Chapter 33 Diseases of the Hepatobiliary System
DIAGNOSIS OF LIVER DISEASE
In making a diagnosis, the clinician should first determine that the animal has liver disease and then try to identify the specific cause. Therefore, this chapter first discusses the diagnosis of liver disease, then the individual conditions causing disease. The liver cannot be examined directly in large animals. The signs of liver disease are caused by failure of some of the liver’s many functions, but these signs may not appear in the early stages of disease. Special tests may be needed to detect early damage or minor impairment of function that has not yet produced clinical signs.
LIVER DISEASE VS. LIVER FAILURE
As with many other organs, such as the heart and kidney, the liver can be diseased long before it fails to function. Thus, early cases of liver disease are not apparent to the owner or veterinarian through physical findings alone; such cases usually are detected by finding elevated levels of liver enzymes or bile acids in the serum. Pathologic changes in the liver may include biliary hyperplasia, death of hepatocytes, and fibrosis, and these may occur long before any signs of failure develop. Some functions may fail before others, and the onset of liver failure varies with the species and the disease process involved.
LIVER RESERVE AND REGENERATION
The liver has a large reserve capacity, and close to 80% of it can be removed before regeneration and recovery are no longer possible. It also has a remarkable capacity to regenerate. Regeneration can occur in areas receiving portal blood, but most cell division takes place in Rappaport zone 1 (the portal area), and the cells are pushed to the central lobular area. The liver undergoes constant repair, and in people it is estimated that the hepatocytes are renewed every 50 to 75 days.1
Normal regeneration does not occur in some cases. Antimitotic agents such as metabolites of pyrrolizidine alkaloids or antineoplastic drugs can prevent cell division. Regeneration may be restricted by connective tissue. Once fibrosis has bridged the various lobules, additional regeneration is impaired because the fibrosis itself perpetuates the condition. Loss of a stroma to build on or lack of portal blood supply also reduces regeneration.
SIGNS OF LIVER DISEASE AND PATHOPHYSIOLOGY
Many signs can be present with liver disease, but no sign is pathognomonic or present consistently. Table 33-1 provides some of the signs that may be present and other possible causes of the same signs.2
Table 33-1 Signs of Liver Disease or Failure
| Sign | Pathogenesis | Other Causes |
|---|---|---|
| Icterus (E) | Failure of uptake, conjugation, or excretion of bilirubin | Massive hemolysis Bile blockage Fasting in the horse |
| Weight loss (E, B) | Energy demand greater than absorbed or metabolized | Poor nutrition, chronic inflammation, parasites, neoplasia, maldigestion, malabsorption |
| Ascites (B) | Portal hypertension and lymph leakage caused by cirrhosis or venoocclusion | Cardiac failure Hypoproteinemia Cushing’s syndrome |
| Change in liver size | Nodular hyperplasia, tumor, cirrhosis Fatty degeneration |
Right-sided heart failure Work hypertrophy Anemia |
| Diarrhea (B) | Bile deficit malabsorption Intestinal edema, portal hypertension |
Gastrointestinal (GI) or systemic disease |
| Pruritus | Retention of bile salts | Dermatologic or central nervous system (CNS) disorders |
| Dermatitis Unpigmented areas |
Hepatogenic photosensitization | Primary phototoxic photosensitization |
| Central nervous system signs Behavioral change, ataxia, dysmetria, circling, stupor, coma, tremors, bellowing |
Hepatic encephalopathy (see text) | Brain diseases Metabolic diseases Toxic diseases |
| Tenesmus (B) | Hepatic encephalopathy | Rectal or colonic disease CNS disease Urogenital disease |
| Rectal prolapse (B) | Tenesmus | Rectal or colonic disease |
| Change in feces color | Bile pigment deficit | Diet |
| Undigested fat | GI disease | |
| Hemorrhage | Failure to synthesize clotting factors II, V, VII, IX, X | Other clotting factor or platelet deficit, trauma, disseminated intravascular coagulation |
| Pain over liver | Inflammation, swelling | Abscesses Traumatic reticulitis |
| Inspiratory stertor (E) | Hepatic encephalopathy | Upper airway obstruction |
| Dyspnea |
E, Frequent sign in horses; B, frequent sign in cattle.
The clinical history is useful in some cases, but consumption of pyrrolizidine alkaloid—containing plants may not be apparent because of the delay between consumption and the onset of clinical signs. Exposure to other hepatotoxins could be detected. Ruminants grazing on land infested with snails are more likely to have liver fluke disease. Administration of horse serum to Equidae 4 to 8 weeks previously will make acute serum hepatitis more likely.
With the exception of pain over the liver elicited with pressure and change in liver size, most signs are related to failure of some function. Liver flukes may cause anemia and hypoproteinemia because of the effect of the parasite and its metabolites. Liver abscesses and other infections may produce signs such as fever and anorexia because of the release of pyrogens and other mediators (caused by the organism and not necessarily related to the liver itself).
Icterus is typically seen in acute liver disease in horses but is not seen in many cases of chronic liver disease, and it is seen less often in ruminants unless biliary blockage occurs. Icterus is caused by failure of uptake, conjugation, or excretion of bilirubin. Excess production caused by hemolysis must also be considered when icterus is present, and is the most common cause of icterus in cattle. Horses are frequently icteric (up to 6 mg/dL unconjugated bilirubin) from anorexia or fasting even when the liver is normal.
Weight loss is a common but nonspecific finding in some cases of chronic liver disease. It may be caused by anorexia or failure of metabolic functions of the liver and is probably not related to impaired fat absorption in the large herbivores. Diarrhea is also seen, especially in cattle with chronic liver disease. It is thought to be related to portal hypertension and increased hydrostatic pressure, although the exact mechanisms are not yet understood. Diarrhea probably is not caused by fat malabsorption or steatorrhea, because the normal herbivore diet contains less than 3% fat.
Ascites is a common finding in calves with liver cirrhosis. The ascites results from portal hypertension caused by venous blockage producing increased hydrostatic pressure and by protein leakage into the peritoneal cavity. Production of hepatic lymph high in protein (>3 g/dL) is increased. Because the liver sinusoids are permeable to plasma proteins, the protein-containing lymph leaks into the interstitial space and then into the peritoneal cavity.3,4 Fluid moves into the abdominal cavity because of both osmotic and hydrostatic forces, according to Starling’s law. The abdominal fluid present with liver disease is a modified transudate, but the protein content may be relatively high (3 to 3.5 g/dL) because of leakage of protein from the liver. Hypoalbuminemia can aggravate the ascites, but if it occurs alone, it more likely will cause intramandibular and brisket edema.
Dermatitis of the white areas may occur because of hepatic photosensitization. The skin of the white areas first becomes erythematous, then thickened with keratin crusts, and finally necrotic. This is caused by the photodynamic agent phylloerythrin, which is formed in the gastrointestinal (GI) tract of herbivores by the bacterial degradation of chlorophyll. After absorption into the portal circulation, phylloerythrin should be conjugated by the liver and excreted into the bile. With cholestasis, the phylloerythrin may be carried to the skin, where it acts as a photodynamic agent.5 After bile duct ligation, the level of phylloerythrin steadily increases.6 Although a small amount is removed by the kidneys, the rate is not fast enough to prevent accumulation in the plasma and the skin. Phylloerythrin in the skin reacts to sunlight and emits energy that causes lesions of the white areas.5
Pruritus is seen in a few cases of liver disease in horses. In people, pruritus is assumed to be caused by bile acid accumulating in the skin when it is not excreted by the liver. This same process may occur in horses, but pruritus is not usually seen in large animals with liver disease.
A change in fecal color is not usually noted in adult herbivores with liver disease because other pigments such as chlorophyll contribute to the color. In young animals with simple digestive tracts, much of the fecal color is from stercobilin, a metabolite of bilirubin. Therefore, with cholestasis, the feces may be a lighter color.
A few signs may be seen terminally in liver disease. Hemorrhage may occur when the clotting factors are not synthesized in adequate amounts. Factors I, II, V, VII, IX, and X are all produced by the liver,1,4 but the disease is usually advanced before a deficit develops.
Tenesmus, often followed by rectal prolapse, is seen in some cattle with liver disease. This may be associated with diarrhea, may be part of hepatic encephalopathy, or may be aggravated by edema of the bowel caused by portal hypertension.
Pharyngeal or laryngeal collapse with loud stertorous inspiratory noises and dyspnea has developed in some cases of hepatic failure, especially in ponies. The exact mechanism for this is not known, but it may also be part of hepatic encephalopathy.7
Horses sometimes develop a terminal hemolytic crisis caused by increased red blood cell (RBC) fragility. This has not been observed in ruminants.
HEPATIC ENCEPHALOPATHY
Hepatic encephalopathy is a neuropsychiatric syndrome caused by hepatic dysfunction or portosystemic shunting of the intestinal blood.8 It is considered a potentially reversible metabolic or neurotransmitter disorder, but it is associated with characteristic (although not specific) lesions in the central nervous system (CNS) such as altered astrocytes.
Signs of hepatic encephalopathy are often subtle and nonspecific. Behavioral changes may be detected by the owner, who is more familiar with the patient’s normal activity. Some docile animals become excitable and difficult to control, whereas other, normally unruly animals may become passive. Depression and incoordination are frequent manifestations, and some animals may walk aimlessly or even head press.9,10 Apparent blindness is seen in some horses, and foot stomping was reported in 7 of 25 cases.11 The animals eventually develop a stupor and may end up in hepatic coma. Yawning may be seen in horses, and ponies may have a stertorous respiratory noise. Ruminants show signs of tenesmus and sometimes vocalize excessively. In humans, hepatic encephalopathy is diagnosed by neuropsychological tests, which are not possible in animals.12
The pathophysiology of hepatic encephalopathy remains undefined and is controversial.10 It occurs when portal blood bypasses the liver, as with congenital shunts in dogs, or with shunts secondary to portal hypertension induced by alcoholic cirrhosis in humans, or when the blood goes through an inadequately functioning liver. How neurologic function is altered has not been determined, but speculation abounds.
It seems plausible to incriminate synergistic neurotoxins that bypass the liver. Most will agree that ammonia level plays a central role in the pathogenesis. Blood ammonia is elevated in most cases of hepatic encephalopathy because the liver is not metabolizing ammonia to urea. Encephalopathy can be precipitated in cirrhotic patients by adding ammonia-generating substances, and there is an increase in cerebrospinal glutamine, the product to which ammonia is detoxified. The astrocytes contain the enzyme glutamine synthetase, which adds a molecule of glutamate to ammonia to form glutamine. The amount of glutamine formed in the brain correlates with the severity of hepatifc encephalopathy.13 However, much higher concentrations of ammonia are needed to induce coma in normal animals, and there is poor correlation between ammonia levels and degree of encephalopathy.11 Four horses with hyperammonemia and encephalopathy without liver disease have been reported.14
Magnetic resonance imaging (MRI) of human cirrhotic patients revealed increased image intensities from the globus pallidus region, probably caused by manganese deposits.12 Serum manganese is increased in these patients, which correlates with the increased MRI intensities and negatively with the neuropsycological tests. This would indicate that manganese is another potential toxin that can enter the brain if not effectively removed from the blood by the liver.
