Chapter 31 Maternal and fetal responses to pregnancy
Introduction
In healthy, non-pregnant women, cyclical variations occur in a range of homeostatic mechanisms across the ovarian cycle, including thirst, fluid balance, appetite, temperature, cardiovascular, haemodynamic, metabolic and respiratory regulation. Following fertilization, luteal changes are accentuated and other physiological and behavioural adaptations unfold in response to distinct neurohormonal interactions that characterize successive phases of pregnancy and lactation. These altered regulatory mechanisms accommodate changing maternal needs and those of the conceptus, during implantation, and embryo formation, while changes occur in a variety of maternal systems to anticipate the distinct needs of the fetus and neonate.
Detailed information on the time course of specific changes and improved understanding of the coordinating factors have increased the potential number of tools to measure key adaptations and assess maternal health. For example, higher plasma concentrations of progesterone and relaxin in early pregnancy are associated with lower mean systolic blood pressure in the second and third trimester, and a positive correlation exists between the extent of plasma volume expansion and fetal growth, reduced risk of pregnancy complications, and pre-term labour (Khraibi et al 2003, Kristiansson & Wang 2001, Steer 2000). Some pre-existing and potential maternal health problems in early pregnancy may be identified through assessment of different hormone levels; and raised body mass index (BMI) (Duvekot et al 1995, Lain et al 2008, Maccarrone & Finazzi-Agro 2004).
This chapter will outline the time course and regulation of adaptive responses of maternal cardiovascular, renal and haemodynamic systems to pregnancy; adaptive responses of maternal hypothalamic–pituitary regulation of prolactin and growth hormone, and adaptive responses of maternal hypothalamic–pituitary–adrenal (HPA) axis to early and late pregnancy (Brunton et al 2008, Douglas 2010). It will conclude by examining the time course and regulation of the adaptive response of the uterus and mammary gland during pregnancy. See also website for more information.
Pulmonary and cardiovascular adaptations
During the infertile cycle, regulation of fluid balance, cardiovascular volume and blood pressure varies during different phases (Chapman et al 1997). This includes some degree of hyperventilation and changing alveolar and arterial tensions of carbon dioxide (CO2) levels influenced by oestrogens and progesterone (see website).
When conception occurs, hyperventilation increases and the magnitude of decline in PCO2 correlates with arterial concentrations of progesterone (Chapman et al 1998, Jensen et al 2005).
During the luteal phase, mean arterial pressure (MAP) declines significantly, leading to a reflexive rise in cardiac output, while blood volume remains constant (Chapman et al 1997, Williams et al 2001). In most studies, systolic blood pressure is slightly raised compared to the follicular phase, while diastolic pressure is 5% lower. Following conception, the largest change in blood pressure occurs before 8 weeks’ gestation. At this point, 80–90% of the total pregnancy-related fall of around 10 mmHg in MAP has already taken place (Duvekot & Peeters 1998). After 8 weeks, MAP continues to fall, reaching its lowest level by 24 weeks (Robson et al 1989). This pregnancy-related decline in MAP is largely due to the fall in diastolic pressure that begins during the luteal phase (Duvekot & Peeters 1998). Systolic blood pressure remains constant until 20 weeks, rising significantly during the second half of pregnancy, while diastolic pressure reaches its lowest point at 24 weeks and rises significantly for the remainder of pregnancy (Mabie et al 1994, Volman et al 2007) (see Figs 31.1 and 31.2).
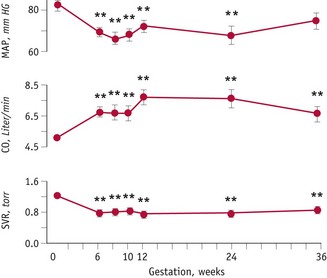
Figure 31.1 Systemic haemodynamic changes before and during pregnancy. Mean arterial pressure (MAP) decreases and cardiac output (CO) increases significantly by 6 weeks’ gestation in association with a decline in systemic vascular resistance (SVR).
(Reproduced with permission from Chapman et al 1998: Fig. 1; 2059.)
