DISORDERS OF BLOOD COAGULATION AND HAEMOSTASIS
The components of the haemostatic system are described on page 629. Although it is convenient to describe separately the intrinsic and extrinsic pathways of blood coagulation, platelet adhesion and aggregation, and activation of the fibrinolytic system, there are numerous points of interaction.
Disturbances of blood coagulation and haemostasis produce excessive haemorrhage or thrombosis, or occasionally both, as in the acquired disorder disseminated intravascular coagulation. As described below and in Table 23.10, the patterns of bleeding differ somewhat between disorders of primary haemostasis (platelet disorders) and defects of blood coagulation. Also, as a general rule, disorders that allow the unchecked generation and deposition of fibrin tend to be associated with thrombosis in the venous circulation (red thrombus), whereas inappropriate platelet activation tends to result in vascular occlusion in arteries and arterioles (white thrombus), although this is by no means a rigid distinction.
Table 23.10 Features that may distinguish bleeding in coagulation defects from that in platelet disorders∗
| Feature |
Platelet defect |
Severe coagulation defect |
| Purpura |
Very common |
Absent |
| Mucosal bleeding |
Common frommouth, gut and nose |
Relativelyuncommon except from urinary tract |
| Joint bleeding |
Absent |
Very common insevere congenitalfactor deficiencies |
| Musclehaematomas |
In response totrauma |
Spontaneous |
| Bleeding aftersurgery |
Immediate |
Often delayedseveral hours |
DISORDERS OF PRIMARY HAEMOSTASIS
Theoretically, primary haemostasis could be defective as a result of platelet abnormalities or defects of the small blood vessels. In fact, vascular disease is rarely the cause of clinically important haemorrhage. Bleeding due to primary haemostatic defects is most commonly secondary to acquired platelet disorders such as thrombocytopenia or disturbance of platelet function.
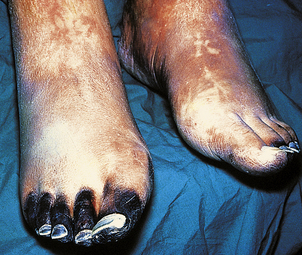
In bleeding due to disorders of primary haemostasis the skin and mucous membranes are especially involved (Table 23.10 and Fig. 23.34).
Thrombocytopenias

Cause spontaneous bleeding when the blood platelet count falls below 20 × 109/l
Due to failure of platelet production or increased destruction/sequestration
When due to production failure, thrombocytopenia is usually accompanied by other evidence of marrow dysfunction: anaemia, leukopenia, leukocytosis or atypical cells
When due to increased destruction, immune mechanisms and disseminated intravascular coagulation are common causes
Although a bleeding tendency results from thrombocytopenia, there must be a substantial reduction in platelet numbers before this occurs. No clinical defect of primary haemostasis occurs with platelet counts greater than 80 × 109/l if they function normally. Increased bleeding after trauma is present with counts of 40–50 × 109/l, but spontaneous skin and mucosal haemorrhage occur only when platelet counts fall to 20 × 109/l. The time to cessation of bleeding from skin incisions increases progressively as the platelet count falls below 80 × 109/l and, when performed in a standardised manner in the bleeding time test, is a good guide to the efficiency of primary haemostasis. The bleeding time is not usually affected by deficiencies of clotting factors because it relies on adequate platelet numbers and function rather than fibrin formation.
Classification
Thrombocytopenia can be conveniently classified according to pathogenesis:
•
failure of platelet production
—
haematological malignancy, including leukaemias, myelodysplasia, myelofibrosis, myeloma and marrow involvement in lymphoma
—
other marrow infiltration, e.g. carcinoma
—
hypoplastic/aplastic anaemia
—
chemotherapeutic agents and occasionally other drugs
—
congenital absence of megakaryocytes
•
increased platelet destruction
—
acute and chronic autoimmune thrombocytopenic purpura
—
drug-induced immune thrombocytopenia
—
neonatal and post-transfusion purpura (alloimmune)
—
massive blood loss and transfusion (dilutional and consumptive)
—
disseminated intravascular coagulation
—
thrombotic thrombocytopenic purpura/haemolytic uraemic syndrome
Where thrombocytopenia is an isolated finding, with normal haemoglobin and white cells, increased platelet destruction is most likely. Failure of platelet production due to a bone marrow abnormality is most commonly associated with a pancytopenia, a leukocytosis or the presence of circulating blast cells in the leukaemias.
Of the causes of thrombocytopenia due to platelet production failure, those due to thiazides, viral infection and congenital megakaryocyte abnormalities are very uncommon. The other disorders resulting in marrow failure have been described earlier.
Autoimmune thrombocytopenic purpura and disseminated intravascular coagulation are the most common disorders in which thrombocytopenia is due to increased destructionor utilisation of platelets.
Autoimmune thrombocytopenic purpura
In autoimmune thrombocytopenic purpura platelets are destroyed in the reticulo-endothelial system, especially the spleen, due to coating with auto-antibody. The disorder is analogous to autoimmune haemolytic anaemia. It occurs in an acute, spontaneously remitting form in children, as a chronic idiopathic state at all ages, and as a drug-induced phenomenon. The acute childhood variety may follow a viral infection. The chronic type is occasionally symptomatic of a disorder such as chronic lymphocytic leukaemia, or lymphoma, may occur in association with other autoimmune disease such as SLE and rheumatoid arthritis, or may present without an associated disorder. It has been recognised that a form of immune-mediated thrombocytopenia occurs in HIV-infected individuals, usually at a point in the disease where there is relative preservation of immune function. Drugs associated with idiopathic thrombocytopenic purpura (ITP) include quinine, heparin and sulphonamides; in most cases, an immune complex mechanism is involved, similar to that in some cases of drug-induced immune haemolytic anaemia.
