Chapter 26 Genetics
Introduction
Genetics is the study of inheritance and variation in both individuals and populations and has its origins in man’s curiosity about reproduction and inheritance, including inquisitiveness about both normal and abnormal developments – for example, why do certain diseases ‘run in families’, whilst others do not?
Virtually all pregnant women will want to know if their baby is within the spectrum of that which is regarded as ‘normal’, and advances in genetics can both help to answer this question and have other great practical benefits in the field of human reproduction: for example, the probability that a child will suffer from a particular genetic disease can be calculated, allowing parents to make informed choices in planning their families; it may be possible, in a few cases, to replace or supplement defective genes so that the diseases they cause can be cured (‘gene therapy’); the potential of stem cells is also on the horizon. Hence, it is important that practitioners in this area are aware of both fundamental principles and relevant applications of genetics. There are many ethical questions associated with genetics and it is also important that these are fully addressed.
Genes, chromosomes and dna
A gene is typically defined as a unit of inheritance. Genes are found on chromosomes, which are long, thread-like structures in the nucleus of the cell; each chromosome is made up of one DNA (deoxyribonucleic acid) molecule with many different proteins associated with it. Many genes are arranged throughout its length in a fixed manner, one after the other (Fig. 26.1).
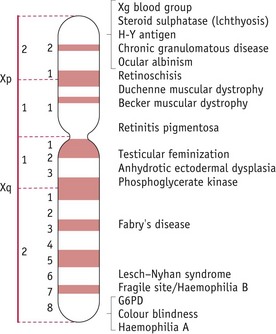
Figure 26.1 Diagrammatic representation of the human X-chromosome showing some of the inherited characteristics carried.
DNA is the major single component of each chromosome and it acts as an information store: all the information necessary for the structure, function, reproduction and development of an organism is stored in a stable and coded form.
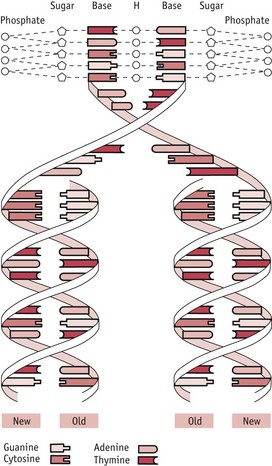
The DNA molecule is a long, thin molecule and consists of two strands wound around each other to form a double helix (Fig. 26.2): it is like a ladder in which the two uprights, whilst still remaining joined together by the rungs, are twisted round each other to form two interweaving spirals. In the DNA molecule, the ‘uprights’ are made of alternating sugar residues and phosphate groups (’sugar–phosphate backbones’) and the ‘rungs’ are composed of organic bases, each rung consisting of two bases. There are four different bases: two purine bases – adenine (A) and guanine (G); and two pyrimidine bases – cytosine (C) and thymine (T). Each ‘rung’ is made up of a pair of bases, one purine and one pyrimidine, and the pairing arrangements in these ‘rungs’ are very specific:
The information stored in DNA needs to be decoded and used to make cellular and organismal components (a process known as ‘gene expression’); most commonly, the stored information in DNA is used to direct the production of proteins. There are thousands of different proteins in the body (for example, collagen in tendons, haemoglobin in red blood cells), each with its own specific metabolic role to play. Any protein is made up of amino acids joined together in a chain. Each protein has its own specific amino acid sequence determining its function.
The encoding of information on a DNA molecule is achieved by specifying the precise order (sequence) of the bases along the molecule’s length. Information is stored in the form of three-base units (codons). Each codon specifies a particular amino acid in a protein. Hence, the sequence of bases in a DNA molecule directly determines the sequence of amino acids in a protein and thus the function of that protein. (Another definition of a gene is, ‘that part of a DNA molecule which contains the series of codons necessary to encode a particular protein’.)
The information stored in DNA is decoded using two processes: transcription and translation. In transcription (which takes place in the nucleus), part of the DNA molecule is used as a template to produce a molecule of messenger RNA (mRNA) and this mRNA then undergoes translation (on the ribosomes in the cytoplasm of the cell) to produce the protein specified ultimately by the DNA (Fig. 26.3).

Ribonucleic acid (RNA) is the other kind of nucleic acid found in the cell. Unlike DNA, it is single-stranded and it also contains the pyrimidine base uracil (U) instead of thymine. There are three different forms of RNA found in the cell, and all of these are involved in the decoding of information from DNA. These forms are: mRNA; rRNA (ribosomal RNA – a major component of the ribosome) and tRNA (transfer RNA – crucial in translation as an ‘adaptor’ molecule between mRNA and protein).
The human genome
The entire genetic complement of a cell or organism is referred to as its genome. Within the nucleus of all nucleated human cells (apart from the gametes – sex cells – ova and spermatozoa), there are 46 chromosomes: thus, the human genome comprises 46 chromosomes. These chromosomes are arranged in 23 (homologous) pairs, and within each of these pairs, one member is derived from the father, the other from the mother. One of these pairs is the sex chromosomes: in the female, this pair consists of two X chromosomes; in the male, it consists of one X and one Y chromosome. The remaining pairs are collectively known as autosomes and the chromosomes in this group are numbered by length, No. 1 being the longest and No. 22 the shortest (see Fig. 26.7).
