Chapter 4 Neutral and acidic nitrogen compounds
Barbiturates
Introduction
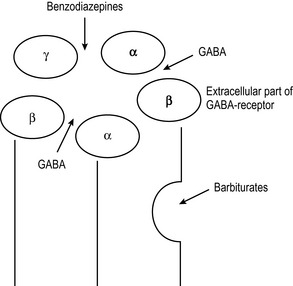
The parent compound of barbiturates was firstly synthesised by Baeyer in 1864. The suffix (al) was added to the names because of the function similarity with the chloral hydrate and the latter is an aldehyde derivative. Most of the barbiturates have been largely replaced with benzodiazepines since the 1960s because of their addictive potential. Barbiturates bind with GABA-receptors. The 3-D structure of GABA-receptors has not been fully experimentally defined yet.
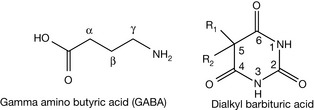
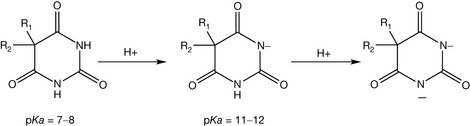
In the 1970s new types of GABA-receptors which could be stimulated by different drugs were discovered and named GABA-B receptors. Furthermore, GABA-C receptors were discovered as well. Hence, the GABA-receptor which was the first one discovered and which is stimulated by barbiturates, and other agents like benzodiazepines, was distinguished by the name of the GABA-A receptor. In the 1980s GABA-receptors were firstly purified and the primary and secondary structures were determined. Barbiturates are derived from barbituric acid. They have a dialkyl barbituric acid form (see Fig. 4.1). They are dibasic acids (i.e. having two pKa values). The first dissociation is the more significant one because it is stronger, pKa = 7–8, while the second dissociation is extremely weak, pKa = 11–12 (Fig. 4.2).
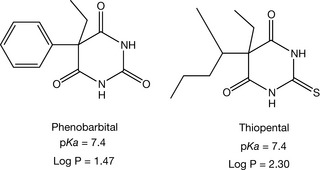
Phenobarbital and thiopental (Fig. 4.3) are still used in clinics. Phenobarbital is reserved for a serious type of epilepsy called status epilepticus, while thiopental is used in general anesthesia.
Physiochemical properties
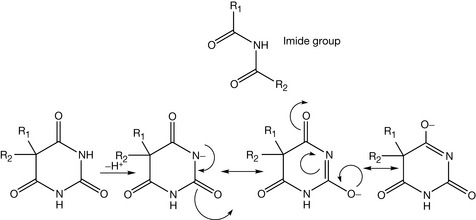
Barbiturates contain an imide group which has an acidic character because its conjugated base is stabilised by electron delocalisation (Fig. 4.4). The conjugation distributes the charge at different atoms in the structure and thus decreases the overall molecular energy. Unsubstituted barbiturates have a pKa approximately 7 and for N-alkylated ones, the pKa is around 8, because the alkyl group is an electron donating group and acidity is promoted by electron withdrawal. Thus barbiturates are weaker acids than acetic acid which has a pKa = 4.8. However, their mid-range pKa values enable barbiturates to be partially ionised at physiological pH (7.4). The ionised form is important to keep the drug soluble in the blood, while the non-ionised form facilitates the crossing of the blood–brain barrier.
Phenobarbital has a pKa = 7.4, calculate the ionised form ratio of it in the blood. (Physiological pH = 7.4.)
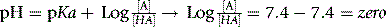
So the ratio is 1:1.
Barbiturates can form sodium and potassium salts (Fig. 4.5). The negative charge is more stable on the large atoms like sulphur or oxygen.
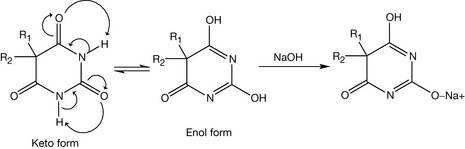
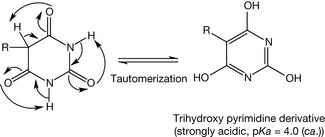
Actually, barbituric acid and 5-alkyl barbituric acid are not pharmacologically active because they tautomerise to the more stable acid derivatives having a pKa of approximately 4 and thus do not fit the receptor. The product is a trihydroxy pyrimidine derivative. It is more stable because the aromaticity of the ring formed is a strong driving force and this form is more thermodynamically favourable (Fig. 4.6).
GABA-A receptors
GABAA is a protein with a pentameric group of subunits (Fig. 4.7). Each subunit has approximately 200 amino acids. There are around four hydrophobic regions which enable the receptor to possess allosteric pockets for interactions, and the hydrophobic region is also important to anchor the protein into the cell membrane. Barbiturates bind with a site different from the site of binding of benzodiazepines, which bind at the active site rather than changing the receptor conformation via an allosteric interaction.