There may be an imbalance of true inhibitory and excitatory neurotransmission. γ-Aminobutyric acid (GABA) is an inhibitory neurotransmitter. The increase in GABAergic tone was originally thought to result from an increase in GABA or its receptor sites. More recently, it has been proposed that neurosteroids with GABA-agonist properties could be involved.15 Ammonia is believed to play a role in the metabolism of GABA in the brain and thus could act synergistically.16 Also, receptor sites for benzodiazepine are increased in the brain of animals with hepatic encephalopathy.17 Benzodiazepine augments the activity of GABA in stimulating the inhibitory neuron and causing sedation.
False neurotransmitters have been proposed to cause the abnormal nerve function. Increased amounts of tryptophan, phenylalanine, and tyrosine in the brain could cause more serotonin, a false neurotransmitter, to accumulate in the brain. Plasma amino acid ratios are altered in horses with pyrrolizidine alkaloid—induced liver disease.18 Concentrations of branched-chain amino acids (valine, isoleucine, leucine) are decreased, and the concentration of aromatic amino acids (tyrosine, phenylalanine, free tryptophan) are increased, partially because the liver is not adequately metabolizing the aromatic amino acids. The amino acids are transported across the blood-brain barrier by a common transport system, so they compete for entry into the CNS. It has been suggested that higher levels of aromatic amino acids in the CNS would lead to the formation of increased amounts of inhibitory neurotransmitters or to the alteration of catecholamine or monoamine neurotransmitters, such as GABA or L-glutamate.
LABORATORY TESTS AND LIVER-DERIVED SERUM ENZYMES
No single test can consistently confirm or rule out the presence of liver disease. Only serum concentrations of γ-glutamyltransferase, globulins, and alkaline phosphatase were found significantly different in horses with or without histologic evidence of liver disease in one study.19 A combination of tests, including serum enzymes, total bile acids, and liver biopsy, may be needed.
Some blood constituents may be altered because of failure of certain metabolic functions of the liver, but none of these changes is specific for liver disease. Blood glucose is sometimes slightly lower in severe liver disease, especially in young animals, possibly because of decreased gluconeogenesis, but none of 28 adult cases in one study had low blood glucose concentrations, and in fact, 14 cases had hyperglycemia.11 Blood ammonia levels can increase fourfold or more in some toxic liver diseases because the urease needed to convert ammonia to urea is found only in the liver. For the same reason, blood urea nitrogen (BUN) may decrease, especially in the terminal phases. In the later stages, the blood-clotting factors may be diminished, with delayed partial thromboplastin time (PTT) and other clotting times.
Terminally, serum albumin concentration may decrease. A large amount of protein synthesis takes place in the liver; all the plasma proteins except γ-globulins are produced by the liver. The amount of these proteins in the blood, however, depends not only on the rate of synthesis but also on the rate of removal. The albumin half-life in cattle is about 16.5 days; in the horse, 19.4 days; and in sheep, 14 days.20 Therefore, serum albumin is reduced mainly in chronic liver disease. With liver damage, the synthesis of α-globulins and β-globulins is increased so that the total plasma protein concentration is invariably normal or elevated, but the albumin/globulin (A/G) ratio may be decreased. The healthy liver has a large reserve for protein synthesis, and lost protein can be replenished. Less than 5% of horses with hypoalbuminemia had liver disease in a group from our teaching hospital. In one study, 18% of the horses with chronic liver disease and 6% of those with acute liver disease had albumin concentrations below the reference value.21 In a British study, only 6 of 37 liver disease cases were hypoalbuninemic, and none of these was hypoproteinemic.11 Therefore, it is assumed that hypoalbuminemia is not a common feature in horses with liver disease, and that hypoproteinemia is rare.
Amino acid ratios are altered in liver disease; the short, branched-chain amino acids are decreased, whereas the aromatic amino acids are increased.18
LIVER ENZYMES
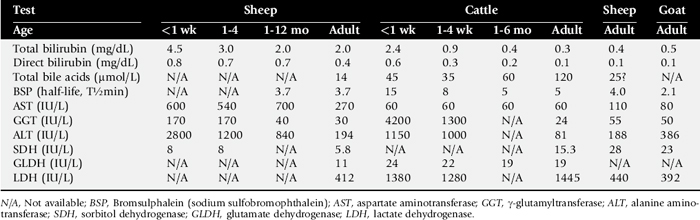
A number of enzymes are compartmentalized in the hepatocyte or in bile duct epithelium. This compartmentalization is useful in holding insoluble molecules close to the enzymes for chemical reactions. Hepatocyte damage may result in release of the enzymes into the circulation, and cholestasis may cause increased release from bile epithelium. Serum levels of these enzymes therefore may be an indication of hepatocyte integrity or bile excretion. Table 33-2 lists frequently tested enzymes and their characteristics. Serum concentrations for some of these enzymes vary with the age of the animal and sometimes even with type or use. Table 33-3 lists some approximate upper limits of normal for animals of various ages. Normal values, especially for the enzymes, should be established by each laboratory. Chapter 22 provides normal adult values from one laboratory.
Table 33-2 Liver-Derived Enzymes
| Enzyme | Specificity | Problems |
|---|---|---|
| GGT | Liver | High in young animals from colostrum |
| Kidney* | ||
| Pancreas | ||
| ALP | Liver | Not specific |
| Bone | ||
| Intestine | ||
| Macrophages | ||
| Placenta | ||
| SDH | Liver | Not elevated in chronic disease; short life; not stable |
| GLDH | Liver | Not elevated in chronic disease |
| AST (SGOT) | Liver | Not specific |
| Muscle | ||
| Heart | ||
| ALT (SGPT) | Liver | Low concentration in cattle and horses; not a good indicator in large herbivore |
| LDH | None unless isoenzymes | Not elevated in chronic disease; short life; not specific |
| OCT | Liver | Analysis not routinely available |
| Ar | Liver | Analysis not routinely available |
GGT, γ-Glutamyltransferase; ALP, alkaline phosphatase; SDH, sorbitol dehydrogenase; GLDH, glutamate dehydrogenase; AST, aspartate aminotransferase; ALT, alanine aminotransferase; LDH, lactate dehydrogenase; OCT, ornithine carbamoyltransferase; Ar, arginase.
* Elevated in urine, not blood.
γ-Glutamyltransferase (GGT) is a frequently tested and fairly specific enzyme that is almost invariably elevated in chronic liver disease. It is found mainly in the biliary tract and indicates biliary damage (flukes) or hyperplasia (pyrrolizidine alkaloid toxicity or aflatoxicosis). GGT also is present in pancreas, mammary gland, lung, kidney tubules, and other duct epithelium; but serum levels usually are not elevated with renal disease because the enzyme is lost in the urine. Serum concentrations are normally higher in neonatal calves (sometimes >4000 IU/L after suckling)22 because the enzyme is concentrated in colostrum.23 GGT levels are also higher in foals than in adult horses, but the higher concentrations may partly be the result of increased production.24 GGT is elevated in many horses with right dorsal displacement of the large colon, possibly caused by transient extrahepatic bile duct obstruction.25 Elevated GGT is the most sensitive indicator of liver disease in the horse.11,26 GGT levels will remain elevated for several weeks.
Alkaline phosphatase (ALP) is usually elevated in chronic liver disease of the horse and is variable in ruminants. ALP can come from other sources, such as bone, intestines, placenta, and macrophages, so it is not specific. Both ALP and GGT are elevated with cholestasis.27 The dehydrogenases, such as sorbitol dehydrogenase (SDH), lactate dehydrogenase (LDH), and glutamate dehydrogenase (GLDH), are found in hepatocytes and are elevated with acute hepatocyte damage, but serum concentrations may return to normal or below normal in chronic liver disease. SDH is liver specific and extremely useful in detecting active hepatocellular necrosis, but it is not as stable as some of the enzymes. LDH is found in many tissues other than liver; thus it is not specific unless isoenzymes are determined.
EXCRETION TESTS FOR FUNCTION
Because the liver excretes a number of endogenous compounds and foreign substances injected into the animal, the rate of excretion or clearance of these substances can be used to test the excretory function of the liver. Bilirubin itself can be used and, if elevated above normal, would indicate liver failure, bile blockage, or excess production from hemolysis. In the horse, bilirubin is also increased during fasting in animals without liver disease.28 With liver damage in the horse or ruminant, most of the retained bilirubin is unconjugated (indirect reacting), and the direct-to-total ratio is usually less than 0.3. With bile blockage or intrahepatic cholestasis, the direct-to-total ratio may be greater than 0.3 in the horse or 0.5 in cattle.
Bilirubin is the main bile pigment and is produced from heme, 75% of which comes from RBCs.29 When the erythrocytes are broken down, heme is converted first to biliverdin and then to bilirubin in the macrophage system. This unconjugated bilirubin is insoluble and must be bound to albumin for transfer, and this bound bilirubin is not removed by the kidneys. Unconjugated bilirubin is taken up by the hepatocytes with cytosolic binding proteins within the hepatocyte. In the hepatocyte, some bilirubin is conjugated to the diglucuronide, but in the horse, more than half the bilirubin in bile is conjugated with glucose. Conjugated (direct reacting) bilirubin is water soluble, and some will enter the general circulation; if concentration is sufficiently high, it will be filtered by the kidneys into the urine. Conjugated bilirubin is secreted into the bile canaliculi by an energy-dependent transport process. In most species, this is the rate-limiting step, but this may not be true in the large herbivores. Conjugated bilirubin passes into the intestine through the bile ducts, and if they are blocked, both conjugated and unconjugated bilirubin will increase in the plasma. In the intestinal tract, bilirubin is converted to urobilinogen by anaerobic bacteria. Some urobilinogen is absorbed and reexcreted by the liver, but a small fraction will pass the normal liver and be excreted in the urine. Therefore, with complete biliary blockage, there will be no urobilinogen in the urine, and with hemolysis, there may be increased urobilinogen in the urine. Urobilinogen is not very stable in the urine; thus analysis must be done within 1 or 2 hours, or the amount detected will be erroneously low.
Fasting decreases the efficiency of plasma bilirubin removal in all species, but the horse shows a greater rise in plasma bilirubin, often reaching a plateau two to three times the normal state. This increase is caused by a decrease in removal of bilirubin by the hepatic transport and not by an increase in its production.30,31
Serum total bile acid concentration is a good test of liver function. The concentration of bile acids in the serum will be increased if there is hepatocyte damage, blockage of bile flow, or shunting of portal blood to the systemic circulation, bypassing the liver. Bile acids are synthesized by the liver from cholesterol. Cholic and chenodeoxycholic acids are the primary bile acids that are conjugated with amino acids before excretion into the bile. Only conjugated bile acids are present in the intestine, are soluble, and form micelles with fat because of their detergent properties. Most of the bile acids excreted in the bile are resorbed by an active transport system in the ileum and carried by the portal circulation back to the liver for reexcretion. In most species, more than 95% of the bile acids are resorbed and recirculated through the enterohepatic circulation.32 The daily synthesis of bile acids is much less than the daily requirement, and one study in ponies showed that the bile acids secreted each day are about 38 times the total pool.33
Many simple-stomached animals have a postprandial increase in serum bile acid because of the release of bile from the gallbladder during eating and subsequent resorption by the ileum. This does not seem to be important in the horse, which has no gallbladder, or in cattle, in which no relationship to feeding could be found.34Table 33-3 lists upper limits of normal for liver function tests in large animals of various ages. In cattle there is an hour-to-hour fluctuation that could be as much as 60 μM/L.34 In the horse, serum concentration above 14 μM/L would indicate liver damage, bile blockage, or shunting.35,36 Serum total bile acid concentration is likely a more sensitive indicator of liver disease in the horse than serum bilirubin; only 8 of 34 horses with liver disease had elevated bilirubin, but 31 of 37 had elevated bile acids.11 In adult cattle, because of the hour-to-hour variations, bile acid concentration on a single sample would have to be above 126 μM/L in beef cattle and 88 μM/L in dairy cattle to be an indication of liver disease.37 Levels are lower (<64 μM/L) in calves less than 6 months old. Serum bile acid concentration is a very specific test that has a high positive predictive value.38
A number of dyes are excreted primarily by the liver; sulfobromophthalein (Bromsulphalein, BSP) is used most often. In large animals, BSP clearance (half-time) is used much more than the retention test. In the test, 500 to 1000 mg (∼2 mg/kg) of BSP is injected intravenously. Blood samples are taken before the injection and two to four times 5 to 12 minutes after the injection (i.e., at 5, 7, 9, and 11 minutes). These samples are analyzed for color produced by BSP, and a half-life is determined by plotting the points on semilog paper. Normal horses have a BSP half-life of less than 3.5 minutes, and for ruminants it is less than 5 minutes. This may be increased in very young animals (see Table 33-3) and in pregnant females. Other dyes, such as indocyanine green, have some advantage (less renal excretion than BSP) but at present are too costly for use in large animals.
