following pathways similar to that represented by the primary track in Figure 6.2. Nevertheless, it provides a useful framework for discussing some of the newer genomics-based approaches. A useful account of the various genetic models that have been developed in different organisms for the identification of new drug targets, and elucidating the mechanisms of action of existing drugs, is given by Carroll and Fitzgerald (2003).
Trawling the genome
The conventional route to new drug targets, starting from pathophysiology, will undoubtedly remain as a standard approach. Understanding of the disease mechanisms in the major areas of therapeutic challenge, such as Alzheimer’s disease, atherosclerosis, cancer, stroke, obesity, etc., is advancing rapidly, and new targets are continually emerging. But this is generally slow, painstaking, hypothesis-driven work, and there is a strong incentive to seek shortcuts based on the use of new technologies to select and validate novel targets, starting from the genome. In principle, it is argued, nearly all drug targets are proteins, and are, therefore, represented in the proteome, and also as corresponding genes in the genome. There are, however, some significant caveats, the main ones being the following:
• Splice variants may result in more than one pharmacologically distinct type of receptor being encoded in a single gene. These are generally predictable from the genome, and represented as distinct species in the transcriptome and proteome.
• There are many examples of multimeric receptors, made up of non-identical subunits encoded by different genes. This is true of the majority of ligand-gated ion channels, whose pharmacological characteristics depend critically on the subunit composition (Hille, 2001). Recent work also shows that G-protein-coupled receptors often exist as heteromeric dimers, with pharmacological properties distinct from those of the individual units (Bouvier, 2001). Moreover, association between receptors and non-receptor proteins can determine the pharmacological characteristics of certain G-protein-coupled receptors (McLatchie et al., 1998).
Despite these complications, studies based on the appealingly simple dogma:
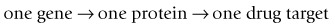
have been extremely productive in advancing our knowledge of receptors and other drug targets in recent years, and it is expected that genome trawling will reveal more ‘single-protein’ targets, even though multiunit complexes are likely to escape detection by this approach.
How might potential drug targets be recognized among the 25 000 or so genes in the human genome?
Several approaches have been described for homing in on genes that may encode novel drug targets, starting with the identification of certain gene categories:
• ’Disease genes’, i.e. genes, mutations of which cause or predispose to the development of human disease.
• ’Disease-modifying’ genes. These comprise (a) genes whose altered expression is thought to be involved in the development of the disease state; and (b) genes that encode functional proteins, whose activity is altered (even if their expression level is not) in the disease state, and which play a part in inducing the disease state.
• ’Druggable genes’, i.e. genes encoding proteins likely to possess binding domains that recognize drug-like small molecules. Included in this group are genes encoding targets for existing therapeutic and experimental drugs. These genes and their paralogues (i.e. closely related but non-identical genes occurring elsewhere in the genome) comprise the group of druggable genes.
On this basis (Hopkins and Groom, 2002), novel targets are represented by the intersection of disease-modifying and druggable gene classes, excluding those already targeted by therapeutic drugs. Next, we consider these categories in more detail.
Disease genes
The identification of genes in which mutations are associated with particular diseases has a long history in medicine (Weatherall, 1991), starting with the concept of ‘inborn errors of metabolism’ such as phenylketonuria. The strategies used to identify disease-associated genes, described in Chapter 7, have been very successful. Examples of common diseases associated with mutations of a single gene are summarized in Table 7.3. There are many more examples of rare inherited disorders of this type. Information of this kind, important though it is for the diagnosis, management and counselling of these patients, has so far had little impact on the selection of drug targets. None of the gene products identified appears to be directly ‘targetable’. Much more common than single-gene disorders are conditions such as diabetes, hypertension, schizophrenia, bipolar depressive illness and many cancers in which there is a clear genetic component, but, together with environmental factors, several different genes contribute as risk factors for the appearance of the disease phenotype. The methods for identifying the particular genes involved (see Chapter 7) were until recently difficult and laborious, but are becoming much easier as the sequencing and annotation of the human genome progresses. The Human Genome Consortium (2001) found 971 ‘disease genes’ already listed in public databases, and identified 286 paralogues of these in the sequenced genome. Progress has been rapid and Lander (2011) contrasts the situation in 2000, when only about a dozen genetic variations outside the HLA locus had been associated with common diseases, with today, when more than 1100 loci associated with 165 diseases have been identified. Notably most of these have been discovered since 2007. The challenge of obtaining useful drug targets from genome-wide association studies remains considerable (Donnelly, 2008).
