13 Cleft lip and palate, and syndromes affecting the craniofacial region
A syndrome is the association of several clinically recognizable signs and symptoms, which can occur together in an affected individual. A large number of syndromic conditions involve the craniofacial region (Gorlin et al, 2001) and these can be broadly subdivided into:
Single gene disorders are the result of specific gene mutations and are inherited according to Mendelian rules, with varying levels of penetrance and expressivity within pedigrees:
Cytogenetics, or the study of chromosomal abnormalities, has also revealed a wide range of physical chromosomal alterations, including variations in both number and structure, which can cause perturbations of gene function and congenital malformations.
Teratogenic agents come in many forms and can include:
Identifying candidate genes for genetic conditions
Elucidating the genetic basis of an inherited condition is not a straightforward task. The human genome contains over 3 billion base pairs within the entire DNA sequence and some genetic disorders can arise from a change in only a single one of these. One of the main obstacles is the location and identification of the causative gene, a process that has become easier with advances in molecular biology and bioinformatics (Box 13.1).
Box 13.1 The human genome project
Publication of the draft human genome sequence (Lander et al, 2001; Venter et al, 2001) has provided an important global resource that will have an effect upon all our lives. Within the human genome there are approximately 30,000 genes distributed across the 23 chromosomes and access to this sequence information has important implications for molecular medicine. In particular, geneticists are now able to identify disease genes and position them within the genome much more easily. A candidate sequence can be entered into sophisticated online browsers and the whole human DNA sequence searched in a matter of minutes. This knowledge is also allowing the development of more rapid and specific tests for the presence of, or susceptibility to certain genetic diseases; which will lead to earlier diagnosis and hopefully treatment of these conditions. In addition, knowledge gained from the human genome will allow progress to be made in therapeutics, with the design of drugs that function at the molecular level and target the specific causes of the disease rather than simply controlling the effects. Finally, the sequence is also an invaluable resource for biologists, providing valuable insight into human evolution and diversity.
For single-gene disorders, positional cloning aims to identify, or at least localize, the chromosomal region where a candidate disease gene may reside. Geneticists use markers within the genome that can be tracked through members of an affected pedigree and provide linkage to the candidate region for a particular condition:
Modern geneticists use DNA polymorphisms as markers. These are identifiable sequence variations situated at specific positions within the genome. By identifying those most closely associated with a disease locus, a region of DNA at a specific point within the genome that might harbor the candidate gene for a particular disease is identified. Once the region has been narrowed down in this way, specific genes within the locus can be investigated.
Unfortunately, many disorders are multifactorial and do not behave in a simple Mendelian manner. These conditions have a more complex aetiological basis, being the combined product of:
A good example of a multifactorial condition that can affect the craniofacial region is nonsyndromic cleft lip and palate. For these conditions, populations rather than family pedigrees usually must be investigated, and more complex methods of genetic mapping are required. For this reason, there has been less success in elucidating the basis of multifactorial conditions than that of single-gene disorders.
Cleft lip and palate
Clefts involving the lip and/or palate (CLP) or isolated clefts of the palate (CP) are the commonest congenital anomaly to affect the craniofacial region in man (Fraser, 1970). They represent a complex phenotype and reflect a failure of the normal mechanisms involved during early embryological development of the face. In human populations CLP and CP can be broadly subdivided into:
Classification
A number of formal classifications have been described for CLP and CP; however, the clinical presentation of these conditions is so variable that a specific description of each individual case is more useful (Fig. 13.1).
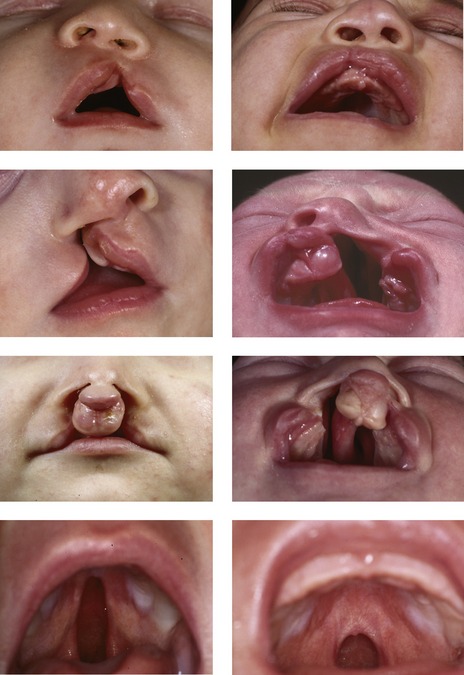
Figure 13.1 Orofacial clefting.
Unilateral cleft lip (top row), unilateral cleft lip and palate (second row), bilateral cleft lip and palate (third row), isolated cleft palate (bottom row).
Epidemiology
Aetiology
Whilst a number of causative genes have been identified for different types of syndromic CLP and CP (Table 13.1), the aetiology and pathogenesis of nonsyndromic forms are poorly understood. This is a reflection of the multifactorial nature of these conditions, being the result of genetic and environmental interactions affecting development of the face at specific time points during embryogenesis. It has been suggested that up to fourteen different genetic loci may be involved in nonsyndromic CLP, which means that very large and genetically pure sample sizes are required to identify specific causative genes (Lidral & Murray, 2004).

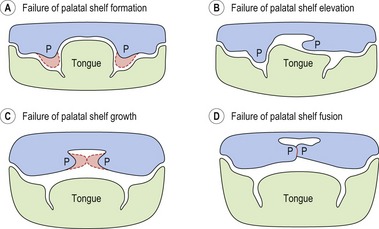
At the embryological level, perturbations in a variety of mechanisms during facial development are known to cause clefting (Fig. 13.2). A growing number of mutant mouse strains that exhibit CP (and to a lesser extent, CLP) have now been generated and these continue to provide a host of candidate genes for the human condition (Gritli-Linde, 2007; Jiang et al, 2006).
