10 Immunity in viral infections
Key points
• Interferons provide a key innate defence to viral infection. Their overall effect is to inhibit viral replication.
• Antibodies act on extracellular virus to prevent establishment of infection. In view of their intracellular replication, viruses are particularly targeted by cell-mediated immunity.
• Many of the symptoms and signs of virus infection reflect immune responses to viral antigens (immunopathology) rather than direct damage due to viral replication.
• Many viral infections induce a temporary immune suppression rendering the host susceptible to bacterial infection, whereas others such as HIV produce a more permanent effect.
• There are many vaccines that protect against viral infections. Smallpox has been eradicated by vaccination and other eradications are feasible.
The host response to an invading virus depends on the characteristics of the infectious agent and where it is encountered. In many cases viral infections are sub-clinical, that is, symptomless. A vast array of host defence mechanisms work in a concerted way to protect the individual from viruses and to eliminate them if an infection occurs. In several instances, virus-induced immune responses may have immunopathological consequences.
The response to viral infections
Interferons
At the time of the discovery of interferon in 1957, the term was used to identify a factor produced by cells in response to viral infection that protected other cells of the same species from attack by a wide range of viruses. It is now clear that this activity is mediated by members of a family of regulatory proteins.
In man, as in a number of other species, there are three main interferons:
1. α-Interferon (IFN-α), produced mainly by peripheral blood mononuclear cells
2. β-Interferon (IFN-β), produced predominantly by fibroblasts
3. γ-Interferon (IFN-γ), a lymphokine produced in response to a specific antigenic signal.
IFN-α and IFN-β are known as type 1 interferons and are considered part of the innate defences whereas IFN-γ is referred to as type 2 interferon.
There is only one gene for IFN-β and one for IFN-γ, but there are at least 18 different IFN-α genes coding for 14 functional proteins. All the IFN-α genes are closely related and clustered on chromosome 9, close to the IFN-β gene; the IFN-γ gene is on chromosome 12. The production of interferons is under strict inductional control. IFN-α and IFN-β are produced in response to the presence of viruses and certain intracellular bacteria. Double-stranded RNA may be the important inducer acting through Toll-like receptors (see p. 112) to signal the production of type 1 interferons. IFN-γ, which has an extensive role in the control of immune responses, is produced by antigen-activated T lymphocytes and natural killer (NK) cells (see pp. 127–8).
To exert their biological effects these molecules must interact with cell surface receptors. IFN-α and IFN-β share a common receptor, whereas IFN-γ binds to its own specific receptor. After binding to the cell surface receptors, interferons act by rapidly and transiently inducing or upregulating some cellular genes and downregulating others. The overall effect is to inhibit viral replication and activate host defence mechanisms.
The antiviral activity is mediated by the interferon released from a virus-infected cell binding to a neighbouring cell and inducing the synthesis of antiviral proteins (Fig. 10.1). Interferons are extremely potent in this function, acting at femtomolar (10−15M) concentrations. They can inhibit many stages of the virus life cycle – attachment and uncoating, early viral transcription, viral translation, protein synthesis and budding. Many new proteins can be detected in cells exposed to interferon, but major roles have been proposed for two enzymes that inhibit protein synthesis: 2′,5′-oligoadenylate synthetase (2,5-A synthetase) and a protein kinase. The activity of both of these enzymes is dependent on double-stranded RNA (dsRNA) provided by viral intermediates in the cell. The protein kinase is responsible for the phosphorylation of histones and the protein synthesis initiation factor eIF2. This leads to the inhibition of protein synthesis within interferon-stimulated cells as a result of inhibition of ribosome assembly. The 2,5-A synthetase is strongly induced in human cells by all three types of interferon and forms 2′,5′-linked oligonucleotides of adenosine from adenosine triphosphate (ATP). These oligonucleotides activate a latent cellular endonuclease that degrades both messenger and ribosomal RNA, with a resultant inhibition of protein synthesis. The requirement for the presence of dsRNA for the full expression of these responses safeguards uninfected cells from the damaging effects of the enzymes. Apart from these well characterized changes, many other changes occur in cells treated with interferons. Some viral proteins can inhibit the interferon response.