Each chromosome carries a specific and particular set of genes and thus each individual carries two copies of any one gene, one copy derived from each parent. Any gene has different forms (or alleles). Classically, a gene has two different alleles. Different alleles produce differences in the appearance (or phenotype) of the individual. For example, the colour of the iris in the eye is controlled by a single gene which has two alleles – one allele causes the iris to be pale (blue or grey), the other allele causes it to be dark (brown, green or black).
With respect to any one gene, therefore, an individual can have two copies of one allele, two copies of the other allele (arrangements known as homozygous) or one copy of each (heterozygous). (Thus, for the hypothetical gene A with two alleles – A and a – the combinations AA, aa and Aa are possible.) Clearly, an individual who is homozygous for one allele would have the phenotype associated with that particular allele, but what of heterozygous individuals? In this case, one allele typically ‘overrides’ the other, an effect described as dominance of one allele over the other allele (which is known as the recessive allele).
One outcome from the Human Genome Project is that it is now known that the human genome contains a total of some 20,000 genes (Klug et al 2009).
Cell division
There are two different types of cell division:
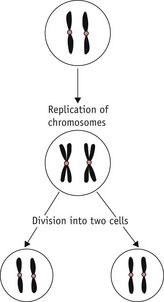
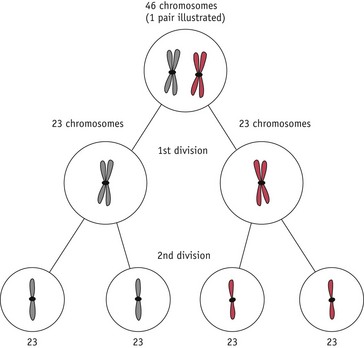
Mitosis occurs extensively during prenatal development to increase cell numbers, in the adult in tissues where cells are routinely lost (such as skin) and in some of the repair processes that occur following tissue damage. Meiosis only occurs during the production of gametes in the gonads. The reduction in chromosome number is essential if sexual reproduction is to take place without doubling the amount of genetic material in each generation. Note that some rearrangement of genetic material can occur during meiosis; this is an important part of the genetic variation that is an essential feature of sexual reproduction (Fig. 26.6).
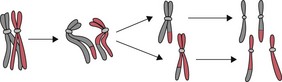
Copying (replication) of DNA occurs during both mitosis and meiosis and it is very important that this occurs accurately, so that errors are not introduced into the DNA, with possible consequent changes in the structure and function of encoded proteins. One very important way in which this is ensured is by the ‘semi-conservative’ mechanism of DNA replication: a parental DNA molecule separates into its two component strands and each of these is then used as a template for the assembly of a new strand. The strict rules of base pairing (A always pairing with T, G with C) ensure that each new double-stranded molecule is an exact copy of the parent molecule (See Fig. 26.2; Russell 2006).
Chromosomal analysis and anomalies
The study of chromosomes is called cytogenetics. Chromosomes can be isolated from accessible adult tissues (such as lymphocytes from the blood) or from amniotic fluid or chorionic villi when information about the embryo/fetus is required. The process of examining the chromosomes is referred to as karyotyping and it can provide very valuable information about the genome of the cell (Fig. 26.7A,B).
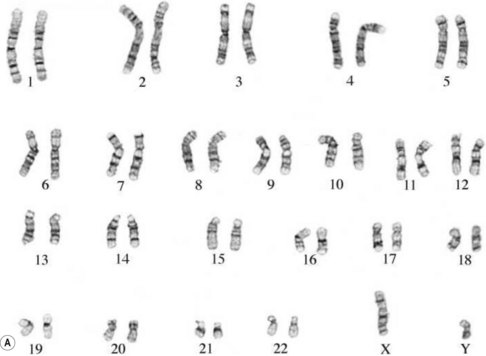
If there is an abnormal number of chromosomes present after fertilization (often due to abnormal events during meiosis), or if there is some alteration in structure, the pregnancy may result in miscarriage or an abnormal fetus. Generally, very major abnormalities (for example, involving whole or large pieces of chromosomes) are likely to have very serious effects. It has been shown that the majority of severe chromosomal abnormalities are not compatible with normal development. Embryos with such defects die at an early stage of development – for example, approximately 15% of pregnancies terminate in spontaneous abortion, with about 50% of these being chromosomally abnormal (Lockwood 2000).
