Chapter 26 Central and peripheral nervous systems
COMMON CLINICAL PROBLEMS FROM CENTRAL AND PERIPHERAL NERVOUS SYSTEM DISEASE
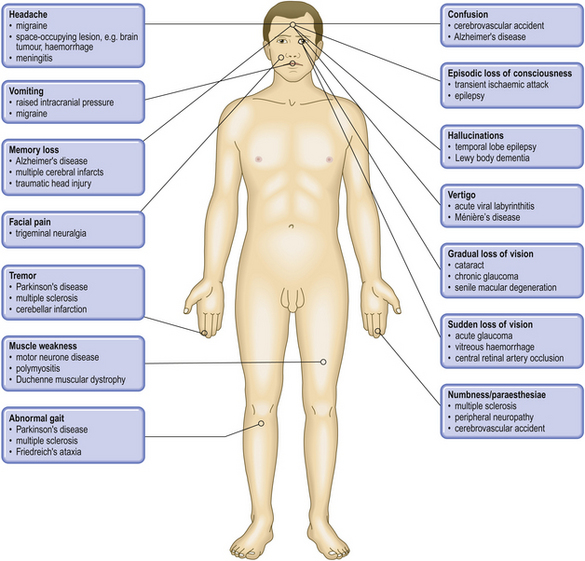
Pathological basis of neurological signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Headache | |
| Neck stiffness | |
| Coma or impaired consciousness | Metabolic, e.g.: Brainstem lesions, e.g.: Cerebral hemisphere lesions, e.g.: |
| Dementia | Loss of limbic or cortical neurones due to ischaemia, toxic injury or neurodegenerative disease, e.g. Alzheimer’s disease |
| Epileptic fits | Paroxysmal neuronal discharges, either idiopathic or emanating from a focus of cortical disease or damage |
| Abnormal reflexes | |
| Muscle deficit | |
| Disease directly or indirectly affecting function of: | |
| Sensory impairment and/or paraesthesiae | Disease directly or indirectly affecting function of: |
| Visual field defects or blindness | Disease involving the eyes, optic nerves and pathway or visual cortex (e.g. cataracts, tumours (intrinsic or extrinsic to optic neural pathway), inflammation or demyelination in the optic pathway, retinopathy, ischaemia) |
| Tinnitus and/or deafness | Impaired transmission of sound through external meatus (e.g. wax) or through middle ear ossicles, or disease affecting the organ of Corti or the auditory nerve |
CENTRAL NERVOUS SYSTEM
NORMAL STRUCTURE AND FUNCTION
The central nervous system (CNS) is the most anatomically complex system in the body, able to function both as a self-contained unit and as the control unit that co-ordinates the activities of the peripheral nervous system (PNS), skeletal muscle and other main organ systems.
The CNS is composed of three principal structures: the brain, brainstem and spinal cord. The brain comprises two hemispheres which are joined by a band of white matter fibres known as the corpus callosum. The grey matter known as the cerebral cortex is located on the outer surface of the hemispheres, and is composed of six layers of neurones. The cerebral cortex is divided into four anatomical regions: the frontal, temporal, parietal and occipital lobes. Each of these has distinct functions, which are summarised in Figure 26.1. The white matter beneath the cerebral cortex is composed of axons which connect the cortical neurones with neurones in other grey matter regions, including the opposite hemisphere. In the centre of the hemispheres there is a complex series of grey matter nuclei known as the basal ganglia, the thalamus and the hypothalamus. Their principal functions are summarised in Table 26.1. The cerebellum is located at the posterior surface of the brainstem, to which it is connected by white matter fibre bundles. The cortex of the cerebellum lies on its outer surface, but its structure is different from that of the cerebral cortex. The function of the cerebellum is summarised in Table 26.1.
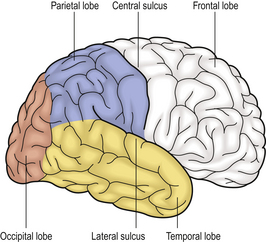
Fig. 26.1 Location and function of the lobes of the cerebral cortex. The frontal lobe is responsible for voluntary movement, intellect, personality and memory. Sensation is appreciated in the parietal cortex, which also has a major role in reading, speech and writing. The temporal lobe has an important role in memory, mood and hearing; the main function of the occipital lobe is vision.
Table 26.1 Functions of the basal ganglia, thalamus, hypothalamus and cerebellum
| Structure | Functions |
|---|---|
| Basal ganglia | |
| Thalamus | |
| Hypothalamus | |
| Cerebellum |
The brainstem contains many ascending and descending white matter fibre bundles which connect the spinal cord to the brain; however, it also contains many nuclei, including cranial nerves 3–12, the substantia nigra, the respiratory centre and the vomiting centre. The spinal cord is largely composed of ascending and descending white matter fibre bundles, such as the corticospinal pathways (descending motor fibres) and the posterior columns (ascending sensory fibres). The grey matter of the spinal cord is located in the centre, and contains several groups of neurones, including the anterior horn cells, which are the lower motor neurones supplying all the skeletal muscle in the trunk and limbs. Motor nerve roots leave the anterior spinal cord to form peripheral motor nerves; sensory nerves from the skin, joints and organs enter the spinal cord by the posterior nerve roots, and then pass into the ascending posterior columns.
Despite the structural and functional complexities of the CNS, the constituent cells can be divided into just five main groups:
Neurones
Neurones are the structural and functional units of the CNS, generating electrical impulses that allow rapid cell–cell communication at specialised junctions known as synapses (Fig. 26.2). Many millions of neurones are present, arranged in layers within the cortex on the surface of the cerebellum and the cerebral hemispheres. Groups of functionally related neurones within the subcortical grey matter are known as nuclei (Table 26.1). Neurones are highly specialised post-mitotic cells which cannot be replaced after cell death. They are subject to unique metabolic demands, having to maintain an axon (which may be up to 1m in length) by intracellular transport. This makes neurones particularly vulnerable to a wide range of insults, principally hypoxia and hypoglycaemia.
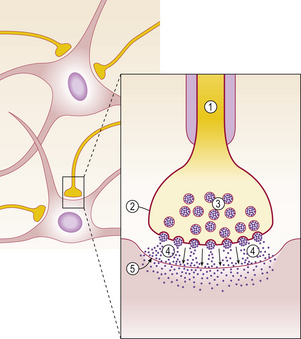
Fig. 26.2 Neuronal signal transmission at synapses. The transmission of the action potential down the axon (1) to the presynaptic membrane (2) results in opening of Ca ion channels, producing an influx of calcium. The subsequent phosphorylation of calcium-binding proteins allows the synaptic vesicles (3) to bind to the presynaptic membrane and release their neurotransmitter contents into the synaptic cleft (4). The neurotransmitters diffuse across the synaptic cleft and bind to receptors in the post-synaptic membrane (5), causing membrane depolarisation and eventually the formation of another action potential.
Neurones contain ion channels within the cell membrane that can be opened by either changing the voltage across the membrane or by the binding of a chemical (neurotransmitter) to a receptor in or near the ion channel. In the resting state, the neuronal cell membrane is relatively impermeable to ions. Opening of the ion channels allows an influx of sodium ions which depolarises the membrane, forming an action potential which is transmitted rapidly down the axon by saltatory conduction. Cell to cell transmission occurs at the synapse (Fig. 26.2). The commonest excitatory neurotransmitter in the CNS is glutamate. Excessive release of glutamate under certain conditions such as cerebral ischaemia and epilepsy can result in excitotoxic neuronal cell death.
Neurones, or nerve cells, vary considerably in size and appearance within the CNS. All possess a cell body, axons and dendrites.
The cell body or perikaryon is easily seen by light microscopy (Fig. 26.3). It contains neurofilaments, microtubules, lysosomes, mitochondria, complex stacks of rough endoplasmic reticulum, free ribosomes and a single nucleus with a prominent nucleolus. Some groups of neurones contain the pigment neuromelanin and are readily identifiable with the naked eye as darkly coloured nuclei, e.g. in the substantia nigra.
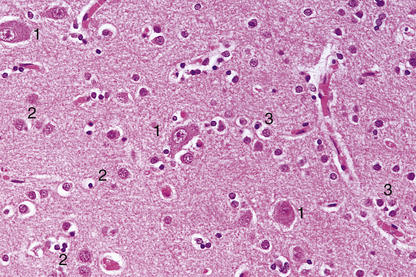
Fig. 26.3 Normal cerebral cortex. Figure shows the normal arrangement of neurones (1), astrocytes (2), oligodendrocytes (3) and capillaries in the cerebral cortex. Although the neuronal perikarya are visible, the cytoplasm of glial cells is best demonstrated by using special histological techniques.
Axons and dendrites are the neuronal processes that convey electrical impulses from and towards the perikaryon respectively. These processes vary enormously in size and complexity, and may be difficult to identify on routine microscopy.
Glia
Glia are specialised supporting cells of the CNS comprising four main groups:
Astrocytes are process-bearing cells which are poorly visualised by light microscopy (Fig. 26.3) unless special staining techniques are used. They perform several important roles:
Oligodendrocytes are the most numerous cells in the CNS. On light microscopy, they are visible as darkly staining nuclei located around neurones and nerve fibres (Fig. 26.3). The most important function of oligodendrocytes is the synthesis and maintenance of myelin in the CNS.
Ependymal cells form the single-cell lining of the ventricular system and the central canal of the spinal cord. They are short columnar cells that bear cilia on the luminal surface. Ependymal cells may participate in the absorption and secretion of cerebrospinal fluid (CSF).
Choroid plexus cells secrete CSF and contain large quantities of mitochondria, rough endoplasmic reticulum and Golgi apparatus within the cytoplasm. They form a cuboidal epithelial covering over the ventricular choroid plexus, and bear atypical microvilli.
Microglial cells
Microglia belong to the macrophage/monocyte system of phagocytic cells. They are normally quiescent, and inconspicuous on light microscopy, but are of major importance in reactive states, for example in inflammatory and demyelinating disorders.
Connective tissue
Connective tissue in the CNS is confined to two main structural groups: the meninges and perivascular fibroblasts.
The meninges comprise the pia, arachnoid and dura mater, and the arachnoidal granulations which are the main sites of CSF absorption. The meninges are composed of fibroblast-like cells which also extend around meningeal and cerebral blood vessels.
Blood vessels
Blood vessels in the CNS are similar in structure and function to those elsewhere in the body, with the important exception of the capillaries. The capillaries within the CNS differ from most other capillaries in several respects:
These special structural features are important constituents of the blood–brain barrier: this is a functional unit which restricts the entry and exit of many substances—including proteins, ions, non-lipid-soluble compounds and drugs—to and from the CNS.
REACTIONS OF CNS CELLS TO INJURY
 Axonal damage results in central chromatolysis in neuronal perikarya, with anterograde degeneration of the damaged axon
Axonal damage results in central chromatolysis in neuronal perikarya, with anterograde degeneration of the damaged axonNeurones
Neurones can undergo various reactive changes to cell injury:
Central chromatolysis is a distinctive reaction which usually occurs in response to axonal damage (Fig. 26.4). This reaction is maximal at around 8 days following axonal damage, and is accompanied by increased RNA and protein synthesis, suggestive of a regenerative response.
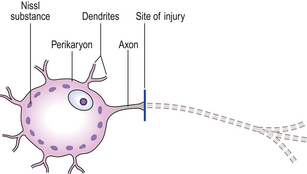
Fig. 26.4 Response to axonal injury: central chromatolysis and anterograde degeneration. Following axonal injury, the perikaryon swells and the nucleus migrates peripherally. The Nissl substance is dispersed to the periphery of the perikaryon, hence the term ‘central chromatolysis’. Anterograde degeneration of the axon occurs distal to the site of injury.
Anterograde degeneration occurs as a result of axonal transection, and is usually accompanied by central chromatolysis (Fig. 26.4). Degeneration of the distal part of the axon will occur following its separation from the intact perikaryon, e.g. by transection. Within 4 days, the distal segment degenerates and becomes fragmented. The myelin sheath surrounding the axon also fragments, but this usually occurs only after axonal degeneration is established. Axonal and myelin debris is then phagocytosed by macrophages, which often remain around the site of injury for several months. Attempts at axonal regeneration do not occur to a significant extent in the CNS.
Atrophy of neurones occurs in many slowly progressive degenerative disorders, e.g. motor neurone disease. Such neurones appear shrunken, and often contain excess lipofuscin pigment. Trans-synaptic atrophy occurs in neurones following loss of the main afferent connections, e.g. in neurones of the lateral geniculate body following damage to the optic nerve or retina.
Astrocytes
Astrocytes undergo hyperplasia and hypertrophy following almost all forms of CNS damage, in a response known as ‘reactive gliosis’. Gliotic tissue is translucent and firm, often forming a limiting barrier to sites of tissue damage, for example at the edge of a cerebral infarct.
DYSFUNCTION OF THE CNS
Diseases of the CNS impair the highly complex integration that is necessary for normal neurological function. The resulting clinical abnormalities can often indicate the anatomical basis of the lesion in the CNS, and this can be investigated in greater detail by imaging of the CNS by MRI scanning. The pathological basis box (p. 749) gives an introduction to some common clinical abnormalities and their pathological basis in the CNS.
INTRACRANIAL SPACE-OCCUPYING LESIONS
Focal brain swelling may be due to inflammatory, traumatic, vascular or neoplastic lesions, and is often accompanied by oedema in the adjacent tissueIntracranial space-occupying lesions may result from a variety of causes, but all share one common feature: an expansion in volume of the intracranial contents. Such brain swelling may be either diffuse or focal.
Diffuse brain swelling
Diffuse brain swelling denotes a generalised increase in the volume of the brain which results from either vasodilatation or oedema.
Vasodilatation
Vasodilatation in the brain occurs following changes in the calibre of intracerebral vessels that cause an increase in cerebral blood volume resulting in brain swelling. This occurs particularly in response to hypercapnia and hypoxia, but may also result from failure of the normal vasomotor control mechanisms, for example in severe head injuries.
Oedema
Oedema in the brain is defined as an abnormal accumulation of fluid in the cerebral parenchyma that produces an increase in cerebral volume. Cerebral oedema can be classified into three main types:
In many instances, cerebral oedema occurs due to a combination of mechanisms; for example, both vasogenic and cytotoxic mechanisms are involved in ischaemia. Cerebral oedema frequently accompanies focal lesions in the brain, thereby exaggerating the mass effect.
Focal brain swelling
Focal lesions of many types can produce an increase in cerebral volume, for example cerebral abscesses, intracranial haematomas and intrinsic neoplasms. Many extrinsic intracranial lesions, for example subdural haematomas and meningiomas, exert a mass effect within the cranial cavity and so act as space-occupying lesions.
Consequences of intracranial space-occupying lesions
The consequences of intracranial space-occupying lesions may be:
Raised intracranial pressure
Raised intracranial pressure is an invariable consequence of enlarging intracranial lesions, as there is very little space within the rigid cranium to accommodate an expanding mass. Initially, however, there is a phase of spatial compensation, made possible in three ways:
Once this phase is passed, there is a critical period in which a further increase in the volume of the intracranial contents will cause an abrupt increase in intracranial pressure. The characteristic clinical signs and symptoms of raised intracranial pressure and their likely causes are:
Intracranial shift and herniation
Intracranial shift and herniation are the most important consequences of raised intracranial pressure due to space-occupying lesions. They usually occur following a critical increase in intracranial pressure, which may inadvertently be precipitated by withdrawing CSF at lumbar puncture. Lumbar puncture is therefore contraindicated in any patient with raised intracranial pressure and a suspected intracranial space-occupying lesion to avoid the risk of precipitating a potentially fatal brainstem herniation.
Lateral shift of the midline structures is a common early complication of intracranial space-occupying lesions. However, patients with acute lateral displacement of the brain due to a hemispheric mass show a depressed level of consciousness even in the absence of an intracranial herniation. The clinical features are summarised in Table 26.2.
Table 26.2 Clinical consequences of intracranial herniation
| Site of herniation | Effect | Clinical consequence |
|---|---|---|
| Transtentorial | ||
| Foramen magnum | Brainstem compression and haemorrhage | |
| Acute obstruction of CSF pathway |
Herniations occur at several characteristic sites within the cranial cavity, depending on the site of the space-occupying lesion (Fig. 26.5). Transtentorial herniation is frequently fatal because of secondary haemorrhage into the brainstem (Fig. 26.6). This is a common mode of death in patients with large intrinsic neoplasms or intracranial haemorrhage.
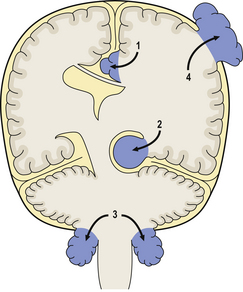
Fig. 26.5 Sites of intracranial herniation. Space-occupying lesions in the cerebral hemispheres may cause herniation of the cingulate gyrus under the falx cerebri (1) or of the hippocampal uncus and parahippocampal gyrus over the tentorium cerebelli (2). Cerebellar tonsillar herniation through the foramen magnum (3) can occur with lesions in the cerebrum or cerebellum. A swollen brain will herniate through any defect in the dura and skull (4).
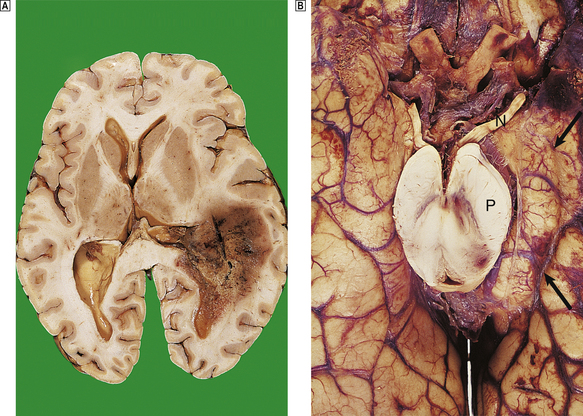
Fig. 26.6 Herniation effects in the brain.  A large haemorrhagic neoplasm (glioblastoma) is present in the right cerebral hemisphere, causing shift of the midline structures to the left and compression of the right lateral ventricle.