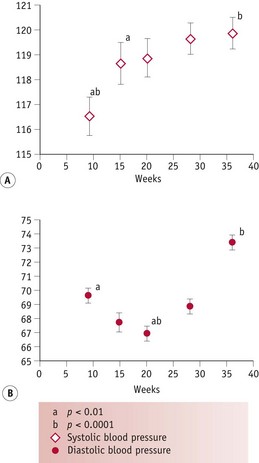
Figure 31.2 Changes in (A) systolic and (B) diastolic blood pressure from 8 weeks’ gestation
(Reproduced from Kristiansson & Wang 2001; Figs 1 and 2: 15, with kind permission of OUP. www.oup.com)
Adaptations in fluid regulation
Osmotic thresholds for thirst and vasopressin secretion decline during the luteal phase (see website) leading to a comparable reduction in plasma osmolality, through a fall in plasma sodium and its associated anions, primarily chloride and bicarbonate (Chapman et al 1997, Vokes et al 1988). When conception occurs, plasma osmolality continues to decline to 8–10 mOsmol/kg below mid-follicular values by 10 weeks’ gestation, and this new setpoint for thirst and vasopressin secretion is maintained throughout pregnancy and labour (Chapman et al 1998, Davison et al 1981). These changes are stimulated by a primary renal and systemic vasodilation, which creates a fall in total vascular resistance (Fig. 31.3).
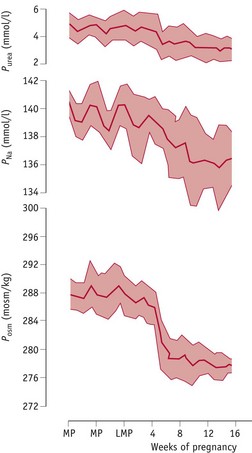
Figure 31.3 Mean values (±SD) for plasma urea (Purea), sodium (PNa) and osmolality (Posm) measured at weekly intervals during the infertile cycle and following conception to 16 weeks’ gestation
(Reproduced with permission from Baylis & Davison 1998; Fig. 10.17; 284.)
In the absence of any increase in plasma volume, the post-ovulatory fall in renal and systemic vascular resistance initiates a decline in ventricular afterload. This stimulates a reflex rise in cardiac output followed by a significant expansion in plasma volume by 6 weeks’ gestation, increasing rapidly to around 45–50% of non-pregnant values by 36 weeks (Chapman et al 1998). The fall in systemic vascular resistance from the luteal phase stimulates renal sodium and water retention, by activating fluid-retaining components of the renin–angiotensin–aldosterone system (RAAS) while blunting its vasoconstrictive effects (Chapman et al 1997, Chidambaram et al 2002, Sealey et al 1987).
When conception occurs, plasma renin activity, aldosterone and atrial natriuretic peptide (ANP) increase significantly by 6 weeks’ gestation (Chapman et al 1997, 1998, Sealey et al 1985). Despite higher basal activity of the RAAS during pregnancy, the early fall in plasma sodium sets a lower response threshold, and renal reactions to natriuretic stimuli like ANP and vascular responses to vasoconstrictor components, notably vasopressin and angiotensin II, are significantly blunted, as occurs during the luteal phase.
These responses are largely mediated by rising levels of hCG, oestrogens and progesterone (Fig. 31.4). Oestrogens stimulate hepatic synthesis of angiotensinogen (renin substrate) and promote formation of angiotensin-(1-7) [ANG-(1-7)] over angiotensin II (ANG II). ANG-(1-7) opposes the pressor effects of ANG II by releasing nitric oxide, bradykinin and prostacyclin (Valdes et al 2001, Zhang et al 2001). Rising levels of hCG in early pregnancy simultaneously attenuate the pressor effects of ANG II while progesterone counteracts its vasoconstrictive actions by decreasing mean arterial pressure. Progesterone also stimulates aldosterone synthesis, promoting water retention, while the natriuretic actions of progesterone are inactivated in the kidneys (Hermsteiner et al 2002, Quinkler et al 2001, Szmuilowicz et al 2006). Together, these modifications of the RAAS sustain volume expansion without an attendant increase in blood pressure (Duvekot & Peeters 1998, Nakamura et al 1988, Sudhir et al 1995).
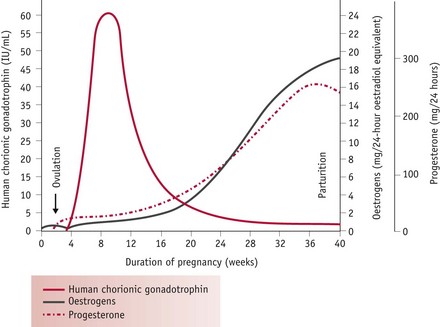
Figure 31.4 Patterns of release of human chorionic gonadotrophin, progesterone and oestrogens from ovulation to birth
(Reproduced with permission from Blackburn 2007:100.)