Barbiturates structure–activity relationships
The barbiturate structure resembles that of hydantoins and dialkyl acetyl urea derivatives which are also neuroactive agents (Fig. 4.8). It is clear that the dialkyl substitution at position 5 of barbiturates is essential for their pharmacological activity. Also, if one or two hydrogens are left at position 5 of the barbiturate, the barbiturate will be converted to the trihydroxy pyrimidine derivative, as discussed earlier.
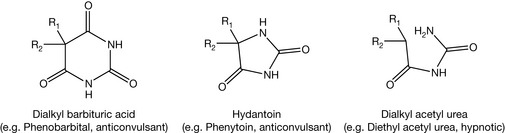
Figure 4.8 Barbiturate structure resembles that of the hydantoins and dialkyl acetyl urea derivatives.
Substitution of carbonyl oxygen with sulphur atom increases the partition coefficient of the drug as in the case of thiopental. Increasing the lipophilicity of the drugs usually decreases their rate of elimination and prolongs their duration of action, but this is not the case for barbiturates. Long alkyl groups at position 5 of barbiturates increase the lipophilicity of barbiturates and this will increase the liver microsomal penetration of the drugs and consequently their metabolism. The lipophilic barbiturates also pass through the blood–brain barrier easily and this will achieve a rapid onset of action. Thiopental is quite lipophilic (log P = 2.3), so it is expected to have a short duration of action and it is used in general anaesthesia because of this character. Its pharmacological effect usually lasts to a maximum 30 minutes.
Metabolism of barbiturates
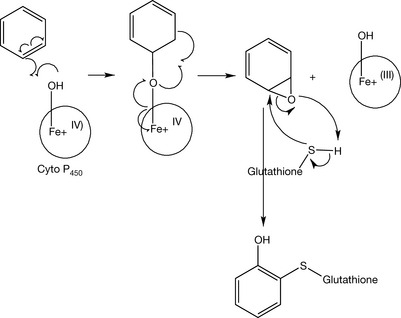
The main site of metabolism in the barbiturate structure is at the dialkyl substituents at position 5. They thus provide a good illustration of the types of metabolism which can occur to alkyl groups. Compounds containing a benzene ring, for example phenobarbital, are susceptible to epoxidation in phase I metabolism and to glutathione conjugation in phase II metabolism (Fig. 4.9).
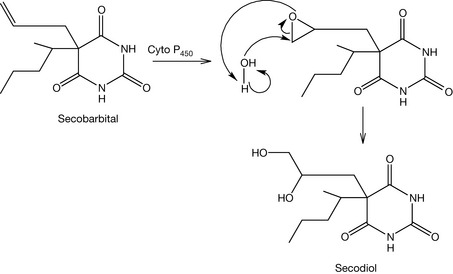
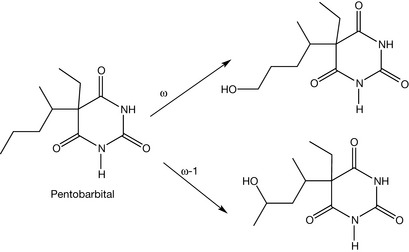
Compounds containing an allyl group are susceptible to either olefinic or allylic oxidation (Figs 4.10, 4.11). Olefinic oxidation is mediated with an epoxidation step like the metabolism of secobarbital to secodiol. Allylic oxidation does not produce epoxide intermediates. The generated alcohol is conjugated with glucuronic acid in phase II metabolism in order to increase the compound hydrophilicity and consequently the urinary excretion. An example of this type of metabolism is the metabolism of hexobarbital. Saturated alkyl groups are also susceptible to the liver metabolism, mainly by ω oxidation (for the terminal carbon) or by ω-1 oxidation (for the carbon just before the terminal one). Pentobarbital is a good example of a barbiturate which undergoes this kind of oxidation (Fig. 4.12).
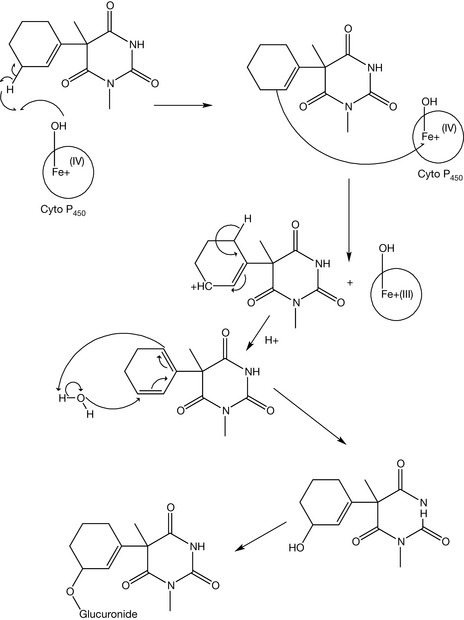
N-dealkylation is also possible. An example of this is the removal of the methyl at position 3 of hexobarbital. The heterocyclic ring of the barbiturates does not usually participate in their metabolism. However, cleavage of the (1–6) bond is possible and it produces malonyl urea derivatives.
