Chapter 24 Antifungal chemotherapy
Introduction
Fungal microorganisms constitute one of the largest groups of living organisms on the planet, with around 75 000 species having been described and an estimated additional 1.5 million fungal species awaiting discovery. Although some fungal species are found in an aquatic environment most are terrestrial. The vast majority of fungi are saprophytic, i.e. they secure food by the extracellular digestion of dead organic matter, such as plants. In nature, the mineralisation of organic matter by fungi plays a pivotal role in ensuring a sustainable ecosystem. These eukaryotic microorganisms are extensively utilised within industry to provide a number of important chemicals or products, e.g. alcohol and bread (Saccharomyces cerevisiae), vitamin B2 (Ashbya gossypii), the enzyme glucose oxidase used in diabetic test strips for monitoring glucose (Aspergillus niger), the enzyme rennin used in coagulation of milk during cheese making (Mucor miehei). In addition, many of the secondary metabolites originating from fungi have proved invaluable in the treatment of human infectious disease, e.g. griseofulvin (Penicillium griseofulvum) and β-lactam antibiotics such as penicillins and cephalosporins (Penicillium chrysogenum and Cephalosporium acremonium, respectively).
Incidence of fungal infections
Despite the large number of known fungal species, only around 300 species are recognised as being true or potential human pathogens. Most of these are opportunistic fungal pathogens responsible for relatively benign superficial or cutaneous infections (mycoses) in healthy humans, e.g. athlete’s foot (Trichophyton species). There are, however, a relatively small number of pathogenic or opportunistic fungi that can cause life-threatening systemic mycoses, e.g. aspergillosis (Aspergillus species). In the past two decades, the incidence of these systemic mycoses has steadily risen due to advances in medical technology such as:
Antifungal chemotherapy
Fungi, being eukaryotic cells, possess many of the cellular features observed in host mammalian cells. During the development of the early antifungal agents, limited knowledge of the fundamental differences between fungal and mammalian cell function meant that researchers were often trying to exploit relatively minor differences in fungal and mammalian biochemical pathways. As a result, early antifungal agents often had limited selectivity and/or an appreciable degree of host toxicity. Pertinent examples will be discussed later in the chapter. In recent years, an increased understanding of basic fungal and mammalian cell function has allowed pathways that are unique to fungi to be identified, and these are currently being exploited in the development of antifungal agents with high potency and specificity but with low host toxicity.
Inhibitors of DNA/RNA synthesis
The only drug used clinically in this class is flucytosine.
Flucytosine
This drug is largely active against yeasts and some Aspergillus species. Flucytosine is a pyrimidine analogue that is actively taken up into cells via the cell membrane enzyme cytosine permease, a natural transporter of cytosine, adenine, guanine and hypoxanthine (Fig. 24.1). Intracellular flucytosine is then converted by the enzyme cytosine deaminase into the antimetabolite 5-fluorouracil, thus it acts as a prodrug (the mechanism of its action is discussed in Ch. 21). The 5-fluorouridine triphosphate metabolite of 5-fluorouracil results in the formation of abnormal RNA, ultimately leading to disruption in protein synthesis. The 5-fluorodeoxyuridine monophosphate metabolite inhibits thymidylate synthase, an essential enzyme required for the formation of thymidine, one of the pyrimidine bases of DNA (see Ch. 21). The selective toxicity of flucytosine against fungal cells results from the absence or limited expression of cytosine deaminase within mammalian cells, which means that its toxicity is selective for these cells.
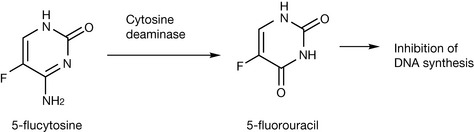
Figure 24.1 Selective conversion of flucytosine to the anti-metabolite 5-fluorouracil by fungal enzymes.
Intrinsic (natural) resistance or the development of resistance (acquired) to flucytosine during therapy is common; therefore susceptibility testing is recommended before and during therapy. Flucytosine is usually administered in conjunction with amphotericin B in the treatment of systemic candidiasis, cryptococcosis and torulopsosis. The combination of flucytosine and amphotericin B is synergistic. However, most filamentous fungi are resistant to flucytosine since they do not express cytosine permease or cytosine deaminase.
Description and preparations
Flucytosine is a white to off-white, crystalline powder, with a solubility of 1 g in 67 mL water at 25°C. Prolonged storage of aqueous flucytosine solutions at temperatures higher than 25°C can result in flucytosine decomposing to 5-fluorouracil, whilst storage below 18°C may result in drug precipitation. It has a low octanol/water partition coefficient. Flucytosine has two pKa values of 2.9 (protonation of weak primary amine group attached to the ring) and 10.71 (deprotonation of amide nitrogen in position 1). Typical preparations include:
Pharmacokinetics
Flucytosine is usually administered intravenously over 20 to 40 minutes. Protein binding is low (approximately 2–4%) and volume of distribution is high (0.7–1 L/kg) with cerebrospinal fluid (CSF) concentrations comparable to serum concentrations. Approximately 90% of a drug dose is excreted unchanged in the urine. The drug has a half-life of 3–6 hours in adult patients with normal renal function. Patients with renal impairment should be given smaller doses which can be estimated according to their creatinine clearance. Only a small percentage of flucytosine is metabolised to 5-fluorouracil by the body, with an area under the curves (AUC) ratio of 5-fluorouracil to flucytosine of 4% being recorded. This may, however, explain why high doses of intravenous flucytosine can result in bone marrow suppression. Oral bioavailability of flucytosine is high, with bioavailability greater than 80% reported. Gastrointestinal microflora is capable of converting flucytosine to fluorouracil and it has been speculated that the oral route may therefore be associated with a greater risk of bone marrow toxicity.
Fungal resistance to flucytosine
The most common mechanism of intrinsic resistance is a decreased uptake of flucytosine by the fungus by a reduction in cytosine permease activity, which results in reduced concentrations of drug entering into the cell. The development of resistance to flucytosine during therapy may be a result of reduced expression of, or a deficiency in, an enzyme at any step in the intracellular metabolism of flucytosine. However, the most common resistance mechanisms observed in clinical isolates are due to either reduced cytosine deaminase or URPTase activity.
Microtubule inhibitors
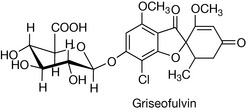
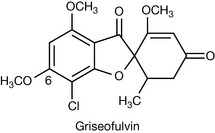
Griseofulvin (Fig. 24.2) is the only drug in this class.