A large haemorrhagic neoplasm (glioblastoma) is present in the right cerebral hemisphere, causing shift of the midline structures to the left and compression of the right lateral ventricle.  Transtentorial herniation at the base of the brain. A prominent groove surrounds the displaced parahippocampal gyrus (arrow). The adjacent 3rd nerve (N) is compressed and distorted and the ipsilateral cerebral peduncle (P) is distorted with small areas of haemorrhage.
Transtentorial herniation at the base of the brain. A prominent groove surrounds the displaced parahippocampal gyrus (arrow). The adjacent 3rd nerve (N) is compressed and distorted and the ipsilateral cerebral peduncle (P) is distorted with small areas of haemorrhage.
Epilepsy
Seizures (fits) may be focal or generalised (p. 775), and are particularly common in patients with raised intracranial pressure due to cerebral abscesses and neoplasms.
Hydrocephalus
Hydrocephalus is a particularly common complication of space-occupying lesions in the posterior fossa that compress and distort the cerebral aqueduct and fourth ventricle (p. 759).
Systemic effects
The systemic effects of raised intracranial pressure are of major clinical importance, as they may result in a life-threatening deterioration in an already ill patient. These are thought to result from autonomic imbalance and overactivity as a result of hypothalamic compression and include:
CNS TRAUMA
CNS damage in non-missile injuries may occur as primary damage (immediate) or secondary damage (after the injury)In the UK, 200–300 per 100000 population present to hospital each year with head injuries, most of which are due to road traffic accidents and falls. Head injuries can be classified according to their aetiology: missile and non-missile (blunt) injuries. The latter are more common.
Missile injury to the brain
Missile injuries to the brain are typically caused by bullets or other small objects propelled through the air. Three main types of injury are recognised:
Non-missile injury to the brain
Non-missile injuries to the brain range from relatively minor injuries with spontaneous improvement (as in concussion injuries), to severe injuries that are rapidly fatal. These injuries occur most commonly in road traffic accidents (55%) and falls (35%), when rotational forces acting on the brain may be accompanied by impact-related forces. The latter often result in a skull fracture, but it is important to note that around 20% of fatal head injuries occur without a fracture. The types of brain damage occurring in non-missile injuries may be classified as either primary or secondary.
Primary brain damage
Primary brain damage occurs at the time of injury. There are two main forms: focal damage and diffuse axonal injury.
Focal damage
The commonest type of focal damage is contusions. These often occur at the site of impact, particularly if a skull fracture is present. Contusions are commonly asymmetrical and may be more severe on the side opposite the impact—the ‘contrecoup’ lesions (Fig. 26.7). Movement of the brain within the skull brings these areas into contact with adjacent bone, resulting in local injury. Large contusions may be associated with an intracerebral haemorrhage, or accompanied by cortical lacerations. Healed contusions are represented by wedge-shaped areas of gliosis and cortical rarefaction which are yellow–brown due to the presence of haemosiderin.
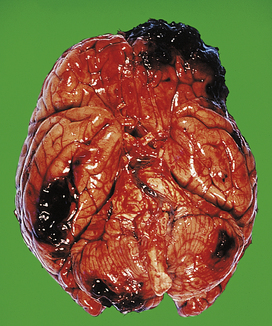
Fig. 26.7 Head injury: contusions and haematomas. A severe blow to the frontal bone has resulted in contusions and haematomas in the frontal lobes. ‘Contrecoup’ contusions are present in the parietal lobes, and in the cerebellum.
Other forms of focal damage, e.g. tears of cranial nerves, pituitary stalk or brainstem, occur less frequently.
Diffuse axonal injury
This type of damage occurs as a result of shearing and tensile strains on neuronal processes produced by rotational movements of the brain within the skull. It often occurs in the absence of a skull fracture and cerebral contusions. Two main components exist:
Modern neuropathological techniques reveal that diffuse axonal injury occurs in almost all fatal head injuries and may occur to a lesser degree in milder injuries (e.g. concussion).
Secondary brain damage
Secondary brain damage occurs as a result of complications developing after the moment of injury. These complications often dominate the clinical picture, and are responsible for death in many cases:
Table 26.3 Mechanisms and clinical manifestations of traumatic intracranial haemorrhage
| Site | Mechanism | Clinical manifestations |
|---|---|---|
| Extradural space | Skull fracture with arterial rupture, e.g. middle meningeal artery | Lucid interval followed by a rapid increase in intracranial pressure |
| Subdural space | Rupture of venous sinuses or small bridging veins due to torsion forces | |
| Subarachnoid space | Arterial rupture | Meningeal irritation with a rapid increase in intracranial pressure |
| Cerebral hemisphere |
Outcome of non-missile head injury
Most patients with minor head injuries make a satisfactory recovery. However, only 20% of survivors of severe head injuries make a good recovery, while 10% remain severely disabled. Important causes of persisting debility are:
Spinal cord injuries
Spinal cord injuries account for the majority of hospital admissions for paraplegia and tetraplegia. Over 80% occur as a result of road traffic accidents; most of the patients are males under 40 years of age. Two main groups of injury are recognised clinically: open injuries and closed injuries.
Open injuries
Open injuries cause direct trauma to the spinal cord and nerve roots. Perforating injuries can cause extensive disruption and haemorrhage, but penetrating injuries may result in incomplete cord transection which can be manifested clinically as the Brown–Séquard syndrome (hemisection of the cord resulting in an upper motor neurone lesion and loss of position and vibration sense on the affected side with loss of pain and temperature sense on the contralateral side, below the level of the injury—see Fig. 26.20).
Closed injuries
Closed injuries account for most spinal injuries and are usually associated with a fracture/dislocation of the spinal column which is usually demonstrable radiologically. Damage to the cord depends on the extent of the bony injuries and can be considered in two main stages:
Complications and outcome
Late effects of cord damage include:
The outcome of cord injuries depends mainly on the site and severity of the cord damage. Patients with incomplete lesions in the cauda equina have an almost normal life expectancy, while patients surviving a high cervical lesion have a much higher morbidity and mortality.
SPINAL CORD AND NERVE ROOT COMPRESSION
The principal causes of spinal cord and nerve root compression are:
The commonest causes of subacute or chronic nerve root and cord compression are intervertebral disc prolapse and spondylosis.
Intervertebral disc prolapse
Intervertebral disc prolapse (Ch. 25) occurs in two main ways:
In both instances, a tear in the annulus fibrosus allows the soft nucleus pulposus to herniate posteriorly. This usually takes place in a lateral direction, causing nerve root compression. Central herniation is less common, but can cause direct cord damage and may also compress the anterior spinal artery, resulting in infarction. Disc prolapse occurs most commonly at the C5/C6 and L5/S1 levels; nerve root compression in the latter results in sciatica.
Spondylosis
Spondylosis due to osteoarthritis (Ch. 25) of the vertebral column occurs commonly with age. It affects around 70% of adults over 40 years of age, and is usually accompanied by degenerative disc disease. It is characterised by bony outgrowths, known as osteophytes, on the upper and lower margins of the vertebral bodies. These may encroach upon the spinal canal or intervertebral foramina to produce nerve root pain which is exacerbated by movement.
HYDROCEPHALUS
Two main groups: primary hydrocephalus, usually accompanied by increased intracranial pressure; and secondary hydrocephalus, compensatory to loss of cerebral tissueThe cerebrospinal fluid (CSF) is secreted by the choroid plexus epithelium in an active process which carefully regulates its biochemical composition. In adults, the total volume of CSF is around 140ml; this volume is renewed several times daily (Fig. 26.8).
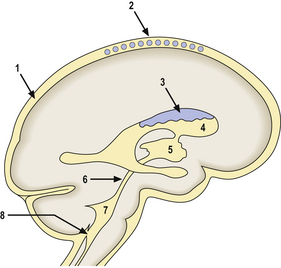
Fig. 26.8 Sites of obstruction in the cerebrospinal fluid (CSF) pathway. The circulation and absorption of CSF in the subarachnoid space (1) and arachnoid granulations (2) is readily impaired by inflammatory exudate and organising haemorrhage. CSF production in the choroid plexus (3) and flow through the lateral ventricles (4) and third ventricle (5) may be obstructed by intracranial or intraventricular neoplasms. The relatively narrow spaces of the cerebral aqueduct (6), and the fourth ventricle (7) and its exit foramina (8), are commonly obstructed by neoplasms, haemorrhage or inflammatory exudate.
CSF resorption occurs primarily at the arachnoid villi. Hydrocephalus is the term used to describe any condition in which an excess quantity of CSF is present in the cranial cavity. These conditions can be considered in two main groups:
Primary hydrocephalus
Primary hydrocephalus includes any disorder in which the accumulation of CSF is usually accompanied by an increase in intracranial pressure. It can be due to:
Obstructive hydrocephalus
Obstructive hydrocephalus is by far the commonest form; it may be either congenital or acquired.
Congenital hydrocephalus
Congenital hydrocephalus occurs in around 1 per 1000 births and occasionally may be so marked as to enlarge the fetal head considerably and interfere with labour. The more severe forms may be diagnosed antenatally by ultrasonography. Congenital malformations, for example Arnold–Chiari malformation (see Fig. 26.22), are the principal causes of congenital hydrocephalus. A few cases in males are due to an X-linked disorder that results in aqueduct stenosis. Aqueduct stenosis is more commonly due to acquired disorders, for example viral infections, which affect both sexes.
Acquired hydrocephalus
Acquired hydrocephalus can result from any lesion that obstructs the CSF pathway (Fig. 26.8). Expanding lesions in the posterior fossa are particularly prone to cause hydrocephalus, as the fourth ventricle and aqueduct are easily obstructed. Some lesions may cause intermittent obstruction, particularly colloid cysts of the third ventricle which may block the foramen of Monro. Obstructive hydrocephalus commonly results from the organisation of blood clot or inflammatory exudate in the CSF pathway following an episode of haemorrhage or meningitis (Fig. 26.9). Intermittent pressure hydrocephalus is thought to result from defective CSF absorption at the arachnoid villi.
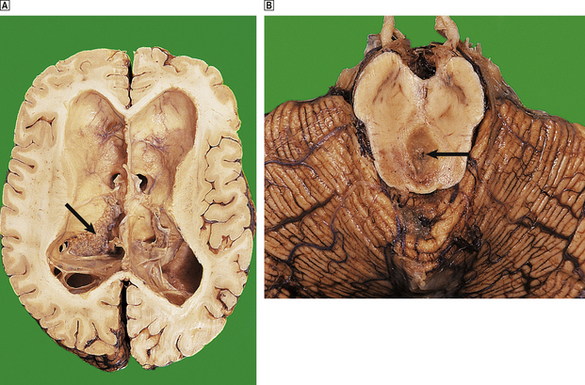
Fig. 26.9 Longstanding hydrocephalus. The lateral ventricles are very dilated and contain a prominent choroid plexus (arrow). The overlying white and grey matter are atrophic. Fibrous adhesions are present in the ventricles posteriorly, suggestive of previous infection. In the same case, the cerebral aqueduct in the midbrain is completely obliterated by glial tissue as a consequence of a previous viral infection (arrow). This has resulted in obstructive hydrocephalus.
Secondary hydrocephalus
In secondary or compensatory hydrocephalus the increase in CSF volume occurs following a loss of brain tissue, for example cerebral infarction or atrophy, so that overall there is no increase in either intracranial volume or intracranial pressure (see Fig. 26.26).
Complications and treatment
The complications of hydrocephalus can be averted or relieved by the insertion of a ventricular shunt with a one-way valve system to drain CSF into the peritoneum. Untreated patients may suffer irreversible brain damage (Fig. 26.9). Ventricular shunts often need to be replaced in growing children and are prone to become infected with low-virulence bacteria, for example Staphylococcus epidermidis. Infection may result in shunt blockage and exacerbation of symptoms attributable to raised intracranial pressure.
SYRINGOMYELIA
Syringomyelia is an uncommon condition in which a cavity (syrinx) develops within the spinal cord, sometimes extending up into the brainstem (syringobulbia). The cavity is usually situated in the central region of the cord, posterior to the central canal. Syringomyelia occurs most frequently in the cervical region of the cord, and usually extends for several centimetres in a vertical direction. However, extensive cavities involving almost the entire length of the cord have been described. Modern radiological techniques are of great value in delineating the extent of the lesion (see Fig. 26.22).
Syringomyelia can arise in a variety of conditions, which may be considered as follows:
The cavities within the spinal cord in syringomyelia are lined by reactive astrocytes and their fibrillary processes. The CSF composition in syringomyelia is normal.
The clinical manifestations of syringomyelia usually occur in adult life, with:
Surgery can sometimes arrest or alleviate symptoms by decompression or draining the fluid in the cystic cavity.
CEREBROVASCULAR DISEASE
Cerebrovascular disease is the third commonest cause of death in the uk, after heart disease and cancer, and is a major cause of morbidity, particularly in the middle-aged and elderly. The ultimate effect of cerebrovascular disease is to reduce the supply of oxygen to the CNS, resulting in hypoxic damage to cells.
Hypoxic damage to the CNS
Hypoxic damage to the CNS occurs when the blood supply to the brain is reduced (oligaemia) or absent (ischaemia). It may also occur:
The cells most vulnerable to hypoxia are the neurones, which depend almost exclusively on the oxidative metabolism of glucose for energy. Experimental evidence suggests that the early stage of hypoxic neuronal damage (microvacuolation) is reversible; in the final stages, however, the damaged neurones shrink and exhibit nuclear pyknosis and karyorrhexis.
The neurones most vulnerable to hypoxia are those in the third, fifth and sixth layers of the cortex, in the CA1 sector of the hippocampus and in the Purkinje cells in the cerebellum. This pattern of selective vulnerability does not hold true at all ages; in infants, certain brainstem nuclei are also vulnerable. The basis of this selective vulnerability is unknown, but it may relate to differences in neuronal metabolism at these sites. Ischaemic neuronal death is characterised by activation of glutamate receptors, causing uncontrolled entry of calcium into the cell. This may be abolished or reduced in some cases by drugs that block glutamate receptors or calcium channels.
Complete cessation of the circulation, such as may occur following myocardial infarction, results in global cerebral ischaemia. In less severe cases, a critical reduction of cerebral blood flow may result in boundary zone infarcts, which occur in zones between territories supplied by each of the main cerebral arteries.
Stroke
The term stroke denotes a sudden event in which a disturbance of CNS function occurs due to vascular disease. The annual incidence of stroke is 3–5 per 1000 of the general population worldwide, but is much commoner in the elderly. These events can be classified clinically into completed strokes, evolving strokes or a transient ischaemic attack in which the CNS disturbance lasts for less than 24 hours. Transient ischaemic attack is a major risk factor for cerebral infarction; most attacks are due to circulatory changes in the CNS occurring as the result of disease in the heart or extracranial arteries.
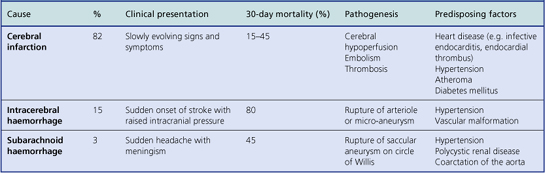
The clinical features of stroke result from focal cerebral ischaemia, and depend on the localisation and nature of the lesion (Table 26.4). Recurrent or multiple strokes often occur in patients with certain risk factors, particularly heart disease, hypertension and diabetes mellitus.
Cerebral infarction
The site and size of a cerebral infarct depend on the site and nature of the vascular lesion. Most infarcts occur within the cerebral hemispheres in the internal carotid territory, particularly in the distribution of the middle cerebral artery. Infarction of the corticospinal pathway in the region of the internal capsule is a common event, resulting in contralateral hemiparesis. Although many infarcts produce clinical symptoms, small infarcts may not result in any apparent neurological disturbance. These micro-infarcts are often found in apparently normal elderly individuals, but are also numerous in the brains of hypertensive patients. Multiple infarcts involving the cerebral cortex may result in dementia (p. 779).
Pathogenesis
The following mechanisms may be responsible for cerebral infarction:
Morphological features
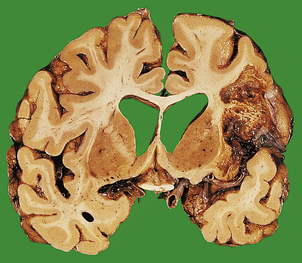
At a very early stage after cerebral infarction, no naked-eye abnormalities are apparent. However, 24 hours after infarction the affected tissue becomes softened and swollen, with a loss of definition between grey and white matter. There may be considerable oedema around the infarct, resulting in a local mass effect. Within 4 days, the infarcted tissue undergoes colliquative necrosis. Histology shows infiltration by macrophages, which are filled with the lipid products of myelin breakdown. Reactive astrocytes and proliferating capillaries are often present at the edge of the infarct. Eventually, all the dead tissue is phagocytosed to leave a fluid-filled cystic cavity with a gliotic wall (Fig. 26.10). Some infarcts are haemorrhagic, possibly due to reflow of blood through anastomotic channels. Anterograde degeneration of nerve fibres occurs distal to the site of infarction, for example in the ipsilateral cerebral peduncle in infarcts involving the internal capsule.
Venous infarction
Venous infarction is a consequence of venous thrombosis in the cranial cavity. This can occur at localised sites, most commonly in the lateral and sagittal sinuses, or as part of a generalised cortical venous thrombosis. Venous thrombosis results in a haemorrhagic infarction of the cerebral cortex and subcortical white matter. It usually occurs secondary to other disease processes, for example local sepsis, dehydration or drugs (e.g. oral contraceptives). Extensive venous infarcts are usually fatal.
Intracranial haemorrhage
Intracerebral and subarachnoid haemorrhage together account for around 18% of strokes. Extradural and subdural haemorrhages usually occur following trauma and are considered in Table 26.3.