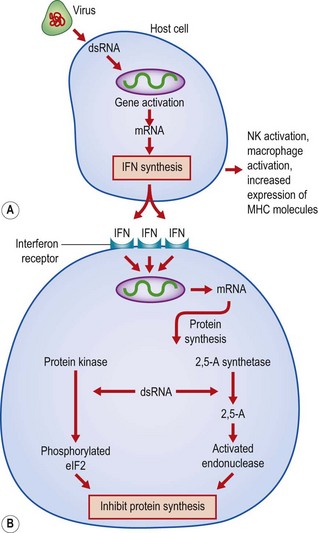
Fig. 10.1 Proposed mechanisms of a induction of synthesis of interferon (IFN)-α and IFN-β, and b production of resistance to virus infection. 2,5-A, 2′,5′-oligoadenylate; dsRNA, double-stranded ribonucleic acid; eIF, eukaryotic initiation factor; MHC, major histocompatibility complex; mRNA, messenger RNA; NK, natural killer cells.
Some effects of interferon are virus-specific. The Mx protein induced in mice by IFN-α/-β is specifically involved in the resistance to influenza virus infection. There is a related protein in human cells, and it is possible that some of the other interferon-induced proteins may confer resistance to specific virus types. Resistance of IFN-γ-treated cells to the parasite Toxoplasma gondii is associated with induction of the enzyme indolamine dioxygenase, which catabolizes the essential amino acid tryptophan. However, interferons have effects on host cell growth and differentiation. Interferons, particularly IFN-α and IFN-β, are potent inhibitors of normal and malignant cell growth. A number of clinical trials have shown that IFN-α is active against some human cancers, especially those of haemopoietic origin.
Interferons are able to modify immune responses by:
• altering the expression of cell surface molecules
• altering the production and secretion of cellular proteins
One of the main ways in which interferons control immune responses is by the induction or enhancement of major histocompatibility complex (MHC)-encoded molecules.
Class I MHC genes are upregulated by all types of interferon, as is the production of β2-microglobulin. IFN-γ induces and increases the expression of MHC class II antigens. In addition, interferons can induce or enhance the expression of Fc receptors and receptors for a number of cytokines. These activities increase the efficiency of antigen recognition and lead to a more effective immune response.
Interferons have also been implicated in the control of B cell responses. When added in vitro or in vivo they can suppress or enhance primary or secondary antibody responses, depending on the dose and time of addition. The regulatory effects seem to be on the B cells themselves, on increased antigen presentation and through an effect on regulatory T cells.
A number of immune effector cells act by killing infected target cells. The cytotoxicity of macrophages, neutrophils, T cells and NK cells is enhanced by interferons. IFN-γ produced by T lymphocytes is capable of activating macrophages to kill intracellular bacteria. This lymphokine has all the activities of the molecule that used to be known as macrophage-activating factor (MAF). NK cells are able to destroy a range of syngeneic, allogeneic and xenogeneic cells in an MHC-unrestricted fashion (see below). All three types of interferon increase NK cell activity in vitro and in vivo, not only by recruiting pre-NK cells to become actively lytic but also by increasing the spectrum of cells lysed. The mechanisms by which interferons make cells cytotoxic are not clear, but it is of interest that interferons can stimulate the production of cytotoxins such as TNF.
TNF has also been reported to have several antiviral activities similar to those of IFN-γ, but working through a different pathway. However, if both TNF and IFN-γ are added together, a synergistic effect is seen. If TNF is added to cells after viral infection, it can lead to their destruction even though the cells are normally resistant to TNF. This effect is also synergistic with IFN-γ. Certain viruses have been shown to trigger the release of TNF from mononuclear cells, and it seems likely that this cytokine is an important host response to viral infections.
Certain cell-mediated reactions are also part of the innate defences against viral infection. NK cells recognize a variety of cell surface molecules and changes in the level of expression of MHC class I molecules on the infected cells leads to expression of lytic activity. A number of viruses, especially those causing latent infections, evade immune recognition by interfering with the MHC class I processing pathway. Cells infected with these viruses do not display MHC class I molecules on their surface and are therefore not recognized by cytotoxic T cells. However, the infected cells are recognized by NK cells. The formation of a close conjugate between the NK cell and the target induces the effector cell to produce molecules that lead to the death of the infected cell by apoptosis. Antibodies that recognize NK cells have been used to deplete these cells in mice. It was found that the treated mice were more susceptible to murine cytomegalovirus than were normal mice. Natural killing may form a first-line defence against viral attack, most importantly by herpesviruses, before the acquired immune response is generated. Natural killing is increased by interferons (both the number of effector cells and their killing potential) and, therefore, these two innate defence mechanisms appear to work together to protect the host from viral infection.