 Self Test 4.2
Self Test 4.2
Predict two metabolic pathways by which the body can detoxify phenobarbital (Fig. 4.3).
Sulphonamides
Introduction
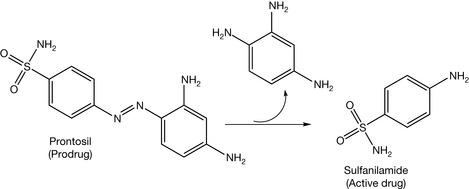
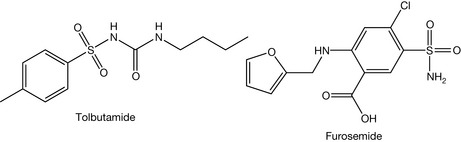
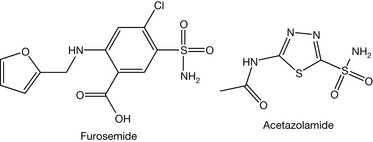
The first reported use of sulphonamides was in 1933. Prontosil was the first agent of the sulphonamide family discovered. Domagk was awarded the Nobel Prize in 1938 because of his discovery of prontosil. Prontosil was active in vivo but not in vitro. This encouraged Trefouel to study this compound. He found that the compound is a prodrug requiring a reductive cleavage by the gut microflora in order to generate the active drug (sulfanilamide, Fig. 4.13). This discovery encouraged scientists to synthesise other sulphonamides. There were more than 4500 sulphonamides synthesised by 1948. However, many of them were not biologically active. Sulphonamides are analogues of para-amino benzoic acid (PABA). The latter is used in folic acid synthesis in microbes. Because of this similarity, sulphonamides block the pathway of the folic acid synthesis. Subsequently, they arrest DNA synthesis and exert a bacteriostatic effect (stopping cell proliferation). Sulphonamides are also used as oral hypoglycaemic agents (tolbutamide, Fig. 4.14) and diuretics (furosemide, Fig. 4.14).
Physiochemical properties
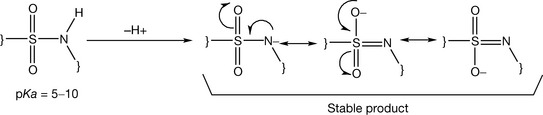
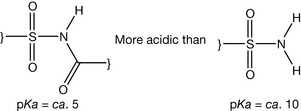
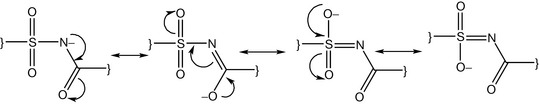
Sulphonamides contain the sulphamido group which has an acidic character because the sulphonyl part is electron withdrawing and the conjugated base is stabilised by electron delocalisation (Fig. 4.15). Substitution of the sulphamido nitrogen with an electron withdrawing group like acetyl or a heterocycle increases the acidity of the sulphamido group (Fig. 4.16). For example, acetyl substitution as in sulfacetamide (Fig. 4.17) produces four resonance forms instead of three, adding additional stability to the ionised form. This means that a further stabilisation of the ionised form occurs and sulfacetamide is one of the most acidic of the sulphonamides. Sulphonamides have also an aniline functionality which is considered as a very weak base (pKa ca. 2) because the lone pair of electrons is involved in the resonance of the aromatic system (Fig. 4.18).
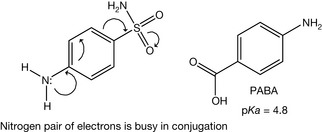
Sulphonamides have to be partially un-ionised to enable penetration of the cell membrane of the microbial cell. Thus the antimicrobial activity is increased with an increase in the pKa value of the drug up to the point where the degree of penetration into the microbial cells is hindered by the extent of low ionisation of the drug in physiological fluids. It was found that the ionised form of a sulphonamide has the antimicrobial activity because of its ability to accumulate inside the microbial cell and its close similarity to PABA (Fig. 4.18) which has an acidic pKa = 4.8 and is totally ionised at the physiological pH. In addition, low pKa sulphonamides (e.g. sulfisoxazole, pKa = 5.0, Fig. 4.19) are fully ionised at physiological pH and are consequently excreted rapidly in the urine. Thus these agents can be used for the urinary tract infections.