Griseofulvin
Dermatophyte fungi are moulds that specifically infect the keratinised layers of the skin by virtue of their ability to degrade keratin. Three genera, Epidermophyton, Microsporum and Trichophyton, are responsible for causing ringworm or tinea infections (dermatophytosis). Griseofulvin is used exclusively for the oral treatment of dermatophyte infections where topical therapies have failed or are inappropriate.
Griseofulvin is produced by various strains of Penicillium griseofulvum. It has a narrow spectrum of antifungal activity, being inactive against other important fungal pathogens such as Candida spp. and Aspergillus spp. It has no other antimicrobial activity.
Griseofulvin appears to be fungistatic, primarily exerting its antifungal activity by interacting with the microtubule proteins, α- and β-tubulin, thereby inhibiting the assembly of functional microtubules that comprise the spindle apparatus of the fungal cell. Disruption of the spindle apparatus inhibits mitosis and results in cell division arrest. The exact binding site of griseofulvin on the microtubules appears to be distinct from drugs such as colchicine and Vinca alkaloids (Ch. 21), and may account for its relatively selective toxicity against fungal cells. However, griseofulvin has been shown to be teratogenic (causing fetal abnormalities) in rats and has induced aneuploidy (abnormal numbers of chromosomes following cell division) in mammalian cells both in vitro and in vivo. For this reason griseofulvin should not be administered to pregnant women or women likely to become pregnant within 1 month of finishing treatment. In addition, males should not father children within 6 months of treatment.
Description and preparations
Griseofulvin is a white to creamy or yellowish white, odourless crystalline powder. It is practically insoluble in water, slightly soluble in methanol and ethanol, soluble in acetone and chloroform, and freely soluble in dimethylformamide and tetrachloroethane. Griseofulvin is stable at room temperature and to light exposure. Typical preparations include:
Pharmacokinetics
The small intestine is the main absorption site of griseofulvin. Oral bioavailability is typically low, being less than 50%, and variable, but can be enhanced by administering griseofulvin with or immediately after a fatty meal. Oral bioavailability is also limited by the rate of drug dissolution and can therefore be enhanced by reducing drug particle size. For this reason most pharmacopoeias specify the particle size of griseofulvin powder, e.g. EP stipulates a particle size range of up to 5 μm in maximum dimension, with an occasional number of larger particles that exceeds 30 μm permitted. Following oral absorption, peak serum levels are observed after 4 hours. In plasma, around 84% of griseofulvin is bound to plasma proteins, predominantly albumin. Griseofulvin has a volume of distribution of 1.2–1.4 L/kg. The terminal half-life is extremely variable between subjects with a range of 9.5–21 hours reported. Griseofulvin is metabolised in the liver, with the major metabolite being 6-desmethylgriseofulvin or its glucuronide derivative. These metabolites have no residual antifungal activity. Around 50% of the metabolite is excreted in the urine with another 30% in the faeces.
 Self Test 24.1
Self Test 24.1A significant proportion of griseofulvin accumulates in keratinous tissues, with concentrations exceeding plasma levels during therapy. However, concentrations rapidly fall after discontinuing therapy due to griseofulvin being excreted in sweat. The resulting high local keratinous tissue concentrations of griseofulvin facilitate its antifungal activity against dermatophyte infections. However, the speed at which griseofulvin accumulates into infected keratinous tissue is dependent on the thickness of keratin at the site of infection and therefore determines the duration of therapy. Infected hair or skin, having a relatively thin keratinous layer, responds to relatively short courses of treatment, typically between 4 and 6 weeks. In contrast, toe- or fingernails, with a thick keratin layer, require prolonged therapy of between 6 and 12 months.
Drugs that interfere with fungal cell membrane function
Sterol components of the fungal cell membrane play an important role in the normal functioning of the cell membrane by modifying its physical integrity. The major cell membrane sterol present varies among eukaryotes. For example, in plants β-sitosterol and stigmasterol are the major membrane sterols whilst in mammalian cells cholesterol is the most common sterol. As a consequence, individual eukaryotic species will possess slightly different biosynthetic pathways (enzymes) to allow manufacture of their preferred sterol. The importance of specific sterols to cell viability has resulted in many pharmaceutical companies directing resources to develop drugs that will specifically inhibit key stages in the fungal sterol biosynthetic pathway that are unique to, or sufficiently different from, the corresponding mammalian pathway. A summary of drugs interfering with fungal sterol biosynthesis is provided in Figure 24.3.
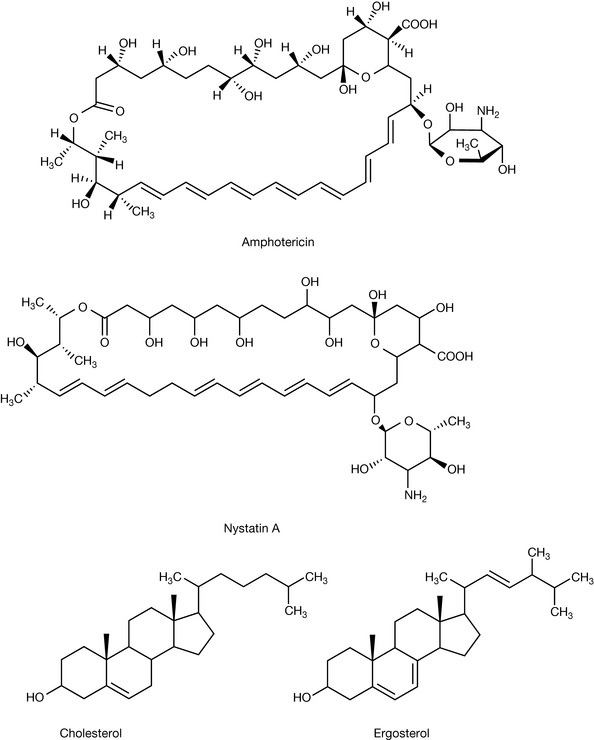
Polyene macrolide antifungal agents
Of the 200 different known polyene macrolides (large-ring lactones), only amphotericin B and nystatin (which is a mixture of related compounds the major component being nystatin A1) have an acceptable toxicity profile for clinical use as antifungal agents. The chemical structures of amphotericin B and nystatin A1 are shown in Figure 24.3.