LIVER BIOPSY
Liver biopsy can be very useful not only in confirming the presence of liver disease, but also in determining or ruling out some etiologies. It is a relatively safe and simple procedure but should be avoided when liver abscesses are suspected. A number of biopsy instruments are available, including aspiration punches, but the Histo-Cut* or TrueCut† and similar notch-cutting needles seem to work quite well. In all species the skin over the biopsy site should be clipped and prepared for aseptic insertion of the needle. A local infiltration of 2% lidocaine helps to reduce the animal’s reaction, but it may still flinch when the pleura and peritoneum are penetrated. A small stab wound is made through the skin at the site of insertion with a No. 11 or a No. 15 Bard-Parker blade. The horse should have a twitch applied or be given chemical sedation if necessary.
The site of skin puncture for liver biopsy in the horse is the right fourteenth intercostal space at the intersection of a line drawn from the tuber coxae to the point of the shoulder. Some operators recommend more cranial insertion (twelfth or thirteenth space), but more lung is penetrated at points farther craniad. If a more cranial position is selected, it should also be more ventral. If the needle is directed slightly craniad and ventrad, it is more likely to remain in the liver parenchyma and not penetrate larger vessels on the visceral surface or pass on through into the right kidney, pancreas, or colon. Ultrasound guidance will increase the chances of obtaining liver specimens without penetrating other organs.
Cattle need to be suitably confined for liver biopsy. A biopsy can often be performed on quiet dairy cows in a stanchion while the tail is elevated (“tailed”). The puncture site in cattle can be located by extending a horizontal line craniad from the middle of the paralumbar fossa. The needle is inserted where this line crosses the eleventh intercostal space on the right side. The lungs do not extend as far caudad as in the horse, so the liver is more exposed. The needle is directed slightly craniad and ventrad, as in the horse.2
Liver biopsy in sheep and goats is slightly more difficult; additional sedation is usually required. A site at the ninth or tenth intercostal space at the level of the ventral end of the last rib has been recommended. The biopsy needle is advanced in a craniomedial direction until the liver is penetrated.
Diffuse or zonal lesions, as seen in most toxic, infectious, and metabolic liver diseases, can usually be diagnosed by liver biopsy. Focal lesions, such as abscesses, granulomas, and neoplasias, as well as liver flukes, are easily missed by liver biopsy. If a liver abscess is suspected, biopsy is contraindicated because of the potential danger of rupturing an abscess. A scoring system has been developed for the prognostic evaluation of equine liver biopsies.39Examining liver biopsy tissue can confirm the presence of liver disease and help establish the type of disease in addition to aiding in the prognosis.
ULTRASOUND EXAMINATION
Ultrasonographic examination of the liver is useful in determining its size, its situation, and the diameter of its vessels. Cholelithiasis, neoplasia, and fibrosis can be diagnosed by ultrasound in the horse.40 Liver abscesses can often be detected, and the circumference of the gallbladder can be determined in cattle.41,42 Parenchymal pattern of the liver is changed with diffuse fibrosis. An ultrasound examination is performed on cattle on the right side in the tenth to twelfth intercostal space. On sheep the examination is done on the right side in the seventh through twelfth intercostal spaces.43 In horses the ultrasonic examination is performed below and caudal to the lung in the eighth to fourteenth intercostal spaces. The liver can also be visualized on the left side of the horse in the lower intercostal spaces. Horses with evidence of liver disease should have a percutaneous liver biopsy regardless of the ultrasonic appearance.40
PROGNOSIS
The severity of clinical signs may be the most useful noninvasive prognostic test in horses with liver disease.44 Indicators of a poor prognosis in liver disease include an albumin concentration less than 2.5 g/dL in horses or an increased globulin level, a prothrombin time (PT) greater than 30% of normal, and greatly elevated GGT and ALP, especially if there is a normal or decreased SDH or GLDH. Marked fibrosis that bridges the liver lobules on histopathology suggests a poor prognosis. Severe pyrrolizidine alkaloid toxicosis carries a particularly grave prognosis because remaining hepatocytes are inhibited from regeneration by mitotic arrest. Terminal clinical signs include development of hemolytic crisis in the horse and marked hepatoencepalopathy in the patient with a fibrotic liver.
INFECTIOUS, TOXIC, AND PARASITIC LIVER DISEASE
ACUTE HEPATITIS IN HORSES
Idiopathic acute hepatic disease (IAHD) is the most common cause of acute hepatitis and hepatic failure in horses.45 IAHD is also known as Theiler’s disease,46 serum hepatitis,47 postvaccinal hepatitis,48 and acute liver atrophy.49 The disease was first recognized by Theiler in large numbers of horses in South Africa following immunization against African horse sickness with simultaneous administration of live virus and hyperimmune equine serum.50 Since then, IAHD has been documented as a potential complication of any equine serum or plasma product used in horses. It is primarily a disease of adult horses51 and recently has been most often associated with the use of either tetanus antitoxin (TAT) or commercial equine plasma.47,49,52-55
Mares appear to be affected with IAHD more frequently than males, with recently parturient, lactating mares at greatest risk.51,54 The seasonal nature of some epidemics of IAHD51,56 (most reported cases occur in summer months) and the observation that some affected horses in outbreaks of IAHD have not been treated with TAT or plasma45,51,56 have led to the concern that a virus is involved, similar to hepatitis B virus in humans, but this remains unproved.46 Alternatively, IAHD has been proposed to be a type III (immune complex—mediated) hypersensitivity reaction.57
The clinical effects of IAHD are associated with signs of acute hepatic failure and include depression, jaundice, inappetence, pica, yawning, photoactive dermatitis, and hepatic encephalopathy.* Fever has been described as absent or rare in horses affected with IAHD.48 Atypical signs that have been reported include progressive weight loss, ventral subcutaneous edema, jugular pulses, ileus, and acute respiratory distress.54,55,59 Intravascular hemolysis has been reported to lead to hemoglobinuria in some terminal cases of IAHD.46 Evidence exists for a subclinical or chronic component in some cases of IAHD.54,60 However, some horses that received TAT have clinicopathologic evidence of liver dysfunction without developing clinical signs, indicating that disease severity may vary.49,54,60
Diagnosis of IAHD is based on the anamnesis, clinical signs, and findings from serum biochemical analysis, hepatic biopsy, and necropsy. High serum levels of unconjugated and total bilirubin and serum bile acids, coupled with high serum activity of GGT, SDH, aspartate aminotransaminase (AST), LDH, and ALP, are indicative of hepatic necrosis in horses. IAHD may be confirmed by hepatic biopsy or postmortem examination of hepatic tissue. Typical histopathologic changes seen in biopsy specimens or obtained at postmortem examination include widespread necrosis of hepatocytes that is most severe in the centrilobular and midzonal areas, with the few living cells confined to the periportal areas.53 The normal architecture of the centrilobular and midzonal areas is replaced by a pale, eosinophilic granular mass in which ghost outlines of necrotic hepatocytes are present.53 A mild inflammatory cell infiltrate is seen in the portal areas, as well as a number of spindle-shaped fibroblastic cells.53 The histopathologic lesion is frequently more advanced than the clinical course of the disease would suggest.53
There is no specific treatment for IAHD.61 Treatment should be aimed at supporting liver function and controlling abnormal behavior. Dietary adjustments are important and should consist of reducing the quantity of protein in the diet while increasing carbohydrate intake.46 Sorghum, milo, or beet pulp, which contains high levels of branched-chain amino acids, can be mixed with molasses to improve palatability and caloric content.46 Diets high in branched-chain amino acids tend to lessen the degree of abnormal behavior associated with hepatic encephalopathy.61 Oral neomycin, lactulose, and mineral oil may be administered to decrease ammonia production and absorption from the GI tract. It may become necessary to sedate horses that display severe signs of hepatic encephalopathy with xylazine or detomidine.61 Continuous intravenous (IV) administration of dextrose and balanced electrolyte solutions to reduce hepatic workload and maintain plasma volume is recommended.60 If spontaneous bleeding occurs at injection sites or at sites of self-inflicted injury, plasma transfusions may be necessary to replace the deficient clotting factors.46 The use of glucocorticoids for treating IAHD is controversial but may be indicated based on some histopathologic evidence of an immune-mediated etiology.57
Although the cause of IAHD is unknown, the administration of TAT to recently parturient, lactating mares is associated with substantial risk for development of fatal IAHD.51,54,60 The susceptibility of postparturient mares to IAHD after TAT administration at foaling is unexplained. Postparturient mares may be the most likely adult horses to receive TAT. Therefore the use of TAT is not without risk, and its use should be restricted to clinical situations necessitating tetanus prophylaxis and in which a history of active tetanus (toxoid) immunoprophylaxis is absent or unknown. The risk of postvaccinal IAHD should be addressed with the owner before administration of TAT.