Several different types of chromosomal abnormality are known:
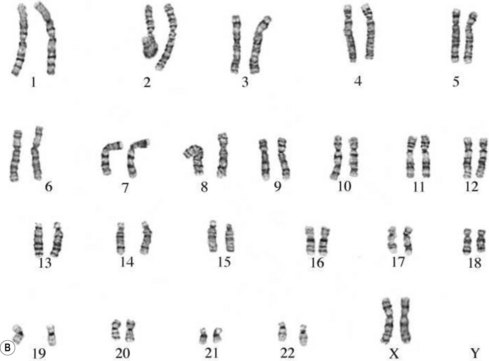
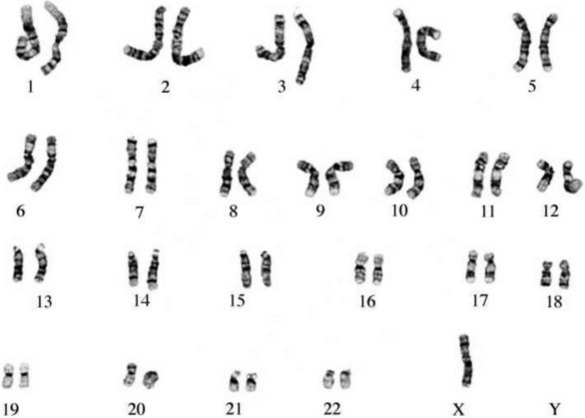
The commonest chromosomal birth anomaly is Down syndrome, where there are 47 chromosomes instead of 46 (termed trisomy 21 – three copies of chromosome 21) (Fig. 26.8A). The incidence is 1.5 per 1000 live births and this figure rises with maternal age (Cummings 2003). There are only two other examples of trisomies that are consistent with life – trisomy 13 and trisomy 18 – and even in these cases, the defects are many and severe, and affected individuals typically survive for only a short time after birth (Fig. 26.8B,C).
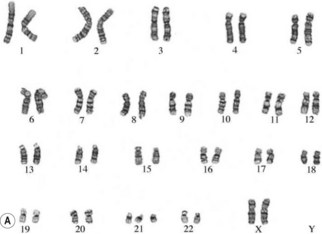


Figure 26.8 A. Karyotype of a female patient with Down syndrome (trisomy 21). B. Karyotype of a male patient with Edward syndrome (trisomy 18). C. Karyotype of a female patient with Patau syndrome (trisomy 13).
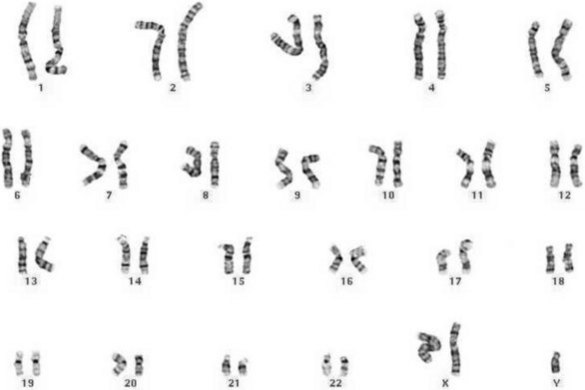
Other common chromosomal anomalies occur among the sex chromosomes; for example, 1.3% of implanted conceptions may carry one X-chromosome without a Y or second X (monosomy X, Turner syndrome – the only viable human monosomy) (Fig. 26.9). Very few of these fetuses survive – about 0.4 per 1000 live-born girls. More common at birth are other anomalies such as XXX (0.65 per 1000 girls), XYY, and XXY or XXXY (1.5 per 1000 boys – Klinefelter syndrome; Cummings 2003) (Fig. 26.10). These sex chromosome anomalies, however, generally produce fewer ill effects than the autosomal anomalies, though these effects can still be serious for the affected individual.
Modes of inheritance
Genetic characteristics (including genetically determined diseases) can be inherited in one of four principal ways:
Note that the usual patterns of dominance and recessiveness do not apply in the cases of genes carried on the sex chromosomes – see below.
Common types of genetic disease include:
Autosomal characteristics/diseases
Most human genetic diseases are due to mutations in autosomal genes, simply because there is much more genetic material in total in the 22 pairs of autosomes than in the single pair of sex chromosomes.
A homozygous person can only transmit one type of allele to any child they have, whilst a heterozygous person (‘carrier’) can transmit either of their two alleles to a child. Which allele a heterozygous parent transmits in any particular case is a random event, so there is a 1 in 2 (50%) chance of the child inheriting either allele. Thus, the following situations can arise:
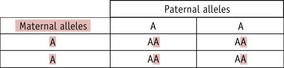
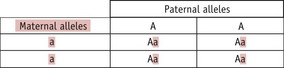
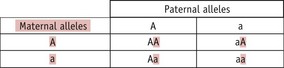
Thus, whilst both autosomal dominant and recessive characteristics/diseases occur in both males and females, dominant characteristics tend to be present in each generation, whilst recessive ones do not. This difference arises because a recessive characteristic can only be seen in the children of a heterozygous parent and either an affected parent or another heterozygous parent. If the particular recessive allele is rare, the probability of this happening is low. In practice, it is often found that parents of an individual affected by a rare recessive characteristic are related to each other (such as first cousins); thus, so-called ‘consanguineous unions’ are more likely to bring together two heterozygous parents.
