Chapter 6 Neurological disorders in the lower extremity
Diabetic peripheral neuropathy
Intermediate dorsal cutaneous nerve
Lambert–Eaton myasthenic syndrome
Peripheral entrapment neuropathies
Neurological disorders in the lower extremity result from disease processes that involve sensory, motor and autonomic nervous systems. They can result from a hereditary or metabolic process, create progressive or static deformity, and be treatable or refractory. Injury at any level within the central or peripheral nervous system is capable of influencing lower extremity function.
THE SPINAL CORD PATHWAYS AND CLINICAL EXAMINATION
The spinal cord is made up of many afferent and efferent pathways. Afferent sensory fibres transmit impulses for striated muscles, joints, skin and subcutaneous tissues. Visceral afferent fibres transmit sensory impulses from smooth muscle, cardiac muscle and glands. These sensory fibres enter the spinal cord in two groups of bundles. The larger medial bundle is composed of medium and larger, more heavily myelinated fibres, whereas the lateral bundle is composed of finely myelinated and unmyelinated small fibres.
Ascending pathways
The dorsal or posterior columns are composed of the fasciculus gracilis and fasciculus cuneatus, representing the incoming medial bundles. They enter the cord just dorsomedial to the tip of the posterior grey columns. The fasciculus gracilis carries impulses from the lower extremity and is made up of afferent nerve fibres from the lower thoracic, lumbar and sacral dorsal roots. These nerve fibres are located in a more medial position within the dorsal columns. The fasciculus cuneatus carries impulses from the upper extremities. Conscious proprioception, light touch, vibratory and position sense are carried within this pathway, receiving input from Meissner corpuscles (light touch), Pacinian corpuscles (vibratory sense) and muscle fibres and Golgi tendon organs (position sense).
These ascending sensory fibres enter the spinal cord and ascend on the same side to the level of the brainstem where they decussate and continue on to the thalamus. Some fibres carrying light touch decussate after ascending only one or two vertebral levels beyond their entry. It is because of this small percentage of ipsilateral fibres that light touch sensation will be spared with a unilateral spinal cord lesion.
Clinical evaluation of vibratory sense is the most sensitive indicator of the integrity of the dorsal columns and may be evaluated in several ways (see Ch. 1). The use of a C (128 Hz) tuning fork is quick and easy, but the method lacks the ability to be reliably duplicated or quantified. The fork is placed on the various dermatomes, and the patient, with eyes closed, is asked to evaluate when vibration stops (Fig. 6.1). A biothesiometer, although not as convenient to use as a tuning fork, can quantify findings and is reproducible. The rate of vibration can be varied, with the instrument displaying a scale of 0–50. As the rate of vibration increases, the readings increase. A patient who does not feel vibration at a setting of 25 is considered to be at risk of neurotrophic injury. Decreased vibratory sense is associated with several disease processes, most notably diabetes mellitus, alcohol abuse, B12 vitamin deficiencies and tabes dorsalis. It decreases with the normal ageing process, and patients over the age of 50 years may have a measurable level of decrease in sensation distally. Care should be taken to separate this loss from that of true dorsal column pathology.
Position sense evaluates the integrity of conscious proprioception. It can be assessed simply by asking the patient, with eyes closed, to determine whether the hallux is dorsiflexed or plantar flexed. Light touch can be evaluated by passing a wisp of cotton over the dermatomes of the foot. The patient should not be ‘tickled’ as this represents evaluation of subliminal pain.
The spinothalamic tracts lie within the anterior lateral aspect of the spinal cord and are composed of A-δ and type C nerve fibres. Pain, temperature and crude touch sensations are carried within this pathway. Sensory fibres enter the spinal cord and ascend no more than one or two vertebral levels before decussating into the contralateral side of the cord. From here, they travel on to the thalamus and are relayed to the cerebral motor cortex.
Evaluation of pain sensation is performed by pricking the patient over the various dermatomes of the extremity with a moderately sharp needle. Any area of decreased sensation should be carefully mapped out and compared from distal to proximal and bilaterally. Temperature may be evaluated with an alcohol-saturated swab. The swab is squeezed to trickle a small amount over the foot. The patient is then asked to identify the cold sensation associated with the evaporation of the alcohol.
The spinocerebellar tract is an extrapyramidal pathway that carries input regarding unconscious proprioception and stereognosis. It remains ipsilateral, the ascending fibres remaining within the cord on the side of entry. Neurons enter the cerebellum via the superior and inferior peduncles.
A cerebellar lesion results in awkwardness and uncoordination of movement. In the lower extremity, the heel-to-shin test is the most reliable clinical indicator of pathology. The patient is placed in a supine position and asked to place the heel of one foot on the contralateral knee or shin and asked to draw the heel distally along the shin. This test should normally display smooth and even movement. Awkwardness or an inability to place the heel on the knee is suggestive of cerebellar disease. Romberg’s test may also be used. The patient is asked to stand with the feet close together and with eyes closed. In the presence of cerebellar pathology the patient will sway.
Descending pathways
The corticospinal tract is the primary motor pathway exiting the cerebral cortex and is responsible for voluntary motor control. It descends from the motor cortex to the brainstem, decussating at the junction of the brainstem and spinal cord and providing contralateral motor control. This tract synapses in the anterior motor horn of the spinal cord. Injuries to this tract result in an upper motor neuron lesion, which characteristically exhibits weakness, hyper reflexia and increased tone (see Table 6.1).
Table 6.1 Damage to upper and lower motor lesions
| Upper motor neuron lesion | Lower motor neuron lesion |
|---|---|
| Spastic paralysis | Flaccid paralysis |
| Hyper reflexia | Hyporeflexia |
| Babinski sign present | Babinski sign absent |
| No fasciculations or fibrillations | Fasciculations and fibrillations |
Evaluation of the voluntary motor system includes the observation of muscle power, bulk and tone, with note taken of any involuntary movements such as fasciculations or tremors, chorea or athetosis. Movement is assessed for smoothness and coordination. Deep tendon reflex responses are evaluated. The presence of an upper motor neuron lesion may be identified by the presence of a pathological reflex response (Fig. 6.2).
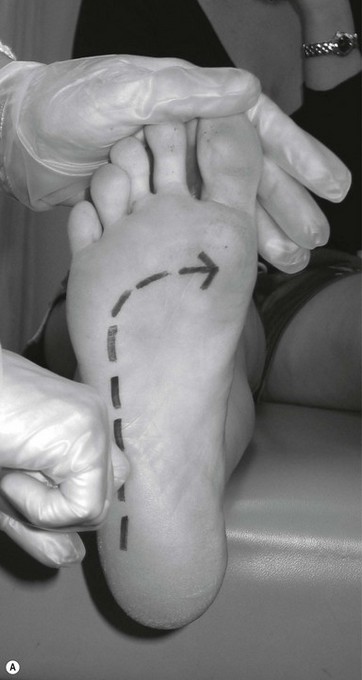
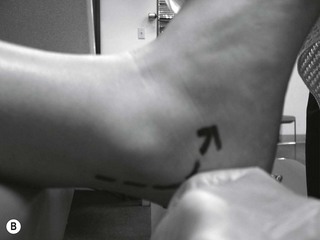
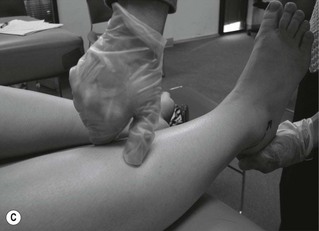
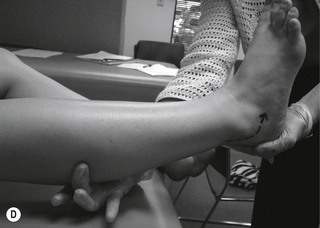
Figure 6.2 Pathological reflex responses. Technique for eliciting the Babinski response (A). Alternative methods include (B) the Chaddock reflex response elicited by stroking behind the fibular malleolus from proximal to distal; (C) the Oppenheim reflex response elicited by stroking the tibial crest using the fingers as callipers from proximal to distal; and (D) the Gordon reflex response, elicited by squeezing the posterior calf. All the reflex responses, when present, will demonstrate flexing and fanning of the lesser digits.
A Babinski sign is virtually pathognomonic for the presence of an upper motor neuron lesion when present beyond the age of 2 years. It is elicited by stroking the lateral aspect of the foot from proximal to distal and then onward across the ball of the foot. If present, the foot will exhibit extension of the great toe with flexion and fanning of the lesser digits. This is a slow response, which occurs over 1–2 seconds, and should not be mistaken for a withdrawal response as seen in a ‘ticklish’ patient. In the presence of a positive withdrawal response, alternative methods to produce a response include the Chaddock, Oppenheim and Gordon reflexes (Fig. 6.2).
Clonus is associated with increased muscle tone and hyper-reflexia, and reflects the presence of a corticospinal tract lesion. Ankle joint clonus is elicited by a quick, vigorous dorsiflexion of the foot with the knee held in flexion. Greater than three beats suggests nerve injury.
Altered deep tendon reflex responses are often associated with lower or upper motor neuron lesions. They also provide information about an intact reflex arc which represents the integration of five components: an intact afferent sensory nerve, a functional synapse at the spinal cord level, an intact motor nerve, an intact and functional neuromuscular junction, and a competent muscle. Abnormalities must be correlated with other aspects of the neurological examination to identify the level of pathology (Table 6.2).
Table 6.2 Percussion responses
| Tendon percussed | Spinal nerve roots | Reflex response |
|---|---|---|
| Biceps brachialis | C5–C6 | Flexion of forearm |
| Triceps brachialis | C7–C8 | Extension of forearm |
| Patellar | L3–L4 | Knee joint extension |
| Achilles | S1–S2 | Ankle joint plantar flexion |
The symmetry of skeletal muscles should be noted and the muscles evaluated for the presence of spasticity or weakness.