Amphotericin B
This drug was initially isolated and characterised from Streptomyces nodosus in 1956. Amphotericin B has broad antifungal activity against a wide range of yeasts and moulds including Aspergillus spp., Mucor spp., Candida spp., Cryptococcus neoformans and Histoplasma capsulatum. It remains the drug of choice in the treatment of life-threatening systemic mycoses. In addition, it also has potent activity against Leishmania protozoa. It has no antibacterial activity.
Description and preparations
Amphotericin B is a yellow to orange, odourless powder. It is practically insoluble in water, ethanol and ether. It is slightly soluble in methanol and soluble in dimethylformamide, dimethylsulphoxide and propylene glycol. It has two pKa values of 5.7 (carboxylic acid functional group) and 10 (amine functional group on aminosugar attached to macrolide ring through a glycoside linkage). At physiological pH it exists as a zwitterion in aqueous solution. The zwitterionic species can form head-to-tail dimers at concentrations above 1 μm and soluble high-order aggregates (micelles) above 10 μm. The seven conjugated double bonds in amphotericin B make the molecule prone to oxidation. Amphotericin B is therefore stored between 2° and 8°C in airtight containers and protected from light. In aqueous solution amphotericin B is inactivated by extremes of pH due to hydrolysis of the glycosidic linkage. It has a high octanol/water partition coefficient. Typical preparations include:
Pharmacokinetics
There is no systemic absorption of amphotericin B following oral administration in the form of a lozenge, tablet or suspension for the treatment of oral, perioral or intestinal fungal infections.
Intravenous administration of the conventional micellar dispersion (Fungizone®) diluted in 5% w/v dextrose is usually performed over 4 to 6 hours and results in amphotericin B becoming widely distributed throughout well-perfused tissues, particularly the heart, lung, liver and spleen. The high molecular weight of amphotericin B precludes its entry into the central nervous system (CNS). Following a single intravenous infusion it has a volume of distribution of 4 L/kg and a half-life of 24–48 hours. Upon prolonged administration, the half-life increases to around 15 days. Amphotericin B is extensively protein bound in the plasma, mainly to β-lipoproteins. Amphotericin B does not appear to be metabolised in vivo and is slowly excreted by the kidneys.
Clinical toxicity of amphotericin B (Fungizone®)
The clinical usefulness of amphotericin B in the treatment of systemic mycoses is limited by its toxicity profile. Acute adverse reactions during and in the immediate post-infusion period include nausea, vomiting, headache, fever and chills. Thrombophlebitis at the infusion site is also a common problem. These acute reactions are dose related and can be ameliorated by pre-infusion administration of paracetamol, chlorphenamine and hydrocortisone. Chronic adverse reactions can include renal, cardiovascular and haematological toxicity. Of these, renal toxicity is the most clinically prevalent with over 80% of all patients receiving amphotericin B exhibiting some renal impairment. The extent of kidney impairment is dose related and often irreversible upon cessation of therapy.
Antifungal activity of amphotericin B
The antifungal activity and toxicity of amphotericin B is intimately related to its chemical structure. Amphotericin B is a surface-active molecule, possessing a hydrophilic (polyhydroxyl backbone on the macrolide ring plus the aminosugar moiety) and hydrophobic side (conjugated heptadiene system on the macrolide ring). Due to this surface activity, amphotericin B in aqueous solution can interact with the sterol component of biological membranes. This interaction is thought to increase membrane permeability through the formation of pores. The increased permeability results in the loss of intracellular protons, cations and small molecules such as amino acids and ultimately causes cell death. In addition, amphotericin B has immunomodulatory activity and can induce oxidative damage. The importance of these two mechanisms to the antifungal activity of amphotericin B remains to be determined.
Mammalian toxicity – molecular mechanisms
The selective toxicity of amphotericin B against fungal rather than mammalian cells is modest and arises from the higher binding affinity of amphotericin B for the main fungal membrane sterol, ergosterol, compared to the main mammalian sterol, cholesterol. This higher affinity is, however, relatively small, approximately 14 times greater for ergosterol compared to cholesterol, and partly explains the narrow therapeutic index of the drug. The similar binding affinities of amphotericin B for both sterols is not surprising given their similar molecular structure (Fig. 24.3).
As previously mentioned, amphotericin B can exist as a number of molecular species in aqueous solution depending on the local concentration of the drug. Upon intravenous administration of Fungizone®, the amphotericin B – deoxycholate micelle – dissociates. This results in high local concentrations of amphotericin B and as a consequence amphotericin B can coexist in the plasma as a number of species: in a monomolecular (uncomplexed) state, as a dimer or as soluble high-order complexes. All of these amphotericin B species are capable of interacting with ergosterol, but only the high-order complexes appear to have appreciable affinity for cholesterol. Therefore the rapid dissociation and release of amphotericin B from Fungizone® exacerbates toxicity by promoting the formation of high-order complexes.
Amphotericin B, when released from Fungizone®, can also associate with the cholesterol component of high density lipoprotein (HDL). Lipoproteins are lipid–protein complexes involved in the circulation and distribution of lipids in the body. The amphotericin B is subsequently transferred from HDL to low density lipoprotein (LDL) by a lipid transfer protein. The resulting amphotericin B-LDL complex can then enter mammalian cells expressing LDL receptors by the process of receptor-mediated endocytosis. Internalised amphotericin B is then capable of interacting with cholesterol present in the cell membrane. Therefore the natural biodistribution of amphotericin B contributes to its clinical toxicity profile.
Drug delivery technology – modulating the clinical profile of amphotericin B
The clinical disadvantages associated with the use of Fungizone® provided the impetus for the development of alternative delivery systems that had increased efficacy but reduced toxicity. Three alternative lipid-based amphotericin B delivery systems have subsequently been developed and introduced into clinical practice.
AmBisome®
This is a sterile lyophilised liposomal formulation. Each vial contains 50 mg amphotericin B, 194 mg hydrogenated soybean phosphatidylcholine, 84 mg distearoylphosphatidylglycerol and 52 mg cholesterol. Upon reconstitution with sterile water for injection, small unilamellar liposomes that have a mean diameter of 80–100 nm are formed. Adult dose: 1–3 mg/kg daily.
Amphocil®
This is a sterile lyophilised colloidal dispersion. Each vial contains 50 or 100 mg amphotericin B and sodium cholesterol sulphate approximately equimolar amount. Upon reconstitution with sterile water for injection it forms a colloidal dispersion. Adult dose: 1–4 mg/kg daily.
Abelcet®
This is a sterile aqueous suspension of a lipid-amphotericin B complex. Each 20 mL vial contains 100 mg amphotericin B, 30 mg/L α-dimyristoylphosphatidylglycerol, 68 mg/L α-dimyristoylphosphatidylcholine. Adult dose: 5 mg/kg daily.