Intracerebral haemorrhage
The commonest cause of intracerebral haemorrhage is hypertensive vascular disease, in which haemorrhages occur most frequently in the basal ganglia (80% of cases), the brainstem, cerebellum and cerebral cortex. Most intracerebral haemorrhages occur in hypertensive adults over 50 years of age. The haematoma acts as a space-occupying lesion, causing a rapid increase in intracranial pressure and intracranial herniation (Fig. 26.11). In survivors, resorption of the haematoma eventually occurs, and a fluid-filled cyst with a gliotic wall is formed. The mortality from spontaneous intracerebral haemorrhage is greater than 80%, and many survivors suffer severe neurological deficit.
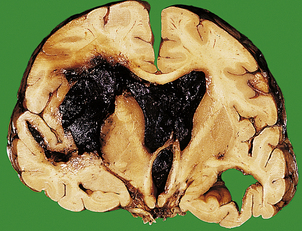
Fig. 26.11 Complications of intracerebral haemorrhage. An intracranial haemorrhage originating in the internal capsule on the left has ruptured into the ventricular system, which is filled with blood. The mass effect of the haematoma has resulted in a shift of adjacent structures to the opposite side.
The pathogenesis of spontaneous intracerebral haemorrhage is not fully understood. For many years, it was thought that most intracerebral haemorrhages in hypertensive patients occurred following rupture of micro-aneurysms on small arterioles, particularly on the lenticulostriate branch of the middle cerebral artery. Recent studies, however, have found that the ruptured vessels are arterioles, which show replacement of smooth muscle by lipids and fibrous tissue (lipohyalimosis), predisposing to rupture. Intracerebral haemorrhage in children and younger adults may occur as a consequence of trauma, or rupture of an arteriovenous malformation. In older adults, haemorrhage into the lobes of the brain may be due to amyloid depostion in the vessel walls (amyloid angiopathy), which is associated with Alzheimer’s disease (p. 779).
Subarachnoid haemorrhage
Subarachnoid haemorrhage usually occurs following rupture of a saccular or ‘berry’ aneurysm on the circle of Willis. Other causes are uncommon, but include trauma, hypertensive haemorrhage, vasculitis, tumours and disorders of haemostasis.
Saccular aneurysms
Saccular aneurysms occur in 1–2% of the general population, but are commoner in the elderly. Most cases of ruptured saccular aneurysm occur between 40 and 60 years of age; males in this age group are affected twice as often as females. Several predisposing factors for saccular aneurysms have been identified.
The role of hypertension in the pathogenesis of these lesions is uncertain, but it does appear that hypertensive patients are more likely to have multiple aneurysms than are normotensive patients. Local vascular abnormalities, such as atheroma, are important in the pathogenesis of saccular aneurysms by altering haemodynamics in affected vessels.
Saccular aneurysms are usually sited at proximal branching points on the anterior portion of the circle of Willis, particularly on the internal carotid, anterior communicating and middle cerebral arteries. Most are less than 10mm in diameter, but some may be partly filled by thrombus, which can obscure their true size on radiological studies (Fig. 26.12). Their pathogenesis is thought to relate to congenital defects in the smooth muscle of the tunica media at the site of an arterial bifurcation, where local haemodynamic factors act to produce a slowly enlarging aneurysm.
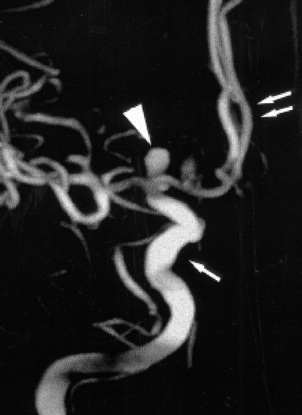
Fig. 26.12 Demonstration of a saccular aneurysm in vivo. This 3D digital subtraction angiogram shows a large grape-like saccular aneurysm (arrowhead) arising at the terminal region of the internal carotid artery (single arrow). The anterior cerebral arteries (double arrows) appear normal.
(Courtesy of Dr D Summers, Edinburgh.)
Clinicopathological features and prognosis
Subarachnoid haemorrhage often presents with the characteristic clinical history of sudden onset of severe headache. Blood accumulates in the basal cisterns and around the brainstem following rupture of a saccular aneurysm. Subarachnoid haemorrhage may be instantly fatal in as many as 15% of cases, with some patients dying later due to rebleed at the site of rupture, or arterial spasm (see below). One-third of survivors are permanently disabled as a consequence of hypoxic brain damage following haemorrhage.
Arterial spasm in the distal cerebral vasculature following rupture causes cerebral ischaemia and infarction, that is often accompanied by brain swelling due to oedema.
Hydrocephalus can occur acutely following rupture as blood accumulates in the basal cisterns, or at a later stage in survivors, where fibrous obliteration of the subarachnoid space or arachnoid granulations may occur.
Systemic hypertension and the CNS
As well as being a major risk factor for stroke, systemic hypertension causes many other changes in the CNS that result in neurological dysfunction:
Spinal cord infarction
Spinal cord infarction is most often due to spinal cord trauma or compression, but may also result from ischaemia following myocardial infarction or aortic dissection. In such cases, the infarct occurs in the mid-thoracic region of the cord, in the distribution of the anterior spinal artery where the arterial blood supply is relatively poor. These infarcts result in paraplegia with a dissociated sensory loss, as the posterior columns are spared. Infarcts in the territory of the posterior spinal artery are very rare.
Intracranial haemorrhage in neonates
Intracranial haemorrhage in neonates has a markedly different pathology from intracranial haemorrhage in adults (Table 26.5). Haemorrhage from the subependymal germinal matrix is particularly important, and is the major cause of death in premature neonates.
Table 26.5 Intracranial haemorrhage in neonates
| Site | Pathogenesis | Complication |
|---|---|---|
| Rupture of veins (birth trauma) | ||
| Subdural space | ||
| Subarachnoid space | Capillary or arterial rupture | Hydrocephalus |
| Subependymal germinal matrix | Prematurity, hypoxia (hyaline membrane disease) | Intraventricular haemorrhage |
| Venous congestion | Venous infarction in white matter (periventricular leukomalacia) | |
| Arterial spasm | Hydrocephalus |
Cerebrovascular malformations
Three main types of vascular malformation occur in the CNS:
Arteriovenous malformations are clinically the most important; these usually consist of an irregular plexus of dilated thick-walled vessels in the superficial grey matter of the cerebral hemispheres or spinal cord. Cerebral lesions may be associated with epilepsy (p. 775), or may rupture to result in a subarachnoid or intracerebral haemorrhage. Cavernous angioma and capillary telangiectasis may also be associated with epilepsy, but are often clinically unapparent.
CNS INFECTIONS
Bacterial infections
Leptomeningitis is the commonest form of bacterial infection in the CNS; it occurs most frequently in children and the elderly CSF in bacterial meningitis contains many neutrophil polymorphs and bacteria; the fluid has high protein and low glucose concentrationsThe CNS is normally sterile but, once bacteria gain access to it, spread of infection can occur rapidly, resulting in widespread meningitis. Bacteria gain access to the CNS by three main routes:
Bacterial meningitis
The clinical term ‘meningitis’ usually refers to inflammation in the subarachnoid space involving the arachnoid and pia mater, i.e. leptomeningitis. However, inflammation of the meninges may involve predominantly the dura mater (pachymeningitis).
Pachymeningitis
Pachymeningitis is usually a consequence of direct spread of infection from the bones of the skull following otitis media or mastoiditis, and is a well-recognised complication of skull fracture. Common bacterial pathogens include Gram-negative bacilli from the middle ear, alpha or beta haemolytic streptococci from paranasal sinuses, or mixed organisms, often with Staphylococcus aureus, from skull fractures. An epidural or subdural abscess may then occur.
Epidural abscess
This is the result of suppuration between the dura mater and the skull or vertebral column. Epidural abscesses can act as space-occupying lesions, and usually require surgical drainage and antibiotic therapy before healing by fibrosis can occur.
Subdural abscess
In contrast to the above, a subdural abscess is seldom a localised lesion, as pus can readily spread in the subdural space over the cerebral hemispheres to form a subdural empyema. Involvement of subdural vessels may result in cerebral cortical thrombophlebitis and arteritis with infarction. Spontaneous resolution is rare, and surgical drainage and antibiotic therapy are usually required before healing can occur.
Leptomeningitis
Leptomeningitis (‘meningitis’) is frequently a result of blood-borne spread of infection, particularly in children, but many cases arise from direct spread of infection from the skull bones. The most important organisms are:
Tuberculosis and syphilis are considered separately on pages 766–767.
Following successful vaccination programmes, bacterial meningitis due to Haemophilus influenzae is now rare. Vaccines are now also available for subgroups A and C of Neisseria meningitidis, and for Streptococcus pneumoniae. Meningococcal meningitis is the commonest variety of bacterial meningitis; it is now ususally due to the subgroup B meningococcus, which can occur as sporadic cases or as an epidemic outbreak in small communities. The organism is spread in droplets from asymptomatic nasal carriers; the carriage rate in small communities may reach over 25%. The organism reaches the CNS by haematogenous spread, and the onset of the symptoms of meningitis may follow a short history of upper respiratory tract infection. A petechial rash may herald the onset of disseminated intravascular coagulation accompanied by adrenal haemorrhage (Waterhouse–Friderichsen syndrome), which is often fatal. Vigorous antibiotic therapy is essential: incomplete or inappropriate therapy can be fatal or may result in a chronic meningitis with marked meningeal thickening.
Diagnosis and complications of bacterial meningitis
Examination of the CSF by lumbar puncture is essential in each case; the main CSF changes in the CNS infections are listed in Table 26.6. The CSF in bacterial meningitis usually contains many organisms, although these are sometimes detected only on culture. In fatal cases, pus is present in the cerebral sulci and around the base of the brain, extending down around the spinal cord (Fig. 26.13).
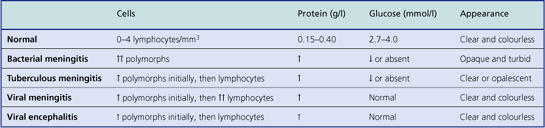
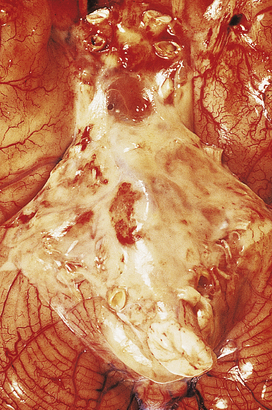
Fig. 26.13 Bacterial meningitis: basal exudate. In this example of pyogenic meningitis due to Escherichia coli, a dense acute inflammatory exudate is present around the brainstem, cerebellum and adjacent structures at the base of the brain. Obstruction of the fourth ventricle exit foramina resulted in acute hydrocephalus in this case.
The meningeal and superficial cortical blood vessels are congested, often with small foci of perivascular haemorrhage. The CSF is usually turbid, even in the ventricles, which often show signs of acute inflammation with fibrin deposition. Common complications of bacterial meningitis are:
Cerebral abscess
A cerebral abscess usually develops from an acute suppurative encephalitis following:
Abscess formation in the brain, as in other tissues, occurs when pus formation is accompanied by local tissue destruction (Fig. 26.14). A pyogenic membrane is formed, and the abscess develops a capsule composed of granulation tissue, and reactive astrocytes and their fibrillary processes. The adjacent brain is markedly oedematous, containing a perivascular inflammatory infiltrate of lymphocytes and plasma cells. Cerebral abscesses frequently enlarge and become multiloculate.
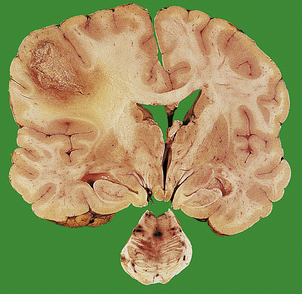
Fig. 26.14 Cerebral abscess: space-occupying lesion. A large abscess in the left parietal lobe is surrounded by oedematous white matter. This has acted as an expanding lesion and displaced the midline structures to the right. Death in this case resulted from a transtentorial brainstem herniation, with a characteristic haemorrhage in the central pons.
The clinical presentation is similar to that of acute bacterial meningitis, but focal neurological signs, epilepsy and fever are common manifestations. Abscesses act as space-occupying lesions and it is important to remember that a lumbar puncture must never be performed as an initial investigation on a patient with a suspected cerebral abscess (or other space-occupying lesion) as this may precipitate a fatal intracranial herniation. Antibiotic therapy is useful in the treatment of abscesses at an early stage, but once a capsule has formed surgical aspiration or excision is usually necessary. Complications of cerebral abscesses include:
Tuberculosis
Tuberculous infection of the CNS is always secondary to infection elsewhere in the body; the lungs are the commonest site. CNS involvement takes two main forms: tuberculous meningitis and tuberculomas.
Tuberculous meningitis
Tuberculous meningitis is usually the result of haematogenous spread from a primary or secondary complex in the lungs. Rarely, it can result from direct spread of infection from a spinal vertebral body to the meninges. The resulting meningitis is characterised by a thick gelatinous exudate which is most marked around the basal cisterns and within cerebral sulci. The exudate often contains grey tubercles adjacent to blood vessels. The findings in the CSF are listed in Table 26.6. On microscopy, the tubercles are seen to consist of granulomas with central caseation in which giant cells may be scanty or absent.
Patients usually present with signs and symptoms of a subacute meningitis, occasionally accompanied by isolated cranial nerve palsies. However, sometimes the clinical features are entirely non-specific and the diagnosis is made only following a lumbar puncture. This disorder is frequently fatal and requires intensive antituberculous chemotherapy.
Tuberculomas
Tuberculomas are uncommon in the UK, but are still encountered in patients originating from some other countries (particularly in Asia). These lesions consist of focal areas of granulomatous inflammation with caseation, and are surrounded by a dense, fibrous capsule. Tuberculomas occur most frequently in the cerebellum and present with signs and symptoms of raised intracranial pressure; features of meningitis are rarely present. As with pyogenic cerebral abscesses, surgical excision may be required.
Viral infections
CNS involvement in HIV infection is common and often accompanied by other viral, bacterial or parasitic infectionsCNS infection by viruses can occur by the following mechanisms:
Certain viruses exhibit neurotropism—a tendency to spread specifically to the CNS from the initial site of infection, for example poliovirus from the gut. Viruses can cause neurological dysfunction either as a result of viral multiplication within cells of the CNS, or as a result of an immunological response to a viral infection (acute disseminated encephalomyelitis; see below). The former mechanism is much more common.
Viral meningitis
Although acute in onset, viral meningitis is usually clinically less severe than bacterial meningitis. In most instances, the viruses reach the CNS by haematogenous spread. Common organisms are:
Characteristic changes are present in the CSF (Table 26.6) and serology or PCR techniques are often used to confirm the diagnosis.
Viral meningitis is characterised by infiltration of the leptomeninges by mononuclear cells (lymphocytes, plasma cells and macrophages), along with perivascular lymphocytic cuffing of blood vessels in the meninges and superficial cortex.
Viral encephalitis
Infection of the brain is a well-recognised complication of several common viral illnesses. Most cases are mild, self-limiting conditions, but others, such as rabies and herpes simplex type I infections, result in extensive tissue destruction and are often fatal. Herpes simplex encephalitis is the commonest variety of acute viral encephalitis in the UK. Despite these differences in severity, all viral infections of the brain and spinal cord produce similar pathological changes in the CNS:
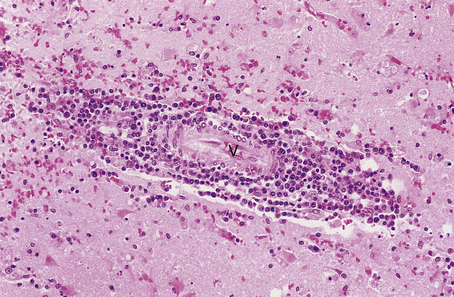
Latent viral infections
Herpes zoster
Herpes zoster results from reactivation of latent varicella zoster virus within sensory ganglia in the CNS, the infection having been established following chickenpox in childhood. Reactivation (resulting in shingles) usually occurs during periods of intercurrent illness or immunosuppression, particularly in the elderly. Acute inflammation of the sensory ganglion (usually a thoracic dorsal root ganglion or the trigeminal ganglion) is accompanied by pain and hyperalgesia along the nerve distribution, followed by erythema and vesicle formation.
Involvement of the ophthalmic division of the trigeminal nerve may result in blindness as a consequence of corneal ulceration and scarring.
Progressive multifocal leukoencephalopathy
Progressive multifocal leukoencephalopathy results from CNS infection by the JC papovavirus. Most cases occur in immunosuppressed patients. The virus produces a cytolytic infection of oligodendrocytes, resulting in demyelination in the white matter. The disease is uniformly fatal.
Antenatal viral infections
The commonest viruses to infect the CNS in utero are cytomegalovirus and rubella virus; the latter is becoming less common following immunisation in schoolgirls. Both viruses cause a necrotising encephalomyelitis resulting in developmental malformations and microcephaly, particularly when infection has occurred during the first trimester of pregnancy.
Persistent viral infections
Persistent viral infections are extremely rare diseases in which infection of the CNS occurs in early life, with neurological disease occurring years later.
Subacute sclerosing panencephalitis
This uncommon disease usually affects children aged 7–10 years and is characterised by a progressive neurological deficit with dementia, myoclonus and focal signs leading to death. Subacute sclerosing panencephalitis is caused by the measles virus, which is usually acquired before the age of 1 year. Large numbers of measles viral inclusion bodies are present within neurones, and high titres of measles antibody can be detected in the CSF. The pathogenesis of this prolonged disorder is not fully understood.
Human immunodeficiency virus (HIV) infection
The CNS is commonly involved in HIV infection both in the acquired immune deficiency syndrome (AIDS) and in pre-AIDS stages. The mechanisms by which HIV gains access to the CNS are uncertain; many research workers believe that the virus is carried across the blood–brain barrier in monocytes or macrophages (the ‘Trojan horse’ theory). Once in the CNS, the virus appears to reside predominantly in microglial cells and multinucleate cells of the macrophage/microglial type (Fig. 26.16). Evidence for direct infection of nerve cells and other glia is not fully established and awaits further research.