PERIPHERAL NERVE INJURY
The peripheral nerve comprises axons from the sensory, motor and autonomic nervous systems. These fibres are surrounded by different layers of connective tissue and packaged to form mixed nerves. Each individual axon is surrounded by the endoneurium, a loose connective tissue covering that serves at the blood–nerve barrier. It is made up of a thin inner layer that surrounds the Schwann cells and ‘dips’ into the nodes of Ranvier. Its outer layer does not dip. These axons are then bundled up into fascicles or funiculi, which are held together by the perineurium. The perineurium comprises many connective tissue layers, with perineural cells held together by tight junctions (Burnett & Zager 2004). These junctions provide the barrier against infectious agents. Groups of fascicles are then packaged together by the epineurium, which is made up of collagen and elastin fibres. It is connected loosely to the surrounding structures and provides the peripheral nerve with flexibility. Peripheral nerves undertake a long journey on their way to the lower extremity and are subject to injury at many levels along their course. The ability of a nerve to recover from injury is dependent on the extent to which the nerve is compromised. Two classification systems exist to help identify nerve injury and predict the probability of nerve repair.
Seddon’s (1943) classification is based on the pathophysiological changes that occur within the injured nerve. Neuropraxia represents a transient loss of conductivity. It frequently occurs with mild compression. There is no actual disruption of the neurofibrils. Full recovery is made within a few days to weeks. Axonotmesis represents axonal nerve damage within the structural framework of the peripheral nerve – the nerve sheath remains intact. Axons distal to the nerve injury undergo degeneration, with subsequent regeneration within the intact neural tubes. Recovery is generally at a rate of 1 mm/day and growth can be evaluated using Tinel’s sign. Neurotmesis represents disruption of the structural framework of the nerve, involving the nerve sheath and the axons contained within it. Regeneration of the nerve is not possible and may result in a ‘stump’ neuroma.
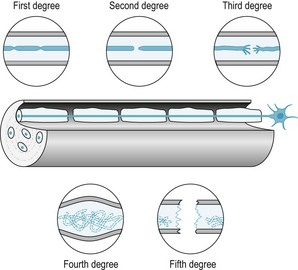
Sunderland (1990) developed a classification system based on an ascending order of the severity of the injury. First-degree nerve injury represents a conduction deficit within an intact axon. There may be some demyelination and the patient may experience an ‘irritable’ stage, with pain, paraesthesias and hyperaesthesias; however, recovery is complete. In second-degree nerve injury, the axon is severed within an intact endoneural sheath. The nerve will regenerate with no residual conduction anomalies. Third-degree nerve injury represents damage to both the axons and fascicles. Degeneration of axons occurs with compromise of the internal structure of the fascicles. Regeneration of the nerve does occur, but healing is unpredictable, with residual motor and/or sensory defects. Pain with this type of nerve injury may be persistent. Fourth-degree nerve injury occurs when the axon, endoneurium and perineurium are disrupted. The nerve trunk becomes a tangled mess of nerve parts. Regeneration is impossible and a neuroma in continuity develops. Complete transaction of the nerve results in fifth-degree injury. Recovery cannot occur without surgical repair (Fig. 6.3).
PERIPHERAL ENTRAPMENT NEUROPATHIES
The peripheral nerves, as they course to their destiny in the lower extremity, are subject to numerous types of injury. Aetiologies include gradual constriction of anatomical structures about the nerve, chronic compression of the nerve against an unyielding fibrous or skeletal structure and external trauma resulting in oedema or increased compartmental pressures. Clinically, these injuries present insidiously, with gradual development of sensory and motor changes and pain referred along the distribution of the involved nerve. Electromyography and nerve-conduction studies are helpful in identifying the location of the lesions and aid in the confirmation of the diagnosis.
Saphenous nerve entrapment occurs very rarely. Arising from the lumbar plexus, it is the largest and longest sensory branch of the femoral nerve. It supplies sensation to the skin over the medial aspect of the thigh, leg and foot. It courses with the femoral artery in the femoral triangle, and then descends and dives medially under the sartorius muscle. The terminal portion of the nerve courses inferiorly with the greater saphenous vein, ending at the level of the first metatarsal head. Entrapment can occur where the nerve exits the subsartorial canal just proximal to the knee joint and exhibits loss of cutaneous sensation along the distribution of the nerve. As there are no motor branches, there is no concomitant muscular weakness or diminished deep tendon reflex responses.
The posterior tibial nerve is the anterior division of the sciatic nerve arising from the sacral plexus. It is a mixed nerve providing motor, sensory and autonomic innervation to structures of the superficial and deep posterior compartments of the lower leg. The nerve courses inferiorly, passing underneath the flexor retinaculum, where it bifurcates into the medial and lateral plantar nerve. It also gives off a small medial calcaneal branch at this level. As the medial plantar nerve enters the vault of the foot, it courses with the medial plantar artery to supply motor innervation to only four plantar muscles: the abductor hallucis, flexor hallucis brevis, flexor digitorum brevis and first lumbricale. The lateral plantar nerve, after entering the vault of the foot, courses laterally, working its way over the heel and plantar lateral aspect of the foot. It provides motor innervation to all remaining intrinsic plantar muscles and sensory innervation to the lateral aspect of the fourth and all of the fifth toe (Fig. 6.4).
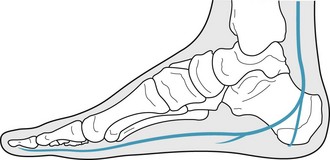
Figure 6.4 Branches of the posterior tibial nerve. The medial calcaneal, medial plantar and lateral plantar branches of the posterior tibial nerve are illustrated.
Proximal tarsal tunnel syndrome is a result of entrapment of the posterior tibial nerve or its branches, typically occurring under the flexor retinaculum. Many factors may contribute to the impingement of this nerve. Excessive subtalar joint pronation is the most common underlying aetiology, creating a narrowing of the tarsal canal. With weight bearing, the tarsal tunnel becomes narrowed. Tarsal tunnel syndrome occurs as the nerve becomes impinged or compressed between the osseous architecture of the foot and surrounding soft-tissue structures. Other aetiologies include entanglement of the nerve within the septal attachments of the flexor retinaculum, or compression from an enlarged abductor hallucis muscle belly, enlarged navicular tuberosity or os tibiales externum. Ischaemic injury or vascular insufficiency may compromise the blood flow to the posterior tibial nerve. Varicosities within the tarsal tunnel, when engorged, may produce tarsal tunnel syndrome. Tarsal tunnel syndrome has also been linked with hypothyroidism and diabetes mellitus.
Clinical presentation, regardless of the underlying aetiology, is a symptom complex of tingling, burning and numbness along the plantar aspect of the foot. These symptoms may be reproduced with percussion of the posterior tibial nerve at the level of the flexor retinaculum. The patient typically has a flexible flat foot with concomitant gastrocnemius–soleus equinus. Electrodiagnosis is helpful; however, studies may remain normal, despite the presence of entrapment. Diagnosis is frequently made on clinical findings alone.
Distal tarsal tunnel syndrome reflects the isolated entrapment of the medial or lateral plantar nerves. The medial plantar nerve becomes compressed between the navicular tuberosity and the abductor hallucis muscle belly, and may be referred to as ‘jogger’s foot’. The first branch of the lateral plantar nerve, referred to as Baxter’s nerve, may become entrapped as it courses laterally between the abductor hallucis and quadratus plantae muscles on its way to the abductor digiti quinti muscle belly. Baxter’s neuritis may present as infracalcaneal heel pain, a history of insidious onset and the persistence of symptoms at rest.
Irritation of a plantar intermetatarsal nerve may lead to the development of a neuroma, an enlargement of the nerve at the level of the metatarsal heads. Symptoms may be described as tingling, burning or numbness and radiate distally into the digits. High-heeled or tight-fitting footwear exacerbates the discomfort. Clinically, direct palpation of the nerve or compression of the metatarsals (Mulder’s sign) will reproduce the patient’s symptoms; the digits innervated by the intermetatarsal nerve may appear separated from each other (Sullivan’s sign). Any of the plantar intermetatarsal nerves may be involved, but the third is most common (Table 6.3).
Table 6.3 Results of nerve trunk irritation
| Involved nerve | Nomenclature |
|---|---|
| Medial plantar digital proper | Joplin’s neuroma |
| First plantar intermetatarsal nerve | Houser’s neuroma |
| Second plantar intermetatarsal nerve | Heuter’s neuroma |
| Third plantar intermetatarsal nerve | Morton’s neuroma |
| Fourth plantar intermetatarsal nerve | Islen’s neuroma |
Treatment for an intermetatarsal space neuroma includes patient education regarding the use of proper fitting footwear, mechanical, orthotic control to minimise subtalar joint pronation, and the use of metatarsal ‘cookie’ pads, which help to spread the metatarsal heads. Injections into the intermetatarsal space of anaesthetic and glucocorticoids or the use of a 4% alcohol sclerosing agent may be of benefit. Surgical resection of the nerve may be indicated when conservative management fails.
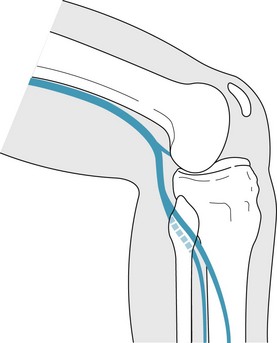
The common peroneal nerve branches laterally from the sciatic nerve trunk within the popliteal fossa. It becomes superficial at this level and winds inferiorly and laterally around the head of the fibula. It then divides into the superficial peroneal nerve within the lateral compartment of the calf and the deep peroneal nerve within the anterior compartment of the calf. The common peroneal nerve is very vulnerable to compression injuries (Fig. 6.5). Neuropraxia can occur simply from crossing one’s legs. Iatrogenic injury may occur secondary to positioning on the operating room table or the placement of a below-knee cast where the proximal edge impinges on the nerve with knee joint flexion. Blunt trauma to the area or traction on the nerve from an inversion ankle injury can cause pathology. Clinical findings include muscular weakness of the lateral and anterior compartments, creating a drop foot deformity. Nerve-conduction and electromyographic (EMG) studies help to confirm the diagnosis.