If two parents are themselves unaffected and have a child with a recessive characteristic/disorder, it indicates that both of them must be heterozygous. There may then be concern about whether any subsequent child they may have will also be affected. In such a case, it is important to appreciate that the risk of any one particular child suffering from the recessive condition is always 1 in 4 or 25% (Fig. 26.13).
Further problems arise with diseases in which the disease process is either very variable in its severity (for example, myotonic dystrophy) or does not become apparent until later in life (such as Huntington’s disease). In the first case, it is possible for people to have and transmit the disease without suffering any serious ill effects themselves (although subtle signs of such diseases can often be detected); in the second, many sufferers have had their families before they realize they carry a mutant allele. In either case, parents may unknowingly pass on mutant alleles to their children and thus have several affected children.
Examples of recessively inherited autosomal conditions include phenylketonuria, galactosaemia and cystic fibrosis; examples of dominantly inherited autosomal conditions include achondroplasia, Marfan syndrome and Huntington’s disease.
Sex-linked characteristics/diseases
Sex-linked conditions are those for which the relevant genes are carried on the sex chromosomes. They are more complex in their inheritance patterns than are autosomal characteristics because of the genetic difference between men and women.
Obviously, there are genes on both the X chromosome and the Y chromosome and hence both X-linked and Y-linked characteristics are known. There are very few genes on the Y chromosome (which is very small) and there are thus very few Y-linked characteristics. As a result of this, Y-linked characteristics are seldom considered and the terms ‘X-linked’ and ‘sex-linked’ are often used interchangeably. Note that the genes carried on the two sex chromosomes are quite different.
Only one of the two X chromosomes is active in any cell in a woman’s body, the other is inactive (a ‘Barr body’). Which X is inactive in any one cell is random and differs throughout the cells of the woman’s body.
A very important consequence of the difference in sex chromosome complement between men and women is that men will always exhibit a characteristic resulting from the allele that they carry on their X chromosome since they do not have a second X chromosome. With respect to women, the situation for X-linked characteristics is very similar to that for autosomally determined characteristics: any one woman can be homozygous dominant, homozygous recessive or heterozygous for any gene on the X chromosome. Thus, a woman will only exhibit a recessive X-linked characteristic if she is homozygous recessive. This means that such a woman must be the child of a father who exhibits that characteristic and either a ‘carrier’ mother or a mother who also exhibits that characteristic, and, since these combinations of parents are very rare, such women, too, are very rare.
Both X-linked dominant and recessive characteristics/diseases are known and their patterns of inheritance can be summarized as follows:
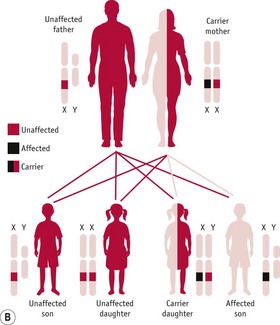
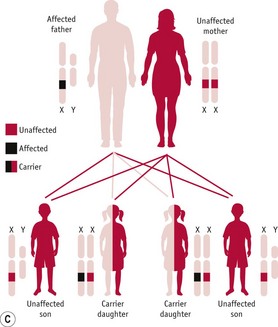
Figure 26.14 A. Pedigree diagram of X-linked recessive inheritance. B. Diagram of X-linked recessive inheritance with carrier mother. C. Diagram of X-linked recessive inheritance with affected father.
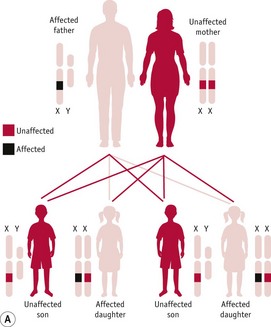
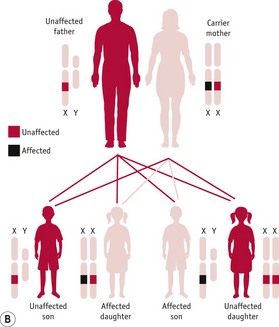
Figure 26.15 A. Diagram of X-linked dominant inheritance with affected father. B. Diagram of X-linked dominant inheritance with affected mother.
Among the best-known examples of X-linked recessive diseases are Duchenne muscular dystrophy, red/green colour-blindness, and the haemophilias. There are only a few X-linked dominant diseases, one example being hypophosphataemic (vitamin D-resistant) rickets.
It is important to note that, in any generation, new mutations can arise during meiosis and, clearly, the above inheritance patterns will then not be applicable.
Polygenic and multifactorial characteristics
Many inherited characteristics/diseases are influenced by several genes rather than just one or two. Such characteristics are said to be polygenic (‘many genes’), and the details of their inheritance patterns are often difficult to define precisely owing to the interplay between the different alleles of the various genes involved (Cummings 2003).