Each of these formulations changes the in vivo pharmacokinetics of amphotericin B with the net result of minimising exposure of the kidneys to it and thus reducing the incidence of toxicity, particularly nephrotoxicity. The lipid-based delivery systems also enhance clinical efficacy by promoting the delivery of amphotericin B to organs where fungal infections commonly reside, e.g. liver and lungs. The severity and incidence of acute adverse reactions with these lipid-based formulations is also less compared to Fungizone®. The exact biodistribution of amphotericin B when administered as a lipid-based formulation appears to be formulation dependent. The reduced toxicity of the lipid-based formulations also appears to be a result of the slow release of amphotericin B from the formulations following administration. This ensures that low concentrations of amphotericin B are maintained in the plasma, thereby minimising the formation of the high-order complexes of amphotericin B that are especially toxic to mammalian cells. In addition, some of the delivery systems, e.g. AmBisome®, appear to reduce toxicity by restricting the formation of amphotericin B-LDL complexes.
Comparative studies of the lipid-based amphotericin B formulations with Fungizone® would appear to indicate that they are less potent on a weight-for-weight basis compared with amphotericin B. However, with the reduced toxicity profile of the lipid formulations, higher doses of amphotericin B can be tolerated by the patient, thereby offsetting any loss in drug potency experienced due to its formulation in a lipid delivery system.
Nystatin
This polyene macrolide antibiotic is isolated from Streptomyces nourseri and S. albidus. Nystatin is considered too toxic for parenteral administration. It is composed of a number of tetraene compounds (compounds that contain four conjugated double bonds), the principle component being nystatin A1 (Fig. 24.3) which has a very similar structure to amphotericin B. Its use is mainly reserved for the treatment of oral, perioral, intestinal, vulvovaginal or cutaneous Candida spp. infections.
Description and preparations
Nystatin is a yellow to light brown hygroscopic powder with an odour characteristic of cereals. The activity of nystatin is unusual in that it is expressed in units. A number of similar standards coexist with the European Pharmacopoeia stating that potency is not less than 4400 IU per milligram. It is insoluble in ethanol, chloroform and ether. It is very slightly soluble in water, slightly soluble in methanol, and soluble in dimethylformamide. Nystatin is hygroscopic and prone to oxidation. It should be protected from light and stored between 2° and 8°C in an airtight container. Typical preparations include:
All nystatin preparations are well tolerated by patients. Very occasionally nausea, vomiting and diarrhoea are reported following oral administration and skin irritation following topical application. Commercial development of a parenteral liposomal formulation of nystatin is in progress for the treatment of systemic mycoses and it is anticipated that this will provide similar clinical benefits to those observed with AmBisome.
Azole antifungal drugs
This is the largest group of antifungal agents and they can be subdivided on the basis of their chemical structure into imidazoles and triazoles. The mode of action of all azole antifungals is identical in that they interfere with fungal sterol biosynthesis by inhibiting fungal cytochrome P450-Erg11p (also known in the nomenclature as lanosterol 14-α-demethylase or Cyp51p) which catalyses the reaction shown in Figure 24.4. This depletes the fungal plasma membrane of ergosterol, an essential membrane sterol, and concomitantly leads to the accumulation of 14-α-methylated precursors (the exact precursors reflect the unique biosynthetic pathway of individual fungal species). The net result is an alteration of plasma membrane fluidity and functional disruption of important membrane-bound enzymes such as chitin synthase and arrest of cell growth (fungistatic action) or cell death (fungicidal action).
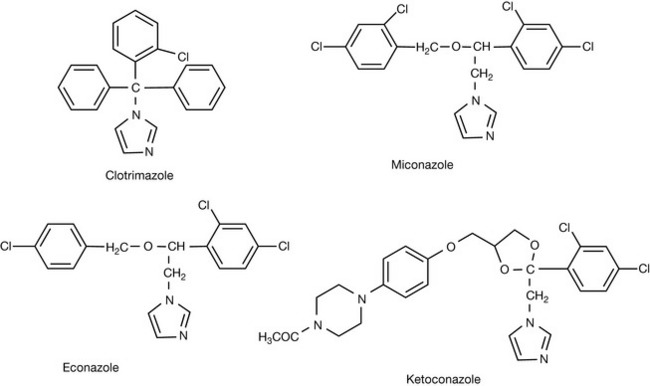
Extensive research has been undertaken in order to understand the structure–activity relationship for the interaction between azole antifungals and cytochrome P450-Erg11p so that optimal activity can be realised against a wider spectrum of fungal pathogens and to address the issue of emerging (acquired) resistance to some of the pre-existing azole antifungal agents in current clinical usage.
At the active site within the cyp450 lies a protoporphyrin IX ring containing iron (III) where cyclic oxidation/reductions are performed. The azole antifungal agent has the ability to bind to this active site such that it prevents access, binding and subsequent transformation of the endogenous, 14-α-methylated sterol ligand.
The imidazole or triazole rings of the antifungal drugs orientate themselves perpendicular to the plane of the protoporphyrin ring, with the lone pair of electrons of the N3 or N4 atoms in the imidazole or triazole rings, respectively, becoming coordinated with the iron atom. The remainder of an individual azole molecule can interact with hydrophobic and aromatic residues within the substrate access channel of the apoprotein. As there is slight variation in the conformation of the active site and in the amino acid composition of the apoprotein between fungal species, this controls the potency and spectrum of antifungal activity of a given azole.
Inhibition of the Cyp450-Erg11p by an azole is non-competitive with respect to the endogenous substrate, leading to a greater disruption of the sterol metabolic pathway than that resulting from a competitive inhibitor. Correspondingly, as cyp450 is not a single enzyme, but consists of a family of closely related isoforms, this explains why all of the azoles, to a greater or lesser extent, can also interact with mammalian cyp450 enzymes, resulting in toxicity.
Imidazoles
Clotrimazole
Clotrimazole (Fig. 24.4) was the first compound of its class and was investigated in the early 1970s as a parenteral agent for the treatment of systemic mycoses. However, initial trials were discouraging. It became apparent that the primary limitation of clotrimazole administered by the parenteral route was that it induced the hepatic microsomal enzymes involved in its own metabolism. Therefore clotrimazole blood levels quickly became undetectable (i.e. below clinically active concentrations) soon after initiating therapy.
