Pharmacology of Local Anesthetics
Local anesthetics, when used for the management of pain, differ from most other drugs commonly used in medicine and dentistry in one important manner. Virtually all other drugs, regardless of the route through which they are administered, must ultimately enter into the circulatory system in sufficiently high concentrations (e.g., attain therapeutic blood levels in their target organ[s]) before they can begin to exert a clinical action. Local anesthetics, however, when used for pain control, cease to provide a clinical effect when they are absorbed from the site of administration into the circulation. One prime factor involved in the termination of action of local anesthetics used for pain control is their redistribution from the nerve fiber into the cardiovascular system.
The presence of a local anesthetic in the circulatory system means that the drug will be transported to every part of the body. Local anesthetics have the ability to alter the functioning of some of these cells. In this chapter, the actions of local anesthetics, other than their ability to block conduction in nerve axons of the peripheral nervous system, are reviewed. A classification of local anesthetics is shown in Box 2-1.
Pharmacokinetics of Local Anesthetics
When injected into soft tissues, local anesthetics exert pharmacologic action on blood vessels in the area. All local anesthetics possess a degree of vasoactivity, most producing dilation of the vascular bed into which they are deposited, although the degree of vasodilation may vary, and some may produce vasoconstriction. To some degree, these effects may be concentration dependent.1 Relative vasodilating values of amide local anesthetics are shown in Table 2-1.
TABLE 2-1
Relative Vasodilating Values of Amide-Type Local Anesthetics
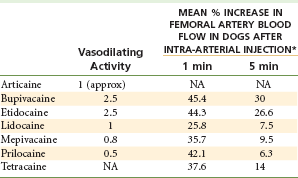
*Each agent injected rapidly at a dose of 1 mg/0.1 mL saline.
Modified from Blair MR: Cardiovascular pharmacology of local anaesthetics, Br J Anaesth 47(suppl):247-252, 1975.
Ester local anesthetics are also potent vasodilating drugs. Procaine, the most potent vasodilator among local anesthetics, is occasionally injected clinically to induce vasodilation when peripheral blood flow has been compromised because of (accidental) intra-arterial (IA) injection of a drug (e.g., thiopental)2 or injection of epinephrine or norepinephrine into a fingertip or toe.3 IA administration of an irritating drug such as thiopental may produce arteriospasm with an attendant decrease in tissue perfusion that if prolonged could lead to tissue death, gangrene, and loss of the affected limb. In this situation, procaine is administered IA in an attempt to break the arteriospasm and reestablish blood flow to the affected limb. Tetracaine, chloroprocaine, and propoxycaine also possess vasodilating properties to varying degrees but not to the degree of procaine.
Cocaine is the only local anesthetic that consistently produces vasoconstriction.4 The initial action of cocaine is vasodilation followed by an intense and prolonged vasoconstriction. It is produced by inhibition of the uptake of catecholamines (especially norepinephrine) into tissue binding sites. This results in an excess of free norepinephrine, leading to a prolonged and intense state of vasoconstriction. This inhibition of the reuptake of norepinephrine has not been demonstrated with other local anesthetics, such as lidocaine and bupivacaine.
A significant clinical effect of vasodilation is an increase in the rate of absorption of the local anesthetic into the blood, thus decreasing the duration and quality (e.g., depth) of pain control, while increasing the anesthetic blood (or plasma) concentration and its potential for overdose (toxic reaction). The rates at which local anesthetics are absorbed into the bloodstream and reach their peak blood level vary according to the route of administration (Table 2-2).
TABLE 2-2
Time to Achieve Peak Blood Level
| Route | Time, min |
| Intravenous | 1 |
| Topical | 5 (approximately) |
| Intramuscular | 5-10 |
| Subcutaneous | 30-90 |
Oral Route
With the exception of cocaine, local anesthetics are absorbed poorly, if at all, from the gastrointestinal tract after oral administration. In addition, most local anesthetics (especially lidocaine) undergo a significant hepatic first-pass effect after oral administration. After absorption of lidocaine from the gastrointestinal tract into the enterohepatic circulation, a fraction of the drug dose is carried to the liver, where approximately 72% of the dose is biotransformed into inactive metabolites.5 This has seriously hampered the use of lidocaine as an oral antidysrhythmic drug. In 1984, Astra Pharmaceuticals and Merck Sharp & Dohme introduced an analog of lidocaine, tocainide hydrochloride, which is effective orally.6 The chemical structures of tocainide and lidocaine are presented in Figure 2-1.

Topical Route
Local anesthetics are absorbed at differing rates after application to mucous membrane: In the tracheal mucosa, absorption is almost as rapid as with intravenous (IV) administration (indeed, intratracheal drug administration [epinephrine, lidocaine, atropine, naloxone, and flumazenil] is used in certain emergency situations); in the pharyngeal mucosa, absorption is slower; and in the esophageal or bladder mucosa, uptake is even slower than occurs through the pharynx. Wherever no layer of intact skin is present, topically applied local anesthetics can produce an anesthetic effect. Sunburn remedies (e.g., Solarcaine, Schering-Plough HealthCare Products, Inc., Memphis, Tenn) usually contain lidocaine, benzocaine, or other anesthetics in an ointment formulation. Applied to intact skin, they do not provide an anesthetic action, but with skin damaged by sunburn, they bring rapid relief of pain. A eutectic mixture of local anesthetics lidocaine and prilocaine (EMLA) has been developed that is able to provide surface anesthesia of intact skin.7
Injection
The rate of uptake (absorption) of local anesthetics after parenteral administration (subcutaneous, intramuscular, or IV) is related to both the vascularity of the injection site and the vasoactivity of the drug.
IV administration of local anesthetics provides the most rapid elevation of blood levels and is used clinically in the primary management of ventricular dysrhythmias.8 Rapid IV administration can lead to significantly high local anesthetic blood levels, which can induce serious toxic reactions. The benefits to be accrued from IV drug administration must always be carefully weighed against any risks associated with IV administration. Only when the benefits clearly outweigh the risks should the drug be administered, as is the case with pre-fatal ventricular dysrhythmias such as premature ventricular contractions (PVCs).9
Distribution
Once absorbed into the blood, local anesthetics are distributed throughout the body to all tissues (Fig. 2-2). Highly perfused organs (and areas), such as the brain, head, liver, kidneys, lungs, and spleen, initially will have higher anesthetic blood levels than less highly perfused organs. Skeletal muscle, although not as highly perfused as these areas, contains the greatest percentage of local anesthetic of any tissue or organ in the body because it constitutes the largest mass of tissue in the body (Table 2-3).