The superficial peroneal nerve, also known as the musculocutaneous nerve, courses inferiorly between the peroneal muscles. It becomes superficial, piercing through the fascia, approximately 10 cm superior to the tip of the lateral malleolus. It is at this point that it may become entrapped. Symptoms may be reproduced with dorsiflexion and eversion of the ankle joint against resistance and direct percussion of the nerve where it exits the fascia.
The superficial peroneal nerve then continues distally to bifurcate into the intermediate and medial dorsal cutaneous nerves (Fig. 6.6). The intermediate dorsal cutaneous nerve provides sensory innervation to the majority of the dorsal aspect of the foot. At the level of the ankle joint it rests approximately 1 cm anterior to the lateral malleolus, just medial to the sinus tarsi. It is extremely susceptible to damage, with inversion ankle injuries and with ankle arthroscopy, by virtue of its proximity to the classic anterolateral portal. The medial dorsal cutaneous nerve sends its branches to the medial aspect of the first ray and the second intermetatarsal space. It may be compressed by footwear, often at the level of the first metatarsal cuneiform joint. It is also vulnerable to laceration during surgical procedures addressing the base of the first metatarsal or the first cuneiform.
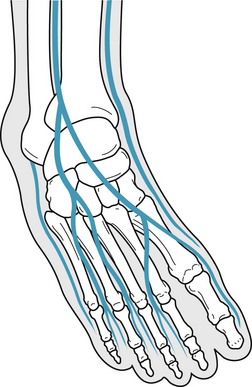
Figure 6.6 Bifurcation of the superficial peroneal nerve into the medial and intermediate dorsal cutaneous nerves.
The deep peroneal nerve, also known as the anterior tibial nerve, courses inferiorly within the anterior muscular compartment. At the level of the ankle joint the nerve anatomically flattens and rests between the extensor hallucis longus and extensor digitorum longus tendons, and it is at this location that the nerve becomes damaged. Entrapment of the deep peroneal nerve is known as anterior tarsal syndrome. Aetiological factors include exostoses of the tarsal bones and tight or ill-fitting shoes, particularly ski boots and high-topped shoes, which demonstrate a maximum point of contact at the dorsal talonavicular joint. Patients will present with a complaint of paraesthesias over the dorsal aspect of the foot and numbness within the first intermetatarsal space. Percussion of the nerve at the level of the ankle joint and provocative testing of ankle joint plantar flexion with concomitant dorsiflexion of the digits will reproduce symptoms. There may be motor weakness of the extensor hallucis longus. Nerve-conduction and EMG studies may confirm the diagnosis.
The sural nerve arises from branches of both the tibial and common peroneal nerves and originates inferior to the popliteal fossa. It courses inferiorly between the heads of the gastrocnemius muscle bellies to rest posterior to the fibular malleolus, continuing distally along the lateral aspect of the foot. Injury commonly occurs at the level of the ankle joint, iatrogenically with a surgeon’s ‘slip of the hand’ or from fibrosis secondary to an inversion ankle injury.
HEREDITARY MOTOR AND SENSORY NEUROPATHIES
The hereditary motor and sensory neuropathies represent a slowly progressive degenerative process of the peripheral motor nerves and the spinocerebellar tracts. This genetically transmitted disorder is divided into two major types: the demyelinating form and the neuronal form. The demyelinating form is characterised by degeneration of the posterior columns, loss of anterior horn cells and degeneration of the spinocerebellar tracts. This form demonstrates slowed nerve-conduction velocities and hypertrophic nerve changes. The neuronal form is characterised by axonal degeneration of the peripheral nerve. The prevalence of these disorders is approximately 1 in 2500 individuals.
Charcot–Marie–Tooth disease type I is also referred to as peroneal muscular atrophy. It is an autosomal-dominant, inherited disorder that results from a mutation in the gene coding for peripheral myelin protein-22 on chromosome 17. Unstable myelin is synthesised and breaks down, resulting in segmental demyelinisation of the peripheral nerves and conduction deficits. To attempt repair, Schwann cells proliferate and lay down defective myelin, which subsequently breaks down, and a vicious cycle develops, resulting in hypertrophy of the peripheral nerves resembling an ‘onion’ bulb.
The onset of symptoms usually occurs between the ages of 5 and 15 years with a presenting complaint of difficulty in walking, muscle cramps and paraesthesias in the legs. Signs of classic lower motor neuron disease develop initially in the peroneal and intrinsic muscle groups of the calf and foot. As the motor weakness progresses, the normal agonist–antagonist relationship between the peroneus longus and brevis and the anterior and posterior tibialis muscles is lost, resulting in the development of a high-arched or cavus-appearing foot. Clawed toe deformities develop from the inability of the intrinsic muscles to stabilise the digits against ground reactive force (Figs 6.7, 6.8). Flaccid paralysis develops, frequently with fascicular twitching in the wasting muscles. Deep tendon reflex responses are decreased or completely absent. Lower extremities are observed as slender legs with plump thighs and are often described as ‘an inverted champagne bottle’ or ‘ostrich legs’.
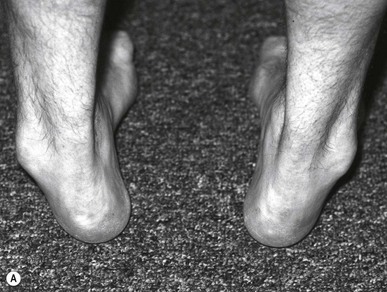
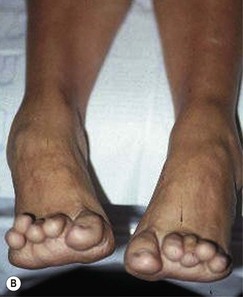
Diagnosis is suggested by the presence of weakness and atrophy of the peroneal muscle group along with a positive family history. Nerve-conduction studies reveal diffuse and uniform slowing of nerve conduction in both sensory and motor nerves. Nerve biopsy is seldom needed but reveals the typical onion-bulb formation comprising Schwann cells and their processes. There is no effective medical management of this demyelinating process. Early in the disease process, passive stretching and strengthening exercises and splinting of unopposed muscle groups may be somewhat helpful but cannot deter the progression of the disease. Advanced cases require surgical intervention to achieve joint immobilisation.
Roussy–Levy syndrome is also known as hereditary areflexic dystasia (Auer-Grumbach et al 1998). Transmitted as an autosomal-dominant trait, symptoms generally develop in early childhood. It is characterised by the presence of an essential tremor and is otherwise very similar clinically to Charcot–Marie–Tooth disease. Patients present with a sensory ataxia or poor judgement of movement, distal muscular atrophy of the peroneal muscles and, frequently, a kyphoscoliosis.
Dejerine–Sottas disease is also known as Charcot–Marie–Tooth disease type III or as hypertrophic polyneuritis. It is transmitted via autosomal-recessive inheritance with onset of symptoms occurring in early infancy. The histological picture and clinical course is much more severe than Charcot–Marie–Tooth type I, resulting in delayed motor milestones, poor walking and an inability to run. Additional findings include sensory conduction deficits with paraesthesias and lightning-like pains in the lower extremities. There is slowly progressive distal muscular weakness with peroneal atrophy, resulting in a cavus foot deformity.
Refsum’s disease is also known as Charcot–Marie–Tooth disease type IV. This extremely rare, autosomal-recessive disorder presents clinically as a triad of retinitis pigmentosa, peripheral neuropathy and cerebellar ataxia. It is the result of a gene mutation that prevents the enzymatic degradation of phytanic acid, an exogenous fatty acid found in chlorophyll, resulting in elevated serum and tissue levels that are associated with neurotoxicity. Onset generally occurs within the first years of life, with lower extremity manifestations of progressive distal neuropathy leading to cavus foot deformity, foot drop and cerebellar ataxia. Dietary control and plasmaphoresis help attenuate the progression of the disorder.
SPINAL RADICULOPATHIES
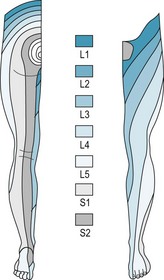
Radiculopathy is defined as pathology pertaining to the spinal nerve roots and is secondary to irritation, inflammation or trauma. The fourth and fifth lumbar and the first and second sacral spinal cord nerve roots provide the majority of the innervation to the lower extremity. These nerves exit the spinal column through the vertebral foramina, where they are subject to impingement by bony spurs, tumours and herniated vertebral discs. Radicular pain may present as focal irritation of a nerve root, or ‘local’, as visceral pain secondary to nerve root irritation, as ‘referred’ pain, which follows the distribution of the nerve involved, or as ‘radicular’ pain.
Radicular pain is a common lower extremity complaint frequently involving the fifth lumbar or first sacral nerve distribution – the junction of the flexible spine upon the fixed sacrum. It must be differentiated from peripheral nerve pathology. Symptoms associated with nerve root irritation include a complaint of ‘pseudoclaudication’. This discomfort is often unilateral, radiating from the buttock to the thigh or leg, exacerbated by standing or walking and described by the patient as pain, numbness or weakness. Sensory and motor loss occurs along the distribution of the nerve root. Deep tendon reflex responses are hyporeflexic or absent (Table 6.4).
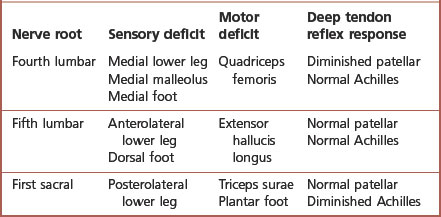
Elevation of the leg with the hips and knees in full extension should reproduce symptoms; the distribution of these symptoms should identify the involved nerve root. The patient may walk with small steps, keeping knees semi-flexed to prevent stretching the nerve root. The spine should be evaluated for abnormal lumbar lordosis, thoracic kyphosis or scoliosis. Evaluation should include imaging studies to determine the nature of the nerve irritation. Treatment is directed toward the underlying aetiological factor and minimising the inflammation, both pharmacologically and biomechanically. The use of functional orthoses, with or without a heel lift, may benefit patients with a leg-length discrepancy.