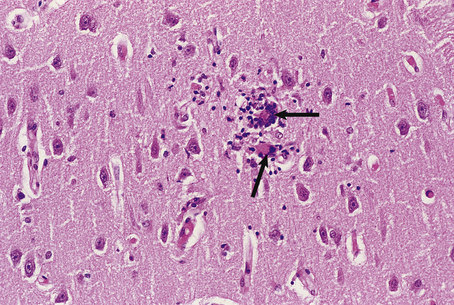
Fig. 26.16 Giant cell encephalitis in AIDS. The giant cells (arrows) in the cerebral cortex are derived from macrophages which are infected with HIV and express viral proteins on the cell surface.
Patients with HIV infection frequently present with neurological abnormalities and at the time of death at least 80% of AIDS patients have CNS pathology resulting from:
Other organisms important in infecting immunosuppressed patients are listed on page 770. Dementia may occur in the absence of overt immunodeficiency (i.e. AIDS); diagnosis can then be made by serology on the blood, or by PCR analysis of CSF.
Prion diseases
Prion diseases are a group of rare transmissible neurodegenerative disorders also known as spongiform encephalopathies. One of these disorders, kuru, was at one time restricted geographically to a small number of islands in the East Indies and appeared to result from ritualistic endocannibalism; eating the brain of an infected individual resulted in the onset of the disease many years later. The disease is now virtually extinct.
Creutzfeldt–Jakob disease
Creutzfeldt–Jakob disease (CJD) usually presents in adult life as a rapidly progressive dementia often accompanied by myoclonus, visual abnormalities and ataxia. It occurs as a sporadic disorder in 1–2 in 1 000 000 per year worldwide; familial and iatrogenic (see below) forms occur more rarely. No specific treatment is available and the disease is uniformly fatal.
In 1968, the disease was found to be transmissible to primates, and further studies have found the infectious agent to be of very small size and highly resistant to heat, ultraviolet light and most chemicals. Its precise nature is as yet unknown. Increasing evidence supports the prion hypothesis, which states that the agent is composed entirely of a modified host protein, prion protein, which accumulates in the brain. Cases of iatrogenic human–human transmission of CJD have been recorded, attributed to implantation of intracerebral electrodes, corneal or dura mater grafts and, most recently, the administration of growth hormone extracted from human pituitary glands. These are, however, rare occurrences and the source of infection in most cases is unknown.
The brain from affected individuals often shows widespread cerebral cortical atrophy. Microscopy of the cortex shows a loss of neurones and a reactive proliferation of astrocytes. Numerous small vacuoles are present within neuronal and astrocytic processes, hence the term spongiform encephalopathy (Fig. 26.17). No inflammatory reaction occurs in this group of disorders.
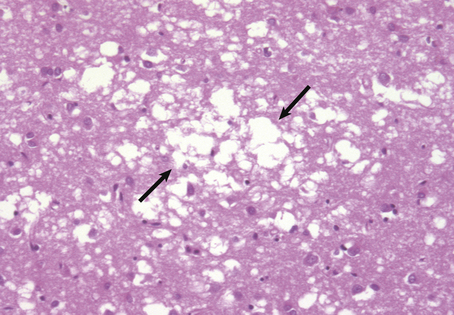
Fig. 26.17 Creutzfeldt–Jakob disease. The cerebral cortex shows a characteristic spongiform vacuolation (arrows) accompanied by neuronal loss and reactive astrocytosis.
Variant Creutzfeldt–Jakob disease
A new variant form of CJD was identified in the UK in 1996, affecting young patients (average age 28 years). This new disease appears to result from the transmission of the bovine spongiform encephalopathy (‘mad cow’ disease) agent to humans, probably via contaminated beef products. Over 160 cases of variant CJD have been identified in the UK so far, including three cases that were transmitted by blood transfusion from infected donors, but the likely number of future cases is uncertain.
Acute disseminated encephalomyelitis
Acute disseminated encephalomyelitis is an infrequent complication of measles, mumps and rubella infections, and may also occur following vaccination for smallpox and rabies. The onset of the disease is sudden, usually occurring 5–14 days after the initial infection or inoculation. This appears to be a T-cell-mediated delayed hypersensitivity response to a protein component of myelin, but the mechanism of sensitisation is unknown. The prognosis is good, with a complete recovery in 90% of cases.
Acute haemorrhagic leukoencephalitis is a related but more severe disorder which is accompanied by immune complex deposition in cerebral vessel walls and is usually rapidly fatal.
Fungal infections
Fungal infections of the nervous system are relatively uncommon; most occur as a consequence of haematogenous spread from the lungs, but direct spread of infection from the nose and paranasal sinuses also occurs. In the UK, most fungal infections of the CNS occur in immunosuppressed patients, but some organisms, for example Cryptococcus neoformans, are capable of producing disease in humans in the absence of any predisposing illness. Cryptococcal infection usually presents as a subacute meningitis in which the inflammatory reaction is often remarkably mild.
Opportunistic fungal infections with Candida albicans and Aspergillus fumigatus are usually accompanied by pulmonary infection. Both organisms may cause meningitis with haemorrhage due to vascular invasion, and characteristically produce multiple cerebral abscesses.
Mucormycosis is a rare fungal infection that particularly affects uncontrolled diabetics, producing a granulomatous mass in the paranasal sinuses that extends to involve directly the skull and frontal lobes. Vascular involvement is also common with this organism, resulting in cerebral infarction.
Parasitic infections
Parasitic infections of the CNS are uncommon except in countries in which human parasites are endemic. The most frequently encountered organisms are:
Infections in immunosuppressed patients
CNS infections are common in immunosuppressed patients, whatever the nature of the underlying disease. The main varieties are:
Many of these infections prove fatal, and a diagnosis is often difficult to establish prior to death. Multiple infections are not uncommon, particularly in the acquired immune deficiency syndrome (AIDS).
DEMYELINATING CONDITIONS
In the CNS, most axons and dendrites are ensheathed in myelin, which is formed from complex folds of oligodendrocyte cell membranes. CNS myelin differs slightly in structure and composition from peripheral myelin, but serves essentially the same functions:
Most of the myelin in the CNS is located in the white matter, but neuronal processes in the grey matter are also surrounded by myelin.
Primary demyelination in the CNS occurs in several conditions where the myelin sheath is destroyed but the axons remain intact. Primary axonal damage results in the breakdown of myelin around damaged axons, a process referred to as secondary demyelination. Whenever myelin breakdown occurs, the debris is phagocytosed by macrophages. Intact myelin is rich in cholesterol and phospholipids, but following phagocytosis it is transformed into droplets of neutral lipids (mainly cholesterol esters).
Multiple sclerosis
Multiple sclerosis is the leading non-traumatic cause of neurological disability in young adults in the Europe and the USA. It is most prevalent in populations living at latitudes remote from the equator; the prevalence is particularly high in northern Europe, but is low in the tropics (Table 26.7). Individuals who migrate from a high-prevalence to a low-prevalence area after the age of 15 years remain at high risk; the disease risk is lower following migration at an earlier age. Studies of twins have shown a higher incidence of concordance in monozygous than in dizygous twins. Recent genetic studies have found an association between multiple sclerosis and the interleukin-2 and -7 receptor alpha genes, and the human leukocyte antigen (HLA) genetic locus.
Table 26.7 Geographical variance in the prevalence of multiple sclerosis
| Area | Crude prevalence per 100 000 population |
|---|---|
| North-east Scotland | 144 |
| Northumberland, England | 50 |
| North Italy | 20 |
| Israel | 13 |
| Mexico | 1.5 |
Multiple sclerosis appears to be an autoimmune disorder, triggered by an environmental factor (e.g. a virus) in a genetically susceptible host. The therapeutic use of corticosteroids and cytokines, such as beta-interferon, which modulate the immune response, has reduced the frequency of disease relapse and progression in some patients.
Clinical features
Most cases present between 20 and 40 years of age. The disease is slightly more common in females than in males, and the onset is usually characterised by the sudden development of a focal neurological deficit which spontaneously recovers. The relative incidences of initial manifestations are:
The disease follows a characteristic relapsing and remitting course. Recovery from each episode of demyelination (relapse) is usually incomplete, and a progressive clinical deterioration ensues. The effects of demyelination may be detected electrophysiologically as delays in the latencies of visual and auditory evoked responses because demyelinated axons conduct nerve impulses more slowly than normal. CSF analysis in multiple sclerosis shows oligoclonal bands of IgG, which is synthesised by plasma cells in the CNS. The progress of the disease is variable. Some patients (particularly children) follow a rapidly progressive course, while others may survive for over 20 years with only minor disability. Most patients die as a result of urinary tract infections, chest infections or pressure sores rather than during an acute episode of demyelination.
Morphological features
The primary abnormalities in multiple sclerosis are confined to the CNS; the peripheral nervous system is not involved. Patients with multiple sclerosis have numerous demyelinated plaques in the brain and spinal cord (Fig. 26.18), often closely related to veins and venules. In early lesions, the plaques are soft and pink with ill-defined boundaries. Histologically, there is myelin breakdown and phagocytosis by macrophages. Oedema is usually present, suggesting a local defect in the blood–brain barrier. Perivascular cuffing with inflammatory cells (plasma cells and T-lymphocytes) is widespread in the acute plaque. The plasma cells synthesise immunoglobulins, which can be detected in the CSF (see above). T-lymphocytes have also been identified at the edges of acute plaques.
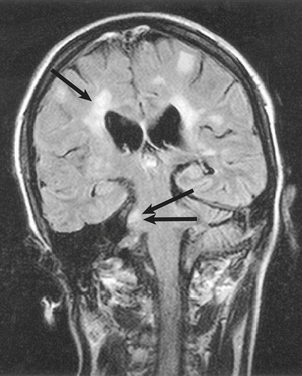
Fig. 26.18 Multiple sclerosis: demonstration of demyelination in vivo. Coronal MRI image showing the typical appearances of multiple sclerosis plaques, particularly in a periventricular location (single arrow) and in the right middle cerebellar peduncle (double arrow).
(Courtesy of Dr D Summers, Edinburgh.)
As myelin breakdown eventually subsides, a reactive gliosis is established, giving rise to a chronic plaque. These lesions consist of sharply defined, grey, lucent areas of demyelination in which oligodendrocytes are scarce or absent. The inflammatory infiltrate also subsides, sometimes leaving small numbers of perivascular lymphocytes at the edge of chronic plaques (Fig. 26.19). Although it appears that oligodendrocytes have the capacity to proliferate in plaques, successful remyelination of established plaques probably never occurs. Axonal damage begins early in multiple sclerosis and correlates with the inflammatory activity in the white matter, contributing to progressive neurological debility.
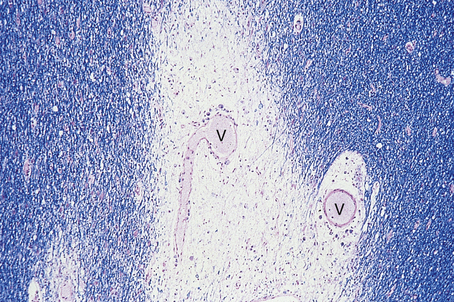
Fig. 26.19 Multiple sclerosis: chronic plaque. The chronic plaque consists of a sharply defined area of myelin loss (which appears pale in this preparation) containing fibrillary astrocytes. A few lymphocytes and macrophages are present around blood vessels (V) in the plaque. Normal myelinated white matter appears blue.
Miscellaneous demyelinating conditions
Leukodystrophies
Although included as demyelinating conditions, it is known that most leukodystrophies result from a failure to synthesise normal myelin (sometimes called ‘dysmyelination’). Two of these disorders—metachromatic leukodystrophy and Krabbe’s globoid cell leukodystrophy—are due to inherited lysosomal enzyme deficiencies, and can be diagnosed antenatally. Others, such as adrenoleukodystrophy, are the result of an inherited abnormality in lipid metabolism, while in others the cause is unknown.
Metabolic disorders
In central pontine myelinolysis, which occurs most frequently in alcoholism and malnutrition, myelin breakdown occurs in the central brainstem and cerebrum. Its pathogenesis is unknown, but some cases appear to result from the rapid alterations in serum sodium levels.
METABOLIC DISORDERS
Hypoglycaemia
The brain is critically dependent on a continuous supply of oxygen and glucose; hypoglycaemia (most often occurring in patients with diabetes mellitus) can result in irreversible neuronal damage and neuronal cell death unless relieved rapidly. Affected patients usually lapse into a coma, and may never recover full neurological function.
CNS toxins
The CNS can be affected by a large number of substances that act as toxins.
Methanol and ethanol
Both methanol and ethanol are toxic to the CNS. Acute poisoning with methanol can result in sudden death with multiple haemorrhagic lesions in the cerebral hemispheres, while chronic ingestion results in degeneration of neurones, e.g. in the retina, where loss of ganglion cells is accompanied by optic nerve atrophy. Ethanol can cause a wide range of CNS disorders (Table 26.8).
Table 26.8 Consequences of excessive ethanol intake on the CNS
| Disease | Features | Mechanism |
|---|---|---|
| Fetal alcohol syndrome (maternal alcoholism) | Direct toxicity | |
| Acute intoxication | Direct toxicity | |
| Cerebral and cerebellar atrophy | Neuronal loss | Direct toxicity |
| Nutritional disorders | Wernicke’s encephalopathy | Deficiency of vitamin B1 |
| Hepatocerebral syndromes | Hepatic toxicity with secondary effects on CNS | |
| Demyelinating disorders | Central pontine myelinolysis | Electrolyte disturbances |
Drugs
Drugs affecting the CNS can be considered in two main groups:
Drugs affecting CNS development include phenytoin and trimethadione, which can cause microcephaly and other congenital abnormalities following maternal ingestion. Drugs affecting the mature CNS include vincristine, which may cause axonal neuropathy.
Metals and industrial chemicals
Metals and industrial chemicals capable of affecting the CNS are listed in Table 26.9.
Table 26.9 Metal and industrial chemical toxins affecting the CNS
| Metal/Chemical | Source | Clinical manifestations of toxicity |
|---|---|---|
| Aluminium | Dialysis water from mains | Progressive encephalopathy in patients undergoing renal dialysis |
| Manganese | Mines | Degeneration of basal ganglia |
| Lead (inorganic) | Paint and petrol fumes | Encephalopathy in children; peripheral neuropathy |
| Mercury | ||
| Acrylamide monomer | Construction industry | Encephalopathy and peripheral neuropathy with axonal degeneration |
| Hexacarbon compounds | Solvents | ‘Giant axonal neuropathy’ affecting the CNS and peripheral nerves |
| Organophosphates | Insecticides | Anticholinesterase activity and distal axonopathy in CNS and peripheral nerves |
Deficiency states
In the developed countries of the world, the commonest deficiency states affecting the CNS are those involving vitamins, e.g. in chronic alcoholism. Elsewhere, the lack of an adequate food supply is responsible for a range of abnormalities that are still poorly understood in terms of their effects on the developing and mature CNS.
Malnutrition
Severe malnutrition may result in irreversible brain damage, particularly if it occurs in infancy during periods of CNS myelination, as the lack of normal myelin development cannot be reversed at a later date. Malnutrition later in life, e.g. kwashiorkor (Ch. 7), may result in encephalopathy and ultimately lead to coma. The underlying mechanisms in these events are uncertain, but may result from severe electrolyte disturbances.
Vitamin deficiency
The major vitamin deficiency states (Ch. 7) affecting the nervous system are shown in Table 26.10. The most important of these are discussed below.
Table 26.10 Major vitamin deficiency states affecting the nervous system
| Vitamin | Deficiency state |
|---|---|
| A | Benign intracranial hypertension (rare) |
| B1 | Wernicke–Korsakoff syndrome |
| B2 | Peripheral neuropathy, ataxia, dementia |
| B6 | Convulsions in infants |
| B12 | Weakness and paraesthesiae in the lower limbs |
| C | Scurvy |
| E | Weakness, sensory loss, ataxia, nystagmus |
Vitamin B1 (thiamine)
Vitamin B1 deficiency is particularly common in chronic alcoholics and in patients with longstanding diseases of the upper gastrointestinal tract. Deficiency results in Wernicke’s encephalopathy, which presents clinically with memory impairment, ataxia, visual disturbances and peripheral neuropathy. This disorder is often accompanied by Korsakoff’s psychosis, in which case the term Wernicke–Korsakoff syndrome is used. Wernicke’s encephalopathy is characterised by perivascular haemorrhages in the region of the fourth ventricle and aqueduct, particularly in the mammillary bodies. Fibrillary gliosis occurs in longstanding cases, when the affected structures appear shrunken. The pathogenesis of the lesions is uncertain.
Vitamin B12 (cyanocobalamin) deficiency
Vitamin B12 deficiency is an important condition that can result from a variety of disorders. The pathogenesis of the CNS damage is unknown; impairment of CNS amino acid and fatty acid metabolism has been implicated. In severe cases, there is extensive degeneration of the posterior columns and lateral corticospinal tracts in the spinal cord (Fig. 26.20); this process is referred to as subacute combined degeneration of the spinal cord. The cerebral hemispheres are involved to a lesser extent. If replacement therapy is commenced at an early stage, the degenerative process is reversible. Longstanding cases show irreversible axonal damage accompanied by a reactive fibrillary gliosis.
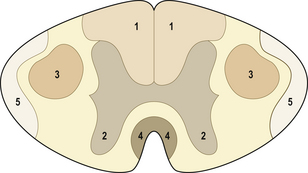
Fig. 26.20 Sites of degenerations in the spinal cord. 1. Dorsal columns, involved in subacute combined degeneration, Friedreich’s ataxia and tabes dorsalis. 2. Anterior horn cells, involved in motor neurone disease and spinomuscular atrophy. 3. Lateral corticospinal tracts, involved in motor neurone disease, subacute combined degeneration and Friedreich’s ataxia. 4. Ventral corticospinal tracts, involved in motor neurone disease. 5. Spinocerebellar tracts, involved in Friedreich’s ataxia.