Routine administration of TAT to parturient mares should be strongly discouraged, and the routine use of active tetanus immunoprophylaxis should be reemphasized. Although the prevalence of serum hepatitis associated with administration of commercial plasma appears to be low in the horse, it should be considered an uncommon risk that can have a fatal outcome.55 IAHD may be observed sporadically or may affect a group of horses. Recognition of IAHD in one horse should necessitate careful observation of horses on the same premises for either clinical or serum biochemical signs of IAHD.60 Based on recent case reports, it appears that not all cases of IAHD are clinically apparent and that screening for its presence utilizing routine serum biochemical analysis is useful in detecting subclinical cases of IAHD.54,55,60
BLACK DISEASE
Definition and Etiology
Black disease (infectious necrotic hepatitis) is a disease of grazing animals, primarily sheep, resulting most often in sudden death.62-65 It is caused by toxins produced by the bacterium Clostridium novyi type B. C. novyi occurs in soils and is normally present in the digestive tracts and livers of animals grazing affected pastures. Disease occurs only when there is sufficient liver damage to provide the anaerobic environment required for growth of the organism and subsequent toxin production. In practice, the liver insult is almost always caused by larval migration of the common liver fluke, Fasciola hepatica. Therefore, this is a seasonal disease related to the liver fluke cycle and occurring in animals typically infested by F. hepatica. Other liver parasites, such as Fascioloides magna, Dicrocoelium dendriticum, and Cysticercus tenuicollis, are occasionally implicated, as is damage to the liver by biopsy or trauma that is severe enough to result in localized anaerobiosis in the liver, but these cases are rare. Such a scenario should be considered when sudden deaths occur at times flukes are not usually migrating or in families such as Equidae that are not normal hosts for F. hepatica. The disease occurs worldwide where liver flukes are present. The C. novyi strains that cause black disease produce at least three potent exotoxins: the alpha-toxin, which is the classical lethal toxin; the beta-toxin, which has both lethal necrotizing and hemolytic lecithinase activities; and the zeta-toxin, which is hemolytic.66
Pathophysiology
C. novyi type B is widely distributed in soils, and its spores are continuously ingested and shed in feces of grazing animals. Some of these organisms cross the intestinal mucosa and become disseminated throughout the animal’s mononuclear macrophage system, including the Kupffer’s cells of the liver. When localized anaerobic conditions occur in the liver, as with migration of liver fluke larvae, these resident spores may germinate and enter a vegetative state. As they proliferate, they release their toxins and create enlarging zones of coagulation necrosis in the liver. The exotoxins produced will enter the general circulation, where they cause damage to neurons, vascular endothelium, and other vital cells and tissues, eventually causing sudden death of the animal.
Clinical Signs
Clinical black disease consists almost entirely of animals found dead. In the unlikely event that an affected animal is recognized before its demise, the signs are nonspecific. The animal will often be off to itself and will appear depressed, anorexic, and possibly in respiratory distress. Temperature is elevated initially, 40° to 41° C (104° to 106° F), but declines before death. Unlike closely related bacillary hemoglobinuria, or “redwater,” affected animals do not show red urine or bleeding from nose or rectum. Diagnosis of clinical black disease can often be accomplished at necropsy or with the help of simple laboratory tests. History will include sudden death, usually in an endemic area during warmer weather, when fluke transmission is active. Flock or herd vaccination will be either overdue or absent. It is also helpful to know the history of fluke infestation on the farm and the timing of fluke control measures, if any have been taken. Time of death should be ascertained as closely as possible. The animal is usually presented in lateral recumbency without signs of struggle. It will be severely bloated, even if death has occurred quite recently, and gives the impression of a carcass in a more advanced state of decomposition than timing would suggest.
Necropsy Findings
Postmortem lesions are often obscured by rapid putrefaction of the tissues. Skinning the animal reveals engorgement and hemorrhage of subcutaneous blood vessels, resulting in “black” discoloration, thus the name of the disease. Once the body cavity is entered, blood-tinged abdominal, thoracic, and pericardial fluids are present. Urine is grossly normal. Subendocardial and subepicardial hemorrhages are present. There may be hemorrhages on serosal and pleural surfaces, but these are not always present. Tissues, especially solid organs such as liver and kidney, appear to be in a state of autolysis much more advanced than time of death would suggest. In addition to the presence of migrating fluke channels, the liver is swollen and congested and has one to several small, pale areas of coagulative necrosis. These lesions are typically along the diaphragmatic surface of the liver, have hyperemic borders, and extend for a variable distance into the hepatic tissue. Careful slicing of the organ may be required to find these lesions.64
Diagnosis
If necropsy does not confirm a diagnosis of black disease, or if there is need for additional documentation, a simple Gram stain of an impression smear taken from the margin of the liver lesion or from any area of a liver without lesions will reveal numerous large, gram-positive rods typical of the clostridia. Because clostridial organisms proliferate rapidly after death, such findings must always be interpreted carefully, with consideration of time elapsed between death and necropsy. Further diagnostic measures include anaerobic culture and isolation of C. novyi type B, identification of the specific toxins, and fluorescent antibody identification of the organism. Characteristic histopathologic changes may also be seen in fixed sections of liver associated with necrotic lesions. Impression smears obtained from the liver lesions are ideal for the fluorescent antibody identification of C. novyi type B. Specific toxin identification is impractical for most laboratory settings but is the confirming procedure of choice.
Treatment and Prevention
Treatment of black disease is rarely undertaken because of its acute to peracute presentation, with animals usually found dead. C. novyi is highly sensitive to penicillin and the tetracyclines, but toxin production is usually too far advanced for antibiotics to be of value. If used, they should be administered at high doses, intravenously if possible for most rapid onset (20,000 IU/kg crystalline penicillin or 5 mg/kg oxytetracycline), followed by intramuscular (IM) or IV doses at appropriate intervals. Supportive IV and/or intraluminal fluid therapy should be initiated to correct dehydration. Antiserum is not available. Care should be exercised in handling affected animals because stress may result in sudden death. Chances of success are small. In the event of an “outbreak,” vaccination should be initiated immediately, along with mass administration of penicillin or tetracycline, preferably in a long-acting form.
A good prevention program requires consideration of both the pathophysiology and immunology of the disease and the natural history of the liver fluke (F. hepatica) that is so intimately involved in expression of black disease. Efforts to clear soils of the offending organism are unrewarding. However, it is recommended that carcasses of animals dying from black disease be burned, buried deeply, or removed from the premises.
Commercial bacterin/toxoids against C. novyi are available in combination with other clostridial vaccines. These products are generally safe and highly efficacious. Duration of protection is short, however, and should not be relied on more than 5 to 6 months. Field experience suggests that monovalent Clostridium haemolyticum vaccine is more effective than combination products in high-risk herds, and efficacy may vary among commercially available combination vaccines. Animals vaccinated when less than 3 to 4 months old require revaccination at weaning. Local reactions at the site of subcutaneous vaccination are common but primarily of cosmetic concern. Intramuscular vaccination should be avoided because it frequently results in permanent damage to muscle tissue, with a negative impact on meat marketing. Timing of vaccination must be determined by the local climate and liver fluke season. In more severe climates where the fluke season is relatively short, a single annual injection before fluke transmission season may suffice. In more moderate climates with longer periods of fluke exposure, a second injection about 5 months later may be required.
Control of liver fluke infestation is closely linked to prevention of black disease. Control measures include pasture or range management; control of water sources; limiting access to streams, canals, ponds, and marshes; and strategic treatment of animals with products effective against flukes. Appropriate disposal of infected carcasses is important. This is a complex subject, and the reader should refer to the section on liver flukes for more detail.
Bacillary Hemoglobinuria (“redwater”)
Definition and Etiology
Bacillary hemoglobinuria (redwater, icterohemoglobinuria) is a disease causing sudden death in cattle and other ruminants and, rarely, horses. It is caused by the toxins of Clostridium haemolyticum (Clostridium novyi type D). This organism is closely related to the etiology of black disease, in which toxin production is closely linked to bacteriophage infection of the clostridial organisms.67 The major biologically active toxins in C. novyi type D (C. haemolyticum) are the beta-toxin, which is a phospholipase C and has both lethal necrotizing and hemolytic lecithinase properties; the zeta-toxin, which is a tropomyosinase; and the theta-toxin, which is a lipase. Prominent actions of the toxins induce localized hepatic necrosis and intravascular hemolysis. C. haemolyticum occurs in soils, and its spores are routinely found in the livers and passed in the feces and urine of healthy animals grazing affected pastures. Disease occurs only when there is sufficient insult to the liver to provide the anaerobic conditions required for bacterial growth and toxin production. In almost all cases the liver insult is caused by migration of Fasciola hepatica (common liver fluke) larvae.68-70 As in black disease, Fascioloides magna, Dicrocoelium dendriticum (the lancet liver fluke), and Cysticercus cellulosae have also been occasionally implicated in the disease, as has liver biopsy or trauma severe enough to result in bruising of the liver.71 Redwater is then, for all practical purposes, a seasonal disease occurring at the time of larval fluke migration.
Bacillary hemoglobinuria is a regionalized disease. It has been reported that C. haemolyticum is limited to alkaline soils, but the disease is also endemic in regions with acid soils. The factors affecting distribution of the disease are not well understood. Even in areas where it is common, distribution is erratic, with some farms severely affected, whereas others nearby are disease free. The disease is expanding from areas where it has traditionally been seen, probably in large part because of the shipment of cattle carrying C. haemolyticum and F. hepatica.
Pathophysiology
Spores of C. haemolyticum are ingested by susceptible animals, cross the intestinal mucosa, and are transported to the liver and other organs, probably within the phagosomes of cells of the mononuclear macrophage series. The spores can persist for long periods in the liver within Kupffer’s cells. Any localized area of anaerobiosis, as caused by migrating fluke larvae, allows these spores to germinate and proliferate. Release of toxins from the vegetative cells further increases the anaerobic environment, favoring accelerated bacterial proliferation, toxin production, and hepatic necrosis. Absorption of toxins into the circulatory system rapidly leads to intravascular hemolysis, icterus, hemoglobinuria, and death.
Clinical Signs
In most cases of bacillary hemoglobinuria, animals are found dead. In the rare cases in which disease is recognized antemortem, signs include malaise (animal by itself, “humped up,” reluctant to move), anorexia, and fever of 40° to 41° C (104° to 106° F), declining as death approaches. Breathing may be rapid and shallow, and blood or blood-tinged froth may be present in the nostrils. Rectal bleeding or bloody feces may also be observed. The eponymous sign is passage of dark-red, “port wine”–colored urine (hemoglobinuria), but this sign is seen relatively infrequently. Blood is thin and watery and coagulates slowly. Mucous membranes are pale and icteric. Severity of signs increases as the disease progresses.
Bacillary hemoglobinuria is usually diagnosed at necropsy as a cause of sudden death. Historical concerns include occurrence in or origination of the animal from an endemic area, season of the year, last known sighting of the live animal, approximate time of death (and time from estimated death to necropsy), and farm history of vaccination and liver fluke control. Frequently the animal has been seen apparently healthy 12 to 24 hours before death. Herd vaccination is either nonexistent or overdue. The animal is usually in lateral recumbency, severely bloated, and without signs of struggle. Blood is often present in the nostrils, mouth, rectum, or vagina. The carcass appears to be in an advanced state of decomposition, even when it is actually quite fresh. A tentative diagnosis can be made on the basis of history and observation of the carcass.
Necropsy Findings
On closer examination, membranes and tissues are icteric. Skinning the animal reveals numerous subcutaneous petechial and ecchymotic hemorrhages, edema, and sometimes emphysema. There will be copious amounts of red-tinged abdominal and thoracic fluid. Hemorrhages are present on all serosal surfaces. Dark-red urine (or traces) is present in the bladder. Lymph nodes are congested and usually hemorrhagic. There may be hemorrhage into the bowel lumen. The spleen is enlarged. The tracheobronchial tree is usually filled with blood-tinged froth or foam. Lungs show hemorrhages, edema, and frequently emphysema. Pericardial fluid is blood tinged, and hemorrhages are present on both the epicardium and the endocardium. Solid organs such as liver and kidney appear to be in advanced stages of autolysis, even in fresh carcasses. The confirming (pathognomonic) lesion is the so-called ischemic hepatic “infarct,” which has a zone of hyperemia at its interface with viable liver tissue. This area of coagulative necrosis, sometimes partially liquefied at its center, can reach up to a 30-cm diameter and have a very irregular outline.69 Unlike a classic infarct, the lesion in bacillary hemoglobinuria results from the progressive enlargement of the focus of coagulative necrosis caused by the bacterial toxins. Any thrombosis seen in the lesion is secondary, with the vasculature being included in the necrotic process, along with other hepatic tissue.71 A thin coat of fibrin may cover the capsule of the liver where it overlies the necrotic lesion.