CHARACTERISTICS OF CEREBELLAR LESIONS
In the lower extremity, the cerebellum controls unconscious posture, balance and the coordination of voluntary movements. Anatomically, it is divided into three lobes. The paleocerebellar, or anterior, lobe is responsible for unconscious posture, balance and proprioception. The flocculonodular, or middle, lobe is vestibular, controlling unconscious equilibrium. The neocerebellar, or posterior, lobe receives input from the cerebral motor cortex and coordinates voluntary skilled movements. Afferent input to the cerebellum is received from the cerebral cortex, vestibular tracts and the spinocerebellar tract, an ipsilateral spinal cord pathway relaying input from the lower extremities. Efferent output is then initiated from the cerebellar nuclei, integrated with input from the red nucleus and basal ganglia and relayed to the motor cortex. Any disease process, trauma or physiological insult to the elements regulating cerebellar activity may result in impairment of the coordination of volitional movements.
Clinically, cerebellar pathology in the lower extremity is recognised by classic, uncoordinated ataxic gait. Dysynergy is a component of cerebellar disease. Voluntary movements cannot be performed smoothly due to the lack of normal coordination between muscular agonists, antagonists and synergists. This is referred to as decomposition of movement. It is recognised by the utilisation of accessory muscles, a wide arc of motion when the patient attempts to reach a goal, easily tired muscles, or asthenia and hyporeflexia.
1Other elements of cerebellar ataxic gait include abnormal timing and coupling of movements known as dysrhythmia concomitant with the inability to gauge distance, speed, strength and velocity of movement known as dysmetria. Patients have a tendency to ‘overshoot’ a desired point or to stop before it is reached. Excessive rebound and delay in the initiation or cessation of movement results from faulty postural fixation of the limbs. An intention tremor exists, with initiation of voluntary movement recognised by an oscillating frequency that varies and often intensifies as the goal is neared. Finally, speech is dysarthric, sounding slurred, jerky or explosive in nature (i.e. syllabic).
Cerebellar gait is recognised as a wide-based gait with a slow, jerky and irregular cadence. Stride length and foot placement vary from step to step and the patient may frequently lose his or her balance. The foot will contact the ground in two phases – heel strike followed by toe contact. This gait pattern has a characteristic ‘double tap’ sound. The patient will undergo numerous postural ‘adjustments’ and may tend to favour one side if the cerebellar lesion is unilateral.
Treatment for cerebellar ataxia is generally palliative. Measures should be taken to increase stability in ambulation. The use of a quad cane or forearm crutch, functional or accommodative orthoses and physical therapy to augment muscular coordination and strength can be very effective.
Friedreich’s ataxia, also known as hereditary spinocerebellar ataxia, is the most common of the inherited ataxias. It is transmitted by an autosomal-recessive gene defect carried on the pericentric region of chromosome 9. This gene codes for the protein frataxin, which is normally present within the mitochondria of the nervous system, heart and pancreas and required for oxidative phosphorylation and iron homeostasis. The cerebellum, spinocerebellar tract, corticospinal tract and posterior columns are all involved in the evolution of this disease process, which is characterised by the progressive loss of voluntary muscle coordination, obstructive cardiac hypertrophy and the possibility of diabetes mellitus.
Symptoms associated with Friedreich’s ataxia generally begin between the ages of 5 and 15 years. Early clinical findings include an early loss of vibratory and position sense, loss of the Achilles and patellar deep tendon reflex responses, and a positive Babinski sign. Patients may have delayed motor milestones and a tendency to stagger and fall. Voluntary coordination progressively deteriorates, resulting in classic ataxic gait with a wide stance, multiple postural corrections and a steppage pattern. Of patients with Friedreich’s ataxia, 80–90% will develop a slowly progressive thoracic scoliosis, 50% will develop cardiac disease and 10% will develop diabetes mellitus.
Lower extremity findings reveal distal muscle weakness that is greatest in the peroneal muscle group. This results in the development of a cavus foot type with flexion contracture of the digits. Gait is unsteady, with a wide base. Most patients become non-ambulatory approximately 15 years after the onset of the disease process. Death usually occurs due to cardiac failure. There is no known cure for this disease and medical management is generally palliative.
CHARACTERISTICS OF BASAL GANGLIA LESIONS
The basal ganglia are a collection of nuclei located deep to the white matter within the brain. Upon receiving input from the premotor cortex regarding a planned movement, efferent output from the basal ganglia will then control this movement. These ganglia, therefore, control intentional movement. Lesions will result in the presence of awkward, unintentional or involuntary movements.
The ganglia that play a role in movement include the caudate, putamen and substantia nigra. The caudate and putamen (which collectively are termed the striatum) receive the majority of their input from the cerebral cortex, sending it on to the globus pallidus. The primary neurotransmitters of the striatum are acetylcholine and γ-aminobutyric acid (GABA). The globus pallidus then ‘outputs’ this information to the thalamus, exerting an essentially inhibitory effect on the thalamus. Degeneration of the caudate and putamen results in the development of choreic, dance-like movements or athetotic, snake-like movements. The globus pallidus also receives input from the subthalamic nucleus. Injury to this nucleus will result in hemiballismus or flailing movements of an arm and leg.
The substantia nigra, which produces dopamine, an essential neurotransmitter for movement, communicates directly with the caudate and putamen. Deterioration of this nucleus results in the symptoms of parkinsonism.
The effect of the basal ganglia on the thalamus is inhibitory. For example, if one needs to sit still, the basal ganglia will inhibit movements other than those associated with postural reflexes. On the other hand, when one needs to move, the basal ganglia will inhibit unnecessary postural reflexes. Disorders of the basal ganglia are classified as hypokinetic or hyperkinetic dyskinesias.
Hyperkinetic dyskinesias
Huntington’s chorea
This disorder was first described by George Huntington in 1872. ‘Chorea’ is literally taken from the Greek word ‘to dance’. It is a chronic, progressive, degenerative, central nervous system disease occurring in adulthood. It is transmitted by autosomal-dominant inheritance and is characterised by choreic involuntary movement, progressive dementia, and psychiatric and behavioural disturbances. Nervous system pathology is generally confined to the brain, with advanced cases demonstrating extensive atrophy of the cerebral cortex, basal ganglia and cerebellum. Diagnosis is by the appropriate clinical presentation and the demonstration of caudate atrophy on magnetic resonance imaging.
Huntington’s chorea is prevalent throughout the world, occurring in all ethnic and racial groups with an incidence of 5–10 per 100 000. The disease does not typically manifest until after the age of 40 years, although there is a juvenile variant that has an onset at under 25 years of age. Symptoms result from a selective loss of nuclei in the caudate and putamen. Gradual development of choreic movements and mental deterioration are the predominant clinical manifestations, which progressively worsen over 15–20 years and result in death.
There is no specific pharmacological treatment that will attenuate the disease process, so management is directed towards minimising symptomatology. Haloperidol may be effective in minimising the irregular movements in the extremities or facial muscles. Patients may also benefit from the use of antidepressant medications.
Sydenham’s chorea or St Vitus’ dance
Sydenham’s chorea is the most important form of chorea in childhood. It is one of the five major diagnostic criteria for rheumatic fever as described by Jones. It may occur up to 6 months following tonsillitis or pharyngitis caused by a group A β-haemolytic streptococcal infection. It is thought to be the result of autoantibodies that target certain areas of the basal ganglia.
Sydenham’s chorea is more common in girls, with a peak incidence at 8 years of age. The onset of symptoms is generally insidious, and may first be recognised by the presence of facial grimacing slowly progressing to involuntary flinging movements and sudden jerks, which may be more prevalent in the upper extremities. Movements are exacerbated when the patient attempts to control them and disappear while sleeping. Diagnosis is predominantly clinical and from past medical history. Serological testing may reveal elevated acute-phase reactants and the presence of streptococcal antibody titres. Cerebrospinal fluid will demonstrate increased serum glucose and leukocytosis.
Treatment must initially be directed toward the underlying aetiology (i.e. the streptococcal infection). Penicillin is still considered the drug of choice, with the use of erythromycin in penicillin-allergic individuals, and must be administered for a minimum of 10 days. Management of the chorea is palliative and usually resolves in 3–6 weeks. In rare instances, when the symptoms are severe or become chronic, valproic acid, carbamazepine or haloperidol may be effective.
Hypokinetic dyskinesias
Parkinsonism
Parkinsonism was first described by James Parkinson in 1817 as the ‘shaking palsy’. It is a distinctive symptom complex characterised by tremor, muscular rigidity, bradykinesia and characteristic alterations of posture and attitude of the extremities, and is sometimes referred to as paralysis agitans. The aetiology is unknown; theories include exposure to unidentified environmental toxins, generation of free radicals and, perhaps, very rarely, inheritance.
Prevalence is roughly 1% of the population in the USA, with a mean age of onset of 55–60 years. Approximately 50 000 new cases are diagnosed each year and this does not appear to be changing with time. The course of the disease is slow, with gradual progression. With appropriate treatment life expectancy will approach the norm.
The major pathological feature is the loss of dopaminergic neurons in the substantia nigra. Via direct and indirect pathways these neurons modulate thalamic input to the cerebral motor cortex. Up to 80% of the neurons are lost before the reduced excitation of the cerebral cortex demonstrates clinical signs of parkinsonism.
Clinical features include the insidious onset of tremor, rigidity, bradykinesia and disturbances in gait and posture. A resting, ‘pill-rolling’ tremor of 4–6 Hz is a common presenting sign and is the initial complaint in 70–75% of cases. It usually begins unilaterally, affecting one hand or, less often, one foot. Rigidity is demonstrated by the stiffness and slowness of movement. A cog-wheeling phenomenon may be superimposed on the rigidity and is more prominent in the extremities. As the rigidity progresses, the patient acquires a stooped posture, with the head tilted forward and the arms flexed at the wrists and elbows, and walks with a shuffling gait. Bradykinesia is observed as slowness, with muscular fatigue on voluntary movement, and may progress to akinesia, which is recognised as a lack or poverty of movement. Loss of facial expression is noted early in the disease, leading to a monotonous, stuttering, ‘deliberate’ speech pattern. As the disease process continues, voluntary muscle fatigue leads ultimately to a ‘masked facies’ and disabling postural difficulties.