The value of information about disease genes in better understanding the pathophysiology is unquestionable, but just how useful is it as a pointer to novel drug targets? Many years after being identified, the genes involved in several important single-gene disorders, such as thalassaemia, muscular dystrophy and cystic fibrosis, have not so far proved useful as drug targets, although progress is at last being made. On the other hand, the example of Abl-kinase, the molecular target for the recently introduced anticancer drug imatinib (Gleevec; see Chapter 4), shows that the proteins encoded by mutated genes can themselves constitute drug targets, but so far there are few instances where this has proved successful. The findings that rare forms of familial Alzheimer’s disease were associated with mutations in the gene encoding the amyloid precursor protein (APP) or the secretase enzyme responsible for formation of the β-amyloid fragment present in amyloid plaques were strong pointers that confirmed the validity of secretase as a drug target (although it had already been singled out on the basis of biochemical studies). Secretase inhibitors reached late-stage clinical development as potential anti-Alzheimer’s drugs in 2001, but none at the time of writing have emerged from the development pipeline as useful drugs. Similar approaches have been successful in identifying disease-associated genes in defined subgroups of patients with conditions such as diabetes, hypertension and hypercholesterolaemia, and there is reason to hope that novel drug targets will emerge in these areas. However, in other fields, such as schizophrenia and asthma, progress in pinning down disease-related genes has been very limited. The most promising field for identifying novel drug targets among disease-associated mutations is likely to be in cancer therapies, as mutations are the basic cause of malignant transformation and real progress is being made in exploiting our knowledge of the cancer genome (Roukos, 2011). In general, one can say that identifying disease genes may provide valuable pointers to possible drug targets further down the pathophysiological pathway, even though their immediate gene products may not always be targetable. The identification of a new disease gene often hits the popular headlines on the basis that an effective therapy will quickly follow, though this rarely happens, and never quickly.
In summary, the class of disease genes does not seem to include many obvious drug targets.
Disease-modifying genes
In this class lie many non-mutated genes that are directly involved in the pathophysiological pathway leading to the disease phenotype. The phenotype may be associated with over- or underexpression of the genes, detectable by expression profiling (see below), or by the over- or underactivity of the gene product – for example, an enzyme – independently of changes in its expression level.
This is the most important category in relation to drug targets, as therapeutic drug action generally occurs by changing the activity of functional proteins, whether or not the disease alters their expression level. Finding new ones, however, is not easy, and there is as yet no shortcut screening strategy for locating them in the genome.
Two main approaches are currently being used, namely gene expression profiling and comprehensive gene knockout studies.
Gene expression profiling
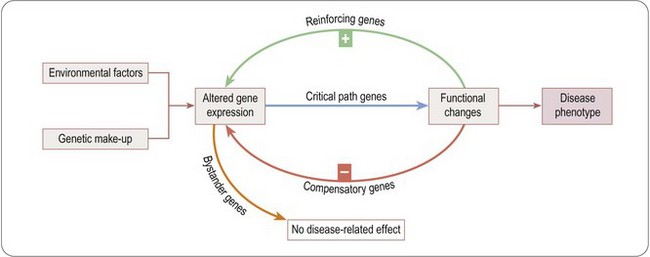
The principle underlying gene expression profiling as a guide to new drug targets is that the development of any disease phenotype necessarily involves changes in gene expression in the cells and tissues involved. Long-term changes in the structure or function of cells cannot occur without altered gene expression, and so a catalogue of all the genes whose expression is up- or down-regulated in the disease state will include genes where such regulation is actually required for the development of the disease phenotype. As well as these ‘critical path genes’, which may represent potential drug targets, others are likely to be affected as genes involved in secondary reinforcing of compensatory mechanisms following the development of the disease phenotype (which may also represent potential drug targets), but many will be irrelevant ‘bystander genes’ (Figure 6.3). The important problem, to which there is no simple answer, is how to eliminate the bystanders, and how to identify potential drug targets among the rest. Zanders (2000), in a thoughtful review, emphasizes the importance of focusing on signal transduction pathways as the most likely source of novel targets identifiable by gene expression profiling.