Treatment
A child born with orofacial clefting will require complex long-term treatment, depending upon the severity of the cleft, and there may be lifelong implications for those individuals unfortunate enough to be affected. The principle objectives of treatment are to establish:
If these objectives are achieved, they maximize the chances of an affected child growing up and developing normally within their social environment.
The clinical management of children born with clefting is most effective when carried out by a fully integrated team, in a centralized unit that treats a high number of patients (Box 13.2). The modern cleft team therefore includes a number of key members, in addition to other specialists who may be involved with long-term care (Table 13.2).
Box 13.2 Providing care for patients with orofacial clefting in the United Kingdom
In the late 1980s, some concern was raised amongst healthcare professionals regarding the quality of care being provided for children born with CLP or CP in the UK. This was based principally upon the outcome of two studies:
This concern led to the establishment of a Clinical Standards Advisory Group (CSAG) national investigation into cleft care in the UK. This study reported upon clinical outcome in a total of 457 5- and 12-year-old children affected by nonsyndromic unilateral CLP (Sandy et al, 1998). On the basis of this investigation, the CSAG Cleft Lip and Palate Committee made a number of recommendations regarding the future provision of cleft care in the UK:
These recommendations reflected the findings that high-quality clinical outcome in cleft care was primarily associated with centralization of services; providing clinical operators that treated large numbers of patients the correct environment for high-quality training and the infrastructure to establish effective clinical audit and intercentre comparison. Inevitably, implementation of these recommendations has required a considerable reduction in the number of centres and personnel providing cleft care in the UK, a feat that has not been achieved without some opposition.
Table 13.2 Members of the modern cleft team
Birth
Giving birth to a child affected by a cleft can be a distressing experience for the parents, particularly if this condition has not been diagnosed in utero (Fig. 13.3). A multitude of emotions can occur, including shock, anger, guilt, grief and even rejection. It is important that adequate support is given to the parents and that a bond is quickly established between the parents and child.
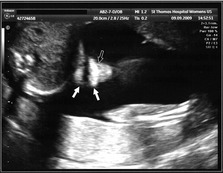
Figure 13.3 Lip development can be monitored using ultrasound scanning. The external nares of the nose (outlined arrow) and both lips (white arrows) can be seen on this 16 week scan. The baby is on its side and development of these structures is normal.
A baby born with CLP may experience difficulty in feeding at birth. CP produces an open communication between the oral and nasal cavities. Suckling can be slow because the baby will have difficulty generating adequate intraoral pressure and milk can be lost through the nose before it is swallowed. It is important to establish an effective feeding regime as soon as possible:
Presurgical orthopedics
A period of active presurgical orthopedic alignment of the cleft alveolar segments is occasionally carried out in the neonate to reduce the size of the cleft defect and facilitate surgical repair. Specialized facial strapping or orthodontic plates (Fig. 13.4) are used, which can be passive or active and help mould or reposition the divided facial and maxillary segments. In particular, these plates have been used for:
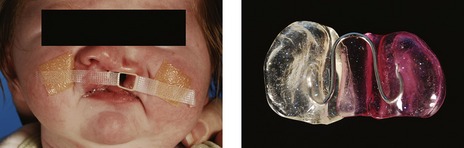
Figure 13.4 Orthopedic cleft appliance to approximate the lip segments prior to repair (left panel) and intraoral orthopedic appliance to approximate the palatal shelves.
Courtesy of Christoph Huppa.
Presurgical orthopedic treatment is usually carried out at the discretion of the operating surgeon. There is currently little substantive evidence to suggest that any of these techniques provide long-term benefit for the dental arch relationship or facial appearance and their use remains controversial.
Surgical repair of cleft lip and palate
A number of individual surgical techniques for repairing the embryonic deficits associated with both the lip and palate have been described. However, evaluating which technique, sequence or timing will provide optimum results is difficult and currently no true consensus for any of these criteria exists (Roberts-Harry & Sandy, 1992; Sandy & Roberts-Harry, 1993).
Early surgery does allow the child to establish good orofacial function as soon as possible and this is particularly important for the development of normal speech. However, surgical repair can be associated with scarring in the maxillary region, which can produce growth deficiencies in all three planes of space:
It is clear from comparative studies that facial growth is compromised in operated cleft subjects when compared with those from unoperated samples, particularly those that have undergone palatal repair (Mars & Houston, 1990). The goal of surgical correction is to minimize any potential growth discrepancy, whilst maximizing the aesthetic and functional outcome.
Lip repair
Surgical repair of cleft lip is usually carried out between 3 and 6 months of age as a single procedure (Fig. 13.6), the exact age being dictated by surgeon preference. Classically, the rule of ‘tens’ has been used, with surgery only taking place once the child is at least 10 weeks old, 10 pounds in weight, and having a haemoglobin level of 10%. However, waiting until these criteria are achieved can delay surgery and it has been argued that this can cause problems with both parent–infant bonding and early growth and development. Indeed, advances in neonatal care and paediatric anaesthesia have made it possible to perform cleft surgery during the neonatal period, although there is currently no clear evidence to suggest that this is particularly advantageous (Schendel, 2000).
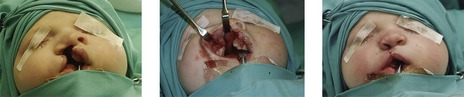
Bilateral cleft lip is also repaired with a single procedure, involving simultaneous correction of the lip, nose and alveolus (Mulliken, 2000). The upper lip orbicularis oris muscle must be freed from each lateral cleft element and repaired in the midline anterior to the premaxilla.