Patients with advanced disease present with a classic shuffling gait. Due to the deterioration of postural reflexes patients develop a forward or backward lean. To correct a forward lean, festination occurs. This is a gait pattern characterised by sudden, short, shuffling steps that become progressively shorter and faster. To correct a backward lean, retropulsion occurs. This is characterised by rapid backward steps.
Classic treatment for parkinsonism is pharmacological intervention directed at providing dopamine with levodopa and carbidopa, or the use of dopamine agonists such as pergolide that stimulate the dopamine receptors. Other medications may be used to address individual movement deficits. Surgical management has also become a treatment option, with procedures directed toward destruction of specific sites within the basal ganglia, stimulation of the thalamus and even fetal cell transplants.
Lower extremity management is essentially palliative. The patient should be encouraged to participate in a regular, moderate exercise programme. Physical therapy modalities that address balance compensation may also help improve ambulation.
CEREBRAL PALSY
Cerebral palsy is a chronic, non-progressive disorder affecting motor dysfunction and, in 60% of patients, mental status. Generally becoming evident in the second year of life, underlying aetiologies are varied and the specific insult is frequently unidentifiable. Prenatal injury is often idiopathic. It may be secondary to hereditary disorders, gestational diabetes, erythroblastosis fetalis, toxaemia and even the presence of more than one fetus in the womb. Natal injury is the most frequent cause, and this encompasses any insult that occurs during birth or within the first week of life. Prematurity, anoxia and respiratory distress are common factors. Postnatal injury includes any insult occurring after the first week of life and up to the second birthday. Aetiologies include infection, poisoning, seizures and trauma. Some form of cerebral palsy occurs in up to 7.5 of every 1000 live births.
Cerebral palsy may involve the pyramidal tracts, extrapyramidal tracts or a mixture of both. Pyramidal tract or spastic cerebral palsy is the most common type, accounting for up to 70% of cases. It represents a lesion within the cerebral cortex demonstrating the characteristics of an upper motor neuron lesion, and may be further categorised by areas of the body affected (Table 6.5).
| Type of cerebral palsy | Area of injury | Clinical involvement |
|---|---|---|
| Spastic: 50–70% | Motor cortex lesion | |
| Monoplegia | One limb involved | Spastic movement of arm or leg |
| Diplegia/paraplegia | Two limbs involved | Spastic movements of arms or legs |
| Quadriplegia | Four limbs involved | Spastic movement of all four limbs |
| Hemiplegia | One side affected | Ipsilateral involvement of arm and leg |
| Double hemiplegia | Both sides affected | Spastic movement is not symmetrical |
| Athetoid: 10–20% | Basal ganglia lesion | Uncontrolled, uncoordinated movements |
| Ataxic: 5–10% | Cerebellar lesion | Incoordination of movement or balance |
| Mixed: 10% | Combination of lesions | Spastic/athetoid most common |
Early clinical signs of spastic cerebral palsy include a change in muscle tone, asymmetry of movement where there is greater involvement of one side or limb, and delayed motor milestones such as sitting up, crawling and walking. The spastic limbs are usually thinner and smaller, presenting with hyper reflexia and clonus. The Babinski sign is usually present in postnatal cases. In the lower extremity, spasticity of the hip flexors and adductors results in internal hip rotation; involvement of the knee flexors and plantar extensors results in toe walking and a cavo varus foot deformity. Arms are held adducted at the shoulders and flexed at the elbows and wrists. Patients demonstrate what is classically described as a ‘scissored’ gait. Treatment is directed towards physical and occupational therapy. Bracing is of some benefit in preventing adaptive contracture. The use of the exotoxin derived from Clostridium botulinum (Botox) may be beneficial. It exerts its effect at the neuromuscular junction, preventing the release of acetylcholine. The result is temporary relaxation of the spastic muscles, creating a transient flaccid paralysis. Within a few months these muscles develop new acetylcholine receptors and paralytic effects are reversed. Surgical intervention may be used to release muscular contractures and restore some normal agonist–antagonist balance between involved muscle groups.
Athetotic cerebral palsy is the result of a basal ganglion lesion. It is characterised by slow, uncontrolled, writhing movements, which may be continuous or intermittent. There is a slow, serpentine movement of the arms and legs interposed on postures of flexion with supination and extension with pronation. These limb movements are accompanied by rotatory movements of the neck. Dyskinesia subsides while sleeping. An increased incidence of athetotic cerebral palsy has been linked to postnatal kernicterus.
Ataxic cerebral palsy occurs secondary to cerebellar dysfunction. It is characterised by an inability to control the rate, range, direction and force of fine motor movements. Typically, there is a balance disturbance that is compensated for by a wide base of gait. There is an intention tremor with the initiation of intentional movement.
Mixed cerebral palsy represents more than one type of motor lesion. Most commonly it is a combination of spastic and athetotic movements. Treatment for the extrapyramidal involvement in cerebral palsy is directed towards the underlying movement disorder.
AUTONOMIC NERVOUS SYSTEM
The autonomic nervous system is primarily responsible for the regulation of peripheral organ systems. Efferent nerves affect the rate and strength of cardiac, smooth and vascular muscle contraction, endocrine and exocrine secretions of glands, as well as visual accommodation and papillary size. It receives afferent input regarding respiratory and vasomotor reflexes from these various systems. It is under the master control of the hypothalamus, which coordinates the automatic reflexes dictating homeostasis.
The system comprises two divisions:, the sympathetic and parasympathetic nervous systems. Both systems are made up of preganglionic axons that synapse with postganglionic fibres innervating the effected organ system. Preganglionic motor neurons of the sympathetic system arise from the intermediolateral columns of the spinal cord between T1 and L2 (the thoracolumbar region). The fibres then pass into the paravertebral sympathetic ganglia, which are organised into two chains running parallel to and on either side of the spinal cord, where they travel up or down the chain for considerable distances before synapsing. This first synapse is cholinergic. Postganglionic fibres continue on to innervate peripheral organs. The second synapse is adrenergic, with the exception of the sweat glands.
The parasympathetic motor nerves originate in the medulla oblongata. The cell bodies of this system occupy a position in the intermediolateral columns of the spinal cord at levels S2 through S4 and cranial nerves three (pupil and ciliary body constriction), seven (tearing and salivation), nine (salivation) and ten (vagus). This is known as the ‘craniocaudal’ region. The synapses of the parasympathetic nervous system are typically close to or within the viscera. The majority of both synapses in this system are cholinergic.
Sympathetic nervous system dysfunction in the lower extremity
Clinical signs of sympathetic nervous system dysfunction in the lower extremity are recognised by visible changes in colour, temperature and hydration of the extremity. Overactivity results in vasoconstriction and hyperhidrosis, resulting in a cyanotic-appearing, cool, clammy extremity. Underactivity or a lack of sympathetic regulation causes vasodilatation and anhidrosis, identified by the presence of bounding pulses, erythema and significant xerosis. With prolonged dysfunction trophic changes persist, muscle bulk is lost and demineralisation of bone may occur.
Hyperhidrosis represents increased activity of the sympathetic nervous system. The hands are generally affected to a greater extent than the feet. Patients with hyperhidrosis are more prone towards tinea, cutaneous diphtheroid infections and bromhidrosis. Treatment with astringents such as glutaraldehyde and antiperspirant creams with aluminium salts is generally effective. Anhidrosis results from complete loss of sympathetic input. Spinal cord transaction above the level of T7 will produce total loss of sweating in both the upper and lower extremities.
Raynaud’s disease represents vasomotor instability mediated by the sympathetic nervous system. It occurs most frequently in women aged between 18 and 40 years. When it is associated with an identifiable underlying aetiology, as with many collagen vascular diseases, it is referred to as Raynaud’s phenomenon. Clinically, Raynaud’s disease is characterised by a ‘triphasic’ colour response of pallor, cyanosis and rubor. Pallor occurs initially as a result of constriction of the small cutaneous vessels. Capillaries and venules then dilate, resulting in sluggish slow blood flow resulting in cyanosis. Reflex hyperaemia then occurs, a process that may be quite painful and results in rubor. Patients may complain of pain, paraesthesias and stiffness of the fingers and toes. Symptoms may be exacerbated by cold environs.
Diagnosis is made on the basis of clinical presentation and elimination of any underlying disorder. A complete blood count (CBC) including a differential rheumatoid profile and erythrocyte sedimentation rate may be quite useful. It will identify the presence of a collagen vascular disease and help to rule out a haematological disorder, and lead or arsenic exposure. Patients should be educated regarding potential triggering factors and taught to minimise anxiety and stress that may cause sympathetic nervous system overactivity. Treatment of the vasomotor instability is palliative, directed toward ‘insulating’ the digit. The use of several pairs of socks or the application of petroleum jelly may prevent heat loss. Nitroglycerine pastes applied to the base of the digits and drug therapy to decrease peripheral vasoconstriction can be effective.
Acrocyanosis occurs secondary to an overactive sympathetic nervous system, although the exact aetiology is unknown. It is characterised by patchy cyanosis at the distal portions of the extremities. It is differentiated from Raynaud’s disease by the persistence of cyanosis. It has a greater incidence in women during the winter months. Symptoms include swelling, decreased touch, heat, cold and pain perception, paraesthesias and hyperhidrosis. It is managed by patient education and measures directed at minimising cold exposure.
Familial dysautonomia is another disorder of autonomic nervous system dysfunction. It is also known as Riley–Day syndrome and is characterised by a complete indifference to pain. It is inherited as an autosomal-recessive trait and occurs in individuals of Mediterranean descent. The most distinctive clinical feature is the absence of tears with emotional crying. A weak suckling response and misdirected swallows lead to feeding difficulties and an increased risk of aspiration pneumonia. Patients may experience a dysautonomic crisis with psychological or physiological stress, experiencing vomiting, tachycardia, hypertension and emotional lability. Classic findings include hyperhidrosis, orthostatic hypotension and an incomplete distribution of indifference to all forms of pain. Mentality is often dulled, and deep tendon reflexes are hyporeflexic or absent. A cavus foot type and the presence of trophic ulcers are frequent findings in the lower extremity. Treatment is palliative. Accommodative footwear should be used to off-load osseous prominences and maintain a normal distribution of the weight-bearing load throughout the gait cycle. Good wound care is essential.