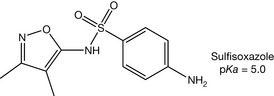
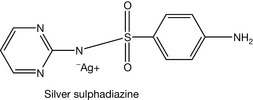
Most sulphonamides are well absorbed orally except for a few of them, e.g. sulphadiazine. Such agents are reserved for gastrointestinal tract infections or for skin infections. Silver sulphadiazine (Fig. 4.20) is poorly absorbed through the skin so it is used as a topical antimicrobial. Sulphonamides with relatively high pKa values are quite lipophilic at the physiological pH. This increases their binding with plasma protein and also facilitates their accumulation in adipose tissue. Consequently, this prolongs the duration of action of these agents.
Mechanism of action
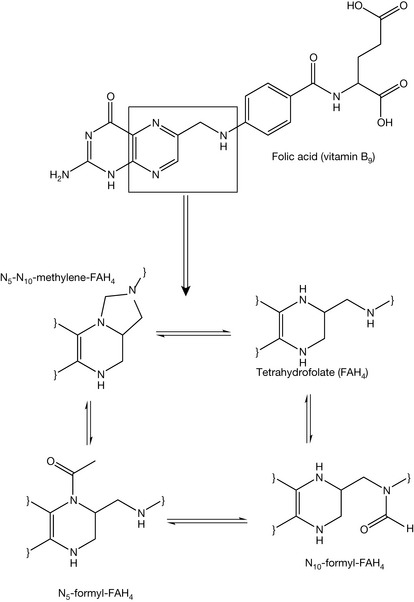
In mammalian cells, folic acid can not be synthesised de novo but it should be taken from dietary sources. While most microbes are not able to take folic acid from the surrounding environment, they instead synthesise it de novo. Folic acid produces derivatives of tetrahydrofolate (FAH4), playing an essential role in the biochemical reactions involving a one carbon transfer process such as methylation of uracil (Fig. 4.21). Lack of folic acid disturbs many pathways, such as nucleic acid synthesis and amino acid synthesis. Impairing the synthesis of DNA stops cell division and consequently cell multiplication.
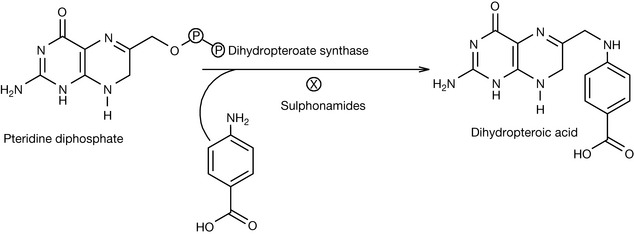
Sulphonamides resemble PABA, so they block dihydropteroate synthase (Fig. 4.22), which is an important enzyme in the folic acid synthesis pathway inside the microbial cell. Microbes can resist sulphonamides by increasing the synthesis of PABA, decreasing the permeability of their membranes to sulphonamides or by promoting active efflux of sulphonamides out of the microbial cell. For resistant species, sulphonamides might be mixed with other antimicrobial agents such as trimethoprim. The combination of sulfamethoxazole and trimethoprim produces a synergism (the antimicrobial activity of the combination is more than the additional activity of two independent antimicrobials). Trimethoprim can block the dihydrofolate reductase enzyme in the folic acid pathway which catalyses the conversion of dihydrofolate (FAH2) to tetrahydrofolate (FAH4) while sulfamethoxazole competes with PABA. The combination therapy is suitable for more resistant microbes.
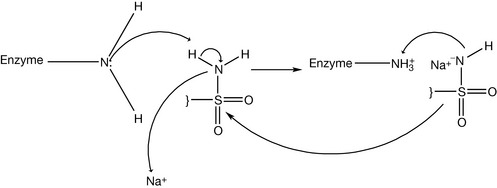
Some of the sulphonamides can be used as diuretics. The mechanism of their action relates to carbonic acid excretion. Carbon dioxide generated from catabolic processes is carried to the lung and then removed by exhalation. However, part of the carbon dioxide is still dissolved in the blood. The dissolved carbon dioxide produces carbonic acid and its conjugated base (i.e. bicarbonate). This mixture of the weak acid and its conjugate base is one of the important buffer systems in the blood. The dissolved carbon dioxide is excreted in the urine. The processes of the conversion of carbon dioxide to carbonic acid and then to bicarbonate are reversible processes, so there is no big thermodynamic favouritism for one of them over the others (Fig. 4.23). These conversions occur normally with a reasonable speed if no catalytic enzyme is available. However, the carbonic anhydrase enzyme speeds up the conversion process. Carbonic anhydrase is abundant in the epithelial tissue of the proximal part of the renal filtration units (i.e. the tubules). The activity of the enzyme increases the concentration of the ionised species (bicarbonate and protons). This process increases the osmotic pressure of the urine filtrate and the urine volume if there is no secretion and reabsorption processes in the proximal tubules counteract this effect. There is electrical voltage across the urinary tubules because the difference in the electrolyte distribution. Differences in the electrolyte distribution generate an electrical field force for negative ions such as bicarbonate to be reabsorbed by the body and for protons to be secreted in the urine (Fig. 4.24). If carbonic anhydrase is inhibited, then most of the dissolved carbon dioxide can reach the distal tubules without being converted into carbonic acid. In the distal tubules there is no significant absorption and secretion except for sodium and water. So the dissolved carbon dioxide slowly becomes hydrated and produces bicarbonate and protons. This increases the osmotic pressure of the urine in the distal tubules and decreases water reabsorption by the body. Consequently, the volume of the urine is increased and diuretic action is established. Carbonic anhydrase forms carbonic acid with a help of a metal which can be considered a co-factor (Fig. 4.25). Sulphonamide can block the carbonic anhydrase enzyme by the reaction between the sulphamido group and the enzyme amine functionality (Fig. 4.26). Acetazolamide (Fig. 4.27) is an example of sulphonamide having a diuretic effect.