The interferon response is rapid and helps to protect the host until acquired responses develop. Interferons induce a febrile response and this may also be important in inhibiting viral growth in some infections with viruses that have a low ceiling temperature for growth.
Acquired immunity
The response to viral antigens is almost entirely T cell dependent. Immunodeficiencies involving T cells are always characterized by markedly enhanced susceptibility to viral infections. However, this tells us little about the effector mechanisms involved, as T cells are required for both antibody production and cytotoxic reactions.
The viral epitopes to which the immune system responds have been studied to give an insight into the mechanisms involved in the host response to these pathogens and also to aid in the development of better vaccines. The recognition of viral antigens is similar to that for all foreign material. B cells and immunoglobulin are able to combine with exposed epitopes, and processed viral fragments presented in the context of MHC molecules are recognized by T cells.
Antigen-specific B cells can act as antigen-presenting cells and thereby generate an immune response. B cells present antigen to T cells and in return are stimulated by growth and differentiation molecules. Intramolecular help may explain hapten–carrier effects. The uptake of an intact virion means that the B cell is able to present peptides derived from internal proteins to T cells (Fig. 10.2). Thus, a B cell that is specific for a surface antigen can receive help from a T cell specific for another molecule as long as it is present within the same particle – intrastructural help.
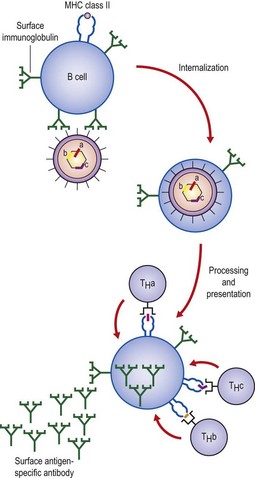
Fig. 10.2 Intrastructural T cell help. A B cell that is specific for a surface component of a virus binds the virus through its receptor, surface immunoglobulin. The virus contains three potential T cell epitopes (a, b and c) within its nucleocapsid. The entire virus can be internalized, and all viral polypeptides will be processed and fragments re-expressed in association with MHC class II molecules (presentation) on the B cell surface.
In most cases, exogenous virus proteins (i.e. those derived from an extracellular virus and taken into a cell) are presented in the context of MHC class II molecules and stimulate TH cells. Cells that are supporting viral replication express virus-derived peptides in association with MHC class I molecules (the endogenous pathway). The fact that some endogenously produced viral proteins are presented in the context of MHC class II molecules suggests that there can be some overlap between the MHC class I and class II pathways.
Humoral immunity
There are several ways in which antibody against viral components can protect the host. Antibodies cannot enter cells, and therefore are ineffective against latent viruses and those that spread directly from cell to cell. They do, however, bind to extracellular viral epitopes. These epitopes can be on intact virions or on the surface of infected cells. The binding of antibody to free virus can inhibit a number of processes essential to virus replication. Antibodies can block binding to the host cell membrane, and thus stop attachment and penetration. The immunoglobulins IgG and IgM have this important function in serum and body fluids, and IgA can neutralize viruses by a similar mechanism on mucosal surfaces. Antibody can also work at stages after penetration. Uncoating, with its release of viral nucleic acid into the cytoplasm, can be inhibited if the virion is covered by antibody.
Antibody can also cause aggregation of virus particles, thus limiting the spread of the infectious particles and forming a complex that is readily phagocytosed. Complement can aid in the neutralization process by opsonizing the virus or directly lysing enveloped viruses. In certain cases complement alone can inactivate viruses. Some retroviruses have a protein that can act as a receptor for C1q, and other viruses have been reported to activate the alternative pathway. In some infections, viral proteins remain on the surface of the cell after entry or become associated with the cell membrane during replication. Antibodies against these molecules can cause cell lysis by the classical pathway, but an intact alternative pathway is necessary to amplify the initial triggering by the antibody-dependent pathway. In certain situations antibody-mediated reactions are not always of benefit (see below). Antibodies are also capable of modulating or stripping viral antigens from the cell surface, allowing the infected cell to avoid destruction by other effector mechanisms.