TABLE 2-3
Percentages of Cardiac Output Distributed to Different Organ Systems

Adapted from Mohrman DE, Heller LJ: Cardiovascular physiology, ed 7, New York, 2010, Lange Medical Books/McGraw-Hill.
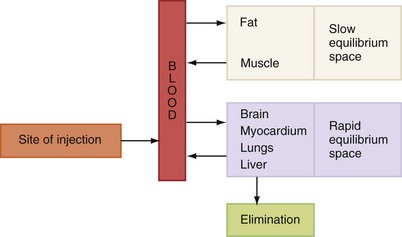
Figure 2-2 Pattern of distribution of local anesthetics after absorption. (Redrawn from Wildsmith JAW, Armitage EN, McClure JH: Principles and practice of regional anesthesia, ed 3, Edinburgh, 2003, Churchill Livingstone.)
The plasma concentration of a local anesthetic in certain target organs has a significant bearing on the potential toxicity of the drug. The blood level of the local anesthetic is influenced by the following factors:
1. Rate at which the drug is absorbed into the cardiovascular system
2. Rate of distribution of the drug from the vascular compartment to the tissues (more rapid in healthy patients than in those who are medically compromised [e.g., congestive heart failure], thus leading to lower blood levels in healthier patients)
3. Elimination of the drug through metabolic or excretory pathways
The latter two factors serve to decrease the blood level of the local anesthetic.
The rate at which a local anesthetic is removed from the blood is described as its elimination half-life. Simply stated, the elimination half-life is the time necessary for a 50% reduction in the blood level (one half-life = 50% reduction; two half-lives = 75% reduction; three half-lives = 87.5% reduction; four half-lives = 94% reduction; five half-lives = 97% reduction; six half-lives = 98.5% reduction) (Table 2-4).
TABLE 2-4
Half-Life of Local Anesthetics
| Drug | Half-life, hours |
| Chloroprocaine* | 0.1 |
| Procaine* | 0.1 |
| Tetracaine* | 0.3 |
| Articaine† | 0.5 |
| Cocaine* | 0.7 |
| Prilocaine† | 1.6 |
| Lidocaine† | 1.6 |
| Mepivacaine† | 1.9 |
| Ropivacaine† | 1.9 |
| Etidocaine† | 2.6 |
| Bupivacaine† | 3.5 |
| Propoxycaine* | NA |
All local anesthetics readily cross the blood–brain barrier. They also readily cross the placenta and enter the circulatory system of the developing fetus.
Metabolism (Biotransformation, Detoxification)
A significant difference between the two major groups of local anesthetics, the esters and the amides, is the means by which the body biologically transforms the active drug into one that is pharmacologically inactive. Metabolism (or biotransformation or detoxification) of local anesthetics is important because the overall toxicity of a drug depends on a balance between its rate of absorption into the bloodstream at the site of injection and its rate of removal from the blood through the processes of tissue uptake and metabolism.
Ester Local Anesthetics
Ester local anesthetics are hydrolyzed in the plasma by the enzyme pseudocholinesterase.10 The rate at which hydrolysis of different esters occurs varies considerably (Table 2-5).
TABLE 2-5
| Drug | Rate of Hydrolysis, µmol/mL/hr |
| Chloroprocaine | 4.7 |
| Procaine | 1.1 |
| Tetracaine | 0.3 |
The rate of hydrolysis has an impact on the potential toxicity* of a local anesthetic. Chloroprocaine, the most rapidly hydrolyzed, is the least toxic, whereas tetracaine, hydrolyzed 16 times more slowly than chloroprocaine, has the greatest potential toxicity. Procaine undergoes hydrolysis to para-aminobenzoic acid (PABA), which is excreted unchanged in the urine, and to diethylamine alcohol, which undergoes further biotransformation before excretion (Fig. 2-3). Allergic reactions that occur in response to ester local anesthetics usually are not related to the parent compound (e.g., procaine) but rather to PABA, which is a major metabolic product of many ester local anesthetics.
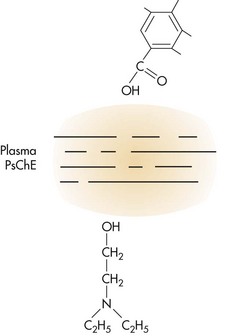
Figure 2-3 Metabolic hydrolysis of procaine. PsChE, Pseudocholinesterase. (From Tucker GT: Biotransformation and toxicity of local anesthetics, Acta Anaesthesiol Belg 26[Suppl]: 123, 1975.)
Approximately 1 of every 2800 persons has an atypical form of pseudocholinesterase, which causes an inability to hydrolyze ester local anesthetics and other chemically related drugs (e.g., succinylcholine).11 Its presence leads to prolongation of higher local anesthetic blood levels and increased potential for toxicity.
Succinylcholine is a short-acting muscle relaxant commonly employed during the induction phase of general anesthesia. It produces respiratory arrest (apnea) for a period of approximately 2 to 3 minutes. Then plasma pseudocholinesterase hydrolyzes succinylcholine, blood levels fall, and spontaneous respiration resumes. Persons with atypical pseudocholinesterase are unable to hydrolyze succinylcholine at a normal rate; therefore the duration of apnea is prolonged. Atypical pseudocholinesterase is a hereditary trait. Any familial history of difficulty during general anesthesia should be carefully evaluated by the doctor before dental care commences. A confirmed or strongly suspected history, in the patient or biological family, of atypical pseudocholinesterase represents a relative contraindication to administration of ester-type local anesthetics.
There are absolute and relative contraindications to the administration of drugs. An absolute contraindication implies that under no circumstance should the drug in question be administered to this patient because the possibility of potentially toxic or lethal reactions is increased, whereas a relative contraindication means that the drug in question may be administered to the patient after careful weighing of the risk associated with use of the drug versus the potential benefit to be gained, and if an acceptable alternative drug is not available. However, the smallest clinically effective dose always should be used because the likelihood of adverse reaction to this drug is increased in this patient.