Polygenic inheritance should not be confused with situations where several genetic disorders can cause the same effect, for example, blindness. There are many different inherited conditions that cause blindness, but most of them are simple autosomal recessive or X-linked conditions with only one mechanism operating.
If environmental factors, such as diet, as well as genetic ones are important in determining the precise nature of a physical characteristic or disease process, the characteristic/disorder is said to be multifactorial in origin – examples of such disorders include neural tube defects and many cases of cleft lip and/or palate. Most characteristics (for example, height) and diseases (such as atherosclerosis – a very common and serious disease of larger arteries) are, in fact, multifactorial in origin, often involving several genes and several environmental factors. It can prove very difficult to determine the precise risk status of any one individual for any particular condition in such cases owing to the large number of different contributory factors involved and the great range of ways in which these can vary.
Origin of genetic diseases
In considering inherited disease, the problem arises as to why such conditions exist. New mutational events occur frequently – for example, because of errors in DNA replication during cell division, or exposure to mutagenic environmental agents, such as various forms of ionizing radiation (e.g. X-rays, ultraviolet light) or mutagenic chemicals (e.g. complex organic molecules in tobacco smoke).
Single new mutations can produce a dominant autosomal disease or a sex-linked disorder. Recessive disorders, however, cannot be explained so simply because both parents must carry the same mutation for the condition to occur. Rare recessive diseases may be explained by the lack of natural selection against a mutation that causes the heterozygous individual no ill effects, thus allowing the mutation to remain until, eventually, two people carrying the same mutation meet and produce a child who then suffers from the particular disease. There are, however, a number of relatively common recessive autosomal diseases (for example, sickle cell anaemia, cystic fibrosis) for which explanations of their high frequency of occurrence must be found. In general terms, such high frequencies are thought to be explained by the existence of some selective advantage that is peculiar to the ‘carrier’ (heterozygous individual). In some cases, the precise nature of this advantage has been determined. For example, in the case of sickle cell anaemia (in which the mutation occurs in the gene for one of the components of the gas transport protein haemoglobin), whilst those people who suffer from this disease (homozygous mutants) often die prematurely, those who carry one normal allele and one mutant (’sickle cell’) allele show an increased resistance to malaria compared with people carrying two copies of the normal allele. Thus, in malarial areas, as the heterozygous person enjoys a selective survival advantage, the mutant allele is selected for and achieves a high frequency (Cummings 2003). In other cases, the precise nature of the advantage remains to be elucidated – it could be that carriers have some advantage in sexual competition, or that gametes carrying such alleles are somehow preferred, but evidence to support these hypotheses has yet to be produced.
Cummings (2003), Russell (2006) and Klug et al (2009) provide excellent, more detailed coverage of all the above material. Also, see the website for further reference to diagrams, animations and other supporting material.
Assessment of embryo and fetus
Our ability to usefully assess the genetic make-up of the embryo and fetus has increased with further understanding of genetic influences on disease. Parental and social expectations of normal/perfect babies appear greater than ever before. Those involved in providing care now need to be aware of developments in this field to enable them to provide appropriate up-to-date information to women and their partners.
The process of screening and diagnosing genetic disorders has advanced significantly in recent years, making it seem easier to assess fetal normality earlier in pregnancy. Prenatal screening and diagnosis need to be differentiated. The Department of Health (2007) and NICE (2008) have laid down national standards for prenatal screening tests that should be offered to all pregnant women. Screening tests usually identify particular individuals or specific populations at increased risk and may just show the probability of a problem with the fetus. In most cases, further specific diagnostic tests are required to give a more definitive diagnosis of fetal abnormality.
Genetic counselling/advice
Women and their partners in a higher-risk category are faced with some challenging decisions early in pregnancy, or before conception. These individuals will need opportunities to seek accurate, relevant, up-to-date information and time to consider the options available. Specialists trained in genetic counselling may be involved in the process, with the midwife having input in a facilitative and supportive role.
The role of genetic counselling is to facilitate effective decision-making, enabling deliberation on the internal and external factors that need to be considered for individuals to make the right choice for them. Examples of internal factors may include personal values and beliefs, previous experiences, and expectations of the present pregnancy or subsequent pregnancies, whilst external factors may relate to cultural values, religious beliefs, family and friend influences, economics, education, and social circumstances. It cannot be assumed when partners are involved that both parties will hold similar views and beliefs about the situation which may threaten their expectations of the baby.
Pre-conception counselling may be conducted in cases of a known or suspected family history of genetic disorders, or a previous pregnancy affected by a genetic disorder (such as cystic fibrosis [CF]). This will allow informed decisions to be made regarding family planning. (It is important to note, however, that this pre-conception approach cannot provide absolute certainty that any child will be disease-free. This is because, firstly, many diseases – such as CF – can be caused by a large number of different mutations and it is both technically demanding and very resource-intensive to screen for all of these. Secondly, even if the parents’ somatic DNA is shown to be free of mutations, there is always the possibility that a new mutation could arise during gamete formation in one or both parents.) However, even with this caveat, this application of molecular genetic research has proved to be of great benefit to families with a history of genetic disease.