Palate repair (palatoplasty)
The timing of palate repair represents a balance between maximizing the positive effects of early palate closure on feeding and speech development, whilst minimizing the potentially negative effects of inhibited maxillary growth and development as a result of surgical scarring.
Currently, repair of CP is normally undertaken between 9 and 12 months of age and usually involves a palatoplasty to move tissue towards the midline, with or without some lengthening of the palate to improve the posterior soft palate seal (Fig. 13.7).

Speech and language
Following repair of the palate, a speech and language therapist monitors speech development closely. Velopharyngeal insufficiency (VPI) is the result of an inadequately functioning soft palate, which may be unable to lift and produce a good seal with the posterior pharyngeal wall. VPI can produce:
The main problem is a lack of mobility in the soft palate, secondary to scarring from the palate repair. In combination with the dental abnormalities, malocclusion and hearing difficulties, which are all often seen in cleft children, VPI can also produce poor articulation, which results in significant difficulties with speech.
VPI is diagnosed by speech assessment and confirmed with videofluoroscopy or nasoendoscopy. Treatment involves surgery combined with speech and language therapy. Surgery is usually carried out as soon as VPI is diagnosed, around the age of 4 before the child begins school, and generally involves either a re-repair of the soft palate or pharyngoplasty. Pharyngoplasty aims to reduce hypernasality by narrowing the velopharyngeal space.
Middle ear disease
Otitis media is also a common finding in children with CP, disruption to the muscles of the soft palate affecting function of the eustachian tube. This can reduce the acuity of their hearing, causing further potential adverse effects on the development of speech and language. It is important that an audiologist monitors these children and if necessary, tympanostomy tubes (or grommets) are placed by an ear–nose–throat (ENT) surgeon.
Dental care during the deciduous dentition
It is important that a program of preventative dentistry is established during early dental development, particularly as many children affected by clefting are vulnerable to caries. Dietary advice should be provided, good oral hygiene established and fluoride supplementation instituted, if necessary.
There is occasionally delay in the eruption of deciduous teeth adjacent to the cleft and the deciduous lateral incisor can be absent, hypoplastic or even duplicated. Crossbites can occur in the buccal segments; however, orthodontic treatment is rarely indicated in the deciduous dentition.
Dental care during the mixed dentition
As the permanent teeth begin to erupt, crossbites affecting both the incisor and molar dentitions can occur and their severity often reflects the degree of disruption that has occurred to maxillary growth and development as a result of previous surgery. The maxillary incisors can also be crowded, rotated and tilted, particularly those adjacent to the cleft (Fig. 13.8).
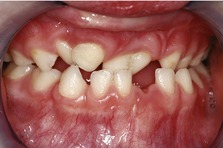
Dental preventative measures should continue during the mixed dentition; in particular, the first molars should be fissure sealed and monitored to ensure these teeth do not become carious. It is particularly important in cleft cases to avoid the potential occlusal complications associated with early loss of deciduous teeth or first molars.
Alveolar bone grafting
The presence of a residual bony defect in the maxillary alveolus of children affected by complete clefts is a deformity associated with a number of functional and aesthetic problems, which can affect both the occlusion and local orofacial region:
Alveolar or secondary bone grafting involves placing cancellous bone, usually harvested from the iliac crest, directly into the maxillary alveolar defect. This procedure is normally carried out at around 8–10 years of age, prior to eruption of the permanent canine, when root formation of this tooth is around two-thirds complete. A period of orthodontic treatment is usually required prior to graft placement to expand the collapsed maxillary arch and create surgical access, maximizing the amount of bone that can be placed (Fig. 13.9). This expansion is often achieved with a tri- or quadhelix appliance, followed by a period of retention with a palatal arch. During this phase of orthodontic treatment, some alignment of the maxillary incisors can be achieved by either extending the arms of the helix or using a simple fixed appliance, but care needs to be taken not to move any teeth into the cleft site where there is no bone, and if this is a concern, these teeth should be aligned after the bone graft.

Figure 13.9 Upper anterior occlusal radiographs of a right-sided unilateral CLP prior to alveolar bone grafting (ABG), following orthodontic expansion and post ABG. Note that the UR2 and UR3 are beginning to erupt following the graft.
Courtesy of Shivani Patel.
Alveolar bone grafting has made a significant contribution to the oral rehabilitation of children with cleft palate (Table 13.3) (Bergland et al, 1986). Traditionally, orthodontic treatment was aimed at tooth alignment followed by expansion of the maxillary arch to create space in the region of the cleft for the placement of a denture or bridge. This approach was associated with a number of significant disadvantages:
Table 13.3 Advantages of alveolar bone grafting
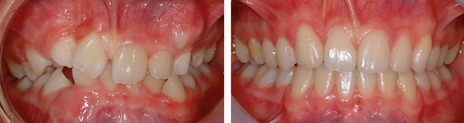
Grafting cancellous bone into the cleft allows teeth to erupt into this region and facilitates tooth movement, which means that orthodontic tooth alignment and space closure can be achieved (Fig. 13.10). Moreover, the timing of alveolar bone grafting means that it does not interfere with growth in the width and length of the anterior maxilla, because this is essentially complete by 8 years of age; whilst vertical development of the maxilla would appear to continue normally after the insertion of a bone graft.