Amide Local Anesthetics
The biotransformation of amide local anesthetics is more complex than that of the esters. The primary site of biotransformation of amide local anesthetics is the liver. Virtually the entire metabolic process occurs in the liver for lidocaine, mepivacaine, etidocaine, and bupivacaine. Prilocaine undergoes primary metabolism in the liver, with some also possibly occurring in the lung.12,13 Articaine, a hybrid molecule containing both ester and amide components, undergoes metabolism in both the blood and the liver.14
The rates of biotransformation of lidocaine, mepivacaine, etidocaine, and bupivacaine are similar. Therefore liver function and hepatic perfusion significantly influence the rate of biotransformation of an amide local anesthetic. Approximately 70% of a dose of injected lidocaine undergoes biotransformation in patients with normal liver function.5 Patients with lower than usual hepatic blood flow (hypotension, congestive heart failure) or poor liver function (cirrhosis) are unable to biotransform amide local anesthetics at a normal rate.15,16 This slower than normal biotransformation rate results in higher anesthetic blood levels and increased risk of toxicity. Significant liver dysfunction (American Society of Anesthesiologists Physical Status classification system [ASA] 4 to 5) or heart failure (ASA 4 to 5) represents a relative contraindication to the administration of amide local anesthetic drugs (Table 2-6). Articaine has a shorter half-life than other amides (27 minutes vs. 90 minutes) because a portion of its biotransformation occurs in the blood by the enzyme plasma cholinesterase.17
TABLE 2-6
Lidocaine Disposition in Various Groups of Patients
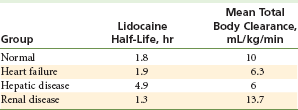
Data from Thomson PD, et al: Lidocaine pharmacokinetics in advanced heart failure, liver disease, and renal failure in humans, Ann Intern Med 78:499–513, 1973.
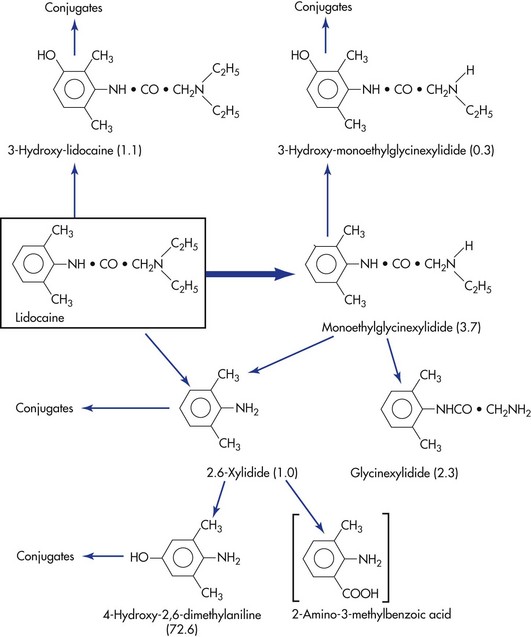
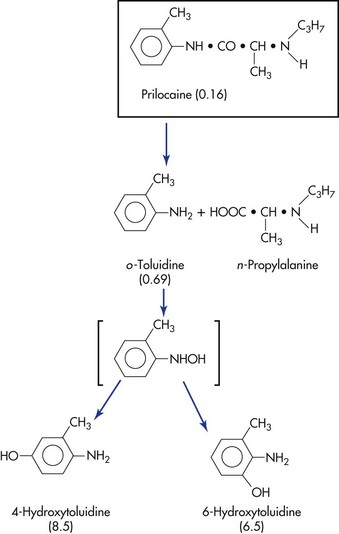
The biotransformation products of certain local anesthetics can possess significant clinical activity if they are permitted to accumulate in the blood. This may be seen in renal or cardiac failure and during periods of prolonged drug administration. A clinical example is the production of methemoglobinemia in patients receiving large doses of prilocaine.18,19 Prilocaine, the parent compound, does not produce methemoglobinemia, but orthotoluidine, a primary metabolite of prilocaine, does induce the formation of methemoglobin, which is responsible for methemoglobinemia. When methemoglobin blood levels become elevated, clinical signs and symptoms are observed. Methemoglobinemia is discussed more fully in Chapter 10. Another example of pharmacologically active metabolites is the sedative effect occasionally observed after lidocaine administration. Lidocaine does not produce sedation; however, two metabolites—monoethylglycinexylidide and glycine xylidide—are thought to be responsible for this clinical action.20
The metabolic pathways of lidocaine and prilocaine are shown in Figures 2-4 and 2-5.
 Development of Local Anesthetic Agents: Timeline
Development of Local Anesthetic Agents: Timeline

Excretion
The kidneys are the primary excretory organ for both the local anesthetic and its metabolites. A percentage of a given dose of local anesthetic is excreted unchanged in the urine. This percentage varies according to the drug. Esters appear only in very small concentrations as the parent compound in the urine because they are hydrolyzed almost completely in the plasma. Procaine appears in the urine as PABA (90%) with 2% unchanged. Ten percent of a cocaine dose is found in the urine unchanged. Amides usually are present in the urine as the parent compound in a greater percentage than the esters, primarily because of their more complex process of biotransformation. Although the percentages of parent drug found in urine vary from study to study, less than 3% lidocaine, 1% mepivacaine, and 1% etidocaine is found unchanged in the urine.
Patients with significant renal impairment may be unable to eliminate the parent local anesthetic compound or its major metabolites from the blood, resulting in slightly elevated blood levels and therefore increased potential for toxicity. This may occur with the esters or amides and is especially likely with cocaine. Thus significant renal disease (ASA 4 to 5) represents a relative contraindication to the administration of local anesthetics. This includes patients undergoing renal dialysis and those with chronic glomerulonephritis or pyelonephritis.
Systemic Actions of Local Anesthetics
Local anesthetics are chemicals that reversibly block action potentials in all excitable membranes. The central nervous system (CNS) and the cardiovascular system (CVS) therefore are especially susceptible to their actions. Most of the systemic actions of local anesthetics are related to their blood or plasma level in the target organ (CNS, CVS). The higher the level, the greater will be the clinical action.
Centbucridine (a quinoline derivative) has proved to be five to eight times as potent a local anesthetic as lidocaine, with an equally rapid onset of action and an equivalent duration.21,22 Of potentially great importance is the finding that it does not adversely affect the CNS or CVS, except in very high doses.
Local anesthetics are absorbed from their site of administration into the circulatory system, which effectively dilutes them and carries them to all cells of the body. The ensuing blood level of the local anesthetic depends on its rate of uptake from the site of administration into the circulatory system (increasing the blood level), and on the rates of distribution in tissue and biotransformation (in the liver), processes that remove the drug from the blood (decreasing the blood level) (see Fig. 2-2).
Central Nervous System
Local anesthetics readily cross the blood–brain barrier. Their pharmacologic action on the CNS is seen as depression. At low (therapeutic, nontoxic) blood levels, no CNS effects of any clinical significance have been noted. At higher (toxic, overdose) levels, the primary clinical manifestation is a generalized tonic–clonic convulsion. Between these two extremes exists a spectrum of other clinical signs and symptoms. (See Box 2-2, “Preconvulsive Signs and Symptoms of Central Nervous Toxicity.”)