Viral infections, particularly those caused by entero-viruses, are frequent and severe when humoral immunity is impaired, as in certain inherited immunodeficiency states. In Bruton-type deficiencies, poliomyelitis may develop after vaccination with the live virus vaccine; meningo-encephalitis, caused by echovirus and coxsackie-virus, may also be seen. In many situations, viruses seem able to escape the humoral defence mechanisms. Some viruses become latent (e.g. herpesviruses) and are reactivated despite the presence of circulating antibody, as they can pass directly from cell to cell. Other escape mechanisms include antigenic variation in which the antigenic structure of the virus (e.g. influenza type A) changes so that antibodies formed to the previous strain are no longer effective.
In viral infections the efficiency of antibody depends largely on whether the virus passes through the bloodstream outside host cells to reach its target organ. Poliovirus crosses the intestinal wall, enters the bloodstream to cause a cell-free viraemia, and passes to the spinal cord and brain where it replicates. Small amounts of antibody in the blood can neutralize the virus before it reaches its target cells in the nervous system.
In comparison, in viral diseases such as influenza and the common cold the viruses do not pass through the bloodstream. These infections have a short incubation period, their target organ being at the site of entry into the body, namely the respiratory mucous membranes. In this type of infection a high level of antibody in the blood is relatively ineffective in comparison with its effect on blood-borne viruses. In this case the antibody must be present in the mucous secretions at the time of infection. There are very low levels of IgG or IgM in secretions, but IgA has been shown to be responsible for most of the neutralizing activity present in nasal secretions against rhinoviruses and other respiratory tract viruses.
One consequence of this is that conventional immunization methods using killed virus or viral subunits, which produce high levels of circulating antibody, are unlikely to be effective against viruses that attack the mucous membranes. Some considerable effort is being directed at developing methods for stimulating local production of IgA in the mucous membranes themselves. Live virus vaccines are effective in this respect, and the intranasal administration of a live-attenuated influenza virus vaccine is an attempt to overcome this problem. The high degree of immunity provided by the oral polio vaccine is due in part to locally produced antibody in the gut neutralizing the virus before it attaches to cell receptors to cause infection. The presence of IgA against polio has been demonstrated in faeces, duodenal fluid and saliva. There is evidence to suggest that parenteral administration of a killed virus may give rise to a secretory IgA response if the individual has previously been exposed to the virus or has received an oral-attenuated vaccine. So there appears to be a link between the systemic immune system and local mucosal immunity.
Humoral immunity does play a major protective role in polio and a number of other viral infections, and is probably the predominant form of immunity responsible for protection from reinfection. Passively administered antibody can protect against several human infections, including measles, hepatitis A and B, and chickenpox, if given before or very soon after exposure. Immunity to many viral infections is lifelong. This may occur because antibodies are boosted by occasional re-exposure to the virus.
Cell-mediated immunity
The destruction of virus-infected cells is an important mechanism in the eradication of virus from the host. Antibody can neutralize free virions, but once these agents have entered the cell other strategies are employed. The destruction of an infected cell before progeny particles are released is an effective way of terminating a viral infection. For this process to occur the immune system must recognize the infected cell, and various types of effector cell have evolved to mediate these processes.
As viral proteins are synthesized within the cell some of these molecules are processed into small peptides. These endogenously produced antigen fragments become associated with MHC class I molecules, and this complex is then transported to the cell surface where it acts as the recognition unit for cytotoxic T (TC) lymphocytes. Most TC cells have a receptor that binds to fragments of the virus sitting in the cleft of an MHC class I molecule. This T cell also has the CD8 molecule on its surface. Some TC cells are restricted in their recognition of antigen by MHC class II molecules and therefore have the CD4 molecule. Once these TC cells have bound to the infected cell they release molecules that induce apoptosis.