The methods available for gene expression profiling are described in Chapter 7. DNA microarrays (‘gene chips’) are most commonly used, their advantage being that they are quick and easy to use. They have the disadvantage that the DNA sequences screened are selected in advance, but this is becoming less of a limitation as more genomic information accumulates. They also have limited sensitivity, which (see Chapter 7) means that genes expressed at low levels – including, possibly, a significant proportion of potential drug targets – can be missed. Methods based on the polymerase chain reaction (PCR), such as serial analysis of gene expression (SAGE; Velculescu et al., 1995), are, in principle, capable of detecting all transcribed RNAs without preselection, with higher sensitivity than microarrays, but are more laborious and technically demanding. Both technologies produce the same kind of data, namely a list of genes whose expression is significantly altered as a result of a defined experimental intervention. A large body of gene expression data has been collected over the last few years, showing the effects of many kinds of disturbance, including human disease, animal models of disease and drug effects; some examples are listed in Table 6.2. In most cases, several thousand genes have been probed with microarrays, covering perhaps 20% of the entire genome, so the coverage is by no means complete. Commonly, it is found that some 2–10% of the genes studied show significant changes in response to such disturbances. Thus, given a total of about 25 000 genes in man, with perhaps 20% expressed in a given tissue, we can expect several hundred to be affected in a typical disease state. Much effort has gone into (a) methods of analysis and pattern recognition within these large data sets, and (b) defining the principles for identifying possible drug targets within such a group of regulated genes. Techniques for analysis and pattern recognition fall within the field of bioinformatics (Chapter 7), and the specific problem of analysing transcription data is discussed by Quackenbush (2001) and Butte (2002), and also Lander (2011). Because altered mRNA levels do not always correlate with changes in protein expression, protein changes should be confirmed independently where possible. This normally entails the production and validation of an antibody-based assay or staining procedure.
Table 6.2 Examples of gene expression profiling studies, showing numbers of mRNAs affected under various conditions
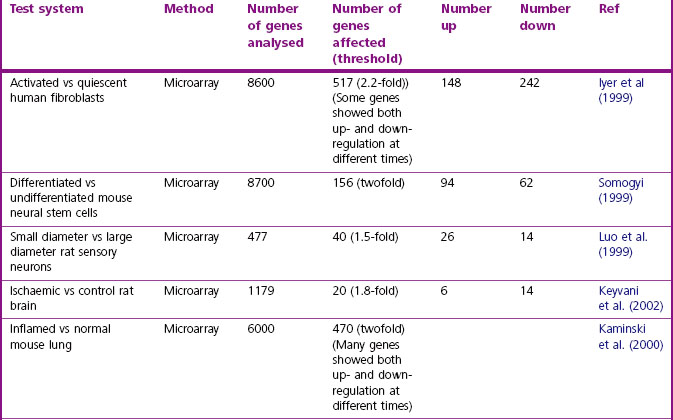
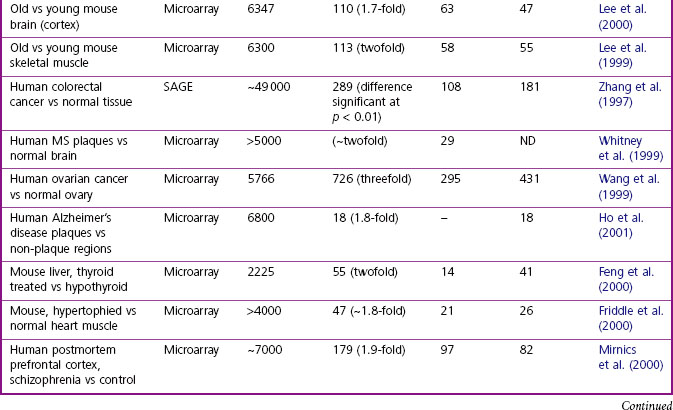

Identifying potential drug targets among the array of differentially expressed genes that are revealed by expression profiling is neither straightforward nor exact. Possible approaches are:
• On the basis of prior biological knowledge, genes that might plausibly be critical in the pathogenesis of the disease can be distinguished from those (e.g. housekeeping genes, or genes involved in intermediary metabolism) that are unlikely to be critical. This, the plausibility criterion, is in practice the most important factor in identifying likely drug targets. As genomic analysis proceeds, it is becoming possible to group genes into functional classes, e.g. tissue growth and repair, inflammation, myelin formation, neurotransmission, etc., and changes in expression are often concentrated in one or more of these functional groups, giving a useful pointer to the biological pathways involved in pathogenesis.