Anticonvulsant Properties
Some local anesthetics (e.g., procaine, lidocaine, mepivacaine, prilocaine, even cocaine) have demonstrated anticonvulsant properties.23,24 These occur at a blood level considerably below that at which the same drugs produce seizure activity. Values for anticonvulsive blood levels of lidocaine are shown in Table 2-7.25
TABLE 2-7
Anticonvulsive Blood Levels of Lidocaine
| Clinical Situation | Lidocaine Blood Level, µg/mL |
| Anticonvulsive level | 0.5-4 |
| Preseizure signs and symptoms | 4.5-7 |
| Tonic–clonic seizure | >7.5 |
Procaine, mepivacaine, and lidocaine have been used intravenously to terminate or decrease the duration of both grand mal and petit mal seizures.23,26 The anticonvulsant blood level of lidocaine (about 0.5 to 4 µg/mL) is very close to its cardiotherapeutic range (see following). It has been demonstrated to be effective in temporarily arresting seizure activity in a majority of human epileptic patients.27 It was especially effective in interrupting status epilepticus at therapeutic doses of 2 to 3 mg/kg when given at a rate of 40 to 50 mg/min.
Mechanism of Anticonvulsant Properties: Epileptic patients possess hyperexcitable cortical neurons at a site within the brain where the convulsive episode originates (called the epileptic focus). Local anesthetics, by virtue of their depressant actions on the CNS, raise the seizure threshold by decreasing the excitability of these neurons, thereby preventing or terminating seizures.
Preconvulsive Signs and Symptoms
With a further increase in the blood level of the local anesthetic to above its therapeutic level, adverse reactions may be observed. Because the CNS is much more susceptible than other systems to the actions of local anesthetics, it is not surprising that the initial clinical signs and symptoms of overdose (toxicity) are CNS in origin. With lidocaine, this second phase is observed at a level between 4.5 and 7 µg/mL in the average normal healthy patient.* Initial clinical signs and symptoms of CNS toxicity are usually excitatory in nature (see Box 2-2).
All of these signs and symptoms, except for the sensation of circumoral and lingual numbness, are related to the direct depressant action of the local anesthetic on the CNS. Numbness of the tongue and circumoral regions is not caused by CNS effects of the local anesthetic.28 Rather it is the result of a direct anesthetic action of the local anesthetic, which is present in high concentrations in these highly vascular tissues, on free nerve endings. The anesthetic has been transported to these tissues by the CVS. A dentist treating a patient might have difficulty conceptualizing why anesthesia of the tongue is considered to be a sign of a toxic reaction when lingual anesthesia is commonly produced after mandibular nerve blocks. Consider for a moment a physician administering a local anesthetic into the patient’s foot. Overly high blood levels would produce bilateral numbing of the tongue, as contrasted with the usual unilateral anesthesia seen after dental nerve blocks.
Lidocaine and procaine differ somewhat from other local anesthetics in that the usual progression of signs and symptoms just noted may not be seen. Lidocaine and procaine frequently produce an initial mild sedation or drowsiness (more common with lidocaine).29 Because of this potential, the U.S. Air Force and the U.S. Navy ground airplane pilots for 24 hours after receipt of a local anesthetic.30
Sedation may develop in place of the excitatory signs. If excitation or sedation is observed during the first 5 to 10 minutes after intraoral administration of a local anesthetic, it should serve as a warning to the clinician of a rising local anesthetic blood level and the possibility (if the blood level continues to rise) of a more serious reaction, including a generalized convulsive episode.
Convulsive Phase
Further elevation of the local anesthetic blood level leads to signs and symptoms consistent with a generalized tonic–clonic convulsive episode. The duration of seizure activity is related to the local anesthetic blood level and is inversely related to the arterial partial pressure of carbon dioxide (pCO2) level.31 At a normal pCO2, a lidocaine blood level between 7.5 and 10 µg/mL usually results in a convulsive episode. When carbon dioxide (CO2) levels are increased, the blood level of local anesthetic necessary for seizures decreases while the duration of the seizure increases.31 Seizure activity generally is self-limiting, because cardiovascular activity usually is not significantly impaired, and redistribution and biotransformation of the local anesthetic continue throughout the episode. This results in a decrease in anesthetic blood level and termination of seizure activity, usually within 1 minute.
However, several other mechanisms also at work unfortunately act to prolong the convulsive episode. Both cerebral blood flow and cerebral metabolism are increased during local anesthetic-induced convulsions. Increased blood flow to the brain leads to an increase in the volume of local anesthetic being delivered to the brain, tending to prolong the seizure. Increased cerebral metabolism leads to a progressive metabolic acidosis as the seizure continues, and this tends to prolong the seizure activity (by lowering the blood level of anesthetic necessary to provoke a seizure), even in the presence of a declining local anesthetic level in the blood. As noted in Tables 2-8 and 2-9, the dose of local anesthetic necessary to induce seizures is markedly diminished in the presence of hypercarbia (see Table 2-8) or acidosis (see Table 2-9).31,32
TABLE 2-8
Effects of pCO2 on the Convulsive Threshold (CD100) of Various Local Anesthetics in Cats
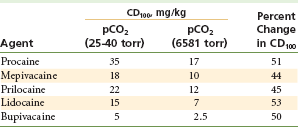
Data from Englesson S, Grevsten S, Olin A: Some numerical methods of estimating acid-base variables in normal human blood with a haemoglobin concentration of 5 g/100 cm3, Scand J Lab Clin Invest 32:289–295, 1973.
TABLE 2-9
Convulsant Dose (CD100) and Acid-Base Status*
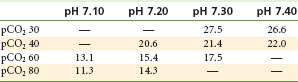
*Intravenous lidocaine 5 mg/kg/min, cats; doses in mg/kg.
From Englesson S: The influence of acid-base changes on central nervous toxicity of local anaesthetic agents, Acta Anaesthesiol Scand 18:88–103, 1974.
Further increases in local anesthetic blood level result in cessation of seizure activity as electroencephalographic (EEG) tracings become flattened, indicative of a generalized CNS depression. Respiratory depression occurs at this time, eventually leading to respiratory arrest if anesthetic blood levels continue to rise. Respiratory effects are a result of the depressant action of the local anesthetic drug on the CNS.
Mechanism of Preconvulsant and Convulsant Actions: It is known that local anesthetics exert a depressant action on excitable membranes, yet the primary clinical manifestation associated with high local anesthetic blood levels is related to varying degrees of stimulation. How can a drug that depresses the CNS be responsible for the production of varying degrees of stimulation, including tonic–clonic seizure activity? It is thought that local anesthetics produce clinical signs and symptoms of CNS excitation (including convulsions) through selective blockade of inhibitory pathways in the cerebral cortex.32-35 de Jong states that “inhibition of inhibition thus is a presynaptic event that follows local anesthetic blockade of impulses traveling along inhibitory pathways.”36
The cerebral cortex has pathways of neurons that are essentially inhibitory and others that are facilitatory (excitatory). A state of balance normally is maintained between the degrees of effect exerted by these neuronal paths (Fig. 2-6). At preconvulsant local anesthetic blood levels, observed clinical signs and symptoms are produced because the local anesthetic selectively depresses the action of inhibitory neurons (Fig. 2-7). Balance then is tipped slightly in favor of excessive facilitatory (excitatory) input, leading to symptoms of tremor and slight agitation.