Lysosomal storage diseases
Lysosomal storage diseases are uncommon inherited disorders characterised by a deficiency of various lysosomal enzymes that results in the accumulation of stored material in cells (Ch. 7). The CNS is involved in many lysosomal storage disorders (Table 26.11).
Table 26.11 Examples of lysosomal storage diseases affecting the CNS
| Disease | Example | Enzyme deficiency |
|---|---|---|
| Sphingolipidosis | ||
| Mucopolysaccharidosis | Hurler’s disease | Alpha-l-iduronidase |
| Glycogenosis | Pompe’s disease | Acid maltase |
| Ceroid lipofuscinosis | Batten’s disease | Lysosomal peptidases and esterases |
Hepatic encephalopathy
Hepatic encephalopathy may occur in patients with liver damage, due to a variety of agents. Encephalopathy in severe cases may progress to coma and result in permanent CNS damage in survivors. Increased levels of ammonia in the blood are associated with encephalopathy, possibly interfering with the function of certain neurotransmitters, such as gamma aminobutyric acid. The commonest cause of hepatic encephalopathy is alcoholic liver disease.
Wilson’s disease
Wilson’s disease, a disorder of copper metabolism, is inherited as an autosomal recessive condition. In some patients, liver disease is severe (Ch. 16) and may result in hepatic encephalopathy. In others, neurological signs predominate with tremor, rigidity and chorea; these abnormalities result from a marked loss of neurones in the basal ganglia, particularly the putamen. Deposition of copper in the cornea results in the characteristic Kayser–Fleischer ring.
EPILEPSY
Epilepsy occurs where an individual suffers repeated seizures due to paroxysmal neurological dysfunction caused by abnormal discharges from neurones in the brain. Epilepsy is one of the commonest serious neurological conditions, with around 350000 affected patients in the UK. Epilepsy can be classified according to the type of seizure, each of which is associated with different forms of brain pathology:
CONGENITAL ABNORMALITIES
Malformations of the CNS occur in 3–4 per 1 00 000 live births. The severe varieties cause considerable morbidity and mortality, but many of these abnormalities are of little clinical significance and may be detected only in later life as an incidental finding. Some of the known causes of CNS malformations in humans are:
In many cases, the underlying causes are unknown. The most frequent malformations are the neural tube defects and posterior fossa malformations.
Neural tube defects
Neural tube defects are the commonest and most important congenital abnormalities of the CNS, occurring in 2–3 per 100000 live births. Failure of the neural tube to close at 28 days’ gestation, or damage to its structure after closure, can be detected in utero by ultrasonography. In 90% of cases, the level of alpha-fetoprotein in the maternal serum and amniotic fluid is increased; this investigation is often used as a screening procedure.
Both cranial and spinal involvement may occur; the term spina bifida is often used for the latter, when the CNS malformation is usually accompanied by rachischisis—failure of the vertebral laminae to develop. The major types of spinal involvement are illustrated in Figure 26.21.
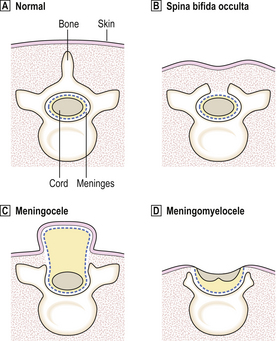
Fig. 26.21 Neural tube defects: spinal involvement. Normal arrangement. Spina bifida occulta: vertebral defect with a normal cord and meninges. The overlying skin is intact.  Meningocele: the meningeal sac is usually covered by intact skin, but rupture of the sac may occur following birth.
Meningocele: the meningeal sac is usually covered by intact skin, but rupture of the sac may occur following birth.  Meningomyelocele: the skin overlying the sac frequently ruptures, exposing the abnormal meninges and spinal cord.
Meningomyelocele: the skin overlying the sac frequently ruptures, exposing the abnormal meninges and spinal cord.
Neural tube defects occur most frequently in the lumbosacral region. The more severe forms result in a considerable neurological deficit with paraplegia and absence of sphincter control. The musculature of the lower limbs undergoes neurogenic atrophy, and meningitis and urinary tract infections are common. Hydrocephalus occurs in cases with an accompanying Arnold–Chiari malformation. These factors account for the generally poor prognosis in severe cases, even after early surgical repair of the spina bifida.
Cranial involvement
Encephalocele and cranial meningocele usually occur in the occipital region, with herniation of the posterior cerebral hemispheres and their coverings respectively through a defect in the skull.
Anencephaly is the commonest of the neural tube defects. It is thought to occur when the developing brain is exposed to amniotic fluid as its coverings fail to develop. The calvaria is usually absent, but the base of the skull is thickened and partly covered by a mass of vascular granulation tissue. The anterior pituitary gland is present but hypoplastic, and several associated visceral and limb abnormalities have been described. The condition is fatal and often results in spontaneous abortion.
Posterior fossa malformations
Arnold–Chiari malformation
Arnold–Chiari malformation is a complex disorder involving the cerebellum, brainstem and spinal cord, and is the commonest congenital malformation in the posterior fossa. It is often associated with a meningomyelocele. The main features are illustrated in Figure 26.22.
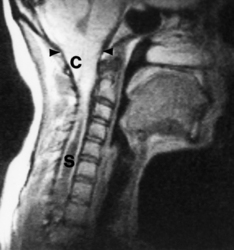
Fig. 26.22 Arnold–Chiari malformation: brain and cord abnormalities. The cerebellar tonsils (C) are displaced downwards from the shallow posterior fossa below the level of the foramen magnum (arrowheads). The brainstem is elongated and a syrinx (S) is present in the spinal cord commencing at the level of the third cervical vertebral body.
(Magnetic resonance image: courtesy of Professor B S Worthington, Nottingham.)
These abnormalities result in an obstructive hydrocephalus, usually of the communicating type (p. 759). There is no entirely satisfactory explanation for the Arnold–Chiari malformation.
Dandy–Walker malformation
Dandy–Walker malformation is the second most important congenital abnormality affecting the posterior fossa. The cerebellar hemispheres are of normal size, but the vermis is absent or hypoplastic. The fourth ventricle is markedly distended and forms a cyst-like structure. This results in obstructive hydrocephalus, which may be detectable antenatally by ultrasonography. The aetiology and pathogenesis of this malformation are unknown.
Other congenital malformations
Many other congenital malformations affecting the CNS have been described. Without being comprehensive, these can be considered in the following broad groups.
Agenesis and dysgenesis
Agenesis and dysgenesis may involve almost any structure within the CNS, but the commonest sites affected are the corpus callosum and the olfactory bulbs and tracts (arhinencephaly). These lesions may occur in isolation or in association with other malformations, for example agenesis of the corpus callosum with the Dandy–Walker malformation, and arhinencephaly with holoprosencephaly, a complex disorder of forebrain diverticulation.
Disorders of cell migration and corticogenesis
Failure of neuronal migration during CNS development results in a number of structural disorders, of which the most important are:
Destructive lesions
Destructive lesions occur most frequently in the developing CNS as a consequence of maternal infections and hypoxia. Extensive destruction of tissue may result in microcephaly, but focal lesions may also occur, such as ulegyria, with severe loss of neurones in the cerebral cortex.
Phakomatoses
Phakomatoses are a group of autosomal dominant inherited neurocutaneous disorders that result in CNS malformations. Important members of this group include neurofibromatosis, tuberous sclerosis and von Hippel–Lindau syndrome.
Chromosomal abnormalities
Chromosomal abnormalities frequently result in mental retardation. CNS malformations are often present in such cases, and are sometimes of sufficient severity to cause permanent disability or death. The best characterised of these disorders include Down’s syndrome, the principal features of which are:
AGE-RELATED CHANGES IN THE CNS
A wide variety of changes has been described in the CNS of normal elderly adults. The extent of these changes often relates to the age of the person, but there is considerable variation from one individual to another.
Brain weight progressively reduces from normal values of around 1450g in males and 1300g in females at about 40 years of age; these values decline more rapidly after the age of 60. This loss of brain substance appears to occur at an earlier age in females than in males, and is most evident in the white matter of the cerebral hemispheres. Ventricular enlargement (compensatory hydrocephalus, p. 759) is a variable finding in elderly brains and can readily be detected in life by CT scanning. On average, the volume of the ventricular system increases from 35ml in young adults to 60 ml in those over 60 years.
SYSTEM DEGENERATIONS
Considerable overlap of clinical and pathological features, but several well-defined entities exist, e.g. Parkinson’s disease and motor neurone diseaseSeveral degenerative conditions affecting the CNS are characterised by the progressive loss of certain groups of functionally related neurones and their associated pathways. These conditions can be considered as system degenerations; these disorders may occur in isolation or as part of a multiple systems degeneration. Many of these disorders have a genetic basis and are associated with nucleotide triplet repeat expansions in the relevant genes. It is therefore important to establish a diagnosis to allow genetic counselling of an affected family. Considerable overlap of both the clinical and pathological features occurs in this group of conditions, but several well-defined examples exist (Table 26.12).
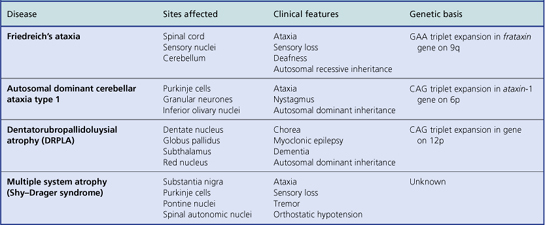
Motor neurone disease
This disorder affects 5 in 100000 of the population, occurring most often in males over the age of 50 years; 5% of cases are familial, some of which have a mutation in the Cu/Mn superoxide dismutase gene on chromosome 21q. Three main disease patterns are recognised clinically:
Most patients die 3–5 years after diagnosis due to respiratory difficulties or the complications of immobility. Examination of the CNS shows loss of motor neurones (in patterns corresponding to the clinical groups listed above) and corticospinal pathway degeneration with reactive gliosis. Occasional surviving motor neurones contain filamentous cytoplasmic inclusions of unknown aetiology; in a minority of cases these inclusions may be widespread in the cerebral cortex and are associated with dementia.
Parkinson’s disease
Parkinson’s disease is characterised clinically by tremor, bradykinesia and rigidity, which usually become manifest between the ages of 45 and 60 years, affecting 1% of the population over 60. Similar clinical features may occur in unrelated conditions, such as cerebrovascular disease or phenothiazine drug therapy. This disorder results in a progressive loss of pigmented neurones in the substantia nigra (Fig. 26.23), the locus ceruleus and several other brainstem nuclei. Surviving neurones at these sites contain round eosinophilic inclusions—Lewy bodies (Fig. 26.24)—containing neuronal proteins, including alpha-synuclein. Lewy body inclusions may occasionally be widespread throughout the brain, particularly in the cerebral cortex, resulting in dementia due to ‘diffuse Lewy body disease’.
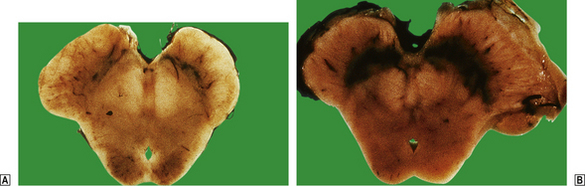
Fig. 26.23 Idiopathic Parkinson’s disease. The pigmented neurones in the substantia nigra within the midbrain degenerate and die off in Parkinson’s disease, giving a pale appearance in comparison to an age-matched normal control.
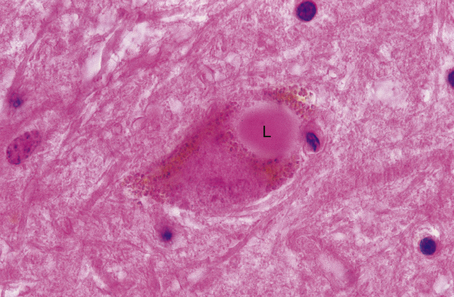
Fig. 26.24 Idiopathic Parkinson’s disease. A surviving pigmented neurone in the substantia nigra contains an intracytoplasmic rounded eosinophilic inclusion known as a Lewy body (L), which contains aggregates of alpha-synuclein.
The neurones of the substantia nigra synthesise dopamine, which acts as an inhibitory neurotransmitter at their axonal projection sites in the basal ganglia (putamen and globus pallidus). Loss of the pigmented neurones results in a relative deficiency of dopamine in the basal ganglia that can be overcome by replacement therapy, for example with l-dopa. This often relieves the clinical symptoms of the disease, but a permanent cure is not yet possible.
A disorder similar to Parkinson’s disease can be produced experimentally by the administration of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). This compound has also produced a severe disorder resembling Parkinson’s disease in intravenous drug addicts when it is present as a contaminant in synthetic heroin preparations. Chronic exposure to environmental toxins, including pesticides, is a risk factor for Parkinson’s disease, and recent genetic studies have identified mutations in the alpha-synuclein gene in some families with an autosomal dominant form of Parkinson’s disease.
DEMENTIA
Dementia has been defined clinically as an acquired global impairment of intellect, reason and personality without impairment of consciousness. Emotional lability and memory dysfunction are prominent manifestations, implying a cerebral cortical disorder. Most patients with dementia exhibit both gross and histological abnormalities within the cerebral cortex, although some rarer causes of dementia appear to involve mainly subcortical structures. A variety of disorders affecting the CNS can result in dementia:
These disorders may be considered in two main categories:
The commonest cause of dementia in Western countries is Alzheimer’s disease (at least 70% of cases), followed by Lewy body dementia and vascular dementia. It is important to establish the cause of dementia in each patient, as in some cases an effective treatment is available. In other cases, dementia may be due to an inherited disorder, in which case genetic counselling is required for the affected family. The major causes of organic dementia are discussed below.
Alzheimer’s disease
Alzheimer’s disease accounts for well over 70% of all cases of dementia in adults. In the UK, it is thought to affect 5% of people over the age of 65 years, rising to 15% of those over 80. As the number of elderly people in the population increases, so there is a concomitant increase in the number of patients suffering from Alzheimer’s disease; this has been termed the ‘silent epidemic’. Females are affected almost twice as frequently as males. Most cases occur sporadically, although a small proportion are inherited as an autosomal dominant disorder. Several gene loci are involved in familial cases, including the amyloid precursor protein (APP) gene on chromosome 21, the presenilin 1 gene on chromosome 14 and the presenilin 2 gene on chromosome 1 (Fig. 26.25). There is an increased incidence of sporadic Alzheimer’s disease in individuals with the ApoE e4 genotype on chromosome 19. The clinical presentation usually occurs after the age of 60 years, but a significant subgroup is affected between the ages of 50 and 60 years. The illness lasts from 2 to 8 years; most patients die from inanition and bronchopneumonia.
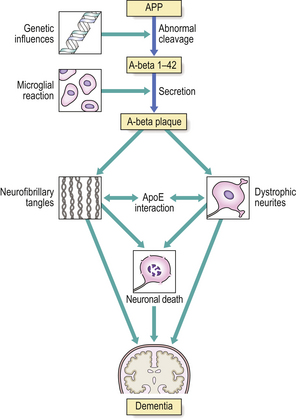
Fig. 26.25 Amyloid cascade pathway in Alzheimer’s disease. This simplified diagram indicates how abnormal processing of the cell surface glycoprotein APP (coded by a gene on chromosome 21) leads to the formation of A-beta plaques, the histological hallmark of Alzheimer’s disease. The mechanisms of interaction with ApoE and neurofibrillary tangle formation are not fully understood.
Morphological features
The brain is reduced in weight, often to 1100 g, and shows cortical atrophy which is often most marked in the frontal and temporal lobes. There is loss of both cortical grey and white matter, with compensatory dilatation of the ventricular system (secondary hydrocephalus, Fig. 26.26). The cerebellum and spinal cord appear normal.
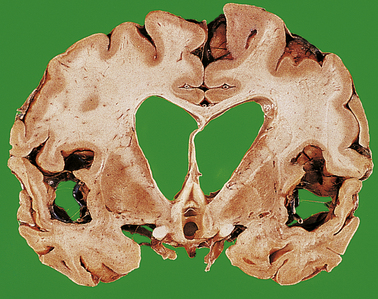
Fig. 26.26 Alzheimer’s disease cortical atrophy. The brain in Alzheimer’s disease shows severe cortical atrophy with narrowing of the gyri and widening of the sulci. White matter loss is accompanied by dilatation of the ventricular system (compensatory hydrocephalus).
The characteristic histological changes in Alzheimer’s disease are most pronounced in the limbic system (amygdala, entorhinal cortex and hippocampus) and cerebral cortex. The severity of these changes correlates with the clinical severity of the dementia. The histological hallmarks of Alzheimer’s disease are A-beta amyloid plaques and neurofibrillary tangles.
A-beta amyloid plaques
A-beta amyloid plaques are best demonstrated in specially stained histological sections of the CNS (Fig. 26.27A). They occur most frequently in the amygdala, hippocampus and cerebral cortex. These plaques can measure up to 200µm in diameter, and in their earliest stages comprise a collection of dilated presynaptic neuronal processes, which contain many organelles, around a diffuse aggregate of amyloid material. As the plaques mature, they enlarge and develop a core of A-beta amyloid protein, which eventually forms the major component of the ‘burnt-out’ plaque. Reactive astrocytes and microglia are usually present at the periphery of the plaque.
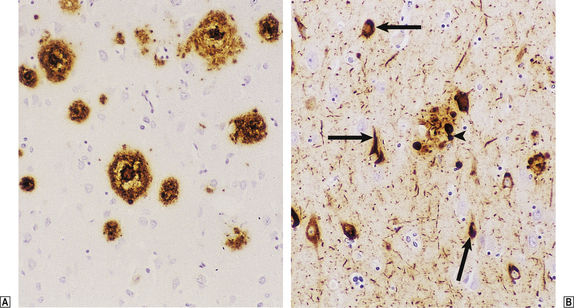
Fig. 26.27 Plaques and tangles in Alzheimer’s disease. The amyloid plaques in Alzheimer’s disease are composed of the A-beta protein, and form irregular rounded masses (brown) in the extracellular matrix in the cerebral cortex. In Alzheimer’s disease, abnormally phosphorylated tau protein (brown) accumulates intracellularly in neurofibrillary tangles (arrows) and in dystrophic neurites (arrowhead).