Renal haemodynamic adaptations
Although the kidneys constitute less than 0.5% of total body weight, in resting non-pregnant adults, blood flow is equal to 25% of cardiac output, reflecting their key role in regulating fluid and electrolyte balance (Stanton & Koeppen 1993). From the mid-luteal phase of the cycle, significant changes occur in renal haemodynamics, as vascular resistance declines, while plasma flow and glomerular filtration rate increase significantly, by 6 weeks’ gestation, compared to values obtained during the mid-follicular phase of the cycle. Minimal renal vascular resistance occurs at 8 weeks’ gestation and this coincides with a peak rise in plasma flow of around 70%, which remains at slightly lower values for the remainder of pregnancy (Baylis & Davison 1998, Chapman et al 1998, Conrad & Novak 2004) (Fig. 31.5).
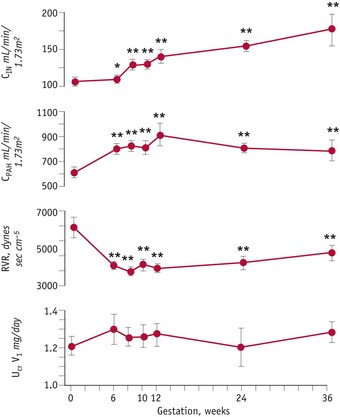
Figure 31.5 Renal haemodynamic changes before and during pregnancy. Changes in glomerular filtration rate, effective renal plasma flow and renal vascular resistance from mid-follicular phase until 36 weeks’ gestation.
(Reproduced with permission from Chapman et al 1998: Fig. 3; 2059.)
These findings suggest that hormonal changes in the corpus luteum, before and after conception, stimulate a primary fall in renal and systemic vascular resistance that initiates a chain of events leading to an early rise in cardiac output, renal sodium and water retention, despite the increase in glomerular filtration rate, leading to an expansion in plasma volume, before any increase in basal metabolic rate (Spaanderman et al 2000). A primary reduction in vascular resistance of non-reproductive organs may be initiated in preparation for the dramatic increase in uteroplacental blood flow during the second and third trimesters.
Considerable evidence suggests that the luteal decline in vascular resistance and plasma osmolality and the subsequent rise in plasma volume and total body water are positive indicators of maternal adaptations to pregnancy and long-term health, because of their strong association with increased fetal growth, reduced perinatal mortality and risk of cardiovascular disease in later life (Churchill et al 1997, Conrad et al 2001, Duvekot et al 1995, Steer et al 1995) (see website).
Hormonal regulation
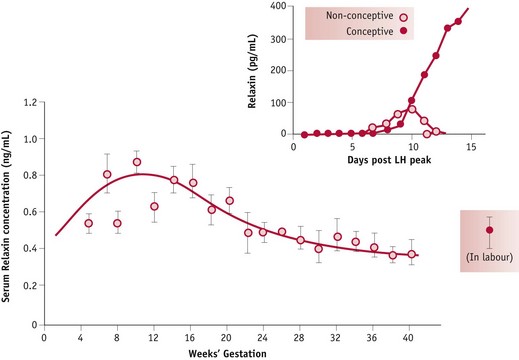
Current findings suggest that the renal and systemic fall in vascular resistance and the rise in cardiac output are stimulated by the post-ovulatory increase in relaxin, oestrogens and progesterone from the corpus luteum followed by a significant rise in adrenomedullin, a long-lasting vasorelaxant, from a variety of tissues by 8 weeks’ gestation (Conrad et al 2001, Di Iorio et al 1999, Hermsteiner et al 2002, Nakamura et al 1988, Novak et al 2001, Sudhir et al 1995).
In pregnancy, relaxin is detectable in the peripheral circulation 6 days after the midcycle LH/FSH surge. By 11 days, concentrations are significantly higher in fertile than in non-fertile cycles and concentrations increase rapidly up to 20 weeks’ gestation (Johnson et al 1991, Stewart et al 1993). Limited human studies have demonstrated that higher plasma concentrations of relaxin and progesterone in early pregnancy are associated with lower mean systolic blood pressure in late pregnancy (Kristiansson & Wang 2001). Recent evidence suggests that while oestrogens have a stimulatory effect on cardiac output and oestrogens and progesterone stimulate systemic and uterine vasodilation, neither hormone seems to influence renal blood vessels, which show a marked degree of dilation following ovulation (Chapman et al 1997, Nakamura et al 1988, Sudhir et al 1995) (Fig. 31.6).