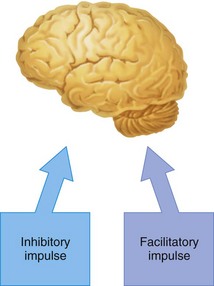
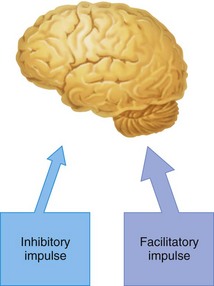
Figure 2-7 In the preconvulsive stage of local anesthetic action, the inhibitory impulse is more profoundly depressed than the facilitatory impulse.
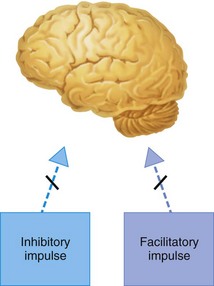
At higher (convulsive) blood levels, inhibitory neuron function is completely depressed, allowing unopposed function of facilitatory neurons (Fig. 2-8). Pure facilitatory input without inhibition produces the tonic–clonic activity observed at these levels.
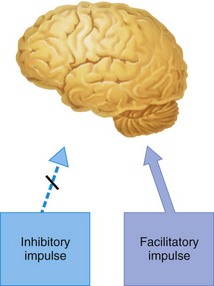
Figure 2-8 In the convulsive stage of local anesthetic action, the inhibitory impulse is totally depressed, permitting unopposed facilitatory impulse activity.
Further increases in anesthetic blood level lead to depression of the facilitatory and inhibitory pathways, producing generalized CNS depression (Fig. 2-9). The precise site of action of the local anesthetic within the CNS is not known but is thought to be at the inhibitory cortical synapses or directly on the inhibitory cortical neurons.
Analgesia
Local anesthetics possess a second action in relation to the CNS. Administered intravenously, they increase the pain reaction threshold and also produce a degree of analgesia.
In the 1940s and 1950s, procaine was administered intravenously for the management of chronic pain and arthritis.37 The “procaine unit” was commonly used for this purpose; it consisted of 4 mg/kg of body weight administered over 20 minutes. The technique was ineffective for acute pain. Because of the relatively narrow safety margin between the analgesic actions of procaine and the occurrence of signs and symptoms of overdose, this technique is no longer in use today.
Mood Elevation
The use of local anesthetic drugs for mood elevation and rejuvenation has persisted for centuries, despite documentation of both catastrophic events (mood elevation) and lack of effect (rejuvenation).
Cocaine has long been used for its euphoria-inducing and fatigue-lessening actions, dating back to the chewing of coca leaves by Incas and other South American natives.38,39 Unfortunately, as is well documented today, prolonged use of cocaine leads to habituation. William Stewart Halsted (1852-1922), the father of American surgery, cocaine researcher, and the first person to administer a local anesthetic by injection, suffered greatly because of an addiction to cocaine.40 In more recent times, the sudden, unexpected deaths of several prominent professional athletes caused by cocaine and the addiction of many others clearly demonstrate the dangers involved in the casual use of potent drugs.41,42
More benign, but totally unsubstantiated, is the use of procaine (Novocain) as a rejuvenating drug. Clinics professing to “restore youthful vigor” claim that procaine is a literal Fountain of Youth. These clinics operate primarily in central Europe and Mexico, where procaine is used under the proprietary name Gerovital. de Jong states that “whatever the retarding effect on aging, it probably is relegated most charitably to mood elevation.”43
Cardiovascular System
Local anesthetics have a direct action on the myocardium and peripheral vasculature. In general, however, the cardiovascular system appears to be more resistant than the CNS to the effects of local anesthetic drugs (Table 2-10).44
TABLE 2-10
Intravenous Dose of Local Anesthetic Agents Required for Convulsive Activity (CD100) and Irreversible Cardiovascular Collapse (LD100) in Dogs
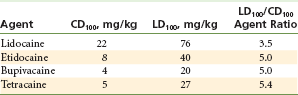
Data from Liu P, et al: Acute cardiovascular toxicity of intravenous amide local anesthetics in anesthetized ventilated dogs, Anesth Analg 61:317–322, 1982.
Direct Action on the Myocardium
Local anesthetics modify electrophysiologic events in the myocardium in a manner similar to their actions on peripheral nerves. As the local anesthetic blood level increases, the rate of rise of various phases of myocardial depolarization is reduced. No significant change in resting membrane potential occurs, and no significant prolongation of the phases of repolarization is seen.45
Local anesthetics produce a myocardial depression that is related to the local anesthetic blood level. Local anesthetics decrease the electrical excitability of the myocardium, decrease the conduction rate, and decrease the force of contraction.46-48
Therapeutic advantage is taken of this depressant action in managing the hyperexcitable myocardium, which manifests as various cardiac dysrhythmias. Although many local anesthetics have demonstrated antidysrhythmic actions in animals, only procaine and lidocaine have gained significant clinical reliability in humans. Lidocaine is the most widely used and intensively studied local anesthetic in this regard.9,29,49,50 Procainamide is the procaine molecule with an amide linkage replacing the ester linkage. Because of this, it is hydrolyzed much more slowly than procaine.51 Tocainide, a chemical analog of lidocaine, was introduced in 1984 as an oral antidysrhythmic drug, because lidocaine is ineffective after oral administration.52 Tocainide also is effective in managing ventricular dysrhythmias but is associated with a 40% incidence of adverse effects, including nausea, vomiting, tremor, paresthesias, agranulocytosis, and pulmonary fibrosis.53,54 Tocainide worsens symptoms of congestive heart failure in about 5% of patients and may provoke dysrhythmias (i.e., is prodysrhythmic) in 1% to 8%.55
Blood levels of lidocaine usually noted after intraoral injection of one or two dental cartridges, 0.5 to 2 µg/mL, are not associated with cardiodepressant activity. Increasing lidocaine blood levels slightly is nontoxic and is associated with antidysrhythmic actions. Therapeutic blood levels of lidocaine for antidysrhythmic activity range from 1.8 to 6 µg/mL.48,56
Lidocaine usually is administered intravenously in a bolus of 50 to 100 mg at a rate of 25 to 50 mg/min. This dose is based on 1 to 1.5 mg/kg of body weight every 3 to 5 minutes and is frequently followed by a continuous IV infusion of 1 to 4 mg/min. Signs and symptoms of local anesthetic overdose will be noted if the blood level rises beyond 6 µg/mL of blood.56
Lidocaine is used clinically primarily in the management of PVCs and ventricular tachycardia. It also is used as a (class-indeterminate) drug in advanced cardiovascular life support and in management of cardiac arrest caused by ventricular fibrillation.57
Direct cardiac actions of local anesthetics at blood levels greater than the therapeutic (antidysrhythmic) level include a decrease in myocardial contractility and decreased cardiac output, both of which lead to circulatory collapse (see Table 2-10).