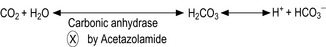
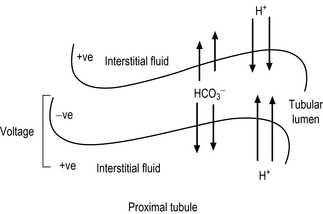
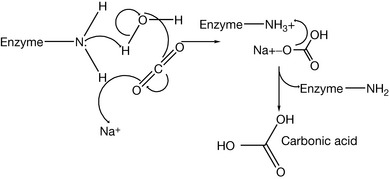
Figure 4.25 Catalytic mechanism of carbonic enzyme in producing carbonic acid from water and carbon dioxide.

The mechanism of action of hypoglycaemic sulphonamides is not fully understood. They have several targets. Maybe the most important ones are in decreasing liver gluconeogenesis and promoting insulin secretion from β-cells in the pancreas. It is proposed that a shortage of insulin decreases the glucose level inside the body’s cells, including the β-cells. The shortage of glucose inside the β-cells decreases energy metabolites so the ATP/ADP ratio is decreased. This closes potassium efflux channels and changes the electrical polarisation of β-cells. This electrical change stimulates calcium channels, allowing calcium influx into the β-cells. Accumulation of calcium in the cell is considered as an internal messenger and induces the exocytosis of insulin vesicles and thus insulin secretion.
Sulphonamides structure–activity relationships
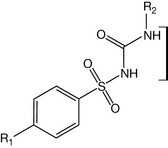
The optimum antimicrobial activity of sulphonamides is obtained wherever the sulphonamide group is in a para position relative to the amino group. The amino group (at N4 position, Fig. 4.28) should remain primary to be active enough for the nucleophilic attack on the pteridin diphosphate.
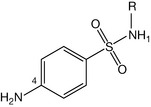
Figure 4.28 N4 nitrogen should remain free for obtaining optimum antimicrobial activity of sulphonamides.
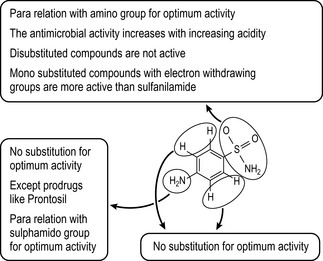
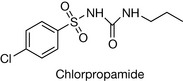
Sulphonamides with unsubstituted ortho and meta positions are more active than ones substituted on these positions because the unsubstituted sulphonamides resemble PABA. The substitution of the sulphamido group at N1 position with an electron withdrawing group such as a heterocycle increases the antimicrobial activity because this increases the acidity of the drug (until pKa ≈ 5.0) and consequently the similarity to PABA in terms of acidity. This information is summarised in Figure 4.29. Sulphonamides which are used as oral hypoglycaemic agents (Fig. 4.30) do not require the substitution at position 4 to be a primary amine. It can be any small group such as methyl or chlorine. However, substitution of large groups at this position decreases the hypoglycaemic activity. It was found that the modification of the first-generation sulphonamides by introducing the carbonyl amido part to obtain a derivative of urea (called sulphonylurea) increased the hypoglycaemic activity. Reserving the distance between two nitrogens of urea gives optimum activity. Activity is greatest if the R2 group is quite lipophilic and larger than R1 (e.g. a propyl or butyl group), e.g. the group is propyl in chlorpropamide (Fig. 4.31). The optimal diuretic action of sulphonamides requires an unsubstituted sulphamido group at the N1 position, e.g. acetazolamide and furosemide (Fig. 4.32).