Reflex sympathetic dystrophy was renamed ‘complex regional pain syndrome type I’ in 1993 by the International Association for the Study of Pain. It is believed to occur as a secondary vasomotor instability that is in some way mediated by the sympathetic nervous system. Frequently associated with a minor form of trauma, such as sprains, soft-tissue wounds, fractures of varied severity, surgical procedures and infection, there is no obvious nerve lesion. Causalgia was renamed ‘complex regional pain syndrome type II’ and is characterised by the same clinical presentation; however, there is an identifiable nerve injury. Sympathetically maintained pain is an additional classification of this type of injury, and is characterised as pain restricted to the distribution of a single nerve. The key finding in all these syndromes is pain out of proportion to the severity of the injury.
The underlying aetiology of complex regional pain syndromes is uncertain and several theories exist to proffer explanation. Doupe in 1944 described a ‘short circuit’ that occurs between the sympathetic nervous system and the nociceptive efferent pathways that occurs after injury. He postulated that, following an injury, the insulation of the nerve is damaged, resulting in disruption of normal nerve impulses. Nerve impulses are then misdirected, resulting in cross-stimulation of afferent pain fibres. Livingston, in 1943, hypothesised that the internuncial neurons within the spinal cord became overstimulated by the afferent input of an irritative nerve lesion. These internuncial neurons, that act as ‘liaisons’ between the afferent and efferent stimulus responsible for mediating reflex responses, then in turn influence skeletal and smooth muscle efferent motor nerves in the spinal cord. Hence, a vicious cycle is established, resulting in the observed symptoms of vasomotor instability, hyperalgesia and impairment of motor function.
Complex regional pain syndrome type I is divided into three stages. The acute stage usually lasts 2–3 months. It is recognised by the presence of severe burning pain along with warmth, swelling and joint stiffness distributed not to a single dermatome or myotome but to an entire region. Symptoms may be exacerbated by limb dependency, emotional stress or physical contact (Fig. 6.9). Radiographs may demonstrate early demineralisation of bone. This is then followed by the dystrophic phase, which includes trophic changes of the skin and more advanced demineralisation of bone.
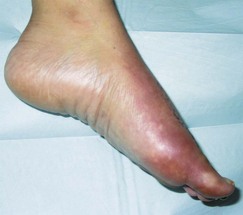
Figure 6.9 Complex regional pain syndrome. Mottled cyanosis with oedema in a 31-year-old female 3 weeks following surgical correction of a bunion deformity. Symptoms were brought on abruptly by placing the foot in a dependent position.
The second or dystrophic stage may last for several months. The warm oedematous phase gives way to a firm, cyanotic, cool extremity. Pain remains the predominant symptom, and this becomes constant, unrelenting and exacerbated by any stimulus. Radiographs demonstrate diffuse osteoporosis, historically heralding the onset of Sudek’s atrophy. Flexion contractures of the digits may begin to develop. The disease process is still reversible at this stage.
This process, without successful treatment, progresses into the atrophic phase, which is irreversible. The pain may diminish, be absent or become intractable. Skin and subcutaneous tissues become atrophic and flexion contractures of the foot become irreversible. Radiographically, the osteoporosis advances and the bone has been characterised as having a ‘ground-glass appearance’.
Incidence is distributed among individuals of all ages, although it is quite uncommon in children under the age of 10 years. Women are up to three times more likely to develop complex regional pain syndrome. Many patients seem to fit a psychological profile – they are overanxious, inquisitive, type A personalities.
Early recognition and treatment of complex regional pain syndrome is critical if there is to be any hope of successful treatment. Diagnosis is generally based on clinical findings along with radiographic changes in the extremity. A triphasic technetium bone scan will demonstrate intense, focal periarticular uptake in the delayed imaging phase. Thermography may also provide diagnostic information. Symptomatic relief in response to a sympathetic nerve block is indicative of sympathetically maintained pain.
Treatment must include aggressive physical therapy in the early phases of the disease process directed towards preventing abnormal joint contractures and muscle wasting. It can include local application of heat, massage, and range of motion and occupational therapy. The extremity should never be immobilised! Sympathetic nerve blocks can be administered on a biweekly basis, directed towards interfering with the aberrant cycling of nerve impulses. Many different classes of medications have been used in the treatment of complex regional pain syndrome; however, none have produced consistent results. The use of non-steroidal anti-inflammatory drugs may be helpful for pain management. The use of narcotic analgesics should be discouraged. Tricyclic antidepressants and anticonvulsants may help with the depression and nerve-mediated pain. Severe cases may require regional anaesthetic blocks or surgical sympathectomy.
DIABETIC PERIPHERAL NEUROPATHY
Peripheral neuropathy is an all too frequent complication of diabetes, affecting the sensory, motor and autonomic neurons of the peripheral nervous system and the organs that these neurons innervate. Hyperglycaemia and the duration of the disease appear to be the primary factors in its development.
Distal symmetrical polyneuropathy is the most common type of neuropathy in diabetics. It develops insidiously and may affect the small sensorimotor nerve fibres, large sensorimotor nerve fibres, or both. Small, unmyelinated C fibres are composed of autonomic and sensory axons that transmit thermal perception and sympathetic function. These are affected early in the disease process. Patients present with prominent paraesthesias and autonomic nervous system dysfunction recognised by the presence of orthostatic hypotension, resting tachycardia and distal anhidrosis.
Large myelinated axons include both motor and sensory nerves. They conduct proprioception, light touch, vibratory and pain sensations. Symptoms of large-fibre involvement include tingling, burning, numbness, allodynia or deep lancinating pain. Sensory ataxia may occur as a result of diminished vibratory and proprioceptive sense. Sensory changes do not always correlate with nerve-conduction deficits. Deep tendon reflex responses are attenuated or absent and there may be distal motor weakness. The neuropathy develops in a length-dependent fashion, progressing from distal to proximal in a ‘stocking and glove’ distribution. Progression of nerve injury leads to the loss of protective threshold or the ability to detect small objects or stimuli, resulting ultimately in the neurotrophic or insensate diabetic foot. This is the cause of diabetic ulceration in up to 85% of patients.
The exact pathophysiology of nerve damage in diabetes remains unclear. A number of theories exist, which include the polyol pathway, microcirculation complications secondary to the stimulation of protein kinase, and the non-enzymatic glycosylation of proteins throughout the body.
The polyol pathway has long been implicated. Peripheral nerve tissue does not require insulin for glucose uptake. Hyperglycaemia results in increased cellular glucose levels within nerve tissue, which require an alternative catabolic pathway to be cleared. Via oxidative reactions, glucose is converted to sorbitol, and sorbitol converted to fructose. Initially it was believed that the accumulation of sorbitol and fructose led to osmotic stress, resulting in nerve injury. However, it is now thought that it is the oxidative stress resulting from the breakdown of intraneural glucose that metabolically compromises neurons and leads to nerve damage. Functional loss of axons seems to occur as a length-dependent loss, resulting in an initial distal neuropathy.
Intracellular hyperglycaemia stimulates the activation of protein kinase C. This enzyme facilitates the transfer of phosphate groups from a donor molecule. Although there are many isoenzymes of protein kinase C, the β-2 form has been implicated as the mediator of microvascular damage. These elevated levels of protein kinase C β-2 result in increased basement membrane matrix protein deposition, leucocyte activation, and smooth muscle proliferation and contraction. This process results in decreased endoneural blood flow, resulting in nerve damage.
Exposure of proteins to high levels of glucose initiates a multistep process, resulting in non-enzymatic glycosylation of these proteins – referred to as advanced glycation end-products (AGEs). Proteins, lipids and nucleic acids are all affected, with a resultant change in metabolic function. The large protein complexes may also be difficult for the body to clear, resulting in AGE accumulation in susceptible tissues. Interaction with collagen in endoneural vessel walls thickens the walls, compromising microcirculation to the nerves.
The wide reach of diabetic neuropathy therefore results in many changes in the lower extremity. Sensory involvement results in a loss of protective threshold and the development of a neurotrophic foot. It is recognised early on as a loss of protective threshold and dorsal column involvement characterised by a loss of vibratory and position sense. This is the most prominent factor in the development of foot ulceration and the clinical path to lower extremity amputation.
Motor involvement affects initially the intrinsic musculature of the foot, leading to what is sometimes referred to as an ‘intrinsic minus’ foot. Atrophy of the intrinsic musculature results in digital contractures, plantar prominence of the metatarsals and abnormal distribution of the normal weight-bearing load with ambulation. In advanced neuropathy a drop foot may develop secondary to anterior compartment muscle wasting in the lower leg. The gastrocsoleus complex, having lost its antagonistic muscle group, then gains mechanical advantage, resulting in ankle joint equinus. This deformity adds further to the weight-bearing load borne by the forefoot, placing the patient at even greater risk of forefoot ulceration.
Autonomic nervous system involvement in the lower extremity results in a profound vasodilatation of all vessels to the lower extremity and sudomotor changes. The foot will present clinically as warm, erythematous and dry. Increased vascular flow to the foot results in demineralisation of bone; it is literally ‘washed away’. This is a major contributing factor to the pathogenesis of Charcot joint disease.
Medical treatment for diabetic peripheral neuropathy must begin with rigid glucose control and patient education regarding the risks and hazards associated with nerve damage. Superficial nerve pain can be managed with capsaicin creams. Deeper nerve pain may be managed with tricyclic antidepressant medications such as amitriptyline or antiseizure medications such as gabapentin. Muscle relaxants may provide relief of deep pain. Disease-modifying drugs that would modulate the pathogenesis of neuropathy are in clinical trials and will open a new frontier in the prevention of neuropathic complications in the diabetic patient.
Treatment in the lower extremity should be directed towards preventing ulceration. Accommodative footwear is indicated in all patients who have lost protective threshold. A laminated plastazote and poron insole provides the ability to offload plantar prominences and provide absorption of abnormal shearing forces. Often an extra-depth shoe will provide ample room for the diabetic foot; however, if severe foot deformities are present, custom-moulded shoes are indicated. Physical therapy is an important adjuvant therapy in the diabetic patient, with treatments directed towards increasing the patient’s balance and muscular strength.