Neurofibrillary tangles
Neurofibrillary tangles are present within the neuronal perikarya and are most easily visualised in specially stained sections (Fig. 26.27B). Affected neurones are most often found in the hippocampus, but may also occur in the cerebral cortex, subcortical grey matter and brainstem nuclei. This form of degeneration consists of a thickening of fibrils within the neuronal cytoplasm, to form a tortuous and elongated corkscrew-like structure. Electron microscopy has shown that each tangle consists of a mass of twisted tubules composed of paired helical filaments, each 10nm in diameter, with a periodic narrowing at 80nm. The major component of these tangles is tau, a microtubule-associated protein, which becomes hyperphosphorylated in Alzheimer’s disease.
Neuronal loss
Neuronal loss is often widespread in the cerebral cortex, but is most severe in the hippocampus.
Amyloid angiopathy
A-beta amyloid may be deposited within the walls of small arterioles and capillaries in the brain in Alzheimer’s disease. This is derived from abnormal cleavage of APP (Fig. 26.25), as for the A-beta amyloid in the plaques.
Neurochemical abnormalities
The functional impairment in Alzheimer’s disease is accompanied by a number of neurochemical abnormalities, the best known of which involve a reduction in cholinergic activity in the cerebral cortex. Treatment with anticholinesterase inhibitors increases levels of acetylcholine in the brain and improves cognitive function in the early stages of the disease.
Aetiology and pathogenesis
Genetic factors are of major importance in the aetiology of Alzheimer’s disease in both familial and sporadic forms. Increasing evidence supports a primary role for A-beta amyloid in the pathogenesis of Alzheimer’s disease. A-beta amyloid is formed as part of the ‘amyloid cascade’ from APP (Fig. 26.25). This explains why individuals with trisomy 21 (Down’s syndrome) develop accelerated Alzheimer-like changes in the CNS as a consequence of their extra APP gene load.
Frontotemporal dementia
This term covers a group of dementias associated with aphasia and behavioural disorders that are characterised by severe atrophy of the frontal and temporal lobes of the brain. This group includes disorders previously referred to as Pick’s disease. Recent research has indicated that the prevalence of frontotemporal dementias is higher than previously thought, perhaps accounting for up to 10% of all cases of dementia. Most cases occur in individuals aged between 35 and 75 years, with some conditions occurring as familial disorders. The pathology is characterised by severe neuronal loss and gliosis in the frontal and temporal cortex, with neuronal inclusions present in some disorders. Treatment is aimed at alleviating symptoms for as long as possible, but death usually occurs within 3–6 years after the onset of symptoms.
Huntington’s disease
The degenerative condition known as Huntington’s disease is inherited as an autosomal dominant disorder. It is uncommon, affecting 4–7 per 100000 in the UK. The disease does not usually become clinically apparent until the fifth decade of life, when the onset of personality change and depression are later accompanied by choreiform movements, jerking and dementia. The gene responsible for Huntington’s disease, the huntingtin gene, has been located on chromosome 4p, allowing an effective means of preclinical and antenatal diagnosis.
The genetic abnormality is an excess number of tandemly repeated CAG nucleotide sequences. The number of repeats influences the age of onset: the more repeats, the earlier the onset.
The gross appearances are illustrated in Figure 26.28. Histology of the caudate nucleus and putamen shows a marked loss of small neurones, accompanied by a reactive fibrillary gliosis. The cerebral cortex in this disorder also shows neuronal loss, but to a variable extent.
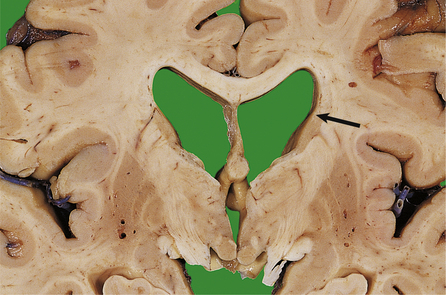
Fig. 26.28 Huntington’s disease: subcortical atrophy. In Huntington’s disease, cerebral atrophy is most marked in the caudate nucleus (arrow), which is markedly narrowed, and the adjacent putamen. These changes are accompanied by compensatory hydrocephalus involving the lateral ventricles.
Neurochemical abnormalities have been identified in this disorder, for example reduced levels of choline acetyltransferase and gamma aminobutyric acid in the basal ganglia. These changes are presumably secondary to the neuronal loss.
Dementia pugilistica
In the ‘punch-drunk’ syndrome seen in boxers, progressive dementia is accompanied by tremor and focal neurological signs. Characteristic findings in the brain are structural abnormalities of the septum pellucidum, thinning of the corpus callosum, degeneration of the substantia nigra and cerebral cortical neurofibrillary tangles. A-beta accumulation has been identified in the brain, leading to the suggestion that brain trauma may play a role in the pathogenesis of Alzheimer’s disease.
PARANEOPLASTIC SYNDROMES
Paraneoplastic syndromes (PNS) are a group of rare neurological disorders that represent non-metastatic complications of malignancy in the nervous system. The commonest of these include limbic encephalitis, resulting in dementia due to inflammatory damage in the hippocampus and limbic system, and the Lambert–Eaton myasthenic syndrome, where damage to the motor end plate in muscle fibres results in weakness. Many PNS are associated with circulating antibodies against antigens expressed in both the tumour (often a small cell lung cancer) and the nervous system, suggesting that PNS may result from a misdirected immune response. Treatment of the underlying tumour often results in clinical improvement from the PNS.
CNS TUMOURS
Present clinically with localising signs due to tissue destruction, or with the non-specific effects of raised intracranial pressurePrimary tumours of the CNS occur in approximately 8–12 per 100000 of the general population. Two main peaks of incidence occur: in the first decade and in the fifth or sixth decades of life. In children, CNS tumours are the second commonest group of neoplasms (after leukaemias), but in adults they rate as the sixth commonest group. The relative incidences of the main groups of primary CNS tumours in adults and in children are shown in Figure 26.29.
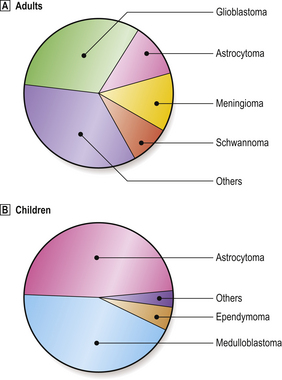
Fig. 26.29 Relative incidence of primary CNS neoplasms. The relative incidences of the commonest primary CNS neoplasms are illustrated for adults and children. Important differences in site also occur in relation to age. In adults, most neoplasms are supratentorial. In children, most arise in the posterior fossa.
Pathogenesis
The pathogenesis of most CNS neoplasms is unknown, but the following factors have been studied:
Classification
As in other organs, tumours of the CNS are classified according to their cellular differentation and presumed cell of origin (Table 26.13).
Table 26.13 Classification of CNS tumours according to presumed cell of origin
| Cell of origin | CNS tumour |
|---|---|
| Glial cells | Astrocytoma, oligodendroglioma, ependymoma, glioblastoma |
| Primitive neuroectodermal cells | Medulloblastoma, neuroblastoma |
| Arachnoidal cell | Meningioma |
| Nerve sheath cells | Schwannoma, neurofibroma |
| Lymphoreticular cells | Lymphoma |
Clinicopathological features
Brain tumours may present clinically in two main ways:
Unlike neoplasms arising in other tissues, primary CNS neoplasms virtually never metastasise to other organs; the reasons for this are not clearly understood. However, infiltration of adjacent tissues both within the nervous system and its coverings (including the skull) is common, for example in meningioma, and seeding to remote parts of the nervous system by the CSF pathway is an important means of spread for certain intrinsic tumours, for example medulloblastomas. Spread by seeding can sometimes occur down a ventriculoperitoneal shunt, resulting in intra-abdominal tumour deposits.
Intrinsic tumours
The commonest group of primary CNS neoplasms are the intrinsic tumours of the brain, which account for all primary CNS neoplasms in children. In adults, intrinsic tumours account for around 65% of primary CNS neoplasms, the majority of which are of glial origin (Fig. 26.29). Intrinsic tumours occur more frequently in male patients.
Astrocytomas
Astrocytomas account for 10% of all primary CNS tumours in adults, but are relatively more frequent in children (Fig. 26.29). They commonly arise in the cerebellum in children, and in the cerebral hemispheres in adults. Astrocytomas are usually classified according to the predominant cell type and degree of differentiation (Fig. 26.30). It is thought that many anaplastic astrocytomas arise as a consequence of dedifferentiation within a pre-existing astrocytic neoplasm. The prognosis for patients with astrocytomas (and gliomas generally) depends on the degree of tumour differentiation, the age of the patient at diagnosis, and the site and size of the neoplasm.
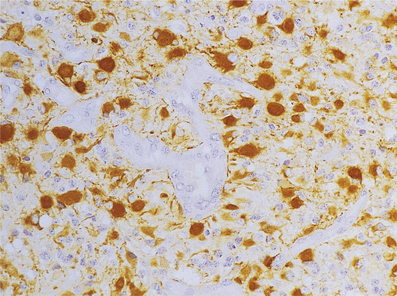
Glioblastoma
Glioblastoma accounts for 30% of all primary CNS tumours in adults, but is extremely rare in children. Most arise in the white matter of the cerebral hemispheres (Fig. 26.6). As its name implies, this neoplasm is characterised histologically by a pleomorphic cell population (Fig. 26.31). Although it is accepted that glioblastomas may arise de novo, it seems likely that many of these neoplasms arise as a consequence of dedifferentiation within a pre-existing astrocytic glioma. Dedifferentiation is accompanied by, or is the result of, a series of genetic events (Fig. 26.32). Mitotic activity in glioblastomas is abundant, and vascular endothelial proliferation is prominent. These features suggest a rapid growth rate; most patients die within 1 year of diagnosis.
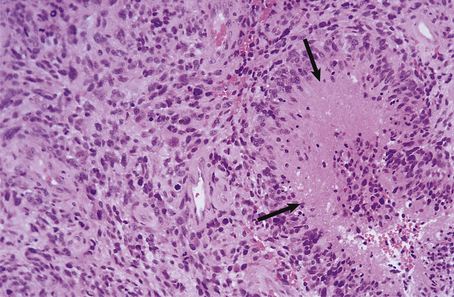
Fig. 26.31 Glioblastoma. Areas of necrosis (arrows) are a characteristic feature of this neoplasm, and are usually surrounded by the nuclei of small malignant cells. The neoplastic cell population is pleomorphic, and also includes multinucleate cells. Vascular endothelial proliferation is another characteristic histological feature.
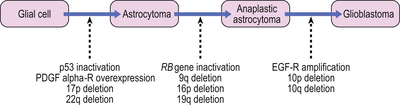
Oligodendroglioma
Oligodendroglioma accounts for 3% of all primary CNS neoplasms in adults, but is rare in children. Oligodendrogliomas are usually ill-defined, infiltrating neoplasms, arising in the white matter of the cerebral hemispheres. Histologically, oligodendrogliomas present a spectrum of appearances which may be graded in a similar manner to astrocytomas. In a well-differentiated tumour, the neoplastic cells are small, rounded and uniform with a clear cytoplasm and prominent cell membrane. Small foci of calcification are common, and a characteristic interweaving vascular pattern is often present. Oligodendrogliomas exhibit a different set of genetic abnormalities to astrocytic gliomas, with losses of chromosomes 1p and 19q.
Ependymoma
An ependymoma arises from an ependymal surface, usually in the fourth ventricle, and projects into the CSF pathway (Fig. 26.33). Most ependymomas are well differentiated, and extensive invasion of adjacent CNS structures is uncommon. A special variant, the myxopapillary ependymoma, occurs in the cauda equina region in adults.
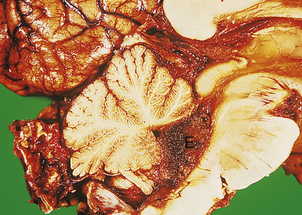
Fig. 26.33 Ependymoma: ventricular obstruction. The ependymoma (E) arising from the lining of the fourth ventricle has almost totally obstructed the CSF pathway and produced obstructive hydrocephalus. This results in characteristic clinical features that are common presenting symptoms for this group of neoplasms.
Choroid plexus papilloma
An uncommon intraventricular papillary growth, choroid plexus papilloma is most often found in a lateral ventricle and usually presents with obstructive hydrocephalus. Although showing little tendency to infiltrate locally, spread via the CSF may occur.
Primitive neuroectodermal tumours
The commonest variety of the primitive neuroectodermal group of tumours is the medulloblastoma, which arises in the cerebellum in children. The growth rate is rapid, and extensive local infiltration is common, often resulting in obstructive hydrocephalus. Meningeal infiltration frequently occurs and CSF seeding is common. As the name implies, these tumours are composed of poorly differentiated neuroepithelial cells which consist of small round nuclei surrounded by a scanty rim of cytoplasm. Mitotic figures are numerous, and evidence of differentiation into mature cell types, such as neurones or glia, is occasionally present. The prognosis for this group of tumours in children has improved in recent years as a consequence of improved treatment with radiotherapy; the 5-year survival rate is around 60%.
Haemangioblastoma
Haemangioblastoma is an uncommon neoplasm arising most often in the cerebellum and forming a well-defined, frequently cystic mass. Histologically, the tumour is composed of blood vessels, separated by stromal cells with clear cytoplasm containing lipid. CNS haemangioblastomas are an important component of von Hippel–Lindau syndrome, an autosomal dominant inherited disease with a genetic locus on chromosome 3p.
Lymphoma
Although an uncommon CNS tumour, there is much current interest in primary CNS lymphomas because of their greatly increased frequency of occurrence in immunosuppressed patients, for example in cardiac and renal transplant patients and in the acquired immune deficiency syndrome (AIDS). Recent studies have implicated the Epstein–Barr virus in the pathogenesis of these neoplasms. Most primary CNS lymphomas are ill-defined masses arising in the white matter or the cerebral hemispheres. Histologically, most are high-grade, non-Hodgkin’s lymphomas of B-cell type. Accordingly, the prognosis is poor and most patients are dead within 2–3 years.
Tumours of neuronal cells
Tumours comprising neuronal elements are rare; they occur most commonly around the region of the third ventricle in children. In gangliocytomas, the neoplastic cells all resemble mature neurones, but gangliogliomas include neoplastic glial cells (usually astrocytic cells).
Miscellaneous cysts
A variety of cystic lesions occur in the CNS which, although not all neoplastic, often present clinically with symptoms and signs similar to those of CNS tumours. Examples include:
Extrinsic tumours
Tumours arising from the coverings of the brain and spinal cord, and from cranial and spinal nerve roots, are less common than intrinsic CNS tumours. Complete surgical removal of extrinsic neoplasms often results in a clinical cure.
Meningiomas
Meningiomas account for around 18% of intracranial neoplasms in adults; female patients outnumber males by 2:1. Meningiomas arise from cells of the arachnoid cap (a component of arachnoid villi). The most frequent sites are the parasagittal region, sphenoidal wing, olfactory groove and foramen magnum. Meningiomas are smooth lobulated masses, which are broadly adherent to the dura. Infiltration of the adjacent dura and overlying bone is not uncommon, but invasion of the brain is exceptionally rare. The brain, however, may be markedly compressed by a meningioma, resulting in considerable anatomical distortion (Fig. 26.34). Histologically, meningiomas display a variety of patterns, the most characteristic of which includes sheets of fusiform cells in a composite solid and whorled pattern. Small foci of calcification (psammoma bodies) are common.
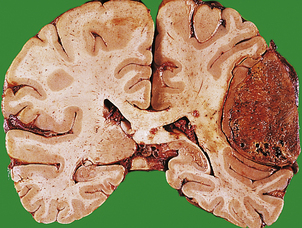
Fig. 26.34 Meningioma: cerebral compression. Meningiomas do not usually invade CNS structures, but may produce clinical manifestations by compression of the adjacent brain. This neoplasm has the lobulated surface characteristic of meningiomas, and is sharply demarcated from the cerebrum.
Occasional meningiomas are frankly malignant and may metastasise outside the CNS, for example to the lung.
Schwannoma
As the name suggests, schwannomas derive from Schwann cells in the nerve sheath of the intracranial or intraspinal roots to sensory nerves. By far the commonest site is the vestibular branch of the 8th cranial nerve in the region of the cerebello-pontine angle; such neoplasms are often known as ‘acoustic neuromas’. As with meningiomas, schwannomas occur most frequently in adults, and are commoner in females. Bilateral 8th nerve tumours commonly occur in patients suffering from neurofibromatosis type 2. Histologically, schwannomas exhibit two main patterns: densely packed spindle-shaped cells with frequent nuclear palisading, and more loosely structured areas with a myxoid stroma which may contain cyst. Malignant change is very uncommon in these tumours.
Neurofibromas
In the CNS, neurofibromas usually arise on the dorsal nerve roots of the spinal cord, and occur most frequently in patients suffering from neurofibromatosis. Unlike schwannomas, neurofibromas are not encapsulated but tend to involve an entire nerve root, producing a localised or diffuse expansion (plexiform neurofibroma). Histologically, neurofibromas consist of a mixture of Schwann cells and fibroblasts, forming bundles of elongated cells with characteristically ‘wavy’ nuclei.
Secondary tumours
The CNS may be involved by other neoplasms in two main ways: compression and invasion, and metastasis.
Compression and invasion
Tumours arising in adjacent organs may compress and invade the CNS, producing localising clinical signs, or presenting as space-occupying lesions. The commonest examples involving the brain are pituitary adenomas, which frequently cause visual impairment due to pressure on the optic chiasm.