Blood and bone marrow changes
Thrombocytopenia is present; severity is variable. Platelet counts of less than 10 × 109/l are not uncommon. Erythrocytes and leukocytes are usually normal. Iron deficiency anaemia may be present due to chronic mucosal bleeding. In the bone marrow there is a non-specific increase in megakaryocyte size and number. It may be possible to detect the auto-antibody in serum by tests analogous to the antiglobulin test used in the investigation of haemolytic anaemias, but poor sensitivity and specificity of these assays limit their clinical utility.
Changes in other organs
Changes in other organs are those of haemorrhage. Bleeding into the skin in the form of purpura is common. Purpura (petechiae) of thrombocytopenic type is due to apparently spontaneous leakage of red cells from capillaries and arterioles in the skin. It is usually most prominent in the skin of the lower legs and feet, suggesting that hydrostatic pressure may play a role. Areas of skin trauma may also be affected. Histological evidence of capillary bleeding may also be present in the serosal linings, mucosae of gastrointestinal and urinary tracts, and the central nervous system. The spleen is usually of normal size or only moderately enlarged, not extending below the costal margin. The sinusoids are congested and splenic follicles reactive. Megakaryocytes may be present in the spleen, a response to the increased platelet turnover.
Clinical features
Clinical features are restricted to excessive haemorrhage. Purpuric rash, skin bruising, epistaxis, menorrhagia and gastrointestinal haemorrhage are common. The presence of mucosal bleeding is relevant in the clinical evaluation of patients with severe thrombocytopenia indicating a more severe bleeding disorder. In severe cases retinal haemorrhage is present and, although it is unusual, fatal intracerebral bleeding is described.
The acute form in childhood is transient and often requires no treatment. Drug-induced ITP responds to withdrawal of the offending medication. Chronic ‘idiopathic’ ITP often responds to immunosuppressive therapy, for example with corticosteroids. Intravenous infusion of a concentrate of normal human IgG prepared from plasma is also often effective. It may act through blockade of the reticulo-endothelial Fc receptors responsible for binding of antibody-coated platelets, or possibly through anti-idiotype activity. Surgical removal of the spleen (splenectomy) may result in long-term remission. Thrombopoietin analogues (discussed above) may also have a role in the treatment of ITP. Prognosis is good.
Other immune thrombocytopenias
The immune thrombocytopenia that is occasionally associated with exposure to the anticoagulant drug heparin is unusual, as it is often accompanied by thrombosis rather than haemorrhage. It results from the development of an immune complex formed between heparin, platelet factor 4 (a peptide secreted from the cytoplasmic granules of stimulated platelets) and the auto-antibody to heparin–platelet factor 4. Binding of the complex to platelet Fc receptors causes platelet activation and consumption with thrombocytopenia. Thrombosis occurs—for example deep vein thrombosis or arterial thrombosis—resulting in myocardial or cerebral infarction. Heparin must be withdrawn and replaced with a non-cross-reacting anticoagulant such as lepirudin or danaparoid.
Neonatal thrombocytopenia due to placental transfer of the platelet-reactive IgG auto-antibody can occur in infants of women with chronic ITP. Also, a condition analogous to rhesus haemolytic disease, due to transplacental passage of an antibody, is occasionally recognised as a cause of neonatal thrombocytopenia—neonatal alloimmune thrombocytopenia (NAIT). The pathogenesis is comparable to that of haemolytic disease of the newborn, the fetus possessing a platelet antigen lacking in the mother, usually HPA1a (p. 653).
Post-transfusion purpura is a very uncommon immune thrombocytopenia typically seen in parous women 10 days or so following transfusion of platelets or occasionally red cell concentrate. It is also due to formation of an allo-antibody to a foreign antigen on transfused platelets, usually HPA1a, although the mechanism by which it results in destruction of the woman’s HPA1a-negative platelets is not clear.
Thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome
In thrombotic thrombocytopenic purpura (TTP) and haemolytic uraemic syndrome (HUS) the dominant features are thrombocytopenia due to platelet consumption in microvascular occlusive platelet plugs and a microangiopathic haemolytic anaemia. Characteristic fragmented erythrocytes—schistocytes—are present on the blood film (Fig. 23.3). Glomerular lesions are characteristic, especially in HUS (Ch. 21), but vascular lesions in other organs, especially the central nervous system, are a feature in TTP. In children, HUS may occur in epidemics, suggesting an infectious origin. When it is associated with an acute haemorrhagic colitis, production of verocytotoxin by Escherichia coli, usually strain O157, from contaminated food has been shown to be the cause.
TTP is usually sporadic, but can be relapsing and may be familial. It appears to be due to platelet aggregation by very high molecular weight multimers of von Willebrand factor. Such multimers are normally secreted by endothelial cells but are rapidly degraded into smaller multimers, which are less reactive with platelets, by a protease called ADAMTS 13 (A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif). In all forms of TTP there is deficiency of ADAMTS 13: in sporadic TTP there is development of an auto-antibody that interferes with the protease activity, whereas in familial TTP there may be a deficiency of the enzyme due to underproduction or abnormalities in its secretion.
Clinically, organ dysfunction due to the microvascular lesions predominates. Renal failure is present. Neurological abnormalities, which may be transient or permanent, including stroke, characterise TTP and distinguish it from HUS. The disease runs a subacute or chronic course. Spontaneous remission is not uncommon in the childhood form, but chronic renal impairment may result. Renal support, including dialysis therapy, may be required in HUS. In TTP, high volume plasma exchange is effective, presumably through removal of auto-antibody and replacement of the cleaving protease in the transfused plasma. Recent data suggest that the therapeutic monoclonal antibody rituximab is of value in the management of TTP due to an auto-antibody.