CHARCOT JOINT DISEASE
Charcot joint disease is also referred to as neuropathic osteoarthropathy. There are two forms: atrophic and hypertrophic. It is a destructive process that can occur very rapidly. The atrophic form, or diabetic osteolysis, is much less common and is characterised by severe bone resorption. The hypertrophic form is characterised by severe osseous proliferation followed by bony coalescence or ‘healing’. The end result is significant foot deformity.
Atrophic joint disease occurs much less frequently. It may be referred to as diabetic osteolysis and is a form of bone resorption thought to be brought on by hyperaemia. Bones are literally ‘washed away’ and there is a characteristic pencilling of the metatarsal heads, with a ‘sucked-candy’ appearance. It is generally localised to the forefoot.
The pathogenesis of hypertrophic Charcot joint disease is most likely multifactorial (Table 6.6); however, two popular theories exist. The ‘neurotraumatic’ theory attributes the bone destruction to the loss of pain and proprioception coupled with repetitive microtrauma. The sensory neuropathy prevents the patient from recognising joint subluxation or the presence of pain associated with it. The ‘neurovascular’ theory suggests that, due to autonomic neuropathy, increased blood flow to the bones results in periarticular osteopenia. This decreased mineralisation coupled with trauma results in joint destruction. Pedal pulses will be bounding, the foot erythematous and warm. Motor neuropathy contributes to the development of structural deformity. Abnormal distribution of the weight-bearing load during gait creates pathological stress throughout the osseous architecture of the foot. This may create microstress fractures. Finally, the non-enzymatic glycosylation of collagen decreases the strength of the joint capsules and ligaments, permitting greater joint subluxation.
Table 6.6 Modified classification system of Eichenholtz
| Stage | Radiographic findings |
|---|---|
| 0 Clinical | Erythema, oedema and increased temperature |
| I Development | |
| II Coalescence | |
| III Reconstruction or consolidation |
Hypertrophic Charcot joint disease (Table 6.7) usually occurs at the midfoot, rearfoot or ankle. Incidence is higher in patients who have had diabetes for an average of 12–18 years and which has been poorly controlled. Patients are often in their fifth or sixth decade of life. Males and females are affected equally. During the development phase there are bony changes consistent with an exaggerated form of osteoarthritis. Clinically, pulses will be bounding, the foot is red, hot, swollen and generally, pain free. Radiographically there is generalised demineralisation, cartilaginous fibrillation and loose-body formation (Fig. 6.10). A Charcot foot can, unfortunately, closely mimic osteomyelitis. As these clinical and radiographic findings mimic infection, osseous malignancy or deep venous thrombosis, the importance of accurate diagnosis cannot be overstated.
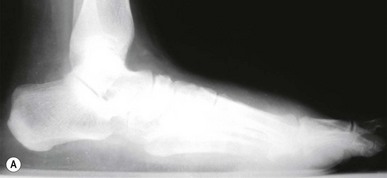
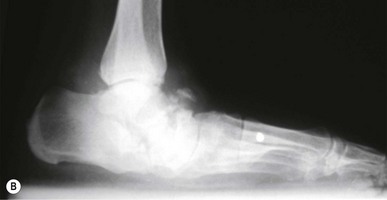
Figure 6.10 (A) Charcot joint disease in a 58-year-old, non-insulin-dependent female. (B) Following surgical correction of a bunion deformity the patient experienced Charcot-mediated collapse of the subtalar joint.
The coalescence stage is characterised by resorption of osseous debris, with early healing of fracture fragments. During the reconstruction phase the foot becomes stable, resulting in well-defined bony contours and ankylosis of involved joints.
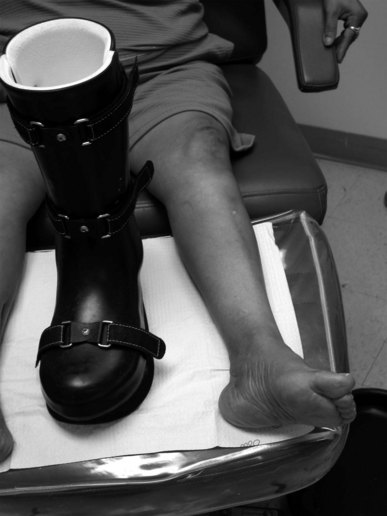
Early recognition is critical to avoid significant foot deformity. Early immobilisation with cessation of weight bearing provides the best clinical outcome. A total contact cast or a Charcot restraint orthotic walker (CROW) boot is used once the oedema associated with the development stage has subsided (Fig. 6.11). A bone stimulator may aid in the reconstructive stage. At the conclusion of this disease process, the patient is left with significant foot deformity. Custom-moulded shoes are required and the possibility of surgical intervention to eliminate bony prominences or fuse subluxed joints must be considered.
DISORDERS OF NEUROMUSCULAR TRANSMISSION
These disorders are associated with weakness and fatigability on exertion. The ‘neurological’ lesion rests within the generation of a motor end-plate potential, which is unable to trigger a muscle response. Although management of these disorders is primarily medical, it is important to differentiate these neuromuscular junction diseases from myopathic diseases.
Myasthenia gravis is an acquired autoimmune disorder, the symptoms of which were first described by Thomas Willis in 1672. The formation of autoantibodies against nicotinic acetylcholine receptors at the neuromuscular junction results in a chronic, progressive weakness of the voluntary skeletal muscles. Involvement may be limited to the external ocular muscles or generalised. The frequency and recognition of myasthenia gravis is increasing, with an annual incidence of 2 per 100 000. The presence of a thymic tumour is a significant predisposing factor, with 30–60% of thymomas associated with myasthenia gravis. Early-onset disease occurs before the age of 40 years, is more common in women and tends to coexist with other autoimmune disorders. Late-onset disease is slightly more common in males, has no thymus enlargement, and has recently shown an increase in frequency. Respiratory failure and aspiration pneumonia are the most severe complications encountered.
The most characteristic feature is painless muscle fatigue exacerbated by exertion. Ptosis and diplopia are the initial presenting symptoms in 50% of patients. These are usually bilateral and asymmetric. Bulbar and facial muscle weakness results in reduced facial expression, with difficulty in swallowing and speaking clearly. Limb weakness is symmetrical, more pronounced proximally and more frequent in the upper extremities. When the lower extremity is affected, the hip flexors, quadriceps and hamstring muscles are affected, the foot plantar flexors and dorsiflexors being less commonly involved. Clinical findings include abnormal fatigability of the limb muscles. Patients may have dyspnoea with only mild to moderate exertion, and experience difficulty climbing stairs, walking or running. Deep tendon reflex responses and sensory findings remain normal throughout the course of the disease.
Diagnostic tests used include the assay of blood serum for the presence of antibodies against the acetylcholine receptors. The presence of these antibodies is diagnostic for the disease, and the degree of elevation of these titres correlates loosely with the severity of the disease. Pharmacological testing may be done with edrophonium, a short-acting acetylcholinesterase inhibitor. Transient improvement of muscle weakness is suggestive of myasthenia gravis. Treatment may include the use of anticholinesterases, prednisolone, plasmapheresis and thymectomy.
Lambert–Eaton myasthenic syndrome is also an acquired autoimmune disorder. The defect in this disease process is with the actual numbers of presynaptic acetylcholine quartals released. The postsynaptic response to acetylcholine is normal. The incidence is equal between men and women, and is strongly associated with underlying tumours, particularly small-cell carcinomas of the lung. Patients will demonstrate weakness and fatigability of the proximal limb and thoracic muscles, and have difficulty with chewing, swallowing and speech. The eye muscles are rarely involved. Diagnosis is confirmed by the presence of antibodies in the serum directed against the calcium channels on the presynaptic side of the motor nerve. Electromyographic findings demonstrate an increase in the amplitude of the action potential following voluntary muscular contraction. Treatment may include the anticholinesterases; however, the effect is not as dramatic as with myasthenia gravis. Guanidine increases the actual numbers of acetylcholine quartals released but carries with it serious side-effects. Management of any underlying malignancy is essential.
There are many congenital myasthenic syndromes, which must be distinguished from the immune-mediated, acquired disorders. There are presynaptic, synaptic and postsynaptic forms, most of which are inherited through autosomal-recessive transmission. Symptoms usually develop by the age of 2 years. The progression and severity of these processes is dependent on the type of myasthenic syndrome.
Auer-Grumbach M, Strasser-Fuchs S, Wagner K. Roussy–Levy syndrome is a phenotypic variant of Charcot–Marie–Tooth syndrome IA associated with a duplication on chromosome 17p11.2. Journal of Neurological Science. 1998;154(1):72-75.
Burnett MG, Zager EL. Pathophysiology of peripheral nerve injury: a brief review. Neurosurgery Focus. 2004;16(5):E1.
Seddon HJ. Three types of nerve injury. Brain. 1943;66:237-288.
Sunderland S. The anatomy and physiology of nerve injury. Muscle and Nerve. 1990;13:771-784.
Feinberg JH, Nadler SF, Krivickas LS. Peripheral nerve injuries in the athlete. Sport and Medicine. 1997;24(6):385-408.
Ochoa J, Danta G, Fowler TJ. Nature of the nerve lesion caused by a pneumatic tourniquet. Nature. 1971;233:265-266.
Ochoa J, Fowler TJ, Gilliatt RW. Anatomical changes in peripheral nerves compressed by a pneumatic tourniquet. Journal of Anatomy. 1972;113:433-455.
Deyo RA, Weinstein JN. Low back pain. New England Journal of Medicine. 2001;344(5):363-370.
Deyo RA, Rainville J, Kent DL. What can the history and physical examination tell us about low back pain? JAMA. 1992;268:760-765.
Malmivaara A, Häkkinen U, Aro T. The treatment of acute low back pain – bed rest, exercises, or ordinary activity? New England Journal of Medicine. 1995;332:351-355.
Hereditary motor and sensory neuropathies
Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Archives of Neurology. 1968;18(6):603-618.
Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259-280.