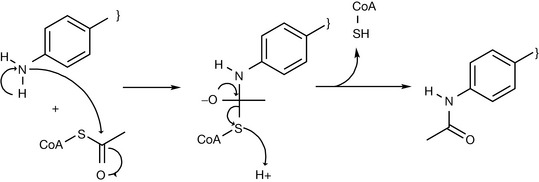
Metabolism of sulphonamides
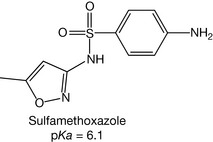
The amino group at position 4 of sulphonamide is a nucleophile and this reactivity can lead to toxicity in the human body. However, the body protects itself by blocking this group with an acetyl group (Fig. 4.33). The acetyl group is produced in the body from catabolic processes such as fat burning. There is a common interindividual variability among people with respect to rate at which they acetylate such groups. There are fast acetylators who detoxify sulphonamides quickly and slow acetylators who are more at risk of sulphonamide toxicity. Acetylation is not like the majority of phase II conjugation reactions since it decreases the water solubility of the compound instead of increasing it because it is a lipophilic group and it converts the polar amine to a less polar amide. This can lead to drug crystallisation in the urine and generates a side effect called crystalluria. This problem can be solved by using sulphonamides with a pKa equal to or less than the urine pH (approximately 6.0) such as sulfisoxazole (pKa = 5.0) or sulfamethoxazole (pKa = 6.1, Fig. 4.34). This means that the acetylated metabolite is significantly ionised at the pH values of urine. For weakly acidic sulphonamides such as sulfanilamide (pKa = 10.4), alkalisation of the urine is required. This can be achieved by administering sodium bicarbonate or another safe alkalising agent.
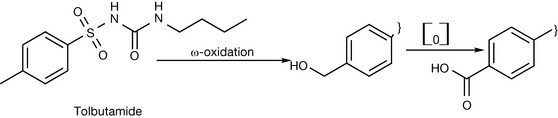
If the amino group is modified by an alkyl group such as methyl in the case of oral hypoglycaemic agents, the alkyl group undergoes ω-oxidation. An example of this is in tolbutamide metabolism (Fig. 4.35). However, substituting the alkyl group at position 4 with a halogen increases the duration of action because it is less susceptible to the liver metabolism. Tolbutamide has a plasma half life of ca. 7 hours whereas chlorpropamide (see Fig. 4.31) has a plasma half life of ca. 35 hours.
Chemical stability of sulphonamides
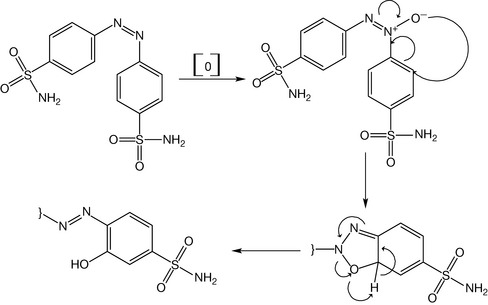
Breakdown of the sulphamido group does not occur in alkaline solutions, but strong acids can degrade it (Fig. 4.36). Sulphonamides are generally photosensitive because of the reactivity of the primary amine at position 4. This group is oxidised under the catalytic action of light to produce diazo dimers.
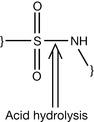
However, the diazo dimer is still susceptible to further oxidation. This leads to the oxidation of the benzene ring (Fig. 4.37). Protecting the sulphonamide solution from exposure to the light is essential; addition of antioxidant as sodium metabisulfite is also important. Sulphonamides undergo a hydrolysis reaction at the amide bond at high temperatures, so sulphonamide aqueous products are not autoclavable unless they are dissolved in other solvents such as propylene glycol. Sterile products of sulphonamides can also be dissolved in previously sterilised water. In addition, aqueous solutions of sulphonamides should be free from carbon dioxide because the dissolved carbon dioxide produces carbonic acid which catalyses the hydrolytic degradation of sulphonamides.
Methyl xanthines
Introduction
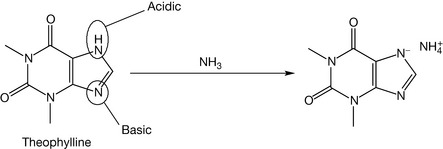
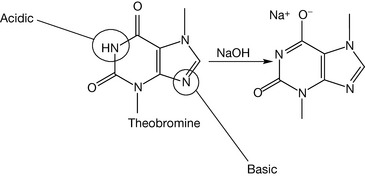
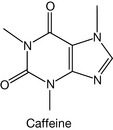
The history of the xanthine alkaloids (Fig. 4.38) goes back to ancient times when their plant sources were discovered. Caffeine was extracted from tea, coffee and cocoa; theophylline was mainly found in tea extract, while theobromine was found in cocoa extract. Methyl xanthines are considered as very weak bases with pKa values <1. They are classified as alkaloids (alkali-like compounds) although they are not really classical alkaloids. For example, theophylline (Fig. 4.39) is a strong enough acid (pKa = 8.8) to be dissociated in a weak base such as ammonia (pKa of ammonia = 9.25). Theobromine (Fig. 4.40) is weaker acid than theophylline and thus it requires stronger bases, such as sodium hydroxide, for it to be ionised. However, caffeine (Fig. 4.41) lacks acidic character because it has no exchangeable protons, so it is not soluble even in sodium hydroxide solution. This makes caffeine more lipophilic than theophylline and thus it has greater action on the CNS, whereas theophylline is used mainly in the peripheral system as a bronchodilator. Caffeine and the other methyl xanthines are much more soluble in hot water than cold water.