Qualitative disorders of platelets
Disorders of platelet function result in excessive bleeding of platelet type and prolonged skin bleeding time, usually in the presence of normal platelet numbers.
Acquired disorders of platelet function
Acquired disorders of platelet function are due to:
•
drugs (e.g. aspirin, clopidogrel, anti-GP IIb/IIIa drugs (abciximab, tirofiban, eptifibatide), anti-inflammatory drugs)
•
metabolic disorders (e.g. uraemia, hepatic failure)
•
myeloproliferative disorders (essential thrombocythaemia, polycythaemia rubra vera)
•
plasma cell disorders (e.g. multiple myeloma, Waldenström’s macroglobulinaemia).
Aspirin and anti-inflammatory drugs block the cyclo-oxygenase enzyme necessary for platelet synthesis of pro-aggregatory thromboxane. Clopidogrel inhibits ADP-mediated platelet activation while abciximab, tirofiban and eptifibatide interfere with glycoprotein IIb/IIIa function. In general any bleeding tendency is mild, but there may be increased skin bruising and bleeding after surgery. Gastric haemorrhage from acute mucosal erosions may be life-threatening. Following percutaneous coronary intervention (PCI) and myocardial infarction, these drugs are being increasingly used in combination with other antithrombotics, thus increasing the bleeding risk. In uraemia and liver failure, platelet interactions with subendothelium are abnormal and bleeding may be severe. In myeloproliferative disease, the clonal defect gives rise to functionally abnormal platelets, and in myeloma platelets become coated with immunoglobulin, which blocks surface receptors and prevents platelet aggregation.
Congenital disorders of platelet function
Hereditary platelet disorders causing life-threatening haemorrhage, such as those where the platelet glycoprotein receptors for von Willebrand factor (Fig. 23.8) (Bernard–Soulier syndrome) or fibrinogen (Glanzmann’s disease) are absent, are extremely rare autosomal recessive diseases. Life-threatening haemorrhage may occur. Mild defects of platelet function, causing easy bruising and bleeding after trauma, are more common. In some, a familial pattern is apparent. Various metabolic disturbances of platelets may be responsible, such as a deficiency of adenine nucleotides due to an abnormality of a type of platelet storage granule known as dense bodies (platelet storage pool deficiency).
Bleeding due to vascular disorders
Vascular disorders do not usually cause serious bleeding. Skin haemorrhage and occasional mucosal haemorrhage may occur. In some disorders, the collagen that supports vessel walls is abnormal. This mechanism probably accounts for the bruising of Cushing’s syndrome and the bruising, mucosal bleeding and perifollicular skin haemorrhages of scurvy (Ch. 7).
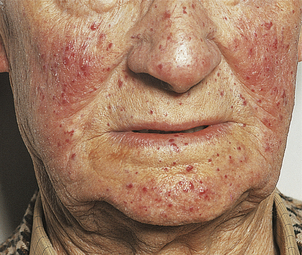
Hereditary haemorrhagic telangiectasia
Telangiectases (microvascular dilatations) accumulate from childhood on mucous membranes, in liver and lungs, and on the skin of hands and face (Fig. 23.45). The condition is inherited as an autosomal dominant trait and involvement of abnormalities in the genes expressing alk-1 or endoglin are described. Nosebleeds and gastrointestinal bleeding may be severe, but bleeding occurs only from telangiectases; coagulation and platelet numbers and function are normal.
Henoch–Schönlein purpura
In Henoch–Schönlein purpura no systemic bleeding tendency is present. It is an immune complex hypersensitivity reaction, usually in children. A rash, superficially similar to thrombocytopenic purpura but with localised oedema causing the lesions to be raised above the skin level, is present on buttocks and lower legs. Arthralgia, abdominal pain and haematuria may occur. It is usually self-limiting. Although the skin rash resembles thrombocytopenic purpura, the platelet count and skin bleeding time are normal.
Platelet disorders causing thrombosis
HUS and TTP have been described; however, the abnormality causing increased platelet reactivity does not lie within the platelet itself in these disorders. Thrombosis is a feature of myeloproliferative disease with thrombocytosis, especially essential thrombocythaemia and polycythaemia rubra vera. Laboratory evidence for increased platelet reactivity can be found in subjects with coronary thrombosis, cerebral thrombosis, diabetes mellitus and other disorders.
Paradoxically, the heparin-induced immune thrombocytopenia that occasionally develops on exposure to this anticoagulant drug may be associated with extensive arterial and venous thrombosis, due to platelet activation by the auto-antibody (p. 659).
DISORDERS OF BLOOD COAGULATION
Coagulation or fibrinolytic disorders may cause thrombosis or haemorrhage
Can be congenital or acquired
Acquired disorders are common, due to anticoagulant drugs, vitamin K deficiency, liver disease and disseminated intravascular coagulation
The most important congenital bleeding disorders are von Willebrand disease and haemophilia
Diseases of the coagulation/fibrinolytic system causing thrombosis as well as those causing haemorrhage are recognised. In practice, the majority of bleeding disorders are acquired and due to anticoagulant drugs or to liver disease, or to clotting factor consumption in disseminated intravascular coagulation. Vitamin K deficiency in the neonatal period has become uncommon due to routine vitamin K administration in neonates; where this is omitted the child is at risk of developing haemorrhagic disease of the newborn which may result in catastrophic intracranial haemorrhage. The severe congenital haemorrhagic diatheses are uncommon but clinically important disorders due to inherited defects of production of a coagulation factor.