Diagnosis
In most cases, diagnosis of redwater can be confirmed at necropsy. Some peracute cases may be less typical, or laboratory confirmation may be needed in certain, unusual cases. A simple Gram-stained impression smear from the liver will reveal numerous typical clostridial organisms. Smears may also be made from spleen, blood, or abdominal fluid, with the same outcome. Postmortem presence of clostridial organisms must be interpreted with caution because they are always present and proliferate rapidly after death. It is important to have an accurate estimate of time of death to determine the significance of these findings. Laboratory confirmation depends on identification of the causative bacterium. Both fresh (refrigerated) and formalin-fixed liver lesions should be submitted. Fluorescent antibody tests on impression smears taken from a liver “infarct” will be positive for C. novyi type D antigens. Extensive biochemical and toxin identification tests are confirmatory but are seldom used if lesions and fluorescent antibody tests are compatible. Histopathology reveals numerous clostridial rods within the hepatic lesion, particularly immediately subjacent to the zone of neutrophils at the advancing margin of the lesion.
Treatment and Prevention
Treatment of bacillary hemoglobinuria is seldom undertaken because of the acute nature of the disease. If there is opportunity for treatment, penicillin at high dosages (at least 20,000 IU/kg IM twice daily) is the antibiotic of choice, although tetracyclines (5 mg/kg IV twice daily or 10 mg/kg IM daily) are acceptable. Because time is critical, initiating treatment with crystalline penicillin IV (20,000 U/kg) is indicated if available. Fluids are given intravenously (IV) or intraruminally to correct dehydration. Because of severe hemolytic anemia, blood transfusion is advisable and should be repeated as necessary. Affected animals must be handled with great care because stress or excitement may result in sudden death. In the rare instance of recovery, hematinics should be given to support RBC regeneration.
An effective prevention program requires consideration of both the pathophysiology and immunology of the disease and the natural history of the liver fluke (F. hepatica) that is so intimately involved in expression of bacillary hemoglobinuria. Efforts to clear soils of the offending organism are unrewarding. However, it is recommended that carcasses of animals dying from bacillary hemoglobinuria be burned, buried deeply, or removed from the premises.
Commercial bacterin/toxoids against C. haemolyticum are available in both monovalent form and in combination with other clostridial vaccines. These products are generally safe and highly efficacious. Duration of protection is short, however, and should not be relied on more than 5 to 6 months. Animals vaccinated when less than 3 to 4 months old require revaccination at weaning. Local reactions at the site of subcutaneous vaccination are common but primarily of cosmetic concern. Intramuscular vaccination should be avoided because it frequently results in permanent damage to muscle tissue, with a negative impact on beef marketing. Timing of vaccination must be determined by the local climate and liver fluke season. In more severe climates where the fluke season is relatively short, a single annual injection before fluke transmission season may suffice. In more moderate climates with longer periods of fluke exposure, a second injection about 5 months later may be required.
Control measures include pasture or range management; control of water sources; limiting access to streams, canals, ponds, and marshes; and strategic treatment of animals with products effective against flukes. This is a complex subject, and the reader should refer to the section on liver flukes for more detail.
HEPATIC FAILURE IN FOALS
Hepatic failure in foals might result from infectious, parasitic, congenital, metabolic, or toxic causes. Iron fumarate is the best documented of the toxic causes.72 Iron toxicity most often occurs when newborn foals are given iron before nursing. Colostral-acquired glutathione or other protective substances may explain the great decrease in iron hepatotoxicity when the iron is administered after colostrum.73 When the iron is given at birth and before colostrum, clinical signs develop 2 to 5 days later. In rare cases, clinical signs may not develop until the foal is older. The initial clinical signs are associated with hepatoencephalopathy and include seizures, marked depression, ataxia, aimless wandering, head pressing, or any sign of abnormal behavior. Icterus is noted in most foals at the time neurologic signs are exhibited, although some foals may die so peracutely that icterus is not noticed. Although not documented in foals, nonsteroidal antiinflammatory drugs (NSAIDs), such as carprofen and mycotoxins, are other potential toxic causes of hepatic failure. Rarely, foals treated with macrolides, trimethoprim-sulfas, or histamine-2 (H2) blockers for pneumonia or diarrhea develop an increase in serum hepatic enzymes, despite the foal improving clinically. With changes in drug treatment, the serum enzymes return to normal, suggesting these cases may have drug-induced hepatopathy.74 Steroid-induced hepatic lipidosis has been observed in foals receiving both prolonged and high doses of corticosteroids.
Other causes of hepatic failure in foals include perinatal herpesvirus infection, leptospirosis, Actinobacillus equuli infection, streptococcal bacteremia, Tyzzer’s disease, other bacterial infections, systemic inflammatory response syndrome (SIRS) and multiple organ system dysfunction, septic portal vein thrombosis (e.g., Rhodococcus equi), chronic neonatal isoerythrolysis, hepatic lipidosis/hyperlipemia in miniature equine foals, cholangitis associated with duodenal ulcer disease, Parascaris equorum migration, congenital anomalies (e.g., atresia of bile duct, portosystemic shunts), and hyperammonemia in Morgan weanlings.
Equine herpesvirus 1 (EHV-1) infection of a near-term fetus may result in the birth of a nonviable foal with hepatic, respiratory, and/or gastrointestinal disease.75 Generally, the clinical condition rapidly declines within 5 days after birth. Affected foals may have severe neutropenia and lymphopenia. Treatment with oral acyclovir (8 to 16 mg/kg every 8 hours) may improve survival rates.76
Leptospirosis was reported to cause jaundice and death in a 10-day-old foal.77 Although Leptospira pomona is known to cause abortion and liver disease (giant cell hepatopathy) in the equine fetus, it is also apparently a rare cause of neonatal liver disease. A. equuli typically causes bacteremic embolic nephritis and acute death in very young (∼3-day-old) foals. It may also cause widespread multifocal hepatitis in foals. Streptococcus zooepidemicus bacteremia may also cause multifocal hepatic abscesses. Tyzzer’s disease is the best-documented cause of bacterial hepatitis in foals, as briefly discussed in the following section. Other bacteria and bacterial toxins may initiate an exaggerated response to sepsis (SIRS), which may result in multiple organ dysfunction, including hepatic failure. Many vasoactive mediators are involved in this process; the hemodynamic system becomes ineffective, and certain organs (e.g., liver) may fail because of hypoxia. Diffuse hepatic necrosis and hepatocellular apoptosis are the characteristic lesions. The clinical signs may be identical to Tyzzer’s disease, except the syndrome may affect a much wider age range. Prompt and aggressive treatment is often successful. Appropriate treatments include broad-spectrum antibiotics, fluids, oxygen, and antioxidants such as dimethyl sulfoxide (DMSO) and acetylcysteine. Bacterial sepsis rarely may cause acute portal vein thrombosis. Variable degrees of hepatic hypoxemia and inflammation accompany the thrombosis. Hepatoencephalopathy does not generally occur in foals, unlike in adult horses, with portal vein thrombosis. Diarrhea may occur in foals from portal hypertension. The thrombus can be seen on ultrasound examination. A complete recovery may occur with long-term antimicrobial therapy.
Foals with severe and prolonged neonatal isoerythrolysis may develop liver failure,78 which may result from a combination of chronic hypoxia and iron overload from multiple transfusions. Levels of conjugated bilirubin are generally high enough to suggest that bile stasis is also present. With severe and persistent hemolysis, bile excretion may become the rate-limiting step in bilirubin clearance. Physical obstruction of bile flow may result from duodenal scarring after a duodenal ulcer,79 congenital biliary atresia, or parasitic obstruction.
Congenital portosystemic shunts occur infrequently in the equine and bovine.80 Clinical signs may not be noted until foals are 2 to 3 months old and begin ingesting large amounts of grain or grass. Waxing and waning signs of encephalopathy are the most common signs in foals. Encephalopathic signs and tenesmus are characteristic of the disease in calves. Elevated plasma bile acids and ammonia levels in foals with encephalopathic signs and normal concentration of hepatic-derived serum enzymes should arouse suspicion of a portosystemic shunt. Shunts may be single or multiple and may be intrahepatic or extrahepatic. Positive-contrast portography is the diagnostic technique of choice. The shunt may also be detected by transrectal portoscintigraphy (in foals) or transabdominal ultrasound examination. Successful medical management followed by shunt ligation has been described in a foal and calf.80,81
Hepatic failure has been seen in Morgan foals.82,83 The onset of clinical signs (depression and weight loss) occurred soon after weaning. Liver enzymes are elevated; and variable degrees of portal and bridging fibrosis with bile duct hyperplasia, karyomegaly, and cytomegaly are often seen on microscopic examination. The disease is fatal and may end with a terminal hemolytic crisis. The cause of the disease is unknown, but it may be inherited.
The best laboratory aids in confirming hepatic failure are abnormally high concentrations of serum bilirubin (both conjugated and unconjugated), ammonia, and prolonged PT. Elevation in serum enzymes that are hepatic specific may indicate hepatic disease. Some foals with toxic hepatic failure have had normal or only modestly elevated SDH levels. GGT levels may be high in normal neonatal foals.84 Plasma glucose concentrations are frequently abnormally low in neonatal hepatic failure. The measurement of bile acids may be useful in determining hepatic dysfunction in foals older than 1 week.85
Treatment of these conditions is discussed later under Therapy of Liver Failure or under the specific condition.
TYZZER’s DISEASE IN FOALS
Tyzzer’s disease is a sporadic, acute focal bacterial hepatitis that occurs in foals 7 to 40 days of age.86 It has been reported in calves and other species.87 The causative organism is Clostridium piliforme, formerly called Bacillus piliformis.88 Multiple strains can infect Equidae.89
In many cases of Tyzzer’s disease, the foal will be found dead with no previous signs of illness. The diagnosis in most cases is made postmortem. Recently, a polymerase chain reaction (PCR) test has been used to diagnose the condition in live foals.90 If detected, clinical signs may include fever, icterus, depression, anorexia, diarrhea, and seizures, none of which is specific for the disease.90,91 Serum chemistry values will show elevated liver enzymes, hyperbilirubinemia, hyperfibrinogenemia, and a severe hypoglycemia. Histopathologic examination of the liver reveals multifocal areas of necrosis in which the organism can be identified with the Warthin-Starry method.
There is one report of successful treatment of a proven case of Tyzzer’s disease in a foal.90 Early antimicrobial therapy and IV fluids to correct the severe hypoglycemia and acidosis, along with antiinflammatory drugs and parenteral nutrition, could be helpful.
CHRONIC ACTIVE HEPATITIS
Definition, Etiology, and Epidemiology
Chronic active hepatitis represents a sustained inflammatory process within the liver. The diagnosis is made histologically when the principal features are infiltration of inflammatory cells into the portal areas, necrosis, and fibrosis.92 Some pathologists do not recognize this as a large animal disease, and although there are few recent reports in the literature, a number of cases fulfill these criteria.