As a consequence of these preliminary studies, the systemic use of clotrimazole as an antifungal agent was abandoned and, instead, its use for the treatment of cutaneous and mucocutaneous infections was pursued. Clotrimazole administered by these routes proved highly successful and it remains a useful clinical agent for the treatment of yeast and dermatophyte infections.
Description and preparations
Clotrimazole is a white to pale yellow crystalline powder. It is a lipophilic substance with a reported logP value of 3.5. It is practically insoluble in water, and soluble in methanol, ethanol, methylene chloride, chloroform and acetone. It should be stored in an airtight container and protected from light. Typical preparations include:
Pharmacokinetics
Less than 1% of clotrimazole enters the systemic circulation following topical application whilst between 3% and 10% of a dose has been reported to do so following vaginal administration. As a result, very little of an applied dose (typically <1%) enters into the systemic circulation. Additionally, with higher systemic doses achieved via vaginal application, the course duration 1–6 days, allied to low blood concentrations, is not enough to induce metabolism.
Miconazole
Like clotrimazole, miconazole was originally developed as an intravenous and oral antifungal agent. The intravenous formulation was discontinued due to anaphylactic reactions associated with the injection vehicle excipient, polyoxyl 35 Castor Oil (Cremophor EL), used to enhance the solubility of miconazole. Unlike clotrimazole, repeated administration does not induce the hepatic microsomal enzymes involved in its own metabolism. Following oral administration, around 20% of a dose is systemically absorbed where it undergoes oxidative O-dealkylation and oxidative N-dealkylation prior to excretion (Fig. 24.5). Approximately 40% of the administered dose is excreted unchanged in the faeces. The faecal route of excretion for metabolites predominates over urinary excretion. Oral administration of miconazole has been observed to interfere with the metabolism of other therapeutic agents including phenytoin, warfarin and quinidine.
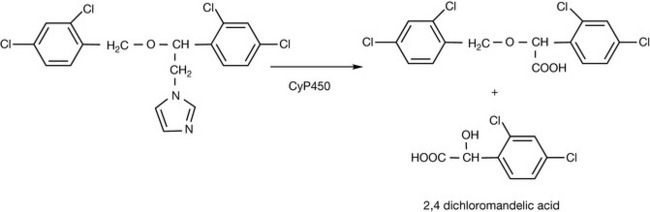
The use of miconazole is now restricted to the treatment of mucocutaneous, intestinal and vulvovaginal fungal infections. A similar range of preparations to clotrimazole are available for the treatment of these infections.
Econazole
Oral and intravenous routes of administration for the treatment of systemic fungal infection using econazole were abandoned when it became apparent that econazole was readily metabolised to inactive compound. Consequently, the clinical uses of econazole have been restricted to topical application like those of clotrimazole and miconazole.
Ketoconazole
When introduced in the early 1980s, ketoconazole (Fig. 24.4) was seen as a useful clinical development as it represented the first imidazole antifungal that had sufficient oral bioavailability to make it useful in the treatment of cutaneous (dermatophytic) and systemic (mycotic) yeasts, with weaker activity against a limited number of moulds (e.g. Aspergillus spp.) infections.
Description and preparations
Ketoconazole is a white to off-white crystalline powder. It is lipophilic with a reported logP value of 3.5. It is practically insoluble in water, sparingly soluble in ethanol, but soluble in methanol and methylene chloride. It should be protected from light.
Pharmacokinetics
Following oral administration, approximately 75% of an administered ketoconazole dose is absorbed. However, there is considerable inter- and intrapatient variation, with oral absorption impaired when gastric acidity is decreased due to clinical pathology, e.g. achlorhydria, or due to concurrent administration of agents that decrease gastric acidity, e.g. antacids, H2-receptor antagonists or proton pump inhibitors, or whether the patient is in a fed or fasted state (absorption is enhanced in the fed state).
Peak plasma levels occur around 2 hours after oral administration, with a terminal half-life of around 8 hours. Ketoconazole is extensively bound (>95%) to plasma proteins, mainly albumin, which results in relatively low free drug concentrations. Therefore, with clinical dosing schedules, ketoconazole tends to be fungistatic rather than fungicidal, which is a potential concern if the patient is immunocompromised. Ketoconazole is widely distributed throughout the body but penetration into CSF is low, excluding its use in the treatment of fungal meningitis.
Ketoconazole is extensively metabolised by the liver following gastrointestinal absorption into several inactive metabolites that are predominantly excreted through bile into the faeces. Only a small fraction of the drug or its metabolites are excreted unchanged in the urine. The major metabolic pathways identified for ketoconazole include oxidation, cleavage, degradation and scission of the imidazole and piperazine rings; oxidative O-dealkylation; aromatic hydroxylation and N-deacetylation. Experimentally, in isolated rat microsomes, the generation of N-deacetyl ketoconazole metabolites has been implicated in the hepatotoxicity that is occasionally associated with ketoconazole therapy. Oxidative attack by flavin-containing monooxygenases on the N-1 position of the N-deacetyl ketoconazole metabolite results in the generation of ring-opened dialdehydes that are cytotoxic.
Ketoconazole also suffers from a number of other clinical disadvantages. At daily doses greater than 400 mg, ketoconazole may reversibly inhibit endogenous steroid synthesis with resultant suppression of testosterone and cortisol and disruption of endocrine function. In addition, ketoconazole extensively interferes with the metabolism of many drugs including warfarin, ciclosporin, HMG-CoA enzyme inhibitors, HIV protease inhibitors and calcium channel blockers to name but a few. These limitations are primarily due to the relative lack of binding specificity that ketoconazole has between mammalian and fungal cyp450 enzymes, and this was an important determinant in the development of newer azole analogues that have greater selectivity and enhanced antifungal activity.