Cardiovascular adaptations
The maternal cardiovascular system undergoes an extensive expansion in response to pregnancy. The first event is decreased vascular resistance of non-reproductive organs, leading to a profound fall in systemic vascular resistance, which reaches its lowest point at the end of the first trimester (Poppas et al 1997). By reducing cardiac afterload, this primary adaptation stimulates an overall rise of approximately 40–50% in cardiac output (Duvekot et al 1995, Volman et al 2007). Longitudinal studies suggest that cardiac output rises significantly during the first trimester, and peaks at 20 to 32 weeks, with no significant changes thereafter, or continues to show further small rises until term (Chapman et al 1997, Desai et al 2004, Duvekot & Peeters 1998, Mabie et al 1994, Volman et al 2007). Measures of the pattern and relative contribution of heart rate (HR) and stroke volume (SV) to increased cardiac output suggest that HR rises progressively until 38 weeks while SV increases significantly by 8 weeks and rises further to reach maximum values between 20 and 37 weeks and declines slightly over the remainder of pregnancy (Desai et al 2004, Robson et al 1989, Volman et al 2007).
Stroke volume increases significantly by 8 weeks, peaks at 16–22 weeks and plateaus or shows further small increases during the third trimester (Volman et al 2007). This represents a rise of 21–22% over pre-pregnancy values. In different studies, HR has been found to increase significantly above pre-pregnancy values by 5 and 16 weeks’ gestation. Data on the remainder of pregnancy suggest that the increase peaks at 31–36 weeks and shows no significant change or a slight decline thereafter. Current evidence indicates that HR may increase by between 11% and 17% over pre-pregnancy values (Capeless & Clapp 1989, Duvekot et al 1993, Robson et al 1989, Volman et al 2007) (see website).
Peripheral arterial vasodilatation
Cardiac output rises in early pregnancy, in response to a fall in systemic vascular resistance that reduces afterload in the myocardial fibres during left ventricular ejection. Decreases in both MAP and total peripheral resistance are evident at 8 weeks’ gestation and reach their lowest point by the middle of pregnancy, before returning to similar or slightly above pre-pregnancy values at term (Desai et al 2004). The decline in peripheral vascular resistance is brought about by early reduced vasomotor tone; remodelling of resistance-sized arteries; relaxation of systemic, renal and pulmonary vascular tone and development of new vascular beds in the placenta during the second trimester (Conrad & Novak 2004, Kelly et al 2004, Schrier & Briner 1991).
The initial fall is induced during the luteal phase by rising levels of oestrogens and progesterone. Progesterone has been shown to reduce muscle tone in the vasculature (Magness & Rosenfeld 1989, Omar et al 1995). When fertilization occurs, the further fall in peripheral vascular resistance creates a state of relative hypovolaemia, causing a reflexive increase in stroke volume and heart rate (Clapp et al 1988, Davison & Noble 1981, Duvekot et al 1993, Schrier & Briner 1991).
The compensatory rise in cardiac output produces a rise in vascular filling state characterized by a rise in left atrial diameter, a rise in glomerular filtration rate and little change in plasma renin, between 5 and 8 weeks’ gestation (Duvekot et al 1995). Studies have demonstrated that the fall in vascular resistance precedes the rise in circulating blood volume during pregnancy. This suggests that systemic vasodilatation is a primary adaptation to pregnancy that initiates a rise in cardiac output and maintains overall tissue perfusion and blood pressure, prior to significant increases in circulating blood volume (Capeless & Clapp 1989, Phippard et al 1986) (Fig. 31.7).
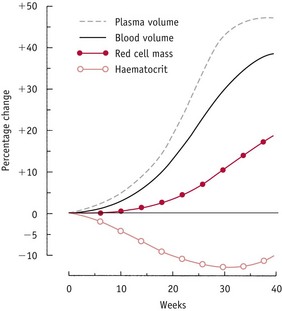
Figure 31.7 Changes in plasma volume, blood volume, red cell mass and haematocrit during normal pregnancy, expressed as a percentage of pre-pregnancy levels.
(Reproduced with permission from Rosso 1990:25.)
Blood volume
The increase in blood volume is composed of a maximum rise of 45–50% in plasma volume and a 20% rise in red cell volume above non-pregnant values. The time course of the increase in plasma volume differs from the rise in red cell mass. Plasma volume begins to increase in the first trimester, increases more rapidly in the second, only slightly during the remainder of pregnancy and is reversed following birth. In contrast, expansion in red cell mass begins in the second trimester and achieves highest increases in the third.