Dental care during the permanent dentition
Once the permanent dentition is established a decision is made regarding the need for orthodontic treatment alone to correct any malocclusion, or a combination of orthodontics and orthognathic surgery. A key factor is the degree of maxillary and midfacial retrusion, but it should be remembered that these patients also exhibit a full range of mandibular growth patterns and mandibular prognathia can also be seen. A period of time monitoring further facial growth may be required before a final decision is made, but if surgery is indicated, presurgical orthodontic treatment will usually begin once facial growth is complete. Occasionally, in cases with severe maxillary retrusion, osteogenic distraction is employed to move the maxilla forwards in a younger, growing patient. This will provide some early improvement in the profile and reduce the size of the jaw movements that will be required for definitive orthognathic surgery in the late teenage years, once facial growth is complete.
In those cases that can be treated with orthodontics alone, there are often a number of specific problems that exist:
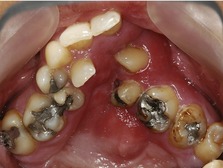
Figure 13.11 Severe crowding secondary to maxillary hypoplasia in a patient with a repaired unilateral CLP.
Orthodontic treatment with fixed appliances is usually indicated in these cases, often in conjunction with some maxillary expansion; but following the same general principles of treatment planning for any malocclusion. Correction of the severely rotated teeth and posterior crossbites often seen in these cases usually requires long-term retention.
Surgical revision
In young adults affected by complete clefts the nasal aesthetics can be poor; in particular, the nose can be asymmetric at the tip and alar base and may require some surgical revision. In addition, revision of any surgical scarring associated with the lip repair may also be required. These procedures are usually carried out following the completion of definitive orthodontic or combined treatment in the late teenage years.
Cleidocranial dysplasia
Cleidocranial dysplasia (CCD; OMIM 119600) is an autosomal dominant skeletal dysplasia characterized by defective ossification of both intramembranous and endochondral bones, in combination with severe dental anomalies. Mutations in the RUNX2 (formerly CBFA1) gene situated on chromosome 6p12-21 have been identified as the cause of CCD (Lee et al, 1997; Mundlos et al, 1997). RUNX2 encodes a transcription factor essential for the terminal differentiation of bone-forming osteoblasts. Mice generated with targeted disruption in Runx2 have a complete absence of bone and they die at birth (Komori et al, 1997; Otto et al, 1997).
In humans, the bones most severely affected are those that develop in membrane:
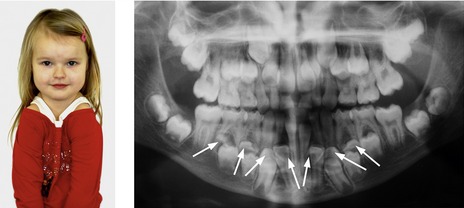
Figure 13.12 Clavicular hypoplasia allowing shoulder approximation (left) and multiple supernumerary teeth (right, arrowed), both features of cleidocranial dysplasia.
The dental features of CCD include a triad of:
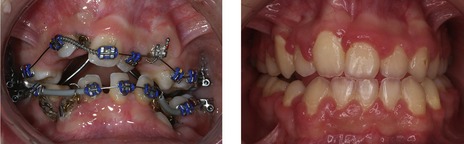
The dental phenotype is highly penetrant and can produce considerable problems, usually manifesting as retention and progressive deterioration of the deciduous dentition with only variable degrees of permanent tooth eruption (Fig. 13.12). Historically, the dental management of CCD has involved either prosthetic replacement of the permanent teeth, with or without judicious tooth extraction, or surgical repositioning and transplantation of affected teeth. A more coordinated approach combines deciduous and supernumerary tooth extraction with surgical exposure and bonding in an attempt to achieve as normal an occlusion as possible (Becker et al, 1997a, b). However, the impacted permanent teeth of individuals affected by CCD can be extremely resistant to orthodontic traction (Fig. 13.13).
Ectodermal dysplasia
The ectodermal dysplasias (ED) represent a heterogeneous group of conditions characterized primarily by defective:
The hypohidrotic or anhidrotic forms of ED are the most common and amongst these, X-linked recessive hypohidrotic ectodermal dysplasia (XLHED; OMIM 305100) is seen most frequently. Affected males have the following clinical features:
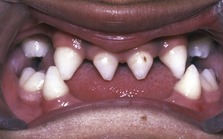
Female carriers in the heterozygous state can also be affected by XLHED, but the clinical features are generally less severe.
An autosomal recessive form of hypohidrotic ectodermal dysplasia (ARHED; OMIM 224900), which is clinically indistinguishable from XLHED, also exists; however, in ARHED affected males and females can exhibit the full clinical spectrum of anomalies.
Autosomal dominant hypohidrotic ectodermal dysplasia (ADHED; OMIM 129490) has also been documented, but this is rare, only being described in a few family pedigrees.
These hypohidrotic forms of ED are caused by disruption to the ectodysplasin (EDA) signalling pathway. This pathway is active in organs that develop via reciprocal signalling between epithelium and mesenchyme and is essential for their normal development (Box 13.3). The EDA signal is initiated by binding of ligand to the EDA type 1 receptor (EDAR). This binding event results in recruitment of an EDAR-associated death-domain protein (EDARRAD). EDARRAD acts as an adapter within the cell cytoplasm, interacting with a number of molecules, which ultimately mediate activation of a signalling pathway called NFkB. XLHED is caused by mutation in the gene encoding the EDA ligand (EDA1) (Xq12-13.1) (Kere et al, 1996), whilst mutation in EDAR can cause both ADHED and ARHED (Monreal et al, 1999).
Box 13.3 What can the mouse tell us about ectodermal dysplasia?
Ectodysplasin signalling pathway.