Congenital clotting factor deficiencies
Deficiencies of most of the coagulation factors have been described, but deficiency of factor VIII (haemophilia A) and factor IX (haemophilia B or Christmas disease), and of von Willebrand factor (von Willebrand disease) are the clinically most important disorders in the group. Haemophilia A and B are X-linked conditions, whereas deficiencies of the other coagulation factors are autosomal recessive conditions. The latter are therefore rare, but are seen more commonly in populations where consanguineous marriage is a feature.
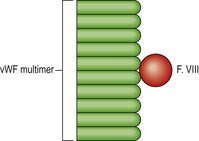
Factor VIII is a co-factor for factor IX allowing activation of factor X and therefore fibrin generation via the intrinsic coagulation pathway (Fig. 23.9); it is probably synthesised predominantly by hepatocytes. In order to circulate in plasma with a normal half-life of 12 hours it requires a carrier protein—von Willebrand factor (vWF) (Fig. 23.46). vWF is a large multimeric protein synthesised and assembled by vascular endothelial cells and megakaryocytes. It has no role in the coagulation cascade but is an essential co-factor for interaction of platelets with exposed subendothelium in primary haemostasis (Fig. 23.8). It has binding sites for factor VIII, platelet glycoprotein Ib and collagen, and thus its role in haemostasis is apparent.
Haemophilia
Haemophilia A and B are identical clinically and pathologically, differing only in the deficient factor. In each case the disorder is due to sex-linked recessively inherited deficiency of the clotting factor, or synthesis of a defective clotting factor. Males are affected. Female carriers have approximately 50% of the normal factor level and may occasionally be mildly clinically affected as a result of the process of lyonisation toward the abnormal X chromosome. Mild, moderate and severe forms of haemophilia are recognised, depending on the residual clotting factor activity (Table 23.11); degree of severity is constant within a kindred. Predictably, from the place of factors VIII and IX in the coagulation mechanism, the APTT is prolonged and the PT is normal
Table 23.11 Classification of haemophilia A
| Category |
Factor VIII % of normal |
Features |
| Severe |
<1 |
Frequent and spontaneoushaemorrhage into jointsand soft tissues from birth Degenerative joint disease |
| Moderate |
1–5 |
Bleeding after trauma,including dental and othersurgical trauma |
| Mild |
>5–30 |
Bleeding after trauma only May be subclinical inmildest form |
The pattern of bleeding is of the coagulation factor deficiency type (Table 23.10). Purpura is not a feature. In severe disease, bleeding from wounds persists for days or weeks. Control can be achieved by coagulation factor replacement by the intravenous route, but this must be administered 12-hourly or by continuous infusion to maintain adequate factor VIII or IX levels. Modern management of severe haemophilia is by prophylaxis administered two or three times per week to convert severe into moderate phenotypes.
Molecular genetics
Molecular genetic studies have recognised a variety of defects in haemophilia, including large deletions of the factor VIII gene, as well as single base changes, which create either a translational stop signal (so-called nonsense mutations), with the consequent synthesis of a truncated protein that is ineffective functionally and rapidly degraded, or single amino acid substitutions that alter the stability and function of the proteins. Inversions in intron 22 and intron 1 are common causes of severe haemophilia A. Large deletions are less common in haemophilia B. Mutations occurring de novo account for a substantial proportion of affected subjects (around 30%).
Clinical features and treatment
Spontaneous haemorrhage into a major joint, especially knees, hips, elbows and shoulders, occurring several times each month is typical of the untreated severe disease. Without factor replacement therapy bleeding continues until the intra-articular pressure rises sufficiently to prevent further haemorrhage. Slow resolution of this exquisitely painful acute haemarthrosis then occurs. Recurrent bleeds within a joint produce massive synovial hypertrophy, erosion of joint cartilage and para-articular bone and changes of a severe osteoarthritis (Ch. 25).
Bleeding into muscles (Fig. 23.47), retroperitoneal tissues and the urinary tract also occurs. Pressure necrosis of adjacent structures such as peripheral nerves may result.
The clinical picture in severe haemophilia is one of recurrent spontaneous haemarthrosis and soft tissue haemorrhage from around 6 months onwards. External bleeding from the urinary tract and epistaxis are also common. By self-administration of clotting factor concentrate at the first symptoms of haemorrhage, or use of prophylactic factor replacement, many of the disabling consequences of haemophilia can now be avoided. The life expectancy in severe haemophilia rose spectacularly as a result of the introduction of coagulation factor concentrates but replacement therapy with these products prepared from pooled human plasma has led to other diseases in subjects receiving such treatment due to virus transmission. Liver disease due to hepatitis C (Ch. 16) is universal in haemophiliacs treated with concentrates prior to the introduction of virucidal preparation using heat and solvents. This disease may be progressive, with changes of cirrhosis and hepatocellular carcinoma eventually ensuing and resulting in the death of a proportion of patients. Many haemophiliac subjects have also contracted hepatitis B, although only 5% of them are persistently HBsAg positive.
Many UK haemophiliacs were infected with HIV and developed the pathological features of the acquired immune deficiency syndrome (AIDS), including Pneumocystis jiroveci pneumonia, toxoplasmosis and systemic candidiasis. Modern virucidally treated factor concentrates appear to be free of hepatitis viruses and HIV, and recombinant factor VIII and IX is now available. The description of transmission of variant Creutzfeldt–Jakob disease (vCJD) by blood transfusion has alerted clinicians to the possibility of passage of this agent by pooled plasma products, although no case has yet been described in an individual with haemophilia.
In around 12% of severe haemophilia A and less than 1% of haemophilia B patients, exposure to therapeutic factor VIII or IX leads to development of antibody to the clotting factor. The presence of such inhibitors makes management of affected individuals very difficult, as infused clotting factor is rendered ineffective by the antibody. Rarely subjects without a congenital bleeding disorder develop comparable clotting factor inhibitors, usually directed against factor VIII. The factor VIII concentration in plasma is markedly reduced and bleeding is often spontaneous and life-threatening in these cases of acquired haemophilia.