Triazoles
Fluconazole
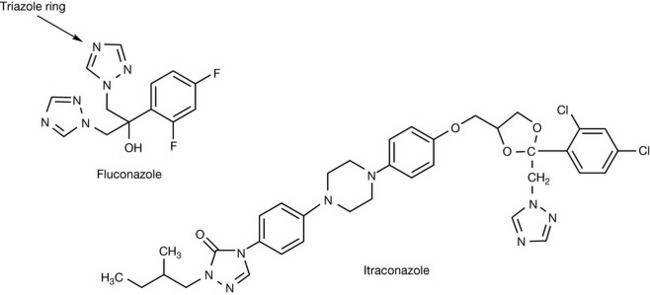
Researchers at Pfizer sought to overcome the limitations of the existing imidazole antifungal compounds by developing analogues of tioconazole that had greater metabolic stability and which were less lipophilic, thereby improving oral bioavailability and delivering high plasma concentrations of unbound drug respectively. After 2 years of extensive research they concluded that the imidazole moiety, which was metabolically vulnerable, had to be replaced with something that had greater metabolic stability. Of the analogues that were synthesised, only one compound, UK-46245, containing a 1,2,4-triazoyl-1-yl group, showed promising activity, with antifungal activity being three times greater than the corresponding imidazole derivative in an in vivo model. Hypothesising that this was due to enhanced metabolic stability, the researchers replaced the metabolically vulnerable hexyl chain with a second triazole group to give compound UK-47265. This compound was a major breakthrough in that it proved to be up to 100 times more potent than ketoconazole, with excellent pharmacokinetic properties. However, during further development, the compound proved to have unacceptable hepatotoxicity and it was also teratogenic in rodents. Subsequent research demonstrated that a 2,4-difluorophenyl derivative, compound UK-49858, was devoid of hepatotoxicity and teratogenicity and retained the excellent antifungal activity and pharmacokinetic profile observed with compound UK-46245. UK-49858 was subsequently renamed as fluconazole (Fig. 24.6). The triazole ring may look strongly basic but, in common with many π-deficient rings (see Ch. 4), it is only very weakly basic with a pKa value <2.
Fluconazole is fungistatic at therapeutic concentrations and has good activity against Candida albicans infections. However, some non-albican Candida species (e.g. C. glabrata, C. parapsilosis, C. krusei and C. tropicalis) are less sensitive to fluconazole. Fluconazole is active against Trichophyton spp, Cryptococcus neoformans, Histoplasma capsulatum, Microsporum spp. and Epidermophyton spp. Fluconazole, however, has no useful activity against moulds such as Aspergillus spp.
Description and preparations
Fluconazole is a white crystalline powder. It is reported to exist in three polymorphic forms and as a hydrate, with form III being used in clinical formulations. Fluconazole has moderate lipophilicity with a reported logP value of 0.5. Fluconazole is slightly soluble in water and propan-2-ol, sparingly soluble in ethanol and chloroform, soluble in acetone and freely soluble in methanol. Typical preparations include:
The adult dose is 50–400 mg once a day with dose and duration of therapy depending upon the clinical indication. Fluconazole has greater specificity than older imidazole compounds such as ketoconazole for fungal cyp450-Erg11p than mammalian cytochrome enzymes, so disruption of endogenous steroid production is not a clinical problem. Fluconazole, however, is still associated with a number of important cyp450-mediated drug interactions with drugs including coumarins, sulphonylureas, ciclosporin and antiretrovirals.
Pharmacokinetics
Fluconazole has good oral bioavailability irrespective of fed or fasted state, with greater than 90% of a dose being absorbed. Peak plasma levels are proportional to dose and are achieved within 1–2 hours of oral dosing. Plasma protein binding, half-life and the volume of distribution at steady-state are 10%, 25–30 hours and 0.55 L/kg, respectively. Fluconazole is found in body fluids such as saliva, breast milk and vaginal secretions at levels comparable to those in the plasma with CSF concentrations around 80% of plasma levels.
Only 11% of a dose is metabolised, with 6.5% being a glucuronide conjugate and 2% an N-oxide. The major route of elimination for fluconazole and its metabolites is renal excretion, with only 2% being excreted in the faeces.
Itraconazole
Itraconazole is a structural analogue of ketoconazole in which the metabolically susceptible imidazole group has been replaced by a triazole moiety and the side chain has been further extended (Fig. 24.6). Its spectrum of antifungal activity is similar to that of fluconazole, but in addition it also has activity against Aspergillus spp. The extended side chain of itraconazole allows it to bind with greater affinity to the apoprotein structure of fungal cytochrome P450-Erg11p than ketoconazole, enhancing antifungal potency and, in parallel, reducing its affinity for mammalian cytochrome P450 enzymes involved in endogenous steroid biosynthesis. However, like ketoconazole, itraconazole still has the capacity to interfere with the metabolism of many important drug classes through interaction with cyp4503A4.
Description and preparations
Itraconazole is an odourless beige crystalline powder with a bitter taste. The racemate mixture of 4 diastereomers (2 enantiomeric pairs) is used in clinical formulations. Itraconazole is very lipophilic with a reported logP value of 5.7. Itraconazole is practically insoluble in water and very sparingly soluble in ethanol. Typical preparations include:
The adult dose is 100–200 mg once or twice a day with dose and duration of therapy depending upon the clinical indication.
Pharmacokinetics
The absorption of itraconazole from the gastrointestinal tract shows similar pH dependency as ketoconazole. The effect of fed and fasted states on oral absorption is formulation dependent with peak plasma levels doubled when capsules are taken with food and lowered by 25% when the oral solution is taken with food. Both formulations, however, have greater than 90% oral bioavailability with time to peak plasma concentration, plasma protein binding, terminal half-life and volume of distribution at steady-state values of 2–5 hours, 99.8%, 20–40 hours and 11 L/kg, respectively. Itraconazole also extensively accumulates in well-perfused organs such as muscle, lung, spleen, liver and kidneys. Itraconazole has unusual pharmacokinetics in that it preferentially accumulates in keratinous tissues of the hair, nails and skin, providing drug concentrations that are four- to tenfold higher than the corresponding plasma concentrations. This permits itraconazole to be administered in a discontinuous or ‘pulsed’ manner for 1 week out of every 4 weeks for 3 months in the treatment of fungal infections of the nails since high, sustained, concentrations are retained within these keratinous tissues. Itraconazole undergoes extensive phase I hepatic metabolism with greater than 30 metabolites identified, many of which are biologically active. The major metabolite is hydroxyitraconazole, where plasma concentrations are two to three times higher than those of itraconazole with comparable antifungal activity. Faecal excretion of itraconazole accounts for between 3% and 18% of the drug, with effectively none undergoing renal excretion. Around 40% of metabolites are excreted in the urine, with the remainder being excreted in the bile.
Interestingly, recent evidence suggests that the stereoisomers of itraconazole are preferentially metabolised to hydroxyitraconazole in humans by CYP3A4 enzymes. However, the resultant impact on itraconazole pharmacokinetics and/or pharmacodynamics and clinical efficacy as an antifungal agent remain to be established. However, the importance of stereochemistry on clinical antifungal activity is not surprising, given that the enzyme is constructed from L-amino-acids.