Box 2-3 summarizes the CNS and cardiovascular effects of increasing local anesthetic blood levels.

Direct Action on the Peripheral Vasculature
Cocaine is the only local anesthetic drug that consistently produces vasoconstriction at commonly employed dosages.4 Ropivacaine causes cutaneous vasoconstriction, whereas its congener bupivacaine produces vasodilation.58 All other local anesthetics produce a peripheral vasodilation through relaxation of smooth muscle in the walls of blood vessels. This leads to increased blood flow to and from the site of local anesthetic deposition (see Table 2-1). Increased local blood flow increases the rate of drug absorption, in turn leading to decreased depth and duration of local anesthetic action, increased bleeding in the treatment area, and increased local anesthetic blood levels.
Table 2-11 provides examples of peak blood levels achieved after local anesthetic injection with and without the presence of a vasopressor.58-60
TABLE 2-11
Peak Plasma Levels Following Local Anesthetic Administration With and Without Vasopressor
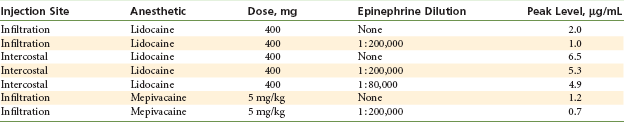
Data from Kopacz DJ, Carpenter RL, Mackay DL: Effect of ropivacaine on cutaneous capillary flow in pigs, Anesthesiology 71:69, 1989; Scott DB, et al: Factors affecting plasma levels of lignocaine and prilocaine, Br J Anaesth 44:1040-1049, 1972; Duhner KG, et al: Blood levels of mepivacaine after regional anaesthesia, Br J Anaesth 37:746–752, 1965.
The primary effect of local anesthetics on blood pressure is hypotension. Procaine produces hypotension more frequently and significantly than does lidocaine: 50% of patients in one study receiving procaine became hypotensive, compared with 6% of those receiving lidocaine.61 This action is produced by direct depression of the myocardium and smooth muscle relaxation in the vessel walls by the local anesthetic.
In summary, negative effects on the cardiovascular system are not noted until significantly elevated local anesthetic blood levels are reached. The usual sequence of local anesthetic–induced actions on the cardiovascular system is as follows:
1. At nonoverdose levels, a slight increase or no change in blood pressure occurs because of increased cardiac output and heart rate as a result of enhanced sympathetic activity; direct vasoconstriction of certain peripheral vascular beds is also noted.
2. At levels approaching yet still below overdose, a mild degree of hypotension is noted; this is produced by a direct relaxant action on the vascular smooth muscle.
3. At overdose levels, profound hypotension is caused by decreased myocardial contractility, cardiac output, and peripheral resistance.
4. At lethal levels, cardiovascular collapse is noted. This is caused by massive peripheral vasodilation and decreased myocardial contractility and heart rate (sinus bradycardia).
5. Certain local anesthetics such as bupivacaine (and to a lesser degree ropivacaine and etidocaine) may precipitate potentially fatal ventricular fibrillation.62,63
Local Tissue Toxicity
Skeletal muscle appears to be more sensitive than other tissues to the local irritant properties of local anesthetics. Intramuscular and intraoral injection of articaine, lidocaine, mepivacaine, prilocaine, bupivacaine, and etidocaine can produce skeletal muscle alterations.64-67 It appears that longer-acting local anesthetics cause more localized skeletal muscle damage than shorter-acting drugs. The changes that occur in skeletal muscle are reversible, with muscle regeneration being complete within 2 weeks after local anesthetic administration. These muscle changes have not been associated with any overt clinical signs of local irritation.
Respiratory System
Local anesthetics exert a dual effect on respiration. At nonoverdose levels, they have a direct relaxant action on bronchial smooth muscle, whereas at overdose levels, they may produce respiratory arrest as a result of generalized CNS depression. In general, respiratory function is unaffected by local anesthetics until near-overdose levels are achieved.
Miscellaneous Actions
Many local anesthetics have been demonstrated to block neuromuscular transmission in humans. This is a result of the inhibition of sodium diffusion through a blockade of sodium channels in the cell membrane. This action normally is slight and usually is clinically insignificant. On occasion, however, it can be additive to that produced by both depolarizing (e.g., succinylcholine) and nondepolarizing (e.g., atracurium, vecuronium) muscle relaxants; this may lead to abnormally prolonged periods of muscle paralysis. Such actions are unlikely to occur in the dental outpatient.
Drug Interactions
In general, CNS depressants (e.g., opioids, antianxiety drugs, phenothiazines, barbiturates), when administered in conjunction with local anesthetics, lead to potentiation of the CNS-depressant actions of the local anesthetic. The conjoint use of local anesthetics and drugs that share a common metabolic pathway can produce adverse reactions. Both ester local anesthetics and the depolarizing muscle relaxant succinylcholine require plasma pseudocholinesterase for hydrolysis. Prolonged apnea may result from concomitant use of these drugs.
Drugs that induce the production of hepatic microsomal enzymes (e.g., barbiturates) may alter the rate at which amide local anesthetics are metabolized. Increased hepatic microsomal enzyme induction increases the rate of metabolism of the local anesthetic.
Specific drug–drug interactions related to the administration of local anesthetics are reviewed in Chapter 10.
Malignant Hyperthermia
Malignant hyperthermia (MH; hyperpyrexia) is a pharmacogenic disorder in which a genetic variant in an individual alters that person’s response to certain drugs. Acute clinical manifestations of MH include tachycardia, tachypnea, unstable blood pressure, cyanosis, respiratory and metabolic acidosis, fever (as high as 42° C [108° F] or more), muscle rigidity, and death. Mortality ranges from 63% to 73%. Many commonly used anesthetic drugs can trigger MH in certain individuals.
Until recently, the amide local anesthetics were thought to be capable of provoking MH and were considered to be absolutely contraindicated in MH-susceptible patients.68 The Malignant Hyperthermia Association of the United States (MHAUS), after evaluating recent clinical research, concluded that in fact no documented cases in the medical or dental literature (over the past 30 years) support the concept of amide anesthetics triggering malignant hyperthermia.69-73
MHAUS maintains a Website with information for both health care providers and patients: www.mhaus.org.