Other coagulation factor deficiencies
Deficiency of factor XII is common. Although the APTT is prolonged, there is no bleeding tendency, as factor XII is not essential for normal coagulation in vivo (p. 616). Factor XI deficiency is a relatively common disorder in some populations. Again, the APTT is prolonged but the bleeding tendency is variable and often subclinical. Recessively inherited deficiencies of factor V, VII, X and XIII are very uncommon but severe bleeding diathesis often presents with umbilical stump or intracranial bleeding in the neonatal period.
von Willebrand disease
von Willebrand disease (vWD) is most commonly a mild bleeding disorder. It is due to synthesis of vWF in reduced amounts or production of functionally abnormal vWF. It is transmitted as an autosomal dominant disorder in most kindred, and epidemiological studies suggest it may be common in mild or subclinical form.
The majority of cases have a quantitative deficiency of vWF (Type 1), but in around 20% a dysfunctional vWF is synthesised and in these the multimeric structure of vWF (Fig. 23.46) is often abnormal, with a reduction in the large multimers (Type 2A and 2B). Types 2M and 2N have a normal multimeric pattern but have abnormalities in binding to platelets and factor VIII respectively. There is marked genetic heterogeneity. The homozygous disease (Type 3) is a serious bleeding diathesis but is extremely uncommon. In the more usual heterozygous form the main manifestations are easy bruising, bleeding after trauma and menorrhagia in females. Haemarthrosis and muscle haematomas are not common features of vWD. The plasma concentration of vWF is reduced, and, because of the requirement for vWF as a carrier protein, factor VIII activity is reduced in parallel; levels of less than 10–20% of normal are unusual, however. The bleeding time is prolonged due to the defect of platelet interaction with exposed subendothelium which arises from a reduced availability of vWF. The APTT is prolonged due to factor VIII deficiency. vWF-rich concentrate prepared from plasma is used for treatment of severe haemorrhage. In many patients the vWF level can be temporarily increased by stimulating its release from endothelial cells by administration of an analogue of vasopressin—desmopressin. This is also effective in mild haemophilia.
Acquired disorders of coagulation
Bleeding due to acquired platelet disorders has been described above. A haemorrhagic diathesis due to coagulation factor deficiency is present in liver disease, disseminated intravascular coagulation and vitamin K deficiency due to immaturity (haemorrhagic disease of the newborn), obstructive jaundice, pancreatic disease or small bowel disease.
Bleeding may also be a feature of therapy with anticoagulant and fibrinolytic drugs and is occasionally due to the development of an acquired inhibitor to factor VIII which develops in isolation or in individuals with B-cell malignancy, autoimmune disease or rarely pregnancy.
Vitamin K deficiency
Vitamin K is obtained from green vegetables and by bacterial synthesis in the gut. It is a fat-soluble vitamin and requires bile for its absorption. Vitamin K is essential for the gamma-carboxylation of clotting factors II, VII, IX and X (and the natural anticoagulants protein C and protein S); in the absence of vitamin K, these factors are released from the liver in an incomplete and inactive form. The PT is particularly prolonged as a result.
A coagulopathy due to vitamin K deficiency occurs when absorption is defective, particularly in obstructive jaundice. In addition, the neonate tends to have vitamin K deficiency due to poor transplacental passage of maternal vitamin K, lack of gut bacteria and low concentrations of the vitamin in breast milk. This exacerbates the inefficient coagulation resulting from low levels of clotting factors secondary to liver immaturity and may produce life-threatening haemorrhage during the first week of life—haemorrhagic disease of the newborn. Vitamin K supplementation corrects the defect.
The oral anticoagulant drug warfarin is a vitamin K antagonist and acts by inhibiting the complete synthesis of coagulation factors II, VII, IX and X. Its use is therefore associated with an increased risk of haemorrhage.
Liver disease
Severe hepatocellular disease is commonly associated with coagulation defects due to failure of clotting factor synthesis, including fibrinogen, and production of abnormal fibrinogen—dysfibrinogenaemia. This is often compounded by thrombocytopenia due to hypersplenism, which complicates portal hypertension, and a qualitative platelet disorder. The skin bleeding time may be prolonged, as is the PT, and often the APTT. Life-threatening haemorrhage may result, particularly from oesophageal varices.
Disseminated intravascular coagulation
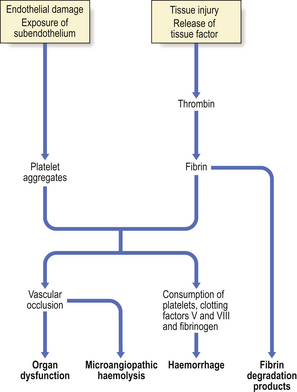
Disseminated intravascular coagulation (DIC) is a common state in which a combination of haemorrhage and thrombosis complicates another disorder. Activation of coagulation leads to the formation of microthrombi in numerous organs and to the consumption of clotting factors and platelets in the process of clot formation, in turn leading to a haemorrhagic diathesis. There are several potential triggers to coagulation activation in DIC (Fig. 23.48) and a wide range of disorders can be complicated by this phenomenon:
•
infection (e.g. septicaemia, malaria)
•
neoplasm (e.g. mucin-secreting adenocarcinoma)
•
tissue trauma (e.g. burns, major accidental trauma, major surgery, shock, intravascular haemolysis, dissecting aortic aneurysm)
•
obstetric complications (e.g. abruptio placentae, retained dead fetus, amniotic fluid embolism, toxaemia)
The pathogenesis of DIC is complex and centres on the enhanced generation of thrombin. Factors that contribute to the process include increased tissue factor expression, suboptimal function of natural anticoagulants, dysregulation of fibrinolysis and the increased availability of anionic phospholipids. Thus, in obstetric disorders, tissue factor release into the maternal circulation from the placenta or fetus, or in amniotic fluid, may trigger coagulation. Many tumours are also rich in procoagulant substances. Septicaemia may also cause coagulation activation by damage to vascular endothelium and induction of tissue factor expression by endothelial cells and monocytes. In liver disease there is reduced clearance of activated clotting factors.