• Anatomical studies can be used to identify whether candidate genes are regulated in the cells and tissues affected by the disease.
• Timecourse measurements reveal the relationship between gene expression changes and the development of the disease phenotype. With many acute interventions, several distinct temporal patterns are detectable in the expression of different genes. ‘Clustering’ algorithms (see Chapter 7; Quackenbush, 2001) are useful to identify genes which are co-regulated and whose function might point to a particular biochemical or signalling pathway relevant to the pathogenesis.
• The study of several different clinical and animal models with similar disease phenotypes can reveal networks of genes whose expression is consistently affected and hence representative of the phenotype (see Emilsson et al. 2008; Schadt 2009).
• The effects of drug or other treatments can be studied in order to reveal genes whose expression is normalized in parallel with the ‘therapeutic’ effect.
Gene knockout screening
Another screening approach for identifying potential drug target genes, based on generating transgenic ‘gene knockout’ strains of mice, as performed by biotechnology company Lexicon Pharmaceuticals (Walke et al., 2001; Zambrowicz and Sands, 2003), aimed to study 5000 genes within 5 years. The 5000 genes were selected on several criteria, including existing evidence of association with disease and membership of families of known drug targets (GPCRs, transporters, kinases, etc.). The group has developed an efficient procedure for generating transgenic knockout mice, and a standardized set of tests to detect phenotypic effects relevant to a range of disease states. In a recent review, Zambrowicz and Sands (2003) present many examples where the effects of gene knockouts in mice produce effects consistent with the known actions and side effects of therapeutic drugs. For example, inactivation of the genes for angiotensin converting enzyme (ACE) or the angiotensin receptor results in lowering of blood pressure, reflecting the clinical effect of drugs acting on these targets. Similarly, elimination of the gene encoding one of the subunits of the GABAA receptor produces a state of irritability and hyperactivity in mice, the opposite of the effect of benzodiazepine tranquillizers, which enhance GABAA receptor function. However, using transgenic gene knockout technology (sometimes dubbed ‘reverse genetics’) to confirm the validity of previously well-established drug targets is not the same as using it to discover new targets. Success in the latter context will depend greatly on the ability of the phenotypic tests that are applied to detect therapeutically relevant changes in the knockout strains. Some new targets have already been identified in this way, and deployed in drug discovery programmes; they include cathepsin K, a protease involved in the genesis of osteoporosis, and melanocortin receptors, which may play a role in obesity. The Lexicon group has developed a systems-based panel of primary screens to look for relevant effects on the main physiological systems (cardiovascular, CNS, immune system, etc.). On the basis of their experience with the first 750 of the planned 5000 gene knockouts, they predicted that about 100 new high-quality targets could be revealed in this way. Programmes such as this, based on mouse models, are time-consuming and costly, but have the great advantage that mouse physiology is fairly similar to human. The use of species such as flatworm (Caenorhabditis elegans) and zebra-fish (Danio rerio) is being explored as a means of speeding up the process (Shin and Fishman, 2002) and more recently studies in yeast have successfully facilitated the discovery of molecules that interact with mammalian drug targets (Brown et al., 2011).