Second-generation triazole antifungals
Voriconazole
Voriconazole resulted from the continuing efforts of Pfizer scientists to retain many of the excellent clinical features of fluconazole whilst extending the spectrum of antifungal activity to include filamentous fungi. Early studies highlighted that the inclusion of a methyl group α to one of the fluconazole triazole groups increases potency against Aspergillus fumigatus. Subsequent experimentation demonstrated that replacement of one of the triazole rings with a 6-membered heterocyclic ring such as pyridine or pyrimidine, whilst retaining a methyl group α to the heterocyclic ring, further enhanced potency against A. fumigatus. The inclusion of fluorinated pyridine or pyrimidine analogues also enhanced antifungal activity, by reducing the susceptibility of the heterocyclic rings to metabolic oxidation, thereby enhancing their half-lives. On the basis of potency and better solubility profile the fluoropyrimidyl analogue was selected for further evaluation. Antifungal testing of individual diastereomers, isolated using a combination of chromatography and recrystallisation following salt formation with the optically active (1R)-10-camphosulfonic acid, indicated that antifungal activity resided almost entirely with the (2R, 3S) enantiomer, compound UK-109496, which was renamed as voriconazole (Fig. 24.7).
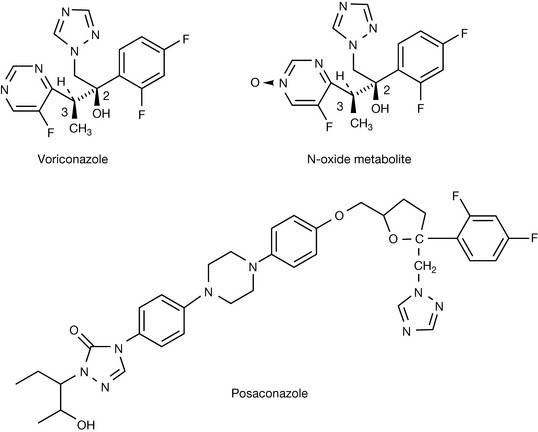
Voriconazole exhibits broad-spectrum activity against a wide range of clinically important fungal infections such as Candida (including fluconazole-resistant species such as C. glabrata and C. krusei), Aspergillus, Scedosporium and Fusarium spp. and atypical pathogens including Acremonium, and Chyrsosporium spp. It has no useful activity against zygomycetes such as Mucor or Rhizopus spp.
Description and preparations
Voriconazole is non-hygroscopic white to light-coloured crystalline powder. Voriconazole is lipophilic with a reported logP value of 2.6. Voriconazole is a weak base that has a maximum aqueous solubility of 2.7 mg/mL at pH 1.2. It is soluble in methanol and ethylacetate. Typical preparations include:
The adult oral dose is 400 mg every 12 hours on the first day of therapy before reducing to 200 mg every 12 hours (or 300 mg every 12 hours if inadequate clinical response) on subsequent days. Tablets should be administered at least 1 hour before, or 1 hour after food, with the oral suspension administered at least 1 hour before or 2 hours after food. Intravenous therapy is initiated at 6 mg/kg every 12 hours on the first day of therapy before reducing to 4 mg/kg every 12 hours on subsequent days. Duration of therapy is dependent upon clinical and mycological response.
Pharmacokinetics
The absorption of voriconazole following oral administration is independent of gastric pH with oral bioavailability being around 96% when taken 1 hour before food. The time to peak plasma is 1–2 hours. During multiple dosing regimens, time to peak plasma and AUC are reduced by 34% and 24%, respectively, when administered with high-fat meals. Plasma protein binding is modest at 58% compared to the rest of the triazole family of antifungal agents and this partly explains the relatively high volume of distribution at steady-state of 4.6 L/kg observed during voriconazole therapy. Voriconazole therefore is extensively distributed in all tissues including the cerebrospinal fluid.
The major route of elimination of voriconazole is hepatic metabolism, followed by renal excretion. Voriconazole is a substrate for the three enzymes, CYP2C19, CYP2C9 and CYP3A4. The major metabolite of voriconazole results from N-oxidation of the fluoropyrimidyl moiety, and accounts for 72% of circulating metabolites (Fig. 24.7). The N-oxide metabolite of voriconazole has no useful antifungal activity. In vivo studies indicate that the polymorphic enzyme CYP2C19 is significantly involved in voriconazole metabolism. Consequently 2–5% of Caucasians and blacks and 15–20% of Asian individuals who are functionally deficient or absent in this enzyme can be expected to be poor metabolisers of voriconazole. In these individuals the AUCs are approximately four times higher than subjects who are homozygous extensive metabolisers. Heterozygous extensive metabolisers have AUCs that are twofold higher than the homozygous extensive metabolisers.
In vitro studies indicate that CYP2C19 and CYP2C9 are high-affinity, low-capacity enzymes, whilst CYP3A4 has a low affinity and high capacity for voriconazole. This explains the observation that voriconazole pharmacokinetics are non-linear due to the fact that it can readily saturate the high-affinity, low-capacity enzymes and this results in a disproportionate increase in bioavailability observed with increasing dose. For example, increasing the oral dose from 200 mg twice a day to 300 mg twice a day has been reported to produce a 2.5-fold increase in the AUC0-∞. The terminal half-life of voriconazole is dependent on dose but has been reported to be around 6 hours following a 200-mg oral dose.
The oral dose of voriconazole does not have to be adjusted in patients who have renal impairment. However, intravenous administration of voriconazole should be avoided in these patients as the carrier vehicle sulfobutyl ether β-cyclodextrin sodium can accumulate in these patients. Dosage adjustment is required in patients who have chronic hepatic impairment. As voriconazole is a substrate for a number of cytochrome P450 enzymes, a number of clinically important drug interactions occur with drugs including ciclosporin, tacrolimus, phenytoin, warfarin, HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors.
Posaconazole
Posaconazole is a structural analogue of itraconazole with an extended spectrum of antifungal activity in which the dichlorophenyl- and dioxolane moieties of itraconazole has been replaced by difluorophenyl- and tetrahydrofuran moieties (Fig. 24.7). Posaconazole is synthesised solely as the (R, R, S, S) enantiomer. In comparative in vitro studies it was the most potent of the clinically available azoles and, unlike other azoles, it also has useful activity against zygomycetes. This extended spectrum of antifungal activity is thought to arise from the long side chain of posaconazole occupying a specific channel within the cypP450-Erg11p that is not utilised by fluconazole or voriconazole.