A number of naturally occurring, spontaneous mouse mutants mimic very accurately the features of human hypohidrotic ED. Tabby, downless and crinkled mutant mice have indistinguishable phenotypes from each other. These mouse mutants have absent or abnormally shaped teeth, a lack of sweat glands, regional alopecia and anomalies in the texture of coat hair; all features of hypohidrotic ED. The genes responsible for each of these mutants have been cloned; Tabby also resides on the X chromosome and is equivalent to human EDA1, downless to EDAR and crinkled to EDARRADD. Indeed, it was only after the positional cloning of crinkled in the mouse that the equivalent gene was identified in humans. Therefore, disruption within the same signalling pathway produces an almost identical phenotype in mouse and human.
Although teeth, hair and sweat glands would at first glance appear to be a very diverse group of structures, their embryonic origins are surprisingly similar. All these appendages develop from interactions between epithelial and mesenchymal tissues and the EDA signalling pathway plays a key role in this process. In the developing tooth Tabby, downless and crinkled have distinct expression domains in different regions of the epithelium. Whilst a lack of EDA signalling results in the hypodontia or anomalies of tooth shape seen in ED, the mouse has also been used to demonstrate that too much EDA signalling can produce the opposite phenotype: supernumerary teeth.
Hemifacial microsomia
Hemifacial microsomia (HFM; OMIM 164210) is a relatively common condition associated primarily with unilateral developmental defects in the orofacial region (Fig. 13.15). HFM occurs in around 1 : 5,600 children and is usually sporadic, although autosomal dominant familial cases have been reported.
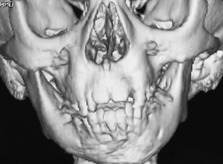
A wide spectrum of clinical features are seen in association with HFM, but a common presentation is:
These skeletal defects characteristically become more pronounced as facial growth progresses.
The soft tissues are also affected in cases of HFM:
Less commonly, CLP, palatal and tongue muscle hypoplasia and velopharyngeal insufficiency are also present. A related condition is Goldenhar syndrome, which incorporates vertebral anomalies and epibulbar dermoids in combination with the features described above.
The aetiology of HFM is not fully understood, but a disruption during early development of the pharyngeal arches is consistent with the phenotype. Haemorrhage of the stapedial artery has been described as one possible mechanism (Poswillo, 1973) and a transgenic mouse line harbouring an insertional mutation within chromosome 10 has been shown to phenocopy the syndrome; this mouse demonstrates microtia, an asymmetric bite and anomalies of the external auditory canal, middle ear and maxilla (Cousley et al, 2002; Naora et al, 1994).
Linkage analysis in human populations has suggested the GOOSECOID (GSC) gene as a potential candidate gene for HFM (Kelberman et al, 2001). GSC encodes a transcription factor strongly expressed in the pharyngeal arches and mice lacking Gsc function have a number of craniofacial defects, including hypoplasia and a lack of coronoid and angular processes in the mandible, and defects in the maxillary, palatine and pterygoid bones (Rivera-Perez et al, 1995; Yamada et al, 1995).
Treacher Collins syndrome
Treacher Collins syndrome or mandibulofacial dysostosis (TCS; OMIM 154500) is a rare autosomal dominant disorder of facial development that occurs in around 1 : 50,000 live births (Fig. 13.16). The regions of the face affected are those derived from pharyngeal arches 1 and 2, but there can be considerable variation in the severity of clinical presentation, even amongst individuals within the same pedigree.

A characteristic facial appearance is common:
Individuals with TCS usually undergo extensive hard and soft tissue reconstruction during the first two decades of life. This treatment is aimed primarily at improving respiratory function and reconstructing the affected soft and hard tissues. The management of such cases requires a dedicated and multidisciplinary team approach (Kobus & Wojcicki, 2006).
A large-scale and collaborative effort identified TCOF1 as the gene mutated in TCS (Dixon et al, 1996). TCOF1 encodes the Treacle nucleolar phosphoprotein thought to have a key role in ribosomal biogenesis, which is essential for normal cell growth and differentiation. Unfortunately, efforts to study TCS using mouse models have been hampered because mice lacking the function of only one Tcof1 allele have a TCS phenotype even more severe than that found in the human, depending upon their genetic background (Dixon & Dixon, 2004). These mice have a compromised production of mature ribosomes in cranial neuroepithelial and neural crest cells, which leads to diminished proliferation and a massive increase in programmed cell death associated with the early neural tube, and a reduction in neural crest cell migration into the early craniofacial region. Therefore, a primary defect in TCS is the production of neural crest cells that populate the first and second pharyngeal arches early in development; cells ultimately responsible for producing much of the facial skeleton (Box 13.4).
Box 13.4 Rescuing a mouse model of Treacher Collins syndrome
Rescue of cell death in the Treacher Collins mouse model by inhibition of p53. Normal neural crest cell proliferation, migration and craniofacial development in the wild type mouse (left). In the Tcof1 mutant, premature death of neural crest cells results in hypoplasia of facial bones and the Treacher Collins phenotype (middle). Inhibition of p53 in the mutant prevents much of the premature death associated with neural crest cells and allows normal facial development to take place.
Reprinted by permission from Macmillan Publishers Ltd: Nature Medicine (McKeown SJ and Bronner-Fraser M (2008). Saving face: rescuing a craniofacial birth defect. Nat Med 14:115–116).
A landmark collaborative study between groups led by Mike Dixon in the UK and Paul Trainor in the USA has recently demonstrated a rescue of the craniofacial defects associated with a mouse model of Treacher Collins syndrome (Jones et al, 2008). This investigation has raised the possibility of future therapeutic prevention of a serious inherited craniofacial birth defect.