’Druggable’ genes
For a gene product to serve as a drug target, it must possess a recognition site capable of binding small molecules. Hopkins and Groom (2002) found a total of 399 protein targets for registered and experimental drug molecules. The 399 targets belong to 130 distinct protein families, and the authors suggest that other members of these families – a total of 3051 proteins – are also likely to possess similar binding domains, even though specific ligands have not yet been described, and propose that this total represents the current limit of the druggable genome. Of course, it is likely that new targets, belonging to different protein families, will emerge in the future, so this number may well expand. ‘Druggable’ in this context implies only that the protein is likely to possess a binding site for a small molecule, irrespective of whether such an interaction is likely be of any therapeutic value. To be useful as a starting point for drug discovery, a potential target needs to combine ‘druggability’ with disease-modifying properties. One new approach is to look at the characteristics of a typical human protein drug target (e.g. hydrophobic, high length, signal motif present) and to then look for these characteristics in a non-target set of proteins so as to identify new potential targets (Bakheet and Doig, 2009).
Target validation
The techniques discussed so far are aimed at identifying potential drug targets within the diversity warehouse represented by the genome, the key word being ‘potential’.
Few companies will be willing to invest the considerable resources needed to mount a target-directed drug discovery project without more direct evidence that the target is an appropriate one for the disease indication. Target validation refers to the experimental approaches by which a potential drug target can be tested and given further credibility. It is an open-ended term, which can be taken to embrace virtually the whole of biology, but for practical purposes the main approaches are pharmacological and genetic.
Although these experimental approaches can go a long way towards supporting the validity of a chosen target, the ultimate test is in the clinic, where efficacy is or is not confirmed. Lack of clinical efficacy causes the abandonment of roughly one-third of drugs in Phase II, reflecting the unreliability of the earlier surrogate evidence for target validity.
Pharmacological approaches
The underlying question to be addressed is whether drugs that influence the potential drug target actually produce the expected effects on cells, tissues or whole animals. Where a known receptor, for example the metabotropic glutamate receptor (mGluR), was identified as a potential target for a new indication (e.g. pain) its validity could be tested by measuring the analgesic effect of known mGluR antagonists in relevant animal models. For novel targets, of course, no panel of active compounds will normally be available, and so it will be necessary to set up a screening assay to identify them. Where a company is already active in a particular line of research – as was the case with Ciba Geigy in the kinase field – it may be straightforward to refine its assay methods in order to identify selective inhibitors. Ciba Geigy was able to identify active Abl kinase inhibitors within its existing compound collection, and hence to show that these were effective in cell proliferation assays.
A variant of the pharmacological approach is to use antibodies raised against the putative target protein, rather than small-molecule inhibitors.
Many experts predict that, as high-throughput screening and automated chemistry develop (see Chapters 8 and 9), allowing the rapid identification of families of selective compounds, these technologies will be increasingly used as tools for target validation, in parallel with lead finding. ‘Hits’ from screening that show a reasonable degree of target selectivity, whether or not they represent viable lead compounds, can be used in pharmacological studies designed to test their efficacy in a selection of in vitro and in vivo models. Showing that such prototype compounds, regardless of their suitability as leads, do in fact produce the desired effect greatly strengthens the argument for target validity. Although contemporary examples are generally shrouded in confidentiality, it is clear that this approach is becoming more common, so that target validation and lead finding are carried out simultaneously. More recently the move has been towards screening enriched libraries containing compounds of a particular chemical phenotype known to, for example, be disposed towards kinase inhibition rather than screening entire compound collections in a random fashion.
Genetic approaches
These approaches involve various techniques for suppressing the expression of specific genes to determine whether they are critical to the disease process. This can be done acutely in genetically normal cells or animals by the use of antisense oligonucleotides or RNA interference, or constitutively by generating transgenic animals in which the genes of interest are either overactive or suppressed.