Physiochemical properties
Methyl xanthines are derivatives of pyrrole and pyridine (Fig. 4.42).
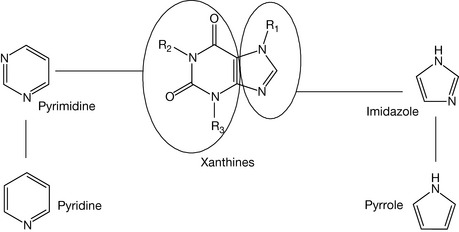
Pyridine is a weakly basic compound (pKa = 5.2). Huckel theory defines an aromatic ring as having (4n+2) pi-electrons where n is 0,1,2,3…. Thus aromatic rings have two pi-electrons but not three also they have six electrons but not five and so on. Pyridine has three pi-bonds so it has six electrons. Consequently, the lone pair of electrons on the nitrogen is not part of the ring sextet (the six pi-electrons). However, pyrrole obviously has two double bonds (4 pi-electrons) so it is impossible to have (n = 0) in the Huckel rule, also if n = 2 then pyrrole should have 10 pi-electrons and these are too much for a small ring. Thus the only possibility is when n = 1 (equivalent to 6 pi-electrons), but pyrrole has only two double bonds so the lone pair of electrons on the nitrogen is part of the ring sextet and it is not available to bond with protons. From this point of view, pyrrole has no basic character. Pyrrole can be considered neutral or a very weak acid because the conjugated base is stabilised by resonance (Fig. 4.43).
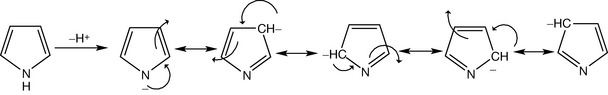
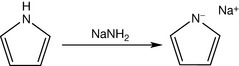
Pyrrole acidity is quite weak and is almost equivalent to the acidity of acetylene. It requires a strongly basic alkali, stronger than sodium hydroxide such as sodamide to be ionised (Fig. 4.44).
Mechanism of action
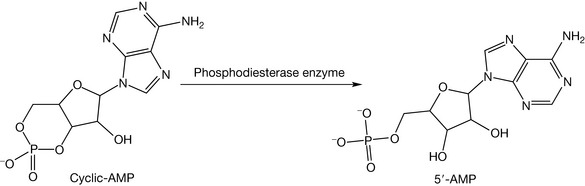
There are several targets for the methylxanthines in the human body. The most common two are phosphodiesterase enzyme inhibition and adenosine receptor inhibition. Some endogenous metabolites and some drugs such as sympathomimetics (e.g. adrenaline) trigger the secretion of intracellular second messengers such as cyclic-AMP in order to complete their pharmacodynamic action with no need for them to enter the cell. Subsequently, cAMP is deactivated by phosphodiesterase (Fig. 4.45) in order to terminate the pharmacological action of the sympathomimetics. Methylxanthines inhibit phosphodiesterase enzyme and consequently prolong the action of cAMP. This explains some of the sympathomimetic-like actions of methylxanthines such as increasing the heart rate. Methylxanthines can also increase cGMP, which is used by the body as a second messenger as well. This is also due to the inhibition of the family of phosphodiesterase enzymes. A second important mechanism of action is the inhibition of adenosine receptors which are found in the brain.
Neutral and acidic heterocycle-containing compounds
Heterocycles such as pyrrole and pyridine derivatives are found commonly in drugs. The main reason for this is their similarity to the benzene ring which enables them to be quite lipophilic, and consequently they are able to penetrate the cell lipid membrane and to pass the blood–brain barrier; this also makes them suitable to be absorbed passively and quickly. They also take some of the characteristics of bases, which gives them some hydrophilicity and enables them to interact with the receptors, to accumulate inside microbial cells, to be distributed well in the blood stream and to be metabolised quickly and easily.
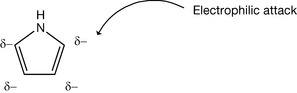
The most important heterocycles are the five- and six-membered rings. The six-membered rings are the derivatives of pyridine which is considered as a basic compound, and its derivatives are basic unless strongly electron withdrawing groups are added to the ring. Pyrrole, as discussed above, is considered either a neutral compound or a very weak acid. It was also mentioned that pyrrole has six pi-electrons according to the Huckel theory. These electrons are distributed to the carbon atoms of the small ring of the pyrrole, so the carbon atoms of pyrrole have greater electron density than those in a benzene ring since there are only five atoms in the ring. Because of this, the pyrrole derivatives are called pi-excessive compounds. This explains the susceptibility of pyrrole to the electrophilic substitution (Fig. 4.46) which is greater than that of benzene.