Jansen GA, Waterham HR, Wanders RJ. Molecular basis of Refsum disease: sequence variations in phytanoyl-CoA hydroxylase (PHYH) and the PTS2 receptor (PEX7). Human Mutation. 2004;23(3):209-218.
Keller MP, Chance PF. Inherited peripheral neuropathy. Seminars in Neurology. 1999;19(4):353-362.
Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot–Marie–Tooth disease type 1A. Brain. 2000;123(7):1516-1527.
Njegovan ME, Leonard EI, Joseph FB. Rehabilitation medicine approach to Charcot–Marie–Tooth disease. Clinical Podiatric Medicine and Surgery. 1997;14(1):99-116.
Pareyson D. Differential diagnosis of Charcot–Marie–Tooth disease and related neuropathies. Neurological Science. 2004;25(2):72-82.
Ouvrier RA, McLeod JG, Conchin TE. The hypertrophic forms of hereditary motor and sensory neuropathy. A study of hypertrophic Charcot–Marie–Tooth disease (HMSN type I) and Dejerine–Sottas disease (HMSN type III) in childhood. Brain. 1987;110(1):121-148.
Bailie DS, Kelikian AS. Tarsal tunnel syndrome. Diagnosis, surgical technique, and functional outcome. Foot and Ankle International. 1998;19(2):65-78.
Boc SF, Hatef J. Space occupying lesions as a cause of tarsal tunnel syndrome. Journal of the American Podiatric Medical Association. 1995;85(11):713-715.
Cimino WR. Tarsal tunnel syndrome: review of the literature. Foot and Ankle. 1990;11(1):47-52.
Dyck PJ, Dyck PJ, Grant IA. Ten steps in characterizing and diagnosing patients with peripheral neuropathy. Neurology. 1996;47(1):10-17.
Goecker RM, Banks AS. Analysis of release of the first branch of the lateral plantar nerve. Journal of the American Podiatric Association. 2000;90(6):281-286.
Hayes DWJr, Mandracchia VJ, Webb GE. Nerve injury associated with plantarflexion–inversion ankle sprains. Clinical Podiatric Medicine and Surgery. 2000;17(2):361-369. vi–vii
Kanbe K, Kubota H, Shirakura K, et al. Entrapment neuropathy of the deep peroneal nerve associated with the extensor hallucis brevis. Journal of Foot and Ankle Surgery. 1995;34(6):560-562.
Leach RE, Purnell MB, Saito A. Peroneal nerve entrapment in runners. American journal of Sports Medicine. 1989;17(2):287-291.
Lorei MP, Hershman EB. Peripheral nerve injuries in athletes. Treatment and prevention. Sports Medicine. 1993;16(2):130-147.
Mahan KT, Rock JJ, Hillstrom HJ. Tarsal tunnel syndrome: a retrospective study. Journal of the American Podiatric Medical Association. 1996;86(2):81-91.
McCluskey LF, Webb LB. Compression and entrapment neuropathies of the lower extremity. Clinical Podiatric Medicine and Surgery. 1999;16(1):97-125. vii
McCrory P, Bell S, Bradshaw C. Nerve entrapments of the lower leg, ankle and foot in sport. Sports Medicine. 2002;32(6):371-391.
McMinn RM, Hutchings RT, Logan BM. Color atlas of foot and ankle anatomy, 2nd edn. Sydney: Mosby-Wolfe; 1996.
Oh SJ, Meyer RD. Entrapment neuropathies of the tibial (posterior tibial) nerve. Neurology Clinics. 1999;17(3):593-615. vii
Poncelet AN. An algorithm for the evaluation of peripheral neuropathy. American Family Physician. 1998:755. 15 February
Sammarco GJ, Chalk DE, Feibel JH. Tarsal tunnel syndrome and additional nerve lesions in the same limb. Foot and Ankle. 1993;14(2):71-77.
Schon LC, Baxter DE. Neuropathies of the foot and ankle in athletes. Clinical Sports Medicine. 1990;9(2):489-509.
Bradley JL, Homayoun S, Hart PE, et al. Role of oxidative damage in Friedreich’s ataxia. Neurochemistry Research. 2004;9(3):561-567.
Delatycki MB, Williamson R, Forrest SM. Friedreich’s ataxia: an overview. Journal of Medical Genetics. 2000;37(1):1-8.
Durr A, Cossee M, Agid Y, et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. New England Journal of Medicine. 1996;335(16):1169-1175.
Ponka P. Hereditary causes of disturbed iron homeostasis in the central nervous system. Annals of the New York Academy of Sciences. 2004;1012:267-281.
Stolze H, Klebe S, Peterson G, et al. Typical features of cerebellar ataxic gait. Journal of Neurology, Neurosurgery and Psychiatry. 2002;73:310-312.
Cummings JL. Behavioral and psychiatric symptoms associated with Huntington’s disease. Advances in Neurology. 1995;65:179-186.
Davutoglu V, Kilinc M, Dinckal H, et al. Sydenham’s chorea-clinical characteristics of nine patients. International Journal of Cardiology. 2004;96(3):483-484.
Dajani AS, Ayoub E, Bierman FZ, et al. Special Report: Guidelines for the Diagnosis of Rheumatic Fever: Jones Criteria, Updated 1992: Special Writing Group of the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young. American Heart Association Circulation. 1993;87(1):302-307.
Hermanowicz N. Management of Parkinson’s disease: strategies, pitfalls, and future directions. Postgraduate Medicine. 110(6), 2001.
Jones TD. Diagnosis of rheumatic fever. JAMA. 1944;126:481-485.
Kirkwood SC, Su JL, Conneally P, Foroud T. Progression of symptoms in the early and middle stages of Huntington disease. Archives of Neurology. 2001;58(2):273-278.
Korn-Lubetzki I, Brand A, Steiner I. Recurrence of Sydenham’s chorea: implications for pathogenesis. Archives of Neurology. 2004;61(8):1261-1264.
Lang AE, Lozano AM. Parkinson’s disease. First of two parts. New England Journal of Medicine. 1998;339(15):1044-1053.
Lang AE, Lozano AM. Parkinson’s disease. Second of two parts. New England Journal of Medicine. 1998;339(16):1130-1143.
Martin JB. Molecular basis of the neurodegenerative disorders. New England Journal of Medicine. 1999;341(18):1407.
Rosenstein LD. Differential diagnosis of the major progressive dementias and depression in middle and late adulthood: a summary of the literature of the early 1990s. Neuropsychology Reviews. 1998;8(3):109-167.
Sapp E, Schwarz C, Chase K, et al. Huntingdon localization in brains of normal and Huntington’s disease patients. Annals of Neurology. 1997;42(4):604-612.
Swedo SE, Leonard HL, Schapiro MB, et al. Sydenham’s chorea: physical and psychological symptoms of St Vitus dance. Pediatrics. 1993;91(4):706-713.
Veasy LG, Wiedmeier SE, Orsmond GS, et al. Resurgence of acute rheumatic fever in the intermountain area of the United States. New England Journal of Medicine. 1987;316(8):421-427.
Gordon N. The role of botulinus toxin type A in treatment – with special reference to children. Brain Development. 1999;21(3):147-151.
Graham HK, Selber P. Musculoskeletal aspects of cerebral palsy – review article. Journal of Bone and Joint Surgery (British). 2003;85-B:157-166.
Grether JK, Nelson KB, Emery ES, Cummins SK. Prenatal and perinatal factors and cerebral palsy in very low birth weight infants. Journal of Pediatrics. 128(3), 1996.
Kuban KC, Leviton A. Cerebral palsy. New England Journal of Medicine. 1994;330(3):188-195.
Schulman LH, Sala DA, Chu MLY, et al. Developmental implications of idiopathic toe walking. Journal of Pediatrics. 1997;130(4):541-546.
Torfs CP, van den Berg B, Oechsli FW, Cummins S. Prenatal and perinatal factors in the etiology of cerebral palsy. Journal of Pediatrics. 1991;118(1):161.
Arnal JF, et al. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell and Molecular Life Science. 1999;55:1078-1087.
Albert SW, Koval KJ, Zuckerman JD. Neuropathic arthropathy: review of current knowledge. Journal of the American Academy of Orthopedic Surgeons. 1996;4:100-108.
Caputo GM, Ulbrecht J, Cavanagh PR, Juliano P. The Charcot foot in diabetes: six key points. American Family Physician. 57(11), 1998.
England JD, Asbury AK. Peripheral neuropathy. Lancet. 2004;363(9427):2151-2161.
Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. Journal of Clinical Investigation. 2003;111(4):431-433.
Moncada S, Higgs A. The L-arginine–nitric oxide pathway. New England Journal of Medicine. 1993;329:2002-2012.
Pittenger GL, Malik RA, Burcus N, et al. Specific fibre deficits in sensorimotor diabetic polyneuropathy correspond to the cytotoxicity against neuroblastoma cells of sera from patients with diabetes. Diabetes Care. 1999;22:1839-1844.
Sommer TC, Lee TH. Charcot foot: the diagnostic dilemma. American Family Physician. 64(9), 2001.
Sumpio BE. Foot ulcers. New England Journal of Medicine. 2000;43(11):787-793.
Vinik AI. Neuropathy: new concepts in evaluation and treatment. Southern Medical Journal. 2002;95(1):21-23.
Wautier JL, Guillausseau PJ. Advanced blycation end products, their receptors and diabetic angiopathy. Diabetes Metabolism. 2001;27:535-542.
Complex regional pain syndrome
Anderson DJ, Fallat LM. Complex regional pain syndrome of the lower extremity: a retrospective study of 33 patients. Journal of Foot and Ankle Surgery. 1999;38:381-387.
Lee KJH, Kirchner JS. Complex regional pain syndrome and chronic pain management in the lower extremity. Foot and Ankle Clinics of North America. 2002;7:409-419.
Maleki J, LeBel AA, Bennet GJ, Schwartzman RT. Patterns of spread in complex regional pain syndrome, type I (reflex sympathetic dystrophy). Pain. 2000;88(3):259-266.
Pittman DM, Gelgrade MJ. Complex regional pain syndrome. American Family Physician. 56(9), 1997.