The etiology of chronic active hepatitis in large domestic animals is not known, but several factors likely are involved. In humans, persistence of the hepatitis B or hepatitis C virus accounts for most cases of chronic hepatitis. In dogs, most cases are reported to be “idiopathic,” even though more cases are reported.93 In horses the histologic diagnosis is often cholangiohepatitis.94 Cholelithiasis and cholangiohepatitis are encountered as forms of liver disease in adult horses and involve ascending infections from the small intestine.95 These are discussed further under Choledocholithiasis, Cholelithiasis, Hepatolithiasis.
Cholangiohepatitis that is not associated with liver flukes also has been confirmed in cattle. Bacterial infection through the portal drainage of the bowel or by ascending infection from the bile duct is a potential cause of cholangiohepatitis. Toxins may play a role.96 Immune-mediated processes are thought to be potential causal factors in humans and may be involved in horses because some respond to corticosteroids, but no definitive work has been done in large animals.
Clinical Signs and Differential Diagnosis
The signs of chronic active hepatitis are similar to those of other types of chronic progressive liver failure, as described previously under diagnosis. Progressive weight loss and depression associated with intermittent fever and icterus may be noted initially. Fever is most consistent with bacterial cholangiohepatitis. Other signs may develop progressively as liver functions begin to fail. Signs of concurrent intraabdominal diseases may be present, and some horses develop peculiar cutaneous lesions at the coronary band or areas of necrotic leathery skin.97 These lesions usually appear as a moist, exfoliative dermatitis caused by an aseptic vasculitis. Differential diagnosis includes pyrrolizidine alkaloid toxicity, bile stones, abdominal abscesses, and other chronic wasting diseases.
Liver-Derived Serum Enzymes and Diagnostic Tests
Serum liver enzyme activities are usually elevated, reflecting active hepatocyte damage. ALP and GGT tend to be greatly elevated in the active stages of the disease process. Serum bile acid concentrations are increased, and the bromosulfophthalein clearance half-life is prolonged. In most cases, serum bilirubin, especially the direct reacting (conjugated) bilirubin, is elevated. A definitive diagnosis is made from histopathologic examination of tissue obtained by liver biopsy (see following discussion). A part of the liver biopsy, tissue should be submitted for bacterial culture and sensitivity. The lesion is described under Necropsy Findings.
Pathophysiology
The exact pathophysiology of chronic active hepatitis is not known. The early stages are associated with inflammation of the bile ducts and the portal areas of the liver. Extension of bacterial infection through the bile duct or through the portal venous drainage may be responsible for the lesion distribution in animals with suppurative cholangiohepatitis. When lymphocytes and plasma cells are predominant in the cellular infiltrate, immune-mediated processes are more likely.
The liver responds by proliferation of bile ducts and bile duct epithelium, which may impair bile excretion. Because hepatocytes are destroyed more rapidly than they are replaced, connective tissue takes their place. Eventually, areas are joined so that the fibrosis itself limits regeneration. At this stage, some cholestasis may occur, along with failure of other metabolic functions. Portal hydrostatic pressures could increase gradually, potentially leading to other clinical signs (e.g., ascites), but this has not been reported in large animals.
Necropsy Findings
Grossly, the liver appears firm and is often pale brown to green in color, and the cut surface may have prominent, irregular markings. Histologically, most of the lesions are present in the periportal areas. Inflammatory cell infiltration consists primarily of mononuclear cells in some cases, whereas a neutrophilic infiltrate that may contain bacteria (often coliforms) is found in others. These infiltrates are believed to indicate the nature of the primary disease process. Biliary hyperplasia may be marked if there is cholangiohepatitis. Loss of hepatocytes and increased fibrous connective tissue may be pronounced in the periportal area.
Treatment and Control
Supportive care is most important in these cases, especially for the maintenance of proper appetite and nutrition (see Chapter 50).98 General measures for treating liver disease apply in these cases (see p. 921). Corticosteroids have been especially useful in horses with a lymphocytic plasmacytic hepatic infiltrate. They act to increase appetite, stabilize cell membranes, and reduce inflammation and connective tissue formation. Initial treatment with dexamethasone at 20 to 40 mg/day for the first 4 to 7 days is followed by a gradual reduction in dosage rate over 2 to 3 weeks, depending on response to therapy.99 Low-level treatment with prednisolone at 400 mg once daily may be required for an additional 2 to 4 weeks. Antibiotics are indicated when a bacterial cholangiohepatitis is suspected on the basis of the histologic features of the liver, the culture of the liver biopsy, and the presence of a persistent, intermittent fever. Although data are limited, enteric organisms are likely to be encountered, and antibiotic therapy should be directed at the likely organism. Bacterial culture and sensitivity from the biopsy specimen can guide appropriate antimicrobial therapy.
Prognosis
The prognosis for cases of chronic active hepatitis in the horse may be more favorable than with other liver diseases, such as toxicities and serum hepatitis. A recent retrospective study reported that five of nine horses with chronic active hepatitis survived.100 Case records at Oregon State University Veterinary Teaching Hospital indicate that about half the cases of chronic active hepatitis with no evidence of Senecio poisoning improved. Liver biopsy and response to therapy are the best guides in formulating a prognosis. The prognosis for improvement and long-term survival is extremely poor in horses that have functional hepatic failure with widespread fibrosis and disruption of normal hepatic parenchyma. The prognosis is fair to good in patients with early (less severe) lesions, particularly those with a lymphocytic plasmacytic cellular infiltrate that responds well to corticosteroids.
PYRROLIZIDINE ALKALOID TOXICITY
Definition and Etiology
Pyrrolizidine alkaloid (PA) toxicity is a chronic, progressive, often-delayed intoxication that results when animals consume plants containing PAs. The condition is manifested by signs of liver failure.101 More than 350 PAs have been identified in over 6000 plant species.102Table 33-4 lists common plants containing PAs.
Table 33-4 Common Plants Containing Pyrrolizidine Alkaloids (PAs)
| Botanical Name (Genus/Species) | Common Name |
|---|---|
| Senecio jacobaea | Tansy ragwort |
| Senecio vulgaris | Common groundsel |
| Senecio douglasii var. longilobus | Threadleaf groundsel |
| Senecio riddellii | Riddell groundsel |
| Senecio trianularis | Tarweed |
| Senecio alpinus | Alpenkreuzkraut (Europe) |
| Amsinckia intermedia | Fiddleneck |
| Crotalaria species | Rattlebox |
| Echium plantagineum, E. lycopsis | Viper’s bugloss, Salvation Jane |
| Heliotropium europaeum | Common heliotrope |
| Symphytum officinale | Comfrey |
| Cynoglossum officinale | Hound’s tongue (houndstongue) |
| Eupatorium maculatum | Bruner’s trumpet |
| Baccharis pteronoides | Yerba de pasmo |
| Borago officinalis | Borage |
| Erechtites species | |
| Trichodesma species |
Clinical Signs and Differential Diagnosis
The clinical signs of PA poisoning are basically those of liver failure, as previously described under Diagnosis of Liver Disease. The most common signs of PA toxicity in the horse are weight loss, slight to moderate icterus, and abnormal behavior (e.g., wandering, ataxia).103 Signs seen less frequently in horses include photosensitization of the white areas and, in rare cases, diarrhea. A few cases in ponies have shown loud, stertorous inspiratory noises, possibly caused by pharyngeal-laryngeal paralysis.7,104 We have seen pruritus in two cases involving horses, but never in cattle. Abortion may occur from ingestion of sublethal doses,105 and PA has been shown to be teratogenic in rats.106 Subtle signs such as poor performance (inability to race up to previous standards) may be seen in horses with pyrrolizidine-induced liver damage before the onset of liver failure. Secondary gastric impaction has been reported in ponies.107
Cattle more frequently show diarrhea, weight loss, tenesmus, prolapsed rectum, and ascites. Calves are much more susceptible than mature cattle. Behavioral changes or subtle neurologic signs may also be seen in cattle, but icterus is uncommon. Differential diagnosis includes other diseases causing liver failure (e.g., aflatoxicosis), as discussed in Chapter 54, and some chronic, debilitating diseases such as gastrointestinal parasites, liver flukes, and Johne’s disease.101
Sheep and goats are more resistant to PA toxicosis but can be affected by certain alkaloids at doses 30 times or more the dose that affects cattle and horses. A consortium of ruminal microbes from sheep degrades PAs found in Seneio jacobaea to less toxic metabolites.108 PA injected directly into the portal vein is toxic in sheep, as is Senecio when put in the abomasum.109 Liver microsomal enzymes in sheep may also play a role in detoxifying PAs110; however, this alone does not seem to account for the differences in susceptibility.111
Clinical Pathology And Diagnostic Tests
The liver-derived serum enzyme activities are elevated during periods of active hepatocyte destruction caused by PA poisoning. Although the dehydrogenases (e.g., SDH, GLDH, LDH) are elevated initially, they may have returned to normal by the time the animal first shows clinical signs of functional failure. Because lesions are largely in the portal region, GGT and ALP tend to be consistently elevated.112,113 In a study of sublethally poisoned horses, AST and the ratio of branched-chain/aromatic amino acids were persistently elevated.105 Bile acids are increased and are increased early in some horses. Serum bile acid concentrations have a good predictive value, and levels above 50 μm/L would be a poor prognostic indicator in the horse.114 Serum protein concentration is usually normal, and only terminally does albumin decrease or blood clotting become altered. Bilirubin, both direct and indirect, tends to be increased in the horse at the later stages of the disease process.
Liver biopsy is useful in arriving at a definitive diagnosis, but other causes of chronic hepatitis (e.g., aflatoxins) may produce a similar histologic appearance. The triad of fibrosis, bile duct proliferation, and megalocytosis is characteristic of PA toxicity. The histologic lesions are described more fully under Necropsy Findings. Some of the changes can be used for prognosis. Modest changes in the hepatocytes and biliary hyperplasia are reversible. Fibrosis bridging portal areas indicates an eventually fatal condition, as does the extensive fibrosis of an end-stage liver.
Feed samples can be examined for PA-containing plants. Cubed or pelleted feeds can be analyzed for PAs, but this is often time-consuming and relatively expensive.* A sulfur-bound pyrrolic metabolite was identified by thin-layer chromatography (TLC) on the hemoglobin of horses exposed to PAs.115
Pathophysiology
There are a number of PAs, and many of the poisonous plants contain four to six different alkaloids. About 50% of the alkaloids are toxic, and some are more toxic than others. The alkaloid concentrations vary slightly among plants from different areas, but there is greater variation in concentrations from different parts of the same plant.116 After absorption the portal circulation carries the alkaloids to the liver, where they are metabolized by microsomal enzymes of the hepatocyte to more toxic pyrroles.117 The pyrroles may cross-link double-strand DNA in a dose-dependent manner.118 The degree of DNA cross-links depends on the concentration of the pyrrol, but not on the base sequence of the oligonucleotide target.119 The cross-linking of DNA produces an antimitotic effect.120 The hepatocytes cannot divide and often become megalocytes as cytoplasm expands without nuclear division. As cells die, they are replaced by connective tissue rather than new hepatocytes. This antimitotic effect may explain why megalocytosis (large hepatocytes and large nuclei) is seen with the disease. Besides cross-linking DNA, pyrroles, which are alkylating agents, may disrupt the hepatocyte in other ways.121 They bind to protein and nucleic acid, thereby inhibiting enzymes and blocking protein synthesis. All these actions may lead to faster death of hepatocytes. With chronic doses, hepatocyte death is more severe in the portal areas (Rappaport zone 1), although some islands of necrosis do occur.112,113 Centrilobular necrosis may be seen with massive doses.