Because of the different pace at which these changes proceed, haemoglobin concentration and haematocrit decline progressively until about 30 weeks’ gestation. From then onwards, this trend is reversed, since increases in red cell volume outstrip plasma volume expansion during the last trimester.
Adaptations in the vascular renin–angiotensin–aldosterone system
Current evidence suggests that plasma volume expansion is primarily stimulated by the decline in total peripheral vascular resistance, which activates multiple changes in different components of the maternal vascular renin–angiotensin–aldosterone system (August et al 1995, Irani & Xia 2008, Joyner et al 2007, Skinner 1993).
Angiotensinogen or renin substrate increases very early in pregnancy and closely mirrors levels of oestrogens in individual women (Skinner 1993). During pregnancy, placental oestrogens are largely converted in a series of enzymatic transformations from dehydroepiandrosterone (DHEA-S) and related androgens in the fetal zone of the adrenal glands and the fetal liver (Pasqualini 2005). When these androgens reach the placenta and membranes, they undergo enzymatic conversion to a form that serves as substrate for the synthesis of oestrogens (Ticconi et al 2006). Rising concentrations of oestrogens in the maternal circulation and the simultaneous fall in mean arterial pressure stimulates renal production of renin, and oestrogens provide the main stimulus for hepatic production of angiotensinogen, which acts as a substrate for renin (Romen et al 1991).
Renin, angiotensins and angiotensin-converting enzymes
Renin is a proteolytic enzyme synthesized and released mainly by specialized smooth muscle cells of afferent arterioles entering the glomeruli of the kidney, in response to low blood pressure, low circulating levels of sodium chloride, a rise in effective renal plasma flow and rising levels of oestrogens (Irani & Xia 2008, Romen et al 1991, Valdes 2001). In pregnancy, plasma renin remains fairly stable between 5 and 10 weeks and measures of active renin show a modest rise after 20 weeks’ gestation (Duvekot et al 1995, Skinner 1993). On reaching the bloodstream, renin cleaves off part of angiotensinogen, triggering an enzymatic cascade that initially forms a biologically inactive peptide called angiotensin I (ANG I). In pregnancy, the next phase involves an oestrogen-induced downregulation of angiotensin-converting enzyme (ACE), which circulates in plasma and is found in most tissues but particularly high activities of the enzyme have been found in the lungs (Valdes et al 2001). This glycoprotein cleaves off part of ANG I to form the biologically active peptide, ANG II. Within the kidneys, ANG II stimulates increased fluid reabsorption in the proximal tubular cells, by enhancing reabsorption of bicarbonate. Within the adrenals, ANG II stimulates cells in the outer zone of the cortex to secrete aldosterone. In the non-pregnant state, ANG II also acts on peripheral arterioles as a potent vasoconstrictor (Skinner 1993).
During pregnancy, the vasodilatory peptide ANG-(1-7) is formed from ANG II, by angiotensin-converting enzyme 2 (ACE2), and from the biologically inactive peptide ANG I in the vasculature (Heitsch et al 2001). Current evidence shows a progressive rise in urinary excretion of ANG-(1-7) from 6 weeks’ gestation (Brosnihan et al 2003, Valdes et al 2001). Although plasma levels of ANG II are double those in the non-pregnant state by the second week of pregnancy, its pressor effects are attenuated by hCG, ANG-(1-7) and by an oestrogen-induced rise in endothelial production of nitric oxide and prostacyclin (Cook & Trundinger 1993, Heitsch et al 2001, Krane & Hamrahian 2007, Skinner 1993, Weiner et al 1994). Oestrogens also inhibit adrenal receptors for ANG II, reducing its stimulatory effect on aldosterone (Wu et al 2003).
Aldosterone, progesterone and deoxycorticosterone
Aldosterone reduces sodium excretion primarily by acting on distal portions of the renal tubules, where it stimulates reabsorption of sodium ions. Plasma levels increase significantly by 12 weeks’ gestation and reach a plateau at 30 weeks that is three to five times higher than non-pregnant values. During the first half of pregnancy, effective renal plasma flow increases by 70–80%, and then declines slightly in the third trimester but remains 50–60% above non-pregnant values, which is greater than occurs in any other physiological state. The resulting increase in glomerular filtration rate increases the sodium load from 20,000 to 30,000 mmol/day (Skinner 1993). In pregnancy, aldosterone plays a key role in stimulating sodium retention (Quinkler et al 2001).