In the majority of cases, midwives become involved when meeting pregnant women. In providing advice to facilitate informed choices, there should be adequate explanations of the processes, risks, benefits, accuracy of the tests (including information on false positives/negatives), outcomes and alternatives for any assessments offered. This should include a focus on how far advanced the pregnancy will be before the test can be completed and how long the results may take to become available (Chapple 2006).
Reflective activity 26.3
Arrange a visit to an ultrasound department, and consider both the information requested by women and their partners, and the information given to them.
What impact did this experience have on your information-giving to women?
The key to successfully supporting women and their partners through this process lies in an awareness of the influence of effective communication skills, acknowledging their values and beliefs. The midwife can facilitate the woman/couple making an autonomous decision by building a trusting relationship and knowing the woman and partner’s expectations and beliefs.
Effective counselling enables couples to believe that they have made the right decisions for them, which includes accepting the consequences. All circumstances need to be handled with sensitivity because some individuals may not have previously considered the possibility of fetal abnormality. Unfortunately, one of the difficulties with effective decision-making in pregnancy is the lack of time available to make significant decisions as gestation continues – especially for women who book late in pregnancy.
Owing to a variety of reasons, such as limited family histories and variations observed in severity of a disease process, it can sometimes be difficult to provide clear guidelines as to inheritance, significance of outcomes and the effect(s) on individuals in any one particular counselling situation. With this in mind, any information offered needs to clarify the gaps in knowledge as well as the advances made in the related field.
Once risk factors have been established, information should be provided on their potential impact on the fetus and baby, including options for treatment, if any, and the nature and course of any possible resultant illnesses. Whilst advances in medicine have considerably altered the outlook for many conditions (for example, persons with cystic fibrosis now have a greatly improved life and health expectancy), it is obviously important that advice is realistic, both highlighting the advances made and stressing the drawbacks currently experienced. Often, referral to specialist support groups may be useful at this time.
Equally, it is essential to discuss the impact of tests on the woman with any significant other people in her life, as influences might be consequential to the responsibility and acceptance of any decisions made.
Once counselled, women (and their partners) will have decisions to make. The choices open to them may include:

The choices are nearly always complex, and consequently it is crucial that accurate knowledge is available to facilitate the best personal decisions. Case scenarios 26.1 and 26.2 on the website illustrate situations that may arise.
Screening for risk indicators
The primary aim of screening for risk indicators/markers is to provide accurate information to facilitate fully informed decisions about proceeding with diagnostic tests to confirm or exclude a genetic disorder. There are various indicators and the utility of these in individual cases may depend on available information, previous history, the duration of gestation, and parental expectations of normality. Midwives have a major role to play in prenatal screening programmes (Chapple 2006).
History taking
History taking has a key role in planning care for the pregnant woman (and partner). It provides detailed information and an opportunity for relationship-building between the woman and the midwife, which, although always important, may be particularly relevant for further decision-making. An accurate, detailed personal, family, medical and obstetric history is essential in identifying risk factors that expose the pregnancy to genetic disorders, which should ideally include relevant details of the biological father of the fetus. Sensitivity to the fact that information is not always available is important, especially where there is no contact with the father or where infertility treatment has been used to achieve the pregnancy.
Consideration of racial and/or geographical origins may also be appropriate (such as with Tay–Sachs disease in Ashkenazi Jews, or haemoglobinopathies in women of Mediterranean, African or Asiatic origin). Situations such as consanguinity should also be examined.
Following the history taking, the midwife and woman/couple should consider the need for further screening. Prior to any decisions, it is essential that the woman is aware that further screening may ultimately lead to a decision about keeping or terminating the pregnancy. For some women, this may be unthinkable and they should be clearly aware of this scenario, whereas for others, having this choice would be essential. Some women may choose to continue with screening anyway, with the view that they would like to know details and prepare for the birth accordingly.
Ultrasound scanning (USS)
NICE (2008) recommends that routine USS is ideally carried out at 10 weeks’ and before 14 weeks’ gestation. For more details about USS, see Chapter 33. In the UK, routine USS in early pregnancy has been used for detection of clinically unsuspected fetal malformations (Neilson 2001). Many units now offer USS at 11–14 weeks’ gestation, which may predict a risk of chromosomal abnormality by measuring the amount of fluid behind the neck of the fetus, specifically the nuchal fold thickness. It is measured in millimetres, computer recorded in conjunction with maternal age, and used to determine a predictive risk rate. Second trimester USS is usually offered to women between 18 and 20 weeks’ gestation for structural anomalies. Diagnostic USS may be carried out in a variety of specific circumstances during pregnancy, such as when the fetus is perceived to be at particularly high risk of malformation.
Biochemical/maternal serum screening
Biochemical screening tests can utilize combination tests to determine risk factors. They can incorporate serum AFPs, unconjugated oestriol and human chorionic gonadatrophin, measured with maternal age and weight in relation to gestational age.