Dysregulated fibrinolytic activity results in the production of plasmin which causes digestion of fibrin and fibrinogen-generating split products (fibrinogen and fibrin degradation products; FDP), which themselves have an anticoagulant and anti-platelet effect and contribute to the haemorrhagic diathesis (Fig. 23.48).
Thrombi, composed of platelets and fibrin, may be found in the microvasculature of brain, lungs, kidneys, heart, spleen and liver. Other organs may also be affected. The distribution of affected organs is variable. Micro-infarcts or more major areas of infarction such as renal cortical necrosis or hepatic necrosis may result. Areas of haemorrhage may also be apparent histologically and on gross examination; any organ may be affected.
The blood platelet count is often low and the PT and APTT prolonged; the fibrinogen concentration may be reduced. Coagulation factors V and VIII are consumed during fibrin generation, and plasma levels may be severely reduced. FDP are present in high concentration. Red cells become damaged as they pass through partially occluded small vessels, and the blood changes of a microangiopathic haemolytic anaemia may be present. The above changes are present in florid DIC. In some cases the course is more chronic and the blood changes considerably more subtle.
Clinical features
These include haemorrhage, which may be torrential, multi-organ failure due to ischaemia, and haemolytic anaemia. Bleeding is from mucous membranes, and into skin, serosal cavities and internal organs. Organ dysfunction may manifest as hepatic or renal failure, neurological disturbance or cardiac and respiratory failure. In some cases a predominantly haemorrhagic picture dominates; in others, thrombotic peripheral ischaemia and gangrene are the major features (Fig. 23.49). Chronic ‘low-grade’ DIC may be only mildly symptomatic. Treatment is largely removal of the underlying cause and clotting factor and platelet replacement. Mortality is high in severe cases.
Coagulation disorders associated with a thrombotic tendency
Familial thrombophilia
Control mechanisms for the prevention of inappropriate fibrin deposition are an important feature of the coagulation system. These mechanisms include the rapid lysis of fibrin by plasmin generated at the site of a thrombus, neutralisation of thrombin by antithrombin, and inactivation of factors Va and VIIIa by activated protein C with its co-factor, protein S (Fig. 23.9). Hereditary defects of these control mechanisms have been described which lead to a life-long tendency to thrombosis—thrombophilia. The thrombosis in these familial thrombophilic states is almost always in the venous system: deep venous thrombosis of the limbs and pulmonary embolism. Thrombotic events rarely manifest before adulthood and usually occur when a second risk factor is also present, commonly pregnancy, exposure to female hormones in the combined oral contraceptive or hormone replacement therapy, immobilisation or surgery. The recognised familial abnormalities associated with such a thrombotic tendency are:
•
antithrombin deficiency resulting in failure of thrombin neutralisation
•
protein C and protein S deficiency resulting in failure of neutralisation of activated factors Va and VIIIa
•
activated protein C resistance, the commonest cause of familial thrombophilia, where a point mutation in the factor V gene leads to synthesis of a factor V variant that has normal procoagulant activity but which is not
inhibited by activated protein C; this variant, called factor V Leiden, is present in 5% or more of northern European subjects
•
a point mutation in the
prothrombin gene resulting in an increased plasma concentration of prothrombin, present in around 2% of northern Europeans
•
dysfibrinogenaemia resulting in abnormal fibrinogen.
Individuals with antithrombin deficiency are heterozygous for the defect. Over 30 mutations leading to deficiency of the protein have been discovered, mostly caused by frameshifts or base changes resulting in a protein that is not secreted or is rapidly removed from the circulation. This type of deficiency is designated ‘Type 1’. In Type 2 a dysfunctional protein is produced due to one of several single base changes that alter the amino acid sequence of the synthesised antithrombin.
Subjects deficient in protein C or protein S are also heterozygous, the homozygous condition producing a severe thrombotic disease often manifesting in the neonate. Type 1 and Type 2 (dysfunctional) defects are also found in protein C deficiency. Several mutations in the genes for protein C and protein S have been identified.
One or more of these genetic thrombophilias, most commonly factor V Leiden, can be found in around 30% of subjects with deep vein thrombosis, and affected family members are also at increased risk of venous thromboembolism compared with the background population.
Acquired prothrombotic states
Acquired coagulation disorders causing thrombosis are also recognised. In systemic lupus erythematosus (SLE) a predisposition to arterial and venous thrombosis is associated with a paradoxical prolongation of the APTT, apparently due to the development of auto-antibodies which interact with phospholipid-bound proteins involved in coagulation activation (‘anti-phospholipid antibodies’). Stroke and deep venous thrombosis are common. These antibodies are also associated with major thrombotic disease in subjects without other evidence of SLE. Women with such antibodies are prone to pregnancy failure due to recurrent miscarriage, possibly secondary to placental thrombosis or poor implantation.The term antiphospholipid syndrome describes those patients with thrombosis or pregnancy failure associated with persistent antiphospholipid antibody. The mechanisms underlying thrombosis in this disorder are not yet known. Long-term therapy with anticoagulant drugs is often indicated, as the risk of recurrent thrombosis is high. The antibodies can be detected in the laboratory through the ability to prolong clotting times in coagulation tests that involve low concentrations of phospholipid, when the term lupus anticoagulant is used, or through binding to negatively charged phospholipid, such as cardiolipin—the anticardiolipin antibody.