In the non-pregnant state, progesterone acts to enhance the excretion of sodium by decreasing its reabsorption in the proximal tubules and by blocking the increased reabsorption of sodium by aldosterone, in the distal tubules. Although progesterone concentrations exceed those of aldosterone at least 50-fold during pregnancy, the renal activity of aldosterone is preserved by the enzymatic conversion of progesterone to a number of different metabolites (Quinkler et al 2001). Current findings suggest that progesterone is effectively converted to various metabolites in the kidneys, which reduces its binding to mineralocorticoid receptors, thereby attenuating its capacity to competitively inhibit aldosterone (Quinkler et al 2001). Some of the increase in progesterone is also converted to deoxycorticosterone (DOC), which acts on the distal tubules to promote the reabsorption of sodium. Rising levels of DOC occur by approximately 8 weeks’ gestation and increase 10–15-fold, to a peak of approximately 100 mg/dL at term, which is higher than aldosterone. However, the salt-retaining properties of this hormone are 30–50 times less potent than aldosterone (Nolten & Rueckert 1981, Skinner 1993) (Fig. 31.8).
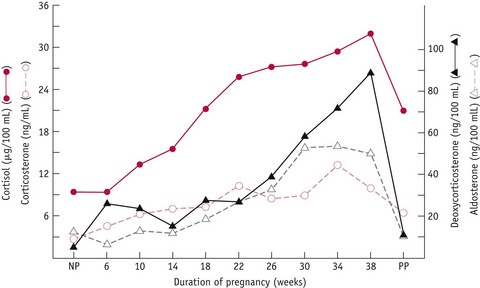
Figure 31.8 Mean adrenocorticosteroid levels in 11 women throughout pregnancy and postpartum (PP) compared to levels in non-pregnant (NP) women.
(Reproduced from Wintour et al 1978:399, with kind permission of OUP. www.oup.com)
Atrial natriuretic peptide
Atrial natriuretic peptide (ANP) is a diuretic, natriuretic and vasodilator hormone, produced in the atrial chambers of the heart. Outside of pregnancy, secretion is primarily stimulated by stretching of the atrial wall that accompanies volume expansion or increases in blood pressure. ANP acts in a variety of ways to decrease fluid intake, promote salt and water excretion, and counter all components of the RAAS (Cootauco et al 2008, Duvekot & Peeters 1998, Kaufman 1995). Within the kidneys, ANP directly inhibits renin production and tubular reabsorption of sodium, and production of aldosterone, in the adrenals. In vitro studies have also shown that ANP produces marked relaxation of vascular smooth muscle and antagonizes the vasoconstrictive effects of ANG II (Cootauco et al 2008, Steegers 1991).
Conflicting evidence currently exists on the pattern of ANP secretion during normal pregnancy and the extent to which its actions are blunted. Plasma levels have been reported to decline from 20 weeks’ gestation, reach lowest levels at 36 weeks and return to non-pregnant values by 12 weeks’ postpartum (Thomsen et al 1993). Other studies have reported a modest rise with advancing gestation, declining at term or at placental separation and then falling significantly by 72 hours postpartum (Lowe et al 1992, Yoshimura et al 1994).
All experimental evidence indicates that atrial, renal and adrenal responsiveness to ANP is blunted during pregnancy. Oestrogens and progesterone reduce ANP receptors in the zona glomerulosa, thereby inhibiting its aldosterone-suppressant effects, while the kidneys display a blunted response to the diuretic effects of both ANP and nitric oxide (Knight et al 2006, Vaillancourt et al 1997). At the same time, the pregnancy-induced rise in nitric oxide blunts the activity of atrial volume receptors, which attenuates the reduction in renal tubular sodium reabsorption, thus facilitating the expansion in extracellular fluid volume (Tam & Kaufman 2002). These regulatory mechanisms persist until late pregnancy, when ANP responsiveness to intravenous volume expansion is enhanced (Lowe et al 1992). Experimental evidence suggests that activation of the cardiac oxytocin system induces a rapid rise in ANP, which may stimulate the rapid diuresis that occurs postpartum (Mukaddam-Daher et al 2002, Yosimura et al 1994).