The Tcof1 heterozygous mouse models human Treacher Collins syndrome; reduced levels of a functional Treacle protein results in abnormal ribosome biogenesis, a lack of proliferation and premature death of precursor neural crest cells. The mechanism underlying this effect appears to be increased abundance of a protein called p53 (an important molecule for cell cycle regulation) secondary to its stabilization and prevention from degradation. If this stabilization of p53 ultimately causes the death of neural crest cells in the Tcof1 embryo—what happens if p53 activity is inhibited? Importantly, could this rescue the phenotype if done at the correct stage of development?
Significantly, by injecting pregnant mice carrying the Tcof1 mutation with a pharmacological inhibitor of p53 function these investigators were able to produce a marked rescue of the craniofacial defects in the newborn mice. This study demonstrates that an understanding of the molecular mechanisms underlying inherited disorders can provide opportunities for therapeutic intervention. However, some caution must be applied in the case of preventing human Treacher Collins syndrome as serious logistical problems still remain. This approach could only be used in families with identified mutations, not those cases arising spontaneously; any treatment would need to begin very early in the pregnancy—in the third week of gestation during neural crest migration; and finally, p53 is a potent tumour suppressor in many tissues and its inhibition can lead to spontaneous tumour development (McKeown & Bronner-Fraser, 2008).
Pierre Robin syndrome
Pierre Robin syndrome or Robin sequence (PRS; OMIM 261800) (Fig. 13.17) occurs in around 1 : 10,000–20,000 and is characterized by a triad of:
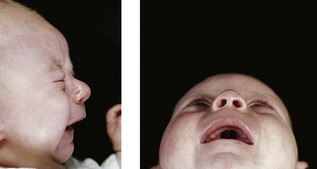
Micrognathia is the primary aetiological factor, an excessively small mandible resulting in the tongue falling downwards and backwards into the pharynx, being compressed between the palatal shelves and preventing their closure. In addition to a large and U-shaped cleft palate, the tongue position can also cause life-threatening respiratory difficulty at birth, obstructing the epiglottis and preventing adequate inhalation of the lungs.
Several theories have attempted to explain why growth and development of the mandible is so restricted in PRS:
It is likely that PRS may also contain a genetic component, as Robin sequence is seen in association with other syndromes: in particular, Stickler syndrome (OMIM 108300), the 22q11 deletion syndromes (see Box 2.1) and some extremely rare conditions featuring this sequence in combination with cardiac, neurological and axial skeletal defects.
In many cases of PRS, the upper airway obstruction presents as a medical emergency at birth, requiring neonatal nasopharyngeal intubation or tracheostomy. However, once the airway is stabilized and a feeding regime put in place, these infants usually thrive; with surgical repair of the cleft palate taking place during the first year of life. It has been suggested that compensatory growth of the mandible may occur during the first 5 years in cases of PRS, but this is controversial. Whilst mandibular skeletal growth certainly does occur in these subjects, there is evidence to suggest that these children are more likely to maintain a class II skeletal relationship (Daskalogiannakis et al, 2001).
Craniosynostosis
The craniosynostoses are a heterogenous group of disorders characterized by premature fusion of the cranial sutures (Fig. 13.18). This can occur in isolation or in association with other anomalies, in a number of well-characterized syndromes (Wilkie, 1997; Wilkie & Morriss-Kay, 2001).

Figure 13.18 CT scans of craniosynostosis.
The top images are of isolated sagittal suture synostosis with calvarial bone growth restricted in a transverse direction. Compensatory growth at the coronal and lambdoidal sutures has resulted in an elongated calvarium. The middle images are of Crouzon syndrome. There is synostosis of the sagittal and coronal sutures and maxillary hypoplasia. The lower images are of Apert syndrome. There is bilateral synostosis of the coronal suture, a wide midline defect in the calvarium and maxillary hypoplasia.
Courtesy of Professor David Rice and Jyri Hukki.
Isolated craniosynostosis
Around 1 : 2,000 children are born with premature fusion of a cranial suture, most commonly the sagittal; but the coronal, metopic and lambdoid sutures can also be affected. These cases usually occur sporadically but can also be familial. The craniofacial features are dependent upon which suture is affected but usually involve distortion of the skull due to excessive compensatory growth in unaffected regions.
Apert syndrome
Apert syndrome (OMIM 101200) is characterized by craniosynostosis, midfacial malformations, symmetrical syndactyly of the hands and feet and mental retardation. This condition can occur sporadically or be inherited in an autosomal dominant manner, but is rare, occurring in around 1 : 65,000. Apert syndrome is characterized by a wide midline calvarial defect, which rapidly closes with bone and a coronal suture that fuses early in infancy. The principle craniofacial features include:
Crouzon syndrome
Crouzon syndrome (OMIM 123500) involves craniosynostosis, ocular proptosis and midfacial malformation, but the limbs are unaffected. It occurs with a similar prevalence to that of Apert syndrome and is also either sporadic or inherited in an autosomal dominant manner. Crouzon syndrome is characterized by the premature fusion of multiple sutures from an early age and is dominated by the following features:
A number of other craniosynostosis syndromes, including Pfeiffer (OMIM 101600) and Saethre-Chotzen (OMIM 101400), exist. The majority of these conditions are associated with mutations in genes encoding receptors for the fibroblast growth factor family of signalling molecules (FGFRs), particularly FGFR2; and increased paternal age is a significant risk factor. These are often gain-of-function mutations, which lead to constitutive activity of the signalling pathway. FGF signalling is important during many stages of embryonic development, but the mutations associated with craniosynostosis clearly have a significant effect during the closely coordinated interactions that take place during formation of membrane bones within the skull.
Oral-Facial-Digital syndromes
The oral-facial-digital syndromes (OFD) represent a heterogeneous group of developmental disorders, which combine both oral and craniofacial abnormalities with anomalies affecting the digits. The best characterized is OFD-1 (OMIM 311200), which is inherited as an autosomal dominant X-linked condition and occurs in around 1 : 50,000 live births; only females are affected because it is lethal in the male heterozygous state.