Many viruses, such as poliovirus and papillomavirus, replicate and produce fully infectious particles inside the cell. These viruses are liberated from the infected cell as it disintegrates. However, other viruses do not wait for the cell to die but are released by a process of budding through the cell membrane. During their replication, virus-encoded molecules (viral antigens) are inserted into the host cell membrane, and the nucleocapsid becomes associated with these molecules. The virus particle finally acquires an envelope as it is released. Such viruses include herpesviruses, alphaviruses, flaviviruses, retroviruses, hepadnaviruses, orthomyxoviruses and paramyxoviruses. Viral antigens often appear on the cell surface very early in the replicative cycle, many hours before progeny virus is liberated. In cells infected with herpes simplex virus, as many as five different viral glycoproteins appear on the cell surface. In other virus infections, such as those caused by poxviruses and papovaviruses, the viral particles are not released by budding, but viral antigens appear on the cell surface. These molecules, including those that are not incorporated into the released virion, can therefore act as signals indicating the presence of virus within a cell. If antibody binds to these cell surface viral antigens, the infected cell can be destroyed by antibody-dependent cell-mediated cytotoxicity. The effector cells have an Fc receptor that recognizes the Fc portion of immunoglobulin bound to viral antigens present on the infected cell surface. This interaction brings the two cells close together, and toxic molecules are released on to the target cell membrane, causing cell death.
CD4+ and CD8+ T cells can produce various lymphokines when stimulated by antigen, including molecules that are active in the elimination of virus (e.g. IFN-γ and TNF), and others that generally increase the effectiveness of the immune system by attracting cells to the site of infection, stimulating the production of more cells and supporting their growth. Macrophages are activated, leading to enhanced microbicidal activities and the production of monokines.
Induction of an immune response
The precise nature of the acquired immune reactions that are generated in response to infection depends to a great extent on the site of infection, type of virus, previous exposure to the agent and the genetic make-up of the host. Both humoral and cell-mediated responses are produced to all infections. The importance of genetic factors is illustrated by the severe X-linked recessive lymphoproliferative syndrome in which fatal infectious mononucleosis results from the unrestricted replication of Epstein–Barr virus, as affected males have reduced numbers of normal lymphocytes.
The virus or viral components entering the peripheral tissues or being produced there are carried to the draining lymph node either free in the lymph or in cells such as Langerhans’ cells. Once in the secondary lymphoid tissues the free virus is taken up by macrophages and processed; viral peptides associate with MHC class II molecules and are transported to the cell surface. Langerhans’ cells that enter the lymph node become interdigitating cells and present viral peptides, associated with MHC class II molecules, on their surface. A TH cell will bind to the peptide–MHC class II complex, expand clonally and produce helper factors. The proportions of the different lymphokines produced determines the type and level of the response generated. B cells will enter the node, and those that bind antigen are stimulated into antibody production. Other T cells will enter the lymph node. If these cells have a receptor that interacts with an antigen fragment–MHC class I complex, they will respond to the growth factors present in the node and proliferate and mature into effector TC cells. After a time, the products of the immune response leave the node to circulate round the body and localize at the site of infection. As the response progresses the pathogen is eliminated, the tissue repaired and memory cells generated. Finally, when all of the antigen has been eliminated, the immune response is terminated. In some instances virus-induced immune responses may have immunopathogical consequences.
Immunopathology
Viruses have evolved a multitude of mechanisms for exploiting weaknesses in the host immune system and avoiding – sometimes actually subverting – immune mechanisms. Some viruses are so successful in avoiding host defences that they persist in the host indefinitely, sometimes in a latent form without producing disease.
One of the most important strategies developed by viruses is to infect cells of the immune system itself. The effect of this is often to disable the normal functioning of the cell type that has been infected. Many common human viruses, including rubella, mumps, measles and herpes viruses, infect cells of the immune system, as does human immunodeficiency virus (HIV). The consequences of viral infection of cells of the immune system have been categorized in two ways:
1. Infections that cause temporary immune deficiency to unrelated antigens and sometimes to the antigens of the infecting virus. It is known that infection with influenza, rubella, measles and cytomegalovirus predisposes to bacterial and other infections. This is sometimes associated with depressed immunoglobulin synthesis and interference with the antimicrobial functions of phagocytes.
2. Permanent depression of immunity to unrelated antigens and occasionally to antigens of the infecting virus. Acquired immune deficiency syndrome is an example of such a disease, where the patient becomes susceptible to otherwise harmless protozoa, bacteria, viruses and fungi.