Integrated (combined) testing
NICE (2008) has recently recommended that all women are offered a combination of tests in the first trimester to determine the risk of the unborn baby having Down syndrome. These tests may not yet be available through all NHS services. The tests comprise nuchal translucency ultrasound scan plus blood tests to measure levels of beta-human chorionic gonadotrophin (β-hCG) and pregnancy-associated plasma protein-A (PAPP-A). These tests should be performed between 11 and 13 weeks and are the most efficient method of detecting Down syndrome (90% as compared to 65% with second trimester serum screening). When necessary, the tests may be repeated between 15 and 20 weeks. The benefit of the ‘combined test’ is that it would reduce the number of women proceeding to invasive tests. Screening by combining maternal age, nuchal scan and blood biochemistry (for maternal serum free β-hCG and PAPP-A) gives a pick-up rate of over 90%. Recent audit figures (NICE 2008) are an impressive 95% with a false positive.
Diagnostic tests
Diagnostic tests are carried out to give definitive results for abnormalities and genetic conditions when required. The procedures usually carry risk factors and it is important that women/partners are fully informed of these and the potential outcomes before making a decision to proceed with the test. Risks include compromising the ongoing pregnancy, pregnancy loss, getting false-positive or false-negative results, and having to face the dilemma of deciding whether to proceed with the pregnancy or choose termination. These choices may be more difficult for some women than for others, depending on their expectations, beliefs and needs for their pregnancy, as well as multiple psychosocial influences.
The purpose of testing is to assess the status of the genome of an individual embryo/fetus. Each individual’s genome is unique (with the exception of that of identical twins). In order to be able to recognize individual sequences of DNA, techniques have been developed to provide methods of analysis for the DNA of an individual. These techniques use genetic probes (which are labelled fragments of DNA) to determine whether a particular DNA sequence is present or absent. Embryonic/fetal genome analysis is only one application of this technology: others include forensic science and paternity disputes.
DNA can be isolated easily from any human cell that is alive and has a nucleus, including buccal cells and skin fibroblasts. The DNA probe is a copy of a relevant sequence that is identifiable and can be used as a ‘probe’ to hybridize a corresponding copy of the sample.
Preimplantation genetic diagnosis (PGD)
Preimplantation genetic screening (PGS) (also known as embryo screening) remains an experimental intervention and comprises procedures that do not look for a specific disease but are used to identify embryos at risk. The technique has to be considered before conception and involves extrauterine assessment of the embryo itself; it can thus provide certainty that the embryo is free of the genetic defects that the particular identification/screening procedure used is able to detect. It has two stages: firstly, in-vitro fertilization techniques are used to create embryos, which are then tested for particular genetic disorders and/or to establish their sex (where the disorder is sex linked). The two major technologies used for single gene cell analysis involve the polymerase chain reaction (PCR – a technique to increase the amount of DNA available for analysis) and fluorescence in-situ hybridization (FISH – the use of fluorescently labelled DNA probes for the detection of single gene defects). It is not feasible to screen for all possible genetic defects and each embryo is screened only for those particular genetic defects likely to occur. After these tests, healthy embryos are implanted into the mother’s uterus using techniques developed for infertility treatments. However, many women fail to achieve a pregnancy after the transfer of good-quality embryos. Following a systematic review of literature to date, there are insufficient data to determine whether PGS is an effective intervention in IVF/ICSI for improving live-birth rates (Twisk et al 2006).
This intervention is fraught with ethical issues, especially from those who believe in the sanctity of human life, and about the numbers of embryos that may perish or become damaged during the process. Currently, PGD appears to be only used for severe/life-threatening disorders and ongoing work is regulated through the independent body HFEA (Human Fertilisation and Embryology Authority) overseeing the use of gametes and embryos in fertility and research (www.hfea.gov.uk).
Chorionic villus sampling (CVS)
CVS is performed to obtain a biopsy of chorionic tissue from the developing embryo (see Ch. 33). The procedure involves placing a fine catheter/forceps by the transcervical or transabdominal route in conjunction with USS. DNA/cytogenetic analysis for chromosomal studies and some biochemical studies can be performed on the tissue biopsy. CVS should not be carried out before 11 weeks’ gestation as a precaution to prevent damage to the fetus.
Amniocentesis
Amniocentesis is performed to obtain a sample of amniotic fluid, which contains cells shed from the surface of the fetus and membranes (see Ch. 33). It can be performed at various stages of gestation but is traditionally undertaken between 16 and 18 weeks’ gestation when maternal age, history or screening indicates a high risk of abnormality. The cells are usually first cultured to determine the fetal karyotype and for detailed DNA analyses (including fetal sexing). The biochemical constituents of the fluid may be tested for non-specific indicators of abnormality (for example, AFPs), for the accumulation of specific metabolites in suspected inborn errors of metabolism, or, in later pregnancy, for fetal assessment. The main limitation of the test at this relatively advanced gestational age is the time taken to culture the cells, with results not being available until around 20 weeks’ gestation (see Ch. 33).