Erythropoiesis
The increase in red cell mass during pregnancy is stimulated by erythropoietin. This glycoprotein hormone is synthesized in ground tissue, in the kidneys and to a lesser extent in the liver. Levels of serum immunoreactive erythropoietin remain at non-pregnant values during the first trimester, begin to rise during the second and reach maximum levels during the third trimester. Within the bone marrow, erythropoietin acts on erythrocyte colony-forming cells. These give rise to increasing numbers of mature erythrocytes within 2 days of increased levels of erythropoietin within the circulation.
At present, the precise mechanisms involved in stimulating erythropoietin during pregnancy remain unclear. The evidence suggests that components of the maternal plasma renin–angiotensin system may be involved. Angiotensinogen and erythropoietin share a number of similarities. Both compete for specific binding to erythropoietin receptors on human bone marrow cells, and bone marrow cells show binding of angiotensinogen that is inhibited by erythropoietin. This evidence suggests that angiotensinogen is a precursor for erythropoietin. While both components are increased in the maternal circulation during pregnancy, the timing and possible interactions between these components and erythropoietin remain to be clarified. A number of pregnancy hormones have stimulatory and inhibitory influences on the actions of erythropoietin. Progesterone partly prevents the inhibitory influence of oestrogen on stem cell utilization of erythropoietin, while placental lactogen and prolactin enhance the stimulatory action of erythropoietin on red cell production. At present, however, the relative significance of these hormonal actions remains unclear (Beguin et al 1990).
Ventilation
Extensive anatomical and functional changes occur in the respiratory system. These accommodate both the progressive increase in gas exchange required by the rising blood volume and the growing space occupied by the uterus. From early pregnancy onwards, the overall shape of the chest alters, by a flaring of the lower ribs that seems to occur independently of any mechanical pressure from the growing uterus. This progressively increases the subcostal angle, from 68 degrees in early pregnancy to 103 degrees at term, and increases the transverse diameter of the chest by approximately 2 cm. Because of the flaring of the lower ribs, the diaphragm rises by a maximum of 4 cm while its contribution to the respiratory effort increases and shows no evidence of being impeded by the uterus. Studies on diaphragmatic movements during respiration – either sitting or lying down – have found them to be larger than in the non-pregnant state. This implies that breathing during pregnancy is more diaphragmatic than costal (de Swiet 1991a, Romen et al 1991) (Fig. 31.9).

Figure 31.9 The ribcage in pregnancy (coloured) and the non-pregnancy state (grey) showing the increased subcostal angle, the increased transverse diameter and the raised diaphragm in pregnancy.
(Reproduced with permission from de Swiet 1991a:88.)
The main functional change that occurs within the lungs is the gradual increase in the amount of air that is inspired or expired with a normal breath. This functional capacity (tidal volume) increases from 500 mL in the non-pregnant state to approximately 700 mL at term. As a result of this change, women breathe more deeply during pregnancy than in the non-pregnant state (Fig. 31.10).
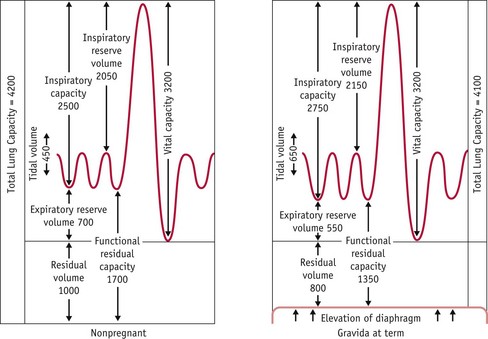
Figure 31.10 Lung volume changes in pregnancy.
(Reproduced with permission from Blackburn 2007:316.)
Since the maximum amount of air that can be expired forcibly after maximum inspiration only increases by 100–200 mL, the increase in tidal volume is produced at the expense of the expiratory reserve volume. This means that a smaller amount of air remains in the lungs at the end of quiet expiration. As less residual air is mixed with the next inspiration of fresh air, this results in lower levels of PCO2 that bring about a reciprocal rise in PO2. PCO2 declines from approximately 39 mmHg in the non-pregnant state to 31 mmHg during pregnancy, while PO2 increases from 93.4 to 101.8 mmHg, over the same period (de Swiet 1991a).
Oxygen consumption
The progressive increases in cardiac output and pulmonary ventilation are proportionately greater than those occurring in maternal and fetal oxygen consumption during pregnancy. Oxygen consumption shows a linear increase with body weight as pregnancy advances, increasing to 38 mL/min (15%) above average values in the non-pregnant state (de Swiet 1991a). It is composed of the overall increase in tissue mass, higher metabolic rate of fetal and placental tissue, along with that of some maternal organs, particularly the heart, lungs and kidneys (Fig. 31.11).