With progressive death of hepatocytes and subsequent fibrosis, liver function begins to fail. The blood supply through the hepatic lobule is disrupted by fibrosis, making regeneration impossible. This becomes a venoocclusive disease, resulting in marked portal hypertension. The increased portal hydrostatic pressure leads to diarrhea and ascites in ruminants. It is unclear why diarrhea and ascites are seen infrequently in horses with PA toxicity.
Epidemiology
PA toxicity is seen wherever plants containing the alkaloids are found. The plants are not very palatable and in most cases are not readily eaten, but animals may eat them when the growth of the toxic plant is so thick that the animal cannot separate it from normal forage, or when other forage is sparse. The plants are toxic in hay, including pelleted and cubed hay. Some herbicides may make the plants more palatable as they begin to die. PAs also survive ensilage; oat silage contaminated with Amsinckia or other PA-containing plants has been responsible for illness in dairy cattle. Seeds of Heliotropium plants have poisoned feedlot cattle.122
The approximate toxic dose of dried Senecio as a percentage of body weight for each species is horses, 5%; cattle, 2% to 5%; goats, 125% to 400%; and sheep, over 150%. Most tansy ragwort contains less than 0.2% PA by weight, but there are reports of much higher levels in some of the other plants.123 Horses were consistently poisoned by greater than 250 mg of total PA per kg body weight.114 The PA in hound’s tongue is extremely toxic to horses.124 This dose does not have to be eaten all at once, since the effects are cumulative. Because signs often are delayed, some animals may not become ill until a year or more after removal from feed sources containing the toxins.113,125 Signs do not occur until hepatocyte loss and replacement by fibrous tissue have caused failure of liver function.
Necropsy Findings
Cattle dying of PA poisoning have ascites and prolapsed rectums and are thin and emaciated. Horses are usually thin and may be icteric. In all species the liver tends to be small, pale brown to yellow, and firm and may appear to be scarred. Hepatic megalocytosis is considered the hallmark of PA poisoning, but it has also been seen in aflatoxicosis and is not always apparent in the earliest stages of PA toxicity. Biliary hyperplasia may occur early in the disease. Nonspecific nuclear changes, such as invaginations of cytoplasm into the nucleus, have been seen.112,113 Isolated hepatocyte necrosis is seen later, and finally, portal or massive generalized fibrosis develops. Once bridging of connective tissue between the portal areas occurs, the disease is fatal. The condition is called a “venoocclusive disease” because perivascular fibrosis sometimes occurs, but this is not a constant or characteristic feature.126 Most of the vascular complications are caused by the generalized fibrosis and remodeling of the liver.
Prognosis
Liver failure may occur with either acute or chronic liver disease; the two must be differentiated for prognosis because end-stage fibrosis caused by PAs has virtually no chance for regeneration and recovery.
No satisfactory treatment for PA poisoning exists. Once obvious clinical signs of liver failure develop, the animal usually dies within 5 to 10 days. Horses with mild clinical signs and reversible histologic lesions survived if they retained an appetite and were not exposed to any more PA-contaminated feed.105 An accurate prognosis concerning mildly affected cases may best be obtained from a combination of consecutive liver biopsies and liver-derived serum enzyme activity,105 as well as from serum bile acid concentration.114 Preventing further exposure to the toxic plants is indicated and may delay or stop the progression of the liver lesions, particularly if an uncontaminated feed source is provided before clinical signs develop.
Treatment
Therapy of liver failure is inappropriate if severe fibrosis has occurred because regeneration is impossible. If the animal still has a reasonable appetite and only modest degrees of fibrosis histologically, treatment may be attempted by providing a low-protein, high-energy diet. The principles of treatment of hepatic disease should be followed (see p. 921).
Prevention and Control
PA poisoning is prevented by keeping susceptible animals from pasture, hay, cubed hay, or seeds that contains PA plants. Senecio vulgaris tends to contaminate mainly first-cutting alfalfa, whereas Amsinckia intermedia is often found in planted fields of oat hay. Senecio can be controlled by cultivation because it is a biennial, or by herbicide spraying in the early, rosette stage. Biologic controls for Senecio jacobaea include the cinnabar moth, Tyria jacobae, and the tansy flea beetle, Longitarsus jacobae. Sheep are sometimes used to graze Senecio-infested pastures to control the weed because they are less susceptible to the poisoning.
OTHER HEPATOTOXINS
The liver is particularly vulnerable to toxic insults because it is the first organ to receive toxins absorbed from the GI tract. Enzymes in the hepatocyte may either activate a toxin or, in some cases, metabolize it before it can cause damage. Most hepatotoxic agents are described in more detail in Chapter 54. Some of the more common liver toxins are listed in Tables 33-5, 33-6, and 33-7, but these are not complete lists because many other plants and chemicals can damage the liver under the right conditions. Once a toxic liver disease has been diagnosed, possible sources of the toxin might be identified from these tables. A more detailed description of the toxin could then be found in Chapter 54.
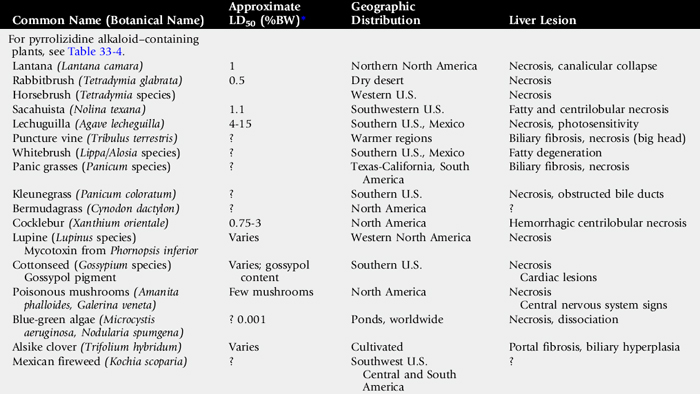

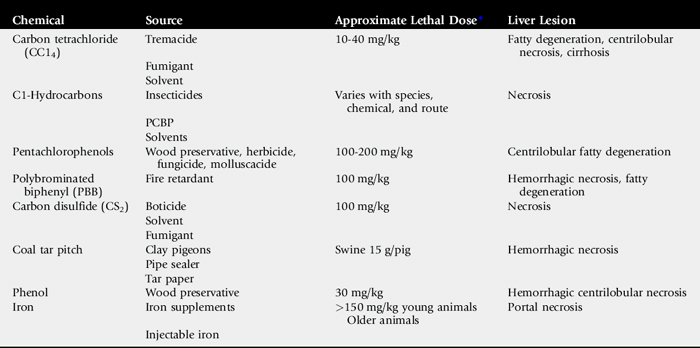
Table 33-7 Drugs Used in Large Animals That Could Damage the Liver
| Drug | Lesion* |
|---|---|
| Carbon tetrachloride | Centrilobular necrosis or fatty degeneration |
| Hexachloroethane | Centrilobular or massive necrosis |
| Carbon disulfide | Necrosis |
| Alcohol | Necrosis/cirrhosis |
| Tetracycline | Fatty degeneration (also renal damage) |
| Clindamycin | Cholestasis, icterus |
| Erythromycin | Cholestasis, icterus |
| Isoniazid | Active hepatitis → cirrhosis/massive necrosis |
| Rifampin | Cholestasis, icterus |
| Nitrofurantoin | Active hepatitis → cirrhosis |
| Anabolic steroids | Cholestasis, icterus |
| Phenothiazine | Cholestasis, icterus tranquilizers |
| Halothane | Active hepatitis → cirrhosis/massive necrosis |
| Fluothane | Active hepatitis/massive necrosis |
| Isoflurane | Active hepatitis/massive necrosis |
| Some diuretics | Cholestasis, icterus |
| Diazepam | Cholestasis, icterus |
| Phenobarbital | Necrosis/cholestasis icterus |
| Aspirin | Active hepatitis → cirrhosis |
| Oil of pennyroyal | Centrilobular necrosis |
| Tannic acid | Centrilobular necrosis, fatty degeneration (renal damage) |
| Dantrolene | Active hepatitis → cirrhosis |
| Copper disodiumedetate | Massive centrilobular necrosis |
| Iron (injectable) | Necrosis, portal, cirrhosis, hemosiderosis |
| Glucocorticoids | Hepatocellular vacuolization |
LIVER FLUKES IN RUMINANTS
The common liver fluke Fasciola hepatica causes a disease of production in ruminants that mimics the production effects and clinical appearance of the GI nematode-parasite complex. The disease often has its maximum economic effect in late fall and winter, when animals are most likely to be under seasonal nutritional stress. F. hepatica is unique among the common helminths of ruminants in that it has an asexual multiplication phase of the life cycle in snail intermediate hosts that is highly sensitive to environmental conditions. F. hepatica is well known for its exponential propagation of infective stages under favorable conditions, sometimes leading to explosive seasonal outbreaks of severe parasitism, especially in sheep. In addition, liver parenchyma migration forms leave necrotic tracts in their wake that are a primary predisposing factor in some areas for acute fatalities caused by Clostridium novyi (black disease) and C. haemolyticum (bacillary hemoglobinuria).127
Epidemiology
The geographic distribution of F. hepatica in the United States is limited mainly to areas in the south-central states and Florida and to the Pacific Northwest, where neutral, well-buffered soils are found and local hydrologic conditions provide suitable habitats for Lymnaea species, the snail intermediate hosts. The important vector species in temperate climates are semiaquatic “mud” snails found in disturbed mud banks and hoofprints in shallow depressions in fields, drainage channels, and temporary water bodies in alluvial river basins, bottomlands, and coastal regions of the southeastern United States. Springs, small streams, seeps, and sloughs serve as habitats in areas of greater terrain relief in the western states. Most infections occur by grazing in and around water in fluctuating habitats that stay wet for more than half the year.
The life cycle development of both the snail host and the parasite in the snail occurs only when temperatures are above 10° C (50° F) and while snail habitats are wet. This leads to a distinct seasonal transmission in the mild, wet winter and spring of the south-central states and Florida and a late spring to fall transmission in the cooler climates of the western states (Figs. 33-1 and 33-2). It takes a minimum of 42 days at 25° C (77° F) or the accumulation of approximately 600 growing-degree days to complete intramolluscan development, with release of cercariae that encyst as infective metacercariae on pasture vegetation (600 growing-degree days = 25° C minus the base temperature of 10° C = 15 growing-degree days × 42 days; an equivalent value for the life cycle is 15° C or 5 growing-degree days × 120 days). Using soil water budget analysis to indicate the length of time habitats are wet and the accumulated sum of growing-degree days over 600, a climate forecast index can be calculated for use in describing the pattern of seasonal transmission at a given site and to provide an indicator of the risk of economic losses in a given climate year. Two weeks of sustained summer heat and drought ends the transmission season by killing pasture metacercariae and forcing snail populations to estivate in soil or perish. In the cooler climates of the western United States, the season ends with drying of springs, seeps, sloughs, and other habitats or with the onset of sustained, cold winter weather. Variation in the annual climate alone may lead to a 100-fold variation in fluke burdens in different climate years (Table 33-8).128
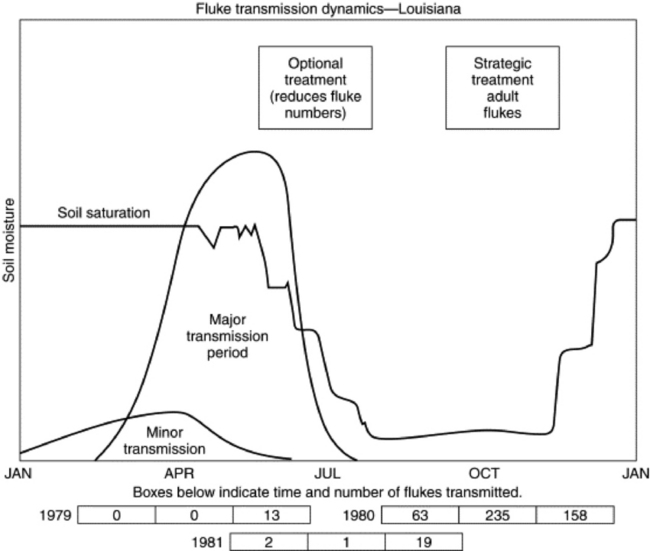
Fig. 33-1 Pattern of Fasciola hepatica transmission typical of the southern United States (based on 10 years of experimental data from Alexandria, Louisiana) and strategic treatment recommendations with adulticidal drugs.