Viruses have also developed other mechanisms to avoid the immune system. These include:
Viruses that cannot enter and replicate within phagocytic cells will be destroyed if they are engulfed by a neutrophil or macrophage. As neutrophils are short-lived cells they do not usually give rise to progeny virus. On the other hand, monocytes and macrophages are long-lived cells and can be responsible for disseminating a virus throughout the body. Viruses that do replicate within macrophages must escape from the phagosome very rapidly before it fuses with the lysosome. Reovirus infection of macrophages is actually helped by the lysosomal enzymes, which initiate ‘uncoating’ of the virus and therefore enhance viral replication.
A virus is relatively safe from immune destruction as long as it remains within the cell and allows only very low or no viral antigen expression on the infected cell membrane. This is what happens in latent infections where herpes simplex or varicella-zoster virus is present in the dorsal root ganglion.
Antibody can actually remove viral antigens from cell membranes, as cross-linking of antigens on the cell surface can lead to their internalization by capping. Here the antigens complexed with the antibody are drawn to one pole of the cell and internalized or shed into the surrounding tissue. Capping occurs on brain cells infected with measles virus in subacute sclerosing panencephalitis. Viruses that move from cell to cell without entering the extracellular fluid can also escape the action of antibodies, as can those passed from cell to cell by cell division.
A number of infections continually shed virus into external secretions, such as saliva, milk or urine. As long as the infected cell forms virus only on the luminal surface of the mucosa, cells of the immune system and antibody will be unable to destroy the infected cell. IgA present in the secretions may neutralize the virus, but this class of antibody does not activate complement efficiently, so the cell will not be lysed. A similar situation applies to epidermal infections with wart virus. The infected cell is keratinized and about to be released from the surface of the body before any virus or viral antigens are produced. The infected cell is therefore isolated from the host’s immune cells.
During the course of an infection various antibodies are formed against different epitopes on a virus. These antibodies are of differing affinities and also stimulate different effector functions. Antibodies against some of the epitopes will neutralize the virus, but other antibodies will be against unimportant epitopes or be of an ineffective isotype that may fail to neutralize the virus, and may actually aid in its infectivity by allowing uptake of virus–antibody complexes via Fc receptors or cause tissue damage through immune complex disease. Soluble antigens liberated from infected cells may ‘mop up’ free antibody so that it can no longer interact and destroy extracellular virus. Whether the small particles present in the serum of patients and carriers with hepatitis B virus infections function in this way is not known, although patients in the prodromal phase may suffer from rash, myalgia and arthralgia. Polyarteritis nodosa and glomerulonephritis can also occur, all suggestive of immune complex formation.
Susceptibility to infection is generally greater in the very young and very old because of a weaker immune response. However, the immunopathology tends to be less severe. In the very young, infections can spread rapidly and prove fatal without the clinical and pathological changes seen in adults. Latent infections are kept under control by the immune system, and in older people the infections show an increased incidence of activation (e.g. zoster, or shingles). Immunological immaturity makes the neonate highly susceptible to many viral infections. Maternally derived antibody provides passive protection for 3–6 months, after which time the infant is at risk of infection; respiratory and alimentary tract infections are frequent.
Physical and physiological differences may also contribute to age-related disease susceptibility. Respiratory infections in old age are probably a bigger problem than in young adults because of weaker respiratory muscles and a poorer cough reflex. In the young the airways are narrower and more easily blocked by secretions and exudate. Infants, because of their low body-weight, show signs of distress from loss of fluid and electrolytes, so that fever, vomiting and diarrhoea tend to be very serious at this time of life. Often the reasons for the differences between infants and adults are not known. Respiratory syncytial virus causes severe illness in the early months of life with croup, bronchiolitis and bronchopneumonia, despite the presence of maternal IgG. In adults the virus usually causes a mild upper respiratory tract infection.
Certain viral infections produce a milder disease in children than in adults (e.g. varicella, mumps, polio myelitis and Epstein–Barr virus infections). Varicella often causes pneumonia in adults, and mumps may involve the testes and ovaries after puberty, giving rise to orchitis and oophoritis. Epstein–Barr virus is excreted in saliva, and in developing countries most individuals are infected early in life, usually asymptomatically. In developed countries where childhood infection is less common, first infection may be delayed to adolescence or early adulthood, when salivary exposure occurs during kissing. In this age group, Epstein–Barr virus infection gives rise to glandular fever. It is not clear why these infections are more severe in adults, but it may be linked to the more powerful immune defences giving rise to immunopathological sequelae in adults.