References
1. Aps, C, Reynolds, F. The effect of concentration in vasoactivity of bupivacaine and lignocaine. Br J Anaesth. 1976;48:1171–1174.
2. Covino, BG. Pharmacology of local anaesthetic agents. Br J Anaesth. 1986;58:701–716.
3. U.S. Food and Drug Administration. Center for Drug Evaluation and Research: Approval letter for phentolamine mesylate, 1998. Available at www.fda.gov/cder/foi/anda/98/40235ap.pdf. [Accessed November 6, 2007].
4. Benowitz, NL. Clinical pharmacology and toxicology of cocaine. Pharmacol Toxicol. 1993;72:1–12.
5. Arthur, GR. Pharmacokinetics of local anesthetics. In: Strichartz, GR, eds. Local anesthetics: handbook of experimental pharmacology, vol 81. Berlin: Springer-Verlag; 1987.
6. Hohnloser, SH, Lange, HW, Raeder, E, et al. Short- and long-term therapy with tocainide for malignant ventricular tachyarrhythmias. Circulation. 1986;73:143–149.
7. Soliman, IE, Broadman, LM, Hannallah, RS, McGill, WA. Comparison of the analgesic effects of EMLA (eutectic mixture of local anesthetics) to intradermal lidocaine infiltration prior to venous cannulation in unpremedicated children. Anesthesiology. 1988;68:804–806.
8. American Heart Association. ACLS provider manual. Dallas, Tex: American Heart Association; 2001. [pp 83–84].
9. Haugh, KH. Antidysrhythmic agents at the turn of the twenty-first century: a current review. Crit Care Nursing Clin North Am. 2002;14:13–69.
10. Kalow, W. Hydrolysis of local anesthetics by human serum cholinesterase. J Pharmacol Exp Ther. 1952;104:122–134.
11. Watson, CB. Respiratory complications associated with anesthesia. Anesth Clin North Am. 2002;20:375–399.
12. Harris, WH, Cole, DW, Mital, M, Laver, MB. Methemoglobin formation and oxygen transport following intravenous regional anesthesia using prilocaine. Anesthesiology. 1968;29:65.
13. Arthur, GR. Distribution and elimination of local anesthetic agents: the role of the lung, liver, and kidneys, PhD thesis. Edinburgh: University of Edinburgh; 1981.
14. Oertel, R, Berndt, A, Kirch, W. Saturable in vitro metabolism of articaine by serum esterases: does it contribute to the resistance of the local anesthetic effect? Reg Anesth. 1996;21:576–581.
15. Nation, RL, Triggs, EJ. Lidocaine kinetics in cardiac patients and aged subjects. Br J Clin Pharmacol. 1977;4:439–448.
16. Thomson, PD, Melmon, KL, Richardson, JA, et al. Lidocaine pharmacokinetics in advanced heart failure, liver disease, and renal failure in humans. Ann Intern Med. 1973;78:499–508.
17. Oertel, R, Rahn, R, Kirch, W. Clinical pharmacokinetics of articaine. Clin Pharmacokinet. 1997;33:617–625.
18. Prilocaine-induced methemoglobinemia—Wisconsin, 1993. MMWR Morb Mortal Wkly Rep. 1994;43:3555–3557.
19. Wilburn-Goo, D, Lloyd, LM. When patients become cyanotic: acquired methemoglobinemia. J Am Dent Assoc. 1999;130:626–631.
20. Strong, JM, Parker, M, Atkinson, AJ, Jr. Identification of glycinexylidide in patients treated with intravenous lidocaine. Clin Pharmacol Ther. 1973;14:67–72.
21. Gupta, PP, Tangri, AN, Saxena, RC, Dhawan, BN. Clinical pharmacology studies on 4-N-butylamino-1,2,3,4,-tetrahydroacridine hydrochloride (Centbucridine), a new local anaesthetic agent. Indian J Exp Biol. 1982;20:344–346.
22. Vacharajani, GN, Parikh, N, Paul, T, Satoskar, RS. A comparative study of Centbucridine and lidocaine in dental extraction. Int J Clin Pharmacol Res. 1983;3:251–255.
23. Bernhard, CG, Bohm, E. Local anaesthetics as anticonvulsants: a study on experimental and clinical epilepsy. Stockholm: Almqvist & Wiksell; 1965.
24. Bernhard, CG, Bohm, E, Wiesel, T. On the evaluation of the anticonvulsive effect of different local anesthetics. Arch Int Pharmacodyn Ther. 1956;108:392–407.
25. Julien, RM. Lidocaine in experimental epilepsy: correlation of anticonvulsant effect with blood concentrations. Electroencephalogr Clin Neurophysiol. 1973;34:639–645.
26. Berry, CA, Sanner, JH, Keasling, HH. A comparison of the anticonvulsant activity of mepivacaine and lidocaine. J Pharmacol Exp Ther. 1961;133:357–363.
27. Walker, IA, Slovis, CM. Lidocaine in the treatment of status epilepticus. Acad Emerg Med. 1997;4:918–922.
28. Chen, AH. Toxicity and allergy to local anesthesia. J Calif Dent Assoc. 1998;26:983–992.
29. Katz, J, Feldman, MA, Bass, EB, et al. Injectable versus topical anesthesia for cataract surgery: patient perceptions of pain and side effects: the Study of Medical Testing for Cataract Surgery Study Team. Ophthalmology. 2000;107:2054–2060.
30. Bureau of Medicine and Surgery. Available at http://navymedicine.med.navy.mil/. [Accessed October 28, 2011].
31. Englesson, S. The influence of acid-base changes on central nervous system toxicity of local anesthetic agents. I. An experimental study in cats. Acta Anaesthesiol Scand. 1974;18:79.
32. Englesson, S, Grevsten, S, Olin, A. Some numerical methods of estimating acid-base variables in normal human blood with a haemoglobin concentration of 5 g-100 cm3. Scand J Lab Clin Invest. 1973;32:289–295.
33. de Jong, RH, Robles, R, Corbin, RW. Central actions of lidocaine-synaptic transmission. Anesthesiology. 1969;30:19.
34. Huffman, RD, Yim, GKW. Effects of diphenylaminoethanol and lidocaine on central inhibition. Int J Neuropharmacol. 1969;8:217.
35. Tanaka, K, Yamasaki, M. Blocking of cortical inhibitory synapses by intravenous lidocaine. Nature. 1966;209:207.
36. de Jong, RH. Local anesthetics. St Louis: Mosby; 1994.