Thrombosis risk is also increased in a range of other conditions, including myeloproliferative diseases (p. 664), cancer, nephrotic syndrome, congestive cardiac failure, atrial fibrillation and paroxysmal nocturnal haemoglobinuria (p. 651).
An increased plasma concentration of homocysteine is also associated with a tendency to thrombosis. The homocysteine level is partly genetically determined, but is also influenced by the dietary content of vitamin B12, folate and pyridoxine (Fig. 23.19). This raises the intriguing possibility of reducing thrombosis risk by dietary manipulation and this approach is currently under investigation.
ANTENATAL DIAGNOSIS OF BLOOD DISORDERS
Several of the more serious haematological disorders can be diagnosed in the fetus. Fetal blood can be obtained from the placenta or umbilical vein with imaging using ultrasound scanning techniques from around the middle of the second trimester. This material can be used for detection of abnormalities of red cells, white cells or platelets and for diagnosis of clotting factor deficiencies. For example, thalassaemia can be identified by measuring relative rates of globin chain synthesis. Alternatively amniotic fluid cells can be obtained by aspiration of the amniotic fluid, and techniques have been developed by which fetal material can be safely obtained by biopsy of chorionic villi, this being performed as early as 9–11 weeks’ gestation.
Fetal DNA can be analysed by restriction endonuclease mapping and RFLP linkage analysis but increasingly direct sequencing for known mutations is used. Using these techniques, first trimester prenatal diagnosis of beta-thalassaemias and of haemophilia has become possible. Gene probes for the diagnosis of red cell enzyme defects are also becoming available.
New non-invasive methods utilising the detection of fetal cells in maternal blood are now starting to be offered in clinical practice.
BLOOD TRANSFUSION
Donor blood, or fractions of blood, can be safely and beneficially administered intravenously. Transfusion of red cells is valuable in the management of some anaemias and in resuscitation after acute haemorrhage, and is essential for the safe performance of many surgical procedures. Other cellular components of blood, especially platelets, can be usefully transfused, for example to treat bleeding in severely thrombocytopenic subjects. Blood plasma is fractionated to provide albumin, immunoglobulin and coagulation factors or can be used as a complete entity as ‘fresh-frozen plasma’. In red cell transfusion it is essential that compatibility is ensured between antigens on the donor erythrocytes and antibodies present in the recipient’s plasma in order to avoid acute haemolysis of the donor cells which may be fatal. The cross-match procedure is used to determine compatibility. The red cells from the donor unit are incubated with the recipient’s serum under a range of conditions that enhance sensitivity to any antibody present. Agglutination or lysis of the red cells indicates the presence of clinically important red cell antibody and that the donor unit is incompatible and cannot be safely administered to the recipient.
Red cell antigens and antibodies
Although there are about 400 red blood cell antigens, most inherited in Mendelian dominant fashion, only a minority are clinically important. An individual lacking a particular antigen may develop an antibody after exposure to red cells carrying that antigen. Exposure occurs by transfusion of red cells or by passage of fetal red cells into the maternal circulation during pregnancy, the fetal cells carrying paternal antigens foreign to the mother. The important clinical consequences of the development of such an ‘immune’ antibody are the development of a haemolytic transfusion reaction on further exposure to red cells carrying the antigen, and haemolytic disease of the newborn due to transplacental passage of maternal IgG antibody against fetal red cell antigens.
The most important ‘immune’ antibody is anti-D, an antibody to the major antigen of the rhesus blood group system (Table 23.12). It is a major cause of haemolytic disease of the newborn.
Table 23.12 The rhesus blood group system
| Allelic genes at closely linked loci code for paired antigens designated C and c, E and e and D. Absence of D is termed d. A set of genes and hence antigens is inherited from each parent and the presence of D determines rhesus ‘positivity’. |
| Genotype |
Rhesus status |
% Frequency (UK) |
|
|
|
|
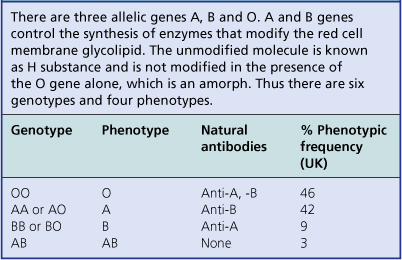
As well as ‘immune’ antibodies, ‘naturally occurring’ antibodies to red cell antigens are also important. In contrast to the IgG immune antibodies, they are predominantly IgM and require no previous red cell antigen exposure. They occur in the ABO blood group system, where naturally occurring anti-A and anti-B are present in subjects whose red cells lack the corresponding antigen (Table 23.13).
In addition to the ABO and rhesus blood group systems the major red cell antigen systems of clinical importance are Kell, Duffy and Kidd, as, with ABO and rhesus, their antibodies are responsible for most cases of haemolytic transfusion reaction.
Clinically important platelet antigens are also recognised. Human platelet antigen 1a (HPA1a) is present on platelets in around 98% of Caucasians. Subjects lacking the antigen are at risk of allo-antibody development from exposure to HPA1a platelets during pregnancy or blood transfusion. Anti-HPA1a is responsible for most cases of post-transfusion purpura and neonatal alloimmune thrombocytopenic purpura (p. 671).
Haemolytic transfusion reactions
Immediate reactions
Massive intravascular haemolysis occurs when complement-activating antibodies, such as anti-A and anti-B, interact with the relevant antigen on transfused red cells. There is typically collapse, with hypotension and pain in the lumbar region. Haemoglobin-stained urine may be passed and oliguric renal failure may ensue. Red cell lysis may trigger disseminated intravascular coagulation. This clinical scenario can develop after transfusion with only a few millilitres of incompatible red cells. Treatment includes immediate interruption of the transfusion, resuscitation with intravenous fluid, immunosuppression with corticosteroid therapy and management of the renal failure. Fatalities still occur. The cross-match procedure should prevent exposure to such incompatible blood. However most cases result from clerical error through mislabelling of the cross-match sample or transfusion to the wrong recipient.