There are also age-related differences in the incidence of infections. It is not surprising that most infections are most common in childhood when the individual is exposed to the micro-organisms for the first time.
Antigenic variation
A micro-organism can avoid the acquired immune response by periodically changing the structure of molecules that are recognized by the host immune system. The immune system selects the variants by not being able to mount an immune response against them before they are shed. The micro-organism will only be able to change a component in a way that does not alter the functioning of the molecule. The molecules involved can be active enzymes, recognition molecules or structural proteins. HIV shows considerable variation in parts of the envelope glycoprotein within a given individual.
The significance of antigenic variation is well illustrated by influenza viruses. Here changes in the surface glycoproteins are linked to the occurrence of epidemics of infection (see Ch. 49).
With influenza virus the infection is localized to the respiratory tract where the principal protection against reinfection is secretory IgA. The virus-specific IgA still present at the mucosal surface a few years after infection can protect against the original infecting virus but may be insufficient to deal with an antigenic variant despite antigenic overlap. Thus, in effect, IgA levels become a selective pressure, which will allow infection by the mutant, and antigenic drift occurs. The very short incubation period (1–3 days) is more rapid than the secondary antibody response so that the IgA levels cannot be boosted in time to abort infection.
Antigenic variation is likely to be an important viral adaptation for overcoming host immunity in long-lived species such as humans where there is a need for multiple reinfection of the same individual if the virus is to survive and the virus is unable to become latent. In shorter-lived animals such as mice and rabbits a susceptible population appears quickly enough to maintain the infectious cycle.
Persistence of virus
Certain viruses give rise to a persistent infection, which is held in check as long as the immune system remains intact.
Chickenpox is a persistent infection characterized by latency in that there is apparent recovery from the original infection but the virus can reappear later in life when a localized eruption, shingles, results. Other herpesviruses, cytomegalovirus and Epstein–Barr virus also persist after infection. If the carrier’s immune system remains intact, there will be no evidence of disease. However, cytomegalovirus causes many problems in immunosuppressed patients. The polyomavirus, JC, usually causes asymptomatic infections, but in the immunosuppressed it has been found in areas of destruction in the central nervous system; the disease is progressive multifocal leucoencephalopathy.
In other persistent infections, the immune system contributes to the pathology of the disease, often over a period of years. Thus, in subacute sclerosing panencephalitis, persistence of measles virus in neurones triggers their destruction by the host’s immune system. Similarly, the chronic active hepatitis seen in those carriers of hepatitis B who continue to produce virions appears to be caused by a TC cell response to viral antigens present in the hepatocyte membrane. In both examples, several years may elapse before symptoms appear.
Vaccines
Natural infection with a virus is an extremely effective means of giving lifelong immunity from the disease. In most cases, where there is one virus type, this means that second attacks are extremely rare. The memory of the immune system ensures that, for these infections, a secondary response can be generated before the virus has time to cause the disease. The level of immunity needed to protect an individual depends on the incubation period of the virus and its life cycle. For viruses with very short incubation periods, a high level of protective immunity must be present before exposure to the infective agent. In the case of a virus with a long incubation period (10–20 days), the immune system has time to generate a protective response.
It is also important to consider the type of immune response that will be protective against different viruses. If antibody gives protection then steps must be taken to ensure that the material to be used for immunization contains the correct epitopes. A denatured antigen will not generate antibodies that can combine with the native virus. Sometimes the chemical treatment of the antigen may destroy important components. Thus the original killed measles virus vaccine did not contain the fusion protein. As a result, vaccinees suffered enhanced disease when exposed to the virus as viral replication and spread could occur in the presence of antibody to the other viral proteins. If T cell immunity is important, the vaccine must be in a form that will give rise to peptides in the correct compartment of the cell to produce antigen fragments in association with MHC-encoded products. It will have to associate with the MHC class II molecules to generate help and with MHC class I molecules to stimulate effector cell formation. For antibody production, T cell help is also required. Therefore, for an antibody response, a killed vaccine may be sufficient but, when T cell immunity is required, a live-attenuated vaccine is needed.
Vaccination has been responsible for the elimination of smallpox and for reducing the incidence of other viral diseases. It should be possible to control many viral diseases, but with some the problem is more difficult. New technologies and a better understanding of the immune system are helping with this task.