In a systematic review of 14 randomized controlled trials, Alfirevic et al (2003) compared safety and accuracy of transcervical and transabdominal CVS with early and second trimester amniocentesis. They concluded that second trimester amniocentesis is safer than transcervical CVS and early amniocentesis, with transabdominal CVS being preferred when an early diagnosis is required. There appeared to be increased risks of spontaneous miscarriage, pregnancy loss and neonatal talipes associated with earlier amniocentesis. Regarding second trimester amniocentesis, there was a non-significant increase in pregnancy loss of 1% compared with a control group (3% versus 2%), with a significant increase noted in spontaneous miscarriages (2.1% versus 1.3%). Results may occasionally be ambiguous, and there is also a small risk of cells failing to grow or contamination by maternal cells.
Fetoscopy
In this technique, the fetus is observed through a fine fibreoptic telescope, during which samples of tissue may be removed under direct vision, for analysis. It may be used to diagnose skin disease, such as epidermolysis bullosa lethalis, by skin biopsy. The technique may be used to perform therapeutic interventions, such as blood transfusion in rhesus incompatibility.
Cordocentesis
This technique is used to obtain a sample of fetal blood to screen for chromosomal abnormalities, haemophilia and haemoglobinopathies (see Ch. 33). It is carried out using USS to guide a fine needle to the base of the umbilical cord, where a sample of fetal blood is extracted for analysis and/or karyotyping.
All screening and diagnostic tests carry some risks, psychological and/or physical; consequently, the decision to proceed or not needs to be carefully measured. The role of the midwife is, therefore, to ensure that couples are given the opportunity to make fully informed autonomous decisions.
Applications of genetics
The development of molecular genetic techniques in the medical field has already yielded many benefits, for example, production of human insulin by genetically modified bacteria for use in diabetes (Russell 2006).
It is important to note that there are potential problems associated with genetic modification and activities are thus closely regulated by statutory bodies (for example, the UK Advisory Committee on Genetic Modification, the Health & Safety Executive). Of particular relevance to midwifery is the fact that the manipulation of human gametes is internationally prohibited. This is to avoid deliberately and directly changing the overall genetic constitution (the ‘gene pool’) of the human species, the consequences of such changes being both unpredictable and irreversible. The only human cells permitted to be genetically modified are the somatic cells (that is, all cells other than the gametes) and modifications to these cells are tightly regulated.
One of the most obvious applications of genetics in midwifery practice is genetic screening of parents and/or embryo (discussed above), but there is also much recent research in genetics from which developments that decrease the morbidity and mortality currently associated with many diseases may arise. In applying this research to clinical situations, the emphasis is on both prevention and cure.
Another obvious potential application of molecular genetic research in medicine is to replace or repair missing or defective alleles with normal ones – ‘gene therapy’. This process involves the insertion of genetic material directly into cells to alter the functioning of those cells (thus, to allow them to produce the normal, functional version of a protein). Success has been achieved in the treatment of some diseases (such as adenosine deaminase deficiency – Klug et al 2009), but there have also been some serious adverse consequences (that is, one fatality during gene therapy treatment – Klug et al 2009). Thus, both technical and safety-related problems remain regarding gene therapy, but this is still an active area of research with some promise.
Another possibility is the range of therapeutic options potentially available based on the use of stem cells – ‘cell-based regenerative therapies’ (National Institutes of Health Stem Cell Information website – http://stemcells.nih.gov), which involves not only complex scientific issues but also complex ethical ones too. The use of stem cells opens up a wide range of novel therapeutic options, but much work remains to be done, with respect to both the scientific and ethical issues, if these too are to be safely and reliably available in the clinical situation.
Media reports about advances in genetic technologies and their applications are both frequent and often dramatic. Consequently, parents/prospective parents may have unrealistic expectations in this area, beyond the current limits of these technologies. These approaches may well prove to be of great benefit in due course, but until both the relevant complex scientific and/or ethical issues are resolved, they remain potential therapeutic options only. It is, therefore, important to be aware of what these technologies both can and cannot achieve when dealing with parents/prospective parents.
Finally, there are resource issues to be considered in relation to these technologies, all of which are resource-intensive. Even when technology is available, consideration will have to be given to the already limited healthcare resources. It may be possible to argue in this context that the potential financial benefits of having children born free from genetic disease outweigh the cost of providing effective life care for a child/adult suffering from the condition. Nonetheless, this will inevitably have to be balanced with the needs of the general population in allocation of funds.
Conclusion
The possibilities for the future are potentially very great and the midwife will need to be aware of these technologies, as parents frequently request information following media coverage. For the midwife, this will include understanding the basis of genetics and the reality of current success in genetic engineering, including the risk:benefit ratios. The message for healthcare professionals is the need to provide appropriate and effective information to enable women and their partners to make informed decisions about the best course of action for them at present.