Antisense oligonucleotides
Antisense oligonucleotides (Phillips, 2000; Dean, 2001) are stretches of RNA complementary to the gene of interest, which bind to cellular mRNA and prevent its translation. In principle this allows the expression of specific genes to be inhibited, so that their role in the development of a disease phenotype can be determined. Although simple in principle, the technology is subject to many pitfalls and artefacts in practice, and attempts to use it, without very careful controls, to assess genes as potential drug targets are likely to give misleading results. As an alternative to using synthetic oligonucleotides, antisense sequences can be introduced into cells by genetic engineering. Examples where this approach has been used to validate putative drug targets include a range of recent studies on the novel cell surface receptor uPAR (urokinase plasminogen-activator receptor; see review by Wang, 2001). This receptor is expressed by certain malignant tumour cells, particularly gliomas, and antisense studies have shown it to be important in controlling the tendency of these tumours to metastasize, and therefore to be a potential drug target. In a different field, antisense studies have supported the role of a recently cloned sodium channel subtype, PN3 (now known to be Nav 1.87) (Porreca et al., 1999) and of the metabotropic glutamate receptor mGluR1 (Fundytus et al., 2001) in the pathogenesis of neuropathic pain in animal models. Antisense oligonucleotides have the advantage that their effects on gene expression are acute and reversible, and so mimic drug effects more closely than, for example, the changes seen in transgenic animals (see below), where in most cases the genetic disturbance is present throughout life. It is likely that, as more experience is gained, antisense methods based on synthetic oligonucleotides will play an increasingly important role in drug target validation.
RNA interference (RNAi)
This technique depends on the fact that short lengths of double-stranded RNA (short interfering RNAs, or siRNAs) activate a sequence-specific RNA-induced silencing complex (RISC), which destroys by cleavage the corresponding functional mRNA within the cell (Hannon, 2000; Kim, 2003). Thus specific mRNAs or whole gene families can be inactivated by choosing appropriate siRNA sequences. Gene silencing by this method is highly efficient, particularly in invertebrates such as Caenorhabditis elegans and Drosophila, and can also be used in mammalian cells and whole animals. Its use for studying gene function and validating potential drug targets is increasing rapidly, and it also has potential for therapeutic applications. It has proved to be a rapid and effective tool for discovering new targets and automated cell assays have allowed the rapid screening of large groups of genes.
Transgenic animals
The use of the gene knockout principle as a screening approach to identify new targets is described above. The same technology is also valuable, and increasingly being used, as a means of validating putative targets. In principle, deletion or overexpression of a specific gene in vivo can provide a direct test of whether or not it plays a role in the sequence of events that gives rise to a disease phenotype. The generation of transgenic animal – mainly mouse – strains is, however, a demanding and time-consuming process (Houdebine, 1997; Jackson and Abbott, 2000). Therefore, although this technology has an increasingly important role to play in the later stages of drug discovery and development, some have claimed it is too cumbersome to be used routinely at the stage of target selection, though Harris and Foord (2000) predicted that high-throughput ‘transgenic factories’ may be used in this way. One problem relates to the genetic background of the transgenic colony. It is well known that different mouse strains differ in many significant ways, for example in their behaviour, susceptibility to tumour development, body weight, etc. For technical reasons, the strain into which the transgene is introduced is normally different from that used to establish the breeding colony, so a protocol of back-crossing the transgenic ‘founders’ into the breeding strain has to proceed for several generations before a genetically homogeneous transgenic colony is obtained. Limited by the breeding cycle of mice, this normally takes about 2 years. It is mainly for this reason that the generation of transgenic animals for the purposes of target validation is not usually included as a stage on the critical path of the project. Sometimes, studies on transgenic animals tip the balance of opinion in such a way as to encourage work on a novel drug target. For example, the vanilloid receptor, TRPV1, which is expressed by nociceptive sensory neurons, was confirmed as a potential drug target when the knockout mouse proved to have a marked deficit in the development of inflammatory hyperalgesia (Davis et al., 2000), thereby confirming the likely involvement of this receptor in a significant clinical condition. Most target-directed projects, however, start on the basis of other evidence for (or simply faith in) the relevance of the target, and work on developing transgenic animals begins at the same time, in anticipation of a need for them later in the project. In many cases, transgenic animals have provided the most useful (sometimes the only available) disease models for drug testing in vivo. Thus, cancer models based on deletion of the p53 tumour suppressor gene are widely used, as are atherosclerosis models based on deletion of the ApoE or LDL-receptor genes. Alzheimer’s disease models involving mutation of the gene for amyloid precursor protein, or the presenilin genes, have also proved extremely valuable, as there was hitherto no model that replicated the amyloid deposits typical of this disease. In summary, transgenic animal models are often helpful for post hoc target validation, but their main – and increasing – use in drug discovery comes at later stages of the project (see Chapter 11).