37. Graubard, DJ, Peterson, MC. Clinical uses of intravenous procaine. Springfield, Ill: Charles C Thomas; 1950.
38. Garcilasso de la Vega: Commentarios reales de los Incas (1609–1617). In: Freud S, ed. Uber coca. Wien: Verlag von Moritz Perles, 1884.
39. Disertacion sobre el aspecto, cultivo, comercio y virtudes de la famosa planta del Peru nombrado coca: Lima, 1794. In: Freud S, ed. Uber coca. Wien: Verlag von Moritz Perles, 1884.
40. Olch, PD, William, S. Halsted and local anesthesia: contributions and complications. Anesthesiology. 1975;42:479–486.
41. Preboth, M. Cocaine abuse among athletes. Am Fam Physician. 1915;62:1850–2000.
42. Harriston, K, Jenkins, S. Maryland basketball star Len Bias is dead at 22. Washington Post. 20 June 1986.
43. de Jong, RH. Local anesthetics, ed 2. Springfield, Ill: Charles C Thomas; 1977. [p 89].
44. Scott, DB. Toxicity caused by local anaesthetic drugs. Br J Anaesth. 1981;53:553–554.
45. Pinter, A, Dorian, P. Intravenous antiarrhythmic agents. Curr Opin Cardiol. 2001;16:17–22.
46. Sugi, K. Pharmacological restoration and maintenance of sinus rhythm by antiarrhythmic agents. J Cardiol. 1999;33(Suppl 1):59–64.
47. Alexander, JH, Granger, CB, Sadowski, Z, et al. Prophylactic lidocaine use in acute myocardial infarction: incidence and outcomes from two international trials. The GUSTO-I and GUSTO-IIb Investigators. Am Heart J. 1999;137:599–805.
48. Cannom, DS, Prystowsky, EN. Management of ventricular arrhythmias: detection, drugs, and devices. JAMA. 1999;281:272–279.
49. Tan, HL, Lie, KI. Prophylactic lidocaine use in acute myocardial infarction revisited in the thrombolytic era. Am Heart J. 1999;137:570–573.
50. Kowey, PR. An overview of antiarrhythmic drug management of electrical storm. Can J Cardiol. 1996;12(Suppl B):3B–8B. [discussion 27B–28B].
51. Slavik, RS, Tisdale, JE, Borzak, S. Pharmacologic conversion of atrial fibrillation: a systematic review of available evidence. Prog Cardiovasc Dis. 2001;44:221–252.
52. Lalka, D, Meyer, MB, Duce, BR, Elvin, AT. Kinetics of the oral antiarrhythmic lidocaine congener, tocainide. Clin Pharmacol Ther. 1976;19:757–766.
53. Perlow, GM, Jain, BP, Pauker, SC, et al. Tocainide-associated interstitial pneumonitis. Ann Intern Med. 1981;94(4 Pt 1):489–490.
54. Volosin, K, Greenberg, RM, Greenspon, AJ. Tocainide associated agranulocytosis. Am Heart J. 1985;109:1392.
55. Bronheim, D, Thys, DM. Cardiovascular drugs. In Longnecker DE, Tinker JH, Morgan GE, Jr., eds.: Principles and practice of anesthesiology, ed 2, St Louis: Mosby, 1998.
56. Kudenchuk, PJ. Advanced cardiac life support antiarrhythmic drugs. Cardiol Clin. 2002;20:19–87.
57. American Heart Association. Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2000;102:8–149.
58. Kopacz, DJ, Carpenter, RL, MacKay, DL. Effect of ropivacaine on cutaneous capillary flow in pigs. Anesthesiology. 1989;71:69.
59. Scott, DB, Jebson, PJR, Braid, DP, et al. Factors affecting plasma levels of lignocaine and prilocaine. Br J Anaesth. 1972;44:1040–1049.
60. Duhner, KG, Harthon, JGL, Hebring, BG, Lie, T. Blood levels of mepivacaine after regional anaesthesia. Br J Anaesth. 1965;37:746–752.
61. Kimmey, JR, Steinhaus, JE. Cardiovascular effects of procaine and lidocaine (Xylocaine) during general anesthesia. Acta Anaesthesiol Scand. 1959;3:9–15.
62. de Jong, RH, Ronfeld, R, DeRosa, R. Cardiovascular effects of convulsant and supraconvulsant doses of amide local anesthetics. Anesth Analg. 1982;61:3.
63. Feldman, HS, Arthur, GR, Covino, BG. Comparative systemic toxicity of convulsant and supraconvulsant doses of intravenous ropivacaine, bupivacaine and lidocaine in the conscious dog. Anesth Analg. 1989;69:794.
64. Zink, W, Graf, BM, Sinner, B, et al. Differential effects of bupivacaine on intracellular Ca2+ regulation: potential mechanisms of its myotoxicity. Anesthesiology. 2002;97:710–716.
65. Irwin, W, Fontaine, E, Agnolucci, L, et al. Bupivacaine myotoxicity is mediated by mitochondria. J Biol Chem. 2002;277:12221–12227.
66. Benoit, PW, Yagiela, JA, Fort, NF. Pharmacologic correlation between local anesthetic-induced myotoxicity and disturbances of intracellular calcium distribution. Toxicol Appl Pharmacol. 1980;52:187–198.
67. Hinton, RJ, Dechow, PC, Carlson, DS. Recovery of jaw muscle function following injection of a myotoxic agent (lidocaine-epinephrine). Oral Surg Oral Med Oral Pathol. 1986;59:247–251.
68. Denborough, MA, Forster, JF, Lovell, RR, et al. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395–396.
69. Gielen, M, Viering, W. 3-in-1 lumbar plexus block for muscle biopsy in malignant hyperthermia patients: amide local anaesthetics may be used safely. Acta Anaesthesiol Scand. 1986;30:581–583.
70. Ording, H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64:700–704.
71. Paasuke, PT, Brownell, AKW. Amine local anaesthetics and malignant hyperthermia (editorial). Can Anaesth Soc J. 1986;33:126–129.
72. Jastak, JT, Yagiela, JA, Donaldson, D. Local anesthesia of the oral cavity. Philadelphia: WB Saunders; 1995. [pp 141–142].
73. Malignant Hyperthermia Association of the United States. Available at www.mhaus.org. [Accessed October 28, 2011].
*ALL chemicals (drugs) have the potential to be poisons, also called toxins. When the resulting blood level is too high, drugs exert negative actions, which we call a toxic reaction or overdose.
*Individual variation in response to drugs, as depicted in the normal distribution curve, may produce clinical symptoms at levels lower than these (in hyperresponders) or may fail to produce them at higher levels (in hyporesponders).