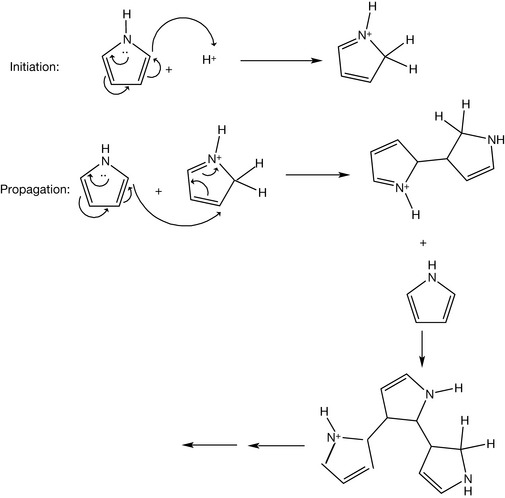
Pyrrole under acidic conditions has a tendency to polymerise instead of forming salts (Fig. 4.47).
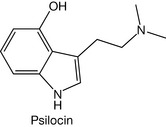
Indole, which is a fusion between pyrrole and benzene, also has the same acidic properties as the pyrrole ring. An example of an indole-containing drug is psilocin (Fig. 4.48).
Replacement of the pyrrole nitrogen with oxygen results in the formation of a furan, which is a neutral compound. Furan polymerises in acidic conditions as does pyrrole. Nitrofurantoin and dantrolene are examples of furan-containing compounds (Fig. 4.49).
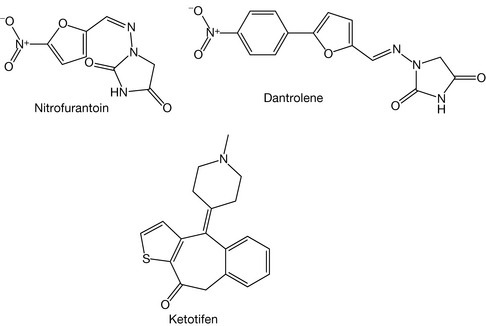
The substitution of the pyrrole nitrogen with a sulphur atom produces thiophene. Thiophene is stable in acidic solution (not polymerised) and is also a neutral compound. Ketotifen is an example of a drug containing thiophene (Fig. 4.49).
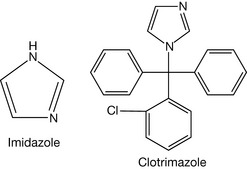
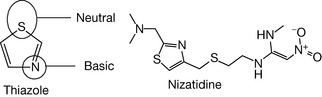
Addition of a nitrogen at the 3 position of the pyrrole results in formation of imidazole (Fig. 4.50). Examples of compounds containing this ring include the imidazole family of antifungal agents, e.g. clotrimazole (Fig. 4.50, see Ch. 24). Substitution of the 3 carbon of thiophene with a nitrogen produces thiazole (Fig. 4.51). The nitrogen of thiazole is still weakly basic (pKa 2.5), weaker than sp2 nitrogen in imidazole because the sulphur is a large atom and thus less effective than nitrogen at committing its lone pair as part of the aromatic sextet. An example of thiazole-containing compound is nizatidine (Fig. 4.51).
Substitution of the 3 carbon of a furan with a nitrogen produces oxazole (Fig. 4.52). The nitrogen is a very weak base (pKa = 0.8) because of the high value of the oxygen electronegativity. Moreover, the substitution of the 2 position of pyrrole with nitrogen forms pyrazole. One nitrogen is considered neutral, while the other nitrogen is considered as a weak base, weaker than imidazole because of the close proximity of the electronegative. Its sp2 nitrogen has a pKa = 2.5.
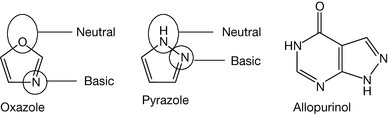
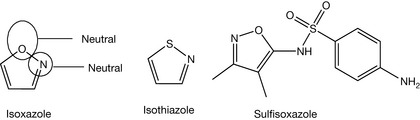
An example of a pyrazole-containing compound is allopurinol (Fig. 4.53). On the other hand, the ortho substitution of furan with nitrogen gives isoxazole, which is considered neutral and found in sulfisoxazole (Fig. 4.53). Isothiazole (Fig. 4.53) is also a neutral compound. Addition of electronegative atoms to imidazole, thiazole, oxazole, isoxazole, pyrazole and isothiazole make them neutral or acidic rings. Figure 4.54 shows drugs containing this type of ring.
 Self Test 4.1
Self Test 4.1