OFD-1 is also characterized by a distinctive facies; there is:
Within the oral cavity, an unusual and marked hyperplasia of the frenula occur, which results in a characteristic pattern of orofacial clefting (Fig. 13.19):
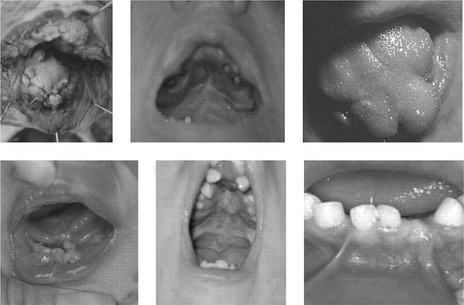
Figure 13.19 Oral abnormalities associated with OFD-1.
These include multiple buccal frenulae, lingual hamartoma; cleft and lobulated tongue, tooth defects and cleft palate (repaired in both middle panels).
Reproduced from Thauvin-Robinet C, Cossée M, Cormier-Daire V, et al (2006). Clinical, molecular, and genotype–phenotype correlation studies from 25 cases of oral–facial–digital syndrome type 1: a French and Belgian collaborative study. J Med Genet 43:54–61. With permission from BMJ Publishing Group Ltd.
The digits also present with a range of anomalies (Fig. 13.20), including:
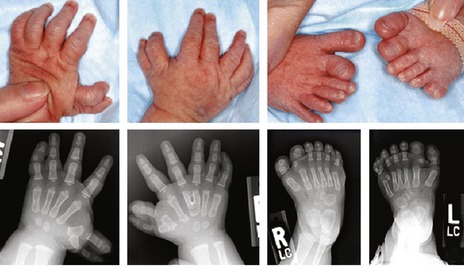
Figure 13.20 Limb abnormalities associated with oral-facial-digital syndrome include anomalies of the hands and feet, including polydactyly, with varying degrees of brachydactyly, syndactyly, and clinodactyly.
Reproduced from Hayes LL, Simoneaux SF, Palasis S, et al (2008). Laryngeal and tracheal anomalies in an infant with oral-facial-digital-syndrome type VI (Váradi-Papp): report of a transitional type. Ped Radiol 38:994–998. With kind permission from Springer Science and Business Media.
The OFD1 gene has been identified and encodes a protein involved in the organization and assembly of primary cilia (Ferrante et al, 2001; Romio et al, 2004), small contractile extensions found on the surface of many cell populations. It is becoming increasingly clear that cilia are important mediators of cell signalling during development and a recently generated mouse model of human OFD-1 suggests that normal cilia formation is defective under this condition (Ferrante et al, 2006).
Holoprosencephaly
Holoprosencephaly (HPE; OMIM 236100) is a clinically heterogeneous and complex developmental defect of the forebrain, in which the cerebral hemispheres fail to split into distinct halves (Muenke and Beachy, 2000). The underlying brain malformation can have a profound affect upon midline development of the face (Fig. 13.21). In the most severe form, the forebrain fails to divide and the face is characterized by cyclopia, with a single eye situated below a rudimentary proboscis and midline clefting of the lip and palate. HPE is one of the commonest causes of embryonic lethality, responsible for up to 1 : 250 miscarriages and seen in around 1 : 15,000 live births.
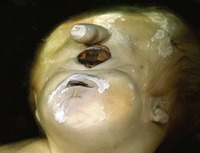
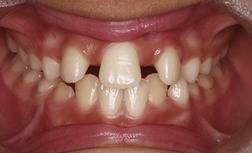
In some cases of HPE, the molecular defects have been identified; HPE-Type 3 (OMIM 142945) is caused by mutation in the sonic hedgehog (SHH) gene. SHH encodes an important signalling molecule, which is expressed in many regions of the embryo, including the prechordal plate (see Fig. 2.5). A lack of signalling in this region leads to a failure of forebrain subdivision and HPE. Mutations in the human SHH gene can also lead to a milder defect in midline facial development, the formation of only a solitary median maxillary central incisor (SMMCI) in the primary or secondary dentition (Fig. 13.22). This can occur as an isolated syndrome (OMIM 147250) or as a manifestation of HPE. Individuals with SMMCI have had offspring with HPE and therefore, SMMCI is a recognized risk factor and can be one of the mildest manifestations in autosomal dominant HPE (Nanni et al, 2001).
Fetal alcohol syndrome
The anomalies associated with the fetal alcohol syndrome (FAS) arise as a direct result of alcohol consumption during pregnancy (Jones et al, 1973). It is estimated that worldwide, FAS affects approximately 1 in 100 newborn children to varying degrees, making it the commonest cause of learning difficulty (O’Leary, 2004). The principle features of FAS include:
The severity of this condition is in proportion to the quantity and timing of alcohol ingestion during gestation. In the most extreme cases, FAS represents a form of HPE. Moreover, whilst the exact mechanisms underlying alcohol teratogenicity are not fully understood, exposure of developing chick embryos to ethanol causes the death of neural crest cell populations in the craniofacial region.
ONLINE MENDELIAN INHERITANCE IN MAN (OMIM). Available at URL http://www.nslijgenetics.org/search_omim.html.
Epstein CJ, Erickson RP, Wynshaw-Boris A. Inborn Errors of Development. Oxford: Oxford University Press; 2004.
Becker A, Lustmann J, Shteyer A. Cleidocranial dysplasia: Part 1—General principles of the orthodontic and surgical treatment modality. Am J Orthod Dentofacial Orthop. 1997;111:28-33.