Summary and conclusions
In the present drug discovery environment most projects begin with the identification of a molecular target, usually one that can be incorporated into a high-throughput screening assay. Drugs currently in therapeutic use cover about 200–250 distinct human molecular targets, and the great majority of new compounds registered in the last decade are directed at targets that were already well known; on average, about three novel targets are covered by drugs registered each year. The discovery and exploitation of new targets is considered essential for therapeutic progress and commercial success in the long term. Estimates from genome sequence data of the number of potential drug targets, defined by disease relevance and ‘druggability’, suggest that from about 100 to several thousand ‘druggable’ new targets remain to be discovered. The uncertainty reflects two main problems: the difficulty of recognizing ‘druggability’ in gene sequence data, and the difficulty of determining the relevance of a particular gene product in the development of a disease phenotype. Much effort is currently being applied to these problems, often taking the form of new ‘omic’ disciplines whose role and status are not yet defined. Proteomics and structural genomics are expected to improve our ability to distinguish druggable proteins from the rest, and ‘transcriptomics’ (another name for gene expression profiling) and the study of transgenic animals will throw light on gene function, thus improving our ability to recognize the disease-modifying mechanisms wherein novel drug targets are expected to reside.
Aronoff DM, Oates JA, Boutaud O. New insights into the mechanism of action of acetaminophen: its clinical pharmacologic characteristics reflect its inhibition of the two prostaglandin H2 synthases. Clinical Pharmacology and Therapeutics. 2006;79:9–19.
Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25:451–457.
Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nature Reviews Neuroscience. 2001;2:274–286.
Brown AJ, Daniels DA, Kassim M, et al. Pharmacology of GPR-55 in yeast and identification of GSK494581A as a mixed-activity glycine transporter subtype 1 imhibitor and GPR55 agonist. Journal of Pharmacology and Experimental Therapeutics. 2011;337:236–246.
Butte A. The use and analysis of microarray data. Nature Reviews Drug Discovery. 2002;1:951–960.
Buysse JM. The role of genomics in antibacterial drug discovery. Current Medicinal Chemistry. 2001;8:1713–1726.
. Model organisms in drug discovery. Carroll PM. Fitzgerald K. Chichester: John Wiley; 2003.
Chandrasekharan NV, Dai H, Roos KL, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proceedings of the National Academy of Sciences of the USA. 2002;99:13926–13931.
Davis JB, Gray J, Gunthorpe MJ, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187.
Dean NM. Functional genomics and target validation approaches using antisense oligonucleotide technology. Current Opinion in Biotechnology. 2001;12:622–625.
. Therapeutic drugs. Dollery CT. Edinburgh: Churchill Livingstone; 1999.
Donnelly P. Progress and challenges in genome-wide association studies. Nature. 2008;456:728–731.
Drews J, Ryser S. Classic drug targets. Nature Biotechnology. 1997;15:1318–1319.
Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428.
Feng X, Jiang Y, Meltzer P, et al. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Molecular Endocrinology. 2000;14:947–955.
Friddle CJ, Koga T, Rubin EM, et al. Expression profiling reveals distinct sets of genes altered during induction and regression of cardiac hypertrophy. Proceedings of the National Academy of Sciences of the USA. 2000;97:6745–6750.
Fundytus ME, Yashpal K, Chabot JG, et al. Knockdown of spinal metabotropic receptor 1 (mGluR1) alleviates pain and restores opioid efficacy after nerve injury in rats. British Journal of Pharmacology. 2001;132:354–367.
Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proceedings of the National Academy of Sciences of the USA. 2001;98:4746–4751.
Hannon GJ. RNA interference. Nature. 2000;418:244–251.
Harris S, Foord SM. Transgenic gene knock-outs: functional genomics and therapeutic target selection. Pharmacogenomics. 2000;1:433–443.
Hemby SE, Ginsberg SD, Brunk B, et al. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Archives of General Psychiatry. 2002;59:631–640.
Hille B. Ion channels of excitable membranes, 3rd ed. Sunderland, MA: Sinauer; 2001.
Ho L, Guo Y, Spielman L, et al. Altered expression of a-type but not b-type synapsin isoform in the brain of patients at high risk for Alzheimer’s disease assessed by DNA microarray technique. Neuroscience Letters. 2001;298:191–194.