Metastasis
The CNS is a common site for metastases, which may occur by haematogenous or direct spread. The commonest neoplasms to metastasise to the CNS are carcinomas of the breast, bronchus, kidney and colon, and malignant melanomas. Metastases often occur at the boundary between grey and white matter (Fig. 26.35) and may present as space-occupying lesions with or without focal signs. Metastatic carcinoma sometimes infiltrates the subarachnoid space producing ‘carcinomatous meningitis’. Patients with this condition present with the symptoms of subacute meningitis, often with multiple cranial nerve palsies. Metastatic deposits within the spinal cord are uncommon, but extradural metastases occur frequently and may present with paraplegia. CNS involvement occurs commonly in acute leukaemias and non-Hodgkin’s lymphomas (Ch. 22) with infiltration of the subarachnoid space and parenchyma. The prognosis for patients with a metastatic neoplasm within the CNS is extremely poor.
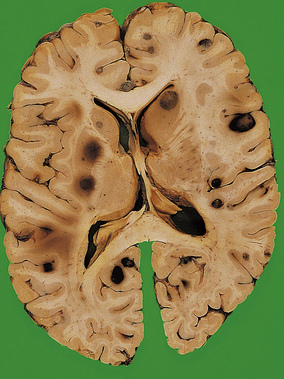
PERIPHERAL NERVOUS SYSTEM
NORMAL STRUCTURE AND FUNCTION
Peripheral nerves may be involved in many diseases, but because they can undergo only a limited number of pathological changes, it is important to consider their normal structure and general pathology before specific disorders are mentioned.
On histology, nerve fibres can be divided into two main groups: myelinated and non-myelinated.
Myelinated fibres
Myelinated fibres range in diameter from 2 to 17µm, with myelin sheaths proportional in thickness to the diameter of the axon. Myelin is formed by the compaction of cell membranes from multiple Schwann cells along the length of the axon, to form a lamellar structure with a periodicity of 14nm. The node of Ranvier is the site where adjacent Schwann cells meet and where their myelin sheaths terminate. This arrangement allows the rapid transmission of electrical impulses by saltatory conduction, up to 10 m/s in the largest fibres.
REACTIONS TO INJURY
Although peripheral nerves may be involved by many disease processes, for example vasculitis or amyloidosis, nerve fibres exhibit only two basic reactions to disease:
These reactions may occur in combination in some peripheral neuropathies; this is usually referred to as combined or mixed degeneration.
Axonal degeneration
Degeneration and loss of axons in peripheral nerves occurs by two main processes: Wallerian degeneration and distal axonal degeneration. Loss of axons in peripheral nerves results in the reduction in amplitude of the conducted impulse, which can be identified on nerve conduction studies.
Wallerian degeneration
Damage to the neuronal body, for example anterior horn cells, spinal nerve roots or nerve trunks, results in degeneration of the axon distal to the site of the injury. In myelinated fibres, this is accompanied by the secondary breakdown of myelin around the degenerate axons (Fig. 26.36). This process is similar to anterograde degeneration in the CNS but occurs more rapidly.
Fig. 26.36 Teased fibre preparations of peripheral nerves in Wallerian degeneration. These show a characteristic fragmentation of the myelin sheath (which appears dark) around the damaged axons.
Regeneration commences 3–4 days following injury; the regenerating axonal sprouts grow at 2–3mm/day. This is accompanied by central chromatolysis in the neuronal perikaryon, and remyelination by Schwann cells. If axonal regeneration and remyelination are successful, the re-innervation of the target organ, for example a motor end-plate of muscle, may occur. Re-innervation is hindered or prevented by factors that inhibit nerve growth, for example ischaemia or cytotoxic drugs, or disrupt the continuity of the perineurium, for example haematoma or scar tissue.
Distal axonal degeneration
The neuronal cell body is responsible for the maintenance of the axon, which often extends for a considerable distance from the perikaryon. When neuronal metabolism is disrupted, the axon often begins to degenerate at its distal end. This form of degeneration is known as a ‘dying-back’ process or distal axonopathy. It usually also results in secondary breakdown of the myelin sheath at the affected site. Axonal regeneration may occur if normal neuronal metabolism is restored before extensive degeneration occurs.
Distal axonal degeneration occurs in various conditions, including vitamin E and B1 deficiencies, acute porphyria, isoniazid and hexacarbon neuropathies.
Segmental demyelination
In segmental demyelination, the continuity of the axon is maintained, but the myelin sheath is broken down over various segments corresponding to the internodes. This results in a marked slowing of impulse conduction along the nerve fibres, detectable on nerve conduction studies.
Primary segmental demyelination
Primary segmental demyelination occurs when damage to Schwann cells results in breakdown of the myelin sheath which they normally maintain. The myelin debris is eventually phagocytosed and digested by reactive macrophages. This can occur in many conditions, such as ischaemia, inherited metabolic disorders such as leukodystrophies, and the neuropathy of diphtheria.
Allergic segmental demyelination
Allergic segmental demyelination occurs when myelin sheaths are stripped and broken down by activated macrophages in the presence of lymphocytes. This mechanism is thought to operate in the Guillain–Barré syndrome. Remyelination of affected segments of nerve can occur in both allergic and primary segmental demyelination. Schwann cells in the affected internodes undergo mitosis within a few days following the injury, after which remyelination can commence.
Hypertrophic neuropathy
Certain chronic peripheral neuropathies for example leukodystrophies and hereditary sensorimotor neuropathy type III, are characterised by hypertrophic peripheral nerves. These are often thickened, with a distinctive ‘onion-bulb’ appearance on microscopy due to the concentric proliferation of Schwann cells around axons in response to repeated segmental demyelination and remyelination.
PERIPHERAL NEUROPATHY
Peripheral nerve disorders are often classified clinically according to the distribution of the lesions:
Additional classifications include the predominant nerve fibre types involved, that is motor, sensory, autonomic or mixed. Many peripheral neuropathies are of a mixed type. Nerve biopsy will in some cases show diagnostic features, for example in amyloid neuropathies or polyarteritis nodosa, but in many cases the aetiology of the neuropathy is not apparent on histology.
TUMOURS AND TUMOUR-LIKE CONDITIONS
Traumatic neuroma
Traumatic neuroma is not a neoplasm but a reactive proliferation of Schwann cells and fibroblasts that occurs at the proximal severed end of a peripheral nerve. Traumatic neuromas contain disordered fascicles of twisted axons, and may produce severe pain (e.g. ‘phantom limb’ pain after amputation) until excised.
Schwannoma
Schwannomas are benign neoplasms that resemble their CNS counterparts histologically (p. 786).
Neurofibroma
Neurofibromas are a common manifestation of neurofibromatosis (see Phakomatoses, p. 776), when they may occur at multiple sites in large numbers and produce gross deformities.
Other tumours
Malignant peripheral nerve sheath tumours are rare; they occur most often in patients with neurofibromatosis, when they sometimes arise from a pre-existing neurofibroma. These neoplasms behave as sarcomas and are frequently fatal. Ganglion cell tumours occasionally arise from autonomic ganglia, particularly in the sympathetic chain. Phaeochromocytomas and neuroblastomas are discussed in Chapter 17.
SKELETAL MUSCLE
Diagnosis of skeletal muscle disorders requires clinicopathological liaison, and cannot be made on muscle biopsy histology alone Three groups of skeletal muscle disorders: neurogenic disorders, myopathies and disorders of neuromuscular transmission Neurogenic disorders and myopathies commonly occur in both children and adults; many of the latter are inherited, e.g. muscular dystrophies Neurogenic disorders may result from lesions affecting motor neurones, nerve roots or peripheral nervesThe diagnosis of skeletal muscle diseases requires multidisciplinary investigation, often involving neurologists, neurophysiologists, neuropathologists, biochemists and geneticists. Muscle biopsy histology can contribute much important information, but it cannot alone be relied upon for a diagnosis. The innervation of muscle can be studied by electromyography and motor nerve conduction studies. Muscle fibres contain the enzyme creatine phosphokinase (CPK), which is released into the blood following muscle fibre damage; its measurement in serum is widely used in the investigation of muscle diseases.
Normal muscle consists of densely packed, uniformly sized myofibres (40–80µm diameter in adults) with peripheral nuclei (Fig. 26.37). The terminal axons supplying each fibre can also be studied using histochemical techniques, but the investigation of motor end plates and subcellular organelles requires electron microscopy. Muscle diseases can be classified clinically and pathologically into three main groups:

Fig. 26.37 Skeletal muscle histology. Normal muscle fibres within the fascicles are of relatively uniform size and are closely packed, with little intervening tissue. The nuclei are at the periphery of each fibre. Histochemical preparation for myosin ATPase demonstrating the normal random mosaic pattern of fibre types within the fascicle (type 1 fibres are dark, type 2a pale and type 2b intermediate in colour). Histochemical preparation for myosin ATPase in a case of chronic spinal muscular atrophy showing a loss of the normal mosaic pattern, with fibre type 2b predominance and grouping.
NEUROGENIC DISORDERS
Neurogenic muscle diseases all result from damage to the muscle innervation. This can occur as a consequence of lesions affecting the motor neurones in the spinal cord and brainstem, motor nerve roots or peripheral motor nerves. The denervated muscle fibres undergo atrophy and are eventually reduced to small clusters of nuclei with very little surrounding cytoplasm. Re-innervation may occur in some longstanding disorders, for example peripheral motor neuropathies, producing the histological appearance of fibre type grouping (Figs 26.37 and 26.38). In progressive disorders, for example motor neurone disease, the anterior horn cells responsible for re-innervation also eventually degenerate, resulting in atrophy of all the fibres in an affected muscle. Four main groups of disorders are responsible for neurogenic muscle disease:
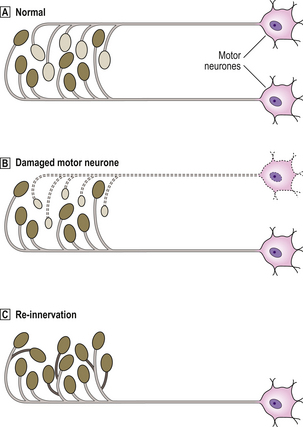
Fig. 26.38 Skeletal muscle: effects of denervation. In normal muscle, the two main fibre types are distributed in a mosaic pattern. The muscle fibre type is determined by its innervation from a motor neurone. A single motor neurone can supply many muscle fibres. Damage to a single motor neurone or its axon results in neurogenic atrophy of muscle fibres; each affected fibre is of the same fibre type. The atrophied denervated fibres can be re-innervated by axons from other motor neurones supplying adjacent fibres. This process can change the fibre type of the re-innervated muscle fibres, resulting in fibre type grouping with loss of the normal mosaic arrangement (see also Fig. 26.37C).
Motor neurone disease
Motor neurone disease is a progressive degenerative disorder affecting principally the anterior horn neurones in the spinal cord. This results in denervation atrophy (Fig. 26.38), fasciculation and weakness in affected muscles.
Spinal muscular atrophy
Spinal muscular atrophy is one of the commonest autosomal recessive disorders, occurring in 1 in 10000 live births. It results from homozygous loss of the survival motor neurone gene 1, and presents in four main forms, which represent allelic variants:
Peripheral neuropathies
Peripheral neuropathies involving motor nerves often present clinically with muscle wasting and weakness, accompanied by sensory loss in a ‘glove and stocking’ distribution. In chronic neuropathies, for example hereditary sensorimotor neuropathies, there is usually evidence of denervation and re-innervation on muscle biopsy.
Miscellaneous spinal cord disorders
There are a number of miscellaneous spinal cord disorders involving the anterior horn cells or the ventral motor nerve roots, for example poliomyelitis, syringomyelia and degenerative diseases of the vertebral column (osetoarthritis and prolapsed intervertebral discs).
MYOPATHIES
The main primary diseases of skeletal muscle may be classified as follows:
Muscular dystrophies
The muscular dystrophies form a group of inherited disorders that result in the progressive destruction of muscle fibres. The muscle innervation is normal in most cases. The most important examples are discussed below.
Duchenne dystrophy
Duchenne dystrophy is an X-linked disorder affecting 1 in 3000–5000 live male births. Approximately one-third of cases represent new mutations. The gene for this disorder has been located to the p21 region of the X chromosome. The gene product, dystrophin, is a protein normally present at the interface between the cytoplasm and the muscle cell membrane. Gene deletions in Duchenne dystrophy result in a deficiency of dystrophin in muscle fibre membranes, causing muscle fibre damage by disruption of the cell membrane, leading to uncontrolled entry of calcium into the cell. Further understanding of the genetic defects in this disorder will allow for a fuller knowledge of its pathogenesis and hence potential for treatment.
The disease usually presents between 2 and 4 years of age, with proximal muscle weakness and pseudohypertrophy of the calves. The serum creatine phosphokinase (CPK) is elevated in the early stages of the disease, and is sometimes also elevated in female carriers. Most patients die before the age of 20, usually of the cardiomyopathy that occurs as part of this condition.
The characteristic biopsy findings are abnormal variation in the diameter of the muscle fibres, with many fibres showing hyaline degeneration or necrosis, with attempts at regeneration (Fig. 26.39). Partial or complete absence of dystrophin can be demonstrated by immunohistochemistry or western blotting of muscle biopsies. Eventually, as the muscle fibre destruction progresses, the muscle is almost totally replaced by fat and connective tissue.
Fig. 26.39 Muscle biopsy in Duchenne muscular dystrophy. Several enlarged densely staining hyaline fibres with numerous small necrotic fibres are present throughout. There is an increased quantity of fibrous and adipose connective tissue (top left) which contributes to the muscular pseudohypertrophy noted clinically in this disease. Compare with Fig. 26.37A.
Becker dystrophy
An X-linked disorder, Becker dystrophy exhibits many similarities to Duchenne dystrophy, but the onset occurs at a later age and the progress of the disease is slower, many patients surviving into adult life. Genetic studies indicate that this disorder is an allelic variant of Duchenne dystrophy, involving deletions in the p21 region on the X chromosome.
Limb girdle dystrophy
The group of disorders known as limb girdle dystrophy are inherited as autosomal recessive conditions. The onset can occur in childhood or adult life, usually with weakness in the pelvic girdle and, later, the shoulder girdle. The progress of the disease is variable, many patients surviving with only mild to moderate disability. Muscle biopsy shows the typical dystrophic features of fibre destruction and regeneration, but to a lesser degree than occurs in Duchenne dystrophy.
Facioscapulohumeral dystrophy
Facioscapulohumeral dystrophy is an autosomal dominant disorder, the genetic locus for which is chromosome 4q35. This disease usually presents in children and young adults with weakness of the face and shoulder girdle. The rate of progress is slow, and many patients survive with only mild disability. Muscle biopsy shows the features of a slowly progressive dystrophy, in which focal lymphocytic infiltration is occasionally present.
Myotonic dystrophy
Myotonic dystrophy is also an autosomal dominant condition, the gene for which has been localised to chromosome 19. The genetic abnormality is an unstable CTG repeat sequence in a cAMP-dependent protein kinase. It usually presents between 20 and 30 years of age with weakness and wasting of facial, limb girdle and proximal limb muscles. Myotonia (persistence of contraction after voluntary effort has ceased) is common in the involved muscles, and patients usually exhibit a number of systemic disorders, including cataract, balding, gonadal atrophy and diabetes mellitus. Characteristic changes are found on electromyography. Muscle biopsy shows dystrophic changes, in which many fibres contain internal nuclei and exhibit a variety of cytoskeletal abnormalities.
Inflammatory myopathies
Muscle can be involved in a variety of infections, most of which are accompanied by a characteristic inflammatory reaction. The infecting organisms may be:
Several systemic inflammatory disorders frequently involve muscle, including sarcoidosis, systemic lupus erythematosus (SLE) and polyarteritis nodosa.
Polymyositis and dermatomyositis
Polymyositis is the commonest inflammatory muscle disorder, occurring most frequently in adults; females are affected more often than males. It may be associated with collagen vascular diseases (Ch. 25), for example systemic lupus erythematosus, or malignancies, for example bronchial carcinoma. Patients usually present with weakness, pain and swelling of proximal muscles. Dermatomyositis is a microangiopathy affecting skin and muscle, where complement deposition causes capillary lysis and muscle ischaemia. The serum CPK is usually elevated in the early stages of both diseases, and characteristic changes are usually present on electromyography.
Histology shows muscle fibre necrosis with phagocytosis of degenerate fibres by macrophages. T-lymphocytes are usually present within the endomysium and around blood vessels. Evidence of muscle fibre regeneration can usually be found, and fibre atrophy may be a striking feature in some cases, particularly in the perifascicular fibres in cases of childhood dermatomyositis.
The muscle fibre damage results from immunological injury by clonally expanded CD8 T-lymphocytes and macrophages. The mechanism of antigen sensitisation is unknown. Treatment with immunosuppressive drugs, such as corticosteroids and azathioprine, is beneficial in many cases.
Inclusion body myositis
Inclusion body myositis is most frequent in elderly patients and clinically resembles polymyositis. Its aetiology is unknown, but affected muscles show inflammation and fibre necrosis associated with small filamentous intracellular inclusions and vacuoles. Unlike polymyositis, it responds poorly to corticosteroids and azathioprine.
Congenital myopathies
Congenital myopathies are uncommon; many of them occur as inherited disorders. Most cases present with hypotonia and floppiness in infancy; these features may prove fatal in severe cases. The diagnosis depends largely on the muscle biopsy appearances, which are thought to reflect delayed development and maturation of the muscle fibres, for example centro-nuclear myopathy, or congenital fibre type disproportion. Hypotonia in infancy is a common manifestation of muscle disease, but may be due to other disorders, including cerebral palsy, hypothyroidism and Down’s syndrome.
Metabolic myopathies
Muscle involvement occurs in many inherited metabolic disorders, such as glycogenosis, carnitine deficiency and mitochondrial disorders. Most of these exhibit other systemic manifestations, for example stroke-like episodes and lactic acidosis in mitochondrial cytopathies. Other metabolic disorders involving muscle include:
Toxic myopathies
Many drugs, for example corticosteroids and penicillamine, can produce muscle damage, which is usually reversible on withdrawal. One of the commonest toxins to affect skeletal muscle is ethanol. Two main patterns of damage are recognised:
DISORDERS OF NEUROMUSCULAR TRANSMISSION
Two main conditions occur in this group: myasthenia gravis and Lambert–Eaton myasthenic syndrome.