Becker A, Shteyer A, Bimstein E, et al. Cleidocranial dysplasia: Part 2—Treatment protocol for the orthodontic and surgical modality. Am J Orthod Dentofacial Orthop. 1997;111:173-183.
Bergland O, Semb G, Abyholm FE. Elimination of the residual alveolar cleft by secondary bone grafting and subsequent orthodontic treatment. Cleft Palate J. 1986;23:175-205.
Cousley R, Naora H, Yokoyama M, et al. Validity of the Hfm transgenic mouse as a model for hemifacial microsomia. Cleft Palate Craniofac J. 2002;39:81-92.
Daskalogiannakis J, Ross RB, Tompson BD. The mandibular catch-up growth controversy in Pierre Robin sequence. Am J Orthod Dentofacial Orthop. 2001;120:280-285.
Delaire J. Theoretical principles and technique of functional closure of the lip and nasal aperture. J Maxillofac Surg. 1978;6:109-116.
Dixon MJ. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 1996;12:130-136.
Dixon J, Dixon MJ. Genetic background has a major effect on the penetrance and severity of craniofacial defects in mice heterozygous for the gene encoding the nucleolar protein Treacle. Dev Dyn. 2004;229:907-914.
Ferrante MI, Giorgio G, Feather SA, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68:569-576.
Ferrante MI, Zullo A, Barra A, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38:112-117.
Fraser FC. The genetics of cleft lip and cleft palate. Am J Hum Genet. 1970;22:336-352.
Gorlin RJ, Cohen MMCJr, Hennekam RCM. Syndromes of the Head and Neck. New York: Oxford University Press; 2001.
Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301:309-326.
Jiang R, Bush JO, Lidral AC. Development of the upper lip: Morphogenetic and molecular mechanisms. Dev Dyn. 2006;235:1152-1166.
Jones KL, Smith DW, Ulleland CN, et al. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet. 1973;1:1267-1271.
Jones N, Lynn ML, Gaudenz K, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125-133.
Kelberman D, Tyson J, Chandler DC, et al. Hemifacial microsomia: progress in understanding the genetic basis of a complex malformation syndrome. Hum Genet. 2001;109:638-645.
Kere J, Srivastava AK, Montonen O, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996;13:409-416.
Kobus K, Wojcicki P. Surgical treatment of Treacher Collins syndrome. Ann Plast Surg. 2006;56:549-554.
Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755-764.
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921.
Lee B, Thirunavukkarasu K, Zhou L, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307-310.
Lidral AC, Murray JC. Genetic approaches to identify disease genes for birth defects with cleft lip/palate as a model. Birth Defects Res A Clin Mol Teratol. 2004;70:893-901.
McKeown SJ, Bronner-Fraser M. Saving face: rescuing a craniofacial birth defect. Nat Med. 2008;14:115-116.
Mars M, Houston WJ. A preliminary study of facial growth and morphology in unoperated male unilateral cleft lip and palate subjects over 13 years of age. Cleft Palate J. 1990;27:7-10.
Mars M, Plint DA, Houston WJ, et al. The Goslon Yardstick: a new system of assessing dental arch relationships in children with unilateral clefts of the lip and palate. Cleft Palate J. 1987;24:314-322.
Monreal AW, Ferguson BM, Headon DJ, et al. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet. 1999;22:366-369.
Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 2000;10:262-269.
Mulliken JB. Repair of bilateral complete cleft lip and nasal deformity—state of the art. Cleft Palate Craniofac J. 2000;37:342-347.
Mundlos S, Otto F, Mundlos C, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773-779.
Nanni L, Ming JE, Du Y, et al. SHH mutation is associated with solitary median maxillary central incisor: a study of 13 patients and review of the literature. Am J Med Genet. 2001;102:1-10.
Naora H, Kimura M, Otani H, et al. Transgenic mouse model of hemifacial microsomia: cloning and characterization of insertional mutation region on chromosome 10. Genomics. 1994;23:515-519.
O’Leary CM. Fetal alcohol syndrome: diagnosis, epidemiology, and developmental outcomes. J Paediatr Child Health. 2004;40:2-7.
Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765-771.
Poswillo D. The aetiology and surgery of cleft palate with micrognathia. Ann R Coll Surg Engl. 1968;43:61-88.
Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973;35:302-328.
Rivera-Perez JA, Mallo M, Gendron-Maguire M, et al. Goosecoid is not an essential component of the mouse gastrula organizer but is required for craniofacial and rib development. Development. 1995;121:3005-3012.
Roberts-Harry D, Sandy JR. Repair of cleft lip and palate: 1. Surgical techniques. Dent Update. 1992;19:418-423.
Romio L, Fry AM, Winyard PJ, et al. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol. 2004;15:2556-2568.
Sandy J, Williams A, Mildinhall S, et al. The Clinical Standards Advisory Group (CSAG) Cleft Lip and Palate Study. Br J Orthod. 1998;25:21-30.
Sandy JR, Roberts-Harry D. Repair of cleft lip and palate: 2. Evaluation of surgical techniques. Dent Update. 1993;20:35-37.
Schendel SA. Unilateral cleft lip repair—state of the art. Cleft Palate Craniofac J. 2000;37:335-341.
Shaw WC, Dahl E, Asher-McDade C, et al. A six-center international study of treatment outcome in patients with clefts of the lip and palate: Part 5. General discussion and conclusions. Cleft Palate Craniofac J. 1992;29:413-418.
Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304-1351.
Wilkie AO. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 1997;6:1647-1656.
Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2:458-468.
Yamada G, Mansouri A, Torres M, et al. Targeted mutation of the murine goosecoid gene results in craniofacial defects and neonatal death. Development. 1995;121:2917-2922.