From Malone JP et al: Prev Vet Med 3:131, 1985.

Fig. 33-2 Pattern of Fasciola hepatica transmission typical of the U.S. Pacific Northwest in natural rainfall and irrigated zones (based on experimental data from Langolis, Oregon) and strategic treatment recommendations with adulticidal drugs.
From Rickard LG et al: Vet Parasitol 41:45, 1992.
Table 33-8 Spring and Autumn Forecast Index Values and Observed Fasciola risk at Dean Lee Research Station, Alexandria, Louisiana
The amount of snail habitat present on individual farms related to pasture wetness conditions, such as premises with low-lying, heavy clay soils with a high water table, is also an important consideration in evaluating the need for fluke control. This factor has also been shown to lead to a 100-fold variation in the risk of fluke losses in the same year on different premises in otherwise ecologically similar areas.129 The snail habitat area on most enzootic livestock operations typically is only 1% to 5% of the total land area; operations with over 5% habitat may be associated with a high risk of liver fluke losses.
Clinical Signs and Pathophysiology
Subclinical production effects of F. hepatica include reduced rate of gain and feed efficiency in growing stock. Economic effects have been experimentally demonstrated in calves on a marginal nutritional plane in the first 5 to 6 months after infection, resulting in an 8% loss in the rate of gain with a mean of 40 flukes and a 28% loss with 140 flukes. In cattle it is estimated that herd economic losses are “negligible” with a burden of less than 10 flukes per animal, “possible” at 10 to 40 flukes, and “probable” with over 40 flukes; clinical disease is seen with a burden of more than 200 flukes. In cow-calf operations flukes often act in concert with periods of nutritional stress and concurrent heavy GI nematode infections, causing further reduction of body condition, poor milking ability, and slower return to estrus. This translates to reduced reproductive efficiency, prolonged calving interval, and lightweight calves at weaning.130
In cattle, flukes may lead to chronic disease or, in rare cases, subacute disease. When clinical signs occur, the typical population “overdispersion” (individual variation in parasite burdens) is manifested first in the 10% to 20% minority of the herd that harbor most of the parasite numbers. Signs may consist of weight loss, emaciation, depression, anorexia, rough hair coat, anemia, hypoproteinemia, submandibular edema, and, in rare cases, mild icterus. The anemia associated with F. hepatica infections is more than can be accounted for by blood feeding by flukes. Evidence indicates that depression anemia and biliary hyperplasia result from high levels of proline, a product of fluke metabolism.131 Cattle are able to mount a protective immune response, with partial acquired resistance to F. hepatica, beginning at 5 to 6 months after initial exposure.132 This and the short lifespan of flukes in cattle typically lead to a linear reduction of fluke numbers, with few surviving by the end of 1 year. Sheep and goats are more susceptible, and fatal acute disease with ascites, abdominal hemorrhage, pallor, and icterus can occur in association with massive entry to the bile ducts at 6 to 10 weeks after infection by 1000 to 5000 or more migrating immature forms from the liver parenchyma. Subacute disease has been associated with a burden of more than 800 flukes acquired over time. Chronic clinical disease in sheep with submandibular edema, ascites, and emaciation has been associated with fluke burdens above 200.133
Necropsy Findings
Necropsy findings attributable to migrating young flukes are caused by tortuous tunnels of coagulative necrosis that organize and fibrose, ultimately leading to a diffusely fibrotic liver parenchyma, especially in the ventral lobe, which in severe cases undergoes marked atrophy. Fibrous tags may result from fibrinohemorrhagic deposits left by large numbers of flukes at liver penetration 3 to 4 days after infection. Inflammatory events associated with penetration of the bile ducts at 6 to 8 weeks after infection are especially pathogenic. Once in the bile ducts, flukes grow rapidly from 1 mm to 2.5 cm or more and induce a proliferative cholangiohepatitis. The spines and suckers of flukes erode and denude the bile duct epithelium, leading to a fibrosed, thickened duct wall that is irregularly dilated and stenotic and begins to calcify in cattle (but not sheep) at about 20 weeks after infection. The bile becomes darkly discolored and laden with regurgitated fluke ingesta, plasma proteins, and inflammatory cells. The extent of pathologic change is generally proportional to the fluke burdens of current or recurring previous infections.133
Diagnosis
Fecal sedimentation methods are the standard means of diagnosing liver flukes. A reusable commercial kit based on sieve-sedimentation of 2-g samples (Flukefinder, Visual Differences, Moscow, Idaho) reduces sample processing time by half and is suitable for use by practitioners for quantitative examination of an individual animal or of 10- to 15-animal herd composite samples. For herd or lot evaluations in cattle, egg counts of less than one egg per 2 g (EP2g) and 25% prevalence 2 to 4 months after the transmission season ends have a “low probability” of economic losses; 1 to 3 EP2g indicates “possible” economic loss; and 10 EP2g or more indicates “high probability” of heavy infections and economic loss. It is important to differentiate F. hepatica eggs from the eggs of Paramphistomum species, a nonpathogenic “rumen fluke” often found in the same herds in both the southern and the western enzootic areas. Flukecides used against F. hepatica are ineffective against rumen flukes, and egg counts thus persist after treatment. Paramphistomum eggs are gray (rather than amber), slightly smaller, and more pointed at the operculum end than F. hepatica. Paramphistomum egg counts can be used as a general indicator of the probable risk of F. hepatica in the absence of control in the southern states, because Paramphistomum flukes are known to be transmitted by the same snail vector in that region.130
Diagnostic enzyme-linked immunosorbent assays (ELISAs) have been developed to detect serum antibodies and coproantigen in the feces of infected animals134 but have not yet found wide use outside of research or diagnostic laboratories. Blood and serum clinicopathologic results reflect the anemia, hypoproteinemia, and mild eosinophilia caused by F. hepatica. Pathologic changes in the liver bile ducts and parenchyma are reflected by an elevation in serum liver enzymes (e.g., GGT, GLDH).
Treatment and Control
Prevention is the key to control in foundation herds and flocks because even low numbers of eggs shed can multiply asexually in snails and lead to significant infection rates during the following transmission season. A single routine fall or late fall-winter treatment after the end of the transmission season with a highly effective drug is recommended to remove adult flukes before winter stress and to prevent egg shedding and snail infection in the next season (see Figs. 33-1 and 33-2). Most of the pathogenic and economic effects of flukes in cattle are reported to occur within the first 5 to 6 months after the major exposure period and may be related to metabolic products associated with the rapid growth and heavy egg production phases of the life cycle. This, coupled with linear loss of heavy fluke burdens in cattle and the onset of effective immunity at 20 weeks after exposure, suggests the value of early flukecide treatment, within 2 to 3 months after the transmission season ends. In some herds an optional curative treatment in spring may be needed in very high-risk years or on high-risk premises, or as a second treatment to remove flukes acquired during late, extended transmission on irrigation pastures or wet coastal areas in the west.
Flukecidal drugs available in the United States are effective against mature flukes in bile ducts (albendazole, 10 mg/kg; closulon, 2 mg/kg). Closulon at 7 mg/kg has added efficacy against juvenile flukes over 6 weeks old in the bile ducts. The optimum time to treat should be based on the estimated susceptibility of mature fluke populations (>12 weeks old after end of transmission season) but early enough to remove flukes while they are most pathogenic (<6 months old). Triclabendazole, which is widely used in other countries, is more than 90% effective against migrating flukes over 2 weeks old and 99% effective against mature flukes and can be effectively given just after transmission ends. Worldwide, a number of other drugs are available, including oxyclozanide, niclofolan, bithionol, and hexochlorophene (80% to 99% of adult flukes), nitroxanil, closantel (>90% adult and 50% to 90% juvenile bile duct flukes), rafoxinide (>50% late migratory flukes, >90% juvenile and adult bile duct flukes), and diamphenathide (>90% migratory flukes, 50% to 80% bile duct flukes).135
The economic benefit of routine flukecide treatment of feedlot calves from enzootic areas in the United States has not been consistently demonstrated industry-wide because of the high variability of fluke burdens in animals from enzootic areas, or because lots are often of mixed origin. Fluke-related losses would be expected to be mainly absorbed by stocker operations, where lightweight calves are typically placed on small grain pastures for extended periods before feedlot, a common practice in the south-central states. The economic return of treatment at feedlot should be evaluated on a case-by-case basis because significant losses may occur if the lot originates from the same premises and the history suggests a high risk of recent, heavy infection rates, such as in favorable climate years or use of irrigated pastures in fluke areas. Fecal egg counts on 10 to 15 randomly selected animals may aid in herd evaluation. An example case of the explosiveness of the life cycle is the greatly reduced feedlot performance and 100% liver condemnations of a lot from a stocker operation in the Pacific Northwest. The animals originated from irrigated pastures where some habitats stayed wet year-round, and untreated calves from fluke areas were allowed to contaminate pastures with eggs, which translated to infection later in the grazing season and to the next stocker group rotated onto the premises.
OTHER FLUKES
Fascioloides magna, the large American liver fluke, may infect cattle and sheep that graze common areas with deer, the natural host. The life cycle is similar to that of Fasciola hepatica, but with a wider variety of lymnaeid snail hosts and a broader geographic distribution in the Gulf States, the Great Lakes area, and the Northwest. Cattle are abnormal, dead-end hosts that react with an intense encapsulation response, forming a closed cyst that does not allow escape of eggs and obviates diagnosis by fecal examination. The liver parenchyma and regional lymph nodes have a characteristic diffuse, black pigmentation. The major economic effect of F. magna in cattle is condemnation of livers and other organs, such as lungs, in which aberrant migration sometimes occurs. In sheep and goats, however, F. magna does not encyst and migrates uninterrupted. One or two F. magna flukes kill sheep before the flukes have time to mature, and this parasite limits sheep production in some areas. Albendazole given at high dosage is moderately effective against F. magna.
In tropical regions, F. hepatica is replaced by Fasciola gigantica, a similar species that is somewhat larger, has a longer prepatent period (10 to 12 weeks), and is of somewhat greater pathogenicity. Dicrocoelium dendriticum occurs worldwide but in North America is an unimportant species, mainly limited to areas of central New York, with smaller foci in Pennsylvania, New England, Quebec, and British Columbia. Albendazole (20 mg/kg) and high doses of thiabendazole (150 to 300 mg/kg) are effective treatments.127