Because antibodies to the rhesus system are not complement-fixing, cell lysis occurs in the reticulo-endothelial system and reactions are generally milder, although they can still be life-threatening.
Delayed reactions
Occasionally a low titre antibody is too weak to be detectable in the cross-match and is unable to cause lysis at the time of transfusion. Transfusion of red cells carrying the relevant antigen leads to a gradual increase in the titre of the antibody, developing over a period of a few days. Delayed, gradual red cell lysis occurs, producing anaemia and jaundice; this is a delayed transfusion reaction.
Other adverse effects of transfusion
Most nucleated cells, including leukocytes, carry antigens of the HLA (human leukocyte antigen) or major histocompatibility complex system. Prior exposure to human leukocyte antigens by transfusion or pregnancy may lead to development of antibody capable of causing fever and rigors on subsequent exposure to the antigens present on leukocytes in transfused blood. This can be avoided by using filters to remove donor leukocytes prior to transfusion. Such non-haemolytic transfusion reactions are unpleasant but rarely dangerous. Allergic reactions may also develop in a recipient because of hypersensitivity to a protein present in the donor plasma. Fever, urticaria and oedema may result.
Virus transmission by blood transfusion was previously a major problem but this has been largely overcome by processes of deferral of donors identified as being at increased risk for certain virus infections including the hepatitis viruses and HIV, combined with increasingly sensitive methods of testing donated blood for antibodies, antigens or nucleic acid indicative of infection with these agents. Nevertheless, transmission of viruses for which screening is not possible or feasible, such as parvovirus and cytomegalovirus, does occur and, although transmission rates for the previously mentioned agents are low, transmission can theoretically still occur. Increased foreign travel has led to more donor exposure to other pathogens and has resulted in the need to defer donors returning from parts of the world where infections such as malaria and West Nile fever are endemic. It is likely that the prion responsible for variant Creutzfeld–Jakob disease can be transmitted by transfusion of blood and as a result potential donors who have been previously transfused are now excluded.
Circulatory overload may result from transfusion of excessive volume. Because bank blood is devoid of functioning platelets, transfusion of large volumes can cause thrombocytopenia and haemorrhage.
Repeated red cell transfusion without blood loss, usually in the management of chronic anaemias such as thalassaemia, inevitably leads to tissue iron overload. A unit of blood contains 200mg of iron, in haemoglobin. Although, initially, deposition occurs in reticulo-endothelial tissues without toxic results, iron later accumulates in skin, liver, myocardium and pancreas. Pigmentation, liver cirrhosis (Ch. 16), heart failure and diabetes mellitus are the consequences. Iron chelating compounds, such as desferrioxamine and deferasirox, are administered by subcutaneous infusion to minimise iron accumulation in tissues.
Transfusion-related acute lung injury (TRALI), which presents with non-cardiac pulmonary oedema within 6 hours of transfusion, most commonly of fresh frozen plasma, is an immunological complication of transfusion mediated by anti-leukocyte antibodies. The main intervention is supportive care. Finally, transfusion-associated graft-versus-host disease is seen following the transfusion of competent lymphocytes into an immuno-incompetent recipient. The transfused cells engraft over around 10 days and then mount an immune attack on the donor due to histo-incompatibility which results in fever, skin rash, pancytopenia and liver failure, which is usually fatal. This occurs in patients with congenital and acquired cell-mediated immune dysfunction—for example, severe combined immunodeficiency (SCID), lymphoma, allogeneic bone marrow transplant and the use of purine analogue drugs. It is prevented by supplying these patients with irradiated cellular blood products.
Commonly confused conditions and entities relating to the blood and bone marrow
| Commonly confused |
Distinction and explanation |
| Pernicious and megaloblastic anaemia |
Pernicious anaemia is a specific example of megaloblastic anaemia in which autoimmune gastritis results in loss of intrinsic factor and failure to absorb vitamin B12. Megaloblastic anaemia can also be due to folate deficiency and to other causes of B12 deficiency. |
| Polycythaemia rubra vera and secondary polycythaemia |
Both are characterised by an increased concentration of red blood cells. Polycythaemia rubra vera is a clonal myeloproliferative condition, whereas secondary polycythaemia is usually a physiological response to hypoxia or to inappropriately excessive erythropoietin production. |
| Myelodysplasia, myelofibrosis and myeloproliferative diseases |
The myelodysplastic syndromes are neoplastic bone marrow disorders at the stem cell level, affecting all myeloid lines, with characteristic morphological changes in the marrow and a tendency to progress to acute leukaemia. Myelofibrosis is characterised by marrow fibrosis and the emergence of extramedullary haemopoiesis. Myeloproliferative diseases, a group of disorders with abnormal proliferation of one or more cell lines, including myelofibrosis, chronic myeloid leukaemia, polycythaemia rubra vera and essential thrombocythaemia, are grouped together because intermediate forms exist and progression from one to another is common. |
FURTHER READING
Hehlmann R., Hochhaus A., Baccarani M.. European LeukemiaNet. Chronic myeloid leukaemia. Lancet. 2007;370:342-350.
Hoffbrand A.V., Tuddenham E.G., Catovsky D., editors. Postgraduate haematology, 5th edn, Oxford: Blackwell Publishing, 2005.
Hoffbrand A.V., Pettit J., Moss P., editors. Essential haematology, 5th edn, Oxford: Blackwell Publishing, 2006.
Melo J.V., Barnes D.J.. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441-453.