Myasthenia gravis
Myasthenia gravis, an autoimmune disorder, usually presents in adults aged 20–40 years, with fluctuating progressive weakness involving particularly the ocular, bulbar and proximal limb muscles. Females are affected more often than males. Over 90% of patients have antibodies against acetylcholine receptor proteins which bind to the post-synaptic receptor and block neurotransmission; anti-striated muscle antibodies are present in a smaller proportion of patients. Linkage with various HLA antigens has been demonstrated, for example A1, B7 and DRw3.
The thymus is hyperplastic in over 50% of patients, and a thymoma is present in a further 15%. The thymus appears to be the site of antigen presentation in this disorder, but the mechanism of sensitisation is unknown. Treatment with cholinergic drugs, immunosuppressive agents such as corticosteroids, plasmapheresis and thymectomy may be beneficial.
Lambert–Eaton myasthenic syndrome
Lambert–Eaton myasthenic syndrome is a rare, non-metastatic complication of malignancy (usually small cell carcinoma of the bronchus) which presents with limb girdle and proximal muscle weakness. Acetylcholine release from motor nerve terminals is impaired by the binding of an abnormal IgG class antibody to presynaptic calcium ion channels.
THE EYE
Vascular disease of the retina in hypertension and diabetes mellitus is a common cause of visual impairmentA brief summary of the anatomical features of the eye is given in Figure 26.40. The unique anatomy and function of the eye mean that the clinical and pathological manifestations of eye diseases often present features not encountered elsewhere.
TRAUMA
Damage to the eye can occur following direct or indirect injuries to the globe. The eye is also susceptible to damage by chemicals, for example ammonia, and physical agents, for example heat and irradiation. Direct injuries to the eye are the most important clinically and may be classified according to the site and nature of the damage; in the perforating injuries, the sclera is only partially torn, but complete rupture occurs in penetrating injuries.
Penetrating and perforating injuries result in the most severe form of traumatic damage to the eye. The immediate complications of penetrating injuries include disruption of the globe, with haemorrhage and detachment of the lens and retina. Infection is a common complication, particularly if the missile is composed of organic material. Sympathetic uveitis and ophthalmitis are uncommon delayed complications of penetrating injuries (see below).
VASCULAR DISEASE
Vascular diseases are a major cause of visual impairment in the middle-aged and elderly. Two main categories are recognised: retinal ischaemia and retinal haemorrhages.
Retinal ischaemia
Retinal ischaemia usually occurs due to the occlusion of a blood vessel by atheroma, vasculitis, thrombosis or embolism. If the central retinal artery is involved, the inner two-thirds of the retina will undergo ischaemic degeneration; occlusion of the posterior ciliary artery damages the photoreceptor cells in the outer retinal layers.
Vascular occlusion in the retina causes exudation of plasma from capillaries. This is seen ophthalmoscopically as ‘hard’ exudates, which appear as discrete, well-defined, pale yellow retinal lesions. Ophthalmoscopy may also reveal ‘soft’ or ‘cotton wool’ exudates; these represent microinfarcts of the retina, involving both ganglion cells and nerve fibres. These lesions are most frequently seen in diabetic and hypertensive retinopathies, both of which are accompanied by changes in the retinal vessels that are readily detectable on ophthalmoscopy (Fig. 26.41).
Fig. 26.41 Diabetic retinopathy. Using fundoscopy, multiple haemorrhages and exudates (arrows) are demonstrable throughout the retina in an adult with longstanding diabetes mellitus.
(Courtesy of Mr B A Noble, Leeds.)
Retinal haemorrhages
Retinal haemorrhages may occur in a number of conditions, for example trauma or infection, but are most commonly found in diabetic and hypertensive retinopathy. Two main patterns of haemorrhage are seen on ophthalmoscopy:
Neovascularisation
Neovascularisation is an important response to retinal ischaemia and haemorrhage, resulting in the proliferation of small vessels around the edge of the lesion. As well as proliferating in the retina, these small vessels may penetrate the vitreous fluid, where the lack of supporting tissue renders them prone to rupture and haemorrhage. Neovascularisation can also occur in response to senile macular degeneration, causing a submacular fibrovascular mass which damages the overlying photoreceptor cells and results in loss of central vision.
INFLAMMATORY LESIONS
Micro-organisms, an important cause of ocular inflammation, can gain access to the eye by haematogenous spread from adjacent tissues, for example the paranasal sinuses, or from the external surface of the eye.
Bacterial infections
Bacterial infections can occur at any site within the eye, but are particularly liable to spread to the vitreous fluid and lens, where the local conditions favour growth of organisms. The cellular reactions to infection in the eye are similar to those elsewhere in the body and will not therefore be described in detail.
Inflammation of the uvea and ciliary body leads to exudation of protein and inflammatory cells into the posterior cornea which can be detected on ophthalmoscopy. Local inflammatory changes can result in adhesions within the anterior chamber, causing glaucoma (see below).
Chlamydial infections
Parasitic infections
Acanthamoeba
Acanthamoeba is a free-living protozoan in mains water supplies. It can cause a corneal infection (keratitis) and may invade the eye, particularly in contact lens wearers. Antibiotic therapy is usually effective, although invasive infections are difficult to eradicate.
Toxoplasmosis
In congenital infections with the protozoan Toxoplasma gondii, the organism spreads to numerous sites in the body. Retinal involvement takes the form of chorioretinitis with extensive tissue destruction and microphthalmos in severe cases.
Toxocara canis
Toxocara canis infection is usually acquired in childhood from contact with ova from infected dogs. Ingestion of the ova is followed by liberation of larvae in the stomach and duodenum; the larvae migrate through the body but do not usually mature. A granuloma can develop in the retina around a dead larva, causing visual obstruction which clinically may mimic an intra-ocular neoplasm.
Sarcoidosis
Ocular involvement is often one of the main manifestations of sarcoidosis, along with erythema nodosum and hilar lymphadenopathy. The granulomatous inflammation characteristic of sarcoidosis occurs in three main forms:
Autoimmune disease
This group of uncommon disorders almost always arises as a consequence of ocular injury, particularly perforating wounds. Prompt clinical attention to such injuries has greatly reduced the incidence of these complications.
Lens-induced uveitis
Release of lens protein into the anterior chamber or vitreous (usually as a result of trauma) occasionally causes a giant cell granulomatous reaction involving the lens and uvea. This results from a delayed hypersensitivity reaction following sensitisation to lens antigens.
Sympathetic ophthalmitis
Trauma to one eye with damage to the iris or ciliary body may cause a delayed hypersensitivity reaction following sensitisation to uveal and retinal antigens. This results in a giant cell granulomatous inflammatory response in either the damaged eye or the second eye. Children are particularly susceptible to this uncommon complication.
CATARACT
The normal structure of the lens depends on the integrity of its elastic capsule, the viability of the lens fibre cells, which contain transparent proteins, and a supply of essential metabolites in the fluid.
Cataracts result from the formation of opaque proteins within the lens which usually also results in a loss of lens elasticity. This can occur in:
Mature cataracts can cause severe visual loss, but this can be treated surgically by removal of the affected lens and insertion of a synthetic plastic substitute. Cataracts occasionally cause glaucoma due to mechanical obstruction of the anterior chamber angle, or lens dislocation.
GLAUCOMA
The normal intra-ocular pressure is 11–21mmHg (1.5–2.8 kPa). This pressure depends on:
Glaucoma denotes a group of common disorders in which the intra-ocular pressure is increased to a level that impedes blood supply to the retina, resulting in optic nerve cupping on fundoscopy and ultimately in blindness. The increase in intra-ocular pressure is usually caused by obstruction to the outflow of aqueous fluid, for example at the trabecular meshwork, canal of Schlemm or the drainage angle of the anterior chamber (Fig. 26.40). Glaucoma affects 1–2% of adults under the age of 40 years, rising to 5% over the age of 70 years. It is commoner in black populations of African origin.
Closed-angle glaucoma
Closure of the irideocorneal angle, thus obstructing the drainage of aqueous humour from the anterior chamber, can occur when the iris is in mid-dilatation, particularly in middle-aged or elderly individuals. This results in acute glaucoma, with corneal oedema, congestion and pain. The next commonest cause of closed-angle glaucoma is neovascularisation around the irideocorneal angle (Fig. 26.42) following a variety of disorders, for example haemorrhage, ischaemia or infection.
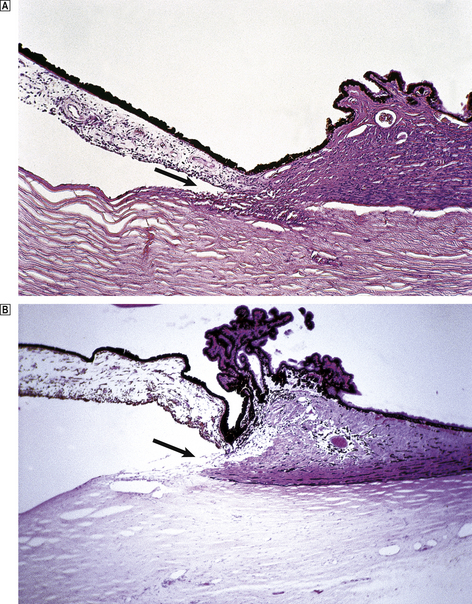
Fig. 26.42 Closed-angle glaucoma: neovascularisation. The aqueous outflow at the anterior chamber angle (arrow) is obstructed by a mass of fibrovascular tissue containing numerous capillaries in contrast to a normal control The resulting increase in intra-ocular pressure caused glaucoma, which eventually necessitated removal of the eye.
Open-angle glaucoma
Open-angle glaucoma can occur as a primary degenerative condition in the elderly, when a progressive accumulation of collagen within the trabeculae and extracellular space of the outflow system increases resistance to the flow of aqueous fluid. This results in a slow increase in intra-ocular pressure which is often manifest clinically as a central visual field defect.
Open-angle glaucoma may also occur due to mechanical obstruction of the outflow system by inflammatory cells, haemorrhage or tumour infiltration. The effects of raised intra-ocular pressure are:
MACULAR DEGENERATION
Macular degeneration is a common cause of visual loss in the elderly in the UK. Progressive damage to the macula, the most sensitive region of the light-sensitive retina (Fig. 26.40), results in a gradual loss of central and detailed vision. Macular degeneration usually occurs over the age of 60 years and is more common in females. However, rare inherited varieties occur, and may affect younger patients. The precise causes of macular degeneration are uncertain. Two main types occur:
OCULAR TUMOURS
A large variety of neoplasms may arise within the eye and its adnexa, tumours in the latter resembling those occurring in the skin, connective tissue and salivary glands. The most important intra-ocular tumours are naevi and malignant melanoma, and retinoblastoma.
Naevi and malignant melanoma
Naevi and malignant melanoma occur most frequently in adults, and derive from the melanocytes of the uveal tract. Naevi are benign melanocytic lesions akin to those commonly occurring in skin. Malignant melanomas occur as a solitary mass in one eye, usually arising in the posterior choroid. The neoplasm often grows rapidly to form an intra-ocular mass that causes extensive retinal detachment and secondary glaucoma (Fig. 26.43). Histologically, two main patterns are recognised:
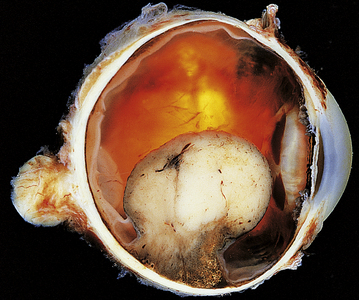
Fig. 26.43 Malignant melanoma: intra-ocular mass. In this eye, a large unpigmented malignant melanoma is arising from the choroid. The neoplasm has caused extensive retinal elevation and detachment.
Melanomas may also arise in the iris; these are associated with a better prognosis and seldom metastasise.
Retinoblastoma
There has been much recent interest in retinoblastoma. It is an uncommon neoplasm, with an incidence in the UK of around 1 per 20000 live births; 5–10% of cases are familial, with affected individuals inheriting a deletion on the long arm of chromosome 13 which always involves the RB tumour suppressor gene on band q14. The same chromosomal abnormality occurs in tumour tissue (but not normal tissue) from patients with sporadic retinoblastomas (Ch. 11).
Children with retinoblastomas present with visual loss, squint or enlargement of the eye, which is occupied by a tumour within the retina. On histology, the neoplasm has the features of a primitive neuroectodermal tumour, in which the cells tend to form rosettes. Local extension along the optic nerve or through the sclera is common, but distant metastases are rare. The results of early enucleation and radiotherapy are good, with a 5-year survival rate of around 90%.
Optic nerve glioma
Optic nerve glioma is a rare neoplasm which occurs most frequently in children and young adults; it is a well-recognised complication of neurofibromatosis and tuberous sclerosis. Patients usually present with progressive visual failure, with proptosis and papilloedema. The histological features are those of a pilocytic astrocytoma. The results of surgery and radiotherapy are good, with over 90% of patients surviving for 5 years or more.
THE EAR
MIDDLE EAR
The middle ear and mastoid air cells of the temporal bone are extensions of the upper respiratory tract, and are lined by ciliated epithelium. Infections in these sites are common, particularly in children, and may occur as part of a generalised upper respiratory tract infection.
Otitis media
Acute otitis media
Acute otitis media may result from primary or secondary bacterial infections; the latter occasionally complicate a viral illness. Acute bacterial otitis media is a suppurative inflammatory process most often caused by Haemophilus influenzae or Streptococcus pneumoniae. The inflammatory exudate can cause the tympanic membrane to bulge and rupture, and may spread to the mastoid air cells, causing acute mastoiditis. This condition usually responds rapidly to antibiotics.
Serous otitis media
Serous otitis media is a non-suppurative process in which fluid accumulates in the middle ear as a consequence of Eustachian tube obstruction. It is an important cause of hearing difficulties in children (‘glue ear’) and can be relieved by removing the Eustachian obstruction, for example in patients with tonsillar hyperplasia.
INNER EAR
The most important clinical manifestations of inner ear disorders are deafness and dizziness. These occur in varying degrees, due to impaired cochlear and vestibular function. Among the most common disorders affecting the inner ear are labyrinthitis, Ménière’s disease and otosclerosis.
Labyrinthitis
Infections of the labyrinth are usually viral: mumps, cytomegalovirus and rubella are the organisms most frequently involved. The inflammatory process usually subsides spontaneously.
Ménière’s disease
Ménière’s disease is an uncommon disorder characterised clinically by attacks of nausea, vertigo, nystagmus, tinnitus and hearing loss. The pathogenesis is unknown, but the disease results in distension of the endolymphatic system in the cochlear duct and saccule. The vestibular membrane of Reissner may rupture, and the distended saccule compresses adjacent structures. The aetiology of Ménière’s disease is uncertain, but similar symptoms may occur in post-infectious labyrinthitis following upper respiratory tract viral infections.
Otosclerosis
Otosclerosis is one of the commonest causes of hearing loss in young adults. It affects females more often than males, and is inherited as an autosomal dominant trait with variable penetrance. The conductive hearing loss results from bone deposition around the stapes footplate, which eventually results in ankylosis. The disease can be treated surgically by stapedectomy.
Commonly confused conditions and entities relating to the central and peripheral nervous systems
| Commonly confused | Distinction and explanation |
|---|---|
| Extradural and subdural haemorrhage | Extradural haemorrhage is usually associated with a skull fracture, resulting in rupture of an artery (e.g. middle meningeal artery); the bleeding is rapid and at relatively high pressure. Subdural haemorrhage is slower venous bleeding leading to formation of a blood clot between the dura and the brain. |
| Saccular aneurysm and microaneurysm | |
| Meningitis and meningism | Meningitis is inflammation of the meninges, whereas meningism is a set of symptoms and signs (headache, photophobia, neck stiffness) indicating meningeal irritation, e.g. by inflammation, subarachnoid haemorrhage. |
| Senile plaques and neurofibrillary tangles | Both occur in the brain in Alzheimer’s disease. Senile plaques are larger and extracellular, with an amyloid protein core. Neurofibrillary tangles are smaller and intracellular and contain tau, a microtubule-associated protein. |
| Multiple sclerosis and systemic sclerosis | Multiple sclerosis is a demyelinating disorder of the nervous system. Systemic sclerosis is a connective tissue disorder also known as scleroderma. Both are probably autoimmune disorders, but there is no other relationship between them. |
Dubowitz V., Sewery C.A.. Muscle biopsy: a practical approach, 3rd edn. London: Saunders; 2007.
Ellison D., Love S., editors. Neuropathology: a reference text of CNS pathology, 2nd edn. 2004 Mosby, Edinburgh
Fuller G., Manford M. Neurology: an illustrated colour text 2nd edn. 2006. Edinburgh: Churchill Livingstone
Graham D.I., Bell J.E., Ironside J.W.. Color atlas and text of neuropathology. London: Mosby-Wolfe; 1995.
Gray F., de Girolami U., Poirier J., editors. Manual of basic neuropathology, 4th edn, Philadelphia: Butterworth-Heinemann, 2004.
Ironside J.W., Moss T., Louis D.N., Lowe J.S., Weller R.O.. Diagnostic pathology of nervous system tumours. Edinburgh: Churchill Livingstone; 2002.
Lee W.R.. Ophthalmic histopathology, 2nd edn. London: Springer; 2002.
Louis D., Ohgaki H., Wiestler O., Cavanee W.K., editors. WHO classification of tumours of the central nervous system, 4th edn, Lyons: IARC, 2007.
Love S., Louis D.N., Ellison D.W., editors. Greenfield’s neuropathology, 8th edn, London: Hodder Arnold, 2008.
National CJD Surveillance Unit, UK: http://www.cjd.ed.ac.uk
Neuroanatomy and Neuropathology on the Internet: http://www.neuropat.dote.hu
Neuropathology: an illustrated interactive course for medical students and residents: htpp://www.neuropathologyweb.org
Neuropathology and Neuroimaging Laboratory (neuropathology slides): http://www.urmc.rochester.edu/neuroslides/index.html
The Whole Brain Atlas: http://www.med.harvard.edu/AANLIB/home.html