Hopkins AL, Groom CR. The druggable genome. Nature Reviews Drug Discovery. 2002;1:727–730.
Houdebine LM. Transgenic animals – generation and use. Amsterdam: Harwood Academic; 1997.
Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nature Reviews. Drug Discovery. 2006;5:821–834.
Iyer VR, Eisen MB, Ross DT, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87.
Jackson IJ, Abbott CM. Mouse genetics and transgenics. Oxford: Oxford University Press; 2000.
Kaminski N, Allard JD, Pittet JF, et al. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proceedings of the National Academy of Sciences of the USA. 2000;97:1778–1783.
Keyvani K, Witte OW, Paulus W. Gene expression profiling in perilesional and contralateral areas after ischemia in rat brain. Journal of Cerebral Blood Flow and Metabolism. 2002;22:153–160.
Kim VN. RNA interference in functional genomics and medicine. Journal of Korean Medical Science. 2003;18:309–318.
Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197.
Lee CK, Klopp RG, Weindruch R, et al. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393.
Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nature Genetics. 2000;25:294–297.
Luo L, Salunga RC, Guo H, et al. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nature Medicine. 1999;5:117–122.
McLatchie LM, Fraser NJ, Main MJ, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339.
Mallet C, Barriere DA, Ermund A, et al. TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS ONE. 2010;5:1–11.
Mirnics K, Middleton FA, Marquez A, et al. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67.
Morphis M, Christopoulos A, Sexton PM. RAMPs: 5 years on, where to now? Trends in Pharmacological Sciences. 2003;24:596–601.
Overington JP, Bissan A-L, Hopkins AL. How many drug targets are there? Nature Reviews. Drug Discovery. 2006;5:993–996.
Phillips MI. Antisense technology, Parts A and B. San Diego: Academic Press; 2000.
Porreca F, Lai J, Bian D, et al. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proceedings of the National Academy of Sciences of the USA. 1999;96:7640–7644.
Quackenbush J. Computational analysis of microarray data. Nature Reviews Genetics. 2001;2:418–427.
Rosamond J, Allsop A. Harnessing the power of the genome in the search for new antibiotics. Science. 2000;287:1973–1976.
Roukos DH. Trastuzumab and beyond: sequencing cancer genomes and predicting molecular networks. Pharmacogenomics Journal. 2011;11:81–92.
Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223.
Shin JT, Fishman MC. From zebrafish to human: molecular medical models. Annual Review of Genomics and Human Genetics. 2002;3:311–340.
Somogyi R. Making sense of gene-expression data. In: Pharma Informatics, A Trends Guide. Elsevier Trends in Biotechnology. 1999;17 (Suppl. 1):17–24.
Velculescu VE, Zhang L, Vogelstein B, et al. Serial analysis of gene expression. Science. 1995;270:484–487.
Walke DW, Han C, Shaw J, et al. In vivo drug target discovery: identifying the best targets from the genome. Current Opinion in Biotechnology. 2001;12:626–631.
Wang Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Medicinal Research Reviews. 2001;21:146–170.
Wang K, Gan L, Jeffery E, et al. Monitoring gene expression profile changes in ovarian carcinomas using cDNA microarray. Gene. 1999;229:101–108.
Weatherall DJ. The new genetica and clinical practice, 3rd ed. Oxford: Oxford University Press; 1991.
Wengelnik K, Vidal V, Ancelin ML, et al. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science. 2002;295:1311–1314.
Whitney LW, Becker KG, Tresser NJ, et al. Analysis of gene expression in mutiple sclerosis lesions using cDNA microarrays. Annals of Neurology. 1999;46:425–428.
Wishart DS, Knox C, Guo AC, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Research. 2008;36:D901–D906.
Zambrowicz BP, Sands AT. Knockouts model the 100 best-selling drugs – will they model the next 100? Nature Reviews. Drug Discovery. 2003;2:38–51.
Zanders ED. Gene expression profiling as an aid to the identification of drug targets. Pharmacogenomics. 2000;1:375–384.
Zhang L, Zhou W, Velculescu VE, et al. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272.