Description and preparations
Posaconazole is white to off-white crystalline powder. Posaconazole is a lipophilic substance with an aqueous solubility of less than 2 μg/mL. Three polymorphic forms of posaconazole have been identified but the three-step synthetic process is controlled to constantly produce form I. Prototype tablet and capsule formulations of posaconazole were abandoned in favour of an oral suspension with superior bioavailability. The ready-to-use oral suspension contains 40 mg/mL which is micronised posaconazole in order to increase its surface area and thus bioavailability. There is at present no parenteral formulation of posaconazole available although a water-soluble ester prodrug (carboxylate ester of posaconazole with γ-butyric acid phosphate) has undergone evaluation.
The adult oral dose is 400 mg every 12 hours with a meal or with 240 mL of a nutritional supplement. In adults who cannot tolerate a meal or a nutritional supplement, the dose should be modified to 200 mg every 6 hours. Duration of therapy is dependent upon clinical and mycological response.
Pharmacokinetics
The absorption of posaconazole following the recommended oral administration schedule is slow with a median time to peak plasma of 5 hours. The effect of food on oral absorption is pronounced, with bioavailability around 2.6 times and 4 times greater when administered with a non-fat meal/nutritional supplement or high-fat meal (≈ 50 g fat), respectively, when compared to the fasted state. The pharmacokinetics of posaconazole remains linear following single or multiple doses of up to 800 mg when taken with a high-fat meal. Steady-state pharmacokinetics are typically achieved after 7–10 days multiple dosing. Posaconazole has a large apparent volume of distribution at steady-state of 1774 L with greater than 98% protein bound, predominantly to serum albumin.
The overall metabolism of posaconazole is limited in comparison to other azole antifungals and predominantly mediated through phase 2 biotransformations via UDP-glucuronosyltransferase pathways. Posaconazole does not appear to be as extensively metabolised as other azole antifungal agents by cytochrome P450 enzymes with oxidative and cleavage products and their subsequent glucuronidation and sulfation derivatives being minor metabolites. These metabolites undergo renal excretion and account for approximately 14% of an administered dose. Over 70% of an administered dose undergoes faecal excretion, with around 66% of the excreted material being posaconazole, presumably due to it being a substrate for intestinal P-glycoprotein.
Posaconazole is also different from other azole antifungal agents in that the plasma concentration of the parent compound exceeds those of its metabolites over its dosing schedule. However, the clinical relevance of this observation has yet to be determined.
No dosage adjustments are required on the basis of age, gender, race or renal function although dosage adjustments may be required in individuals with severe hepatic impairment. However, posaconazole is known to interact with a number of other clinically used drug substances. Posaconazole is an inhibitor of CYP3A4 at clinical dosages resulting in elevated plasma levels of drugs extensively metabolised by this enzyme, e.g. terfenadine, quinidine, HMG-coenzyme A inhibitors, non-nucleoside reverse transcriptase inhibitors and calcium channel blockers. In addition, inhibitors or inducers of UDP-glucuronosyltransferase pathways or P-glycoprotein may result in increased or decreased plasma concentrations of posaconazole, respectively.
Like other clinically available azole antifungal agents, co-administration with H2-antagonists or proton pump inhibitors reduces bioavailability as a consequence of a reduction in gastric acidity.
Ravuconazole
Ravuconazole is currently progressing through clinical trials. It has an identical pharmacophore to voriconazole and as such has a very similar spectrum of antifungal activity to voriconazole. In preclinical models, ravuconazole has demonstrated that it is extensively distributed in tissues, including the CNS, and has an extensive plasma half-life. Oral and parenteral (as a water soluble di-lysine phosphoester prodrug) formulations are being evaluated in phase II studies in humans.
Miscellaneous antifungal drugs
Naftifine (Fig. 24.8) was discovered during random screening for antifungal action and it was found to inhibit ergosterol biosynthesis via blocking epoxidation of squalene, which is an early step in ergosterol biosynthesis. Naftifine was only found to be suitable for topical use; therefore numerous analogues were prepared, resulting in terbinafine (Fig. 24.8) which was found to orally active.
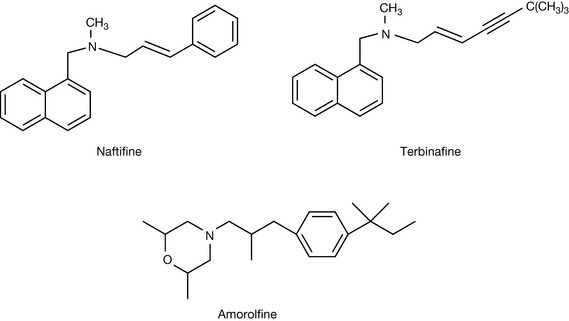
Amorolfine
Amorolfine (Fig. 24.8) is an inhibitor of Δ14 reductase, which is one of the steps in the conversion of lanosterol to ergosterol. It is used topically for the treatment of fungal nail infections.
Drugs that interfere with fungal cell walls
Echinocandins
The fungal cell wall is an obvious therapeutic target due to its absence in mammals. However, the development of effective clinical chemotherapeutic agents that interfere with the synthesis, structure or function of fungal cell walls has been complicated due to diverse interspecies differences in the chemical composition of fungal cells walls, with notable intraspecies variation also observed due to cell wall remodelling in response to changes in environment (e.g. apical hyphal growth in moulds) or reproductive requirements (e.g. budding in yeasts). Major components of all fungal cell walls include polysaccharide biopolymers based on glucose (β-1, 3 and β-1, 6 glucans), mannose (mannans) or N-acetyl-D-glucosamine (chitin) with glycoproteins also present. Each of these components contributes to the overall architecture and function of the fungal cell wall. The first class of antifungal agents to enter into clinical practice that interfere with the fungal cell wall were the echinocandins.
The echinocandins are amphiphilic cyclic hexapeptides with an N-linked acyl lipid side chain. Original members of the class were isolated from a number of Aspergillus species. Early studies undertaken with echinocandin B established that these natural products had potent activity against Candida species but had the significant disadvantage of only being active by parenteral administration and causing erythrocyte lysis. This led to analogues of echinocandin B being synthesised in an attempt to identify structure–activity relationships for this class of agent that would retain antifungal activity but have minimal mammalian toxicity. Structural modification was divided into two parts: modification of the peptide nucleus, and modification of the fatty acid side chain. As can be seen from continuing development of drugs in this field, there is a constant battle between microbes and humans.
 Self Test 24.1
Self Test 24.1