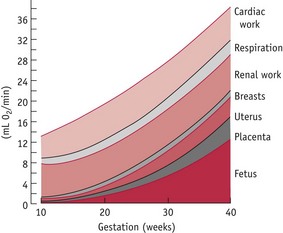
Figure 31.11 Partition of the increased oxygen consumption in pregnancy among the organs concerned.
(Reproduced with permission from de Swiet, 1991a:91.)
The increase in oxygen consumption is facilitated by a 40–50% increase in ventilation and by an 18% increase in the oxygen-carrying capacity of the blood. Because of this relative oversupply of oxygen, higher concentrations are returned to the heart from the venous circulation, making the arteriovenous oxygen difference significantly smaller than in the non-pregnant state. The extent of the arteriovenous oxygen difference is smallest in early pregnancy and does not reach average non-pregnant values until term (de Swiet 1991a).
The increase in ventilation during pregnancy reduces alveolar and plasma concentrations of carbon dioxide. Studies have demonstrated that arterial partial pressure of carbon dioxide (PCO2) is about 30 mmHg in late pregnancy, compared to 39 mmHg during the follicular phase of the cycle. Since fetal PCO2 remains at approximately 41 mmHg, the lower levels in the maternal circulation encourage the diffusion of CO2 from fetal blood, across the placental membranes.
Uterine adaptations
The uterus changes from being a small pelvic organ, with a cavity of around 10 mL and weighing approximately 50 g. By 36 weeks, its weight has increased to an estimated 1100 g, representing almost a 20-fold increase in mass, and its average volume is 5 litres. It is then in contact with the anterior abdominal wall and extends as far as the xiphisternum. During pregnancy, the uterus is a central recipient of the increases in circulating blood volume. In the follicular phase of the cycle, uterine blood flow is approximately 45 mL/min (Burton et al 2009). This changes very little during early pregnancy, but rises sharply from about 20 weeks, to reach approximately 750 mL/min at term, when it receives nearly 20% of total cardiac output (Burton et al 2009, Steer 1991).
Uterine growth is characterized by a highly regulated process of myometrial cell differentiation:
Growth is stimulated by hCG, progesterone, growth factors, oestrogens and oxytocin, and by progressive distension exerted by the fetus, placenta and amniotic fluid, particularly during the third trimester. These factors promote synthesis of structural and contractile proteins (Blackburn 2007, Shynlova et al 2009, Ticconi et al 2006 (see website). During the first few months of pregnancy, growth is accompanied by increasing thickness of the myometrium in the corpus and the fundus. As the organ increases in length from around 12 weeks, the isthmus is gradually formed as an area with reduced density of muscle fibres (Steer 1991).
Myometrial changes
The bundles of smooth muscle fibres within the uterus are arranged in three or four layers embedded in a matrix of connective tissue and ground substance. The former acts as intramuscular tendons while the latter transmits contractile forces from individual cells along the muscle bundle during labour (Blackburn 2007). Two outer layers contain longitudinal and circular fibres that are partly continuous with the supporting ligaments. The middle layers that hold the vascular supply have a criss-cross pattern of fibres that run in all directions. Finally, the inner layer is composed of longitudinal fibres and covers the decidua (Steer 1991) (see website).
The smooth muscle forming the myometrium does not have the precise transverse alignment of thick and thin filaments that characterizes the organization of skeletal fibres. Filaments of smooth muscle are situated in random bundles throughout the cells, and myosin filaments are arranged alongside actin in uninterrupted unidirectional order. In addition to these main contractile filaments, smooth muscle also contains intermediate filaments. These are attached to all areas of cell membrane, which allows them to form networks across the cell. As a result of this organization, contractions can generate force in any direction and also produce a much greater degree of shortening than in skeletal muscle. For most of pregnancy, this action remains local, as few intracellular connections are formed and quiescence is actively maintained, by hormonal mechanisms that regulate the process of differentiation, until the last few weeks of pregnancy (Blackburn 2007, Shynlova et al 2009) (see website).
Conclusion
The adaptations of pregnancy affect all systems of the body, and are regulated by a complex series of neurohormonal interactions between maternal reproductive organ systems and the fetoplacental unit. This chapter has identified the time course of key changes to enable midwives to look for the characteristic features of each trimester.